 Piotr Paluch1,2
Piotr Paluch1,2 Rafal Augustyniak
Rafal Augustyniak Jan Stanek
Jan Stanek- 1Faculty of Chemistry, University of Warsaw, Warsaw, Poland
- 2Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Łódź, Poland
- 3Tallin University of Technology, Tallinn, Estonia
In nuclear magnetic resonance spectroscopy of proteins, methyl protons play a particular role as extremely sensitive reporters on dynamics, allosteric effects, and protein–protein interactions, accessible even in high-molecular-weight systems approaching 1 MDa. The notorious issue of their chemical shift assignment is addressed here by a joint use of solid-state 1H-detected methods at very fast (nearly 100 kHz) magic-angle spinning, partial deuteration, and high-magnetic fields. The suitability of a series of RF schemes is evaluated for the efficient coherence transfer across entire 13C side chains of methyl-containing residues, which is key for establishing connection between methyl and backbone 1H resonances. The performance of ten methods for recoupling of either isotropic 13C–13C scalar or anisotropic dipolar interactions (five variants of TOBSY, FLOPSY, DIPSI, WALTZ, RFDR, and DREAM) is evaluated experimentally at two state-of-the-art magic-angle spinning (55 and 94.5 kHz) and static magnetic field conditions (18.8 and 23.5 T). Model isotopically labeled compounds (alanine and Met-Leu-Phe tripeptide) and ILV-methyl and amide-selectively protonated, and otherwise deuterated chicken α-spectrin SH3 protein are used as convenient reference systems. Spin dynamics simulations in SIMPSON are performed to determine optimal parameters of these RF schemes, up to recently experimentally attained spinning frequencies (200 kHz) and B0 field strengths (28.2 T). The concept of linearization of 13C side chain by appropriate isotope labeling is revisited and showed to significantly increase sensitivity of methyl-to-backbone correlations. A resolution enhancement provided by 4D spectroscopy with non-uniform (sparse) sampling is demonstrated to remove ambiguities in simultaneous resonance assignment of methyl proton and carbon chemical shifts.
1 Introduction
For protein studies by nuclear magnetic resonance (NMR), amide and methyl 1H resonances are the most commonly exploited. The latter ones are particularly convenient due to the magnetic equivalence of three 1H spins (thus threefold sensitivity gain), and enhanced longitudinal relaxation, both caused by fast methyl rotation. In solution NMR, multiple-quantum correlations (HMQC) can be employed to select only the slowly relaxing methyl 1H–13C coherences (Tugarinov et al., 2003). When combined with extreme 1H dilution by deuteration and selective methyl amino acid labeling, the approach allows to study the local dynamics and protein interactions for systems close to MDa molecular weight (MW) (Rosenzweig and Kay 2014; Huang and Kalodimos 2017; Boswell and Latham, 2018).
A prerequisite for the interpretation of NMR data at atomic resolution is a unique, site-specific mapping of chemical shifts to individual atoms. However, conventional resonance assignment strategies for proteins are centered around backbone 1H resonances (Sattler, Schleucher, and Griesinger 1999). Chemical shifts of methyl spins, which are peripheral with respect to backbone, are clearly challenging to assign in a systematic way, particularly if detached methyl sites are the only nondeuterated spins. Tailored experiments were developed to correlate methyl to amide frequencies; however, in addition to long coherence transfers involved and thus intrinsically low sensitivity, they require full 1H occupancy of (detected) amide sites (Tugarinov and Kay 2003b). The increased proton density is detrimental to 1H resolution and sensitivity particularly in large-MW proteins. Alternative strategy relies on correlation to backbone 13C′ and 13Cα spins (Tugarinov and Kay 2003a), which can be accomplished in the absence of amide 1H, but the issue of extended coherence transfer pathway persists.
Mutagenesis is commonly employed to address the methyl assignment issue (Amero et al., 2011). In this approach, single point mutations are introduced, and fingerprint 1H–13C HMQC spectra are recorded and compared for as many samples as methyl-containing amino acids. The major disadvantages are the labor and cost of isotope-enriched compounds needed to prepare typically tens of samples. Other pitfalls are ambiguities due to overlap of 1H–13C cross-peaks, or global chemical shift changes induced by mutations (Gorman et al., 2018).
An orthogonal approach is based on the observation of 1H–1H methyl proximities (NOEs), which can aid the assignment given the presence of the 3D structure determined using other techniques (Gorman et al., 2018). These data are interpreted either by a spectroscopist or, more effectively, by dedicated algorithms intensively developed over the last few years (Schmidt and Güntert 2012, 2013; Pritišanac et al., 2019; Pritišanac, Alderson, and Güntert 2020). The approach depends critically on the NOESY data quality and performs well mostly for methyl 1H spins experiencing a dense network of interactions in rigid regions.
Immobilized proteins, such as in amyloid fibrils, sedimented large-MW aggregates, lipid bilayers, or microcrystals, are amenable to solid-state NMR (ssNMR), since this method does not experience high-MW slow-tumbling limitations characteristic of the solution counterpart (Tycko 2011; Miao and Cross 2013; Baker and Baldus 2014; Ladizhansky 2017; Linser 2017; Mandala, Williams, and Hong 2018; van der Wel 2018; Wiegand 2020). The possibility of detection of methyl 1H resonances at high resolution in ssNMR was investigated already at low magic-angle spinning (MAS) frequencies (up to 20 kHz), when coupled to high to extreme 1H dilution by deuteration (Agarwal et al., 2006; Agarwal and Reif 2008; Asami, Schmieder, and Reif 2010; Asami et al., 2012; Asami and Reif 2013; Mainz et al., 2013). The advent of fast-spinning (above 40 kHz) MAS probes allowed narrow methyl 1H lines at significantly higher proton content, either when labile amide 1H sites are fully reprotonated (Lewandowski et al., 2011; Linser et al., 2011), or when Ile, Leu, and Val residues are also selectively and nonrandomly (100% CH3 or CHD2) protonated at methyl sites (Huber et al., 2011; Agarwal et al., 2014; Andreas, et al., 2015a; Kurauskas et al., 2016; Gauto et al., 2019), or when Leu and Val residues are reverse-labeled in an otherwise deuterated matrix (“proton clouds”) (Sinnige et al., 2014). Quite importantly, Schanda and co-workers recently showed that at 55–57 kHz MAS and B0 = 14.1 T, the 13CH3 isotopomer yields significantly higher sensitivity compared to 13CHD2 labeling, and only at a minor loss of 1H resolution (Kurauskas et al., 2016). Complete elimination of detrimental sensitivity and resolution effects of strong 1H–1H dipole interactions by MAS remains a challenge, particularly in methyl-dense protein regions, and would require yet unavailable MAS rates (above 250 kHz) (Kai Xue et al., 2017; K Xue et al., 2018; Kai Xue et al., 2019, Xue et al., 2020). Nevertheless, even in non-deuterated but relatively small proteins, resolved methyl 1H–13C correlations (of 1H linewidths of about 150 Hz) were obtained with 100–111 kHz MAS and high static magnetic fields (23.5 T) (Andreas et al., 2016).
There are few examples of de novo assignment of methyl 1H resonances in ssNMR, and frequently the assignment has been aided by either correlations to previously available 13C shifts using either dipolar (Agarwal and Reif 2008; Asami and Reif 2012) or scalar-based (Andreas et al., 2015b) (H)CCH-type spectra, or transferred from solution NMR by comparison to relatively uncomplicated 1H, 13C-CP-HSQC (Huber et al., 2011). In an early work, a strategy was proposed that bases on correlation of methyl resonances to backbone 13Cα and C′ spins using (H)CCH-TOBSY experiment, and detection of dilute methyl 1H spins, but was found challenging for Leu and Ile residues due to low efficiency of multi-13C-bond transfers (Agarwal and Reif 2008). A systematic strategy relying on correlations of side-chain 13C to backbone 15N and 1H, originally proposed by Linser for perdeuterated proteins at slow MAS (25 kHz) (Linser 2011), and more recently at fast MAS (55–60 kHz) with rotor-asynchronous MOCCA mixing (Kulminskaya et al., 2016; Vasa et al., 2018), can be readily adapted for residues selectively 100% reprotonated at methyl sites, but their efficiency with respect to methyl-containing spin systems was not investigated in detail. In the case of non-deuterated (“fully protonated”) proteins, such a long coherence transfer encounters sensitivity limitations, and more practical is a “two-hop” strategy in which side-chain 1H and 13C resonances are first correlated to alpha 1H and 13C spins using (H)CCH and H(C)CH experiments (Andreas et al., 2016; Stanek et al., 2016), and subsequently to amide 1H and 15N shifts using a combination of 13Cα–15N–1HN and 15N–13Cα–1Hα correlations (Zhou et al., 2007; Stanek et al., 2016). Both aforementioned approaches were proposed for a general use (for all amino acid residue types), without a particular emphasis on the methyl assignment. In fact, the literature data on non-deuterated microcrystalline protein GB1 (Andreas et al., 2016) show that WALTZ mixing at MAS rate νR = 111 kHz and B0 = 23.5 T leads to an effective 13Cx→13Cα transfer only in two-, pseudo-two-, and three-aliphatic 13C-spin systems (Figures 1A,B), and becomes problematic for terminal spins in pseudo-three (Glu, Gln)- and larger 13C spin systems (Figure 1C). Unfortunately, the latter include valine, leucine, and isoleucine, which additionally suffer from branching of 13C chain at β- or γ-positions (Figure 1D). This raises questions of feasibility of methyl resonance assignment under less sensitivity-favorable sample or hardware conditions, and calls for a careful optimization of 13C mixing scheme at arising new MAS conditions and B0 fields.
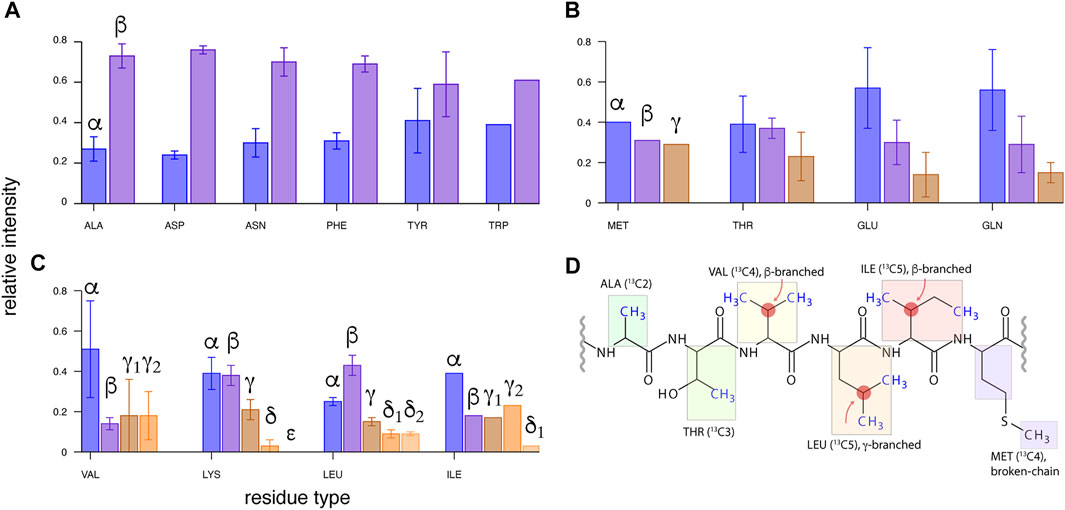
FIGURE 1. Transfer efficiencies from side-chain 13C (β, γ, … ) to 13Cα spin for various residues in microcrystalline U-13C,15N-labeled GB1 protein using 13C WALTZ-16 mixing of 15 ms duration, with MAS of νR = 111 kHz (in a 0.7-mm rotor) and on a 23.5-T spectrometer. Intensity of 13CX-13Cα-1Hα cross-peaks in (H)CCH spectrum (Andreas et al., 2016; Stanek et al., 2016) was normalized to a sum of 1.0 for each residue, and averaged within each amino acid type. The data were grouped into (A) two- and pseudo-two-, (B) three- and pseudo-three-, and (C) four- and five-aliphatic 13C spin systems (disregarding backbone and side-chain carbonyl and aromatic 13C spins). (D) Topologies of 13C side chains for six amino acid residue types containing a methyl group, from left to right: least to most challenging for methyl resonance assignment.
In this work, we critically assess the efficiency of several 13C mixing schemes for methyl 1H and 13C resonance assignment at fast MAS; determine their optimal range of B0, MAS frequency, and 13C RF strength within and beyond currently available regimes; and explore alternative 13C labeling to further boost sensitivity of the approach.
2 Materials and Methods
2.1 Sample Preparation
2,3-13C (99%)-labeled crystalline alanine was purchased from Eurisotop (France) and manually packed into a Bruker 1.3-mm MAS rotor.
N-formylated microcrystalline uniformly 13C,15N-labeled Met-Leu-Phe tripeptide (“fMLF”) was purchased from Giotto Biotech (Sesto Fiorentino, Italy) and manually packed into Darklands OÜ 0.81 mm MAS rotor.
The plasmid coding for the Src homology 3 (SH3) domain (965–1,025) of chicken α-spectrin (gene SPTAN1, Uniprot P07751) in a pET3a vector was a kind gift of Dr. T. Schubeis (High Field NMR Centre in Lyon, France). The insert was subcloned into a modified pET28a vector that resulted in a construct including an N-terminal His6-tag followed by a SUMO solubility tag. This allowed us to express His6-SUMO-SH3 fusion protein.
For preparation of the NMR samples, transformed Escherichia coli BL21 (DE3) cells were grown in M9 D2O media supplemented with 1 g/L of 15NH4Cl (Cortecnet, France) and 3 g/L of (2H,13C)-glucose (Cortecnet, France) as the sole nitrogen and carbon sources, respectively, following the established procedure (Tugarinov, Kanelis, and Kay 2006). Labeling of the Ile, Leu, and Val side chains was achieved by the addition of the amino acid precursors 1 h prior to the induction with 1 mM IPTG. For Leu and Val residues, we used two kinds of precursors that yield continuous 13C chains from methyl (either Cδ or Cγ) to Cα and C′ atoms, provided that 13C-enriched glucose is also employed (Goto et al., 1999; Tugarinov and Kay 2003a). The specific labeling patterns and the origin of particular nuclei are shown in Supplementary Figure S1. SH3 sample with branched side chains of leucine and valine residues (hereafter referred to as “ILV-C5” sample) was prepared using 100 mg/L of 1,2,3,4,4′-13C-3-2H-labeled α-ketoisovaleric-acid (sodium salt, Eurisotop, France, catalogue number CDLM-4418-PK) as a Leu and Val precursor. The corresponding sample with linearized 13C-side chains of Leu/Val residues (referred to as “ILV-C4” sample) required the addition of 100 mg/L 1,2,3,4-13C-3,4′,4′,4′-2H-labeled α-ketoisovaleric acid (sodium salt, Eurisotop, France, catalogue number CDLM-8100-PK). For both samples, 60 mg/L of 1,2,3,4-13C-3,3-2H-labeled α-ketobutyric acid (sodium salt, Eurisotop, France, catalogue number CDLM-4611-PK) was used as the precursor of isoleucine residues with uniform 13C enrichment (Supplementary Figure S1). The cells were grown at 24°C for 18 h after the induction.
The purification protocol of SH3 protein was modified with respect to the original one (Pauli et al., 2000, 2001; Chevelkov et al., 2006). Instead, a standard protocol involving His-trap affinity column and HiLoad 16/60 Superdex 75 gel filtration column (GE Healthcare) was employed. Briefly, we used 50 mM HEPES, 200 mM NaCl, and 1 mM DTT (pH 7.4) supplemented with 20 mM imidazole as a lysis buffer, and the same buffer including additionally 400 mM imidazole was used to elute a protein from the His-trap column. The cleavage of the His6-SUMO tag was achieved with a custom-made Ulp1 protease (Reverter and Lima 2009) in a lysis buffer lacking imidazole. The reaction was monitored with the SDS-PAGE and, once completed, the protein was passed through the His-trap column again to remove SUMO as well as His-tagged Ulp1 protease. Cleaved SH3 protein was dialyzed at 4°C overnight against the Superdex 75 running buffer (20 mM citric acid, 150 mM NaCl, pH 3.5), concentrated, and purified on a gel filtration column.
To obtain solid-state NMR protein samples, fractions containing pure SH3 were pooled, concentrated to approximately 10 mg/mL using Vivaspin 3-kDa cutoff centrifugal concentrators (Sartorius) and extensively dialyzed against 100 mM (NH4)2SO4 adjusted to pH 3.5 with sulfuric acid. Finally, to crystallize the protein, ammonia water solution was added dropwise to reach pH 7.5. Obtained turbid solutions were stored in a refrigerator (4°C) for approximately a week for a slow buildup of microcrystals. Crystallization efficiency was estimated to 85% by a spectrophotometric measurement of protein concentration decrease in the supernatant. The suspension was mixed with 1 M CuNa2EDTA 9:1 v/v (effective cCu2+ = 100 mM), and left for impregnation of SH3 crystals with paramagnetic Cu2+ ions for 3 days. In the case of the SH3 “ILV-C4” sample, TSP was added to the supernatant at an effective concentration of 10 mM for chemical shift calibration reference. About 2 mg of protein was transferred to a Bruker 1.3-mm MAS rotor by 30 min of ultracentrifugation at an average acceleration of 96,500 g. Each Darklands OÜ 0.81-mm MAS rotor was filled with approximately 0.5 mg of protein in five equal parts by stepwise packing in a tailored ultracentrifuge adapter experiencing an average acceleration of 135,000 g for 30 min.
2.2 Nuclear Magnetic Resonance Spectroscopy
The NMR 13C–13C 2D correlation experiments on model compounds (alanine and fMLF) were performed using a standard RF irradiation scheme shown in Figure 2A. A collection of homonuclear mixing schemes was employed as detailed below. For each case, a series of 2D spectra with gradually incremented mixing time was acquired, in the range suggested by literature and spin dynamics simulations, if permitted by probe RF circuity.
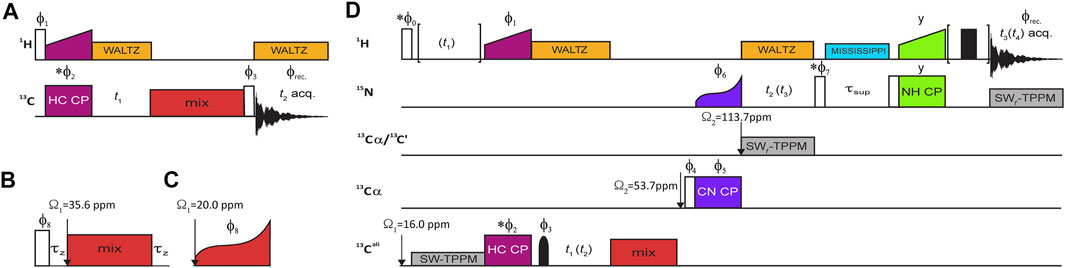
FIGURE 2. RF irradiation schemes of NMR experiments. (A) 13C–13C 2D correlation experiment with mixing two variants of 13C mixing expanded in (B) and (C). All schemes act on the 13C magnetization preceded and followed by a z-filter (τz) (B) with the exception of DREAM (C), which operates on the 13C single-quantum coherence. (D) Pulse diagram for 3D (H)CMET(CC)(CA)NH, with the optional evolution period for methyl 1H coherences enclosed in brackets (for the 4D variant). The optional 1H spin echo immediately before acquisition was only applied for Bruker 1.3-mm probe to eliminate probe 1H background signal. Open and solid rectangles represent 90° and 180° high-power pulses, respectively, and a solid bell shape denotes a methyl 13C-selective Q3 refocusing pulse (Emsley and Bodenhausen 1992) of 942.7 μs duration and peak RF of ν1 = 2.56 kHz. 13C mixing is marked in red, and cross-polarization transfer steps are labeled and color-coded as maroon, purple, and lime green simultaneous irradiation on pairs of respective channels for H→C, C→N, and N→H transfers. 1H low-power WALTZ (Shaka, Keeler, and Freeman 1983) decoupling is marked in orange, while heteronuclear (13C or 15N) frequency-swept TPPM decoupling (Bennett et al., 1995) is marked in gray. MISSISSPPI solvent suppression (Zhou and Rienstra 2008) is marked in cyan. Changes in 13C offset (Ω) are denoted with vertical arrows. Unless indicated otherwise, all pulse phases are set to x. The following phase cycling was employed: ϕ1 = y, ϕ2 = y, ϕ3 = {2 (x), 2 (−x), 2 (y), 2 (−y)}, ϕ8 = {x, −x}, ϕrec. = {x, −x, −x, x, y, −y, −y, y} for 2D experiment with inset (B); ϕ1 = {y, −y}, ϕ2 = {2 (x), 2 (−x)}, ϕ3 = {4 (x), 4 (−x)}, ϕ8 = {2 (x), 2 (−x)}, ϕrec. = {x, −x, x, −x} for 2D DREAM experiment (A + C); ϕ1 = {4 (y), 4 (−y)}, ϕ2 = x, ϕ3 = {x, x, y, y}, ϕ4 = y, ϕ5 = {8 (x), 8 −x)}, ϕ6 = {y, –y}, ϕ7 = x, ϕ8 = -y, ϕrec. = {x, –x, –x, x, –x, x, x, –x, –x, x, –x, x, –x, –x, x} for 3D experiment (D) with recoupling block (B); and ϕ1 = {8 (y), 8 (−y)}, ϕ2 = {4 (x), 4 (−x) }, ϕ3 = {2 (x), 2 (y), 2 (−x), 2 (−y)}, ϕ4 = x, ϕ5 = x, ϕ6 = {y, −y}, ϕ7 = {x}, ϕ8 = {4 (x), 4 (– x)}, ϕrec. = {x, –x, –x, x, –x, x, x, –x, –x, x, –x, x, –x, –x, x} for 3D DREAM experiment (D + C). Pulse with phase marked with an asterisk are involved in quadrature detection according to States-TPPI procedure.
The experiments for site-specific assignment of methyl 13C and 1H resonances in SH3 protein that provide correlations to backbone amide 15N and 1H chemical shifts [3D (H)C(CC)(CA)NH] were straightforwardly adopted from the literature (Linser 2011) and are shown in Figure 2B. Despite long methyl 1H transverse relaxation times observed, the experiments employ 1H–13C cross-polarization (CP) instead of INEPT-type transfer, since the latter would suffer from inefficiency of conversion of anti- to in-phase 13C coherence caused by concurrent evolution of two (passive) 1JCH couplings in CH3 moieties. Implementations of presented experiments for Bruker spectrometers are freely available from the community-based repository Zenodo as detailed below.
For the site-specific evaluation of experimental performance with various 13C mixing schemes, resolution provided by 3D spectra was required, preventing acquisition of mixing time series for SH3 protein. Optimal mixing times were, however, possible to determine using first-increment 1D optimizations prior to 3D data acquisition.
The series of 2D 13C–13C correlation experiments on 2,3-13C-alanine was carried out on a Bruker Avance III spectrometer operating at 1H and 13C frequency of 600.1 and 150.9 MHz, respectively, equipped with a Bruker 1.3-mm H/C/N MAS probehead. The sample has been spun at 55,555 ± 10 Hz and controlled by a Bruker MAS-II unit, without temperature stabilization. 1H, 13C pulses and power during 1H to 13C cross-polarization have been carefully calibrated prior to experiments. Detailed information on pulse lengths, RF amplitude, spectral windows, etc. is provided in Supplementary Table S1.
2D experiments on 13C,15N-fMLF tripeptide and 3D and 4D experiments on two SH3 protein samples were performed on a Bruker Avance III HD spectrometer operating at a 1H, 13C, and 15N frequency of 799.7, 201.1, and 81.0 MHz, respectively, equipped with a 0.81-mm H/C/N/D MAS probehead developed by Ago Samoson’s group (Darklands OÜ, Estonia). NMR data were acquired at two MAS frequencies, νR = 55.5 and 94.5 kHz (98 kHz for fMLF).
3D experiments on “ILV-C5”-labeled SH3 protein were also performed on a Bruker NEO 23.5-T spectrometer (in CRMN Lyon, France) equipped with a Bruker 1.3-mm H/C/N/D MAS probe, and MAS-III and BCU-II spinning and temperature stabilization units.
For the experiments on 13C,15N-fMLF magic angle setting has been set using KBr sample prior to the actual series. In this case, no temperature stabilization device was used. 1H and 13C 90° pulse lengths were carefully calibrated using 1D 13C-detected CP experiment.
Prior to experiments on SH3 samples, a careful magnet shimming was performed to maximize reliability of linewidth measurements. The classical shimming protocol employing adamantane sample is very time-consuming in 0.81-mm MAS rotors; thus, a sample of silicon grease was used first, leading to full-width at half-height (FWHH) of 1H line of 16 Hz at 50 kHz MAS. Subsequently, shim currents were refined using adamantane sample resulting in an FWHH of the low-field 13C resonance of 1.1 Hz at 50 kHz MAS under low-power 1H decoupling.
Magic angle was finely adjusted directly on the protein sample at the target MAS frequency by maximizing the intensity of the 1H signal in the (H)NH 1D experiment followed by a 5-ms 1H spin echo. Sample temperature was stabilized to 20°C using Darklands OÜ VT controller. Thermocouple readout was calibrated to sample temperature by measurement of the 207Pb chemical shift of Pb(NO3)2 at the exactly same cooling gas flow, spinning speed, and thermocouple target temperature. 1H, 15N 90° pulse lengths have been carefully calibrated for each sample and spinning speed using the (H)NH experiment. 13C 90° pulse length was calibrated using the (H)CONH experiment. RF amplitude of hard and soft pulses, as well as of decoupling and recoupling schemes was automatically calculated in a pulse program based on widths and powers of reference high-power 90° pulses. 1H–13C, 13C–15N, and 15N–1H cross-polarization power was optimized directly using the first increment of the (H)C(DIPSI)(CA)NH experiment, and propagated to all 3D (and 4D) experiments at each sample and spinning condition. Fast recycling (0.3 or 0.4 s interscan delay) was applied to improve sensitivity, taking advantage of longitudinal 1H relaxation enhancement by paramagnetic Cu2+ ions (Ganapathy et al., 1981; Wickramasinghe et al., 2007, 2009). Details on NMR data acquisition for fMLF tripeptide and SH3 protein are provided in Supplementary Tables S1, S3, respectively.
B0 was not stabilized in either case, but the field drift was monitored using 1D 1H spectra between experiments and had a negligible effect on data (e.g., a total 11 Hz 1H downfield drift over 10 days on an 18.8-T spectrometer for “ILV-C5” SH3 sample at 94.5 kHz MAS). The stability of CP conditions was monitored using 1D (H)(CA)(N)H and (H)(C)(DIPSI)(CA)(N)H (for proteins) or 13C-detected 1D CP (for fMLF) experiments.
2D and 3D data were Fourier processed in Bruker TopSpin using parameters reported in Supplementary Tables S2, S4. The non-uniformly sampled 4D data were converted using in-house written script bruk2ssa (courtesy of M. Górka), and processed using signal separation algorithm for distortion-free spectral reconstruction (Stanek, Augustyniak, and Koźmiński 2012). All spectra were analyzed in NMRFAM-Sparky (Lee, Tonelli, and Markley 2015).
2.3 Homonuclear 13C Mixing Schemes
The key element determining the performance of experiments shown in Figure 2 is 13C mixing. For this comparative study, we have made a selection of literature RF designs based on the following criteria: (1) 13C should occur primarily between bonded 13C spins by recoupling of 13C–13C scalar (J) or dipolar (D) interactions, (2) RF requirements are acceptable for a typical “fast” MAS probe, and (3) they pose no danger to the hydration of fragile protein samples, e.g., by an extended period of high-power 1H irradiation. For the last reason, we did not consider second-order recoupling schemes suitable for fast MAS, such as MIRROR (Scholz et al., 2008), PARIS (Weingarth et al., 2009), SHANGHAI (Hu et al., 2011), or CORD/CORD-RFDR (Hou et al., 2013; Lu et al., 2015). In the presence of relaxation, the efficiency of these sequences deteriorates with faster MAS, concomitantly to increasing efficiency of averaging of H–H dipolar interactions. Protein deuteration, which is highly recommended for resolution in large proteins, is also clearly incompatible with second-order recoupling mechanism (De Paëpe, Lewandowski, and Griffin 2008) as it prohibitively dilutes the proton interaction network.
2.3.1 TOBSY
TOBSY was designed based on average Hamiltonian theory (AHT) and symmetry properties of particular interactions (Levitt 2007), imposing that chemical shift anisotropy (CSA), isotropic chemical shifts (CS), and dipolar interaction terms are suppressed in the zeroth- and first-order Hamiltonians, and the zeroth-order term stems only from the isotropic scalar interaction (Baldus and Meier 1996; Hardy, Verel, and Meier 2001). In a general design denoted CNνn (Edén and Levitt 1999), a primitive multi-pulse block C is repeated N times over n rotor cycles with the gradual phase incrementation in Δϕ = 2πν/N steps (Figure 3A). Low-power TOBSY schemes suitable for fast MAS employ POST primitive block (90ϕ360ϕ+π270ϕ), and use N = 9, ν = 1, and n = 9p ± 3 (where p is an integer), e.g., C9121, C9124, and C9130, to retain only assumed interaction (Tan et al., 2018). The schemes with particular n differ in robustness to dipolar C–H interactions and susceptibility to CSA (in higher-order AHT terms). TOBSY requires an RF strength of ν1 = 2N/n νR and thus is readily applicable in typical fast MAS probes for n > 18. Here, we employed sequences with n = 24, 30, 33, 39, and 48, with mixing time either varied between 0 and 50 ms (in 2D series) or optimized for maximum transfer (in 3D experiments).
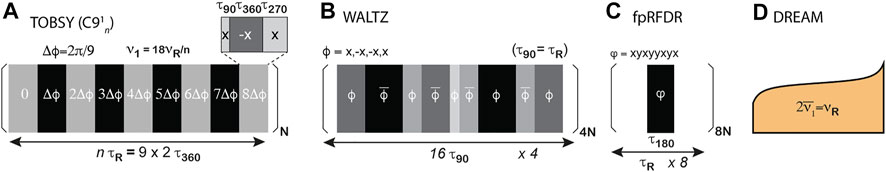
FIGURE 3. RF schemes of 13C mixings compared in this study. (A) TOBSY C91n variants with variable n. (B) WALTZ-16, a representative of TOCSY class. The primitive element is composed of phase-alternated 90, 180, 270, and 360° pulses, depicted as rectangles color-coded from light gray to black. (C,D) Transfer schemes primarily recoupling 13C dipolar interactions: (C) finite-pulse RFDR and (D) DREAM.
2.3.2 TOCSY
Isotropic mixing schemes developed for 13C mixing in solution NMR (Bax, Clore, and Gronenborn 1990; Kay et al., 1993), hereafter referred to using a general term TOCSY, retain only the Hamiltonian term associated with scalar (J) interaction between 13C nuclei (in the presence of the overall tumbling). For solids under MAS, all isotropic interactions are preserved; thus, these RF schemes induce the evolution of JCC interactions while suppressing the evolution of JHC and isotropic 13C chemical shifts. The proper treatment of anisotropic interactions is not ensured at all; however, at sufficiently fast MAS, their contribution is supposedly small. Simple arguments advocated for the use of WALTZ-16 (Shaka, Keeler, and Freeman 1983) mixing synchronized with rotation (τ90 = τR; Figure 3B), and basic properties were verified using 2-13C-spin SIMPSON simulations (Andreas et al., 2016). Classical TOCSY mixing schemes that are robust with respect to large chemical shift offsets, namely, DIPSI-3 (Shaka, Lee, and Pines 1988) and FLOPSY-16 (Kadkhodaie et al., 1991), were also employed here. In solution NMR, TOCSY designs are ranked according to the figure of merit, i.e., the bandwidth with respect to applied RF strength, but such a ranking is of limited relevance here since MAS probes can easily generate sufficient ν1. Although not required, we retained rotor synchronization (ν1 = ¼ νR) in experiments for three selected TOCSY sequences, but tested other RF conditions in spin dynamics simulations. Mixing time was varied (for alanine and fMLF) or optimized in 1D experiments (for proteins) in steps of 188.448, 217.32, and 96.0 τR (assuming τR = τC90) for FLOPSY, DIPSI, and WALTZ, respectively.
2.3.3 RFDR
Finite-pulse 13C RFDR (Bennett et al., 1998) recouples dipolar 13C interactions in the first-order average Hamiltonian by the application of high-power π pulse each rotor cycle (Figure 3C). Although the sequence is known to suffer from dipolar truncation (Griffin 1998), this is not necessarily a disadvantage for intraresidue 13C transfers. RFDR requires ν1 > ½ νR for pulses partially covering the mixing time; in practice, ν1 on the order of νR is preferred. The ratio of τ180/τR determines the scaling factor of dipolar interaction (nominally of about 2 kHz for a bonded 13C–13C spin pair), and in this study, we used ν1 = 150 (for fMLF) and 100 kHz (otherwise). The following is the phase cycle (xy8) of the π pulse: x, y, x, y, y, x, y, x, as suggested previously (Shen et al., 2012). Mixing time was optimized in steps of multiples of 8τR between 0 and 50 ms.
2.3.4 DREAM
DREAM recouples dipolar 13C interactions in the first-order average Hamiltonian using an adiabatic pulse for introducing HORROR condition (ν1 = ½ νR, Figure 3D) (Verel et al., 1998; Verel, Ernst, and Meier 2001). DREAM is commonly employed with slow (Pauli et al., 2001) and fast MAS (Penzel et al., 2015) to trigger 13Cα→13Cβ or 13C′→13Cα coherence transfer, for which RF and offset conditions can be straightforwardly determined. Transfers in multi-spin systems were studied and conditions were optimized at “slow” MAS (Westfeld et al., 2012); however, the proper order of HORROR conditions cannot in general be satisfied due to characteristic chemical shifts in 13C side chains. Additionally, by its nature, DREAM cross-peak intensity is negative with respect to the origin coherence, which potentially causes destructive interferences within a spin system or due to spectral overlaps. The RF shape was defined as usual,
2.4 Spin Dynamics Simulations Using SIMPSON
Spin dynamics simulations have been performed using SIMPSON (version 4.0.0c) (Bak, Rasmussen, and Nielsen 2011; Tošner et al., 2014). Powder averaging has been performed with 1,848 {α, β, γ} Euler angles that describe the orientation of the molecule in the rotor frame. A total of 168 {α, β} angle pairs have been selected using REPULSION algorithms (Bak and Nielsen 1997) and 11 γ angles have been regularly sampled from 0 to 360°. Spin systems have been generated using the SIMMOL package (Bak et al., 2002).
For simulations in the case of a model four-spin system (Hα–Cα–Cβ–Hβ), dipolar spin couplings have been generated based on distances in L-alanine structure (ref. code LALNIN61 in CCS database), and only α and β carbons and protons have been considered. 1JCC and 1JHC couplings have been set to 33 and 145 Hz, respectively. In addition, only one proton from the CH3 group has been considered with the H–C dipolar coupling value reduced by three due to fast rotation around the C–C axis. Other relevant parameters of the spin system are reported in the Supplementary Material.
For simulation in the case of C6 and C5 spin systems, dipolar couplings have been calculated for the geometry of leucine-8 in the X-ray structure of chicken SH3 protein (PDB 1SHG). Experimentally determined chemical shifts in fMLF have been assumed, and CSA parameters were calculated in Gaussian. 1JCC couplings were set to 50 Hz for the 13Cα–13C′ pair and to 33 Hz between aliphatic 13C spins. 2JCC was set to 3 Hz, and longer-distance J couplings were neglected. Examples of SIMPSON input files are provided in the Supplementary Material.
3 Results and Discussion
3.1 Homonuclear Mixing in Model Two-13C Spin Systems
Efficiency of 13C mixing varies in general with MAS frequency νR, RF strength ν1 (if not imposed by νR), B0 field (through scaling of isotropic chemical shift differences and CSA), presence of 1H–13C interactions, and extent of dynamics as well as other design-specific parameters detailed above. To mitigate the complexity of the problem, one usually refers to model two-13C spin systems, such as, e.g., in the acetate anion. Here, we used 2,3-13C2,15N-labeled crystalline alanine as a closer analogue of a protein residue, additionally possessing a methyl group, and avoiding interference with large CSA of the (unlabeled) carbonyl carbon. The efficiency of 13Cβ→13Cα transfer was quantified by observation of cross-peak intensity in a τmix series of 2D 13C–13C spectra at fast MAS (νR = 55.5 kHz) and moderate B0 field (14.1 T).
The results shown in Figure 4 confirm the high efficiency of selected RF designs. Also, fundamental differences between them exhibit the following: TOBSY and TOCSY sequences recouple JCC in a time close to 1/(2J) (scaling factors might apply to the effective coupling constant, e.g., for FLOPSY). RFDR initially shows rapid oscillations with frequency related to a scaled dipolar coupling (DCC ≈ 2 kHz), stabilizes at approximately τmix = 5–10 ms, and then decays due to π pulse imperfections and incoherent effects. DREAM shows a steady buildup, reaches a plateau at τmix ≈ 5 ms, and then decays slowly mostly due to 13C T1ρ-like relaxation. The results are generally consistent with the literature, also confirming the increasing robustness of TOBSY C91n to DHC interactions with increasing n (Figure 4A) (Tan et al., 2018). Although RFDR experimentally showed highest performance, observed differences between considered schemes are rather minor.
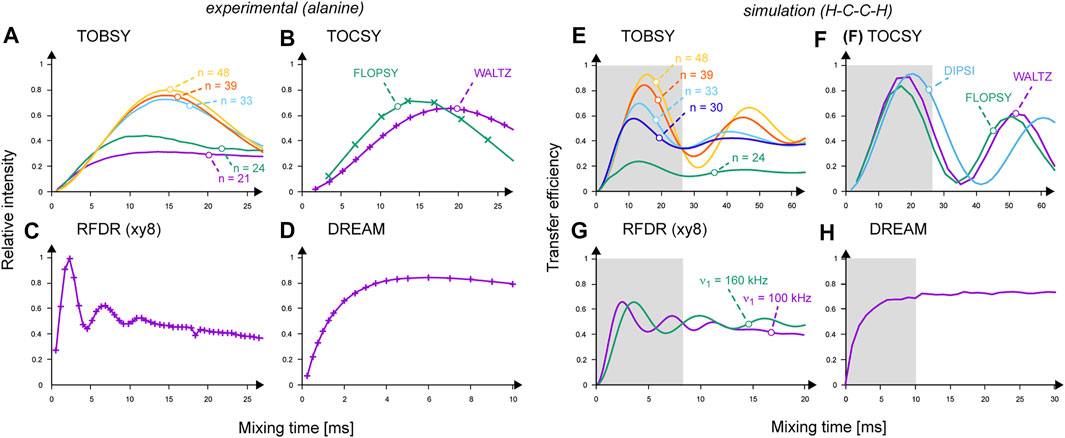
FIGURE 4. (A–D) Performance of 13C mixing schemes observed experimentally in 13C–13C 2D spectra on 2,3-13C2-labeled alanine: (A) TOBSY C91n with n = 21, 24, 33, 39, and 48 are labeled and color-coded as purple, green, cyan, red, and orange curves, respectively; (B) FLOPSY-16 (in green) and WALTZ-16 (in purple); (C) RFDR at ν1 = 100 kHz; (D) DREAM. Cross-peak intensities were normalized to the most intense point in the RFDR series. Experimental points are connected with lines for eye guidance only. (E–H) Buildup of transferred 13C magnetization (absolute transfer efficiency) in a two 13C- and two 1H-spin system simulated in SIMPSON. (E) TOBSY C91n with n = 24, 30, 33, 39, and 48 are labeled and color-coded as green, dark blue, cyan, red, and orange curves, respectively; (F) FLOPSY-16 (in green), DIPSI-3 (in light blue) and WALTZ-16 (in purple); (G) RFDR at ν1 = 100 (in purple) and 160 kHz (in green); (H) DREAM. Gray boxes indicate the mixing time ranges sampled experimentally on alanine.
Spin dynamics simulations are instrumental for the in-depth understanding of complex RF schemes applied under new MAS conditions. Here, we resorted to SIMPSON as a generally agreed simulation platform (Tošner et al., 2014), and attempted first to reproduce experimental results. We modeled alanine using a minimal system consisting of two 13C and 1H spins as described above. Results shown in Figures 4E–H reproduce well the experimental buildup curves, optimal mixing times, and scaling factors of recoupled interactions. Differences in efficiency between TOBSY C91n sequences with n = 48, 39, 33, and 24 (Figure 4E) are more pronounced than in the experiment (Figure 4A). C9148 allows for an almost complete transfer for τmix ≈ 16 ms. For longer mixing times, recoupling of higher-order AHT terms occurs (mostly the cross-terms between 13C isotropic and anisotropic chemical shifts, and DHC interactions), leading to coherence dephasing and decreased intensity of subsequent maximums. Somewhat surprisingly, all TOCSY sequences perform excellently (Figure 4F), despite no deliberate treatment of anisotropic interactions. RFDR performance is relatively worse than in the experiment, and equalizes 13C magnetization between both coupled 13C spins. Supposedly, incoherent effects and B1 field inhomogeneity in the MAS coil affect RFDR to the smallest extent among the considered mixing types. In simulations, DREAM does not show an optimum, but a steadily increasing coherence transfer.
Having validated the simulation platform, we attempted to differentiate 13C mixing schemes with respect to destructive interferences (cross-terms) with interactions that are field-dependent (isotropic and anisotropic CS), and proton-content-dependent (DH-C interactions). All combinations of selectively “activated” interactions were probed, and the effects of most relevant cross-terms are summarized in Supplementary Figure S2. We confirmed that TOBSY C91n sequences with n > 30 are quite robust to DHC, with (JCC, DHC), (DCC, DHC), and (DCC, CSA) being the primary cross-terms responsible for non-ideal performance. We additionally confirmed that TOBSY is robust with respect to an increasing chemical shift difference (or B0 field), e.g., up to ΔΩ = 15 kHz for C9124 at νR = 55 kHz (and v1 = 2/3 νR). Among TOCSY sequences, FLOPSY appeared to be most susceptible to (JCC, DHC) interferences (or, in other words, to the presence of protons). Also, all TOCSY designs are less affected by (DCC, DHC) terms than TOBSY and very robust to CSA (Supplementary Figure S2). Thus, WALTZ and DIPSI are good candidates for high B0 field measurements, and on systems with high proton density. Within the initial buildup, RFDR is extremely robust to all interaction interferences, and in fact even enhanced by recoupling of JCC interactions.
Additionally, we verified the susceptibility of TOBSY and TOCSY sequences to B1 field inhomogeneity, which can be substantial in MAS RF coils [as large as 20% in the active volume (Tošner et al., 2018)]. As shown in Supplementary Figure S3, TOBSY sequences only tolerate maximum 5% RF deviations (or miscalibration), and TOCSY designs, DIPSI in particular, are significantly more robust with this respect.
However, multiple limitations of spin dynamics simulations must be considered when their results are interpreted quantitatively: (1) no incoherent effects (relaxation) are included; (2) structural variability of dipolar interactions, chemical shifts, and CSA are tedious to replicate; and (3) pulse transients, limited short-term rotation stability, and other experimental deficiencies are neglected.
3.2 Homonuclear 13C Transfer Across Multi-Spin Side Chains in Model Systems
Subsequently, we performed an analogous series of 2D 13C–13C correlation spectra on a sample of crystalline tripeptide fMLF, where the leucine residue serves as a realistic and convenient reference multi-spin system containing a methyl group. Spectral excerpts in Figure 5 demonstrate that, while one-bond transfers remain effective in all cases, efficiencies of multi-bond ones are low and differ dramatically between particular RF schemes (see Supplementary Figure S4 for mixing time dependencies). In contrast to the case of alanine, RFDR performs worst in this respect, emphasizing the limited utility of simplistic two-13C spin systems for evaluation of 13C mixing performance.

FIGURE 5. Strips from 2D 13C–13C spectra recorded for fMLF at νR = 55.5 kHz on an 18.8-T spectrometer. Presented are diagonal and cross-peaks originating from Leu 13Cδ1 magnetization (prior to 13C mixing). Spectrum diagonal is indicated with a dashed line. Spectra with a mixing time optimal for Cδ1→ Cα transfer are shown: 34.6, 32.0, 21.4, 25.3, and 36.3 ms for TOBSY C91n, with n = 24, 30, 33, 39, and 48, respectively (A–E); 23.7, 27.4, and 20.7 for FLOPSY-16 (F), DIPSI-3 (G), and WALTZ-16 (H), respectively; and 11.2 and 14 ms for RFDR (ν1 = 150 kHz) (I) and DREAM (J), respectively.
It was noted previously in both solution (Tugarinov and Kay 2003a) and solid-state NMR (Agarwal and Reif 2008) that a combination of a branched 13C chain, large number of 13C spins, and large chemical shift differences makes the leucine spin system extremely challenging for any 13C mixing. In the following, we will deliberately focus on the most demanding Cδ1→Cα transfer as the sensitivity-limiting step, regardless of which side-chain assignment strategy mentioned above [the one-step side-chain to backbone, (H)C(CC)(CA)NH, or the two-step approach using (H)CCH and (H)NCAHA spectra] is employed. (Another potentially relevant transfer Cδ1→C′ is expected to be even more problematic due to a large CSA and distinct chemical shift.)
The entire experimental series was repeated at very fast spinning conditions (νR = 98 kHz) to probe sensitivity of selected mixing schemes to coherent and incoherent effects of dipolar C–H and H–H interactions. Absolute efficiencies of Cδ1→Cα transfer were quantified as described in Supplementary Material, and presented in Figure 6. Strikingly, all RF schemes greatly benefit from increased MAS rate (up to a factor of 4.5–5 for three TOCSY representatives). At νR = 55.5 kHz DREAM performs best, and the most efficient scheme at νR = 98 kHz is DIPSI, with TOBSY C9148 being close to the leaders at both MAS frequencies. In both cases, RFDR and TOBSY C91n with n < 30 remain evident outliers.
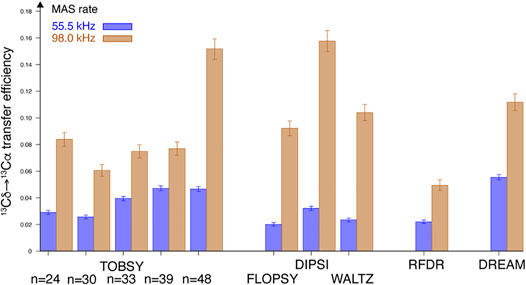
FIGURE 6. Maximum absolute efficiency of Cδ1 → Cα magnetization transfer observed in leucine residue of fMLF at B0 = 18.8 T field and at νR = 55.5 (blue bars, left in each pair) and 98 kHz (tan bars, right in each pair).
Given such a dramatic response to MAS frequency, we attempted to verify capabilities of spin dynamics simulation for reproducing the behavior of a complex, but extremely useful leucine spin system, and possibly extrapolate it to different spinning and field conditions. Unfortunately, despite intense optimization of SIMPSON routines (e.g., the reuse of propagators) and the use of the state-of-the-art high-performance clusters, inclusion of any meaningful number of 1H spins (L ≈ 5–6) turned out unfeasible due to the exponential scaling of required computational resources (4L). Nevertheless, many features of the transfers were quite well reproduced in the exclusively 13C 6-spin system, in particular (1) the optimal Cδ1 → Cα mixing times (Supplementary Figure S5), (2) the shape of build-up curves for individual transfers (Supplementary Figure S6), and (3) poor performance of RFDR (≈5%) at both spinning speeds. The results also corroborate TOBSY C9148 and DIPSI as the performance leaders at νR ≈ 100 kHz (Supplementary Figure S7). The exact quantification of DREAM poses difficulties due to the absence of an optimum in simulations, but efficiency at τmix = 25 ms, as used in the experiments, places this mixing scheme closer to the best than to mediocre designs.
The absence of protons in the spin system used in simulations resulted in almost identical performance of all TOBSY schemes at νR = 100 kHz, which clearly disagrees with the experiments. Likely for the same reason, the experimentally observed relative order of performance of TOCSY sequences was not reproduced. To some extent, one can still rely on the analysis of cross-term importance for four spin system (Supplementary Figure S2) to predict the impact of protons also in leucine residues. However, absolute efficiencies predicted in simulations (Supplementary Figure S7) and observed experimentally (Figure 6) show discrepancies from roughly 25% (for TOBSY C9148 and DIPSI) to more than 100% (e.g., for FLOPSY and WALTZ) at νR = 98–100 kHz, and even larger ones at the lower spinning speed (νR = 55.5 kHz). These disagreements arise not only due to the (unaccounted) coherent effects of 1H–13C interactions, but also due to 13C relaxation, suggesting the ultimate need of experimental verification for a quantitative comparison at specific νR and B0.
Despite apparent limitations, SIMPSON simulations still provide upper limits for transfer efficiencies, which is useful to predict performance trends at different experimental conditions. For example, νR dependence sampled between 30 and 200 kHz (Supplementary Figure S8) shows an optimal MAS range of 80–100 kHz for TOBSY and TOCSY schemes. The penalty observed at νR > 100 kHz is surprising, and in fact related to RF strength (ν1), which was fixed in a constant proportion to νR. To isolate ν1 dependence without traversing through recoupling conditions, we performed additional simulations at constant ν1 = 55.5 kHz and ν1 = ¼ νR, but indirectly varied RF bandwidth of sequences via modulation of resonance frequencies and CSA with the strength of B0 field (Figure 7). At high field (above ν0,H = 800–1,000 MHz), most sequences show insufficient 13C bandwidth, thus decreasing the performance of a multibond transfer. At low field, the transfer efficiency degrades as well, with a very similar behavior to that observed in Supplementary Figure S8 for large rotation rates νR. This suggests that the effect actually relates only to the effective 13C bandwidth, and we confirmed by observation of C′ magnetization that an excessive RF strength is detrimental to 13Cα magnetization due to the concurrent drainage to C′.
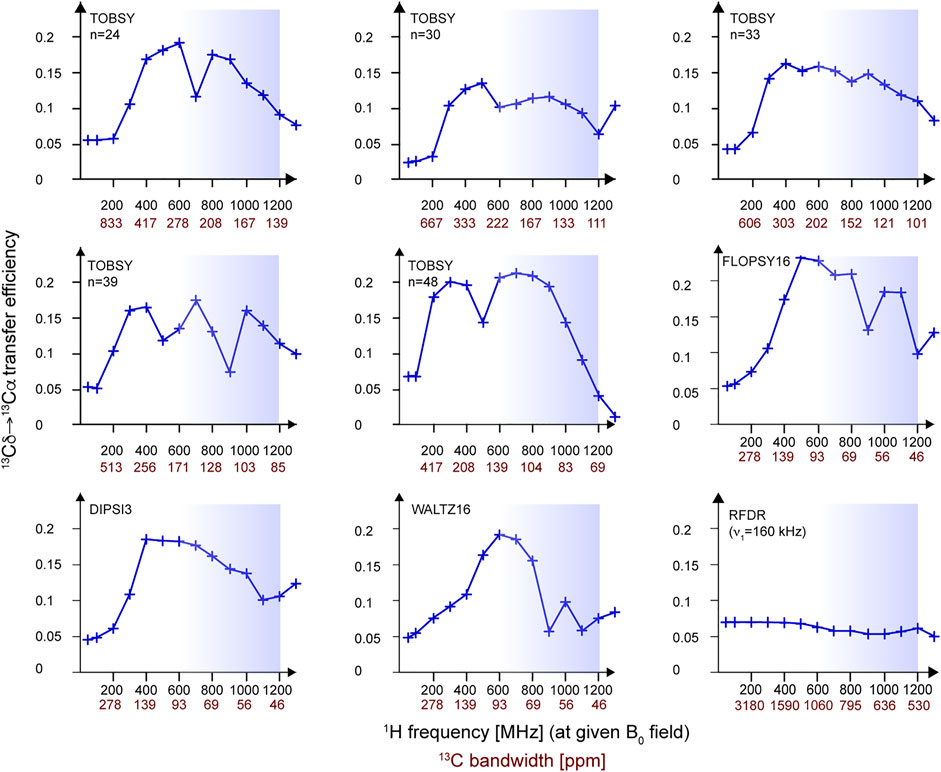
FIGURE 7. Efficiency of Cδ1→Cα transfer in a 13C6 spin system simulated in SIMPSON for a range of magnetic fields B0 (corresponding to 1H frequency between 50 and 1,300 MHz), MAS frequency of 55.5 kHz, and a constant RF strength of ν1 = ¼ νR ≈ 13.9 kHz. Note that, in essence, the dependence on 13C bandwidth (ν1 expressed in 13C ppm) of a particular mixing is investigated here. For each B0 point, a full buildup was performed, and an optimum was picked at the mixing times that are reported in Supplementary Figure S9. All RF schemes except the particularly computationally demanding DREAM were evaluated. Blue boxes indicate the B0 field range available for 1H-detected MAS NMR at present.
This leads to a paradox that increased MAS rates might not necessarily be beneficial if RF strength is bound to νR by the mixing design. Contrary to the case of TOBSY, TOCSY schemes tolerate variable ν1/νR ratios in a wide range below ¼ (Supplementary Figure S10), which gives an additional flexibility for their application at moderate-to-high fields and very fast spinning. The simulation does not account for RF-dependent 13C T1ρ, and very small RF should obviously also be avoided, particularly in systems showing increased microsecond local dynamics.
3.3 Methyl Resonance Assignment in Proteins by Correlation to Backbone Spins
Results obtained for fMLF strongly suggest proton dilution for the effective 13C mixing in extended spin systems in proteins. Fortunately, with fast MAS, this can be accomplished without compromising the occupancy of methyl 1H sites, e.g., by selective labeling of Ile, Leu, and Val residues from suitable precursors coupled to expression in D2O media (Tugarinov and Kay 2004). Importantly, 13C (and 2H)-enriched glucose must be used to preserve the continuity of Ile and Leu 13C side chains (Supplementary Figure S1), since α and β of the former and α and carbonyl carbons of the latter residue are incorporated from the medium rather than from the precursor (Lundström et al., 2007).
To demonstrate the efficacy of methyl 1H assignment, we resorted to a model SH3 domain of α-spectrin in a microcrystalline state. Given the selective 1H labeling, a magnetization transfer from methyl to backbone amide 1H (and not a reverse one) is strongly preferred for the sensitivity reasons. In addition to 3-fold larger occupancy of methyl 1H, the initial polarization is enhanced by fast T1 relaxation of methyl protons, while NMR signal detection is performed on well-dispersed, generally narrower, and, thus, more sensitive amide 1H protons. Naturally, a protocol for complete reprotonation of labile amide 1H sites after expression in D2O is a prerequisite for this approach. While extensive deuteration is not a necessity for proteins as simple as SH3 at fast MAS (>50 kHz) and high field (here 18.8 T), it would certainly be required for resolution and sensitivity reasons in more challenging applications. We employed a slightly adapted literature RF irradiation scheme (shown already in Figure 2D), with various 13C mixings as in the previous cases.
3.3.1 Joint Effect of the Rotation Frequency and RF Strength
For SH3 sample we acquired 3D spectra with site-specific resolution at a single optimized mixing time for each 13C mixing. Two spinning conditions, νR = 55.5 and 94.5 kHz, were applied using the same probe and rotor for the maximum cohesion of the data, and the representative strips are presented in Figure 8 (for νR = 94.5 kHz).
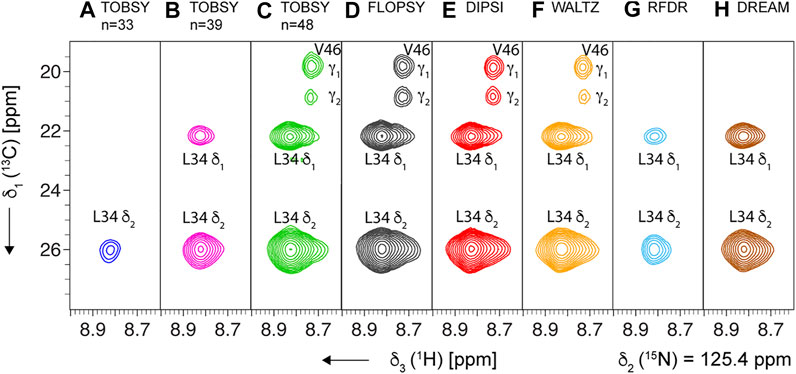
FIGURE 8. Representative strips from 3D (H)C(CC)(CA)NH experiment for SH3 protein recorded at MAS frequency of 94.5 kHz and on an 18.8-T spectrometer, using the following mixing schemes at their optimal mixing time (see Supplementary Table S3): (A–C) TOBSY C91n, with n = 33, 39, and 48, respectively, (D) FLOPSY-16, (E) DIPSI-3, (F) WALTZ-16, (G) RFDR (ν1 = 100 kHz), and (H) DREAM. All planes show the cross-section at δ2 (15N) = 125.4 ppm, and show two cross-peaks for Leu-34 residue (L34 1HN,15N intra-residue correlations to δ1 and δ2 13C). The most intense spectra (panels C–F) additionally show cross-peaks of Val-46 residue due to partial overlap of L34 and V46 1HN and 15N frequencies.
The experiment inherently shows only the transferred signal, thus poses difficulties to the rigorous quantification of 13C transfer efficiency. Nevertheless, we attempted to correct for uneven efficiency of the remaining part of the pulse sequence (notably CP steps) at different MAS conditions by normalizing the cross-peak intensities to those observed in the 3D (H)CANH experiment, which shares majority of the coherence pathway. The observed relative signal-to-noise ratio was averaged over 24 individual strong and resolved correlations. Results shown in Figure 9 (for all residue types) and Supplementary Figure S11 (separately for Val, Leu, and Ile) show a dramatic increase of 13C mixing efficiency with fast MAS (94.5 w. r. t. 55.5 kHz), with a similar effect (a factor of 3.2–3.5 for TOCSY and C9148) to that observed on nondeuterated fMLF. Despite a dilute network of 1H interactions in the SH3 sample, and averaging also over valine and isoleucine residues, the best-performing RF schemes are virtually the same as for the leucine residue in fMLF (Figure 6), namely, DREAM at 55.5 kHz MAS, and DREAM, TOBSY C9148, and three TOCSY sequences at 94.5 kHz MAS. The relevance of 1H–1H and 1H–13C interactions might be due to the fact that, despite partial sample deuteration, the local 1H environment of leucine, valine, and isoleucine residues is quite dense, with only 4 out of 11, 2 of 9, and 7 of 11 protons replaced by deuterons, respectively. As for the overall proton density, 14 ILV residues (out of 62 residues in SH3) contain 81 protons at methyl sites. These, in addition to 116 backbone and side-chain labile protons, yield a moderate rather than a low protonation level (approximately 38%).
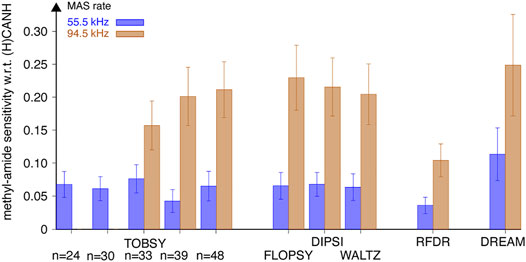
FIGURE 9. Sensitivity (S/N per unit time) of methyl-to-amide correlations in 3D (H)C(CC)(CA)NH spectra normalized to sensitivity of respective intraresidue correlations in 3D (H)CANH spectra, recorded for SH3 protein at a MAS frequency of 55.5 (blue bars, left in each pair) and 94.5 kHz (tan bars, right in each pair) on an 18.8-T spectrometer using various 13C mixing schemes. Relative intensity of 24 cross-peaks [for 6 valine (all except Val-46), 1 isoleucine, and 6 leucine residues] was averaged with equal weights. Data for TOBSY C9124 and C9130 were not collected at νR = 94.5 kHz. The error bars reflect the standard deviation (scatter) of values observed for a set of residues, not the experimental error of average sensitivity.
Compared to fMLF, spins in SH3 protein undergo higher-amplitude dynamics, and certainly there is a larger contribution of incoherent effects, such as leading to 13C T2 and T1ρ relaxation and 1H spin-diffusion. T1ρ relaxation is further suppressed by stronger RF at increased MAS frequency (particularly for DREAM, which effectively spin-locks the 13C coherence), as it is bound to νR for all sequences except RFDR. As for the coherent effects, increased MAS frequency also more effectively averages DCH interactions (i.e., a fast rotation suppresses second-order AHT cross-terms, deleterious, e.g., for TOBSY with low n). Definitely, there is an additional S/N benefit from line narrowing of amide 1H resonances at 94.5 kHz MAS (separable under a few of assumptions), but, as described below, it is estimated to contribute no more than 20% to the overall S/N gain.
In view of the apparent relevance of H–C and H–H interactions in deuterated SH3, additional efficiency gains are expected with the use of high-power 1H decoupling (ν1,H > 3νR) during 13C mixing; however, this likely endangers sample hydration and thus spectral quality. Overall, fast MAS seems to be an attractive route to amplify the signal, and, in our comparison, the gain in efficiency well compensates the 2- to 3-fold sensitivity losses entailed by active volume reduction from a 1.3-mm rotor (optimal for νR ≈ 55 kHz) to a 0.7- or 0.81-mm rotor (νR ≈ 100 kHz). We speculate that for methyl resonance assignment using the presented approach, nondeuterated proteins would benefit from fast MAS to an even larger extent.
3.3.2 Effect of the Static Magnetic Field
Our experiments were performed at a typical high-field spectrometer (18.8 T) used commonly in protein studies by 1H-detected MAS. Stronger B0 fields are increasingly accessible; therefore, it is relevant to explore the performance of 13C mixing at such conditions. For example, for a transition to a 23.5-T field, spin dynamics simulations on a six-13C spin system suggest only a minor and negative impact on transfer efficiency (results in Figure 7 are summarized for two considered B0 fields in Supplementary Figure S12). For experimental verification, we performed a full series of 3D experiments on SH3 protein (from the same crystallization batch) in a 1.3-mm rotor spun at νR = 55.5 kHz on a 1,000-MHz 1H spectrometer. Data were analyzed with a site-specific resolution and averaged over all reliable cross-peaks of Leu, Val, and Ile residues as mentioned above. Also, as before, we normalized the obtained S/N ratios of cross-peaks with respect to peaks in 3D (H)CANH to eliminate the effects of larger Boltzmann polarization, rotor active volume, possibly different sample packing density, and specific RF coil efficiency (see Supplementary Material). Results compared in Figure 10 illustrate that only selected 13C mixing schemes, notably, FLOPSY, RFDR, and DIPSI, actually profit from higher B0 field. Note that for consistency with the data acquired at 18.8 T, we did not optimize RF strength (ν1) for TOCSY sequences (in the range below ¼ νR), despite potential gains in efficiency (see Figure 7 and Supplementary Figure S10). The rationale for observed efficiency changes are again coherent and incoherent effects of H–H and H–C interactions, which evade proper treatment in our spin dynamics simulations.
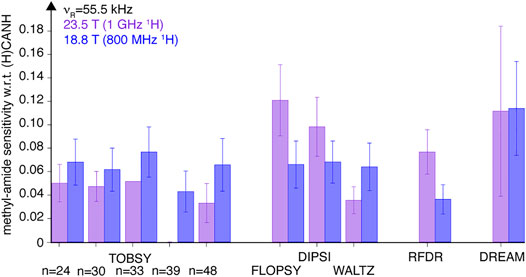
FIGURE 10. Efficiency of various 13C mixing schemes compared between 18.8- and 23.5-T fields [purple (left) and blue (right) bars, respectively]. Presented are sensitivity of correlation peaks in 3D (H)C(CC)(CA)NH spectra of SH3 protein obtained at 55.5 kHz MAS, with respect to sensitivity of correlations in (H)CANH experiment at both conditions. Relative sensitivity is averaged equally over 24 cross-peaks of Val, Leu, and Ile residues. Data for TOBSY C9139 were not collected at 23.5 T field. The error bars reflect the standard deviation (scatter) of values observed for a set of residues, not the experimental error of average sensitivity.
3.3.3 Linearization of the 13C Side Chain
One of the reasons for a low transfer efficiency between methyl and alpha 13C spins using JCC-based isotropic mixing in Leu, Val, and Ile is a branched topology of the 13C chain. A 2-fold degradation of FLOPSY-8 efficiency for Ile residues was identified by Kay and co-workers, and remedied with relay-type (COSY) experiments in which 13C coherence is sequentially transferred with a careful manipulation of individual 13C spins using very frequency selective pulses (Tugarinov and Kay 2003b). This approach is not immediately applicable in MAS NMR, since required transfer delays are prohibitively long compared to 13C T2 lifetimes. For Leu residues, the overlap between chemical shifts of δ and γ 13C precludes the elimination of detrimental passive coupling to the second methyl 13C; thus, in solution NMR, the issue was addressed by a tailored amino acid labeling, in which one of the methyl groups was labeled as 12CD3 (Tugarinov and Kay 2003a). Since the labeling of the α-ketoisovalerate (precursor) is not stereospecific, a 1:1 mixture of Leu (13CH3, 12CD3) and Leu (12CD3,13CH3) isotopomers is obtained (and analogously for valines), entailing a 50% loss of methyl 1H occupancy. Nevertheless, this sensitivity loss was compensated by larger efficiency of complex out-and-back transfer schemes proposed by Kay and co-workers for methyl 1H assignment in solution.
To verify the utility of the linearization of 13C side chains of Leu and Val residues for MAS NMR, we first performed SIMPSON simulations, and compared the behavior of 5-13C spin (linear) and 6-13C spin (branched) spin systems that mimic the leucine residue with respective isotope labeling patterns (Supplementary Figures S13 and Supplementary Figure S14). Indeed, for all J-based isotropic mixing schemes (TOBSY and TOCSY), a 2- to 3-fold efficiency gain is predicted at νR = 100 kHz, and comparable at νR = 55.5 kHz. It is thus expected that, in this case, the 50% loss of initial signal is at least compensated by increased Cδ→Cα (Cγ→Cα for valines) transfer efficiency. Dipolar-based mixing (RFDR and DREAM) marginally profits from the simplification of 13C chain, likely only due to dispersion of 13C magnetization over a smaller number of 13C spins.
The sample of SH3 with linearized side chains of Leu and Val [formally {I (δ1), L (13CH3, 12CD3), V (13CH3,12CD3)} U-(15N, 13C, 2H, 1HN)-labeled, with Ile-13C6, Leu-13C5,Val-13C4, here referred to as “ILV-C4” for brevity] was prepared by following carefully the expression protocol of the previous (“ILV-C5”, in fact Ile-13C6, Leu-13C6, and Val-13C5) sample, but using the 1,2,3,4-13C-3,4′,4′,4′-2H-labeled α-ketoisovalerate as a precursor (Tugarinov and Kay 2003a). The microcrystals were paramagnetically doped with EDTA-chelated CuII ions, and transferred into MAS 0.81-mm rotor with comparable density (see 13C 1D spectra of both samples in Supplementary Figure S15A). As expected, the methyl 1H signal in direct-excitation spectra decreased approximately twice (Supplementary Figure S15B). We repeated the entire series of 3D (H)C(CC)(CA)NH experiments at both spinning frequencies νR = 55.5 and 94.5 kHz, using virtually the same previous experimental settings. Differences in CP efficiency and rotor packing density were compensated by normalization to the respective intraresidue peak intensities in (H)CANH spectra. Finally, for each condition, the relative cross-peak intensities that approximate Cδ (or Cγ)→Cα transfer efficiencies were averaged over 24 correlation peaks. The comparison in Figure 11 provides evidence that transfer efficiency increased well beyond the factor of 2, yielding improved sensitivity for “ILV-C4” sample (with the exception of FLOPSY), and unexpectedly also manifested for dipolar-based mixings, for which a total sensitivity loss was expected based on SIMPSON simulations. Despite the 50% dilution of methyl 1H spins, “ILV-C4”-labeling clearly yields superior results, with DIPSI, WALTZ, and TOBSY C9148 as the methods of first choice. Interestingly, if the stereospecifically labeled 13C4-α-ketoisovalerate was commercially available, S/N ratios could double, corresponding to a further sensitivity gain (i.e., per square root of time) of √2 (accounting for the need for two separate acquisition series).
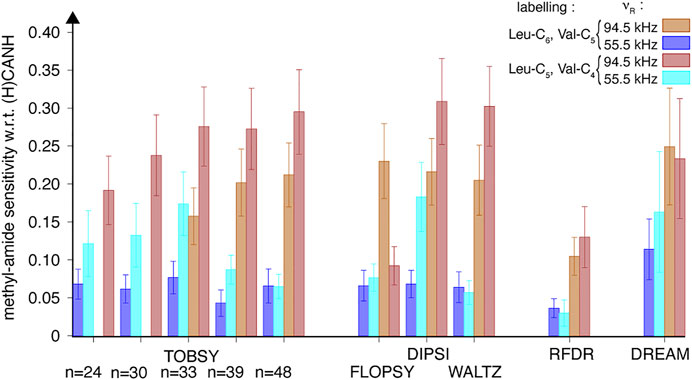
FIGURE 11. Relative efficiency of 13C mixing schemes compared between two SH3 protein samples: with branched (“ILV-C5”, tan filled bars) and linear (“ILV-C4”, red filled bars) 13C spin systems of leucine and valine residues, quantified using 24 cross-peak intensities in 3D (H)C(CC)(CA)NH spectra recorded at a MAS frequency of 94.5 kHz on an 18.8-T spectrometer. Data for 55.5 kHz MAS is shown as blue and cyan bars for “ILV-C5” and “ILV-C4” SH3 samples, respectively. Sensitivity of each cross-peak was normalized to respective intra-residue peak in 3D (H)CANH spectrum at each sample and spinning condition, and subsequently averaged over Val, Leu, and Ile residues.
The unexpectedly high gains observed with “ILV-C4” labeling can be explained by (1) decreased proton density, particularly in the local environments of leucine and valine 13C spin systems, and (2) increased detection sensitivity due to amide 1H line narrowing. The relevance of the first effect on the efficiency of 13C transfer can be appreciated based on the MAS frequency dependence discussed above for the “ILV-C5” sample. Here, we illustrated the effect by measurement of methyl 1H linewidth change upon additional proton dilution, as observed in 1H, 13C-CP-HSQC spectra as cross-peak raw full-width at half-height. Indeed, a 2-fold linewidth reduction is observed at νR = 55.5 kHz (Figure 12A and Supplementary Figure S16A, B), surpassing the effect of faster rotation for the “ILV-C5” sample (the ratio of 1.3 between 55.5 and 94.5 kHz). For the “ILV-C4” sample, both spinning conditions lead to similar linewidths (46 ± 11 Hz) with only few exceptions.
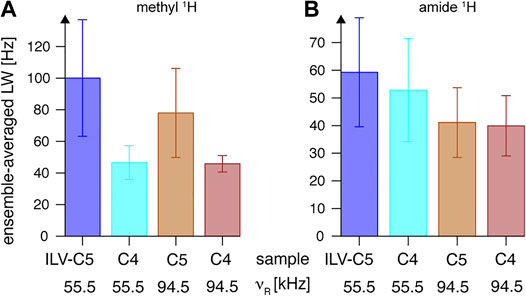
FIGURE 12. Linewidths (full widths at half height) of (A) methyl and (B) backbone amide 1H resonances observed in 2D 13C-1H and 15N–1H CP-HSQC spectra, respectively, for “ILV-C4” (at νR = 55.5 kHz in cyan, and at νR = 94.5 kHz in red) and “ILV-C5” (at νR = 55.5 kHz in blue, and at νR = 94.5 kHz in tan) SH3 samples on an 18.8-T spectrometer. Linewidths observed in each case were averaged over all resonances, and the error bars indicate the standard deviation (the dispersion of values observed for all residues), not the precision of linewidth determination.
The second effect was carefully quantified by 1H amide linewidth measurements in fingerprint 1H, 15N-CP-HSQC spectra (also without application of a window function in 1H dimension). As expected, proton dilution in the methyl side chain of Leu and Val residues has a relatively smaller impact on amide 1H coherence lifetimes (Figure 12B). At νR = 94.5 kHz, both samples show similar linewidths of 40 Hz (see Supplementary Figures S16C,D for a per-peak comparison), but approximately 9% difference is observed at νR = 55.5 kHz. The line-narrowing effect of rotation frequency increase is of a factor of 1.4 and 1.3 for “ILV-C5” and “ILV-C4” samples, respectively. Overall, the use of “ILV-C4” labeling is beneficial for resolution of 1H, 13C methyl resonances, and sensitivity of methyl-to-amide correlations.
3.3.4 Assignment of Protons: 4D Correlations
As shown in Figure 13, even in proteins as small as SH3, a single 3D (13Cmet-edited) spectrum can result in massive ambiguities. For example, V53 γ2, V58 γ2, L8 δ1, and L34 δ1 1H assignment cannot be established from (H)C(CC)(CA)NH spectrum alone, since these methyl sites exhibit close 13C shifts (highlighted by a dashed line in 13C-CP-HSQC, panel C). The joint analysis of a pair of 3D (H)C(CC)(CA)NH and H(C)(CC)(CA)NH spectra could help to correctly match 1H to 13C frequencies within a spin system (given sufficient resolution of each spectrum); however, if both methyl sites in a residue show either close 1H or close 13C shifts, the ambiguity remains (e.g., in Val-58). The stereospecific labeling, i.e., using leucine and valine pro-S-C4 and pro-R-C4 precursors (yet unavailable for the presented approach) would be a costly solution.
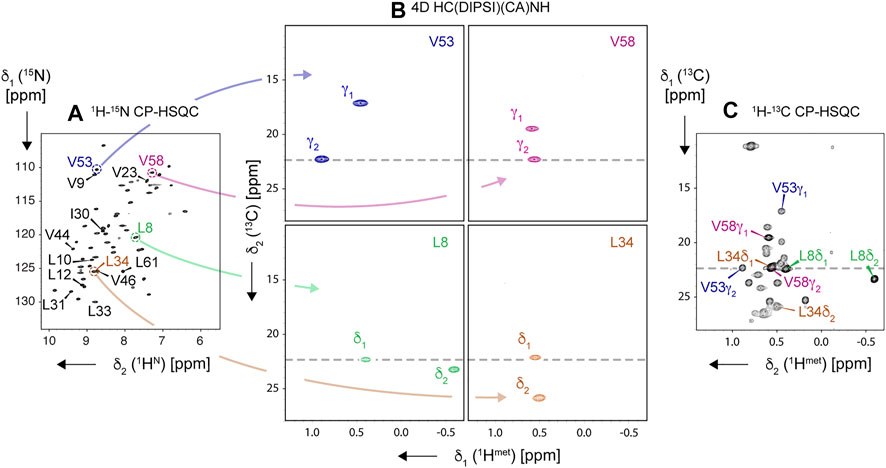
FIGURE 13. Resolving ambiguity of methyl 1H and 13C resonance assignment using 4D HC(DIPSI)(CA)NH experiment, demonstrated on the “ILV-C4” sample of SH3 protein, spun at 94.5 kHz on an 18.8-T spectrometer. Navigation in the 4D spectral matrix is based on peak positions in 2D 1H, 15N-CP-HSQC (A), allowing to display intraresidue pairs of correlations in 1H, 13C-cross sections of the 4D cube (B). Obtained methyl resonance assignments for four representative residues (L8, L34, V53, and V58, color-coded) are readily transferable to 2D 1H, 13C-CP-HSQC spectrum (C). In (A), amide correlations of all Leu, Val, and Ile residues are labeled. All correlations except for residue Val-46 were observed in the 4D experiment.
Alternatively, one can resort to high-resolution 4D spectroscopy with non-uniform sampling, increasingly popular for MAS NMR (Huber et al., 2011; Linser et al., 2011; Paramasivam et al., 2012; Linser et al., 2014; Xiang et al., 2014; Fraga et al., 2017; Sergeyev et al., 2017; Marchanka et al., 2018; Vasa et al., 2018). Indeed, the use of a 4D HC(CC)(CA)NH spectrum (Figure 13B) allows disambiguating the assignment as demonstrated for L34 and V58 spin systems (close 1H shifts), which additionally suffer from overlap of L34 δ1 and V58 γ2 peaks in 1H, 13C-CP-HSQC. Given that a 4D NUS spectrum can be acquired in a similar time to a pair of 3D ones, this approach is recommended for all but the smallest proteins.
In the presented 4D NUS experiment, the average S/N of cross-peaks, normalized to 24 h of acquisition, was 25 ± 7 (Supplementary Figure S17). One can safely interpolate that a minimal S/N ratio of 10 is obtained in 3.8 h for SH3 (62 aa). If we limit the discussion to the class of microcrystalline samples of comparable spectral properties (CP efficiency and linewidths) and crystal packing density, S/N of a single peak scales inversely with the number of amino acids K (to account for the smaller number of molecules in a rotor). Thus, the time needed for the same minimal S/N ratio scales as K2, yielding reasonable acquisition times of 0.42, 1.7, 3.7, 6.7, 10.4, and 15 days needed at 18.8 T for proteins of K = 100, 200, 300, 400, 500, and 600 residues, respectively. It is noteworthy that these times would be approximately 2- and 3.4-fold shorter, respectively, on 23.5- and 28.2-T (1,000 and 1,200 MHz 1H) spectrometers available nowadays (assuming B03/2 scaling of S/N ratio). Our results thus suggest feasibility of site-specific assignment of methyl resonances in sizable proteins for which the crystallization conditions and deuteration protocol have been already established, and can potentially be of great value to further solution or solid-state NMR studies. Whether methyl chemical shifts are readily transferable to other sample conditions is yet to be investigated. However, provided no significant alteration to protein fold, aliphatic 13C should not experience large deviations as demonstrated recently for maltose binding protein (Stanek et al., 2020).
4 Conclusion
We showed that a careful selection of 13C homonuclear mixing allows one to obtain sensitive correlations of methyl-to-amide 1H chemical shifts under fast MAS and high B0 field conditions. For highly deuterated proteins on an 18.8-T spectrometer, the best performance was obtained with DIPSI-3, WALTZ-16, and TOBSY C9148 at νR = 94.5 kHz, and with DREAM at νR = 55.5 kHz. Dramatic improvement of the multi-bond (methyl to alpha 13C) transfer efficiency was observed upon increase of MAS frequency from 55.5 to 94.5 kHz, which is attributed to the suppression of incoherent effects of H–H and H–C dipolar interactions. Further significant performance increase was obtained by the linearization of 13C side chains of leucine and valine residues, with additional gains in resolution of methyl 1H resonances. We demonstrated that unambiguous assignment of methyl 1H and 13C resonances is feasible for microcrystalline proteins by a combination of protein deuteration, paramagnetic T1 relaxation enhancement, suitable 13C isotope labeling, ultra-fast MAS, and 4D spectroscopy with non-uniform sampling. These findings pave the way for efficient assignment complementary to the labor-intensive mutagenesis-based strategy, with protein mass limitations largely mitigated by the favorable scaling of sensitivity in MAS NMR.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://doi.org/10.5281/zenodo.5911897.
Author Contributions
PP and JS designed and performed the research and analyzed data. RA prepared protein samples. M-LO, KV, and AK designed, constructed, and tested the MAS 100-kHz system (probe, rotation, and temperature controllers and rotor tools) under supervision of AS. PP, RA, and JS wrote the manuscript.
Funding
This research was funded by the Polish National Science Centre with grant No. 2019/33/B/ST4/02021. JS and PP thank the Polish National Agency for Academic Exchange for the generous support of Polish Return programme (Contract No. PPN/PPO/2018/1/00098). The computational resources for this work were provided by the Polish Infrastructure for Supporting Computational Science in the European Research Space (PLGRID, grant ID: plgnmrsi2, plgnmrsi3). We gratefully acknowledge financial support from the TGIR-RMN-THC Fr3050 CNRS for the access to the 23.5-T NMR spectrometer in CRMN (Lyon, France).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Tobias Schubeis (CRMN Lyon) for sharing the SH3 plasmid and the original sample preparation protocol. The help of Tanguy Le Marchand in setting up the experiments on the 23.5-T spectrometer (CRMN Lyon) is greatly appreciated. The authors thank Morgane Callon (ETH Zurich) and Alons Lends (CBMN—IECB Bordeaux) for hints on rotor packing. We gratefully acknowledge M.J. Potrzebowski for providing access to the 14.1-T spectrometer at CBMM PAS in Łódź (Poland). The authors wish to express gratitude to Rector of Tallin University of Technology for financial support, and Darklands OÜ for technical support. We would like to thank W. Koźmiński for technical advice and maintenance of NMR system at CNBCh of University of Warsaw.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.828785/full#supplementary-material
References
Agarwal, V., Diehl, A., Skrynnikov, N., and Reif, B. (2006). High Resolution 1H Detected 1H,13C Correlation Spectra in MAS Solid-State NMR Using Deuterated Proteins with Selective 1H,2H Isotopic Labeling of Methyl Groups. J. Am. Chem. Soc. 128 (39), 12620–12621. doi:10.1021/ja064379m
Agarwal, V., Penzel, S., Szekely, K., Cadalbert, R., Testori, E., Oss, A., et al. (2014). De Novo 3D Structure Determination from Sub-milligram Protein Samples by Solid-State 100 kHz MAS NMR Spectroscopy. Angew. Chem. Int. Ed. 53 (45), 12253–12256. doi:10.1002/anie.201405730
Agarwal, V., and Reif, B. (2008). Residual Methyl Protonation in Perdeuterated Proteins for Multi-Dimensional Correlation Experiments in MAS Solid-State NMR Spectroscopy. J. Magn. Reson. 194 (1), 16–24. doi:10.1016/j.jmr.2008.05.021
Amero, C., Asunción Durá, M., Noirclerc-Savoye, M., Perollier, A., Gallet, B., Plevin, M. J., et al. (2011). A Systematic Mutagenesis-Driven Strategy for Site-Resolved NMR Studies of Supramolecular Assemblies. J. Biomol. NMR 50 (3), 229–236. doi:10.1007/s10858-011-9513-5
Andreas, L. B., Jaudzems, K., Stanek, J., Lalli, D., Bertarello, A., Le Marchand, T., et al. (2016). Structure of Fully Protonated Proteins by Proton-Detected Magic-Angle Spinning NMR. Proc. Natl. Acad. Sci. USA 113 (33), 9187–9192. doi:10.1073/pnas.1602248113
Andreas, L. B., Le Marchand, T., Jaudzems, K., and Pintacuda, G. (2015b). High-Resolution Proton-Detected NMR of Proteins at Very Fast MAS. J. Magn. Reson. 253, 36–49. doi:10.1016/j.jmr.2015.01.003
Andreas, L. B., Reese, M., Eddy, M. T., Gelev, V., Ni, Q. Z., Miller, E. A., et al. (2015a). Structure and Mechanism of the Influenza A M218-60 Dimer of Dimers. J. Am. Chem. Soc. 137 (47), 14877–14886. doi:10.1021/jacs.5b04802
Asami, S., and Reif, B. (2012). Assignment Strategies for Aliphatic Protons in the Solid-State in Randomly Protonated Proteins. J. Biomol. NMR 52 (1), 31–39. doi:10.1007/s10858-011-9591-4
Asami, S., and Reif, B. (2013). Proton-Detected Solid-State NMR Spectroscopy at Aliphatic Sites: Application to Crystalline Systems. Acc. Chem. Res. 46 (9), 2089–2097. doi:10.1021/ar400063y
Asami, S., Schmieder, P., and Reif, B. (2010). High Resolution 1H-Detected Solid-State NMR Spectroscopy of Protein Aliphatic Resonances: Access to Tertiary Structure Information. J. Am. Chem. Soc. 132 (43), 15133–15135. doi:10.1021/ja106170h
Asami, S., Szekely, K., Schanda, P., Meier, B. H., and Reif, B. (2012). Optimal Degree of Protonation for 1H Detection of Aliphatic Sites in Randomly Deuterated Proteins as a Function of the MAS Frequency. J. Biomol. NMR 54 (2), 155–168. doi:10.1007/s10858-012-9659-9
Bak, M., and Nielsen, N. C. (1997). REPULSION, A Novel Approach to Efficient Powder Averaging in Solid-State NMR. J. Magn. Reson. 125 (1), 132–139. doi:10.1006/jmre.1996.1087
Bak, M., Rasmussen, J. T., and Nielsen, N. C. (2011). SIMPSON: A General Simulation Program for Solid-State NMR Spectroscopy. J. Magn. Reson. 213 (2), 366–400. doi:10.1016/j.jmr.2011.09.008
Bak, M., Schultz, R., Vosegaard, T., and Nielsen, N. C. (2002). Specification and Visualization of Anisotropic Interaction Tensors in Polypeptides and Numerical Simulations in Biological Solid-State NMR. J. Magn. Reson. 154 (1), 28–45. doi:10.1006/jmre.2001.2454
Baker, L. A., and Baldus, M. (2014). Characterization of Membrane Protein Function by Solid-State NMR Spectroscopy. Curr. Opin. Struct. Biol. 27 (August), 48–55. doi:10.1016/j.sbi.2014.03.009
Baldus, M., and Meier, B. H. (1996). Total Correlation Spectroscopy in the Solid State. The Use of Scalar Couplings to Determine the Through-Bond Connectivity. J. Magn. Reson. Ser. A 121 (1), 65–69. doi:10.1006/jmra.1996.0137
Bax, A., Clore, G. M., and Gronenborn, A. M. (1990). 1H1H Correlation via Isotropic Mixing of 13C Magnetization, a New Three-Dimensional Approach for Assigning 1H and 13C Spectra of 13C-Enriched Proteins. J. Magn. Reson. (1969) 88 (2), 425–431. doi:10.1016/0022-2364(90)90202-K
Bennett, A. E., Rienstra, C. M., Auger, M., Lakshmi, K. V., and Griffin, R. G. (1995). Heteronuclear Decoupling in Rotating Solids. J. Chem. Phys. 103 (16), 6951–6958. doi:10.1063/1.470372
Bennett, A. E., Rienstra, C. M., Griffiths, J. M., Zhen, W., Lansbury, P. T., and Griffin, R. G. (1998). Homonuclear Radio Frequency-Driven Recoupling in Rotating Solids. J. Chem. Phys. 108 (22), 9463–9479. doi:10.1063/1.476420
Boswell, Z. K., and Latham, M. P. (2018). Methyl-Based NMR Spectroscopy Methods for Uncovering Structural Dynamics in Large Proteins and Protein Complexes. Biochemistry 58 (3), 144–155. doi:10.1021/acs.biochem.8b00953
Chevelkov, V., Rehbein, K., Diehl, A., and Reif, B. (2006). Ultrahigh Resolution in Proton Solid-State NMR Spectroscopy at High Levels of Deuteration. Angew. Chem. Int. Ed. 45 (23), 3878–3881. doi:10.1002/anie.200600328
De Paëpe, G., Lewandowski, J. R., and Griffin, R. G. (2008). Spin Dynamics in the Modulation Frame: Application to Homonuclear Recoupling in Magic Angle Spinning Solid-State NMR. J. Chem. Phys. 128 (12), 124503. doi:10.1063/1.2834732
Edén, M., and Levitt, M. H. (1999). Pulse Sequence Symmetries in the Nuclear Magnetic Resonance of Spinning Solids: Application to Heteronuclear Decoupling. J. Chem. Phys. 111 (4), 1511–1519. doi:10.1063/1.479410
Emsley, L., and Bodenhausen, G. (1992). Optimization of Shaped Selective Pulses for NMR Using a Quaternion Description of Their Overall Propagators. J. Magn. Reson. (1969) 97 (1), 135–148. doi:10.1016/0022-2364(92)90242-Y
Fraga, H., Arnaud, C. A., Gauto, D. F., Audin, M., Kurauskas, V., Macek, P., et al. (2017). Solid‐State NMR H–N–(C)–H and H–N–C–C 3D/4D Correlation Experiments for Resonance Assignment of Large Proteins. ChemPhysChem 18 (19), 2697–2703. doi:10.1002/cphc.201700572
Ganapathy, S., Naito, A., and McDowell, C. A. (1981). Paramagnetic Doping as an Aid in Obtaining High-Resolution Carbon-13 NMR Spectra of Biomolecules in the Solid State. J. Am. Chem. Soc. 103 (20), 6011–6015. doi:10.1021/ja00410a003
Gauto, D. F., Estrozi, L. F., Schwieters, C. D., Effantin, G., Macek, P., Sounier, R., et al. (2019). Integrated NMR and Cryo-EM Atomic-Resolution Structure Determination of a Half-Megadalton Enzyme Complex. Nat. Commun. 10 (1), 2697. doi:10.1038/s41467-019-10490-9
Gorman, S. D., Sahu, D., O'Rourke, K. F., and Boehr, D. D. (2018). Assigning Methyl Resonances for Protein Solution-State NMR Studies. Methods 148 (September), 88–99. doi:10.1016/j.ymeth.2018.06.010
Goto, N. K., Gardner, K. H., Mueller, G. A., Willis, R. C., and Kay, L. E. (1999). A Robust and Cost-Effective Method for the Production of Val, Leu, Ile (δ1) Methyl-Protonated 15N-, 13C-, 2H-Labeled Proteins. J. Biomol. NMR 13 (4), 369–374. doi:10.1023/A:1008393201236
Griffin, R. (1998). Dipolar Recoupling in MAS Spectra of Biological Solids. Nat. Struct. Mol. Biol. 5 (7), 508–512. doi:10.1038/749
Hardy, E. H., Verel, R., and Meier, B. H. (2001). Fast MAS Total Through-Bond Correlation Spectroscopy. J. Magn. Reson. 148 (2), 459–464. doi:10.1006/jmre.2000.2258
Hou, G., Yan, S., Trébosc, J., Amoureux, J.-P., and Polenova, T. (2013). Broadband Homonuclear Correlation Spectroscopy Driven by Combined R2nv Sequences under Fast Magic Angle Spinning for NMR Structural Analysis of Organic and Biological Solids. J. Magn. Reson. 232, 18–30. doi:10.1016/j.jmr.2013.04.009
Hu, B., Lafon, O., Trébosc, J., Chen, Q., and Amoureux, J.-P. (2011). Broad-Band Homo-Nuclear Correlations Assisted by 1H Irradiation for Bio-Molecules in Very High Magnetic Field at Fast and Ultra-fast MAS Frequencies. J. Magn. Reson. 212 (2), 320–329. doi:10.1016/j.jmr.2011.07.011
Huang, C., and Kalodimos, C. G. (2017). Structures of Large Protein Complexes Determined by Nuclear Magnetic Resonance Spectroscopy. Annu. Rev. Biophys. 46 (1), 317–336. doi:10.1146/annurev-biophys-070816-033701
Huber, M., Hiller, S., Schanda, P., Ernst, M., Böckmann, A., Verel, R., et al. (2011). A Proton-Detected 4D Solid-State NMR Experiment for Protein Structure Determination. ChemPhysChem 12 (5), 915–918. doi:10.1002/cphc.201100062
Kadkhodaie, M., Rivas, O., Tan, M., Mohebbi, A., and Shaka, A. J. (1991). Broadband Homonuclear Cross Polarization Using Flip-Flop Spectroscopy. J. Magn. Reson. (1969) 91 (2), 437–443. doi:10.1016/0022-2364(91)90210-K
Kay, L. E., Xu, G. Y., Singer, A. U., Muhandiram, D. R., and Forman-Kay, J. D. (1993). A Gradient-Enhanced HCCH-TOCSY Experiment for Recording Side-Chain 1H and 13C Correlations in H2O Samples of Proteins. J. Magn. Reson. Ser. B 101 (3), 333–337. doi:10.1006/jmrb.1993.1053
Kulminskaya, N., Vasa, S. K., Giller, K., Becker, S., Kwan, A., Sunde, M., et al. (2016). Access to Side-Chain Carbon Information in Deuterated Solids under Fast MAS through Non-rotor-synchronized Mixing. Chem. Commun. 52 (2), 268–271. doi:10.1039/C5CC07345F
Kurauskas, V., Crublet, E., Macek, P., Kerfah, R., Gauto, D. F., Boisbouvier, J., et al. (2016). Sensitive Proton-Detected Solid-State NMR Spectroscopy of Large Proteins with Selective CH3 Labelling: Application to the 50S Ribosome Subunit. Chem. Commun. 52 (61), 9558–9561. doi:10.1039/C6CC04484K
Ladizhansky, V. (2017). Applications of Solid-State NMR to Membrane Proteins. Biochim. Biophys. Acta (Bba) - Proteins Proteomics 1865 (11), 1577–1586. doi:10.1016/j.bbapap.2017.07.004
Lee, W., Tonelli, M., and Markley, J. L. (2015). NMRFAM-SPARKY: Enhanced Software for Biomolecular NMR Spectroscopy. Bioinformatics 31 (8), 1325–1327. doi:10.1093/bioinformatics/btu830
Levitt, M. H. (2007). “eMagRes,”. Editors R. K. Harris, and R. L. Wasylishen (Chichester, UK: John Wiley & Sons), 9. doi:10.1002/9780470034590
Lewandowski, J. R., Dumez, J.-N., Akbey, Ü., Lange, S., Emsley, L., and Oschkinat, H. (2011). Enhanced Resolution and Coherence Lifetimes in the Solid-State NMR Spectroscopy of Perdeuterated Proteins under Ultrafast Magic-Angle Spinning. J. Phys. Chem. Lett. 2 (17), 2205–2211. doi:10.1021/jz200844n
Linser, R., Bardiaux, B., Andreas, L. B., Hyberts, S. G., Morris, V. K., Pintacuda, G., et al. (2014). Solid-State NMR Structure Determination from Diagonal-Compensated, Sparsely Nonuniform-Sampled 4D Proton-Proton Restraints. J. Am. Chem. Soc. 136 (31), 11002–11010. doi:10.1021/ja504603g
Linser, R., Bardiaux, B., Higman, V., Fink, U., and Reif, B. (2011). Structure Calculation from Unambiguous Long-Range Amide and Methyl 1H−1H Distance Restraints for a Microcrystalline Protein with MAS Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 133 (15), 5905–5912. doi:10.1021/ja110222h
Linser, R. (2011). Side-Chain to Backbone Correlations from Solid-State NMR of Perdeuterated Proteins through Combined Excitation and Long-Range Magnetization Transfers. J. Biomol. NMR 51 (3), 221–226. doi:10.1007/s10858-011-9531-3
Linser, R. (2017). Solid-State NMR Spectroscopic Trends for Supramolecular Assemblies and Protein Aggregates. Solid State. Nucl. Magn. Reson. 87 (August), 45–53. doi:10.1016/j.ssnmr.2017.08.003
Lu, X., Guo, C., Hou, G., and Polenova, T. (2015). Combined Zero-Quantum and Spin-Diffusion Mixing for Efficient Homonuclear Correlation Spectroscopy under Fast MAS: Broadband Recoupling and Detection of Long-Range Correlations. J. Biomol. NMR 61 (1), 7–20. doi:10.1007/s10858-014-9875-6
Lundström, P., Teilum, K., Carstensen, T., Bezsonova, I., Wiesner, S., Hansen, D. F., et al. (2007). Fractional 13C Enrichment of Isolated Carbons Using [1-13C]- or [2-13C]-Glucose Facilitates the Accurate Measurement of Dynamics at Backbone Cα and Side-Chain Methyl Positions in Proteins. J. Biomol. NMR 38 (3), 199–212. doi:10.1007/s10858-007-9158-6
Mainz, A., Religa, T. L., Sprangers, R., Linser, R., Kay, L. E., and Reif, B. (2013). NMR Spectroscopy of Soluble Protein Complexes at One Mega-Dalton and beyond. Angew. Chem. Int. Ed. 52 (33), 8746–8751. doi:10.1002/anie.201301215
Mandala, V. S., Williams, J. K., and Hong, M. (2018). Structure and Dynamics of Membrane Proteins from Solid-State NMR. Annu. Rev. Biophys. 47 (1), 201–222. doi:10.1146/annurev-biophys-070816-033712
Marchanka, A., Stanek, J., Pintacuda, G., and Carlomagno, T. (2018). Rapid Access to RNA Resonances by Proton-Detected Solid-State NMR at >100 kHz MAS. Chem. Commun. 54 (65), 8972–8975. doi:10.1039/c8cc04437f
Miao, Y., and Cross, T. A. (2013). Solid State NMR and Protein-Protein Interactions in Membranes. Curr. Opin. Struct. Biol. 23 (6), 919–928. doi:10.1016/j.sbi.2013.08.004
Paramasivam, S., Suiter, C. L., Hou, G., Sun, S., Palmer, M., Hoch, J. C., et al. (2012). Enhanced Sensitivity by Nonuniform Sampling Enables Multidimensional MAS NMR Spectroscopy of Protein Assemblies. J. Phys. Chem. B 116 (25), 7416–7427. doi:10.1021/jp3032786
Pauli, J., Baldus, M., Van Rossum, B., De Groot, H., and Oschkinat, H. (2001). Backbone and Side-Chain 13C and 15N Signal Assignments of the α-Spectrin SH3 Domain by Magic Angle Spinning Solid-State NMR at 17.6 Tesla. ChemBioChem 2 (4), 272–281. doi:10.1002/1439-7633(20010401)2:4<272::aid-cbic272>3.0.co;2-2
Pauli, J., van Rossum, B., Förster, H., de Groot, H. J. M., and Oschkinat, H. (2000). Sample Optimization and Identification of Signal Patterns of Amino Acid Side Chains in 2D RFDR Spectra of the α-Spectrin SH3 Domain. J. Magn. Reson. 143 (2), 411–416. doi:10.1006/jmre.2000.2029
Penzel, S., Smith, A. A., Agarwal, V., Hunkeler, A., Org, M.-L., Samoson, A., et al. (2015). Mai-Liis Org, Ago Samoson, Anja Böckmann, Matthias Ernst, and Beat H MeierProtein Resonance Assignment at MAS Frequencies Approaching 100 kHz: A Quantitative Comparison of J-Coupling and Dipolar-Coupling-Based Transfer Methods. J. Biomol. NMR 63 (2), 165–186. doi:10.1007/s10858-015-9975-y
Pritišanac, I., Alderson, T. R., and Güntert, P. (2020). Automated Assignment of Methyl NMR Spectra from Large Proteins. Prog. Nucl. Magn. Reson. Spectrosc. 118-119 (June), 54–73. doi:10.1016/j.pnmrs.2020.04.001
Pritišanac, I., Würz, J. M., Alderson, T. R., and Güntert, P. (2019). Automatic Structure-Based NMR Methyl Resonance Assignment in Large Proteins. Nat. Commun. 10 (1), 1–12. doi:10.1038/s41467-019-12837-8
Reverter, D., and Lima, C. D. (2009). Preparation of SUMO Proteases and Kinetic Analysis Using Endogenous Substrates. Methods Mol. Biolin 497, 225–239. doi:10.1007/978-1-59745-566-4_15
Rosenzweig, R., and Kay, L. E. (2014). Bringing Dynamic Molecular Machines into Focus by Methyl-TROSY NMR. Annu. Rev. Biochem. 83 (1), 291–315. doi:10.1146/annurev-biochem-060713-035829
Sattler, M., Schleucher, J., and Griesinger, C. (1999). Heteronuclear Multidimensional NMR Experiments for the Structure Determination of Proteins in Solution Employing Pulsed Field Gradients. Prog. Nucl. Magn. Reson. Spectrosc. 34 (2), 93–158. doi:10.1016/S0079-6565(98)00025-9
Schmidt, E., and Güntert, P. (2012). A New Algorithm for Reliable and General NMR Resonance Assignment. J. Am. Chem. Soc. 134 (30), 12817–12829. doi:10.1021/ja305091n
Schmidt, E., and Güntert, P. (2013). Reliability of Exclusively NOESY-Based Automated Resonance Assignment and Structure Determination of Proteins. J. Biomol. NMR 57 (2), 193–204. doi:10.1007/s10858-013-9779-x
Scholz, I., Huber, M., Manolikas, T., Meier, B. H., and Ernst, M. (2008). MIRROR Recoupling and its Application to Spin Diffusion under Fast Magic-Angle Spinning. Chem. Phys. Lett. 460 (1), 278–283. doi:10.1016/j.cplett.2008.05.058
Sergeyev, I. V., Itin, B., Rogawski, R., Day, L. A., and McDermott, A. E. (2017). Efficient Assignment and NMR Analysis of an Intact Virus Using Sequential Side-Chain Correlations and DNP Sensitization. Proc. Natl. Acad. Sci. USA 114 (20), 5171–5176. doi:10.1073/pnas.1701484114
Shaka, A. J., Keeler, J., and Freeman, R. (1983). Evaluation of a New Broadband Decoupling Sequence: WALTZ-16. J. Magn. Reson. (1969) 53 (2), 313–340. doi:10.1016/0022-2364(83)90035-5
Shaka, A. J., Lee, C. J., and Pines, A. (1988). Iterative Schemes for Bilinear Operators; Application to Spin Decoupling. J. Magn. Reson. (1969) 77 (2), 274–293. doi:10.1016/0022-2364(88)90178-3
Shen, M., Hu, B., Lafon, O., Trébosc, J., Chen, Q., and Amoureux, J.-P. (2012). Broadband Finite-Pulse Radio-Frequency-Driven Recoupling (Fp-RFDR) with (XY8)41 Super-cycling for Homo-Nuclear Correlations in Very High Magnetic Fields at Fast and Ultra-fast MAS Frequencies. J. Magn. Reson. 223 (October), 107–119. doi:10.1016/j.jmr.2012.07.013
Sinnige, T., Daniëls, M., Baldus, M., and Weingarth, M. (2014). Proton Clouds to Measure Long-Range Contacts between Nonexchangeable Side Chain Protons in Solid-State NMR. J. Am. Chem. Soc. 136 (12), 4452–4455. doi:10.1021/ja412870m
Stanek, J., Andreas, L. B., Jaudzems, K., Cala, D., Lalli, D., Bertarello, A., et al. (2016). NMR Spectroscopic Assignment of Backbone and Side-Chain Protons in Fully Protonated Proteins: Microcrystals, Sedimented Assemblies, and Amyloid Fibrils. Angew. Chem. Int. Ed. 55 (50), 15504–15509. doi:10.1002/anie.201607084
Stanek, J., Augustyniak, R., and Koźmiński, W. (2012). Suppression of Sampling Artefacts in High-Resolution Four-Dimensional NMR Spectra Using Signal Separation Algorithm. J. Magn. Reson. 214 (January), 91–102. doi:10.1016/j.jmr.2011.10.009
Stanek, J., Schubeis, T., Paluch, P., Güntert, P., Andreas, L. B., and Pintacuda, G. (2020). Automated Backbone NMR Resonance Assignment of Large Proteins Using Redundant Linking from a Single Simultaneous Acquisition. J. Am. Chem. Soc. 142 (12), 5793–5799. doi:10.1021/jacs.0c00251
Tan, K. O., Agarwal, V., Lakomek, N.-A., Penzel, S., Meier, B. H., and Ernst, M. (2018). Efficient Low-Power TOBSY Sequences for Fast MAS. Solid State. Nucl. Magn. Reson. 89, 27–34. doi:10.1016/j.ssnmr.2017.11.003
Tošner, Z., Andersen, R., Stevensson, B., Edén, M., Nielsen, N. C., and Vosegaard, T. (2014). Computer-Intensive Simulation of Solid-State NMR Experiments Using SIMPSON. J. Magn. Reson. 246 (September), 79–93. doi:10.1016/j.jmr.2014.07.002
Tošner, Z., Sarkar, R., Becker-Baldus, J., Glaubitz, C., Wegner, S., Engelke, F., et al. (2018). Overcoming Volume Selectivity of Dipolar Recoupling in Biological Solid-State NMR Spectroscopy. Angew. Chem. Int. Ed. 57 (44), 14514–14518. doi:10.1002/anie.201805002
Tugarinov, V., Hwang, P. M., Ollerenshaw, J. E., and Kay, L. E. (2003). Cross-Correlated Relaxation Enhanced 1H−13C NMR Spectroscopy of Methyl Groups in Very High Molecular Weight Proteins and Protein Complexes. J. Am. Chem. Soc. 125 (34), 10420–10428. doi:10.1021/ja030153x
Tugarinov, V., Kanelis, V., and Kay, L. E. (2006). Isotope Labeling Strategies for the Study of High-Molecular-Weight Proteins by Solution NMR Spectroscopy. Nat. Protoc. 1 (2), 749–754. doi:10.1038/nprot.2006.101
Tugarinov, V., and Kay, L. E. (2004). An Isotope Labeling Strategy for Methyl TROSY Spectroscopy. J. Biomol. NMR 28 (2), 165–172. doi:10.1023/B:JNMR.0000013824.93994.1f
Tugarinov, V., and Kay, L. E. (2003a). Ile, Leu, and Val Methyl Assignments of the 723-Residue Malate Synthase G Using a New Labeling Strategy and Novel NMR Methods. J. Am. Chem. Soc. 125 (45), 13868–13878. doi:10.1021/ja030345s
Tugarinov, V., and Kay, L. E. (2003b). Side Chain Assignments of Ile δ1 Methyl Groups in High Molecular Weight Proteins: an Application to a 46 ns Tumbling Molecule. J. Am. Chem. Soc. 125 (19), 5701–5706. doi:10.1021/ja021452+
Tycko, R. (2011). Solid-State NMR Studies of Amyloid Fibril Structure. Annu. Rev. Phys. Chem. 62 (1), 279–299. doi:10.1146/annurev-physchem-032210-103539
van der Wel, P. C. A. (2018). New Applications of Solid-State NMR in Structural Biology. Emerging Top. Life Sci. 2 (1), 57–67. doi:10.1042/ETLS20170088
Vasa, S. K., Singh, H., Rovó, P., Linser, R., and Linser, R. (2018). Dynamics and Interactions of a 29 KDa Human Enzyme Studied by Solid-State NMR. J. Phys. Chem. Lett. 9 (6), 1307–1311. doi:10.1021/acs.jpclett.8b00110
Verel, R., Baldus, M., Ernst, M., Beat, H., and Meier, H. (1998). A Homonuclear Spin-Pair Filter for Solid-State NMR Based on Adiabatic-Passage Techniques. Chem. Phys. Lett. 287 (3–4), 421–428. doi:10.1016/S0009-2614(98)00172-9
Verel, R., Ernst, M., and Meier, B. H. (2001). Adiabatic Dipolar Recoupling in Solid-State NMR: The DREAM Scheme. J. Magn. Reson. 150 (1), 81–99. doi:10.1006/jmre.2001.2310
Weingarth, M., Demco, D. E., Bodenhausen, G., and Tekely, P. (2009). Improved Magnetization Transfer in Solid-State NMR with Fast Magic Angle Spinning. Chem. Phys. Lett. 469 (4), 342–348. doi:10.1016/j.cplett.2008.12.084
Westfeld, T., Verel, R., Ernst, M., Böckmann, A., and Meier, B. H. (2012). Properties of the DREAM Scheme and its Optimization for Application to Proteins. J. Biomol. NMR 53 (2), 103–112. doi:10.1007/s10858-012-9627-4
Wickramasinghe, N. P., Kotecha, M., Samoson, A., Past, J., and Ishii, Y. (2007). Sensitivity Enhancement in 13C Solid-State NMR of Protein Microcrystals by Use of Paramagnetic Metal Ions for Optimizing 1H T1 Relaxation. J. Magn. Reson. 184 (2), 350–356. doi:10.1016/j.jmr.2006.10.012
Wickramasinghe, N. P., Parthasarathy, S., Jones, C. R., Bhardwaj, C., Long, F., Kotecha, M., et al. (2009). Nanomole-Scale Protein Solid-State NMR by Breaking Intrinsic 1H T1 Boundaries. Nat. Methods 6 (3), 215–218. doi:10.1038/nmeth.1300
Wiegand, T. (2020). A Solid-State NMR Tool Box for the Investigation of ATP-Fueled Protein Engines. Prog. Nucl. Magn. Reson. Spectrosc. 117 (April), 1–32. doi:10.1016/j.pnmrs.2020.02.001
Xiang, S., Chevelkov, V., Becker, S., and Lange, A. (2014). Towards Automatic Protein Backbone Assignment Using Proton-Detected 4D Solid-State NMR Data. J. Biomol. NMR 60 (2–3), 85–90. doi:10.1007/s10858-014-9859-6
Xue, K., Motz, C., Asami, S., Decker, V., Wegner, S., Tosner, Z., et al. (2018). Magic-Angle Spinning Frequencies beyond 300 kHz Are Necessary to Yield Maximum Sensitivity in Selectively Methyl Protonated Protein Samples in Solid-State NMR. J. Phys. Chem. C 122, 16437–16442. doi:10.1021/acs.jpcc.8b05600
Xue, K., Sarkar, R., Lalli, D., Koch, B., Pintacuda, G., Tosner, Z., et al. (2020). Impact of Magnetic Field Strength on Resolution and Sensitivity of Proton Resonances in Biological Solids. J. Phys. Chem. C 124 (41), 22631–22637. doi:10.1021/acs.jpcc.0c05407
Xue, K., Sarkar, R., Motz, C., Asami, S., Camargo, D. C. R., Decker, V., et al. (2017). Limits of Resolution and Sensitivity of Proton Detected MAS Solid-State NMR Experiments at 111 kHz in Deuterated and Protonated Proteins. Sci. Rep. 7 (1), 7444. doi:10.1038/s41598-017-07253-1
Xue, K., Sarkar, R., Tosner, Z., Lalli, D., Motz, C., Koch, B., et al. (2019). MAS Dependent Sensitivity of Different Isotopomers in Selectively Methyl Protonated Protein Samples in Solid State NMR. J. Biomol. NMR 73 (10–11), 625–631. doi:10.1007/s10858-019-00274-0
Zhou, D. H., and Rienstra, C. M. (2008). High-Performance Solvent Suppression for Proton Detected Solid-State NMR. J. Magn. Reson. 192 (1), 167–172. doi:10.1016/j.jmr.2008.01.012
Keywords: NMR resonance assignment, methyl groups, solid-state NMR, fast MAS, proton detection, TOCSY, isotope labeling/method
Citation: Paluch P, Augustyniak R, Org M-L, Vanatalu K, Kaldma A, Samoson A and Stanek J (2022) NMR Assignment of Methyl Groups in Immobilized Proteins Using Multiple-Bond 13C Homonuclear Transfers, Proton Detection, and Very Fast MAS. Front. Mol. Biosci. 9:828785. doi: 10.3389/fmolb.2022.828785
Received: 03 December 2021; Accepted: 08 February 2022;
Published: 29 March 2022.
Edited by:
Józef Romuald Lewandowski, University of Warwick, United KingdomReviewed by:
Bernd Reif, Technical University of Munich, GermanyThomas Vosegaard, Aarhus University, Denmark
Copyright © 2022 Paluch, Augustyniak, Org, Vanatalu, Kaldma, Samoson and Stanek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Stanek, amFuc3RhbmVrQGNoZW0udXcuZWR1LnBs
†ORCID: Piotr Paluch, orcid.org/0000-0001-8601-3977; Rafal Augustyniak, orcid.org/0000-0001-6591-7192; Ago Samoson, orcid.org/0000-0001-7757-6356; Jan Stanek, orcid.org/0000-0003-1660-168X