 Shao Wei
Shao Wei- 1Research and Communication Center of Exercise and Health, Xiamen University of Technology, Xiamen, China
- 2Xiamen Cardiovascular Hospital, Xiamen University, Xiamen, China
- 3Key Laboratory of Chemical Biology of Fujian Province, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, China
Despite recent advances in therapies, cardiovascular diseases ( CVDs ) are still the leading cause of mortality worldwide. Previous studies have shown that metabolic perturbations in cardiac energy metabolism are closely associated with the progression of CVDs. As expected, metabolic interventions can be applied to alleviate metabolic impairments and, therefore, can be used to develop therapeutic strategies for CVDs. β-hydroxybutyrate (β-HB) was once known to be a harmful and toxic metabolite leading to ketoacidosis in diabetes. However, the minor metabolite is increasingly recognized as a multifunctional molecular marker in CVDs. Although the protective role of β-HB in cardiovascular disease is controversial, increasing evidence from experimental and clinical research has shown that β-HB can be a “super fuel” and a signaling metabolite with beneficial effects on vascular and cardiac dysfunction. The tremendous potential of β-HB in the treatment of CVDs has attracted many interests of researchers. This study reviews the research progress of β-HB in CVDs and aims to provide a theoretical basis for exploiting the potential of β-HB in cardiovascular therapies.
Introduction
Cardiovascular diseases (CVDs) include a group of heart and blood vessel disorders, ranging from the peripheral artery, coronary artery, cardiac valve, cardiac muscle, and congenital heart diseases to arrhythmias and, ultimately, heart failure (Kalayinia et al., 2018) (Hajar, 2016). Despite the recent advances in clinical therapy, CVDs are still the leading cause of mortality worldwide (Kalayinia et al., 2021) (Chen et al., 2020). Hence, an in-depth molecular mechanistic understanding of CVDs is of great significance.
Recent studies reported that the primary mechanisms underlying the pathology of CVDs are closely relevant to metabolic perturbations (Yurista et al., 2021) (Ussher et al., 2016). As it is well-known, chronic obesity was a pathogenic factor of metabolic imbalance leading to CVDs (Lopaschuk et al., 2007). Obesity causes remarkable changes in cardiac energy metabolism, and the prominent effect is increasing the fatty acid uptake and oxidation by the heart (Lopaschuk et al., 2007). Moreover, fat accumulation can directly contribute to CVDs through reduced insulin sensitivity, impaired insulin production, and decreased glucose uptake in various tissues (Peters et al., 2017) (Wang et al., 2019) (Becker et al., 2017). Reactive oxygen species (ROS) were another potential pathogenic factor for CVDs, and the pathogenic mechanism is associated with metabolic disorders (Roth et al., 2017) (Geesala et al., 2020). As a by-product of cell respiration, ROS result from the metabolism of oxygen and are continuously produced in all aerobic organisms (Wang et al., 2020a). Increased ROS levels can lead to decreased availability of nitric oxide and vasoconstriction, subsequently promoting arterial hypertension (Senoner and Dichtl, 2019). ROS also negatively affect myocardial calcium treatment (Wang et al., 2020a), inducing arrhythmias and cardiac remodeling by facilitating hypertrophic signal transduction and apoptosis (Senoner and Dichtl, 2019). ROS also promote atherosclerotic plaque formation (Bertero and Maack, 2018). Even in the absence of obesity and dynamics of ROS, alterations in substrate metabolism of numerous organs resulting from the onset of CVDs can contribute to metabolic impairments in patients (Ussher et al., 2016). At the same time, CVDs can change the body and myocardial metabolism, usually accompanying the worsening of cardiac function and health outcomes (Ussher et al., 2016). From another perspective, the heart is the organ with the highest energy expenditure and oxidative demand (Bertero and Maack, 2018). The perturbations in cardiac energy metabolism, hence, could be significant contributors to CVDs.
As described earlier, exploring the effects of metabolic interventions on the improvement of cardiac dysfunction will provide a new direction for the treatments of CVDs. β-hydroxybutyrate, the most prominent ketone body, was once deemed to be a harmful and toxic substance, leading to ketoacidosis in patients with diabetes (Møller, 2020). Until now, increasing experimental and clinical research evidences have revealed the therapeutic potentials of β-HB in CVDs (Han et al., 2020). Traditionally, the concept of β-HB is the energy metabolic substrate, representing an alternative fuel source, for oxidative tissues, including the brain, heart, and skeletal muscle, in starvation and carbohydrate shortage (Evans et al., 2017; Monsalves-Alvarez et al., 2020; Wang et al., 2021). Moreover, β-HB is widely linked to various cellular processes by regulating gene transcription (Han et al., 2018), inflammation and oxidative stress (Wang et al., 2020a), cardiac remodeling (Sultan, 1990), and cardiovascular risk factors (Cotter et al., 2013). The roles played by both the metabolic substrate and signal molecule potentially allow β-HB to be used to treat cardiac dysfunctions and other vascular diseases. This study reviews the research progress of the metabolic and signaling effects of β-HB and provides a theoretical basis for exploiting the potentials of β-HB in CVDs therapies.
The Biological Synthesis and Utilization of β -Hydroxybutyrate
β-HB, a chiral molecule with two enantiomers (R/D and S/L), is the most abundant ketone body in mammals, significantly contributing to the dynamic range of ketone body levels (Evans et al., 2017) (Takahashi et al., 2019). The synthesis of β-HB occurs mainly in the liver mitochondrial matrix with a series of enzymes (Mierziak et al., 2021). BDH1, a core enzyme, which catalyzes the final step in the β-HB synthesis, can introduce chiral specificity (Lincoln et al., 1987). Due to the BDH-induced chiral specificity, only R-3-β-HB is produced by normal metabolism and then is readily catabolized into acetyl-CoA and ATP (Newman and Verdin, 2017). Fasting, exercise, caloric restriction, ketogenic diet, and other approaches resulting in endogenous production of β-HB can produce R-3-β-HB rather than S-3-β-HB (Mierziak et al., 2021) (Newman and Verdin, 2017). Thus, β-HB in this review article is mainly referring to R-3-β-HB.
As is well known, the synthesis of β-HB (Figure 1A) begins with the condensation of two acetyl-CoA molecules to form acetoacetyl-CoA in a reaction catalyzed by beta-ketothiolase (Mierziak et al., 2021). The well-established regulation of β-HB synthesis primarily depends on the substrate availability in the form of fatty acids and the expression and activity of the enzyme HMG-CoA synthase (HMGCS2; EC 2.3.3.10) (Newman and Verdin, 2017) (Garber et al., 1974). Robust expression of HMGCS2 is restricted to hepatocytes and colonic epithelium (Chen et al., 2017). The HMGCS2 enzyme catalyzes a fate-committing ketogenic reaction: condensation of β-oxidation–derived acetoacetyl-CoA (AcAc-CoA) and acetyl-CoA to generate HMG-CoA, which is then cleaved by HMGCL to generate acetoacetic acid (AcAc) (Puchalska and Crawford, 2017). AcAc is reduced to β-HB in an NAD-/NADH-coupled near-equilibrium reaction, which is catalyzed by phosphatidylcholine-dependent mitochondrial BDH1 (Cotter et al., 2013). BDH1 modulates mitochondrial redox in the liver and extrahepatic tissues, in which the ratio of AcAc to β-HB is directly proportional to that of mitochondrial NAD + to NADH (Williamson et al., 1967). However, molecular mechanisms precisely controlling the ketogenic rate remain to be addressed in detail (Puchalska and Crawford, 2017).
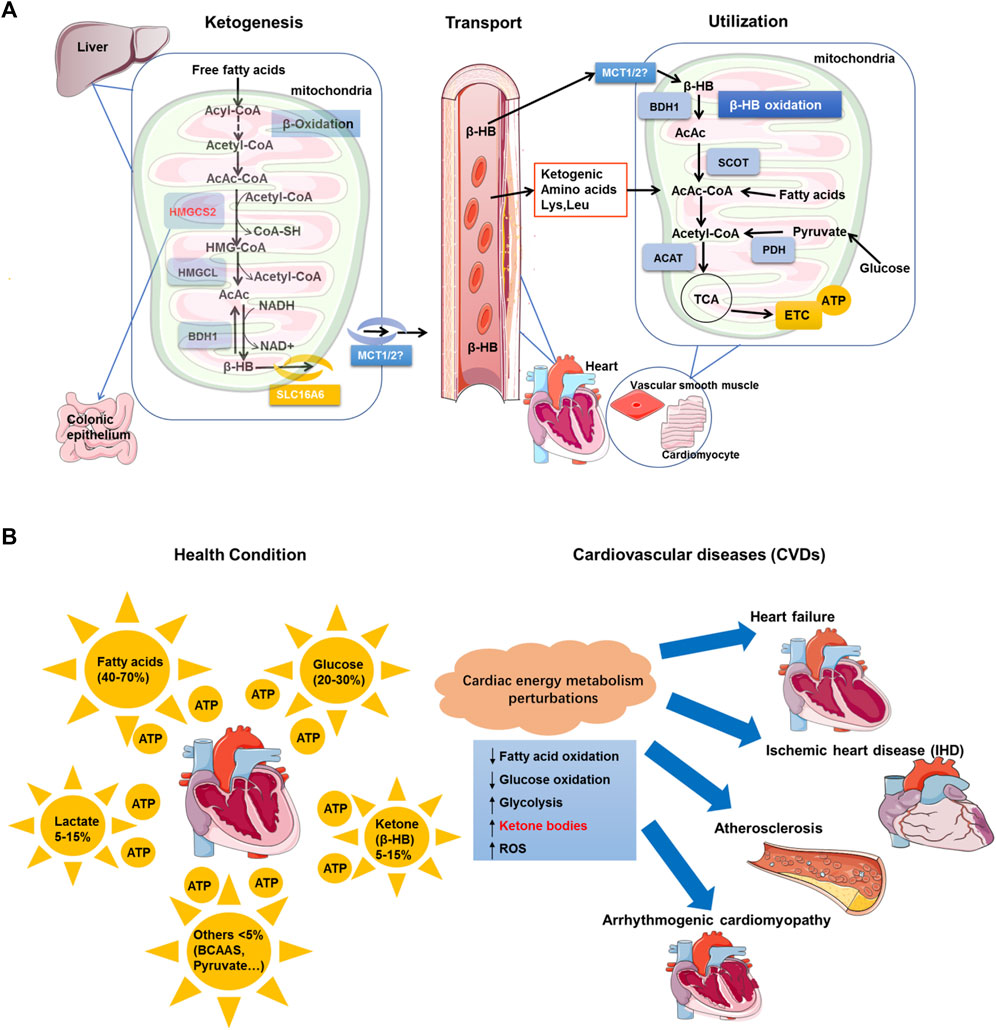
FIGURE 1. Perturbations of cardiac energy metabolism in CVDs and β-HB utilization in cardiomyocytes. (A). Biological synthesis of β-HB mainly occurs in hepatic mitochondria, where the fate-committing enzyme HMGCS2 is expressed. β-HB is synthesized from acetyl-CoA that is derived from β-oxidation. β-HB is transported through the circulatory system. After being transported, β-HB can be oxidized in extrahepatic organs, including the heart and vascular smooth muscle. Most extrahepatic mitochondria lack HMGCS2, while they have abundant enzymes for β-HB utilization, including BDH1, SCOT, and ACAT. In cardiomyocytes, β-HB can generate acetyl-CoA to enter the TCA cycle and electron transport chain (ETC), finally producing ATP. (B). In healthy conditions, fatty acids, glucose, lactate, ketone bodies (principally, β-HB), and other metabolites are substrates for cardiac energy metabolism. The perturbations in cardiac energy metabolism are significant contributors to cardiovascular pathologies, including heart failure, ischemic heart disease (IHD), atherosclerosis, and arrhythmogenic cardiomyopathy. In the case of CVDs, the myocardial fatty acid oxidation rates and glucose oxidation alterations are decreased. On the opposite, the level of myocardial glycolysis, ketone bodies, and ROS is observably increased.
Although the synthesis of β-HB occurs mainly in the liver, its utilization occurs mainly in extrahepatic tissues (Cotter et al., 2014a) (Cotter et al., 2014b). That is because liver cells have a strong enzyme system for synthesizing β-HB but lack the enzyme systems for utilizing β-HB (Puchalska and Crawford, 2017). Nonetheless, the extrahepatic tissues, such as the brain, myocardium, and skeletal muscle have abundant and efficient ketone body–decomposing enzymes, which can break down ketone bodies to regenerate acetyl CoA. Thereafter, acetyl CoA is oxidized for supplying energy (Williamson et al., 1967). In addition, as a polar molecule, β-HB is readily soluble in water and blood. In the blood, β-HB can be transported to extrahepatic tissues, where they primarily undergo terminal oxidation to contribute to the TCA cycle (Cotter et al., 2013). The circulating β-HB level is always related to physiological and pathological conditions. In general, the β-HB level greater than 0.5 mM has been considered a cut-off point for entry into ketosis (Chu et al., 2021). In pathological conditions, such as diabetes, the serum β-HB level can be elevated to as high as 20 mM. In neonates, however, upon fasting, prolonged exercise, or following a ketogenic diet, the serum β-HB level can be increased up to 1–8 mM (Adal et al., 2006) (Harvey et al., 2019). It was previously reported that the β-HB levels in patients with congestive heart failure (CHF) were increased to about 2.67 mM, and the increase of β-HB was in proportion to the severity of cardiac dysfunction (Lommi et al., 1996). Although the increased circulating β-hydroxybutyrate has been reported in a variety of CVDs since the 1990s (Lommi et al., 1996), it took a long time to clarify the mechanism behind this phenomenon.
Previous studies have shown that monocarboxylic acid transporters (MCTs) can transport β-HB out of the mitochondrial membrane and liver cell plasma membrane (Puchalska and Crawford, 2017) (Abdul Kadir et al., 2020). However, the transport of β-HB is less understood, relative to the synthesis and utilization of β-HB. The monocarboxylate transporter SLC16A6 has been identified to be a key transporter for exporting β-HB from the liver (Hugo et al., 2012). Many transporters are present in the cells; however, a small number of them have been characterized. Whether other transporters facilitate either the uptake of β-HB into target tissues or its intracellular movement needs further research. β-HB can also be transported into cardiomyocytes via monocarboxylate transporter 1 (MCT1) and MCT2 and then enter the mitochondria (Puchalska and Crawford, 2017). The uptake and utilization of ketone bodies by cardiomyocytes is a complex process of synergistic action of multiple enzymes (Abdul Kadir et al., 2020), and we will cover this in detail in the following sections.
β -Hydroxybutyrate in Cardiac Energy Metabolism and its Metabolic Effects on Cardiovascular Disease
Given that the heart is the organ with the highest energy expenditure and oxidative demand (Chandler et al., 2004), it can be expected that perturbations in cardiac energy metabolism are significant contributors to the progression of CVDs (Ussher et al., 2016) (Song et al., 2020). In addition to glucose and fatty acids, the heart also oxidizes various substrates, including β-HB, lactate, and amino acids (Kolwicz et al., 2016). Moreover, it has been demonstrated that β-HB can serve as an energy source in the absence of sufficient blood glucose, which is of particular importance during starvation or illness (Puchalska and Crawford, 2017). Thus, the effects of β-hydroxybutyrate on cardiac energy metabolism and cardiovascular disease have interested many researchers.
Cardiac energy metabolism has been well-documented (Lopaschuk et al., 2007) (Abdul Kadir et al., 2020) (Abozguia et al., 2009) (Figure 1B). In healthy conditions, metabolic flexibility is a crucial feature of cardiac energy metabolism (Maack et al., 2018). A healthy heart can derive energy from various circulating substrates, including fatty acids, glucose, amino acids, ketone bodies, and lactate. Fatty acid metabolites contribute to the main ATP production of the heart (>60%) (Qian and Wang, 2020). Interestingly, compared to fatty acids, glucose is less consumed (Lopaschuk et al., 2007) (Murashige et al., 2020). Glycolysis is responsible for only about 5% of the ATP production in the normal oxygenated heart (Abozguia et al., 2009), although low availability, lactate, ketone bodies (prominent, β-HB), and amino acids also contribute to the ATP production of the heart (Abozguia et al., 2009). As indicated by the previous works, the myocardium displays the highest β-HB consumption and oxidizes ketone bodies in proportion to prevailing concentrations at the cost of glucose and fatty acids (Veech, 2004). Furthermore, the metabolic flexibility of a healthy heart is highly dynamic, as demonstrated by its ability to rapidly change the pattern of fuel utilization to adapt to the substrate and hormonal environment (Ussher et al., 2016).
On the contrary, the progression of CVDs is associated with loss of metabolic flexibility of cardiomyocytes (Yurista et al., 2021). The energy deficit is common in cardiomyocytes, and these metabolic alterations depend on the stages of disease pathophysiology (Chandler et al., 2004). Even in the early stages, the transition of energy substrate utilization from fatty acids to glucose also occurs in the structural heart disease (Sack et al., 1996). Moreover, the altered utilization of the energy substrate also plays a key role in the progression of heart failure (HF) (Maack et al., 2018) (Stanley et al., 2005). Intriguingly, in the context of reduced fatty acid oxidation, the failing heart appears to reprogram metabolism to increase reliance on ketone bodies acting as a fuel source (Aubert et al., 2016; Bedi et al., 2016; Horton et al., 2019). In the pathologically remodeled heart (e.g., hypertension or myocardial infarction) and the diabetic heart, the O2 consumed for ATP production during ketone metabolism is more efficient than FAO (Bertero and Maack, 2018). Compared to FAO, ketone body oxidation is energetically efficient, yielding more energy for ATP synthesis per molecule of oxygen invested (the ratio of phosphate/oxygen [P/O]) (Sato et al., 1995) (Kashiwaya et al., 2010) (Veech, 2004). As is known, ischemic heart disease (IHD) remains the leading cause of cardiovascular death globally (GBD 2017 Causes of Death Collaborators, 2015). The myocardial intermediary energy metabolism is significantly altered in IHD (Ussher et al., 2016). In response to the downregulated oxidative metabolism, glycogen breakdown and glycolysis rates are increased, as glycolysis can produce ATP anerobically rapidly (Taegtmeyer et al., 2016). Glucose oxidation rates are also markedly decreased in the ischemic myocardium, whereas glycolytic rates are significantly increased due to the stimulation of glycogenolysis (Wisneski et al., 1987) (Vary et al., 1981). On the other hand, several works reported minimal reliance of a healthy heart on amino acids acting as a source of ATP (Monsalves-Alvarez et al., 2020). Regardless of this, it has been suggested that amino acid metabolism in the heart may also be vital during ischemia (Drake et al., 2012). Atherosclerosis also represents another common form of CVDs. The presence of atherosclerotic plaques in the coronary vessels can trigger a vast majority of IHD. However, whether atherosclerosis can cause changes in myocardial energy metabolism, in general, remains poorly understood (Ussher et al., 2016).
The ketogenic shift seems to be a universal cardiac response to stress. β-HB acting as a fuel is particularly significant in the hypertrophied and failing heart (Birkenfeld et al., 2019). Moreover, in the context of ischemia or reperfusion injury, β-HB also confers potential cardioprotective effects (Wang et al., 2008) (Al-Zaid et al., 2007), possibly due to either the increased mitochondrial abundance in the heart or the upregulation of crucial oxidative phosphorylation mediators (Snorek et al., 2012). It is reported that plasma β-HB and its cardiac utilization increased in patients with arrhythmogenic cardiomyopathy (Song et al., 2020). A series of CVDs with heart failure increase the reliance on ketone bodies for cardiac ATP production and are accompanied by increased circulating β-HB levels in the blood (Lommi et al., 1996) (Abdul Kadir et al., 2020). Previous studies reveal that β-HB competes with other substrates in the heart as the fuel when the availability of fatty acid and carbohydrate is limited (Kolwicz et al., 2016; Abdul Kadir et al., 2020). In the animal model, studies indicate that fatty acid utilization is downregulated and ketone body utilization is upregulated in failing hearts of mice (Aubert et al., 2016). Thus, the shift of energy metabolism to ketone body metabolism is an efficient alternative avenue for oxidative ATP production in CVDs.
Although preliminary interventional and observational studies indicate potential benefits of the metabolic effects of β-HB in the heart, an adverse viewpoint still exists. In cardiomyocytes, an early study reported that β-HB causes concurrent inhibition of glucose metabolism and FAO metabolism, thereby impairing myocardial energy supply (Taegtmeyer, 1994). Nevertheless, recent studies directly measured cardiac β-HB, glucose, and FAO metabolism in mouse models, and the findings are not consistent with those of the previous study (McCommis et al., 2020) (Brahma et al., 2020). Furthermore, increased β-HB levels do not always correlate with positive clinical outcomes in humans. Arrhythmogenic cardiomyopathy (AC) is a severe disease that may cause sudden death, lacking clinical biomarkers. A recent study suggested that the elevated plasma level of β-HB might be a potential predictor of major adverse cardiovascular events (MACEs) and disease progression in patients with AC and their clinically asymptomatic relatives (Song et al., 2020). It was also reported that compared with normal controls, the atrial samples from patients with atrial fibrillation exhibited increased levels of ketone bodies (Xie et al., 2016). The metabolic effects of the increased level of circulating β-HB are not fully understood yet, and more research needs to be performed.
Signaling Effects of β -Hydroxybutyrate on Cardiovascular Disease
In addition to the metabolic effects, β-HB is also a signaling metabolite that regulates cellular signals by targeting diverse biomolecules (Newman and Verdin, 2017) (Puchalska and Crawford, 2017). The primary signaling actions of β-HB (Figure 2) are closely related to cardiovascular diseases, including binding to cell-surface receptors; inhibiting histone deacetylases (HADCs); acting as a substrate for protein posttranslational modification; and modulating potassium flux across the plasma membrane (Mierziak et al., 2021) (Newman and Verdin, 2017).
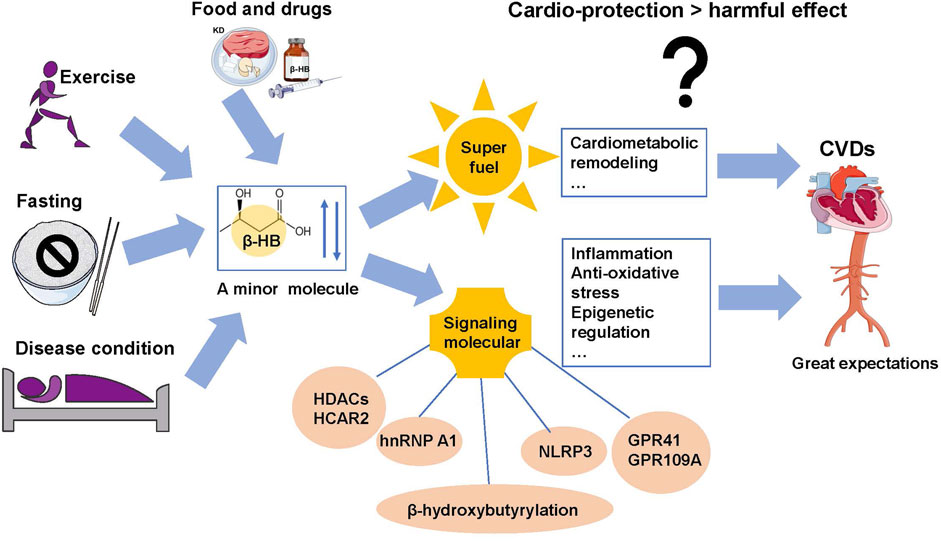
FIGURE 2. Summary of regulation of β-HB levels and its multi-effects on CVDs. Fasting, exercise, ketogenic diet, drugs, and some disease conditions change the endogenous β-HB levels. Beyond their contribution to energy generation, β-HB may exert signaling effects on inflammation, oxidative stress, cell death, and cardiac remodeling that may induce either the prospective cardiovascular protection or harmful effect on CVDs.
As is known, β-HB serves as an endogenous ligand for G protein–coupled receptors (GPRs) (Chu et al., 2021). β-HB can specifically bind to HCAR2 and activate HCAR2 to inhibit lipolysis of adipocytes, which might represent a feedback mechanism for regulating the availability of the fatty acid precursors of ketone body metabolism (Offermanns et al., 2011) (Taggart et al., 2005). To be more specific, HCAR2 activation in neurons can potentiate glutaminergic signaling that helps regulate blood pressure and heart rate (Rezq and Abdel-Rahman, 2016). In addition, β-HB also functions as a ligand for FFAR3 (also known as GPR41) which is another G protein–coupled receptor highly expressed in sympathetic ganglions throughout the body of mice (Nøhr et al., 2015) (Kimura et al., 2011). It was reported that β-HB can suppress sympathetic tone and heart rate through antagonistic action against FFAR3 during fasting (Oshima et al., 2019). Interestingly, it was also reported that β-HB acts as an agonist of FFAR3 and regulates voltage-dependent calcium channels (Won et al., 2013). Further works are required to confirm the regulatory effects of β-HB on FFAR3 functions. Furthermore, it is noteworthy that heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) was a new direct binding target of β-HB. Through hnRNP A1–mediated upregulation of Oct4, β-HB can prevent vascular senescence (Han et al., 2018).
β-HB also plays essential signaling roles in CVDs via inhibiting histone deacetylases (HADCs). HDACs play essential roles in regulating mitochondrial metabolism and function by balancing the acetylation activities of histone acetyltransferases (HATs) (Felisbino and McKinsey, 2018). It was previously reported that in many cardiac pathological conditions, such as heart failure, diabetic heart, and myocardial I/R injury, HDAC activities are significantly elevated (Granger et al., 2008; Aune et al., 2014). Inhibition of HDAC activities, hence, is an effective treatment for cardiac I/R injury and failing heart. As an endogenous inhibitor of Class I HDACs, β-HB possesses significant cardioprotective effects (Hasselbalch et al., 1996). By inhibiting HDAC activities, β-HB upregulates expressions of the Foxo3a and MT2 genes to reduce oxidative stress (Shimazu et al., 2013). In addition, β-HB also has inhibitory activity against HDAC1, which may be related to cardiomyocyte autophagy (Cao et al., 2011; Shimazu et al., 2013). However, whether the cardioprotective effects of β-HB are mediated by autophagy is still unclear. When β-HB was used to inhibit HDAC6, it did not show positive benefit and was even detrimental to cardiomyocytes during I/R injury (Aune et al., 2014). Furthermore, β-HB accumulation induced by a long-term ketogenic diet can inhibit HDAC2 and activate Sirt7 transcription, which is harmful to heart health by promoting cardiac fibrosis (Xu et al., 2021). Given the controversial results in cardio-protection, the complicated relationship between β-HB and HDACs remains to be addressed in detail.
On the other hand , the signaling roles of β-HB in the CVDs is linked to the inflammation. As is known, the inflammatory mechanisms in CVDs are closely associated with NLRP3, which has a vital role in innate immunity and inflammation (Wang et al., 2020b). Significantly, β-HB can function as an endogenous inhibitor of the NLRP3 inflammasome, attenuating inflammatory responses (Yamanashi et al., 2017). It was revealed that inhibition of potassium efflux contributes to the mechanism of β-HB, inhibiting the activation of the NLRP3 inflammasome (Kimura et al., 2011). Mice deficient in NLRP3 are protected from obesity and insulin resistance when fed on a high-fat diet (Yudkoff et al., 2007). The ablation of NLRP3 also has attenuated diabetes and atherosclerosis (Youm et al., 2015) (Duewell et al., 2010). Increasing the circulating β-HB levels can reduce the activation of the cardiac NLRP3 inflammasome in mice with heart failure (Byrne et al., 2020).
In addition, the signaling roles of β-HB in the CVDs also involved antioxidative stress and epigenetic regulation. ROS has negative effects on myocardial calcium treatment, inducing arrhythmias and cardiac remodeling by facilitating hypertrophic signal transduction and apoptosis (Senoner and Dichtl, 2019) (Huynh and Heo, 2019). Oxidation of ketone bodies may also curtail ROS production (Paoli, 2014). Administration of β-HB in mice can prevent liver ROS (Michal et al., 2015). Interestingly, under hypoxia conditions, reducing the serum level of β-HB can improve the excitation–contraction of cardiac cells (Klos et al., 2019). In addition, metabolism-mediated epigenetic changes represent an adapted mechanism for cellular signaling, which has been the historical focus of interest. β-HB can dramatically increase lysine β-hydroxybutyrylation of histone tails, which is an epigenetic marker associated with fasting responses and muscle catabolic states (Cavallucci and Pani, 2021). However, the regulatory mechanism underlying the β-hydroxybutyrate–mediated epigenetic pathway remains unclear, hindering its clinical functional research. Further studies need to be performed. In addition to the several signal roles of β-HB mentioned previously, it is reported that increases in circulating β-HB can mediate CVDs benefiting from the drugs of sodium-glucose cotransporter inhibitors (SGLT2i) (Packer, 2020). However, the mechanisms underlying the cardiovascular benefits of SGLT2i remain elusive, which might be related to the signaling role of β-HB (Kaplan et al., 2018).
Future Perspectives
As summarized earlier, both the roles of the β-HB metabolism and the underlying mechanism are of great significance for the mechanistic understanding of the occurrence and development of pathological cardiac remodeling and for the guidance of clinical treatment. The minor metabolite, β-HB, has both advantages and disadvantages in CVDs. The current research reveals that the advantages of β-HB far outweigh the disadvantages (Sato et al., 1995) (Xu et al., 2021) (Nielsen et al., 2019) (Gormsen et al., 2017). Even though evidence for the beneficial effects of β-HB on cardiovascular disease is rapidly emerging, it is still unclear whether the role of β-HB is essentially adaptive or maladaptive in CVDS. In addition, it is also unclear what makes β-HB a double-edged sword in treating cardiovascular disease. Both the optimal timing and application strategy of β-HB for CVDs treatments are worthy of further exploration.
Author Contributions
SW, LB, WY, ZZ, LD and HC: writing—editing—review and proofreading. All authors listed have made substantial, direct, and intellectual contributions to this work and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31971357) and the National Natural Science Foundation of Fujian Province (No. 2020D022).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AcAc, acetoacetic acid; AC, arrhythmogenic cardiomyopathy; ACAT, acyl-coenzyme a-cholesterol acyltransferase; BDH, beta-hydroxybutyrate dehydrogenases; CVDs, cardiovascular diseases; ETC, electron transport chain; FAO, fatty acid oxidation; FFAR3, free fatty acid receptors 3; GPRs, G protein–coupled receptors; HDAC, histone deacetylase; HF, heart failure; I/R, ischemia/reperfusion; IHD, ischemic heart disease; KD, ketogenic diet; MACEs, major adverse cardiovascular events; MCT, monocarboxylate transporters; NLRP3, nucleotide-binding domain-like receptor protein 3; ROS, reactive oxygen species; SCOT, succinyl-CoA:3-ketoacid-CoA transferase; SGLT2i, sodium–glucose co-transporter 2 inhibitors; β-HB, beta-hydroxybutyrate.
References
Abdul Kadir, A., Clarke, K., and Evans, R. D. (2020). Cardiac Ketone Body Metabolism. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1866 (6), 165739. [published Online First: 2020/02/23]. doi:10.1016/j.bbadis.2020.165739
Abozguia, K., Shivu, G., Ahmed, I., Phan, T., and Frenneaux, M. (2009). The Heart Metabolism: Pathophysiological Aspects in Ischaemia and Heart Failure. Cpd 15 (8), 827–835. [published Online First: 2009/03/12]. doi:10.2174/138161209787582101
Adal, E., Koyuncu, G., Aydın, A., Çelebi, A., Kavunoǧlu, G., and Çam, H. (2006). Asymptomatic Cardiomyopathy in Children and Adolescents with Type 1 Diabetes Mellitus: Association of Echocardiography Indicators with Duration of Diabetes Mellitus and Metabolic Parameters. J. Pediatr. Endocr. Met. 19 (5), 713–726. doi:10.1515/jpem.2006.19.5.713
Al-Zaid, N. S., Dashti, H. M., Mathew, T. C., and Juggi, J. S. (2007). Low Carbohydrate Ketogenic Diet Enhances Cardiac Tolerance to Global Ischaemia. Acta Cardiol. 62 (4), 381–389. doi:10.2143/ac.62.4.2022282
Aubert, G., Martin, O. J., Horton, J. L., Lai, L., Vega, R. B., Leone, T. C., et al. (2016). The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 133 (8), 698–705. [published Online First: 2016/01/29]. doi:10.1161/CIRCULATIONAHA.115.017355
Aune, S. E., Herr, D. J., Mani, S. K., and Menick, D. R. (2014). Selective Inhibition of Class I but Not Class IIb Histone Deacetylases Exerts Cardiac Protection from Ischemia Reperfusion. J. Mol. Cell. Cardiol. 72, 138–145. doi:10.1016/j.yjmcc.2014.03.005
Becker, M., Levings, M. K., and Daniel, C. (2017). Adipose-tissue Regulatory T Cells: Critical Players in Adipose-Immune Crosstalk. Eur. J. Immunol. 47 (11), 1867–1874. [published Online First: 2017/08/30]. doi:10.1002/eji.201646739
Bedi, K. C., Snyder, N. W., Brandimarto, J., Aziz, M., Mesaros, C., Worth, A. J., et al. (2016). Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 133 (8), 706–716. doi:10.1161/circulationaha.115.017545
Bertero, E., and Maack, C. (2018). Metabolic Remodelling in Heart Failure. Nat. Rev. Cardiol. 15 (8), 457–470. [published Online First: 2018/06/20]. doi:10.1038/s41569-018-0044-6
Birkenfeld, A. L., Jordan, J., Dworak, M., Merkel, T., and Burnstock, G. (2019). Myocardial Metabolism in Heart Failure: Purinergic Signalling and Other Metabolic Concepts. Pharmacol. Ther. 194, 132–144. doi:10.1016/j.pharmthera.2018.08.015
Brahma, M. K., Ha, C. M., Pepin, M. E., Mia, S., Sun, Z., Chatham, J. C., et al. (2020). Increased Glucose Availability Attenuates Myocardial Ketone Body Utilization. Jaha 9 (15), e013039. [published Online First: 2020/08/05]. doi:10.1161/JAHA.119.013039
Byrne, N. J., Matsumura, N., Maayah, Z. H., Ferdaoussi, M., Takahara, S., Darwesh, A. M., et al. (2020). Empagliflozin Blunts Worsening Cardiac Dysfunction Associated with Reduced NLRP3 (Nucleotide-Binding Domain-like Receptor Protein 3) Inflammasome Activation in Heart Failure. Circ. Heart Fail 13 (1), e006277. doi:10.1161/CIRCHEARTFAILURE.119.006277
Cao, D. J., Wang, Z. V., Battiprolu, P. K., Jiang, N., Morales, C. R., Kong, Y., et al. (2011). Histone Deacetylase (HDAC) Inhibitors Attenuate Cardiac Hypertrophy by Suppressing Autophagy. Proc. Natl. Acad. Sci. U.S.A. 108 (10), 4123–4128. [published Online First: 2011/03/04]. doi:10.1073/pnas.1015081108
Cavallucci, V., and Pani, G. (2021). The Leucine Catabolite and Dietary Supplement β-Hydroxy-β-Methyl Butyrate (HMB) as an Epigenetic Regulator in Muscle Progenitor Cells. Metabolites 11 (8), 512. [published Online First: 2021/08/27]. doi:10.3390/metabo11080512
Chandler, M. P., Kerner, J., Huang, H., Vazquez, E., Reszko, A., Martini, W. Z., et al. (2004). Moderate Severity Heart Failure Does Not Involve a Downregulation of Myocardial Fatty Acid Oxidation. Am. J. Physiology-Heart Circulatory Physiology 287 (4), H1538–H1543. doi:10.1152/ajpheart.00281.2004
Chen, S.-W., Chou, C.-T., Chang, C.-C., Li, Y.-J., Chen, S.-T., Lin, I.-C., et al. (2017). HMGCS2 Enhances Invasion and Metastasis via Direct Interaction with PPARα to Activate Src Signaling in Colorectal Cancer and Oral Cancer. Oncotarget 8 (14), 22460–22476. doi:10.18632/oncotarget.13006
Chen, X.-F., Chen, X., and Tang, X. (2020). Short-chain Fatty Acid, Acylation and Cardiovascular Diseases. Clin. Sci. 134 (6), 657–676. doi:10.1042/Cs20200128
Chu, Y., Zhang, C., and Xie, M. (2021). Beta-hydroxybutyrate, Friend or Foe for Stressed Hearts. Front. Aging 2, 16. doi:10.3389/fragi.2021.681513
Cotter, D. G., Ercal, B., André d'Avignon, D., Dietzen, D. J., and Crawford, P. A. (2014). Impairments of Hepatic Gluconeogenesis and Ketogenesis in PPARα-Deficient Neonatal Mice. Am. J. Physiology-Endocrinology Metabolism 307 (2), E176–E185. doi:10.1152/ajpendo.00087.2014
Cotter, D. G., Ercal, B., Huang, X., Leid, J. M., d’Avignon, D. A., Graham, M. J., et al. (2014). Ketogenesis Prevents Diet-Induced Fatty Liver Injury and Hyperglycemia. J. Clin. Invest. 124 (12), 5175–5190. doi:10.1172/Jci76388
Cotter, D. G., Schugar, R. C., and Crawford, P. A. (2013). Ketone Body Metabolism and Cardiovascular Disease. Am. J. Physiology-Heart Circulatory Physiology 304 (8), H1060–H1076. doi:10.1152/ajpheart.00646.2012
Drake, K. J., Sidorov, V. Y., McGuinness, O. P., Wasserman, D. H., and Wikswo, J. P. (2012). Amino Acids as Metabolic Substrates during Cardiac Ischemia. Exp. Biol. Med. (Maywood) 237 (12), 1369–1378. doi:10.1258/ebm.2012.012025
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., et al. (2010). Erratum: NLRP3 Inflammasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature 466 (7306), 652. doi:10.1038/nature09316
Evans, M., Cogan, K. E., and Egan, B. (2017). Metabolism of Ketone Bodies during Exercise and Training: Physiological Basis for Exogenous Supplementation. J. Physiol. 595 (9), 2857–2871. [published Online First: 2016/11/20]. doi:10.1113/JP273185
Felisbino, M. B., and McKinsey, T. A. (2018). Epigenetics in Cardiac Fibrosis. JACC Basic Transl. Sci. 3 (5), 704–715. doi:10.1016/j.jacbts.2018.05.003
Garber, A. J., Menzel, P. H., Boden, G., and Owen, O. E. (1974). Hepatic Ketogenesis and Gluconeogenesis in Humans. J. Clin. Invest. 54 (4), 981–989. [published Online First: 1974/10/01]. doi:10.1172/JCI107839
GBD 2017 Causes of Death Collaborators (2015). Global, Regional, and National Age-Sex Specific All-Cause and Cause-specific Mortality for 240 Causes of Death, 1990-2013: a Systematic Analysis for the Global Burden of Disease Study 2013. Lancet 385 (9963), 117–171. doi:10.1016/S0140-6736(14)61682-2
Geesala, R., Issuree, P. D., and Maretzky, T. (2020). The Role of iRhom2 in Metabolic and Cardiovascular-Related Disorders. Front. Cardiovasc. Med. 7, 612808. [published Online First: 2020/12/18]. doi:10.3389/fcvm.2020.612808
Gormsen, L. C., Svart, M., Thomsen, H. H., Søndergaard, E., Vendelbo, M. H., Christensen, N., et al. (2017). Ketone Body Infusion with 3‐Hydroxybutyrate Reduces Myocardial Glucose Uptake and Increases Blood Flow in Humans: A Positron Emission Tomography Study. Jaha 6 (3), e005066. [published Online First: 2017/03/01]. doi:10.1161/JAHA.116.005066
Granger, A., Abdullah, I., Huebner, F., Stout, A., Wang, T., Huebner, T., et al. (2008). Histone Deacetylase Inhibition Reduces Myocardial Ischemia-Reperfusion Injury in Mice. FASEB J. 22 (10), 3549–3560. doi:10.1096/fj.08-108548
Hajar, R. (2016). Framingham Contribution to Cardiovascular Disease. Heart Views 17 (2), 78–81. [published Online First: 2016/08/12]. doi:10.4103/1995-705X.185130
Han, Y.-m., Bedarida, T., Ding, Y., Somba, B. K., Lu, Q., Wang, Q., et al. (2018). β-Hydroxybutyrate Prevents Vascular Senescence through hnRNP A1-Mediated Upregulation of Oct4. Mol. Cell. 71 (6), 1064–1078. [published Online First: 2018/09/11]. doi:10.1016/j.molcel.2018.07.036
Han, Y.-M., Ramprasath, T., and Zou, M.-H. (2020). β-Hydroxybutyrate and its Metabolic Effects on Age-Associated Pathology. Exp. Mol. Med. 52 (4), 548–555. doi:10.1038/s12276-020-0415-z
Harvey, K. L., Holcomb, L. E., and Kolwicz, S. C. (2019). Ketogenic Diets and Exercise Performance. Nutrients 11 (10), 2296. doi:10.3390/nu11102296
Hasselbalch, S. G., Knudsen, G. M., Holm, S., Hageman, L. P., Capaldo, B., and Paulson, O. B. (1996). Transport of D-Glucose and 2-fluorodeoxyglucose across the Blood-Brain Barrier in Humans. J. Cereb. Blood Flow. Metab. 16 (4), 659–666. doi:10.1097/00004647-199607000-00017
Horton, J. L., Davidson, M. T., Kurishima, C., Vega, R. B., Powers, J. C., Matsuura, T. R., et al. (2019). The Failing Heart Utilizes 3-hydroxybutyrate as a Metabolic Stress Defense. Jci Insight 4 (4), e124079. doi:10.1172/jci.insight.124079
Hugo, S. E., Cruz-Garcia, L., Karanth, S., Anderson, R. M., Stainier, D. Y. R., and Schlegel, A. (2012). A Monocarboxylate Transporter Required for Hepatocyte Secretion of Ketone Bodies during Fasting. Genes. Dev. 26 (3), 282–293. doi:10.1101/gad.180968.111
Huynh, D. T. N., and Heo, K.-S. (2019). Therapeutic Targets for Endothelial Dysfunction in Vascular Diseases. Arch. Pharm. Res. 42 (10), 848–861. [published Online First: 2019/08/20]. doi:10.1007/s12272-019-01180-7
Kalayinia, S., Arjmand, F., Maleki, M., Malakootian, M., and Singh, C. P. (2021). MicroRNAs: Roles in Cardiovascular Development and Disease. Cardiovasc. Pathol. 50, 107296. doi:10.1016/j.carpath.2020.107296
Kalayinia, S., Goodarzynejad, H., Maleki, M., and Mahdieh, N. (2018). Next Generation Sequencing Applications for Cardiovascular Disease. Ann. Med. 50 (2), 91–109. [published Online First: 2017/10/14]. doi:10.1080/07853890.2017.1392595
Kaplan, A., Abidi, E., El-Yazbi, A., Eid, A., Booz, G. W., and Zouein, F. A. (2018). Direct Cardiovascular Impact of SGLT2 Inhibitors: Mechanisms and Effects. Heart Fail Rev. 23 (3), 419–437. doi:10.1007/s10741-017-9665-9
Kashiwaya, Y., Pawlosky, R., Markis, W., King, M. T., Bergman, C., Srivastava, S., et al. (2010). A Ketone Ester Diet Increases Brain Malonyl-CoA and Uncoupling Proteins 4 and 5 while Decreasing Food Intake in the Normal Wistar Rat. J. Biol. Chem. 285 (34), 25950–25956. doi:10.1074/jbc.m110.138198
Kimura, I., Inoue, D., Maeda, T., Hara, T., Ichimura, A., Miyauchi, S., et al. (2011). Short-chain Fatty Acids and Ketones Directly Regulate Sympathetic Nervous System via G Protein-Coupled Receptor 41 (GPR41). Proc. Natl. Acad. Sci. U.S.A. 108 (19), 8030–8035. [published Online First: 2011/04/27]. doi:10.1073/pnas.1016088108
Klos, M., Morgenstern, S., Hicks, K., Suresh, S., and Devaney, E. J. (2019). The Effects of the Ketone Body β-hydroxybutyrate on Isolated Rat Ventricular Myocyte Excitation-Contraction Coupling. Archives Biochem. biophysics 662, 143–150. doi:10.1016/j.abb.2018.11.027
Kolwicz, S. C., Airhart, S., and Tian, R. (2016). Ketones Step to the Plate: a Game Changer for Metabolic Remodeling in Heart Failure? Am. Heart Assoc. 133, 689–691. doi:10.1161/CIRCULATIONAHA.116.021230
Lincoln, B. C., Rosiers, C. D., and Brunengraber, H. (1987). Metabolism of S-3-Hydroxybutyrate in the Perfused Rat Liver. Archives Biochem. Biophysics 259 (1), 149–156. [published Online First: 1987/11/15]. doi:10.1016/0003-9861(87)90480-2
Lommi, J., Kupari, M., Koskinen, P., Näveri, H., Leinonen, H., Pulkki, K., et al. (1996). Blood Ketone Bodies in Congestive Heart Failure. J. Am. Coll. Cardiol. 28 (3), 665–672. doi:10.1016/0735-1097(96)00214-8
Lopaschuk, G. D., Folmes, C. D. L., and Stanley, W. C. (2007). Cardiac Energy Metabolism in Obesity. Circulation Res. 101 (4), 335–347. [published Online First: 2007/08/19]. doi:10.1161/CIRCRESAHA.107.150417
Maack, C., Lehrke, M., Backs, J., Heinzel, F. R., Hulot, J.-S., Marx, N., et al. (2018). Heart Failure and Diabetes: Metabolic Alterations and Therapeutic Interventions: a State-Of-The-Art Review from the Translational Research Committee of the Heart Failure Association-European Society of Cardiology. Eur. Heart J. 39 (48), 4243–4254. doi:10.1093/eurheartj/ehy596
McCommis, K. S., Kovacs, A., Weinheimer, C. J., Shew, T. M., Koves, T. R., Ilkayeva, O. R., et al. (2020). Nutritional Modulation of Heart Failure in Mitochondrial Pyruvate Carrier-Deficient Mice. Nat. Metab. 2 (11), 1232–1247. [published Online First: 2020/10/28]. doi:10.1038/s42255-020-00296-1
Michal, P., Eric, B., Fanny, L., Lefebvre, P., and Staels, B. (2015). Ketone Body Therapy Protects from Lipotoxicity and Acute Liver Failure upon Pparα Deficiency. Mol. Endocrinol. 29 (8), 1134–1143. doi:10.1210/me.2014-1383
Mierziak, J., Burgberger, M., and Wojtasik, W. (2021). 3-Hydroxybutyrate as a Metabolite and a Signal Molecule Regulating Processes of Living Organisms. Biomolecules 11 (3), 402. doi:10.3390/biom11030402
Møller, N. (2020). Ketone Body, 3-Hydroxybutyrate: Minor Metabolite - Major Medical Manifestations. J. Clin. Endocrinol. Metab. 105 (9), 2884–2892. [published Online First: 2020/06/12]. doi:10.1210/clinem/dgaa370
Monsalves-Alvarez, M., Morales, P. E., Castro-Sepulveda, M., Sepulveda, C., Rodriguez, J. M., Chiong, M., et al. (2020). β-Hydroxybutyrate Increases Exercise Capacity Associated with Changes in Mitochondrial Function in Skeletal Muscle. Nutrients 12 (7), 1930. [published Online First: 2020/07/03]. doi:10.3390/nu12071930
Murashige, D., Jang, C., Neinast, M., Edwards, J. J., Cowan, A., Hyman, M. C., et al. (2020). Comprehensive Quantification of Fuel Use by the Failing and Nonfailing Human Heart. Science 370 (6514), 364–368. doi:10.1126/science.abc8861
Newman, J. C., and Verdin, E. (2017). β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 37, 51–76. [published Online First: 2017/08/23]. doi:10.1146/annurev-nutr-071816-064916
Nielsen, R., Møller, N., Gormsen, L. C., Tolbod, L. P., Hansson, N. H., Sorensen, J., et al. (2019). Cardiovascular Effects of Treatment with the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 139 (18), 2129–2141. doi:10.1161/Circulationaha.118.036459
Nøhr, M. K., Egerod, K. L., Christiansen, S. H., Gille, A., Offermanns, S., Schwartz, T. W., et al. (2015). Expression of the Short Chain Fatty Acid Receptor GPR41/FFAR3 in Autonomic and Somatic Sensory Ganglia. Neuroscience 290, 126–137. [published Online First: 2015/02/01]. doi:10.1016/j.neuroscience.2015.01.040
Offermanns, S., Colletti, S. L., Lovenberg, T. W., Semple, G., Wise, A., and IJzerman, A. P. (2011). International Union of Basic and Clinical Pharmacology. LII: Nomenclature and Classification of Hydroxy-Carboxylic Acid Receptors (GPR81, GPR109A, and GPR109B). Pharmacol. Rev. 63 (2), 269–290. [published Online First: 2011/04/02]. doi:10.1124/pr.110.003301
Oshima, H., Miki, T., Kuno, A., Mizuno, M., Sato, T., Tanno, M., et al. (2019). Empagliflozin, an SGLT2 Inhibitor, Reduced the Mortality Rate after Acute Myocardial Infarction with Modification of Cardiac Metabolomes and Antioxidants in Diabetic Rats. J. Pharmacol. Exp. Ther. 368 (3), 524–534. doi:10.1124/jpet.118.253666
Packer, M. (2020). Autophagy Stimulation and Intracellular Sodium Reduction as Mediators of the Cardioprotective Effect of Sodium-Glucose Cotransporter 2 Inhibitors. Eur. J. Heart Fail 22 (4), 618–628. doi:10.1002/ejhf.1732
Paoli, A. (2014). Ketogenic Diet for Obesity: Friend or Foe? Ijerph 11 (2), 2092–2107. doi:10.3390/ijerph110202092
Peters, H. P. F., Schrauwen, P., Verhoef, P., Byrne, C. D., Mela, D. J., Pfeiffer, A. F. H., et al. (2017). Liver Fat: a Relevant Target for Dietary Intervention? Summary of a Unilever Workshop. J. Nutr. Sci. 6, e15. [published Online First: 2017/06/21]. doi:10.1017/jns.2017.13
Puchalska, P., and Crawford, P. A. (2017). Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell. Metab. 25 (2), 262–284. doi:10.1016/j.cmet.2016.12.022
Qian, N., and Wang, Y. (2020). Ketone Body Metabolism in Diabetic and Non-diabetic Heart Failure. Heart Fail Rev. 25 (5), 817–822. doi:10.1007/s10741-019-09857-3
Rezq, S., and Abdel-Rahman, A. A. (2016). Central GPR109A Activation Mediates Glutamate-dependent Pressor Response in Conscious Rats. J. Pharmacol. Exp. Ther. 356 (2), 457–466. [published Online First: 2015/12/02]. doi:10.1124/jpet.115.229146
Roth, G. A., Johnson, C., Abajobir, A., Abd-Allah, F., Abera, S. F., Abyu, G., et al. (2017). Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 70 (1), 1–25. [published Online First: 2017/05/22]. doi:10.1016/j.jacc.2017.04.052
Sack, M. N., Rader, T. A., Park, S., Bastin, J., McCune, S. A., and Kelly, D. P. (1996). Fatty Acid Oxidation Enzyme Gene Expression Is Downregulated in the Failing Heart. Circulation 94 (11), 2837–2842. doi:10.1161/01.cir.94.11.2837
Sato, K., Kashiwaya, Y., Keon, C. A., Tsuchiya, N., King, M. T., Radda, G. K., et al. (1995). Insulin, Ketone Bodies, and Mitochondrial Energy. FASEB J. 9 (8), 651–658. doi:10.1096/fasebj.9.8.7768357
Senoner, T., and Dichtl, W. (2019). Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 11 (9), 2090. doi:10.3390/nu11092090
Shimazu, T., Hirschey, M. D., Newman, J., He, W., Shirakawa, K., Le Moan, N., et al. (2013). Suppression of Oxidative Stress by β-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 339 (6116), 211–214. [published Online First: 2012/12/12]. doi:10.1126/science.1227166
Snorek, M., Hodyc, D., Sedivý, V., Durišová, J., Skoumalová, A., Wilhelm, J., et al. (2012). Short-term Fasting Reduces the Extent of Myocardial Infarction and Incidence of Reperfusion Arrhythmias in Rats. Physiol. Res. 61 (6), 567–574. doi:10.33549/physiolres.932338
Song, J. P., Chen, L., Chen, X., Ren, J., Zhang, N. N., Tirasawasdichai, T., et al. (2020). Elevated Plasma β-hydroxybutyrate Predicts Adverse Outcomes and Disease Progression in Patients with Arrhythmogenic Cardiomyopathy. Sci. Transl. Med. 12 (530), eaay8329. doi:10.1126/scitranslmed.aay8329
Stanley, W. C., Recchia, F. A., and Lopaschuk, G. D. (2005). Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 85 (3), 1093–1129. doi:10.1152/physrev.00006.2004
Sultan, A. M. N. (1990). The Effect of Fasting on D-3-Hydroxybutyrate Metabolism in the Perfused Rat Heart. Mol. Cell. Biochem. 93 (2), 107–118. [published Online First: 1990/03/27]. doi:10.1007/BF00226182
Taegtmeyer, H. (1994). Energy Metabolism of the Heart: from Basic Concepts to Clinical Applications. Curr. Probl. Cardiol. 19 (2), 59–113. [published Online First: 1994/02/01]. doi:10.1016/0146-2806(94)90008-6
Taegtmeyer, H., Young, M. E., Lopaschuk, G. D., Abel, E. D., Brunengraber, H., Darley-Usmar, V., et al. (2016). Assessing Cardiac Metabolism. Circ. Res. 118 (10), 1659–1701. [published Online First: 2016/03/26]. doi:10.1161/RES.0000000000000097
Taggart, A. K. P., Kero, J., Gan, X., Cai, T.-Q., Cheng, K., Ippolito, M., et al. (2005). (d)-β-Hydroxybutyrate Inhibits Adipocyte Lipolysis via the Nicotinic Acid Receptor PUMA-G. J. Biol. Chem. 280 (29), 26649–26652. [published Online First: 2005/06/03]. doi:10.1074/jbc.C500213200
Takahashi, Y., Terada, S., Banjo, M., Seike, K., Nakano, S., and Hatta, H. (2019). Effects of β-hydroxybutyrate Treatment on Glycogen Repletion and its Related Signaling Cascades in Epitrochlearis Muscle during 120 Min of Postexercise Recovery. Appl. Physiol. Nutr. Metab. 44 (12), 1311–1319. [published Online First: 2019/05/06]. doi:10.1139/apnm-2018-0860
Ussher, J. R., Elmariah, S., Gerszten, R. E., and Dyck, J. R. B. (2016). The Emerging Role of Metabolomics in the Diagnosis and Prognosis of Cardiovascular Disease. J. Am. Coll. Cardiol. 68 (25), 2850–2870. doi:10.1016/j.jacc.2016.09.972
Vary, T. C., Reibel, D. K., and Neely, J. R. (1981). Control of Energy Metabolism of Heart Muscle. Annu. Rev. Physiol. 43 (1), 419–430. doi:10.1146/annurev.ph.43.030181.002223
Veech, R. L. (2004). The Therapeutic Implications of Ketone Bodies: the Effects of Ketone Bodies in Pathological Conditions: Ketosis, Ketogenic Diet, Redox States, Insulin Resistance, and Mitochondrial Metabolism. Prostagl. Leukot. Essent. Fat. acids 70 (3), 309–319. doi:10.1016/j.plefa.2003.09.007
Wang, K., Dong, Y., Liu, J., Qian, L., Wang, T., Gao, X., et al. (2020). Effects of REDOX in Regulating and Treatment of Metabolic and Inflammatory Cardiovascular Diseases. Oxidative Med. Cell. Longev. 2020, 1–13. [published Online First: 2020/12/08]. doi:10.1155/2020/5860356
Wang, L., Chen, P., and Xiao, W. (2021). β-Hydroxybutyrate as an Anti-aging Metabolite. Nutrients 13 (10), 3420. [published Online First: 2021/10/24]. doi:10.3390/nu13103420
Wang, L., Zhao, R.-p., Song, X.-y., and Wu, W.-f. (2019). Targeting ERβ in Macrophage Reduces Crown-like Structures in Adipose Tissue by Inhibiting Osteopontin and HIF-1α. Sci. Rep. 9 (1), 15762. [published Online First: 2019/11/02]. doi:10.1038/s41598-019-52265-8
Wang, P., Tate, J. M., and Lloyd, S. G. (2008). Low Carbohydrate Diet Decreases Myocardial Insulin Signaling and Increases Susceptibility to Myocardial Ischemia. Life Sci. 83 (25-26), 836–844. doi:10.1016/j.lfs.2008.09.024
Wang, Y., Liu, X., Shi, H., Yu, Y., Yu, Y., Li, M., et al. (2020). NLRP3 Inflammasome, an Immune‐inflammatory Target in Pathogenesis and Treatment of Cardiovascular Diseases. Clin. Transl. Med. 10 (1), 91–106. [published Online First: 2020/06/09]. doi:10.1002/ctm2.13
Williamson, D., Lund, P., and Krebs, H. (1967). The Redox State of Free Nicotinamide-Adenine Dinucleotide in the Cytoplasm and Mitochondria of Rat Liver. Biochem. J. 103 (2), 514–527. doi:10.1042/bj1030514
Wisneski, J. A., Gertz, E. W., Neese, R. A., and Mayr, M. (1987). Myocardial Metabolism of Free Fatty Acids. Studies with 14C-Labeled Substrates in Humans. J. Clin. Invest. 79 (2), 359–366. doi:10.1172/jci112820
Won, Y.-J., Lu, V. B., Puhl, H. L., and Ikeda, S. R. (2013). -Hydroxybutyrate Modulates N-type Calcium Channels in Rat Sympathetic Neurons by Acting as an Agonist for the G-Protein-Coupled Receptor FFA3. J. Neurosci. 33 (49), 19314–19325. [published Online First: 2013/12/07]. doi:10.1523/JNEUROSCI.3102-13.2013
Xie, Z., Zhang, D., Chung, D., Tang, Z., Huang, H., Dai, L., et al. (2016). Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell. 62 (2), 194–206. [published Online First: 2016/04/23]. doi:10.1016/j.molcel.2016.03.036
Xu, S., Tao, H., Cao, W., Cao, L., Lin, Y., Zhao, S.-M., et al. (2021). Ketogenic Diets Inhibit Mitochondrial Biogenesis and Induce Cardiac Fibrosis. Sig Transduct. Target Ther. 6 (1), 54. [published Online First: 2021/02/10]. doi:10.1038/s41392-020-00411-4
Yamanashi, T., Iwata, M., Kamiya, N., Tsunetomi, K., Kajitani, N., Wada, N., et al. (2017). Beta-hydroxybutyrate, an Endogenic NLRP3 Inflammasome Inhibitor, Attenuates Stress-Induced Behavioral and Inflammatory Responses. Sci. Rep. 7 (1), 7677. doi:10.1038/s41598-017-08055-1
Youm, Y. H., Nguyen, K. Y., Grant, R. W., Goldberg, E. L., Bodogai, M., Kim, D., et al. (2015). Ketone Body β-hydroxybutyrate Blocks the NLRP3 Inflammasome-Mediated Inflammatory Disease. Nat. Med. 21, 263–269. doi:10.1038/nm.3804
Yudkoff, M., Daikhin, Y., Melø, T. M., Nissim, I., Sonnewald, U., and Nissim, I. (2007). The Ketogenic Diet and Brain Metabolism of Amino Acids: Relationship to the Anticonvulsant Effect. Annu. Rev. Nutr. 27, 415–430. [published Online First: 2007/04/21]. doi:10.1146/annurev.nutr.27.061406.093722
Keywords: cardiovascular diseases, cardiac energy metabolism, β-hydroxybutyrate, super fuel, signaling metabolite, cardiovascular therapies
Citation: Wei S, Binbin L, Yuan W, Zhong Z, Donghai L and Caihua H (2022) β-Hydroxybutyrate in Cardiovascular Diseases : A Minor Metabolite of Great Expectations. Front. Mol. Biosci. 9:823602. doi: 10.3389/fmolb.2022.823602
Received: 27 November 2021; Accepted: 04 April 2022;
Published: 13 June 2022.
Edited by:
Roberto Carnevale, Sapienza University of Rome, ItalyReviewed by:
Marko Cincović, University of Novi Sad, SerbiaCopyright © 2022 Wei, Binbin, Yuan, Zhong, Donghai and Caihua. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huang Caihua, caihua.huang@foxmail.com; Lin Donghai, dhlin@xmu.edu.cn