
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 08 March 2022
Sec. Genome Organization and Dynamics
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.817735
 Sara H. A. Agwa1*
Sara H. A. Agwa1* Hesham Elghazaly2Mahmoud Shawky El Meteini3
Hesham Elghazaly2Mahmoud Shawky El Meteini3 Yahia A. Yahia4
Yahia A. Yahia4 Radwa Khaled5,6
Radwa Khaled5,6 Aya M. Abd Elsamee7Reham M. Darwish7Shaimaa M. Elsayed7Hala Hafez8
Aya M. Abd Elsamee7Reham M. Darwish7Shaimaa M. Elsayed7Hala Hafez8 Basma S. Mahmoud8Fouda EM9
Basma S. Mahmoud8Fouda EM9 Marwa Matboli10*
Marwa Matboli10*The SARS-CoV-2 pandemic has led to over 4.9 million deaths as of October 2021. One of the main challenges of creating vaccines, treatment, or diagnostic tools for the virus is its mutations and emerging variants. A couple of variants were declared as more virulent and infectious than others. Some approaches were used as nomenclature for SARS-CoV-2 variants and lineages. One of the most used is the Pangolin nomenclature. In our study, we enrolled 35 confirmed SARS-CoV-2 patients and sequenced the viral RNA in their samples. We also aimed to highlight the hallmark mutations in the most frequent lineage. We identified a seven-mutation signature for the SARS-CoV-2 C36 lineage, detected in 56 countries and an emerging lineage in Egypt. In addition, we identified one mutation which was highly negatively correlated with the lineage. On the other hand, we found no significant correlation between our clinical outcomes and the C36 lineage. In conclusion, the C36 lineage is an emerging SARS-CoV-2 variant that needs more investigation regarding its clinical outcomes compared to other strains. Our study paves the way for easier diagnosis of variants of concern using mutation signatures.
The World Health Organization declared COVID-19 as a pandemic on 11 March 2020 since it appeared as a cluster of pneumonia with unknown cause in Wuhan, Hubei Province, China, in December 2019 (Huang et al., 2020) (Wu et al., 2020). The symptoms range from asymptomatic presentations to dizziness, dry cough, fever, and shortness of breath (Mirzaei et al., 2020) and peak at long-term damage in the lungs (Del Rio et al., 2020) and death in many cases.
The world has face huge economic losses due to lockdown restrictions (Verschuur et al., 2021). Non-pharmaceutical interventions (NPI) against the coronavirus helped to reduce its incidences like mask-wearing, personal hygiene (Cowling et al., 2020), and physical distancing (Huang et al., 2021).
SARS-CoV-2 is a positive-sense RNA virus in the order Nidovirales, family Coronaviridae with an approximately 30 kb single-stranded RNA genome (Elena and Sanjuán, 2005) RNA viruses possess a high mutation rate that is higher than their hosts which impacts viral pathogenicity, infectivity, and transmissibility. The SARS-CoV-2 RNA genome encodes 16 non-structural proteins (NSP) and at least 10 structural proteins including spike (S), ORF3a, envelop (E), membrane (M), open reading frame 6 (ORF6), ORF7a, ORF7b, ORF8, nucleocapsid (N), and ORF10 (Cagliani et al., 2020; Yuan et al., 2021).
The severe morbidity and mortality worldwide worried medical and scientific societies and forced them to make intense and rapid strategies for vaccine development (Zhou et al., 2020). After the isolation and sequencing of the SARS-CoV-2 genome, different genetic clades appeared in Hong Kong in the first 2 months after the identification of SARS-CoV-2 including the V, S, and L clades (To et al., 2021), these variants were thought to worsen vaccine potency (Mahase, 2021) and also cause reinfections (Zucman et al., 2021).
Baud et al. supported the hypothesis that the mortality of SARS-CoV-2 changes depending on geographical regions as they reported that the death rate incidence outside of China is three times higher compared to death rates in China (Baud et al., 2020), The different policies in each country influence the infection rates, and herd immunity of different genetic populations is also considered an important factor.
The persistence of COVID-19 accumulates mutations that paralyze the drug development process albeit with the massive efforts of pandemic trapping. Many studies reported specific mutations related to geographical regions: Val483Ala and Gly476Ser are primarily observed in samples from the United States, whereas Val367Phe is found in samples from China, Hong Kong Special Administrative Region, France, and the Netherlands (Ou et al., 2021).
Varying patients’ responses to different variants of SARS-CoV-2 revealed the need to trace the different variants of SARS-CoV-2 and to study their transmissibility and virulence. For instance, some variants were found to be more virulent and transmissible such as Alpha, Delta, Gamma, Kappa, and Omicron (Christie, 2021; Otto et al., 2021).
Identifying mutations and correlating between them help to identify key features of different strains. Correlating significant mutations and relating them to clinical findings aid in highlighting variants of concern that exhibit more virulence and resistance.
Next-generation sequencing (NGS) techniques are the milestone that can easily identify new and virulent mutations which may help in solving the massively widespread and rapid mutation rates of the pandemic. In addition, NGS may help in tracing the mutation rates and the evolutionary clock of the virus. NGS tools also provide lower cost and unbiased methods for detecting pathogens, with high-speed sequencing that can sequence billions of nucleic acid fragments at once and aid in vaccine and antiviral research, phylogenetic analysis, viral transmission tracing, and pathogen evolution monitoring (Udugama et al., 2020; John et al., 2021).
In this study, we aimed to correlate mutations with lineages to identify the hallmarks of identified lineages. This identification may lead to spotlighting the variants of concern. This method of identification may lead to better treatments, vaccine development, better viral diagnostic approaches, risk categorization, and predict the possible future mutation mechanisms in Egypt. In addition, we aimed to highlight the virulence of viral lineages in Egypt by correlating them with our clinical outcomes. This correlation may lead to a better prognosis of specific viral lineages that may help in clinical decisions and reduce the economic burden nationally and internationally.
The study protocol was approved by the Ethical Committee of Ain Shams University, approval number: (FMASU P17a/2020). Samples used in this study were previously ethically approved with informed patients’ consent in an ongoing project. Reports from hospital records were also used.
Between April 2020 and August 2020, nasopharyngeal (NP) and oropharyngeal swabs were gathered from 35 patients positive for SARS-CoV-2. Inclusion criteria included patients with symptoms and those confirmed to be SARS-CoV-2-positive by real-time PCR; weight ≥10 kg; and age ≥3 years old. Based on the fact that all populations are susceptible to SARS-CoV-2 infection, only individuals or family members who did not give consent to participate were excluded. Also, non-Egyptian patients were excluded. Patients inside every group were sub-grouped according to the severity of symptoms: Mild, moderate, and severe based on their criteria for patient selection including age, sex, and the severity of the disease according to the COVID-19 Treatment Guidelines Panel, National Institutes of Health (COVID-19 Treatment Guidelines Panel, 2019). Fever, cough, and weariness are common symptoms of mild infections. Moderate individuals may suffer breathing difficulties or mild pneumonia. Severe cases may experience severe pneumonia, organ failure, and possible death (World Health Organization, 2021).
Oropharyngeal and nasopharyngeal swab samples were collected from hospitalized patients from different places in Egypt (Medany Hospital, Demerdash Hospital, Central Labs, Qalyobeyyah, and Internal Medicine Hospital) as set out in the guidelines of the Ministry of Health and Population in Egypt. Patients had completed a questionnaire that covered age, history of fever and/or respiratory symptoms, traveling history, any underlying lung disease, history of chronic or immune-compromised conditions, and outcome. The records were used retrospectively to assess the patients’ clinical characteristics and severity to categorize their cases into (mild, moderate, or severe).
Samples placed in a centrifuge tube were labeled with the patient unique ID and containing 2 ml of viral transport media (VTM) were agitated vigorously for 10 s using a vortex mixer. VTM was split into two pre-labeled, sterile cryovials with the correct patient ID. One cryovial was immediately placed in a freezer (−80°C), while the other cryovial was used for molecular studies at Medical Ain Shams Research Institute (MASRI) Molecular Genomic Labs.
Viral RNA isolation was performed using a MagMax viral/pathogen nucleic acid isolation kit (ThermoFisher Scientific, Waltham, MA, United States). Real-time reverse transcription-polymerase chain reaction (RT-PCR) was used for simultaneous amplification of four target genes, including nucleocapsid protein (N), and open reading frame 1ab (ORF1ab), ORF3a, and S proteins. COVID-19 detection was done using ProLab/CerTest Biotech ViaSure SARS-CoV-2. The Real-time PCR detection Kit (VS-NCO296T, CerTest Biotec, S.L, Spain, Catalogue number VS-NCO213L) was used in an Applied Biosystems™ 7500 Fast Real-Time PCR System following the cycling and fluorescence acquisition parameters detailed in the manufacturer’s protocol. Five microliters of RNA was isolated from clinical samples and checked for quantity, purity, and quality by a Qubit® 2.0 Fluorometer (Qubit® RNA Assay Kit, Life Technologies, CA, United States) High Sensitivity Kit (Invitrogen, Carlsbad, CA, United States). The RNA was then used in each real-time PCR reaction, with a final volume of 20 µl. Samples were processed with appropriate negative, internal, and positive controls. Samples were run in duplicate. Real-Time Detection Systems analysis was done by Applied biosystem 7500 Real-Time PCR Software v2.0. The cycle threshold value of [C t] below 34 was considered to be positive. Compliance with the WHO-recommended research protocol confirmatory laboratory testing was carried out.
After viral RNA isolation, reverse transcription and cDNA synthesis were completed. After RNA extraction and assessment, RNA was reverse-transcribed using the SuperScript™ VILO™ cDNA Synthesis Kit (Cat. No.11754050; Invitrogen, Grand Island, United States), according to the product protocol. Targets for sequencing were obtained based on the Ion AmpliSeqTM SARS-CoV-2 Panel (ThermoFisher, Waltham, MA, United States). Library preparation was made using the Ion AmpliSeqTM Library Kit Plus (ThermoFisher, Waltham, MA, United States) (Cat. Nos. 4488990). Primer pool 1 and two target amplification reactions were combined and amplicons were partially digested; barcode adapters were ligated and purified using the Ion Xpress™ Barcode Adapters 1–96 Kit (Cat. No. 447451), then libraries were quantified using the Ion Library TaqMan™ Quantitation Kit (Cat. No. 4468802), the Ion 530™ Kit–Chef (Cat. No. A34461), according to the user guide.
The libraries were sequenced on the Ion GeneStudio S5 Series System platform with an Ion AmpliSeq SARS-CoV-2 Research Panel (ThermoFisher Scientific, Waltham, MA, United States) that contains two pools with amplicons ranging from 125 bp to 275 bp in length and includes >99% of the SARS-CoV-2 genome, covering all serotypes. A complete genome (29,903 nucleotides) was assembled, with 0.13% unique mutations to the other viral genomes.
Using BLAST against the NCBI betacoronavirus database, the closest matches were several sequences with a bit score of 33,479, including, for example, isolate SARS-CoV-2/human/USA/VA-DCLS-0556/2020 (99.9%), accession (MT739463). The assembled genome along with the other SARS-CoV-2 genomes obtained and clustered from GISAID was aligned using MAFFT (Katoh et al., 2002).
We used Torrent Suite Software–provided with the Ion AmpliSeq SARS-CoV-2 research panel–for generating de novo full-length sequences from raw samples’ sequences. Sequence genes’ annotations were carried out using the COVID19AnnotateSnpEff plugin as instructed by the provider’s manual.
Phylogenetic analysis was done on all 35 sequences using the MAFFT (version 7) command-line tool (Katoh et al., 2002). The unweighted pair group method with arithmetic mean (UPGMA) was used for constructing the phylogenetic tree, and the iTOL (version 5) online tool was used to visualize it (Letunic and Bork, 2021).
The analysis was made using R (version 3.6.2). Missense mutations were plotted as a matrix against samples. If a mutation is present in a sample, it was given a value of 1. If the sample matched the reference at a site of mutation, it was given a zero value. Spearman’s correlation coefficients were computed for network analysis using the qgraph R package (version 1.6.9) (Epskamp et al., 2012).
Samples were divided into two clusters based on the Euclidean distance between samples. Clustering was plotted using “heatmap.2” under the “gplots” R package (version 2.17.0). Sample grouping was carried out based on the clusters formed into two groups, A and B, based on the genetic variations.
Correlation analyses were made between clinical outcomes and the two clusters. Shapiro-Wilk’s test was used for normality and F-test for homogeneity for every outcome. The most appropriate test was used for every outcome according to the previous assumptions.
We used the Phylogenetic Assignment of Named Global Outbreak Lineages (Pangolin) (version 3.1.5) command-line tool to classify our samples (Rambaut et al., 2020). We used the Sorting Intolerant from Tolerant (SIFT) web server (version 6.2.1) to predict the effect of correlated mutations on the protein function (Sim et al., 2012).
A total of 35 samples were selected based on quality checks comprising 15 men and 20 women during the early months of the pandemic (Table 1).
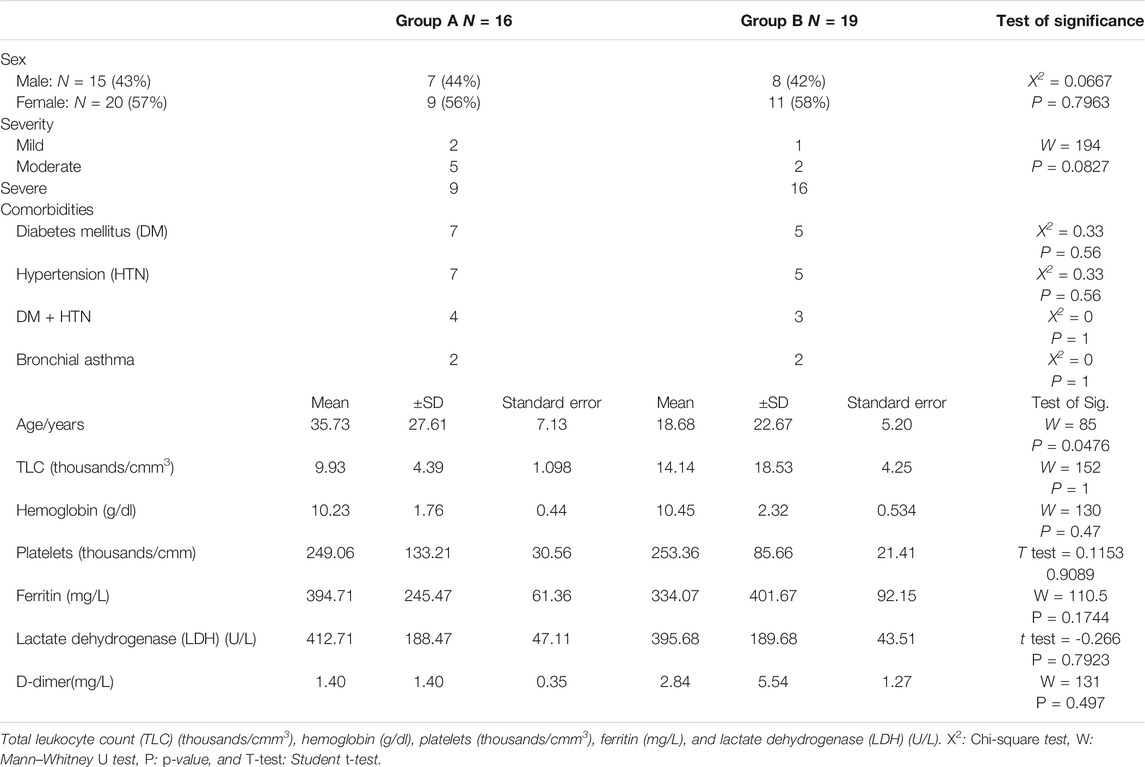
TABLE 1. Group classifications according to gender, severity, and age with clinical outcomes of patients.
Patients’ severity of symptoms was termed mild, moderate, or severe (Table 1) based on their age, sex, and the severity of the disease.
In total, 160 modifications were recorded and distributed across four genomic regions; ORF1ab comprises the longest SARS-CoV-2 gene (approximately 24 kb), corresponding to a polyprotein made up of 16 non-structural proteins (NSP1-16), we found that over 56% of all mutations were recorded in this ORF1ab specifically in positions 2,841, 10,097, 11,083, 17,766, 4,002, 12,534, and 13,536, this was followed by the spike (S) protein in positions 23,403 and 23,593 and nucleocapsid (N) protein in positions 28,881 and 28,908 with the lowest number of variants found in ORF3a coding genes in position 25,563 as represented in Table 2. Moreover, c.2576C > T (p. Asp614Gly) in S was the most abundant missense mutation among samples, found in 29 samples (Table 2).

TABLE 2. Frequent nucleotide and amino acid modifications in analyzed genomes.
Phylogenetic analysis revealed the distinction of the C36 lineage from other lineages forming a clade of 16 leaves (Figure 1).
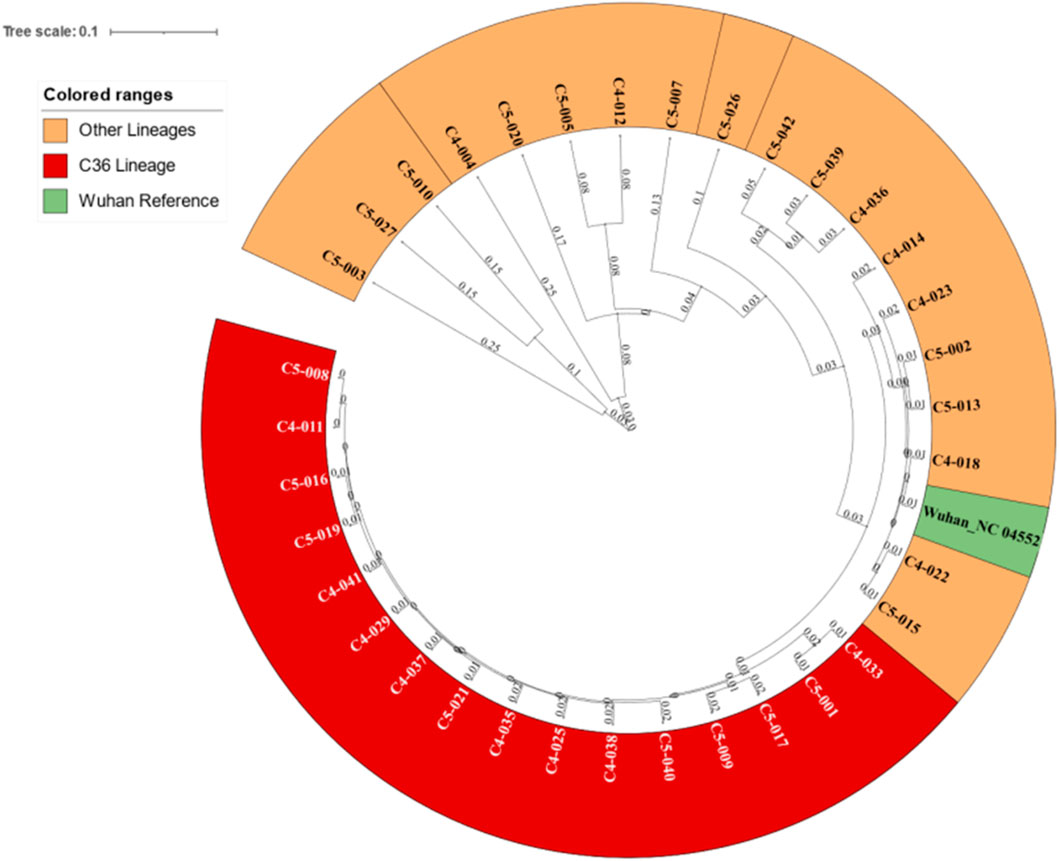
FIGURE 1. Phylogenetic tree for the 35 samples revealing the C36 clade and its distance from other lineages.
The most frequent mutations were from cytosine or guanine to thymidine in all samples (Figure 2A) that represented more than 56% of mutations in all samples with a frequency of 302 mutations (Figure 2C). About 56% of mutations appeared in ORF1ab (Figure 2B).
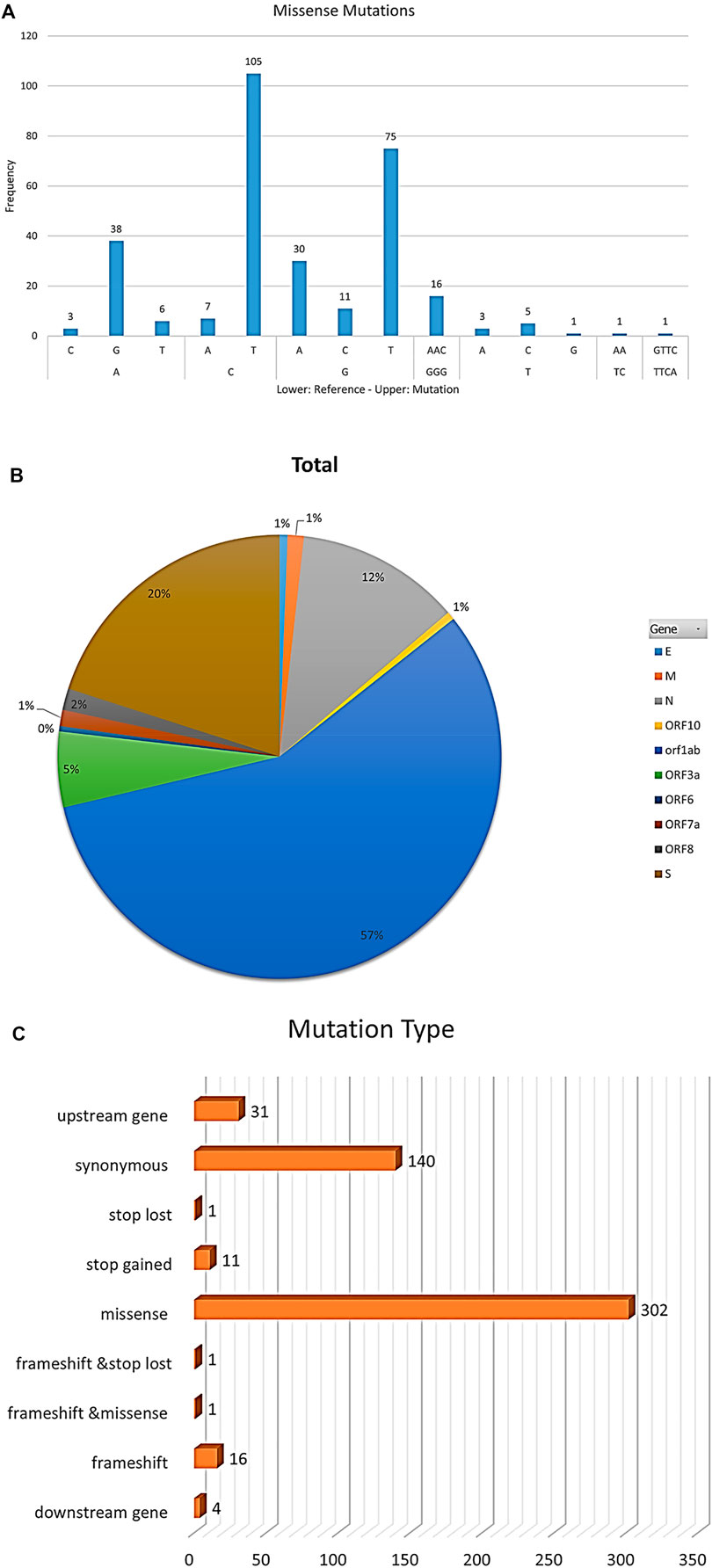
FIGURE 2. The figure represents statistics of mutation frequencies in all samples. (A) Bar plot represents frequencies of nucleotide mutations where the x-axis lower row represents reference nucleotide while the x-axis upper row represents the mutated nucleotide in samples. Frequency is represented on the y-axis. (B) Pie-chart represents mutations’ total frequencies in genes in all samples. (C) Bar plot represents mutations’ total frequencies per mutation type.
Network analysis showed a high positive correlation between seven mutations in Nucleoprotein, spike, and ORF1ab genes, and a high negative correlation between the seven mutations and one mutation in the ORF3a gene (Figure 3). The dendrogram (Figure 4) showed two clades of samples; a clade that carried the 7 correlated mutations was composed of 16 samples (group A); the second clade was composed of 19 samples carrying the negatively correlated mutation (Gln57His) (group B).
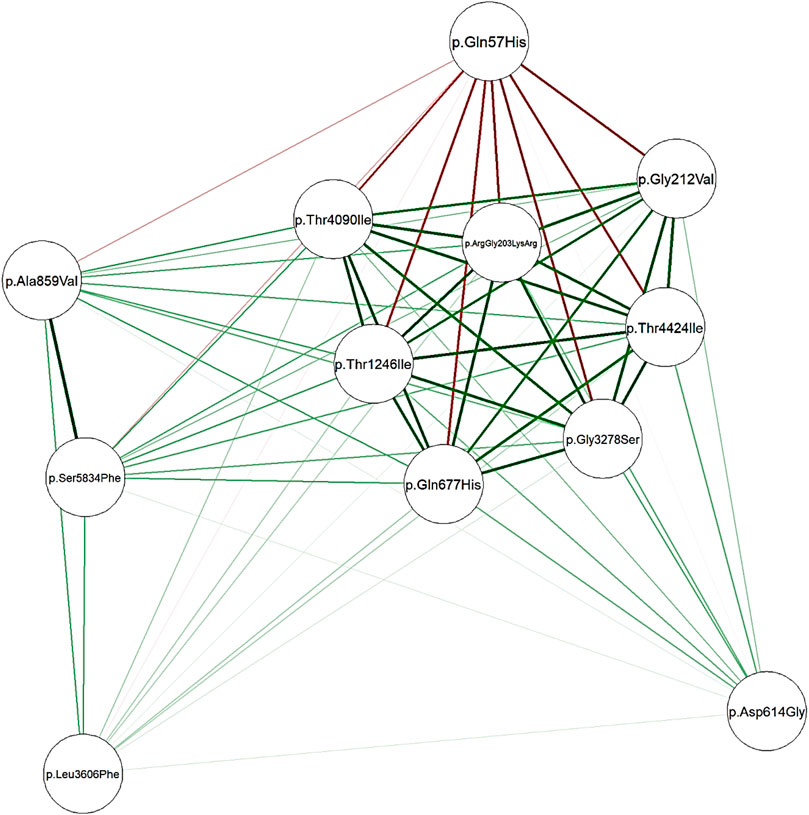
FIGURE 3. Network plotted based on Spearman’s correlation matrix between mutations. Green edges represent a positive correlation coefficient while red edges represent a negative correlation. Intense color represents a higher correlation while the color fades when correlation falls to zero.
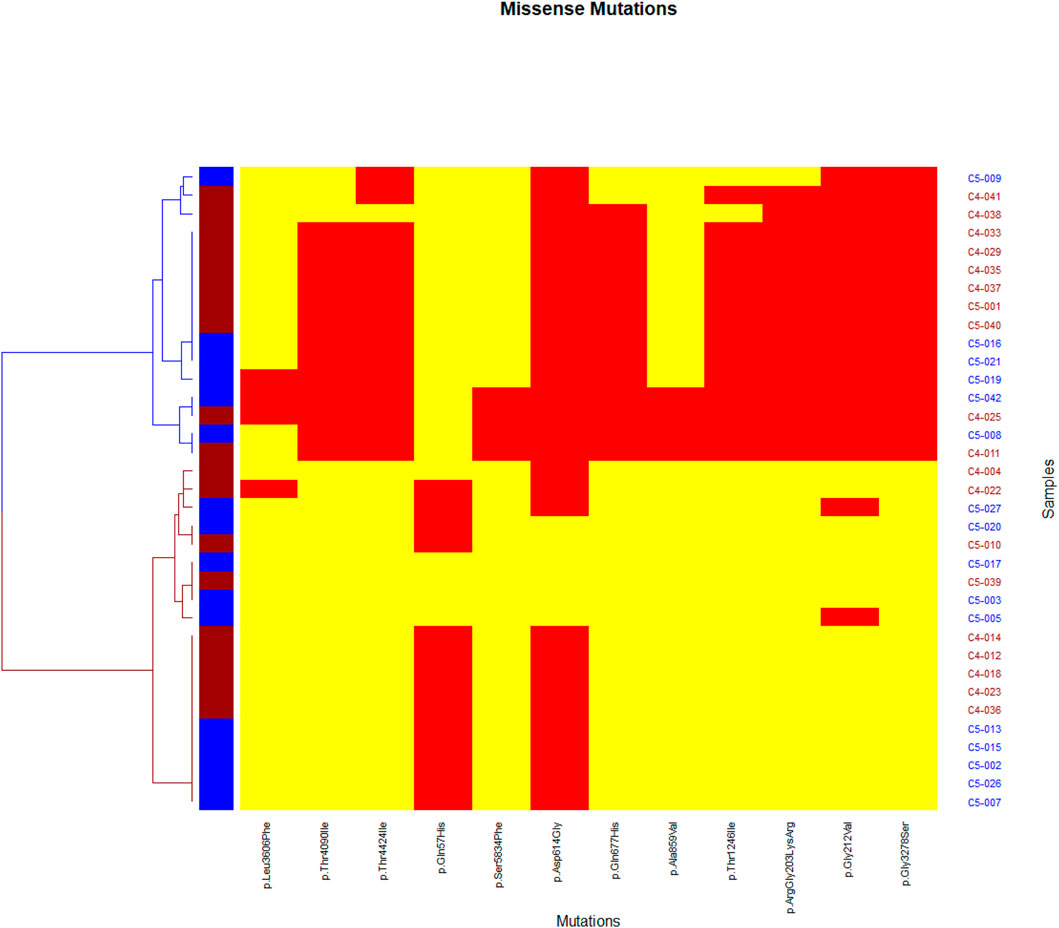
FIGURE 4. Heatmap representing missense mutations on the x-axis and samples on the y-axis. A yellow color indicates the absence of the mutation in the sample while a red color indicates the presence of the mutation. Two clades appear, a blue clade which we considered as a group (A), and a red clade as group (B).
Patients presented with comorbidities such as diabetes mellitus, hypertension, or both were reported. Previously diagnosed asthmatic patients were reported as having a comorbidity as well. Cough was reported in all samples, analyzed using Mann-Whitney’s U test, and no statistically significant difference was observed between the two groups (p-value = 0.4783). The severity of symptoms was reported in all samples (Figure 5), and Mann-Whitney’s U test was used. The two groups showed no statistical significance in the severity outcome (W = 194, p-value = 0.08277), Laboratory outcomes were reported such as (TLC, hemoglobin, platelets, ferritin, lactate dehydrogenase, D-dimer); statistical tests were chosen after testing assumptions such as normality (using Shapiro-Wilk’s test) and homogeneity of variance (using F-test). Based on the prior assumptions, Mann-Whitney U, Student t, and Chi-square tests were used as in Table 1. No statistical significance was found between group A and group B (Table 1).
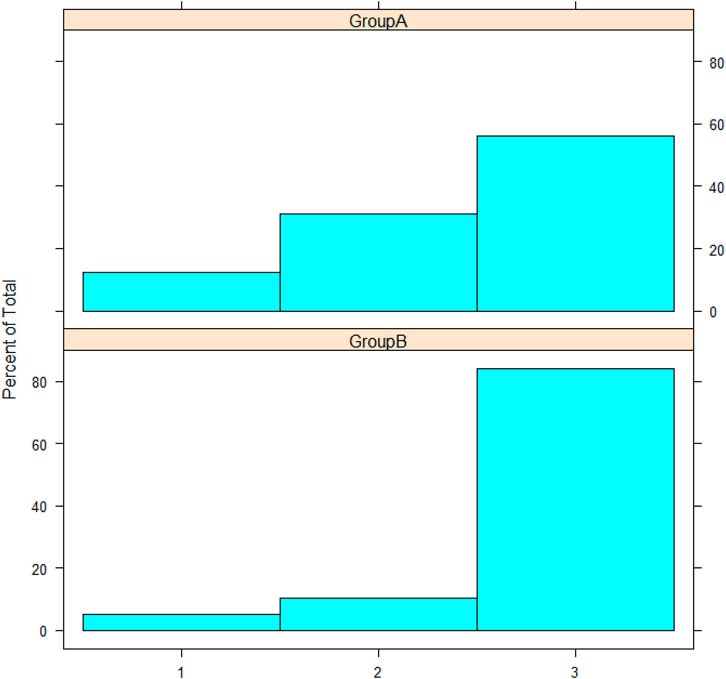
FIGURE 5. The histogram represents severity; the y-axis represents the frequency percentage in each group; the x-axis represents severity as numbers: 1, 2, and 3 for mild, moderate, and severe, respectively.
Phylogenetic analysis revealed 16 sequences under the same clade that were identified as C36 lineages using further analysis (Figure 1).
Group A samples were all classified as lineage C36 according to Pangolin. Group B samples were classified under A and B lineages and their sub-lineages. In group B, the Gln57His mutation at ORF3a was predicted to affect the function of the protein with a high score (0.00). In group A, the Gly204Arg mutation in the nucleocapsid protein and Thr1246Ile and Thr4090Ile mutations in ORF1ab were predicted to affect their proteins with scores of 0.02, 0.00, and 0.00, respectively. However, other correlated mutations on protein function were tolerated according to the SIFT algorithm.
All sequenced data were submitted into the SARS-CoV-2 Global Initiative on Sharing All Influenza Data (GISAID) database as shown in Table 3. In all figures, we used the corresponding abbreviations (Table 3) throughout the study.

TABLE 3. SARS-CoV-2 Egyptian patients’ samples sequenced and deposited with GISAID.
Sequencing using NGS techniques revealed the blurry areas in the SARS-CoV-2 genome that helped us to make panoramic insights about mutation patterns and explain the mounting infectivity of the virus all over the world. Moreover, these techniques helped us to put forward the right explanation of population re-infection and antigenic consequences (Li et al., 2005).
We analyzed the genomic variants of 35 Egyptian patients during the first wave of the pandemic and divided them into two groups after phylogenetic analysis. The first group (B) included all lineages except C36 lineage. While group (A) included only sequences that were classified as the C36 lineage. According to Pangolin, the C36 lineage first appeared in the United States on 13 March, 2020. However, the highest incidence according to the GISAID database is in the Egyptian population. The C36 lineage has been detected in at least 56 countries worldwide (Anderson et al., 2021).
The C36 lineage compromises 34% of all sequenced variants in Egypt, 11% of sequenced variants in Germany, 10% of sequenced variants in the United Kingdom, 7% of sequenced variants in the United States, and 6% of sequenced variants in Denmark until January 2022 according to Pangolin.
Roshdy et al. confirmed the presence of the C36 lineage early in the pandemic and its evolution into several sub-lineages, including C.36.1, C.36.3, and C.36.3.1, circulating across the Egyptian patients’ genome. They also discovered that mutations in this lineage show potential fitness and pathogenicity in the same manner that mutations in Alpha, Beta, Gamma, Delta, and Omicron (variants of concern) do (Roshdy et al., 2022). The spike mutation related to C36 lineage Gln677His in position 23,593 which emerged firstly in the United States confers an advantage in spreading and transmissibility through its position in the S1/S2 boundary upstream furin cleavage site (Hodcroft et al., 2021).
Among the 35 genomes, more than 56% of mutations were missense mutations with a frequency of 302 mutations followed by synonymous mutations with a frequency of 140 mutations and frameshifts with a frequency of 16 mutations (Figure 2C). C > T transitions may be interfered with by cytosine deaminases (Lyons and Lauring, 2017). G > T transversions are more likely to be introduced by oxo-guanine from reactive oxygen species (Li et al., 2006).
Approximately 56% of mutations appeared in ORF1ab, which represents more than two-thirds of the genome, controls viral replication, and consequently, these mutations might affect the replication speed of the virus (Yin, 2020).
The most common variant located in the ORF1ab region was the missense mutation c.9832G > A in region 10,097 that changed glycine amino acid into serine p.Gly3278Ser in 16 of our samples. In group (B), Thr1246Ile and Thr4090Ile mutations in ORF1ab were predicted to affect their proteins with scores of 0.00 and 0.00, respectively, and were considered influential parameters that could be possibly linked to the virus’s speed replication and infectivity that contribute to patient severity status.
The S protein of SARS-CoV-1 and SARS-CoV-2 forms homo-trimers protruding in the viral surface that facilitates viral entry into the host cells via interacting with angiotensin-converting enzyme 2 (ACE2) which is their main receptor expressed in lower respiratory tract cells (Letko et al., 2020) (Bakhshandeh et al., 2021).
Variants in the spike protein domain showed strong evidence of reducing the neutralization sensitivity to convalescent sera and monoclonal antibodies. These variants potentially lessened the protection afforded by the current vaccines that target the spike region. Asn439Lys emerged in Scotland in the spike region and was found to enhance the binding affinity for the ACE2 receptor and reduce the neutralizing activity of some monoclonal antibodies (Thomson et al., 2021) (Greaney et al., 2021) (Wibmer et al., 2021) (Gaebler et al., 2021) (Collier et al., 2021).
We reported that the most frequent modified nucleotides were recorded at position 23,403 in the spike protein c.1841A > G, this missense mutation changed aspartic acid into glycine p.Asp614Gly found in 29 samples (Table 2) (Alouane et al., 2020) (Lobiuc et al., 2021). The p.Asp614Gly mutation firstly appeared in late January in China and rapidly emerged in the global population within a mere 3 months, studies illustrated that the p.Asp614Gly mutation confers a moderate advantage for virus transmissibility, infectivity, replication, and elevated fitness; it may explain the high frequency of infections in the Egyptian population (Hou et al., 2020) (Yurkovetskiy et al., 2020).
Cong et al. studied the N protein and its impact on the coronaviral life cycle by the contribution to helical ribonucleoproteins formation during RNA genome packaging, modulating viral RNA synthesis during replication and transcription, and modifying metabolism in infected people (Cong et al., 2020). Studies showed that N genes are more conserved and stable, with 90% amino acid homology and fewer mutation frequencies throughout time (Dutta et al., 2020). Changes in the N protein charge resulted in enhanced virus replication and ultimately increased infectivity and fitness (Wu et al., 2021). The missense mutation in nucleocapsid phosphoprotein (N) in position 28,881 p. ArgGly203LysArg found in 15 of our patients is already observed in 1,573 samples out of 10,022 SARS-CoV-2 genomes studied from the US, United Kingdom, and Australia (Koyama et al., 2020). The statistical analysis found that the Gly204Arg mutation in nucleocapsid protein which was found in group B in position 28,881 appeared to influence protein with a score of 0.02. Studies showed that Arg203Lys and Gly204Arg are concomitant mutations in the N protein, which are quickly rising in frequency and may be linked to the virus’s infectivity (Zhu et al., 2021). These mutations are found commonly in lineages B.1.1.7 (Alpha) (Caserta et al., 2021; Wu et al., 2021) and P.1 (Gamma) (Faria et al., 2021; Wu et al., 2021). Another mutation p. Gly212Val in position 28,908 was also found in N protein and repeated 18 times.
ORF3a, although it is considered an accessory protein, has a vital role in cell surface localization and allows viral entry within the host and possesses immunogenic properties (Zhong et al., 2003) (Liu et al., 2014). Moreover, ORF3a is involved in ion channel formation and modulates the release of the virus from the host cell (Liu et al., 2014). Majumdar et al. extensively studied the emerged mutations that appeared in the ORF3a protein in silico and related these mutations with high mortality rates for SARS-CoV-2 infection through host immune evasion and extreme cytokine storm through JAK-STAT, chemokine, and cytokine-related pathways (Majumdar and Niyogi, 2020).
Interestingly, our data revealed that the Gln57His mutation at ORF3a affected the function of the protein with a high score (0.00) in group B. Our findings are supported by a study that reports that ORF3a mutation Gln57His leads to a major truncation of the ORF3b protein (Chu et al., 2021).
Zekri et al. previously identified 204 distinct mutations of the Egyptian strains classified under clade B lineage and its sub-lineages, distributed on ORF1ab, S, N, ORF3a, ORF7a, ORF8, M, E, and ORF6. In addition, they found that Asp614Gly was the most frequent mutation appearing in all their samples. Interestingly Asp614Gly also appeared in 83% of our samples (Zekri et al., 2021).
Our data showed no statistical significance in the severity outcome between the studied groups (p-value = 0.08277).
The laboratory tests investigated in this study included LDH, PLT, Hb, D-dimer, serum ferritin, and platelet counts. Other studies reported the influence of SARS-CoV-2 on those parameters. For instance, LDH was reported to increase in severely symptomatic patients to reach 6-fold its normal values (Henry et al., 2020). Serum ferritin and D-dimer were significantly increased in COVID patients and elevated in more virulent cases (Cheng et al., 2020; Hussein et al., 2021). Platelets and total leucocytes declined in COVID patients as reported by Wool and Miller (Wool and Miller, 2021). However, our study reported no significant correlation between the C36 mutation signature and clinical outcomes.
Our study highlights the mutation signature for the C36 lineage over other lineages. The mutation signature proposes seven positively correlated mutations and one negatively correlated mutation. On the other hand, our study reported no significantly correlated clinical outcomes or predisposing comorbidities that hallmark the C36 lineage. Interestingly, C36 tends to affect older patients. However, our clinical findings need more investigation using a larger sample size.
The study was done based on the guidelines of the Declaration of Helsinki, and received approval from the Research Ethics Committee, Faculty of Medicine, Ain Shams University, Egypt, dated 13/5/2020, FWA 000016584.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by the Ethical Committee of Ain Shams University (Ethical comiitte approval number: FMASU P17a/2020. The patients/participants provided their written informed consent to participate in this study.
SA participated in funding acquisition and resources, supervision and original drafting—reviewing and editing. YY shared in Bioinformatic and NGS data analysis, validation, original drafting, methodology. RK shared in data analysis and original drafting, HE, MSE, AMA, RMD, SME, HH, BSM, and FEM shared in data acquisition and formal analysis, validation and reviewing. MM shared in conceptualization, methodology, supervision and reviewing and editing. All authors have read and agreed to the published version of the manuscript.
This study was funded by Ain Shams University, School of Medicine, 2020–3.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alouane, T., Laamarti, M., Essabbar, A., Hakmi, M., Bouricha, E. M., Chemao-Elfihri, M. W., et al. (2020). Genomic Diversity and Hotspot Mutations in 30,983 SARS-CoV-2 Genomes: Moving toward a Universal Vaccine for the "Confined Virus". Pathogens 9 (10), 829. doi:10.3390/pathogens9100829
Anderson, K., Su, A., and Wu, C. (2021). C36 Lineage Report. Retrieved from Outbreak.info: https://outbreak.info/situation-reports?pango=C.36. (Accessed October 29, 2021).
Bakhshandeh, B., Jahanafrooz, Z., Abbasi, A., Goli, M. B., Sadeghi, M., Mottaqi, M. S., et al. (2021). Mutations in SARS-CoV-2; Consequences in Structure, Function, and Pathogenicity of the Virus. Microb. pathogenesis 154, 104831. doi:10.1016/j.micpath.2021.104831
Baud, D., Qi, X., Nielsen-Saines, K., Musso, D., Pomar, L., and Favre, G. (2020). Real Estimates of Mortality Following COVID-19 Infection. Lancet Infect. Dis. 20 (7), 773. doi:10.1016/s1473-3099(20)30195-x
C Caserta, L., Mitchell, P. K., Plocharczyk, E., and Diel, D. G. (2021). Identification of a SARS-CoV-2 Lineage B1.1.7 Virus in New York Following Return Travel from the United Kingdom. Microbiol. Resour. Announc 10 (9), e00097–21. doi:10.1128/MRA.00097-21
Cagliani, R., Forni, D., Clerici, M., and Sironi, M. (2020). Coding Potential and Sequence Conservation of SARS-CoV-2 and Related Animal Viruses. Infect. Genet. Evol. 83, 104353. doi:10.1016/j.meegid.2020.104353
Cheng, L., Li, H., Li, L., Liu, C., Yan, S., Chen, H., et al. (2020). Ferritin in the Coronavirus Disease 2019 (COVID-19): A Systematic Review and Meta-Analysis. J. Clin. Lab. Anal. 34 (10), e23618. doi:10.1002/jcla.23618
Christie, B. (2021). Covid-19: Early Studies Give hope Omicron Is Milder Than Other Variants. Bmj 375, n3144. doi:10.1136/bmj.n3144
Chu, D. K. W., Hui, K. P. Y., Gu, H., Ko, R. L. W., Krishnan, P., Ng, D. Y. M., et al. (2021). Introduction of ORF3a-Q57h SARS-CoV-2 Variant Causing Fourth Epidemic Wave of COVID-19, Hong Kong, China. Emerg. Infect. Dis. 27 (5), 1492–1495. doi:10.3201/eid2705.210015
Collier, D. A., De Marco, A., De Marco, A., Ferreira, I. A. T. M., Meng, B., Datir, R. P., et al. (2021). Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA Vaccine-Elicited Antibodies. Nature 593 (7857), 136–141. doi:10.1038/s41586-021-03412-7
Cong, Y., Ulasli, M., Schepers, H., Mauthe, M., V'kovski, P., Kriegenburg, F., et al. (2020). Nucleocapsid Protein Recruitment to Replication-Transcription Complexes Plays a Crucial Role in Coronaviral Life Cycle. J. Virol. 94 (4), e01925–19. doi:10.1128/JVI.01925-19
COVID-19 Treatment Guidelines Panel (2019). Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. National Institutes of Health. Available at https://www.covid19treatmentguidelines.nih.gov/(Accessed 1 31, 2022).
Cowling, B. J., Ali, S. T., Ng, T. W. Y., Tsang, T. K., Li, J. C. M., Fong, M. W., et al. (2020). Impact Assessment of Non-pharmaceutical Interventions against Coronavirus Disease 2019 and Influenza in Hong Kong: an Observational Study. Lancet Public Health 5 (5), e279–e288. doi:10.1016/s2468-2667(20)30090-6
Del Rio, C., Collins, L. F., and Malani, P. (2020). Long-term Health Consequences of COVID-19. JAMA 324 (17), 1723–1724. doi:10.1001/jama.2020.19719
Dutta, N. K., Mazumdar, K., and Gordy, J. T. (2020). The Nucleocapsid Protein of SARS-CoV-2: a Target for Vaccine Development. J. Virol. 94 (13), e00647–20. doi:10.1128/JVI.00647-20
Elena, S. F., and Sanjuán, R. (2005). Adaptive Value of High Mutation Rates of RNA Viruses: Separating Causes from Consequences. J. Virol 79 (18), 11555–11558.
Epskamp, S., Cramer, A. O., Waldorp, L. J., Schmittmann, V. D., and Borsboom, D. (2012). Qgraph: Network Visualizations of Relationships in Psychometric Data. J. Stat. Softw. 48, 1–18. doi:10.18637/jss.v048.i04
Faria, N. R., Mellan, T. A., Whittaker, C., Claro, I. M., Candido, D., Mishra, S., et al. (2021). Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil. Science (New York, N.Y.) 372 (6544), 815–821. doi:10.1126/science.abh2644
Gaebler, C., Wang, Z., Lorenzi, J. C. C., Muecksch, F., Finkin, S., Tokuyama, M., et al. (2021). Evolution of Antibody Immunity to SARS-CoV-2. Nature 591 (7851), 639–644. doi:10.1038/s41586-021-03207-w
Greaney, A. J., Loes, A. N., Crawford, K. H. D., Starr, T. N., Malone, K. D., Chu, H. Y., et al. (2021). Comprehensive Mapping of Mutations in the SARS-CoV-2 Receptor-Binding Domain that Affect Recognition by Polyclonal Human Plasma Antibodies. Cell Host & Microbe 29 (3), 463–476. doi:10.1016/j.chom.2021.02.003
Henry, B. M., Aggarwal, G., Wong, J., Benoit, S., Vikse, J., Plebani, M., et al. (2020). Lactate Dehydrogenase Levels Predict Coronavirus Disease 2019 (COVID-19) Severity and Mortality: A Pooled Analysis. Am. J. Emerg. Med. 38 (9), 1722–1726. doi:10.1016/j.ajem.2020.05.073
Hodcroft, E. B., Zuber, M., Nadeau, S., Vaughan, T. G., Crawford, K. H. D., Althaus, C. L., et al. (2021). Emergence and Spread of a SARS-CoV-2 Variant Through Europe in the Summer of 2020. Nature 595 (7869), 707–712.
Hou, Y. J., Chiba, S., Halfmann, P., Ehre, C., Kuroda, M., Dinnon, K. H., et al. (2020). SARS-CoV-2 D614G Variant Exhibits Efficient Replication Ex Vivo and Transmission In Vivo. Science 370 (6523), 1464–1468. doi:10.1126/science.abe8499
Huang, B., Wang, J., Cai, J., Yao, S., Chan, P. K. S., Tam, T. H.-w., et al. (2021). Integrated Vaccination and Physical Distancing Interventions to Prevent Future COVID-19 Waves in Chinese Cities. Nat. Hum. Behav. 5 (6), 695–705. doi:10.1038/s41562-021-01063-2
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. The Lancet 395 (10223), 497–506. doi:10.1016/s0140-6736(20)30183-5
Hussein, A. M., Taha, Z. B., Gailan Malek, A., Akram Rasul, K., Qasim Hazim, D., Jalal Ahmed, R., et al. (2021). D-dimer and Serum Ferritin as an Independent Risk Factor for Severity in COVID-19 Patients. Mater. Today Proc. [Epub ahead of print] doi:10.1016/j.matpr.2021.04.009
John, G., Sahajpal, N. S., Mondal, A. K., Ananth, S., Williams, C., Chaubey, A., et al. (2021). Next-Generation Sequencing (NGS) in COVID-19: A Tool for SARS-CoV-2 Diagnosis, Monitoring New Strains and Phylodynamic Modeling in Molecular Epidemiology. Curr. Issues Mol. Biol. 43 (2), 845–867. doi:10.3390/cimb43020061
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 30 (14), 3059–3066. doi:10.1093/nar/gkf436
Koyama, T., Platt, D., and Parida, L. (2020). Variant Analysis of SARS-CoV-2 Genomes. Bull. World Health Organ. 98 (7), 495–504. doi:10.2471/blt.20.253591
Letko, M., Marzi, A., and Munster, V. (2020). Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat. Microbiol. 5 (4), 562–569. doi:10.1038/s41564-020-0688-y
Letunic, I., and Bork, P. (2021). Interactive Tree of Life (iTOL) V5: an Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 49 (W1), W293–W296. doi:10.1093/nar/gkab301
Li, F., Li, W., Farzan, M., and Harrison, S. C. (2005). Structure of SARS Coronavirus Spike Receptor-Binding Domain Complexed with Receptor. Science 309 (5742), 1864–1868. doi:10.1126/science.1116480
Li, Z., Wu, J., and Deleo, C. (2006). RNA Damage and Surveillance under Oxidative Stress. IUBMB Life (International Union Biochem. Mol. Biol. Life) 58 (10), 581–588. doi:10.1080/15216540600946456
Liu, D. X., Fung, T. S., Chong, K. K.-L., Shukla, A., and Hilgenfeld, R. (2014). Accessory Proteins of SARS-CoV and Other Coronaviruses. Antivir. Res. 109, 97–109. doi:10.1016/j.antiviral.2014.06.013
Lobiuc, A., Dimian, M., Gheorghita, R., Sturdza, O. A. C., and Covasa, M. (2021). Introduction and Characteristics of SARS-CoV-2 in North-East of Romania during the First COVID-19 Outbreak. Front. Microbiol. 12, 654417. doi:10.3389/fmicb.2021.654417
Lyons, D. M., and Lauring, A. S. (2017). Evidence for the Selective Basis of Transition-To-Transversion Substitution Bias in Two RNA Viruses. Mol. Biol. Evol. 34 (12), 3205–3215. doi:10.1093/molbev/msx251
Mahase, E. (2021). Covid-19: South Africa Pauses Use of Oxford Vaccine after Study Casts Doubt on Efficacy against Variant. Bmj 372, n372. doi:10.1136/bmj.n372
Majumdar, P., and Niyogi, S. (2020). ORF3a Mutation Associated with Higher Mortality Rate in SARS-CoV-2 Infection. Epidemiol. Infect. 148, e262. doi:10.1017/s0950268820002599
Mirzaei, R., Mohammadzadeh, R., Mahdavi, F., Badrzadeh, F., Kazemi, S., Ebrahimi, M., et al. (2020). Overview of the Current Promising Approaches for the Development of an Effective Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Vaccine. Int. immunopharmacology 88, 106928. doi:10.1016/j.intimp.2020.106928
Otto, S. P., Day, T., Arino, J., Colijn, C., Dushoff, J., Li, M., et al. (2021). The Origins and Potential Future of SARS-CoV-2 Variants of Concern in the Evolving COVID-19 Pandemic. Curr. Biol. 31 (14), R918–R929. doi:10.1016/j.cub.2021.06.049
Ou, J., Zhou, Z., Dai, R., Zhang, J., Zhao, S., Wu, X., et al. (2021). V367F Mutation in SARS-CoV-2 Spike RBD Emerging during the Early Transmission Phase Enhances Viral Infectivity through Increased Human ACE2 Receptor Binding Affinity. J. Virol. 95 (16), e0061721. doi:10.1128/JVI.00617-21
Rambaut, A., Holmes, E. C., O’Toole, Á., Hill, V., McCrone, J. T., Ruis, C., et al. (2020). A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 5 (11), 1403–1407. doi:10.1038/s41564-020-0770-5
Roshdy, W. H., Khalifa, M. K., San, J. E., Tegally, H., Wilkinson, E., Showky, S., et al. (2022). SARS-CoV-2 Genetic Diversity and Lineage Dynamics of in Egypt. medRxiv [Epub ahead of print]. doi:10.1101/2022.01.05.22268646
Sim, N. L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., and Ng, P. C. (2012). SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 40, W452–W457. doi:10.1093/nar/gks539
Thomson, E. C., Rosen, L. E., Shepherd, J. G., Spreafico, R., da Silva Filipe, A., Wojcechowskyj, J. A., et al. (2021). Circulating SARS-CoV-2 Spike N439K Variants Maintain Fitness while Evading Antibody-Mediated Immunity. Cell 184 (5), 1171–1187. e20. doi:10.1016/j.cell.2021.01.037
To, K. K. W., Chan, W. M., Ip, J. D., Chu, A. W. H., Tam, A. R., Liu, R., et al. (2021). Unique Clusters of Severe Acute Respiratory Syndrome Coronavirus 2 Causing a Large Coronavirus Disease 2019 Outbreak in Hong Kong. Clin. Infect. Dis. 73 (1), 137–142.
Udugama, B., Kadhiresan, P., Kozlowski, H. N., Malekjahani, A., Osborne, M., Li, V. Y. C., et al. (2020). Diagnosing COVID-19: The Disease and Tools for Detection. ACS nano 14 (4), 3822–3835. doi:10.1021/acsnano.0c02624
Verschuur, J., Koks, E. E., and Hall, J. W. (2021). Observed Impacts of the COVID-19 Pandemic on Global Trade. Nat. Hum. Behav. 5 (3), 305–307. doi:10.1038/s41562-021-01060-5
Wibmer, C. K., Ayres, F., Hermanus, T., Madzivhandila, M., Kgagudi, P., Oosthuysen, B., et al. (2021). SARS-CoV-2 501Y.V2 Escapes Neutralization by South African COVID-19 Donor Plasma. Nat. Med. 27 (4), 622–625. doi:10.1038/s41591-021-01285-x
Wool, G. D., and Miller, J. L. (2021). The Impact of COVID-19 Disease on Platelets and Coagulation. Pathobiology 88 (1), 15–27. doi:10.1159/000512007
World Health Organization (2021). Covid-19 Symptoms and Severity. Available at https://www.who.int/westernpacific/emergencies/covid-19/information/asymptomatic-covid-19 (Accessed December, 2021).
Wu, F., Zhao, S., Yu, B., Chen, Y.-M., Wang, W., Song, Z.-G., et al. (2020). A New Coronavirus Associated with Human Respiratory Disease in China. Nature 579 (7798), 265–269. doi:10.1038/s41586-020-2008-3
Wu, H., Xing, N., Meng, K., Fu, B., Xue, W., Dong, P., et al. (2021). Nucleocapsid Mutations R203K/G204R Increase the Infectivity, Fitness, and Virulence of SARS-CoV-2. Cell Host & Microbe 29 (12), 1788–1801. e6. doi:10.1016/j.chom.2021.11.005
Yin, C. (2020). Genotyping Coronavirus SARS-CoV-2: Methods and Implications. Genomics 112 (5), 3588–3596. doi:10.1016/j.ygeno.2020.04.016
Yuan, F., Wang, L., Fang, Y., and Wang, L. (2021). Global SNP Analysis of 11,183 SARS‐CoV‐2 Strains Reveals High Genetic Diversity. Transbound Emerg. Dis. 68 (6), 3288–3304. doi:10.1111/tbed.13931
Yurkovetskiy, L., Wang, X., Pascal, K. E., Tomkins-Tinch, C., Nyalile, T. P., Wang, Y., et al. (2020). Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 183 (3), 739–751. e8. doi:10.1016/j.cell.2020.09.032
Zekri, A.-R. N., Easa Amer, K., Hafez, M. M., Hassan, Z. K., Ahmed, O. S., Soliman, H. K., et al. (2021). Genomic Characterization of SARS-CoV-2 in Egypt. J. Adv. Res. 30, 123–132. doi:10.1016/j.jare.2020.11.012
Zhong, N., Zheng, B., Li, Y., Poon, L., Xie, Z., Chan, K., et al. (2003). Epidemiology and Cause of Severe Acute Respiratory Syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. The Lancet 362 (9393), 1353–1358. doi:10.1016/s0140-6736(03)14630-2
Zhou, F., Yu, T., Du, R., Fan, G., Liu, Y., Liu, Z., et al. (2020). Clinical Course and Risk Factors for Mortality of Adult Inpatients with COVID-19 in Wuhan, China: a Retrospective Cohort Study. The lancet 395 (10229), 1054–1062. doi:10.1016/s0140-6736(20)30566-3
Zhu, Z., Liu, G., Meng, K., Yang, L., Liu, D., and Meng, G. (2021). Rapid Spread of Mutant Alleles in Worldwide SARS-CoV-2 Strains Revealed by Genome-wide Single Nucleotide Polymorphism and Variation Analysis. Genome Biol. Evol. 13 (2), evab015. doi:10.1093/gbe/evab015
Keywords: SARS-CoV-2, mutation, NGS, C36 lineage, Egypt
Citation: Agwa SHA, Elghazaly H, El Meteini MS, Yahia YA, Khaled R, Abd Elsamee AM, Darwish RM, Elsayed SM, Hafez H, Mahmoud BS, EM F and Matboli M (2022) Identifying SARS-CoV-2 Lineage Mutation Hallmarks and Correlating Them With Clinical Outcomes in Egypt: A Pilot Study. Front. Mol. Biosci. 9:817735. doi: 10.3389/fmolb.2022.817735
Received: 18 November 2021; Accepted: 08 February 2022;
Published: 08 March 2022.
Edited by:
Jade L. L. Teng, The University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Ruibang Luo, The University of Hong Kong, Hong Kong SAR, ChinaCopyright © 2022 Agwa, Elghazaly, El Meteini, Yahia, Khaled, Abd Elsamee, Darwish, Elsayed, Hafez, Mahmoud, EM and Matboli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sara H. A. Agwa, c2FyYS5oLmFnd2FAbWVkLmFzdS5lZHUuZWc=; Marwa Matboli, ZHJtYXJ3YV9tYXRib2x5QG1lZC5hc3UuZWR1LmVn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.