
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Biosci. , 14 October 2022
Sec. Protein Biochemistry for Basic and Applied Sciences
Volume 9 - 2022 | https://doi.org/10.3389/fmolb.2022.1032177
This article is part of the Research Topic Metalloproteins as Sensors of Gaseous Small Molecules - From Bench to Bed and Beyond View all 6 articles
 Mirelly Cunha da Silva1
Mirelly Cunha da Silva1 Vanessa Maria dos Santos1
Vanessa Maria dos Santos1 Matheus Vinícius B. da Silva1
Matheus Vinícius B. da Silva1 Tereza Cristina M. M. Prazeres2
Tereza Cristina M. M. Prazeres2 Maria do Socorro S. Cartágenes2
Maria do Socorro S. Cartágenes2 Natália Tabosa M. Calzerra3
Natália Tabosa M. Calzerra3 Thyago Moreira de Queiroz1*
Thyago Moreira de Queiroz1*A Disintegrin and Metalloprotease 17 (ADAM17), also called tumor necrosis factor-ɑ (TNF-ɑ) convertase (TACE), is a well-known protease involved in the sheddase of growth factors, chemokines and cytokines. ADAM17 is also enrolled in hypertension, especially by shedding of angiotensin converting enzyme type 2 (ACE2) leading to impairment of angiotensin 1–7 [Ang-(1–7)] production and injury in vasodilation, induction of renal damage and cardiac hypertrophy. Activation of Mas receptor (MasR) by binding of Ang-(1–7) induces an increase in the nitric oxide (NO) gaseous molecule, which is an essential factor of vascular homeostasis and blood pressure control. On the other hand, TNF-ɑ has demonstrated to stimulate a decrease in nitric oxide bioavailability, triggering a disrupt in endothelium-dependent vasorelaxation. In spite of the previous studies, little knowledge is available about the involvement of the metalloprotease 17 and the NO pathways. Here we will provide an overview of the role of ADAM17 and Its mechanisms implicated with the NO formation.
Hyperstimulation of the renin-angiotensin system (RAS), mainly associated with other cardiovascular risk factors, can induce a cascade of deleterious actions, such as an increase in blood pressure (BP) (Melaku, 2018). Hypertension is a multifactorial disease that results from the junction of genetic and environmental factors that present neuroendocrine, hemodynamic, redox and inflammatory components, which combine with each other to induce functional and structural changes in the cardiovascular system. (Prado et al., 2021).
The most active metabolite of the RAS is angiotensin II (Ang-II), which promotes vascular injury and hypertension primarily through an interaction with the Ang-II type 1 receptor (AT1R) (Yang et al., 2017). During RAS hyperactivity, there is evidence of increased activation of AT1R via Ang-II, which promotes translocation of ADAM17 to the cell membrane (Xu et al., 2017; Mukerjee et al., 2019). The ADAM family represents one of the main groups of sheddase proteases, which promotes proteolysis in the extracellular domain of integral membrane protein, controlling the biological activity of membrane proteins (Arribas and Borroto, 2002; Chow and Fernandez-Patron, 2007).
It has been described that ADAM-mediated cleavage may contribute to the tissue remodeling and dysfunction, as well as induction of inflammation. Thus, ADAM is related to the development of cardiovascular diseases (CVD), including atherosclerosis and hypertension, and may be a potential pharmacological target in the treatment of these diseases. (Kawai et al., 2021).
Among members of the ADAM family, ADAM17, also known as tumor necrosis factor (TNF)-α converting enzyme (TACE), acts as a metalloproteinase that cleaves the TNF-α precursor (Black et al., 1997; Kawai et al., 2021). Furthermore, ADAM17 also cleaves the active extracellular ectodomain of ACE2, releasing a soluble form of this enzyme (Figure 1), whose pathophysiology importance is not fully understood. (Melaku, 2018).
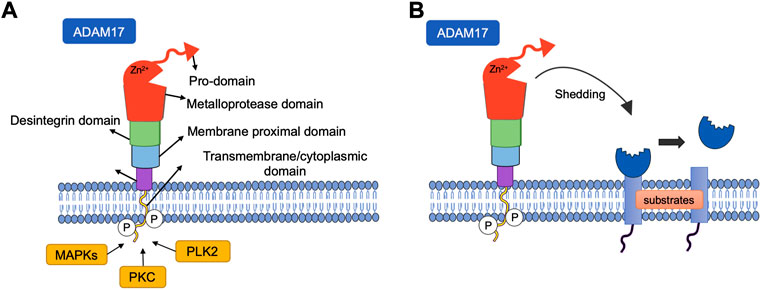
FIGURE 1. (A) Structure of ADAM metallopeptidase domain 17 (ADAM17). The structure of ADAM17 can be divided into six domains, all of which have distinct functions. CANDIS: Conserved ADAM-seventeen Dynamic Interaction Sequence. (B) Shedding of cell surface proteins induced by ADAM17. ADAM17 function is regulated by phosphorylation of the cytoplasmic domain by intracellular kinases such as protein kinase C (PKC), Polo-like kinase 2(PLK2), Mitogen Activated Protein Kinase (MAPKs). Various substrates, including receptors, cytokines and growth factors are targets of this protease, undergoing cleavage and release in the soluble form.
Membrane-bound ACE2 induces the production of Ang-(1–7) from Ang-II in vessel walls and other tissues. The Ang-(1–7) formed is involved in cardioprotective effects mediated by its binding and activation of the MasR (Yang et al., 2017), promoting the release of NO, decreased sympathetic activity and baroreflex dysfunction, as well as reduced BP levels (Xu et al., 2017). Thus, the ACE2-Ang-(1–7)-Mas axis counter-regulates the classic RAS in tissues involved in the maintenance of BP and homeostasis of the cardiovascular system (Rabelo et al., 2011).
ACE2 deficiency is associated with decreased NO bioavailability mediated downregulation of aortic endothelial nitric oxide synthase (NOS) expression (Rabelo et al., 2016). Furthermore, impairment of the ACE2/Ang-(1–7)/Mas pathway is associated with increased reactive oxygen species (ROS) production due to elevation nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) activity, as well as increased NO degradation (Rabelo et al., 2008). It has been shown that ADAM17 knockdown in the brain can restore ACE2 activity and attenuate the development of hypertension, suggesting that ADAM17-induced ACE2 cleavage is involved in the development of neurogenic hypertension (Xia et al., 2013). On the other hand, the inducible NOS (iNOS)/NO/cGMP/PKG pathway has also been shown to activate ADAM17, a mechanism that can limit inflammation (Chanthaphavong et al., 2012).
NO is a free radical, gaseous, lipophilic, and highly reactive (Wu et al., 2021), that induces an important role in the regulation of vascular homeostasis (Cyr et al., 2020). Numerous evidences demonstrate that NO plays a central role in the maintenance of BP. (Ahmad et al., 2018a).
The gaseous molecule is synthesized by three distinct isoforms of the enzyme NOS, each with different functional properties and expression patterns: iNOS, neuronal NOS (nNOS) and endothelial NOS (eNOS). In general, these proteins are involved in the synthesis of NO and L-citrulline from oxygen (O2) and L-arginine. (Cyr et al., 2020). This enzymatic conversion also requires cofactors such as tetrahydrobiopterin (BH4) and NADPH (Ahmad et al., 2018a).
Although eNOS is found primarily in vascular endothelial cells, its expression has been shown in several other cells, such as renal tubular epithelium, platelets, cardiac myocytes and certain neurons in the brain (Cyr et al., 2020). Vasodilator agonists, such as acetylcholine and bradykinin, induce an increase in intracellular calcium and promote eNOS activation, increasing NO production in endothelial cells (Ahmad et al., 2018a). The NO formed in the endothelial cell diffuses into the smooth muscle cell, activating guanylate cyclase (sGC) and producing cyclic guanosine monophosphate (cGMP), which in turn activates protein kinase G (PKG) which mediates intracellular Ca2+ reduction, thus favoring smooth muscle relaxation (Reinero et al., 2021).
The positive regulation of NO/sGC/cGMP pathway in hypertension has been emerged as a promising therapeutic mechanism to drecrease BP (Ahmad et al., 2018a). Reduced bioavailability of NO contributes to vascular dysfunction in hypertension (Li et al., 2015; Wang et al., 2022). The term endothelial dysfunction (ED) is a disorder characterized, anatomically, by intact endothelial cells (Di Daniele et al., 2021), but with alteration in the production of endothelium-derived factors, such as increased synthesis of ROS, abnormal production of pro-inflammatory cytokines, like TNF-α and interleukin-1 (IL-1), increased production of cyclooxygenase (COX) derivatives, such as thromboxane A2 and prostaglandins H2, E2, and F2α (Félétou, 2009; Bernatova, 2014; Kakabadze et al., 2021). In addition to attenuation of endothelium-derived hyperpolarizing factor and decreased production and action of NO.
In general, decreased NO bioavailability in hypertension is associated with decreased synthesis by eNOS or increased inactivation of NO by oxidative stress (Pinheiro et al., 2017). One study has showed a reduced activity and expression of eNOS in spontaneously hypertensive rats (SHR) (from 4 to 14 weeks), but not in normotensive Wistar Kyoto (WKY) control, which may contribute to the development of hypertension (Chou et al., 1998). Furthermore, in this genetic model of hypertension, it was also shown that hyperactivity of NADPH oxidase and the superoxide (O2-) produced are associated with reduced NO-dependent relaxation, ED, and vascular hypertrophy (Zalba et al., 2000).
In some disease conditions, including hypertension, activation of NADPH oxidase, especially NOX1 and NOX2 isoforms, promotes the synthesis of ROS, which causes the oxidation of BH4 to BH2, resulting in eNOS uncoupling (Li et al., 2015; Wu et al., 2021). This uncoupling is characterized by the discrepancy between NO production and eNOS levels, due to changes in the action of this enzyme, with consequent production of the O2- ion instead of NO (Bouloumié et al., 1997; Montezano and Touyz, 2012; Yuyun et al., 2018).
Certain cytokines have been reported to act on endothelial cells, contributing to reduced production and activity of vasodilator mediators, such as NO (Sprague and Khalil, 2009). As cells of the immune system are activated in hypertension, they produce cytokines which act on the adjacent tissue to promote the synthesis of ROS derived from NADPH oxidase (NOX1 and NOX2) (Manea, 2010). In the vasculature, for example, cytokine-stimulated ROS synthesis induces NO inactivation and attenuation of endothelium-dependent relaxation (Griendling et al., 2021).
It has been reported an increase in arginase expression in endothelial cells and acute inflammatory cells, with consequent reduction in L-arginine concentration, the substrate of eNOS (Cyr et al., 2020). Furthermore, TNF-α has also been shown to decrease eNOS mRNA levels in human endothelial cells (Yoshizumi et al., 1993).
TNF-α has already been shown to stimulate ROS production in endothelial cells, smooth muscle cells, and neutrophils (Madge and Pober, 2001; Sprague and Khalil, 2009). On the other hand, the ROS formed activates transcription factors, such as nuclear factor Kappa-B (NF-κB), which will modulate the gene expression of adhesion molecules, chemokines, and pro-inflammatory cytokines (Theofilis et al., 2021), that induce the activation and infiltration of more immune cells, such as macrophages, contributing to target organ damage, characteristic of hypertension (Griendling et al., 2021).
The action of cytokines, such as IL-1β, on macrophages stimulates the production of large amounts of NO by iNOS (Szabo, 1995). Increased expression of iNOS in SHR cultured smooth muscle cells has also been demonstrated (Wu et al., 1996). Large volumes of NO react with O2− to produce peroxynitrite, which can inhibit the activity of eNOS and sGC (Xia and Zweier, 1997; Leo et al., 2015).
It has been shown that the production of NO by iNOS has an effective role in the exacerbation of inflammation, which can promote an increase in leukocyte cytotoxicity during the inflammatory process, and also in the regulation and increase of COX-2, which is the enzyme that produces the mediators of inflammation (Hunter, 2002; Ye et al., 2008).
Blocking the NO production pathway using pharmacological tools such as the NOS inhibitor NG-nitro-L-arginine methyl-ester (L-NAME) has been used to study the importance of NO in hypertension (Ahmad et al., 2018a). Some studies demonstrate that inhibition of NO synthesis through oral administration of L-NAME produces arterial hypertension (Baylis et al., 1992; Seth et al., 2016), as well as cardiac remodeling, such as left ventricular hypertrophy and myocardial fibrosis, in addition to aortic wall thickening and aortic collagen deposition (Bunbupha et al., 2015). Corroborating previous reports, Jin et al. (2017) also demonstrated that L-NAME promoted cardiac dysfunction accompanied by reduced eNOS expression and NO bioavailability (Jin et al., 2017).
An important mechanism of L-NAME-induced hypertension is oxidative stress, since in this model the uncoupling of eNOS is evidenced, leading to an overwhelming generation of vascular O2- (Maneesai et al., 2018). According to Boonprom et al. (2017), the molecular mechanism related to cardiac fibrosis of L-NAME hypertensive rats involves the activation of oxidative stress (Boonprom et al., 2017). It has already been described that the lipid peroxidation was elevated in L-NAME-induced hypertension (Kumar et al., 2010; Kumar et al., 2012). In addition, significantly decreased levels of catalase (Cat), glutathione peroxidase (GPx) and superoxide dismutase (SOD) were evidenced in cardiac and aortic tissue (Sawant and Bodhankar, 2016) and erythrocytes (Kumar et al., 2012) of rats with L-NAME hypertension (Bunbupha et al., 2015).
Pro-inflammatory phenotypic alterations are also described in hypertension induced by NOS inhibition by L-NAME, including increased expression of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) in the vascular wall (Luvarà et al., 1998). Elevated transforming growth factor beta 1 (TGF-β1) plasma levels and renal TGF-β1 expression have also been reported, indicating that the raise in TGP-β1 expression may be related to renal and vascular changes induced by NOS suppression in the L-NAME model (Bunbupha et al., 2021). In this same model, increases in cardiac expression and serum levels of cytokines, such as interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and TNF-α were also demonstrated (Miguel-Carrasco et al., 2008; Bunbupha et al., 2015), as well as the overexpression in vascular and cardiac tissue of the protein iNOS (Bunbupha et al., 2015; Leo et al., 2015).
RAS overactivation may be involved in vascular and renal dysfunction, mediated by hypertension induced by NO biosynthesis inhibition. These abnormalities have been associated with increased serum angiotensin converting enzyme (ACE) activity as well as upregulation of AT1R expression in the kidney (Bunbupha et al., 2021). Research has shown increased ACE activity in both heart and aortic tissue in L-NAME hypertension, demonstrating a relationship between ACE activity and NO synthesis in this model of hypertension (Sawant and Bodhankar, 2016).
In addition, L-NAME administration increases Ang-II levels and promotes cardiac AT1R overexpression, and these effects are associated with left ventricular wall hypertrophy and fibrosis (Sonoda et al., 2017; Gao et al., 2018). In addition, exogenous Ang-II has also been documented to reduce L-arginine transport in aortic endothelial and renal cells of mice, which in turn can induce a reduction in NO availability and support the hypertension development (Rajapakse et al., 2014).
As previously described, NO is involved in several functions, such as regulating BP, vascular tone, smooth muscle proliferation as well as being involved in the inflammatory process (Machha and Schechter, 2011; Shreshtha et al., 2018). An important source of NO in the cardiovascular system is eNOS, but a variety of endogenous and exogenous stressors can alter the production of NO, in addition to increase its degradation (Cyr et al., 2020). These stressors, such as RAS overactivation, oxidative stress and inflammation, contribute to an imbalance that favors an increase of ROS and consequent reduction in NO bioavailability, which promotes endothelial dysfunction and increase in blood pressure (Figure 2).
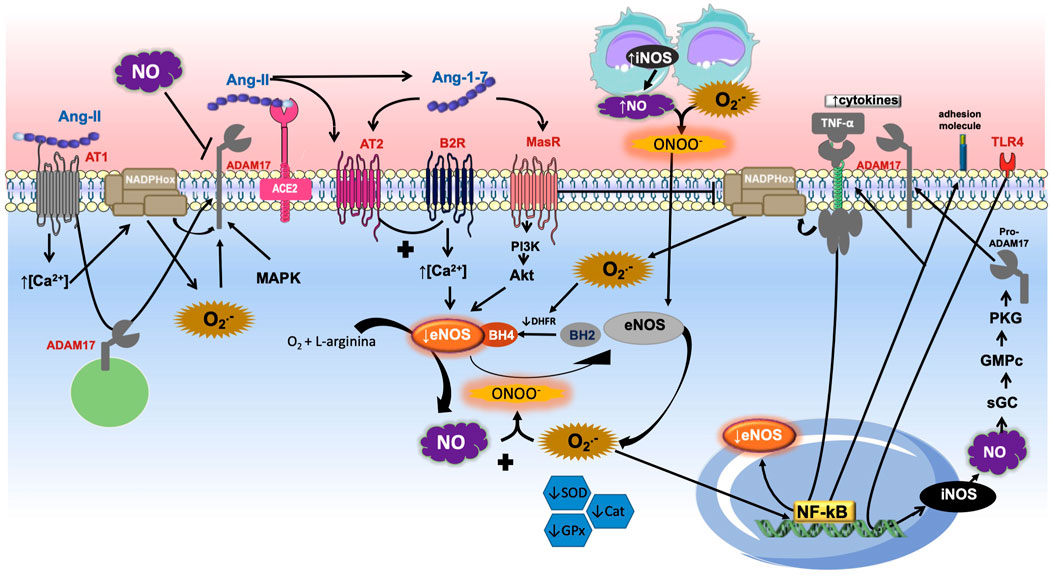
FIGURE 2. Role of ADAM17-mediated shedding and nitric oxide (NO). The Angiotensin-II (Ang-II) binding into Ang-II type 1 receptor (AT1R) induces the calcium concentration increase, which promotes nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) stimulation and increases the production of superoxide anion (O2-), which interacts with NO to produce peroxynitrite. Reactive species oxidize BH4 to BH2 causing eNOS uncoupling. The binding of Ang-II into its receptor causes desintegrin and metalloprotease 17 (ADAM17) moving to the membrane and its activation directly. ADAM17 also can be activated by reactive oxygen species (ROS) and MAP kinase family (MAPK). Once the ADAM17 is in the cell surface it induces shedding of diverse membrane-anchored proteins such as the angiotensin converting enzyme type 2 (ACE2), inhibiting the conversion of Ang-II into Ang-(1–7). The heptapeptide binds into Mas receptor (MasR) which induces PI3K/AKT pathway stimulation and NO formation through endothelial nitric oxide synthase (eNOS). Similarly, Ang-(1–7) is able to promote Ang-II type 2 (AT2R) receptor activation, which stimulates the bradykinin (BK)-NO cascade by bradykinin type 2 receptor (B2R). The increase of inducible NOS (iNOS) promotes NO production, which in the presence of high concentrations of O2- induces peroxynitrite (ONOO−) formation, further contributing to the uncoupling of eNOS. In addition, decreased levels of the endogenous antioxidants superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase (Cat) were observed. The pro-inflammatory binding of tumor necrosis factor α (TNF-α) into its receptor stimulates NADPH oxidase and ROS formation. O2- induces nuclear factor Kappa-B (NF-κB) promotion and decreasing eNOS, activation of adhesion molecules and enhance of TNF-α pathway. This figure also shows that the activation of toll-like receptor 4 (TLR4) stimulates the transcription of iNOS and consequent activation of guanylate cyclase (sGC), cyclic guanosine monophosphate (cGMP), protein kinase G (PKG) pathway. It promotes the maturation and translocation of ADAM17 to cell surface, with consequent shedding of TNF-α receptor and decreasing of inflammation development.
ADAM17 posses a great dissemination and It is involved acting as a sheddase by proteolytic cleavage of several cell surface proteins enroled in the inflammation progression, involving proteins of the signal transduction, control of cell adhesion, and release of growth factors and various cytokines (Hao et al., 2004; Garton et al., 2006; Iwata et al., 2009). The TNF-ɑ, cytokine receptors (IL-6 receptor (IL-6R), macrophage colony stimulating factor I (M-CSFRI) and TNF-receptor I and II (TNF-RI and TNF-RII) are among the most studied cytokines/receptors as targets for ADAM17 shedding (Müllberg et al., 1993; Black et al., 1997; Garton et al., 2001; De Queiroz et al., 2020).
TNF-ɑ has pro-inflammatory effects which are related with activation of TNFR1, and it increased in human serum was associated to be a signal in the hypertension development in patients with chronic kidney disease (Saulnier et al., 2014). The deletion of TNFR1 gen in mice led to a rise in systolic BP in response to Ang-II infusion (Chen et al., 2010). Another study demonstrated that deletion of TNFR1 in subfornical organ ameliorates sympathetic drive and heart failure in rats (Yu et al., 2017), which can be due to inhibition of TNFR2 shedding, leading to TNF-ɑ actions and increasing BP induced by Ang-II (Chen et al., 2010). All together, these findings indicate that selective activation or deletion of TNFR1 contributes to a mechanism that may lower or increase BP, respectively.
Studies have shown that the ADAM17-dependent TNF-ɑ shedding involves a critical molecule called inactive rhomboid protein 2 (RHBDF2, known as iRhom2). This protein is included in the family of rhomboid protease of catalytically inactive serine proteases. In mammals, two iRhoms were identified, iRhom1 and iRhom2. The first one is ubiquitously expressed in many cells and tissues, whereas iRhom2 is assumed to be expressed mostly in cells of immune system (Babendreyer et al., 2020). This protein is enrolled in ADAM17 maturation (Siggs et al., 2012), however the underlying mechanism is still little explored (Li et al., 2017).
The metalloprotease 17 is synthesized with a pro-form with a prevention of the catalytical domain of the sheddase. Once ADAM17 is carried from the endoplasmic reticulum (ER) to the Golgi complex (GC), the pro-domain is cleaved by pro-protein convertases, such as furin. Then, the proteolytically active, mature ADAM17 is carried to the cell surface (Endres et al., 2003). iRhoms are key controllers in this mechanism likewise they are crucial for the transport of ADAM17 from the ER to the GC (Adrain et al., 2012; Mcilwain et al., 2012; Babendreyer et al., 2020).
Alterations evoked by ADAM17-induced cytokines shedding and other membrane proteins, such as ACE2, may lead to endothelium dysfunction (Kawai et al., 2021), which in turn, induces vascular hemodynamics, endothelial damage, barrier dysfunction, and dysregulation of vascular tonus control, thus changing the vascular environment, leading to cardiovascular incidents (Zhang et al., 1995; Zhang et al., 2020; Da Silva et al., 2021).
The blood flow applies a continuous shear stress on the endothelium. Biological levels of shear stress led to a vasorelaxant, anti-thrombotic and anti-inflammatory phenotype in endothelial cells. ED is associated with an inflammatory response of the endothelium (Rajendran et al., 2013). Inflammatory responses may be beneficial to remove pathogens in the tissue or to initiate its repair. Nevertheless, persistent inflammation provides ED, which can produce various CVDs (Davignon and Ganz, 2004).
The relation between inflammation and hypertension have been studied for more than two decades, however the precise pathophysiological mechanism involved in that association is intricate and partly understood.
The activation of bradykinin receptor B1 (B1R)-mediated signaling pathways, a very known peptide inductor of inflammation, is associated in the pathogenesis of many CVDs that are associated with or induced by inflammation (Sriramula, 2020). Studies have showed that activation of B1R in the PVN occasioned elevated neuroinflammation, ROS production and sympathoexcitation, inducing to the development of neurogenic hypertension in deoxycorticosterone acetate-salt (DOCA)-treated mice (Sriramula and Lazartigues, 2017). In addition, our collaborators have demonstrated the evidence for ADAM17-mediated ACE2 shedding in neurons (Xu et al., 2018). For the first time, Parekh and Sriramula (2020) have showed that B1R activation produced an ADAM17-mediated shedding and lower ACE2 activity in neurons. Furthermore, the results support the new concept that B1R is implicated in glutamate-mediated effects on ADAM17 activity and cleavage of ACE2 (Parekh and Sriramula, 2020).
In an interesting study using a mouse model of Ang-II-induced hypertension without smooth muscle ADAM17 participation, caused by gen deletion or systemic pharmacological inhibition of ADAM17, the vascular hypertrophy and perivascular fibrosis were reduced (Takayanagi et al., 2016). The mechanism involved in this effect has been associated to the mediation of ADAM17 in the epidermal growth factor receptor (EGFR) transactivation induced by Ang-II in vascular smooth muscle cells (Forrester et al., 2016; Kawai et al., 2021). Furthermore, in vitro findings have showed that ROS are also able to induce the activation of ADAM17 in platelets, and this effect would illustrate a limiting mechanism for platelet function (Brill et al., 2009). Also, the production of ROS mediated by NADPH oxidase 4 (NOX4) is required to induce ADAM17 expression and following induction of cardiac hypertrophy (Zeng et al., 2013).
Our research group has used an antioxidant approach with the lipoic acid (LA) to demonstrate that LA was able to reduce BP and improves baroreflex sensitivity in renovascular hypertensive rats (Queiroz et al., 2012). In addition, using Neuro2A cells (neuroblastoma cell line) and a DOCA-salt model of hypertension, the antioxidant therapy preserved ACE2 compensatory role by breaking the feedforward cycle between oxidative stress and ADAM17 especially due to the oxidative stress decreasing. Thus, ADAM17 could be a new target to preclude the development of neurogenic hypertension (De Queiroz et al., 2015). Moreover Zeng et al. (2019) documented that NADPH oxidase, such as Nox1/4 subunits inhibitor GKT137831 inhibited hypertensive cardiac remodeling, and it reduced the expression of ADAM17 and pro-inflammatory cytokines such as TNF-α, IL-1β and IL-6 in abdominal artery coarctation-induced hypertensive rats (Zeng et al., 2019).
The findings described in this topic demonstrate the critical role of ADAM17. This metalloprotease needs to be in the active form to induce the shedding of multiple proteins related to inflammation such as cytokines, adhesion molecules and cytokines receptors. The activation and transportation of ADAM17 to the membrane surface is elicited by iRhom proteins, especially iRhom2. In addition, the cytokine TNF-ɑ is recognized to promote inflammation and BP increasing by its binding in TNF-ɑ receptors. On the other hand, It is already known that brain or vascular inflammation promotes increase of BP. This mechanism involves ROS production, ADAM17 activation and ACE2 dysfunction with a injury in Ang-(1–7) formation and deleterious effects, including hypertension development.
In hypertension induced by chronic inhibition of NO synthesis, it has been described that Ang-(1–7) can attenuate BP elevation and target organ damage in L-NAME-treated SHR (Benter et al., 2006). It was also described that activation of the MasR by Ang-(1–7) causes a vasorelaxant effect through the eNOS-NO-cGMP-PKG pathway, which contributes to the decrease in BP in SHR (Zhang et al., 2019). However, ACE2, the Ang-(1–7) producing enzyme, deficiency is associated with lower NO bioavailability through a reduction in aortic eNOS expression (Rabelo et al., 2016).
In addition, the vasoprotective effects of Ang-(1–7) are also related to the maintenance of NO availability due to the reduction in ROS (Heitsch et al., 2001). Benter et al. (2008) observed that Ang-(1–7) promotes inhibition of NADPH oxidase NOX4 and prevents renal vascular dysfunction in diabetic hypertensive rats (Benter et al., 2008). Another study documented that chronic infusion of Ang-(1–7) attenuated the increase in the expression of gp91phox, a catalytic subunit of the NADPH oxidase, in a dose-dependent manner in SHR brain (Jiang et al., 2013). Moreover, indicators of oxidative stress were evidenced in both the aorta and plasma of mice with MasR gene deletion, this was demonstrated by the higher levels of thiobarbituric acid reactive substances, upregulation of gp91phox and reduction in SOD and catalase activities in this animal model (Xu et al., 2008).
It has also been reported that reduced levels of NO can reduce the formation of Ang-(1–7) (Rajapakse et al., 2019). Interestingly, one study suggests that NO is a permissive factor for macula densa renin release (Castrop et al., 2004) and that exogenous (NO donor) and endogenous NO inhibit the ACE from human serum and cultured endothelial cells (Persson et al., 2000). Based on these results, Rajapakse et al. (2019) suggested that NO can act through activating a cardioprotective pathway of the RAS, by increasing the production of Ang-I, but reducing the conversion of Ang-I to Ang-II. In this sense, more Ang-I may be available for the synthesis of Ang-(1–7) (Rajapakse et al., 2019). In addition, studies have showed that NO directly interacts with AT1R, promoting its inhibition as showed in vascular smooth muscle cells (Ichiki et al., 1998; Ahmad et al., 2018a). Also, the increase of the exogenous NO precursor induced an upregulation in the eNOS/NO/cGMP pathway and diminished the Ang-II levels in a model of cardiac hypertrophy in rats (Ahmad et al., 2018b).
A correlation between NO and ADAM17 has been documented, as it was evidenced in research that exogenous NO donor reduces ADAM17 activity, as shown by the decrease of substrates released by ADAM-17 in murine endothelial cells (Bzowska et al., 2009). This reduction in NO-induced ADAM17 activation possibly involves nitrosylation of the thiol group on cysteine residues in the inhibitory prodomain of this enzyme (Zhang et al., 2000; Xu et al., 2016).
The mechanisms of ADAM17 activation still remain controversial, however, some evidence points to ROS, NOX4 and MAP-kinase family proteins as possible activators (Brill et al., 2009; Kuan et al., 2013; Zeng et al., 2013). Some years ago, NO was revealed as an activator of TACE, as shown by Chanthaphavong et al. (2012) that demonstrated the nitric oxide synthase (iNOS) stimulating factors such as lipopolysaccharide (LPS), through the Toll-like four receptor (TLR4), induce iNOS transcription, which culminates in the production of NO in hepatocytes. This lipid-soluble molecule binds to sGC which promotes the formation of cGMP and activation of PKG, which in turn promotes the phosphorylation and activation of ADAM17 (Chanthaphavong et al., 2012).
NO also plays an important role in activating ADAM17. Studies have showed that increases in Ca2+ concentrations can also induce NOS activation and promote NO release, which has the ability to inhibit the cytochrome oxidase enzyme of the electron transport chain and ultimately leads to formation of ROS, activating ADAM17 (Dada and Sznajder, 2011).
The activation of ADAM17 is related with the up-regulation of iRhom2, likewise the phosphorylation of ADAM17 and iRhom2, which are also dependent of NO/cGMP/PKG pathway. These conclusions indicate that the increase of iNOS/NO expression includes the rapid shedding of TNFR1 to limit the TNF-ɑ signaling (Chanthaphavong et al., 2012; Deng et al., 2015). Due to a central and tissue specific regulator of ADAM17, recently iRhom2 has arisen as a novel target protein for a specific inhibition of ADAM17 (Geesala et al., 2020).
TNF-ɑ signaling also has been associated with a growth of salt appetite and sodium reabsorption stimulation in renal tubules by a mechanism that involve the suppressing of NOS, becoming a critical factor to induce hypertension (Ramseyer and Garvin, 2013; Zhang et al., 2014; Rudemiller and Crowley, 2016). In addition, TNF-ɑ knockout mice showed a growth in eNOS formation and prevented an increase in BP of Ang-II-induced hypertension mice in comparison with the wild type animals (Sriramula et al., 2008; Rodriguez-Iturbe et al., 2017).
In addition to NO, other endogenous gaseous mediators have revealed to modulate ADAM17 level/expression, such as the gas hydrogen sulfide (H2S). Studies have showed that H2S eliminates the both mRNA and protein TNF-α-induced TACE expression. H2S was able to prevent TNF-α-induced endothelial damage through a protective mechanism mainly mediated by downregulation of ADAM17 activity with following suppression of soluble TNF-α shedding and the cytokine monocyte chemoattractant protein (MCP-1) release in the endothelial cells medium. These findings show the protective role of H2S, especially as inhibitor of inflammatory and pro-atherogenic processes (Perna et al., 2013).
As mentioned earlier, NO is involved in diverse physiological mechanisms, including the pathways that promote BP reduction. NO releasing can be evoked by MasR activation induced by Ang-(1–7), which is formed, especially, by ACE2, inducing in turn, reduction in BP. This convertase enzyme could be negatively modulated by ADAM17. Studies have demonstrated that NO could inhibit ACE expression and also ADAM17 activity. Conversely, ROS, NOX4 and other MAP kinases were able to activate ADAM17, in addition to Ang-II binding into AT1R. Furthermore, NO produced by iNOS would induce the cGMP/PKG pathway, promoting ADAM17 phosphorylation and its activation. The active form of ADAM17 is able to induce TNFR1 shedding and limit the TNF-ɑ actions in that receptor. Nevertheless, these actions persist controversial (Figure 2).
ADAM17 orchestrates many different signaling pathways linked to hypertension and other CVDs as showed in this review. ADAM17 has showed to induce neointimal hyperplasia in vasculature (Takaguri et al., 2011). The TACE expression is increased in atherosclerosis (Canault et al., 2006) and in the left ventricle after Ang-II infusion (Patel et al., 2014) and that ADAM17 polymorphism is associated with cardiovascular mortality (Morange et al., 2008; Takayanagi et al., 2016). Therefore, the metalloprotease 17 presents itself as a possible therapeutic target to the treatment of hypertension.
Ludwig et al. (2005) identified an ADAM17 inhibitor, named GW280264X, which has showed to block the constitutive release of mIL-6R, CX3CL1/fractalkine, and chemokine C-X-C ligand 16 (Ludwig et al., 2005). Recently, another study has developed a new ADAM17 inhibitor composed of a zinc-binding dithiol moiety, SN-4, which showed a specif binding to ADAM17, avoiding TNF-α shedding (Tateishi et al., 2021). Together, the studies demonstrate possible future TACE inhibitors. These effects would induce a decrease in inflammation and a consequent improvement in hypertension. In addition, inhibition of other ADAM17 activators for instance ROS, as showed by our group using lipoic acid or a SOD mimetic, tempol, were able to prevent the hypertension development in a DOCA-salt hypertension model (De Queiroz et al., 2015).
ADAM17 also triggers BP through a CNS-dependent mechanism. DOCA-salt hypertensive rats exhibited an ADAM17 increase in the hypothalamus, with a consequent reduction in ACE2 expression and activity in the brain, inducing a BP enhance, inflammation and autonomic dysfunction. So, knockdown of ADAM17 in the brain can blunt the development of hypertension (Xia et al., 2013). Furthermore, in the brain of hypertensive patients ADAM17-mediated ACE2 shedding seems to be stimulated by Ang-II, suggesting the participation of ADAM17 in neurogenic hypertension in human (Xu et al., 2017; Kawai et al., 2021).
Studies also have showed limitations to reveal the ADAM17 as a therapeutic target in hypertension. The increase of ADAM17 showed an essential role in the signal transduction for cardiovascular remodeling associated with ER stress however not for hypertension in Ang-II-treated mice (Takayanagi et al., 2016). Other findings have demonstrated that TNF-ɑ has associated with reduction in BP and strong inflammation. These opposite effects probably are due to the biding into two different receptors TNFR1 and TNFR2. Thus, the exact receptor subtype in ADAM17 downstream pathway to induce TNF-ɑ shedding still controversial (De Queiroz et al., 2020).
As shown along this manuscript, the protein iRhom2 acts as a key molecule in the ADAM17 activation and transportation to cell surface membrane. For this reason, the inactive rhomboid protein also becomes a possible pharmacological target to the treatment of hypertension, however more studies are need to address this question.
Here we briefly reviewed the role of ADAM17 shedding and nitric oxide. Both metalloprotease and gaseous molecule have showed central roles in the hypertension increase and promotion of other cardiovascular diseases. NO is the main gaseous molecule released from the vascular endothelium, which induces vasodilation mainly by NO/sGC/cGMP/PKG pathway. ADAM17, also named TACE, is involved in the shedding of many inflammatory cytokines and also in ACE2 cleavage. This last peptide is critical in the conversion of Ang-II into Ang-(1–7), which promotes the anti-hypertensive effects through the binding into MasR or AT2R, equally inducing NO releasing. ADAM17 can be activated by ROS, NOX4 and MAP-kinase family proteins. Furthermore, the activation of ADAM17 is associated with the up-regulation of iRhom2, which is phosphorylated by a NO/cGMP/PKG-dependent mechanism. This last mechanism was observed in hepatocytes, which suggest us to study it in a cardiovascular model of hypertension. Therefore, once ADAM17 is expressed in the membrane, it stimulates the shedding and pro-hypertensive effects. However, the ADAM17 inhibition by ROS decreasing, downregulation of some MAP-kinases, or upregulation of Ang-(1–7)/MasR pathway, inducing NO formation, produces reduction in hypertension development.
NC, TQ, and MC conceived the manuscript and revised it critically. MS, VS, MS, and TP drafted the manuscript and prepared the figures.
This study was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (436,605/2018-0) to TQ. This study was also financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brazil (CAPES)—Finance Code 001.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adrain, C., Zettl, M., Christova, Y., Taylor, N., and Freeman, M. (2012). Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 335, 225–228. doi:10.1126/science.1214400
Ahmad, A., Dempsey, S. K., Daneva, Z., Azam, M., Li, N., Li, P.-L., et al. (2018a). Role of nitric oxide in the cardiovascular and renal systems. Int. J. Mol. Sci. 19, 2605. doi:10.3390/ijms19092605
Ahmad, A., Sattar, M. A., Azam, M., Khan, S. A., Bhatt, O., and Johns, E. J. (2018b). Interaction between nitric oxide and renal α1-adrenoreceptors mediated vasoconstriction in rats with left ventricular hypertrophyin Wistar Kyoto rats. PLoS One 13, e0189386. doi:10.1371/journal.pone.0189386
Arribas, J., and Borroto, A. (2002). Protein ectodomain shedding. Chem. Rev. 102, 4627–4638. doi:10.1021/cr010202t
Babendreyer, A., Rojas-González, D. M., Giese, A. A., Fellendorf, S., Düsterhöft, S., Mela, P., et al. (2020). Differential induction of the ADAM17 regulators iRhom1 and 2 in endothelial cells. Front. Cardiovasc. Med. 7, 610344. doi:10.3389/fcvm.2020.610344
Baylis, C., Mitruka, B., and Deng, A. (1992). Chronic blockade of nitric oxide synthesis in the rat produces systemic hypertension and glomerular damage. J. Clin. Invest. 90, 278–281. doi:10.1172/JCI115849
Benter, I. F., Yousif, M. H., Anim, J., Cojocel, C., and Diz, D. (2006). Angiotensin-(1–7) prevents development of severe hypertension and end-organ damage in spontaneously hypertensive rats treated with L-NAME. Am. J. Physiol. Heart Circ. Physiol. 290, H684–H691. doi:10.1152/ajpheart.00632.2005
Benter, I. F., Yousif, M. H., Dhaunsi, G. S., Kaur, J., Chappell, M. C., and Diz, D. I. (2008). Angiotensin-(1–7) prevents activation of NADPH oxidase and renal vascular dysfunction in diabetic hypertensive rats. Am. J. Nephrol. 28, 25–33. doi:10.1159/000108758
Bernatova, I. (2014). Endothelial dysfunction in experimental models of arterial hypertension: Cause or consequence? Biomed. Res. Int. 2014, 598271. doi:10.1155/2014/598271
Black, R. A., Rauch, C. T., Kozlosky, C. J., Peschon, J. J., Slack, J. L., Wolfson, M. F., et al. (1997). A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 385, 729–733. doi:10.1038/385729a0
Boonprom, P., Boonla, O., Chayaburakul, K., Welbat, J. U., Pannangpetch, P., Kukongviriyapan, U., et al. (2017). Garcinia mangostana pericarp extract protects against oxidative stress and cardiovascular remodeling via suppression of p47phox and iNOS in nitric oxide deficient rats. Ann. Anatomy-Anatomischer Anzeiger 212, 27–36. doi:10.1016/j.aanat.2017.03.007
Bouloumié, A., Bauersachs, J., Linz, W., SchöLkens, B. A., Wiemer, G., Fleming, I., et al. (1997). Endothelial dysfunction coincides with an enhanced nitric oxide synthase expression and superoxide anion production. Hypertension 30, 934–941. doi:10.1161/01.hyp.30.4.934
Brill, A., Chauhan, A. K., Canault, M., Walsh, M. T., Bergmeier, W., and Wagner, D. D. (2009). Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc. Res. 84, 137–144. doi:10.1093/cvr/cvp176
Bunbupha, S., Apaijit, K., Potue, P., Maneesai, P., and Pakdeechote, P. (2021). Hesperidin inhibits L‐NAME‐induced vascular and renal alterations in rats by suppressing the renin–angiotensin system, transforming growth factor-β1, and oxidative stress. Clin. Exp. Pharmacol. Physiol. 48, 412–421. doi:10.1111/1440-1681.13438
Bunbupha, S., Prachaney, P., Kukongviriyapan, U., Kukongviriyapan, V., Welbat, J. U., and Pakdeechote, P. (2015). Asiatic acid alleviates cardiovascular remodelling in rats with L‐NAME‐induced hypertension. Clin. Exp. Pharmacol. Physiol. 42, 1189–1197. doi:10.1111/1440-1681.12472
Bzowska, M., Stalińska, K., Mezyk-Kopeć, R., Wawro, K., Duda, K., Das, S., et al. (2009). Exogenous nitric oxide inhibits shedding of ADAM17 substrates. Acta Biochim. Pol. 56, 325–335. doi:10.18388/abp.2009_2465
Canault, M., Peiretti, F., Kopp, F., Bonardo, B., Bonzi, M. F., Coudeyre, J. C., et al. (2006). The TNF alpha converting enzyme (TACE/ADAM17) is expressed in the atherosclerotic lesions of apolipoprotein E-deficient mice: Possible contribution to elevated plasma levels of soluble TNF alpha receptors. Atherosclerosis 187, 82–91. doi:10.1016/j.atherosclerosis.2005.08.031
Castrop, H., Schweda, F., Mizel, D., Huang, Y., Briggs, J., Kurtz, A., et al. (2004). Permissive role of nitric oxide in macula densa control of renin secretion. Am. J. Physiol. Ren. Physiol. 286, F848–F857. doi:10.1152/ajprenal.00272.2003
Chanthaphavong, R. S., Loughran, P. A., Lee, T. Y., Scott, M. J., and Billiar, T. R. (2012). A role for cGMP in inducible nitric-oxide synthase (iNOS)-induced tumor necrosis factor (TNF) α-converting enzyme (TACE/ADAM17) activation, translocation, and TNF receptor 1 (TNFR1) shedding in hepatocytes. J. Biol. Chem. 287, 35887–35898. doi:10.1074/jbc.M112.365171
Chen, C. C., Pedraza, P. L., Hao, S., Stier, C. T., and Ferreri, N. R. (2010). TNFR1-deficient mice display altered blood pressure and renal responses to ANG II infusion. Am. J. Physiol. Ren. Physiol. 299, F1141–F1150. doi:10.1152/ajprenal.00344.2010
Chou, T.-C., Yen, M.-H., Li, C.-Y., and Ding, Y.-A. (1998). Alterations of nitric oxide synthase expression with aging and hypertension in rats. Hypertension 31, 643–648. doi:10.1161/01.hyp.31.2.643
Chow, F. L., and Fernandez-Patron, C. (2007). Many membrane proteins undergo ectodomain shedding by proteolytic cleavage. Does one sheddase do the job on all of these proteins? IUBMB life 59, 44–47. doi:10.1080/15216540600879087
Cyr, A. R., Huckaby, L. V., Shiva, S. S., and Zuckerbraun, B. S. (2020). Nitric oxide and endothelial dysfunction. Crit. Care Clin. 36, 307–321. doi:10.1016/j.ccc.2019.12.009
Da Silva, G. M., Da Silva, M. C., Nascimento, D. V. G., Lima Silva, E. M., Gouvêa, F. F. F., De França Lopes, L. G., et al. (2021). Nitric oxide as a central molecule in hypertension: Focus on the vasorelaxant activity of new nitric oxide donors. Biol. (Basel) 10, 1041. doi:10.3390/biology10101041
Dada, L. A., and Sznajder, J. I. (2011). Mitochondrial Ca2+ and ROS take center stage to orchestrate TNF-α-mediated inflammatory responses. J. Clin. Invest. 121, 1683–1685. doi:10.1172/JCI57748
Davignon, J., and Ganz, P. (2004). Role of endothelial dysfunction in atherosclerosis. Circulation 109, Iii27–32. doi:10.1161/01.CIR.0000131515.03336.f8
De Queiroz, T. M., Lakkappa, N., and Lazartigues, E. (2020). ADAM17-Mediated shedding of inflammatory cytokines in hypertension. Front. Pharmacol. 11, 1154. doi:10.3389/fphar.2020.01154
De Queiroz, T. M., Xia, H., Filipeanu, C. M., Braga, V. A., and Lazartigues, E. (2015). α-Lipoic acid reduces neurogenic hypertension by blunting oxidative stress-mediated increase in ADAM17. Am. J. Physiol. Heart Circ. Physiol. 309, H926–H934. doi:10.1152/ajpheart.00259.2015
Deng, M., Loughran, P. A., Zhang, L., Scott, M. J., and Billiar, T. R. (2015). Shedding of the tumor necrosis factor (TNF) receptor from the surface of hepatocytes during sepsis limits inflammation through cGMP signaling. Sci. Signal. 8, ra11. doi:10.1126/scisignal.2005548
Di Daniele, N., Marrone, G., Di Lauro, M., Di Daniele, F., Palazzetti, D., Guerriero, C., et al. (2021). Effects of caloric restriction diet on arterial hypertension and endothelial dysfunction. Nutrients 13, 274. doi:10.3390/nu13010274
Endres, K., Anders, A., Kojro, E., Gilbert, S., Fahrenholz, F., and Postina, R. (2003). Tumor necrosis factor-alpha converting enzyme is processed by proprotein-convertases to its mature form which is degraded upon phorbol ester stimulation. Eur. J. Biochem. 270, 2386–2393. doi:10.1046/j.1432-1033.2003.03606.x
Félétou, M. (2009). Calcium‐activated potassium channels and endothelial dysfunction: Therapeutic options? Br. J. Pharmacol. 156, 545–562. doi:10.1111/j.1476-5381.2009.00052.x
Forrester, S. J., Kawai, T., O'brien, S., Thomas, W., Harris, R. C., and Eguchi, S. (2016). Epidermal growth factor receptor transactivation: Mechanisms, pathophysiology, and potential therapies in the cardiovascular system. Annu. Rev. Pharmacol. Toxicol. 56, 627–653. doi:10.1146/annurev-pharmtox-070115-095427
Gao, Y., Wang, Z., Zhang, Y., Liu, Y., Wang, S., Sun, W., et al. (2018). Naringenin inhibits NG-nitro-L-arginine methyl ester-induced hypertensive left ventricular hypertrophy by decreasing angiotensin-converting enzyme 1 expression. Exp. Ther. Med. 16, 867–873. doi:10.3892/etm.2018.6258
Garton, K. J., Gough, P. J., Blobel, C. P., Murphy, G., Greaves, D. R., Dempsey, P. J., et al. (2001). Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J. Biol. Chem. 276, 37993–38001. doi:10.1074/jbc.M106434200
Garton, K. J., Gough, P. J., and Raines, E. W. (2006). Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J. Leukoc. Biol. 79, 1105–1116. doi:10.1189/jlb.0106038
Geesala, R., Issuree, P. D., and Maretzky, T. (2020). The role of iRhom2 in metabolic and cardiovascular-related disorders. Front. Cardiovasc. Med. 7, 612808. doi:10.3389/fcvm.2020.612808
Griendling, K. K., Camargo, L. L., Rios, F. J., Alves-Lopes, R., Montezano, A. C., and Touyz, R. M. (2021). Oxidative stress and hypertension. Circ. Res. 128, 993–1020. doi:10.1161/CIRCRESAHA.121.318063
Hao, L., Du, M., Lopez-Campistrous, A., and Fernandez-Patron, C. (2004). Agonist-induced activation of matrix metalloproteinase-7 promotes vasoconstriction through the epidermal growth factor-receptor pathway. Circ. Res. 94, 68–76. doi:10.1161/01.RES.0000109413.57726.91
Heitsch, H., Brovkovych, S., Malinski, T., and Wiemer, G. (2001). Angiotensin-(1-7)–stimulated nitric oxide and superoxide release from endothelial cells. Hypertension 37, 72–76. doi:10.1161/01.hyp.37.1.72
Hunter, R. P. (2002). Nitric oxide, inducible nitric oxide synthase and inflammation in veterinary medicine. Anim. Health Res. Rev. 3, 119–133. doi:10.1079/ahrr200246
Ichiki, T., Usui, M., Kato, M., Funakoshi, Y., Ito, K., Egashira, K., et al. (1998). Downregulation of angiotensin II type 1 receptor gene transcription by nitric oxide. Hypertension 31, 342–348. doi:10.1161/01.hyp.31.1.342
Iwata, M., Silva Enciso, J. E., and Greenberg, B. H. (2009). Selective and specific regulation of ectodomain shedding of angiotensin-converting enzyme 2 by tumor necrosis factor alpha-converting enzyme. Am. J. Physiol. Cell Physiol. 297, C1318–C1329. doi:10.1152/ajpcell.00036.2009
Jiang, T., Gao, L., Zhu, X.-C., Yu, J.-T., Shi, J.-Q., Tan, M.-S., et al. (2013). Angiotensin-(1–7) inhibits autophagy in the brain of spontaneously hypertensive rats. Pharmacol. Res. 71, 61–68. doi:10.1016/j.phrs.2013.03.001
Jin, S., Teng, X., Xiao, L., Xue, H., Guo, Q., Duan, X., et al. (2017). Hydrogen sulfide ameliorated L-NAME-induced hypertensive heart disease by the Akt/eNOS/NO pathway. Exp. Biol. Med. 242, 1831–1841. doi:10.1177/1535370217732325
Kakabadze, K., Megreladze, I., Khvichia, N., Mitagvaria, N., Kipiani, N., Dumbadze, M., et al. (2021). Some aspects of role of nitric oxide in the mechanisms of hypertension (experimental study). Cardiol. Res. 12, 16–24. doi:10.14740/cr1172
Kawai, T., Elliott, K. J., Scalia, R., and Eguchi, S. (2021). Contribution of ADAM17 and related ADAMs in cardiovascular diseases. Cell. Mol. Life Sci. 78, 4161–4187. doi:10.1007/s00018-021-03779-w
Kuan, T. C., Chen, M. Y., Liao, Y. C., Ko, L., Hong, Y. H., Yen, C. Y., et al. (2013). Angiotensin II downregulates ACE2-mediated enhancement of MMP-2 activity in human cardiofibroblasts. Biochem. Cell Biol. 91, 435–442. doi:10.1139/bcb-2013-0031
Kumar, S., Prahalathan, P., and Raja, B. (2012). Syringic acid ameliorates L-NAME-induced hypertension by reducing oxidative stress. Naunyn. Schmiedeb. Arch. Pharmacol. 385, 1175–1184. doi:10.1007/s00210-012-0802-7
Kumar, S., Saravanakumar, M., and Raja, B. (2010). Efficacy of piperine, an alkaloidal constituent of pepper on nitric oxide, antioxidants and lipid peroxidation markers in L-NAME induced hypertensive rats. Int. J. Res. Pharm. Sci. 1, 300–307.
Leo, M. D., Kandasamy, K., Subramani, J., Tandan, S. K., and Kumar, D. (2015). Involvement of inducible nitric oxide synthase and dimethyl arginine dimethylaminohydrolase in Nω-nitro-L-arginine methyl ester (L-NAME)-induced hypertension. Cardiovasc. Pathol. 24, 49–55. doi:10.1016/j.carpath.2014.09.002
Li, Q., Yon, J.-Y., and Cai, H. (2015). Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 33, 1128–1136. doi:10.1097/HJH.0000000000000587
Li, X., Maretzky, T., Perez-Aguilar, J. M., Monette, S., Weskamp, G., Le Gall, S., et al. (2017). Structural modeling defines transmembrane residues in ADAM17 that are crucial for Rhbdf2-ADAM17-dependent proteolysis. J. Cell Sci. 130, 868–878. doi:10.1242/jcs.196436
Ludwig, A., Hundhausen, C., Lambert, M. H., Broadway, N., Andrews, R. C., Bickett, D. M., et al. (2005). Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb. Chem. High. Throughput Screen. 8, 161–171. doi:10.2174/1386207053258488
Luvarà, G., Pueyo, M. E., Philippe, M., Mandet, C., Savoie, F. O., Henrion, D., et al. (1998). Chronic blockade of NO synthase activity induces a proinflammatory phenotype in the arterial wall: Prevention by angiotensin II antagonism. Arterioscler. Thromb. Vasc. Biol. 18, 1408–1416. doi:10.1161/01.atv.18.9.1408
Machha, A., and Schechter, A. N. (2011). Dietary nitrite and nitrate: A review of potential mechanisms of cardiovascular benefits. Eur. J. Nutr. 50, 293–303. doi:10.1007/s00394-011-0192-5
Madge, L. A., and Pober, J. S. (2001). TNF signaling in vascular endothelial cells. Exp. Mol. Pathol. 70, 317–325. doi:10.1006/exmp.2001.2368
Manea, A. (2010). NADPH oxidase-derived reactive oxygen species: Involvement in vascular physiology and pathology. Cell Tissue Res. 342, 325–339. doi:10.1007/s00441-010-1060-y
Maneesai, P., Bunbupha, S., Potue, P., Berkban, T., Kukongviriyapan, U., Kukongviriyapan, V., et al. (2018). Hesperidin prevents nitric oxide deficiency-induced cardiovascular remodeling in rats via suppressing TGF-β1 and MMPs protein expression. Nutrients 10, 1549. doi:10.3390/nu10101549
Mcilwain, D. R., Lang, P. A., Maretzky, T., Hamada, K., Ohishi, K., Maney, S. K., et al. (2012). iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 335, 229–232. doi:10.1126/science.1214448
Melaku, L. (2018). Angiotensin-converting enzyme 2 and its potential protective effect upon heart. Arch. Med. Health Sci. 6, 238. doi:10.4103/amhs.amhs_44_17
Miguel-Carrasco, J. L., Mate, A., Monserrat, M. T., Arias, J. L., Aramburu, O., and Vázquez, C. M. (2008). The role of inflammatory markers in the cardioprotective effect of L-carnitine in L-NAME-induced hypertension. Am. J. Hypertens. 21, 1231–1237. doi:10.1038/ajh.2008.271
Montezano, A. C., and Touyz, R. M. (2012). Reactive oxygen species and endothelial function–role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 110, 87–94. doi:10.1111/j.1742-7843.2011.00785.x
Morange, P. E., Tregouet, D. A., Godefroy, T., Saut, N., Bickel, C., Rupprecht, H. J., et al. (2008). Polymorphisms of the tumor necrosis factor-alpha (TNF) and the TNF-alpha converting enzyme (TACE/ADAM17) genes in relation to cardiovascular mortality: The AtheroGene study. J. Mol. Med. 86, 1153–1161. doi:10.1007/s00109-008-0375-6
Mukerjee, S., Gao, H., Xu, J., Sato, R., Zsombok, A., and Lazartigues, E. (2019). ACE2 and ADAM17 interaction regulates the activity of presympathetic neurons. Hypertension 74, 1181–1191. doi:10.1161/HYPERTENSIONAHA.119.13133
Müllberg, J., Schooltink, H., Stoyan, T., Günther, M., Graeve, L., Buse, G., et al. (1993). The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 23, 473–480. doi:10.1002/eji.1830230226
Parekh, R. U., and Sriramula, S. (2020). Activation of kinin B1R upregulates ADAM17 and results in ACE2 shedding in neurons. Int. J. Mol. Sci. 22, E145. doi:10.3390/ijms22010145
Patel, V. B., Clarke, N., Wang, Z., Fan, D., Parajuli, N., Basu, R., et al. (2014). Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell. Cardiol. 66, 167–176. doi:10.1016/j.yjmcc.2013.11.017
Perna, A. F., Sepe, I., Lanza, D., Capasso, R., Zappavigna, S., Capasso, G., et al. (2013). Hydrogen sulfide reduces cell adhesion and relevant inflammatory triggering by preventing ADAM17-dependent TNF-α activation. J. Cell. Biochem. 114, 1536–1548. doi:10.1002/jcb.24495
Persson, K., Whiss, P. A., Nyhlén, K., Jacobsson-Strier, M., Glindell, M., and Andersson, R. G. (2000). Nitric oxide donors and angiotensin-converting enzyme inhibitors act in concert to inhibit human angiotensin-converting enzyme activity and platelet aggregation in vitro. Eur. J. Pharmacol. 406, 15–23. doi:10.1016/s0014-2999(00)00647-6
Pinheiro, L. C., Tanus-Santos, J. E., and Castro, M. M. (2017). The potential of stimulating nitric oxide formation in the treatment of hypertension. Expert Opin. Ther. Targets 21, 543–556. doi:10.1080/14728222.2017.1310840
Prado, A. F., Batista, R. I., Tanus-Santos, J. E., and Gerlach, R. F. (2021). Matrix metalloproteinases and arterial hypertension: Role of oxidative stress and nitric oxide in vascular functional and structural alterations. Biomolecules 11, 585. doi:10.3390/biom11040585
Queiroz, T. M., Guimarães, D. D., Mendes-Junior, L. G., and Braga, V. A. (2012). α-lipoic acid reduces hypertension and increases baroreflex sensitivity in renovascular hypertensive rats. Molecules 17, 13357–13367. doi:10.3390/molecules171113357
Rabelo, L. A., Alenina, N., and Bader, M. (2011). ACE2–angiotensin-(1–7)–Mas axis and oxidative stress in cardiovascular disease. Hypertens. Res. 34, 154–160. doi:10.1038/hr.2010.235
Rabelo, L. A., Todiras, M., Nunes-Souza, V., Qadri, F., Szijártó, I. A., Gollasch, M., et al. (2016). Genetic deletion of ACE2 induces vascular dysfunction in C57bl/6 mice: Role of nitric oxide imbalance and oxidative stress. PloS one 11, e0150255. doi:10.1371/journal.pone.0150255
Rabelo, L. A., Xu, P., Todiras, M., Sampaio, W. O., Buttgereit, J., Bader, M., et al. (2008). Ablation of angiotensin (1-7) receptor Mas in C57Bl/6 mice causes endothelial dysfunction. J. Am. Soc. Hypertens. 2, 418–424. doi:10.1016/j.jash.2008.05.003
Rajapakse, N. W., Giam, B., Kuruppu, S., Head, G. A., and Kaye, D. M. (2019). Impaired l-arginine-nitric oxide pathway contributes to the pathogenesis of resistant hypertension. Clin. Sci. 133, 2061–2067. doi:10.1042/CS20190851
Rajapakse, N. W., Karim, F., Straznicky, N., Fernandez, S., Evans, R. G., Head, G. A., et al. (2014). Augmented endothelial‐specific L‐arginine transport prevents obesity‐induced hypertension. Acta Physiol. 212, 39–48. doi:10.1111/apha.12344
Rajendran, P., Rengarajan, T., Thangavel, J., Nishigaki, Y., Sakthisekaran, D., Sethi, G., et al. (2013). The vascular endothelium and human diseases. Int. J. Biol. Sci. 9, 1057–1069. doi:10.7150/ijbs.7502
Ramseyer, V. D., and Garvin, J. L. (2013). Tumor necrosis factor-α: Regulation of renal function and blood pressure. Am. J. Physiol. Ren. Physiol. 304, F1231–F1242. doi:10.1152/ajprenal.00557.2012
Reinero, M., Beghetti, M., Tozzi, P., Segesser, L. K. V., Samaja, M., and Milano, G. (2021). Nitric oxide–cGMP pathway modulation in an experimental model of hypoxic pulmonary hypertension. J. Cardiovasc. Pharmacol. Ther. 26, 665–676. doi:10.1177/10742484211014162
Rodriguez-Iturbe, B., Pons, H., and Johnson, R. J. (2017). Role of the immune system in hypertension. Physiol. Rev. 97, 1127–1164. doi:10.1152/physrev.00031.2016
Rudemiller, N. P., and Crowley, S. D. (2016). Interactions between the immune and the renin-angiotensin systems in hypertension. Hypertension 68, 289–296. doi:10.1161/HYPERTENSIONAHA.116.06591
Saulnier, P. J., Gand, E., Ragot, S., Ducrocq, G., Halimi, J. M., Hulin-Delmotte, C., et al. (2014). Association of serum concentration of TNFR1 with all-cause mortality in patients with type 2 diabetes and chronic kidney disease: Follow-up of the SURDIAGENE cohort. Diabetes Care 37, 1425–1431. doi:10.2337/dc13-2580
Sawant, S. H., and Bodhankar, S. L. (2016). Flax lignan concentrate ameliorates nω-nitro-L-arginine methyl ester (L-NAME)-Induced hypertension: Role of antioxidant, angiotensin-converting enzyme and nitric oxide. Pharmacologia 7, 157–169. doi:10.5567/pharmacologia.2016.157.169
Seth, M. K., Hussain, M. E., Pasha, S., and Fahim, M. (2016). Effects of a novel ACE inhibitor, 3-(3-thienyl)-l-alanyl-ornithyl-proline, on endothelial vasodilation and hepatotoxicity in l-NAME-induced hypertensive rats. Drug Des. devel. Ther. 10, 1533–1542. doi:10.2147/DDDT.S77761
Shreshtha, S., Sharma, P., Kumar, P., Sharma, R., and Singh, S. (2018). Nitric oxide: It’s role in immunity. J. Clin. Diagnostic Res. 12, 1–5. doi:10.7860/JCDR/2018/31817.11764
Siggs, O. M., Xiao, N., Wang, Y., Shi, H., Tomisato, W., Li, X., et al. (2012). iRhom2 is required for the secretion of mouse TNFα. Blood 119, 5769–5771. doi:10.1182/blood-2012-03-417949
Sonoda, K., Ohtake, K., Uchida, H., Ito, J., Uchida, M., Natsume, H., et al. (2017). Dietary nitrite supplementation attenuates cardiac remodeling in l-NAME-induced hypertensive rats. Nitric Oxide 67, 1–9. doi:10.1016/j.niox.2017.04.009
Sprague, A. H., and Khalil, R. A. (2009). Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 78, 539–552. doi:10.1016/j.bcp.2009.04.029
Sriramula, S., Haque, M., Majid, D. S., and Francis, J. (2008). Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 51, 1345–1351. doi:10.1161/HYPERTENSIONAHA.107.102152
Sriramula, S. (2020). Kinin B1 receptor: A target for neuroinflammation in hypertension. Pharmacol. Res. 155, 104715. doi:10.1016/j.phrs.2020.104715
Sriramula, S., and Lazartigues, E. (2017). Kinin B1 receptor promotes neurogenic hypertension through activation of centrally mediated mechanisms. Hypertension 70, 1122–1131. doi:10.1161/HYPERTENSIONAHA.117.09744
Szabo, C. (1995). Alterations in nitric oxide production in various forms of circulatory shock. New Horiz. 3, 2–32.
Takaguri, A., Kimura, K., Hinoki, A., Bourne, A. M., Autieri, M. V., and Eguchi, S. (2011). A disintegrin and metalloprotease 17 mediates neointimal hyperplasia in vasculature. Hypertension 57, 841–845. doi:10.1161/HYPERTENSIONAHA.110.166892
Takayanagi, T., Forrester, S. J., Kawai, T., Obama, T., Tsuji, T., Elliott, K. J., et al. (2016). Vascular ADAM17 as a novel therapeutic target in mediating cardiovascular hypertrophy and perivascular fibrosis induced by angiotensin II. Hypertension 68, 949–955. doi:10.1161/HYPERTENSIONAHA.116.07620
Tateishi, H., Tateishi, M., Radwan, M. O., Masunaga, T., Kawatashiro, K., Oba, Y., et al. (2021). A new inhibitor of ADAM17 composed of a zinc-binding dithiol moiety and a specificity pocket-binding appendage. Chem. Pharm. Bull. 69, 1123–1130. doi:10.1248/cpb.c21-00701
Theofilis, P., Sagris, M., Oikonomou, E., Antonopoulos, A. S., Siasos, G., Tsioufis, C., et al. (2021). Inflammatory mechanisms contributing to endothelial dysfunction. Biomedicines 9, 781. doi:10.3390/biomedicines9070781
Wang, Q., Zhang, C., Yang, C., Sun, Y., Chen, K., and Lu, Y. (2022). Capsaicin alleviates vascular endothelial dysfunction and cardiomyopathy via TRPV1/eNOS pathway in diabetic rats. Oxid. Med. Cell. Longev. 2022, 6482363. doi:10.1155/2022/6482363
Wu, C.-C., Hong, H.-J., Chou, T.-C., Ding, Y.-A., and Yen, M.-H. (1996). Evidence for inducible nitric oxide synthase in spontaneously hypertensive rats. Biochem. Biophys. Res. Commun. 228, 459–466. doi:10.1006/bbrc.1996.1682
Wu, Y., Ding, Y., Ramprasath, T., and Zou, M.-H. (2021). Oxidative stress, GTPCH1, and endothelial nitric oxide synthase uncoupling in hypertension. Antioxid. Redox Signal. 34, 750–764. doi:10.1089/ars.2020.8112
Xia, H., Sriramula, S., Chhabra, K. H., and Lazartigues, E. (2013). Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ. Res. 113, 1087–1096. doi:10.1161/CIRCRESAHA.113.301811
Xia, Y., and Zweier, J. L. (1997). Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc. Natl. Acad. Sci. U. S. A. 94, 6954–6958. doi:10.1073/pnas.94.13.6954
Xu, J., Mukerjee, S., Silva-Alves, C. R., Carvalho-Galvão, A., Cruz, J. C., Balarini, C. M., et al. (2016). A disintegrin and metalloprotease 17 in the cardiovascular and central nervous systems. Front. Physiol. 7, 469. doi:10.3389/fphys.2016.00469
Xu, J., Sriramula, S., and Lazartigues, E. (2018). Excessive glutamate stimulation impairs ACE2 activity through ADAM17-mediated shedding in cultured cortical neurons. Cell. Mol. Neurobiol. 38, 1235–1243. doi:10.1007/s10571-018-0591-8
Xu, J., Sriramula, S., Xia, H., Moreno-Walton, L., Culicchia, F., Domenig, O., et al. (2017). Clinical relevance and role of neuronal AT1 receptors in ADAM17-mediated ACE2 shedding in neurogenic hypertension. Circ. Res. 121, 43–55. doi:10.1161/CIRCRESAHA.116.310509
Xu, P., Costa-Goncalves, A. C., Todiras, M., Rabelo, L. A., Sampaio, W. O., Moura, M. M., et al. (2008). Endothelial dysfunction and elevated blood pressure in MAS gene-deleted mice. Hypertension 51, 574–580. doi:10.1161/HYPERTENSIONAHA.107.102764
Yang, G., Chu, P.-L., Rump, L. C., Le, T. H., and Stegbauer, J. (2017). ACE2 and the homolog collectrin in the modulation of nitric oxide and oxidative stress in blood pressure homeostasis and vascular injury. Antioxid. Redox Signal. 26, 645–659. doi:10.1089/ars.2016.6950
Ye, Y., Lin, Y., Manickavasagam, S., Perez-Polo, J. R., Tieu, B. C., and Birnbaum, Y. (2008). Pioglitazone protects the myocardium against ischemia-reperfusion injury in eNOS and iNOS knockout mice. Am. J. Physiol. Heart Circ. Physiol. 295, H2436–H2446. doi:10.1152/ajpheart.00690.2008
Yoshizumi, M., Perrella, M. A., Burnett, J., and Lee, M. E. (1993). Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ. Res. 73, 205–209. doi:10.1161/01.res.73.1.205
Yu, Y., Wei, S. G., Weiss, R. M., and Felder, R. B. (2017). TNF-α receptor 1 knockdown in the subfornical organ ameliorates sympathetic excitation and cardiac hemodynamics in heart failure rats. Am. J. Physiol. Heart Circ. Physiol. 313, H744–H756. doi:10.1152/ajpheart.00280.2017
Yuyun, M. F., Ng, L. L., and Ng, G. A. (2018). Endothelial dysfunction, endothelial nitric oxide bioavailability, tetrahydrobiopterin, and 5-methyltetrahydrofolate in cardiovascular disease. Where are we with therapy? Microvasc. Res. 119, 7–12. doi:10.1016/j.mvr.2018.03.012
Zalba, G., Beaumont, F. J., José, G. S., Fortuno, A., FortuñO, M. a. A., Etayo, J. C., et al. (2000). Vascular NADH/NADPH oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension 35, 1055–1061. doi:10.1161/01.hyp.35.5.1055
Zeng, S. Y., Chen, X., Chen, S. R., Li, Q., Wang, Y. H., Zou, J., et al. (2013). Upregulation of Nox4 promotes angiotensin II-induced epidermal growth factor receptor activation and subsequent cardiac hypertrophy by increasing ADAM17 expression. Can. J. Cardiol. 29, 1310–1319. doi:10.1016/j.cjca.2013.04.026
Zeng, S. Y., Yang, L., Yan, Q. J., Gao, L., Lu, H. Q., and Yan, P. K. (2019). Nox1/4 dual inhibitor GKT137831 attenuates hypertensive cardiac remodelling associating with the inhibition of ADAM17-dependent proinflammatory cytokines-induced signalling pathways in the rats with abdominal artery constriction. Biomed. Pharmacother. 109, 1907–1914. doi:10.1016/j.biopha.2018.11.077
Zhang, F., Tang, H., Sun, S., Luo, Y., Ren, X., Chen, A., et al. (2019). Angiotensin-(1-7) induced vascular relaxation in spontaneously hypertensive rats. Nitric Oxide 88, 1–9. doi:10.1016/j.niox.2019.03.007
Zhang, J., Patel, M. B., Griffiths, R., Mao, A., Song, Y. S., Karlovich, N. S., et al. (2014). Tumor necrosis factor-α produced in the kidney contributes to angiotensin II-dependent hypertension. Hypertension 64, 1275–1281. doi:10.1161/HYPERTENSIONAHA.114.03863
Zhang, Y., Murugesan, P., Huang, K., and Cai, H. (2020). NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 17, 170–194. doi:10.1038/s41569-019-0260-8
Zhang, Z. G., Chopp, M., Bailey, F., and Malinski, T. (1995). Nitric oxide changes in the rat brain after transient middle cerebral artery occlusion. J. Neurol. Sci. 128, 22–27. doi:10.1016/0022-510x(94)00216-b
Keywords: endotehlium, hypertension, inflammation, nitric oxide, metalloprotease, oxidative stress
Citation: da Silva MC, dos Santos VM, da Silva MVB, Prazeres TCMM, Cartágenes MdSS, Calzerra NTM and de Queiroz TM (2022) Involvement of shedding induced by ADAM17 on the nitric oxide pathway in hypertension. Front. Mol. Biosci. 9:1032177. doi: 10.3389/fmolb.2022.1032177
Received: 30 August 2022; Accepted: 04 October 2022;
Published: 14 October 2022.
Edited by:
Shiliang Tian, Purdue University, United StatesReviewed by:
Cristina Espinosa-Diez, University of Pittsburgh, United StatesCopyright © 2022 da Silva, dos Santos, da Silva, Prazeres, Cartágenes, Calzerra and de Queiroz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thyago Moreira de Queiroz, dGh5YWdvLnF1ZWlyb3pAdWZwZS5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.