 Vivek Sarohi
Vivek Sarohi Sanchari Chakraborty
Sanchari Chakraborty Trayambak Basak
Trayambak Basak- 1School of Biosciences and Bioengineering, Indian Institute of Technology (IIT)- Mandi, Himachal Pradesh, India
- 2BioX Center, Indian Institute of Technology (IIT)- Mandi, Himachal Pradesh, India
Extracellular matrix (ECM) plays a critical role in maintaining elasticity in cardiac tissues. Elasticity is required in the heart for properly pumping blood to the whole body. Dysregulated ECM remodeling causes fibrosis in the cardiac tissues. Cardiac fibrosis leads to stiffness in the heart tissues, resulting in heart failure. During cardiac fibrosis, ECM proteins get excessively deposited in the cardiac tissues. In the ECM, cardiac fibroblast proliferates into myofibroblast upon various kinds of stimulations. Fibroblast activation (myofibroblast) contributes majorly toward cardiac fibrosis. Other than cardiac fibroblasts, cardiomyocytes, epithelial/endothelial cells, and immune system cells can also contribute to cardiac fibrosis. Alteration in the expression of the ECM core and ECM-modifier proteins causes different types of cardiac fibrosis. These different components of ECM culminated into different pathways inducing transdifferentiation of cardiac fibroblast into myofibroblast. In this review, we summarize the role of different ECM components during cardiac fibrosis progression leading to heart failure. Furthermore, we highlight the importance of applying mass-spectrometry-based proteomics to understand the key changes occurring in the ECM during fibrotic progression. Next-gen proteomics studies will broaden the potential to identify key targets to combat cardiac fibrosis in order to achieve precise medicine-development in the future.
1 Introduction
Cardiovascular diseases (CVDs) remain the scourge of humanity causing a high number of morbidities and mortalities on a global scale. CVD refers to a broad range of disease manifestations affecting the heart, and subsequently, the vascular system (Murtha et al., 2017). Recent reports from American Heart Association (AHA) indicated ≈17.6 million (95% CI, 17.3–18.1 million) deaths globally were associated with CVD in 2016 accounting for an increase of 14.5% (95% CI, 12.1%–17.1%) deaths within a decade (from 2006) (Benjamin et al., 2019). Heart failure (HF) has remained one of the major contributors to the global burden of CVD. HF refers to the significant decline of the cardiac output (pumping capability of the organ) leading to dysfunction of the organ. According to NHANES data, in between 2009 and 2012, an estimated 5.7 million adults ≥20 years of age had HF and the prevalence increased to an estimated 6.2 million from 2013 to 2016 (Virani et al., 2020). According to the National Heart Failure Registry (India), almost 1.3 million (1% of the population) people get affected by HF annually (ICC NHFR Report, 2022). The prevalence of heart failure has risen over time among elder individuals (Savarese and Lund, 2017). Developed countries have a 1–2% incidence of heart failure but heart failure incidence in people older than 70 years has increased to 10%. It has only a 35% survival rate for patients after their first diagnosis (BLEUMINK et al., 2004). Interestingly, HF patients in India are much younger and have higher morbidity and mortality compared to the Western world (Chaturvedi et al., 2016; Dokainish et al., 2017). Clinical management of HF is quite challenging due to the lack of a precise therapeutic regimen. The cost of heart transplantation is still beyond the reach of the general population. Despite rigorous glycemic control, symptomatic lipid-lowering drugs, ion-channel blockers, and common use of renin–angiotensin–aldosterone system (RAAS) inhibitors, the prevalence of HF progression has remained steady, a fact that emphasizes the need for better therapies to stop HF progression (Domanski et al., 2003; Granger et al., 2003; Klotz et al., 2007). One of the most common pathophysiological characteristics that underpin most CVDs predisposing to heart failure is cardiac fibrosis. Cardiac fibrosis is a condition in which fibrous proteins (collagens, elastin, fibronectin, etc.) get excessively deposited in the extracellular matrix (ECM) of heart tissues (Tian et al., 2017). This deposition subsequently leads to stiffness, overgrowth, and scarring in the heart tissues resulting in heart failure (Weber and Brilla, 1991). ECM remodeling resulting in cardiac fibrosis is one of the significant processes involved in the progression of most of the CVDs causing HF. Cardiac fibrosis has emerged as one of the major causes of death due to tissue fibrosis accounting for almost ∼800,000 deaths per year globally (Hinderer and Schenke-Layland, 2019). Cardiac fibrosis is characterized by the development and relentless progression of fibrosis in the myocardium compartments in response to multiple pathogenic stimuli including oxidative stress, hypertension, hypoxia, and inflammation. Assembling the knowledge of intracellular signaling driving the excessive production of ECM components along with the biochemical remodeling in the extracellular space will provide a better understanding of the cardiac fibrosis mechanism. The exact composition of ECM differs in different tissues of different organs (McCabe et al., 2022). Since the ECM composition is varied in different tissues; it is of sheer importance to obtain an in-depth analysis of the myocardium matrisome to understand the pathophysiology of cardiac fibrosis. High-throughput techniques are required to perform in-depth ECM analysis. Next-gen sequencing (NGS) and proteomics are two techniques that are capable of performing in-depth analysis; although NGS-based workflows (mainly RNA-Seq analysis) provide a vivid snapshot of the gene expression during cardiac fibrosis (Faita et al., 2012; McLellan et al., 2020). On the contrary, the expressional abundances, post-translational modifications (hydroxylations, glycosylations, etc.), proteolytic processing, protein assembly network (crosslink analysis) of the core, and associated ECM proteins can only be depicted with the mass-spectrometry (MS)-based approaches (Barallobre-Barreiro et al., 2012; Terajima et al., 2014; Basak et al., 2016; Sarohi et al., 2022b). Mass-spectrometry (MS)-based proteomics has been useful in identifying primary sequences, protein–protein interactions, and PTMs (Aebersold and Mann, 2003). Conventional proteomics analysis of ECM matrisomes is filled with several challenges (Chang et al., 2016). A very high abundance of collagens present in ECM increases the dynamic range in detecting low-abundant ECM proteins. Furthermore, the insoluble nature of the ECM matrisome, highly post-translationally modified peptides (of collagens and elastin), and the presence of crosslinked peptides add more challenges to generate MS-amenable peptides (Basak et al., 2016; Chang et al., 2016; Hedtke et al., 2019; McCabe et al., 2022). Recently, modified biochemical methods for ECM proteomics sample preparation, varied fragmentation methods with high-resolution MS, and optimized bioinformatics workflows have made significant advancements to analyze ECM in-depth (Garcia-Puig et al., 2019; Taha and Naba, 2019; McCabe et al., 2021).
In this review, we summarize the role of different ECM components during cardiac fibrosis progression leading to heart failure. Furthermore, we highlight the importance of applying next-gen proteomics (NGP) to understand the key changes occurring in the ECM during fibrotic progression. NGP will provide greater molecular-level details regarding the different activated pathways and biochemical processes during cardiac fibrosis leading to the broadening of the potential to identify key targets to combat cardiac fibrosis.
2 Extracellular components altered during cardiac fibrosis
The extracellular matrix (ECM) has a tissue-specific structure and function. Cardiac muscle is primarily composed of terminally differentiated tissue-specific cells such as cardiomyocytes, fibroblasts, etc. (Figure 1). These cells contribute to the maintenance and deposition of cardiac-specific ECM. The cardiac ECM is a dynamic, robust, and functionally versatile component that forms the non-cellular part of the cardiac muscle. It acts not only as an architectural scaffold that supports the cardiac cells but also actively participates in the development and differentiation of cardiac and vascular cells (Bloksgaard et al., 2018) (Jane-Lise et al., 2000).
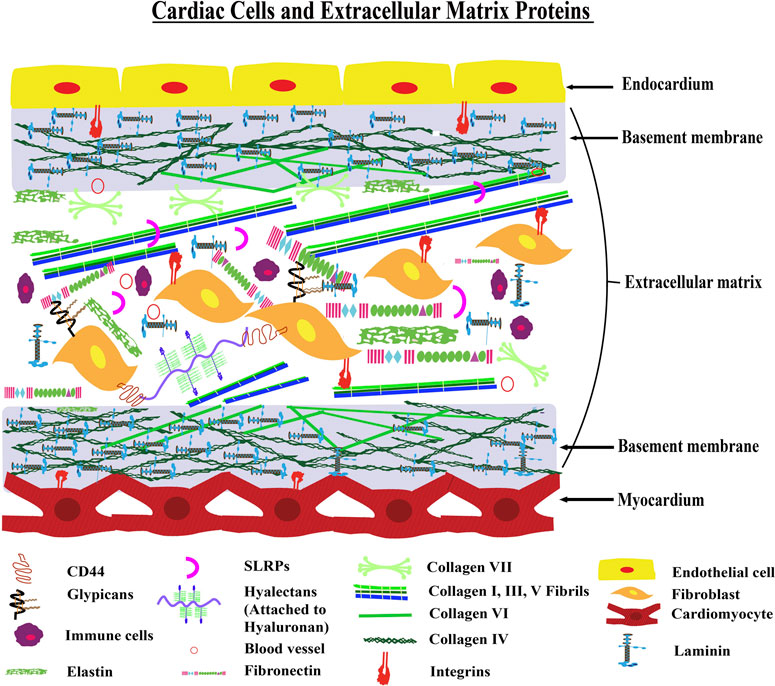
FIGURE 1. Architecture of Cardiac ECM—The figure shows a model transverse section of the myocardium and endocardium layers of the heart. The myocardium layer contains cardiac muscle cells with a basement membrane and the endocardium layer is made up of endothelial cells with a basement membrane. There is an interstitial matrix between the myocardium and endocardium layers. This interstitial space contains fibroblast and extracellular matrix proteins including collagens, elastin, and fibronectin. Different inflammatory cells are also present in the interstitial matrix conditionally.
2.1 Collagens
The primary source for the synthesis and deposition of collagens, one of the important components of the myocardium ECM, is the cardiac fibroblast (Villarreal et al., 2009; Hinderer and Schenke-Layland, 2019). There are 44 types of collagen chains present in the human proteome (Nauroy et al., 2018a). A recent proteomic study has shown that collagens have tissue-specific distinct expressional patterns present in the ECM (McCabe et al., 2022). However, collagen I is the most abundant protein in the ECM of almost every tissue in the human body including the myocardium (McCabe et al., 2022). Another proteomic study was done by Barallobre-Barreiro et al.; they identified 19 collagen chains in the human heart. They found the level of collagens (COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, COL6A1, and COL14A1) to be elevated in ischemic and non-ischemic heart disease patients compared to healthy humans (Barallobre-Barreiro et al., 2021a). Interestingly, single-nucleotide polymorphism in basement membrane collagen (COL4A1 and COL4A2) genes has been found to be associated with a higher risk of coronary artery disease (Steffensen and Rasmussen, 2018). In addition to collagen IV, collagen V was also found to be associated with cardiac diseases (Yokota et al., 2020). Yokota et al. performed a tandem mass tag (TMT)-based quantitative proteomic study of cardiac tissues from infarcted mice hearts and clearly depicted that the collagen (COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, COL5A3, COL6A1, COL6A2, and COL6A3) abundances were increased in the infarcted area. They also found that the deletion of collagen V resulted in a paradoxical increase in the size of post-infarction scars in mice. However, the absence of collagen V causes the scar to be less stiff. This less stiffness in the scar is sensed by mechanosensitive integrins. Then, integrin-signaling promotes transdifferentiation of fibroblast into myofibroblast leading to increased scar size. This indicates that collagen V regulates the scar size during post-MI in mice via an integrin-dependent manner (Yokota et al., 2020). Collagen I and III make up almost 95% of the total collagen present in the ECM-maintaining tissue structure (Di Lullo et al., 2002; Gagliano et al., 2009; Fedarko, 2014). They provide structural support by transmitting mechanical forces (contractile) throughout the myocardium to maintain cardiac muscle architecture during a regular cardiac cycle. Collagen I and III also contribute to the visco-elastic properties of the myocardium (Bishop, 1998). The increased accumulation of collagens I–III that occurs in the hypertensive heart and HF has been associated mostly with fibrosis (Weber et al., 1994a). A recent report suggested that even macrophages can contribute to fibrosis by producing excessive collagens, suggesting a secondary source for collagen production in the myocardium (Simões et al., 2020). The myocytes and endothelial cells are lined over basal lamina (basement membrane-BM) primarily composed of Type IV and VI collagens. Collagen-IV, the main component of BM and a highly post-translationally modified protein, plays a major role in the cell–matrix interaction thereby regulating cellular differentiation, migration, proliferation, adhesion, and signaling cascade. The signaling cascade is executed via the cell–ECM interaction through cell-surface receptors (integrins) with arginine, glycine, and aspartic acid (RGD) sequences present in the triple-helical domain. Col-IV and Col-VI expressions have been shown to be increased in pressure-overloaded rat myocardium leading to cardiac insufficiency by alterations of cell–ECM interactions (Kostin, 2000), (Chapman et al., 1990). More importantly, collagen post-translational modifications (PTMs) could also play a significant role in cell–ECM interactions (Myllyharju, 2008; Jürgensen et al., 2011; Pokidysheva et al., 2014; Stawikowski et al., 2014; Sipila et al., 2018; Rappu et al., 2019).
2.2 Collagen modifications
Collagen biosynthesis involves heavy post-translational modifications (PTMs). Proteomics studies have shown that these PTMs have a role in the stability, function, and communication of collagen I with other ECM components and cell-surface receptors to regulate cell adhesion, migration, and cardiac regeneration (Pokidysheva et al., 2014; Basak et al., 2016; Merl-Pham et al., 2019; Sarohi et al., 2022b). Injury and scars are non-repairable in the mammalian heart due to a lack of regeneration capability (Cutie and Huang, 2021). Contrasting to human cardiac tissues, zebrafish have the ability to regenerate heart tissue (Chen W. C. W. et al., 2016; Sánchez-Iranzo et al., 2018). In an interesting proteomic study done by Sarohi et al., they detected 36 collagen chains in zebrafish heart. The levels of collagen PTMs were found to be altered during the regeneration process in the zebrafish heart (Sarohi et al., 2022b). This hints toward the role of collagen PTMs in the zebrafish heart-regeneration process. Col-IV is more post-translationally modified compared to Col-I. The two amino acids which are majorly modified during collagen biosynthesis are proline and lysine (Myllyharju, 2005; Salo and Myllyharju, 2021). Proline can be either modified into 3-hydroxyproline (3-HyP) or 4-hydroxyproline (4-HyP). Prolyl 3-hydroxylation is catalyzed by prolyl 3-hydrolases. Prolyl 3-hydroxylase 1 (P3H1), P3H2, P3H3, and P3H4 isoforms are present in mammals (Vranka et al., 2004). On the other hand, prolyl 4-hydroxylation is catalyzed by prolyl 4-hydroxylases. Prolyl 4-hydroxylase subunit alpha-1 (P4HA1), P4HA2, and P4HA3 isoforms are involved in prolyl 4-hydroxylation of collagen chains (Salo and Myllyharju, 2021). While 4-HyP provides stability to the collagen structure (Salo and Myllyharju, 2021), the significance of the site-specific presence of 3-HyP in collagen chains has still remained elusive. Lack of 3-HyP in the collagen can cause abnormality in tissue function, embryonic growth, myopia, etc. (Cabral et al., 2007; Homan et al., 2014; Pokidysheva et al., 2014; Hudson et al., 2015). Lysine modifications are of diverse types in collagens. Lysine can be catalyzed to hydroxylysine (HyK) by procollagen-lysine, and 2-oxoglutarate 5-dioxygenases (PLODs) (Salo and Myllyharju, 2021), also known as lysyl hydroxylases (LHs). PLOD1, PLOD2, and PLOD3 isoforms are present in mammals. HyK serves as the primary substrate for the O-glycosylation modifications on collagen chains. Hydroxylation of lysines is essential for the assembly of a functional collagen structure. Lack of the lysyl-hydroxylation ability causes muscular abnormalities (Takaluoma et al., 2007). Hydroxylated lysines can be O-glycosylated to galactosyl-hydroxylysine (G-HyK) and glucosylgalactosyl-hydroxylysine (GG-HyK). O-glycosylation of HyKs is catalyzed by enzymes called procollagen galactosyltransferase 1 (COLGALT1) also known as GLT25D1 and COLGALT2 (GLT25D2) (Hennet, 2019; Salo and Myllyharju, 2021). Prolines and lysines are modified in large numbers in collagens. The role of the lysyl and prolyl-hydroxylation and O-glycosylation (G-HyK and GG-HyK) sites in collagens is poorly understood during fibrosis progression. Proteomics provides broader opportunities to identify these PTMs in a site-specific manner in order to study the roles of these PTMs during cardiac fibrosis (Merl-Pham et al., 2019; Sarohi et al., 2022b). Apart from hydroxylation and O-glycosylation, lysine can also be modified to allysines by oxidation. This reaction is catalyzed by lysyl oxidase enzymes. Lysyl oxidase (LOX) is a copper-dependent enzyme that oxidizes peptidyl lysine to peptidyl a-aminoadipic-γ-semialdehyde that begins the cross-linking of collagen and elastin in the extracellular matrices (Smith-Mungo and Kagan, 1998). LOX family is comprised of classical LOX and four other LOX-like enzyme isoforms— LOXL1, LOXL2, LOXL3, and LOXL4 (Molnar et al., 2003). Lu et al. showed that inhibition of LOX reduced myocardial fibrosis and cardiac remodeling whereas transforming growth factor-β (TGF-β) induced LOX activity and augmented cardiac fibrosis and heart failure (Lu et al., 2019). Ohmura et al. experimentally proved that the effect of cardiac hypertrophy agonist angiotensin II in the rat increases the LOXL1 mRNA by almost two-fold promoting enhanced myocardial stiffness (Ohmura et al., 2012). Increased crosslinking of collagens during fibrosis is possibly one of the main mechanistic phenomena involved in the progression of heart failure. Yang et al. showed that LOXL2 is up-regulated in the cardiac interstitium during cardiac fibrosis and found abundant levels in the serum of heart-failure patients (Yang et al., 2016). They have also shown that antibody-mediated inhibition of LOXL2 attenuates cardiac fibrosis and dysfunctions (Yang et al., 2016). LOXL3 has also been found to be associated with spinal development. Loss of LOXL3 can cause perinatal death (Zhang et al., 2015). The roles of LOXL3 and LOXL4 in cardiac fibrosis are not clearly understood to date. Recently, Busnadiego et al. experimentally showed that LOXL4 was involved in ECM deposition in endothelial cells during vascular remodeling induced by TGF-β (Busnadiego et al., 2013). These enzymes of the lysyl-oxidase family have emerged as attractive targets for tissue fibrosis. Recently, Santamaria et al. have shown that the LOX family members are increased post-MI and correlated with the accumulation of matured collagen in the infarct zone (González-Santamaría et al., 2016). Thus, understanding the functional role of these enzymes in ECM remodeling during cardiac fibrosis is of utmost importance. Targeted MS-based approaches (such as multiple reaction monitoring-MRM) can be developed to assess the levels of all these five forms of LOX family enzymes at once (Freue and Borchers, 2012). Developing this kind of assay will help achieve isoform specificity in a disease-specific manner. An increase of LOX family enzymes hints toward increased crosslink in the myocardium leading to more stiffened myocardium. Thus, the levels of these LOX-dependent crosslinks may be evaluated in greater detail in several animal models of cardiac fibrosis progression. Furthermore, circulating crosslink (allysine-mediated crosslinks), the measurement may also serve as a potential surrogate marker to assess the stage and severity of cardiac fibrosis among patients.
2.3 Fibronectin
Fibronectins (FN) are insoluble glycoproteins secreted by fibroblasts and are associated with the basal lamina of ECM (Schwarzbauer, 1991). Similar to Col-IV, they have binding domains (arginine, glycine, aspartic acid, or RGD motif) for cell-surface receptors, ECM proteins, and proteoglycans. The expression of FN is influenced by mechanical or local factors such as growth factors. The expressions of FN isoform mRNA EIIIB and EIIIA are preceded by an increased expression of TGF-β (Villarreal and Dillmann, 1992) followed by the deposition of collagens I-III in a fibrotic scar (Contard et al., 1991). Fibronectin, deposited in the tissue scar during heart failure (Oliviéro, 2000) is one of the ECM markers for cardiac fibrosis. Genetic inhibition of the FN in mice regulates the excessive deposition of collagens and transdifferentiation of myofibroblasts in the I/R heart failure mice model (Valiente-Alandi et al., 2018). In hypertrophied cardiomyocytes, deletion of FN improves survival (Konstandin et al., 2013).
2.4 Laminin
Laminin is a major structural glycoprotein of the basal lamina of cardiomyocytes. Laminin binds with transmembrane receptors present on the sarcolemma of cardiomyocytes and connects with collagen IV, perlecan, nidogen, and fibronectin (Tryggvason, 1993). Laminin transduces the interaction between the cytoskeleton of cardiomyocytes and the remaining is of the basal lamina of cardiomyocytes. It forms the structural component of the basal membrane and connects with extracellular domains and the cytoskeleton or myofibrils. It has different functional domains that bind collagen IV, nidogen, proteoglycans, and transmembrane receptors (Tryggvason, 1993). Merosin (Laminin 2) is abundant in cardiac-striated muscles. Deficiency of Laminin two results in breakage of cytoskeleton-extracellular matrix linkage leading to muscular dystrophy and dilated cardiomyopathy (Campbell, 1995). Sarcolemma properties in failing hearts may be contributed by an imbalance in the decreased or steady-state level of the laminin α2 chain and increased myocyte size (Lundgren et al., 1988). It is also proven that laminin and not fibronectin, is essential for the survival of adult cardiomyocytes in culture conditions (Lundgren et al., 1988). Furthermore, laminin α4 deficiency results in cardiac dysfunction and cardiac vessel dilation leading to heart failure. Laminin deficiency causes loss of interaction between actin of cardiomyocyte and ECM resulting in apoptosis (Wang et al., 2006). Structurally important laminin is essential for cardiac functioning. Impairment in laminin can contribute to the development of cardiac diseases.
2.5 Metalloproteinases
Matrix metalloproteinases (MMPs) are zinc-dependent enzymes catalytically active in the extra-cellular space (Gonzalez-Avila et al., 2019). MMPs are responsible for increasing or inhibiting matrix degradation which is a critical regulator of ECM remodeling during cardiac fibrosis (Frangogiannis, 2017). There are six types of MMPs found in ECM: gelatinases (MMP-2 and 9), collagenases (MMP-1, 8, 13, and 18), stromelysins (MMP-3, 10, and 11), matrilysins (MMP-7), membrane-type MMPs (MMP-14, 15, 16, and 17), and other unspecified MMPs. Collagenase and gelatinase MMPs play a crucial role in collagen cleavage in ECM (Frangogiannis, 2017). Collagenases (MMP-1) cleave collagens in fragments that result in the formation of substrates for other less-specific proteases, such as gelatinases (MMP-2). Less-specific MMPs are responsible for the degradation of type IV collagen and fibronectin (Shapiro, 1998). MMP-1 was found to attenuate cardiac fibrosis in suprarenal aortic-banding mice model (Foronjy et al., 2008). A study done on coronary artery-ligated rat revealed that inhibition of MMP-9 improves cardiac function by increasing autophagic flux and regulating ECM remodeling in post-MI chronic heart failure (Nandi et al., 2020). The level of MMP-14 was found to be elevated in swine models with left ventricle fraction shortening compared to the healthy controls (Plavelil et al., 2020). MMPs are regulated by a class of proteins called tissue inhibitors of metalloproteinases (TIMPs). TIMPs are the regulators of cardiac remodeling and myocardial ECM homeostasis. Four types of TIMPs: TIMP-1, TIMP-2, TIMP-3, and TIMP-4 are found in ECM and play a crucial role in cardiac fibrosis progression (Kandalam et al., 2011), (Kassiri et al., 2009; Takawale et al., 2017), (Yarbrough et al., 2014). A study performed on an experimental mice model of angiotensin II and pressure overload-induced cardiac fibrosis showed that TIMP-1 deficiency reduces cardiac fibrosis in mice (Takawale et al., 2017). A disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) is a metalloproteinase enzyme. ADAMTS has catalytic action on procollagen type I, II, III, and IV propeptides. However, the exact role of ADAMTS in the cardiac function is elusive. Interestingly ADAMTS-deficient mice showed exaggeration in the cardiac hypertrophy induced by pressure overload and angiotensin-II (Ang-II) (Wang et al., 2017). This observation indicates that ADAMTS has a role in cardiac hypertrophy regulation.
2.6 Cartilage intermediate layer protein
CILP is a secreted matricellular protein that has gained significant interest in the context of cardiac fibrosis recently. CILP has two isoforms—CILP1 and CILP2. CILP (CILP1) was mainly identified as a protein in the mid-zone of the articular cartilage (Lorenzo et al., 1998a). Lorenzo et al. first identified and named the protein on the basis of its immunolocalization in 1998. Although the precursor protein of CILP is a single polypeptide chain, it gets cleaved upon or before secretion by a furin-like protease into two fragments: N-terminal CILP and C-terminal CILP (Lorenzo et al., 1998b). The expression of CILP mRNA can be induced by TGF-β (Mori et al., 2006) whereas CILP-1 binds with TGF-β and inhibits the TGF-β-mediated pathways (Seki et al., 2005). It also acts as an antagonist of IGF-1 (Johnson et al., 2003). CILP is associated with cartilage structure and cartilaginous diseases such as Luber Disc Disease (LDD) (Seki et al., 2005). Masuda et al. found that mRNA expression of CILP was higher in adult (3–4 years old) porcine compared to young (7–10 days old) porcine (Masuda et al., 2001). Lorenzo et al. showed that CILP-1 gene expression was up-regulated in early-stage osteoarthritis (Lorenzo et al., 2004). In contrast to CILP-1, Bernardo et al. showed that CILP-2 gene expression was down-regulated. Previously, CILP was only considered as a protein involved in cartilage, but in 2012, Barallobre-Barreiro found in a proteomic analysis that CILP-1 is also involved in cardiac ECM remodeling (Barallobre-Barreiro et al., 2012; Van Nieuwenhoven et al., 2017). In 2018, Zhang et al. tried to experimentally establish that CILP-1 helps to reduce cardiac fibrosis through the TGF-β signaling pathway (Zhang et al., 2018). But recently, a study involving TMT-based proteomics showed that CILP-1 promotes cardiac fibrosis after MI interfering through the mTORC-1 pathway (Zhang et al., 2021). Thus, the role of CILP-1 during cardiac fibrosis has been established. Further exploration of the role of CILP-1 in different pathologies related to cardiac fibrosis is required for a better understanding of the function of CILP-1 in cardiac pathologies.
2.7 Transglutaminase
Transglutaminases are in the Ca2+-dependent enzyme family that involve post-translation modification of proteins where isopeptide crosslinking is formed due to transamidation of glutamine residue (Greenberg et al., 1991). Among the other transglutaminases, tissue transglutaminase-2 (TGM2) is mostly abundant in cardiomyocytes, macrophages, and vascular cells (Sane et al., 2007) and up-regulated in various fibrotic diseases (Benn et al., 2019). Multimerization of latent TGF-β binding proteins (LTBPs) is essential for the proper activation of profibrotic cytokine TGF-β in fibrosis and the process is enhanced by transglutaminase-2 (Troilo et al., 2016). Transglutaminase-2 participates in myofibroblast activation through the TGF-β pathway and plays a critical role in various fibrotic diseases including lung, kidney, and heart diseases. Wang et al. showed that inhibition of transglutaminase-2 attenuates cardiac fibrosis in Ang-II model (Wang et al., 2018).
2.8 Proteoglycans associated with cardiac fibrosis
Proteoglycans are densely glycosylated proteins. In the glycosylation process, glycosaminoglycan (GAG) chains are attached to serine residues of proteoglycans. Proteoglycans have distribution in the plasma membrane, extracellular matrix, and intracellular structure of cells (Frevert and Wight, 2006). In the extracellular matrix, proteoglycans have a quite complex distribution. Proteoglycans form complexes with collagen and hyaluronan (Frevert and Wight, 2006). On the basis of size, structure, and localization, proteoglycans are classified into four groups (AD et al., 2010).
2.8.1 Syndecans: cell-surface proteoglycans
Syndecans are proteoglycans present on the surface of cells. The syndecan group has four transmembrane receptors. The extracellular domain of these receptors interacts with growth factors (such as TGF-β, Ang-II, etc.) available in ECM (Couchman, 2003). In cardiac ECM, syndecan-1 and syndecan-4 play a significant role in the healing of MI and protection from injury and dysfunction. Syndecan-1 plays a cardioprotective role by enhancing MMP-2 and MMP-9 activities and inhibiting leukocyte migration and trans-endothelial adhesion (Vanhoutte et al., 2007). The cardioprotective role of syndecans-4 has been observed in many experiments. Without syndecans-4, endothelial cell proliferation tube, formation, and fibroblast impairment occur but overexpression of syndecans-4 impairs cardiac function (Matsui et al., 2011). However, in cardiac-remodeling experiments, syndecan-1 and syndecan-4 have been also shown to play other roles. Higher concentrations of extracellular syndecans-1 exaggerate fibrosis by increasing the expression of CCN-2 resulting in enhanced collagen deposition during angiotensin II-induced pressure overload in mice (Schellings et al., 2010). Overexpressed syndecan-4 increases cardiac rupture after MI by disrupting the granulation tissue formation (Matsui et al., 2011).
2.8.2 Hyalectans
Hyalectans are proteoglycans with lectin-binding properties. Hyalectans have the ability to bind with a different kind of GAG with no sulfate called hyaluronan. Versican is a proteoglycan of the hyalectan group (Iozzo and Murdoch, 1996). It is ubiquitously expressed in the human body. Studies on versican function in cardiac tissues showed that it is useful in the cardiac ECM during cardiac development (Cooley et al., 2012). Versican has the ability to bind with many components of ECM and inflammatory factors present in the ECM including fibulin (1 and 2), collagen I, hyaluronan, tenascin, fibronectin, and various chemokines (Wu et al., 2005). Studies on versican showed that versican has an important role in modulating inflammatory responses. This indicates that versican may have some role in the pathophysiology of many cardiac conditions (Kawashima et al., 2000).
2.8.3 Perlecan: basement membrane proteoglycans
Perlecan is a proteoglycan of the basement membrane. It is predominantly found in vascular ECM. Perlecan has both pro-angiogenic and anti-angiogenic properties (Mongiat et al., 2003; Iozzo et al., 2009). Fibroblast growth factor-2 (FGF-2) mediates the functions of perlecan. Perlecan plays a crucial role in cardiac development (Zhou et al., 2004; Sasse et al., 2008). A deficiency of perlecan in mice impairs cardiac development and is also critical for maintaining stability in the adult heart upon injury (Sasse et al., 2008). The role of perlecan in the pathophysiology of cardiac diseases is elusive.
2.8.4 Small leucine-rich proteoglycans
Small leucine-rich proteoglycans (SLRPs) are proteoglycans with small molecular weights (36–42 KDa). SLRPs are rich in leucine. In cardiac ECM, SLRPs have interactions with various cytokines, growth factors, cell-surface receptors, ECM proteins, collagens, TLRs, epidermal growth factor receptors and insulin growth factor receptors, low-density lipoprotein receptors, and TGF-β. The ability of SLRPs to interact with several components indicates the involvement of SLRPs in a wide range of cellular functions and pathophysiologic responses, including inflammation, collagen fibril assembly, fibrosis, cell transdifferentiation, and atherosclerosis (Cabello-Verrugio et al., 2012) (Hildebrand et al., 1994). SLRP decorin works as an antagonist of TGF-β. Expression of the decorin gene in spontaneously hypertensive rats reduces the profibrotic TGF-β expression resulting in a reduction in fibrosis. Individuals having aortic stenosis show increased expression of SLRP osteoglycin and it is strongly correlated with the left ventricular mass (Merline et al., 2009). This correlation signals that SLRPs have some role in cardiac remodeling and SLRPs can be further explored to investigate their exact role in cardiac remodeling.
2.9 Glycoproteins associated with cardiac fibrosis
Glycoproteins are proteins glycosylated with oligosaccharides at amino side chains. Glycoproteins are widely present in prokaryotic and eukaryotic cells. In cardiac ECM, glycoproteins play a non-structural role. The following glycoproteins are associated with cardiac remodeling.
2.9.1 Thrombospondins
Thrombospondin (TSP) is a family of five glycoproteins. Thrombospondins have anti-angiogenic properties. Thrombospondins are subdivided into two groups. Trimeric TSP-1 and TSP-2 are in the first group and pentameric TSP-3, TSP-4, and TSP-5 are in the second group (Iruela-Arispe et al., 1993). TSPs are only secreted during injury or cardiac development. During cardiac development, TSP-1 is expressed. TSP-2 is expressed in connective tissue whereas TSP-3, TSP-4, and TSP-5 are organ-specific and are expressed in the brain, cartilage, lung, and the nervous system. TSP-1 and 2 play significant roles in cardiac pathology. TSP-1 plays a protective role after MI by inhibiting angiogenesis and facilitating the activation of TGF-β. Deletion of TSP-1 in canine and murine-reperfused infarction models resulted in a prolonged post-infarction inflammatory response and extensive post-infarction remodeling (Frangogiannis et al., 2005). TSP-1 is also a potent angiostatic mediator in the context of the diabetic heart. TSP-1 can mediate vascular rarefaction by enhancing angiopoietin-2 expression in diabetic hearts (Gonzalez-Quesada et al., 2013). TSP-2 is required for maintaining the integrity of the myocardial matrix during a pressure overload condition. It protects the cardiac matrix during pressure overload induced by Ang-II infusion by inhibiting the MMP activity (Schroen et al., 2004). TSP-2 has protective effects on the ageing heart; it activates pro-survival Akt signaling and inhibits MMP activity (Swinnen et al., 2009).
2.9.2 Tenascins
Tenascins are multimeric glycoproteins present in the extracellular matrix. Their repeated units make different structures similar to hepta repeats, epidermal growth factor (EGF)-like repeats, fibronectin type III repeats, and one globular domain which matches with fibronectin. Three types of tenascins are present in humans. Tenascin-R and tenascin-C are present in a large number of developing tissues including the nervous system (Chiquet-Ehrismann, 1995). Tenascin-X mRNA and proteins are expressed in the heart and skeletal muscle. Tenascin-X binds with heparin, fibronectin, laminin, or collagens. Tenascin-X contributes to the stability of ECM of the cardiac tissues (Matsumoto et al., 1994). Rabinovitch and Coll did an in vitro experiment. The experiment proposed a functional paradigm of ECM-dependent cell survival where metalloproteinases (MMPs) up-regulate or down-regulate TN-C causing EGF-R clustering and EGF-dependent growth or apoptosis, respectively (Jones and Jones, 2000). In vitro, TN-C mRNA and protein accumulation are induced by mechanical stress in neonatal rat cardiomyocytes in an amplitude-dependent manner. Stimulation occurs through a nuclear factor-kappa B-dependent and an angiotensin II-independent mechanism (Yamamoto et al., 1999).
2.9.3 Secreted protein acidic and rich in cysteine
SPARC is a non-structural glycoprotein of the ECM. SPARC was first detected in bones so they are also known as osteonectin (Termine et al., 1981) SPARC shows a high affinity toward collagen and calcium. During infarct healing, SPARC facilitates collagen crosslinking (Schellings et al., 2009). This crosslinking can be detrimental if it is induced by pressure overload and ageing and it can lead to diastolic dysfunction (Komatsubara et al., 2003). SPARC contains properties of the de-adhesion (Murphy-Ullrich, 2001). It interacts and modulates the activity of various growth factors, such as FGF-2, vascular endothelial growth factor, platelet-derived growth factor, insulin-like growth factor-I, and TGF-β which are crucially involved in tissue repair, angiogenesis, and fibrosis (Motamed et al., 2002) (Francki et al., 1999).
2.9.4 Osteopontin
Osteopontin is a matricellular glycoprotein. Osteopontin expression is highly increased during injury (Wang and Denhardt, 2008). The integrin-binding site in osteopontin is activated by the thrombin (Waller et al., 2010). Osteopontin contains a calcium-binding domain. Levels of osteopontin are found elevated in the experimental models of cardiac fibrosis and hypertrophy. The osteopontin expression is also found high in MI, valvular disease, and ageing. Osteopontin increases collagen deposition and reduces chamber dilation post-MI (Trueblood et al., 2001).
2.9.5 Periostin
Periostin is a matricellular glycoprotein. Periostin is involved in the epithelial–mesenchymal transition, healing, and cardiac development. Periostin has elevated expression in the case of myocardial injury. It can interact with integrin. Periostin contributes to cardiac fibrosis; it stimulates fibroblasts and collagen deposition in response to injury (Ladage et al., 2013).
2.10 CCN protein family
CCN family consists of six members. It is named CCN because of its three members: cysteine-rich protein 61, connective tissue growth factor, and nephroblastoma-overexpressed protein. Earlier, these were considered as growth factors, but later, studies found them working as cell–extracellular matrix adhesion modulators because of their interactions with proteoglycans, integrins, growth factors, and cytokines (Ladage et al., 2013). The CCN level increases in response to myocardial injury, especially, the connective tissue growth factor (CTGF) increases (Ohnishi et al., 1998). The expression of CTGF is elevated in response to MI and pressure overload. CTGF aids in fibrotic, angiogenic responses, and TGF-β signaling. CCN5 has contrasting effects. CCN5 reduces the fibrotic response and hypertrophy by affecting the TGF-β signaling (Yoon et al., 2010).
2.11 Membrane receptors
Cell-surface receptors are very important for the communication of ECM components with cellular components. Cell-surface receptors play a pivotal role in the regulation or progression of cardiac fibrosis and heart failure. Cell-surface receptors such as integrins, discoidin domain receptors (DDR), Fc receptor gamma (FcRy), and toll-like receptors (TLR) are associated with the fibrotic response (Haudek et al., 2008; Borza et al., 2018; Katare et al., 2020; Yokota et al., 2020). Integrins are important for the mechanotransduction (Valencik et al., 2006). Integrin plays a pivotal role in cardiac fibrotic responses (Chen C. et al., 2016). The integrin can sense mechanical stress in the ECM and induce a fibrotic response (Yokota et al., 2020). In addition to integrin, DDR can also induce a fibrotic response. DDR is one of the main targets for combating fibrosis. It is one of the key players involved in the fibrotic response (Borza et al., 2018; Moll et al., 2019). TLR mediates inflammatory pathways. TLR activation has also been found to increase cardiac fibrosis in rats (Katare et al., 2020). Cell-surface receptors can also regulate cardiac fibrosis. It was found that FcRy regulates fibrotic response after binding with serum amyloid p (SAP). Deletion of FcRy in mice led to failure in inhibiting the development of cardiac fibrosis and heart failure by SAP administration (Haudek et al., 2008). However, FcRy also mediates the pathway of activation of platelets by collagen. This platelet activation pathway leads to the neointima formation in the left femoral arteries of mice (Konishi et al., 2002).
2.12 Next-gen proteomics approaches for better understanding the ECM during cardiac fibrosis
Dysregulated ECM remodeling results in the development of cardiac fibrosis. Figure 2 shows the ECM proteins altered during cardiac fibrosis. MS-based proteomics has been utilized in many studies done on cardiac ECM (Suna et al., 2018; Garcia-Puig et al., 2019; Barallobre-Barreiro et al., 2021b). A proteomics and transcriptomics study performed to understand the role of calcitonin on atrial fibrosis showed that the absence of calcitonin increases atrial fibrosis and overexpression of calcitonin prevents atrial fibrosis. One more interesting observation from this study was that the transcriptomic study of human atrial fibroblast showed little change after calcitonin exposure, but the proteomics study covered calcitonin exposure in greater detail and extensive alteration in ECM proteins and pathways related to fibrosis was observed by proteomics (Moreira et al., 2020). This study highlights the significance of proteomics in understanding cardiac fibrosis. Proteomics studies have been significant in deciphering the role of cardiac-ECM remodeling during cardiac fibrosis. The role of ECM remodeling in heart failure, myocardial infarction (MI), atrial fibrogenesis, aortic dilation, post-stent neointima formation, ischemia/reperfusion injury, and zebrafish heart regeneration has been studied with proteomics (Barallobre-Barreiro et al., 2012; 2021b; Fava et al., 2018; Suna et al., 2018; Daseke et al., 2019; Garcia-Puig et al., 2019; Moreira et al., 2020; Chalise et al., 2021). Along with these cardiac ECM protein-level studies, one in-depth site-specific collagen PTM study has also been done on the zebrafish heart (Sarohi et al., 2022b). Although cardiac ECM remodeling has been studied by proteomics in various cardiovascular disease conditions, identification and quantitation of the total ECM proteins have remained a challenge. Human matrisome has 274 core matrisome proteins and 753 matrisome-associated proteins (Nauroy et al., 2018b). However, these 1,027 proteins are not yet identified in any proteomic-based cardiac ECM study. NGP can be utilized to increase the depth of cardiac matrisome coverage with optimized ECM enrichment methods in conjunction with high-resolution MS. Quantitative proteomic analysis of alteration in total ECM proteins and PTMs with NGP will provide deeper insights into the progression of ECM remodeling during cardiac fibrosis. It will also help in identifying key ECM proteins/PTMs specifically altered during cardiac fibrosis, which can further be explored as a biomarker for early diagnosis of cardiac fibrosis.
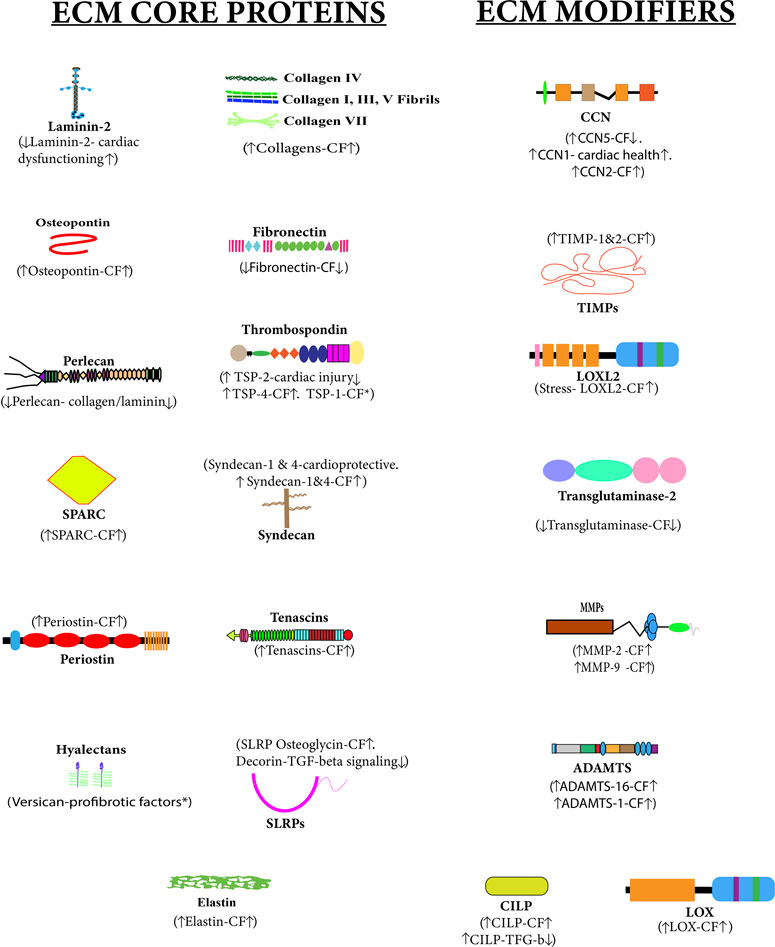
FIGURE 2. Altered cardiac matrisome during cardiac fibrosis—extracellular matrix (ECM) core proteins and ECM-modifier proteins associated with cardiac fibrosis are shown in this figure. The upside arrow (↑) shows up-regulation, and the downside arrow (↓) shows down-regulation. There is a direct or inverse correlation between the level of some ECM proteins during cardiac fibrosis (CF). Versican and TSP1 have been reported to have pro-fibrotic activities.
3 Is fibrosis always bad?
Fibrosis is generally associated with a decline in tissue functionality leading to organ failure. However, inherently, it is a fundamental repair response of a failing heart to restore the function of an injured tissue (Diegelmann and Evans, 2004). However, dysregulation in the degree of inflammation and repair may lead to diseased conditions through the development of tissue-scarring (White and Mantovani, 2013). Fibrosis has remained one of the main pathophysiological mechanisms of many CVDs. ECM deposition is a general repair response to any injury/insult or stress in the cardiac tissues (Diegelmann and Evans, 2004). ECM gets deposited in the damaged tissue to prevent further tissue damage and to initiate the reparation/healing process. In the initial phase of the healing process, collagens and other ECM proteins get deposited excessively in the cardiac ECM. Then, regeneration of tissue starts and excessive collagen with other ECM proteins get cleared from the tissue. So, excessive ECM protein deposition plays a significant role in the healing/reparation process of the tissues. This phenomenon has been vividly documented in some fishes and urodeles as they have evolved to master cardiac regeneration. However, dysregulation in the healing/repair process leads to irreversible excessive ECM protein deposition, which results in fibrosis. In fibrosis, ECM proteins are not cleared from the tissue and this excessive deposition causes the formation of scar tissue. Surprisingly, mammals do not have the capability to utilize the fibrotic response as a healing mechanism. On the contrary, Zebrafish can regenerate the injured/amputated heart. It is important to note that in zebrafish, excessive ECM proteins get deposited in the cardiac tissues, but it does not result in pathological fibrosis. As the regeneration proceeds, the excessively deposited ECM proteins are cleared from the cardiac tissues and the heart comes back to its normal state. Recently, our group has shown site-specific PTM changes on different collagen chains during the zebrafish heart-regeneration (Sarohi et al., 2022b). Analyzing the ECM protein repertoire and their post-translational modifications (such as glycosylations, hydroxylations, and crosslinking) may provide key insights into the possibilities of mammalian cardiac regeneration.
4 Development of cardiac fibrosis
4.1 Fibrotic response starts with the initiating phase with elevated levels of circulating and myocardial pro-fibrotic growth factors and cytokines (Kong et al., 2014a). In the effective phase, pro-fibrotic growth factors and cytokines bind to their receptors and lead to the activation of specific signaling pathways. This results in the transformation of cardiac fibroblasts (CFs) into myofibroblasts, which expresses the highly contractile protein a-SMA and produces different matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) to regulate the homeostasis of ECM (Kong et al., 2014a; Frieler and Mortensen, 2015). Cardiac fibrosis occurs during ischemic heart disease, MI, hypertrophic cardiomyopathy, diabetic cardiomyopathy, and dilated cardiomyopathy (Hinderer and Schenke-Layland, 2019; Sarohi et al., 2022a). Replacement/reparative fibrosis occurs in MI after cardiomyocyte loss. Cardiac fibrosis can be of four types as shown in Figure 3.
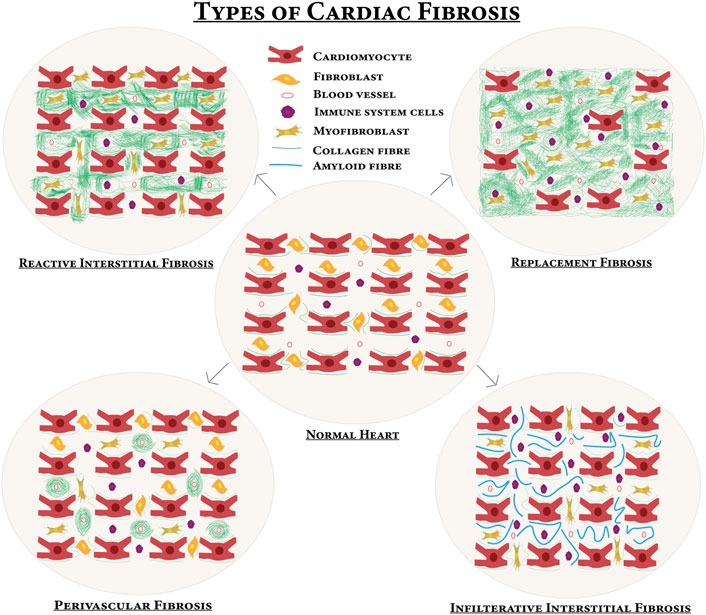
FIGURE 3. Different types of cardiac fibrosis compared with normal myocardium—upon certain profibrotic stimulations, the normal heart turns into a fibrotic one. In reactive interstitial fibrosis, the collagen is increased but the viability of the myocardium persists. In replacement fibrosis, cardiomyocytes are replaced by fibrosis and cardiomyocytes die, and then extensively produced collagen occupies the space of the dead cardiomyocytes and the myocardium fails to perform the contractile function. Infiltrative interstitial fibrosis is found in amyloidosis or Anderson–Fabry disease and there is inflammatory cell infiltration. A new matrix is produced around blood vessels in perivascular fibrosis.
The diagnosis of cardiac fibrosis has remained a challenge in the clinical setting. Biomarkers are usually based on an elevated cardiac collagen deposition that can be a significant feature/sign for many if not every cardiovascular diseases. Pro-collagen types I and III and their propeptides that reflect collagen synthesis (Weber et al., 1994b) and collagen turnover can be investigated by detecting products of matrix metalloproteinase action on collagen in the blood. From the serum collagen peptide concentrations, (i) biomarkers of collagen synthesis: PINP (pro-collagen type I N-terminal propeptide), PICP (procollagen type I C-terminal propeptide), and PIIINP (procollagen type III N-terminal propeptide); and (ii) a biomarker of collagen degradation: ICTP (collagen type I C-terminal telopeptide) (Prockop and Kivirikko, 1995; Querejeta et al., 2004; Fernández-Real et al., 2012; Flevari et al., 2012; López et al., 2012). It is confirmed that PICP is a promising fibrosis marker that is both minimally invasive and reproducible (Klappacher et al., 1995; Martos et al., 2009; Massoullié et al., 2019). The levels of MMP-2 and MMP-9 are also found to be elevated in cardiac fibrosis condition (Medeiros et al., 2019). Not only protein biomarkers but MRI-based techniques (T1, T2 relaxation times, proton density, Gadolinium CMR Enhancement Technique, Gadolinium-Enhanced CMR of Myocardial Fibrosis (MI), and Myocardial T1 mapping) are also quite useful in the detection of the cardiac fibrosis (Judd et al., 1995; Kim et al., 1996; Messroghli et al., 2004; Croisille et al., 2006; Schelbert et al., 2015). However, cardiac fibrosis-specific biomarker has yet not been delineated.
4.1 Transdifferentiation of cardiac fibroblast into myofibroblast
The following pathways induce fibroblast transdifferentiation and collagen deposition leading to cardiac fibrosis. Many recent studies have been directed toward understanding the source, development, and activation of fibroblasts into myofibroblasts that can be targeted to develop better anti-fibrotic therapies.
4.1.1 Smad-dependent pathway
The Smad-dependent pathway operates in response to TGF-β. TGF-β binds with the TGF-β type II receptor and then the TGF-β type II receptor activates the TGF-β type I receptor. The activated TGF-β type I receptor activates Smad2 and Smad3. Smad4 forms a heterotrimeric complex with Smad2 and Smad3 (Figure 4). This heterotrimeric complex translocates into the nucleus and binds with Smad-binding elements (SBE). It recruits the CREB-binding protein or p300 to enhance the transcription of profibrotic genes. Smad3 null mice study confirmed the role of Smad signaling in cardiac fibrosis (Bujak et al., 2007). Smad6 and Smad7 have an inhibitory effect on TGF-β type I receptor, they can even induce the degradation of TGF-β type I receptor. Smads also modulate the expression of microRNAs. Modulation of microRNA expression by Smads is on both the post-translational level and the transcriptional level (Blahna and Hata, 2012).
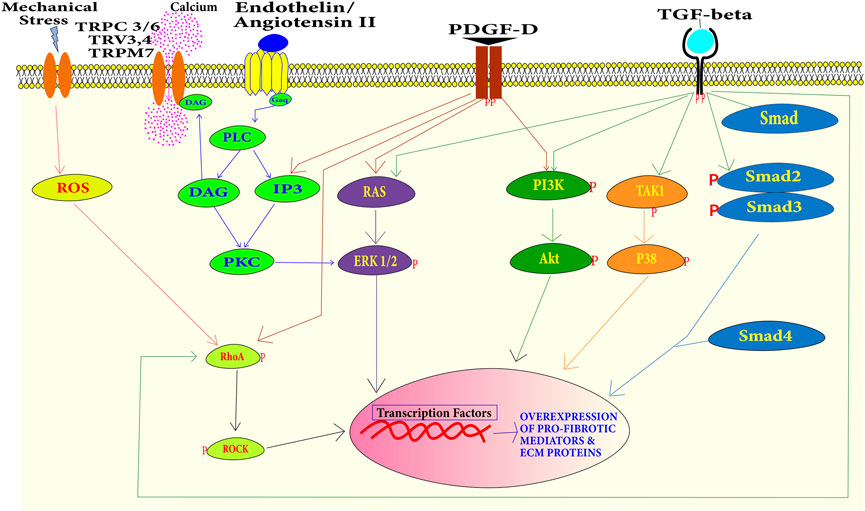
FIGURE 4. Summary of the signaling pathways during cardiac fibrosis–the binding of profibrotic factors including TGF-β, Ang-II, PDGF, endothelin-1, and mechanical stress has cascading effects. Activation of Smads leads to the transcription of excessive ECM proteins and pro-fibrotic mediator proteins. Transcription of these myofibroblast phenotypic factors can also be activated by PI3K/Akt, TAK1/P38 MAPK, RAS/Erk MAPK, and RhoA/ROCK pathways.
4.1.2 Smad-independent pathways
Cardiac fibrosis can also be developed without the involvement of Smads (Figure 4). TGF-β can induce signaling independent of Smads as well. A study reported that MAPK can be activated in response to TGF-β in Smad null cells. This indicates that MAPK activation is independent of Smad signaling (Engel et al., 1999). TGF-β binding to TGF-β type II receptor activates it. This activated receptor activates TGF-β-activated kinase 1 (TAK1). TAK1 activation leads to the activation of p38 (Wang et al., 1997). Extracellular-regulated protein kinases (ERK) can also get activated independently of Smad through small GTPase Ras under growth factor stimulation. In a study on Marfan mice ERK, it was found to induce cardiac fibrosis (Rouf et al., 2017). MAPK and ERK can also activate the Smad-signaling pathway. ERK can up-regulate the TGF-β expression and p38 can activate a Smad interacting transcriptional factor Activating Transcriptional Factor-2 (ATF2) (Massagué, 2000). Phosphoinositide-3 kinase (PI3K) can also be activated through the growth factor receptor. Activated PI3K can activate the Protein Kinase B, well-known as Akt. Activated Akt induces mTOR downstream signaling. This signaling eventually leads to the transcription of profibrotic genes and the development of cardiac fibrosis and excessive scarring (Aoyagi and Matsui, 2011). Activated RhoA activates the Rho-associated coiled–coil-containing kinases (ROCKs). RhoA can be activated by a growth factor stimulus or by reactive oxygen species. ROCKs are downstream mediators of the effects of activated RhoA. Along with regulating the actin cytoskeleton, ROCK phosphorylate targets are important for remodeling including myosin light chain (MLC) and myosin-binding subunit (MBS) on MLC phosphatase (MLCP). Up-regulation of ROCK leads to cardiac remodeling (Shimizu and Liao, 2016). The Wingless/int1 (WNT) signaling pathway also plays a role in the development, proliferation, and differentiation of cardiac cells (Foulquier et al., 2018).
4.1.3 Smad linker phosphorylation
Phosphorylation of the linker region of Smads modulates the Smad-dependent TGF-β pathway. Smads have three regions—MH1, linker, and MH2 (Burch et al., 2011). Canonical Smad-dependent TGF-β signaling works through the phosphorylation of the MH2 region (C-terminal) of Smads (Euler-Taimor and Heger, 2006). However, the linker region of Smads can also be phosphorylated by kinases activated by both Smad-dependent and -independent pathways. Activated p38 MAPK, ERK, Jnk, TAK1, ROCK, calcium-calmodulin dependent (CAM) kinase, glycogen synthase kinase-3 (GSK3), and cyclin-dependent kinase (CDK) can phosphorylate the linker region of Smads (Burch et al., 2011; Kamato et al., 2020). Initially, it was found that phosphorylation of the linker region prevents the translocation of Smads into the nucleus and inhibits the Smad-dependent TGF-β signaling pathways. But later, it was found that Smads phosphorylated at the linker region which can be translocated into the nucleus and stimulate gene expression (Kamato et al., 2020). A study performed on human mesenchymal stem cells showed that interleukin-1β (IL-1β) delays the TGF-β signaling by Smad linker phosphorylation mediated by TAK1 (van den Akker et al., 2017). Smad linker phosphorylation also contributes to cardiac pathophysiologies. Smad2 linker phosphorylation can stimulate the synthesis of biglycan and hyperelongation of the GAG chains (Burch et al., 2010; Rostam et al., 2016). Hyperelongated GAG chains facilitate the binding of lipoproteins onto the walls of blood vessels (Rostam et al., 2016). Lipoprotein accumulation on the blood-vessel walls results in atherosclerosis leading to heart failure (Rostam et al., 2016). Phosphorylation on the linker and C-terminal region of the Smads mediate different cellular functions. Region-specific phosphorylation of Smads exerts dynamic complexity in gene expression. Phosphorylation of Smads can be potentially targeted for the management of cardiovascular diseases.
4.3 NGP approaches for understanding the development of cardiac fibrosis
Transdifferentiation of cardiac fibroblast into myofibroblast upon stimulation through various profibrotic factors is a key process in the development of cardiac fibrosis. Both Smad-dependent and Smad-independent pathways involve the cascading downstream processes. The major proteins of these pathways are regulated by the process of phosphorylation (Figure 4). These pathways are well-studied in fibroblast transdifferentiation. However, the immunoblotting technique is widely used to detect the activation of various components of these pathways by detecting the phosphorylated epitope of a particular protein in a signaling cascade. Although immunoblot is a reliable and gold-standard technique, only a limited number of candidates (only those proteins with specific phosphorylation antibody availability) involved in a pathway can be evaluated. In a fibroblast that is transdifferentiating into myofibroblast, there can be different profibrotic pathways active in real-time that are cumulatively contributing to the transdifferentiation process. A more global approach is required to map these real-time complex signaling cascades. Dissecting and mapping the dynamic kinome across all the samples at the same time requires more rigorous, specific MS-based approaches to biologically and technically relevant data. These challenges can only be overcome by utilizing MS-based proteomics (Humphrey et al., 2018; Savage and Zhang, 2020; Liu et al., 2021). The field of phosphoproteomics has emerged greatly in the last decade (Lundby et al., 2013; Sharma et al., 2014; Alam et al., 2015; Riley and Coon, 2016; Steger et al., 2016; Tan et al., 2017; Kuzmanov et al., 2020a; Wu et al., 2020; Sims et al., 2022). It has contributed significantly to biological research. Lundby et al. studied the β-adrenergic receptor signaling in murine heart using phosphoproteomics (Lundby et al., 2013). β-Adrenergic receptors signaling increases the heart rate by increasing cardiomyocyte contractility. They treated mice with β-adrenergic receptors agonist and antagonist. They found that activation of the potassium ion channel by phosphorylation was associated with increased heart rate (Lundby et al., 2013). Kuzmanov et al. screened for potential targets to reduce cardiac fibrosis utilizing phosphoproteomics in hypertrophic cardiomyopathy patients and mice models of cardiac fibrosis. They found GSK-3 to be a consistent mediator of fibrosis and the inhibition of GSK-3 resulted in reduction of cardiac fibrosis (Kuzmanov et al., 2020b). Optimized phosphorylation enrichment followed by NGP can provide finer molecular level signaling details about the total kinome (phosphoproteome) of the transdifferentiating myofibroblast. Analysis of total phosphoproteome can decipher the phosphorylated components of the different active pathways. This can also be used in studying the synergistic and antagonistic effects of different pathways. Analysis of phosphoproteome can also help in the identification of phosphoproteins highly contributing to the development of cardiac fibrosis. These phosphoproteins can be targeted to regulate cardiac fibrosis.
5 Major contributors of cardiac fibrosis
The development of cardiac fibrosis is a complex process. Cellular damage, inflammation, drugs, toxins, oxidative stress, and mechanical stimulus such as pressure overload may trigger transdifferentiation of fibroblasts into specialized cells termed as myofibroblasts. The transdifferentiation of fibroblast to myofibroblast can be formed due to direct stimulants from the inflammatory cells and circulatory stimulants from the blood vessels (Lee et al., 1995; Testa et al., 1996; Campbell and Katwa, 1997; Bujak et al., 2007). But, transdifferentiation can also be stimulated from endothelial, epithelial, and cardiomyocyte cells (Hay, 1995; Thiery, 2002; Zeisberg et al., 2007). These cells under certain conditions secrete profibrotic agents that either stimulate fibroblast directly or first stimulate immune system cells which then stimulate fibroblast to proliferate into myofibroblast. These triggers can be in the form of signaling molecules such as TGF-β1, fibroblast growth factor (FGF), endothelin-1, and cytokines such as IL-1, IL-6, and TNF-α (Schroer and Merryman, 2015). The myofibroblast cells not only arise from fibroblasts (resident or bone-marrow source) but can also arise from epithelial-to-mesenchymal transitions (EMT) and endothelial-to-mesenchymal transitions (EndMT) (Kong et al., 2014b). These contributors can be targeted to combat cardiac fibrosis (Figure 5).
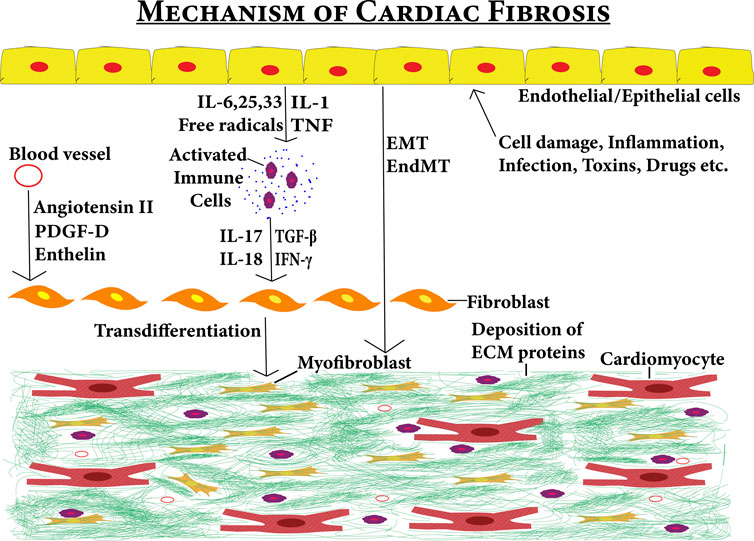
FIGURE 5. Fibrotic niche contributed by several stimuli–the figure shows the various factors contributing to the development of cardiac fibrosis. Effects of various stimuli (cell damage, inflammation, infections toxins, and drugs) on epithelial/endothelial cells can induce cardiac fibrosis either directly via EMT/End MT pathways or indirectly by first activating immune system cells via cytokines. Then, the activated immune system cells secrete profibrotic cytokines leading to the transdifferentiation of cardiac fibroblasts into myofibroblasts. The circulation system also contributes to cardiac fibrosis by entering the profibrotic factors into the cardiac tissues. These profibrotic factors also induce cardiac fibroblasts to myofibroblast transdifferentiation leading to cardiac fibrosis.
5.1 ECM processing and deposition
Collagens I and III are majorly synthesized by myofibroblasts which are then processed to attain their mature form due to the action of lysyl oxidase. Mature collagen chains cross-link together and form a mesh of fiber. Collagen I which contributes to rigidity is observed to be deposited in higher concentrations. This leads to increased tension in the ventricular wall (Segura et al., 2014). Stimulating factors from myofibroblasts promote further transdifferentiation of fibroblasts and lead to more ECM remodeling (Bonnans et al., 2014). Other factors such as ageing and metabolic dysfunctional conditions such as those seen in diabetes can also contribute to the exacerbated response of collagen deposition (Prabhu and Frangogiannis, 2016).
5.2 Immune and inflammatory cell participation
In normal conditions, basal levels of progenitor cells are maintained which gets differentiated into a specialized form under stressed conditions. Examples of such cells are macrophages that are involved in ECM remodeling and those that differentiate from monocytes. During cardiac remodeling, two subsets of macrophages differentiated by heterogeneous markers can be observed (Frangogiannis, 2019). M1 and M2 belong to the macrophage subpopulation associated with a distinct role in remodeling. M1 macrophages have pro-inflammatory action while M2 macrophages have anti-inflammatory and cardioprotective action. The polarization of M1 macrophages from a monocyte is mediated by cytokines TNF-α and interferon-gamma (IFN-γ) secreted from the type 1 T-helper (Th1) cells (Frangogiannis, 2019; Viola et al., 2019; Kim et al., 2021). The bacterial lipopolysaccharide (LPS) is also a potent mediator of M1 polarization (Kim et al., 2021). The pro-inflammatory cytokines TNF-α, IL-1α, IL-1β, IL-6, IL-12, and IL-23 are secreted by M1 macrophages (Voll et al., 1997; Kim et al., 2021). M1 macrophages are discerned to be engaged in proteolytic and signaling activities (secretion of TNF, IL-1, or ROS) throughout the inflammatory stage, specifically in the infarcted area. M1 macrophages mediate the removal of dead cells and ECM debris. Cytokines secreted from M1 macrophages gradually increase the infarct size (Duncan et al., 2020; Chen et al., 2022). Contrastingly, M2 macrophages are involved in drafting fibroblasts to the injury site and secreting pro-fibrotic mediators such as IL-10, TGF-β, and platelet-derived growth factor (PDGF) (Frangogiannis, 2019; Viola et al., 2019; Kim et al., 2021). The M2 macrophages initiate an anti-inflammatory response to drive the healing processes. M2 macrophage polarisation from the monocyte is mediated by IL-4 and IL-13 secreted by Th2 cells (Voll et al., 1997; Viola et al., 2019). A study performed on a mice model of doxorubicin-induced heart failure showed that administration of M2-like macrophages improved heart function and reduced cardiomyocyte apoptosis (Liu et al., 2022). Shiraishi et al. showed that the population of M2-like macrophages was increased in infarcted areas in mice hearts (Shiraishi et al., 2016). The selective depletion of M2-like macrophages in mice resulted in frequent cardiac rupture, reduced collagen fibril formation in infarcted areas, and decreased tissue repair post-MI (Shiraishi et al., 2016). MMP-28 facilitates M2 macrophage activation. The deletion of MMP-28 in mice resulted in MI-induced ventricular dysfunction and rupture. Collagen synthesis was also reduced in MMP-28 mice post-MI (Ma et al., 2013). These studies indicate that M2 macrophages are required to facilitate the healing process in cardiac tissues. Certain pro-fibrotic and inflammatory functions are also mediated by mast cells (Levick Goldspink, 2014). The contents secreted by mast cells such as histamine can act as proliferative agents for fibroblasts and stimulate their activation during pulmonary fibrosis (Veerappan et al., 2013). It also participates in the process of cardiac remodeling (Levick Goldspink, 2014). T-cells are well-known to be associated with heart failure progression. Nevers et al. showed that heart failure samples from transverse aortic constriction (TAC) mice and non-ischemic patients had increased affinity and higher adhesion for activated vascular endothelium. Furthermore, T-cell recruitment can promote pathological cardiac remodeling during heart failure (Nevers et al., 2015, 2017). Immune cells and cytokines secreted by the immune cells majorly contribute to ECM remodeling. Cytokines leading to dysregulated ECM remodeling can be therapeutically targeted and cardioprotective cytokines can be further explored for better management of cardiovascular diseases.
5.3 Epithelial-to-mesenchymal transition and endothelial-to-mesenchymal transition
EMT is a type of cellular switching proven to be very important in biological programs. The expression of epithelial markers is lost during this process of losing polarity and adhesion properties (Hay, 1995; Thiery, 2002). Meanwhile, the expressions of mesenchymal markers such as smooth muscle a-actin (SMA) and fibroblast-specific proteins emerge (Zeisberg et al., 2007). Additionally, phenotypes corresponding to migration and transdifferentiation are attained. Zeisberg et al. explored that one of the origins of the fibroblast is the transition from endothelial cells by performing the lineage analysis in the murine model of cardiac fibrosis which is extended to prove that it promotes the progression of the same. Although it is shown that TGF-β in the same model triggers EndMT through Smad3 transcriptional activity, on the other hand, the recombinant form of human BMP-7 i. e rhBMP-7 reduces EndMT (Zeisberg et al., 2007). It was found that under a diabetic milieu, endothelin-1 production increased by vascular endothelial cells contributing to exacerbated cardiac fibrosis through EndMT (Widyantoro et al., 2010). Furthermore, an elevated level of endothelin-1 has been found to be associated with cardiac fibrosis progression among diabetic patients (Takahashi et al., 1990; Widyantoro et al., 2010; Sen et al., 2012; Russo and Frangogiannis, 2016). EMT and EndMT are the processes that accelerate the progression of fibrosis. Fibrosis progression is reduced by inhibiting these processes (Widyantoro et al., 2010). These processes can be therapeutically targeted for inhibition of the progression of cardiac fibrosis.
5.4 Blood vessel: An important ECM content source
Angiotensin II is a major protein hormone that acts on the central nervous system causing blood vessels to constrict. This aids in maintaining fluid balance and blood pressure in the body (Lee et al., 1995; Campbell and Katwa, 1997). In the case of cardiac fibrosis, angiotensin II is seen to up-regulate TGF-β1 in fibroblast and myofibroblast in vivo and in vitro conditions which consequently affect collagen production (Kupfahl et al., 2000).
5.5 Inflammatory cytokines
Inflammatory factors can be released by various cardiac cells. The presence of inflammatory factors correlates with cardiac dysfunction. Elevated levels of IL-1, TNF-α, and IL-6 indicate the severity of cardiac diseases in heart patients (Testa et al., 1996). Overexpression of TNF-α in the heart results in cardiac fibrosis, eventually leads to dilated cardiomyopathy (Kuhota et al., 1997). IL-25,33 activates inflammatory cells. IL-1 and IL-6 demonstrate pleiotropic roles in fibrosis; these cytokines contribute to fibroblast transdifferentiation. IL-6 is found to aid in Ang-II-induced cardiac fibrosis (Ma et al., 2012). The role of IFN-γ in cardiac fibrosis is unclear. It can have a pathological or protective role in cardiac fibrosis (Levick and Goldspink, 2014). IL-17 inhibits MKP-1 (MAPK phosphatase-1) and activates p38 MAPK and ERK 1/2, thus it contributes to cardiac fibroblast transdifferentiation and migration (Valente et al., 2012). Overexpression of IL-18 is found to aggravate the remodeling and fibrosis in the heart (Xing et al., 2010).
5.6 NGP approaches for identifying potential targets for therapeutics of cardiac fibrosis
The aforementioned factors are majorly known for contributing to the development and progression of cardiac fibrosis. Not just the matrisome and phosphoproteome, but cardiac tissue-based total proteome studies by NGP can also greatly help in assessing the major contributors of fibrosis and even identify unreported contributors. NGP analysis of total tissue lysate can provide deeper insights into the alteration in cellular and extracellular proteins/PTMs during cardiac fibrosis. These altered proteins/PTMs can be further screened for their specificity during cardiac fibrosis. The role of cardiac fibrosis-specific protein and site-specific PTMs can be determined during cardiac fibrosis. These proteins/PTMs can then be explored as potential targets for the treatment/reversal of cardiac fibrosis which is not curable as of now.
6 Available treatment of cardiac fibrosis
Pharmaceutical agents are generally used in the treatment and control of cardiac fibrosis. Mechanical devices are used to support heart functions in patients with severe cardiac fibrosis or during high-dose treatment with pharmaceutical agents.
6.1 Angiotensin-converting enzyme inhibitors
Experimentally, angiotensin-convertase enzyme inhibitors are found to block cardiac remodeling and reverse the remodeling of cardiac tissues (Klotz et al., 2007). Ang-II is generated by proteolytic cleavage of Ang-I by ACE. Ang-II is a profibrotic agent (Kawano et al., 2000; Díez, 2004). Because ACE inhibitors block the Ang-II formation, they are used in the treatment of cardiac fibrosis.
6.2 Angiotensin receptor blockers
Angiotensin receptor blockers prevent vasoconstriction and are used for the treatment of cardiac fibrosis. Prolonged treatment with ARB can reverse the remodeling in a patient having cardiac fibrosis (Granger et al., 2003). ARBs can be prescribed to patients who have ACE inhibitor intolerance.
6.3 Beta-adrenergic receptor (β) blockers
High levels of catecholamines significantly contribute to the progression of cardiac diseases. Beta-adrenergic receptor (β) blockers temporarily reduce the blood pressure and progression of heart diseases. β-blockers are helpful in improving the systolic function and reducing the dimensions of the left ventricle. Studies on β-blockers reveal that they increase the survival rate of heart patients (Domanski et al., 2003).
6.4 Aldosterone antagonists
A high expression of aldosterone in the body causes mineral imbalance. Aldosterone increases the level of sodium in the body and decreases water and potassium levels in the body. Aldosterone receptors are highly expressed in cases of heart failure. Treatment with aldosterone receptors improves cardiac function and reduces remodeling in patients having cardiac fibrosis (Pitt et al., 1999).
6.5 Mechanical assistive devices
6.5.1 Left ventricle-assist devices
Mechanical devices are used to assist in the cardiac function. LVADs reduce the progression of cardiac diseases. Prolonged use of LVAD with pharmaceutical agents promotes reverse remodeling (Ambardekar and Buttrick, 2011).
6.5.2 Cardiac resynchronization therapy
The biventricular pacemaker device is used in CRT. CRT improves cardiac function, reduces dilation of the ventricle, and promotes reverse remodeling (St. John Sutton et al., 2009).
6.6 Novel and emerging therapies
Phosphodiesterase type 5 (PDE5) inhibitors are also used in the treatment of cardiac fibrosis to aid other therapies (Guazzi et al., 2011). PDE5 inhibitors improve the diastolic function. Some microRNAs have cardioprotective natures, these can be utilized for the control and treatment of cardiac fibrosis (Kukreja et al., 2011). Table 1 summarizes the potential drugs for cardiac fibrosis under different phases of clinical trials in the United States (2020–2021).
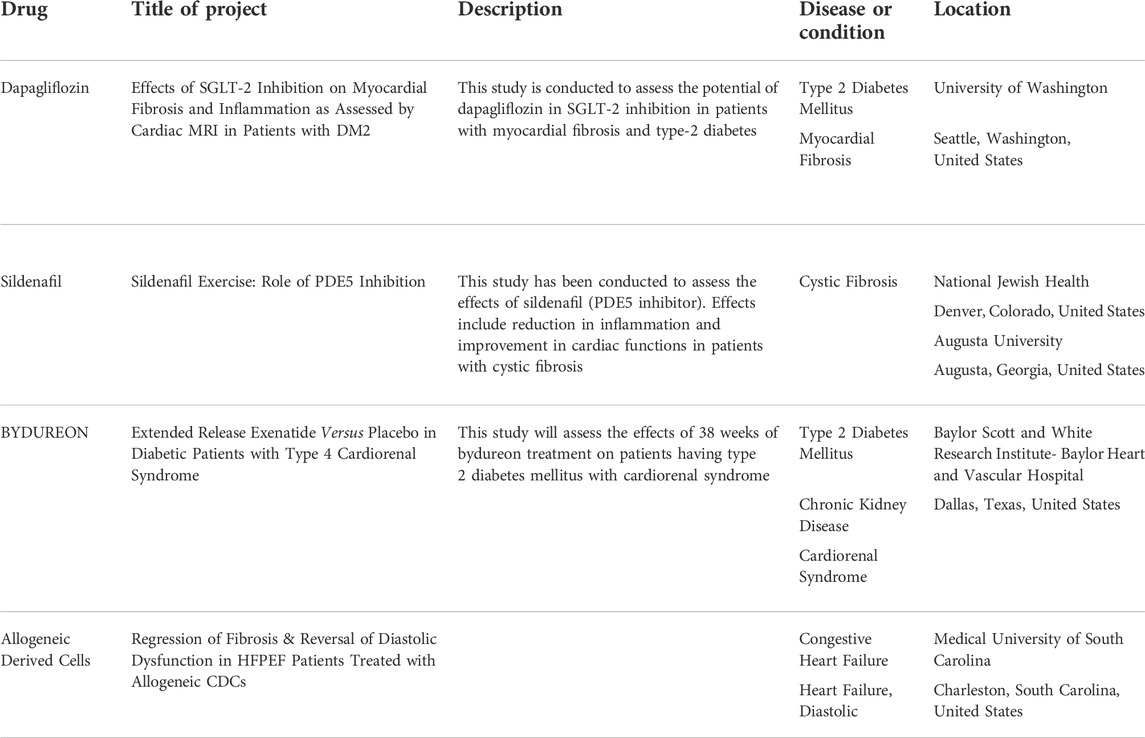
TABLE 1. Drugs and biological agents undergoing trials in the United States in 2020–2021 (ClinicalTrials.gov, 2021).
7 Conclusion
Dissecting the ECM proteome during cardiac fibrosis is highly important to delineate the complex process of ECM remodeling-mediated scar-tissue formation. Exploring the ECM protein repertoire with mass-spectrometry has remained a challenge mostly because of its insoluble nature. In the recent past, sample-prep methods have been optimized for enriching ECM samples (Lindsey et al., 2018; Barallobre-Barreiro et al., 2021a; McCabe et al., 2021, 2022). Simultaneously, enormous advances (in resolution and sensitivity) in next-gen mass-spectrometry capabilities have increased the depth of analysis. For the last few decades, immunohistochemistry-based staining of collagens (mostly fibrillar) has remained as the gold standard for diagnosing tissue fibrosis. However, proteomics holds the key to discovering new potential biomarkers for cardiac fibrosis patients. Following the success of MudPiT technology in proteomics, recently advanced 4D proteomics have been developed (Yang et al., 2022). Retention time, mass-to-charge ratio, and intensity of the peaks are the three dimensions of MS analysis in ion chromatogram, now the fourth dimension of ion mobility spectrometry (IMS) has been introduced into proteomics (Meier et al., 2021; Yang et al., 2022). In 4D proteomics, peptides are firstl separated in nanoLC and they are again separated in the gas phase in IMS. These two different types of separations provide a higher resolution resulting in higher sensitivity in the identification of peptides (Meier et al., 2021; Yao et al., 2021; Yang et al., 2022). Co-eluting and fusion peptides can also be accurately analyzed by IMS-MS. The in-depth analysis in proteomics is also greatly affected by the data acquisition mode. Conventionally, dynamic exclusion in the data-dependent acquisition (DDA) limits the identification of low-abundant proteins and site-specific PTMs. In several scenarios, the identification of PSMs (peptide–spectrum matches) at the MS2 level becomes a huge challenge. This leads to the difficulty in the accurate quantitation of low-abundant and co-eluting peptides and peptides with similar mass-to-charge ratios. Recent developments with data-independent acquisition (DIA) are a better approach for the quantitation of proteins and PTMs. In DIA, a targeted extraction of the MS2 ions aligned with its precursor improves the sensitivity and specificity of quantitation. Combining the resolution of IMS-MS with DIA is the next Frontier in the proteomics (Soni, 2022) approach to substantially dig deeper with greater sensitivity and specificity to expand the myocardium-specific ECM protein repertoire along with site-specific post-translational modifications. Furthermore, the application of NGP-based ECM proteomics in in vitro and in vivo models of cardiac fibrosis will not only broaden the mechanistic understanding but may yield potential candidates to tinker with ECM remodeling to combat cardiac fibrosis (Figure 6).
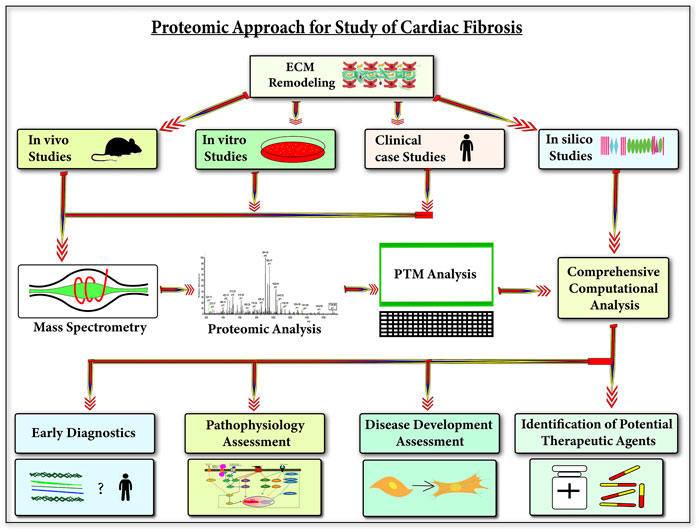
FIGURE 6. Next-gen proteomics applications to understand cardiac fibrosis—this figure shows the applications of understanding ECM during cardiac diseases using next-gen proteomics. Utilizing next-gen proteomics (protein and PTM analysis) on three biological study types (animal models, cell line models, and clinical human cases) with in silico studies can provide an in-depth understanding of the role of cardiac ECM in the development of cardiac diseases. This understanding will be significantly beneficial in the early diagnosis of cardiac diseases, pathophysiology, and development assessment of cardiac diseases and also in the identification of potential therapeutic agents for cardiac diseases.
Author contributions
VS and SC worked on summarizing the recent knowledge about progression of cardiac fibrosis. VS summarized the applications of next-gen proteomics in studying cardiac fibrosis. VS prepared all the figures. VS and SC wrote the initial drafts of the manuscript. TB conceptualized the overall structure of the review and finalized the manuscript.
Funding
This study was funded by the Seed grant (IITM/SG/TB/75) from IIT-Mandi.
Acknowledgments
The Seed grant (IITM/SG/TB/75) from IIT-Mandi to TB is acknowledged for this work. VS and SC acknowledge the HTRA fellowship (MHRD, Govt. of India) for the doctoral program.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ad, T., Ss, S., Gn, T., and Nk, K. (2010). Proteoglycans in health and disease: Novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 277, 3904–3923. doi:10.1111/J.1742-4658.2010.07800.X
Aebersold, R., and Mann, M. (2003). Mass spectrometry-based proteomics. Nature 422, 198–207. doi:10.1038/nature01511
Alam, M. M., Solyakov, L., Bottrill, A. R., Flueck, C., Siddiqui, F. A., Singh, S., et al. (2015). Phosphoproteomics reveals malaria parasite Protein Kinase G as a signalling hub regulating egress and invasion. Nat. Commun. 6 (1 6), 7285–7315. doi:10.1038/ncomms8285
Ambardekar, A. V., and Buttrick, P. M. (2011). Reverse remodeling with left ventricular assist devices: A review of clinical, cellular, and molecular effects. Circ. Heart Fail. 4, 224–233. doi:10.1161/CIRCHEARTFAILURE.110.959684
Aoyagi, T., and Matsui, T. (2011). Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 17, 1818–1824. doi:10.2174/138161211796390976
Barallobre-Barreiro, J., Didangelos, A., Schoendube, F. A., Drozdov, I., Yin, X., Fernández-Caggiano, M., et al. (2012). Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation 125, 789–802. doi:10.1161/CIRCULATIONAHA.111.056952
Barallobre-Barreiro, J., Radovits, T., Fava, M., Mayr, U., Lin, W. Y., Ermolaeva, E., et al. (2021a). Extracellular matrix in heart failure: Role of ADAMTS5 in proteoglycan remodeling. Circulation 144, 2021–2034. doi:10.1161/CIRCULATIONAHA.121.055732
Barallobre-Barreiro, J., Radovits, T., Fava, M., Mayr, U., Lin, W. Y., Ermolaeva, E., et al. (2021b). Extracellular matrix in heart failure: Role of ADAMTS5 in proteoglycan remodeling. Circulation 144, 2021–2034. doi:10.1161/CIRCULATIONAHA.121.055732
Basak, T., Vega-Montoto, L., Zimmerman, L. J., Tabb, D. L., Hudson, B. G., and Vanacore, R. M. (2016). Comprehensive characterization of glycosylation and hydroxylation of basement membrane collagen IV by high-resolution mass spectrometry. J. Proteome Res. 15, 245–258. doi:10.1021/ACS.JPROTEOME.5B00767
Benjamin, E. J., Muntner, P., Alonso, A., Bittencourt, M. S., Callaway, C. W., Carson, A. P., et al. (2019). Heart disease and stroke statistics-2019 update: A report from the American heart association. Circulation 139, e56–e528. doi:10.1161/CIR.0000000000000659
Benn, M. C., Weber, W., Klotzsch, E., Vogel, V., and Pot, S. A. (2019). Tissue transglutaminase in fibrosis — More than an extracellular matrix cross-linker. Curr. Opin. Biomed. Eng. 10, 156–164. doi:10.1016/J.COBME.2019.06.003
Bishop, J. E. (1998). Regulation of cardiovascular collagen deposition by mechanical forces. Mol. Med. Today 4, 69–75. doi:10.1016/S1357-4310(97)01193-3
Blahna, M. T., and Hata, A. (2012). Smad-mediated regulation of microRNA biosynthesis. FEBS Lett. 586, 1906–1912. doi:10.1016/J.FEBSLET.2012.01.041
Bleumink, G., Knetsch, A., Sturkenboom, M., Straus, S., Hofman, A., Deckers, J., et al. (2004). Quantifying the heart failure epidemic: Prevalence, incidence rate, lifetime risk and prognosis of heart failure: The rotterdam study. Eur. Heart J. 25, 1614–1619. doi:10.1016/j.ehj.2004.06.038
Bloksgaard, M., Lindsey, M., and Martinez-Lemus, L. A. (2018). Extracellular matrix in cardiovascular pathophysiology. Am. J. Physiol. Heart Circ. Physiol. 315, H1687-H1690–H1690. doi:10.1152/ajpheart.00631.2018
Bonnans, C., Chou, J., and Werb, Z. (2014). Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell. Biol. 15, 786–801. doi:10.1038/nrm3904
Borza, C. M., Pozzi, A., and Plosa, E. J. (2018). Discoidin domain receptor 2, a potential therapeutic target in lung fibrosis. Am. J. Respir. Cell. Mol. Biol. 59, 277–278. doi:10.1165/RCMB.2018-0161ED
Bujak, M., Ren, G., Kweon, H. J., Dobaczewski, M., Reddy, A., Taffet, G., et al. (2007). Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 116, 2127–2138. doi:10.1161/CIRCULATIONAHA.107.704197
Burch, M. L., Yang, S. N. Y., Ballinger, M. L., Getachew, R., Osman, N., and Little, P. J. (2010). TGF-β stimulates biglycan synthesis via p38 and ERK phosphorylation of the linker region of Smad2. Cell. Mol. Life Sci. 67, 2077–2090. doi:10.1007/s00018-010-0315-9
Burch, M. L., Zheng, W., and Little, P. J. (2011). Smad linker region phosphorylation in the regulation of extracellular matrix synthesis. Cell. Mol. Life Sci. 68, 97–107. doi:10.1007/s00018-010-0514-4
Busnadiego, O., González-Santamaría, J., Lagares, D., Guinea-Viniegra, J., Pichol-Thievend, C., Muller, L., et al. (2013). LOXL4 is induced by transforming growth factor β1 through Smad and JunB/Fra2 and contributes to vascular matrix remodeling. Mol. Cell. Biol. 33, 2388–2401. doi:10.1128/MCB.00036-13
Cabello-Verrugio, C., Santander, C., Cofré, C., Acuña, M. J., Melo, F., and Brandan, E. (2012). The internal region leucine-rich repeat 6 of decorin interacts with low density lipoprotein receptor-related protein-1, modulates Transforming Growth Factor (TGF)-β-dependent signaling, and inhibits TGF-β-dependent fibrotic response in skeletal muscles. J. Biol. Chem. 287, 6773–6787. doi:10.1074/jbc.M111.312488
Cabral, W. A., Chang, W., Barnes, A. M., Weis, M., Scott, M. A., Leikin, S., et al. (2007). Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 39, 359–365. doi:10.1038/NG1968
Campbell, K. P. (1995). Three muscular dystrophies: Loss of cytoskeleton-extracellular matrix linkage. Cell. 80, 675–679. doi:10.1016/0092-8674(95)90344-5
Campbell, S. E., and Katwa, L. C. (1997). Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J. Mol. Cell. Cardiol. 29, 1947–1958. doi:10.1006/jmcc.1997.0435
Chalise, U., Becirovic-Agic, M., and Lindsey, M. L. (2021). Neutrophil crosstalk during cardiac wound healing after myocardial infarction. Curr. Opin. Physiol. 24, 100485. doi:10.1016/J.COPHYS.2022.100485
Chang, C. W., Dalgliesh, A. J., López, J. E., and Griffiths, L. G. (2016). Cardiac extracellular matrix proteomics: Challenges, techniques, and clinical implications. Proteomics. Clin. Appl. 10, 39–50. doi:10.1002/PRCA.201500030
Chapman, D., Weber, K. T., and Eghbali, M. (1990). Regulation of fibrillar collagen types I and III and basement membrane type IV collagen gene expression in pressure overloaded rat myocardium. Circ. Res. 67, 787–794. doi:10.1161/01.res.67.4.787
Chaturvedi, V., Parakh, N., Seth, S., Bhargava, B., Ramakrishnan, S., Roy, A., et al. (2016). Heart failure in India: The INDUS (India ukieri study) study. J. Pract. Cardiovasc. Sci. 2, 28. doi:10.4103/2395-5414.182988
Chen, C., Li, R., Ross, R. S., and Manso, A. M. (2016a). Integrins and integrin-related proteins in cardiac fibrosis. J. Mol. Cell. Cardiol. 93, 162–174. doi:10.1016/J.YJMCC.2015.11.010
Chen, G., Jiang, H., Yao, Y., Tao, Z., Chen, W., Huang, F., et al. (2022). Macrophage, a potential targeted therapeutic immune cell for cardiomyopathy. Front. Cell. Dev. Biol. 0, 908790. doi:10.3389/FCELL.2022.908790
Chen, W. C. W., Wang, Z., Missinato, M. A., Park, D. W., Long, D. W., Liu, H. J., et al. (2016b). Decellularized zebrafish cardiac extracellular matrix induces mammalian heart regeneration. Sci. Adv. 2, e1600844. doi:10.1126/SCIADV.1600844
Chiquet-Ehrismann, R. (1995). Tenascins, a growing family of extracellular matrix proteins. Experientia 51, 853–862. doi:10.1007/BF01921736
ClinicalTrials.gov (2021). ClinicalTrials.gov is a database of privately and publicly funded clinical studies conducted around the world. Available at: https://clinicaltrials.gov/ (Accessed August 26, 2022).
Contard, F., Koteliansky, V., Marotte, F., Dubus, I., Rappaport, L., and Samuel, J. L. (1991). Specific alterations in the distribution of extracellular matrix components within rat myocardium during the development of pressure overload. Lab. Investig. 64, 65–75.
Cooley, M. A., Fresco, V. M., Dorlon, M. E., Twal, W. O., Lee, N. V., Barth, J. L., et al. (2012). Fibulin-1 is required during cardiac ventricular morphogenesis for versican cleavage, suppression of ErbB2 and Erk1/2 activation, and to attenuate trabecular cardiomyocyte proliferation. Dev. Dyn. 241, 303–314. doi:10.1002/dvdy.23716
Couchman, J. R. (2003). Syndecans: Proteoglycan regulators of cell-surface microdomains? Nat. Rev. Mol. Cell. Biol. 4, 926–937. doi:10.1038/nrm1257
Croisille, P., Revel, D., and Saeed, M. (2006). Contrast agents and cardiac MR imaging of myocardial ischemia: From bench to bedside. Eur. Radiol. 16, 1951–1963. doi:10.1007/S00330-006-0244-Z
Cutie, S., and Huang, G. N. (2021). Vertebrate cardiac regeneration: Evolutionary and developmental perspectives. Cell. Regen. 10, 6–10. doi:10.1186/s13619-020-00068-y
Daseke, M. J., Valerio, F. M., Kalusche, W. J., Ma, Y., DeLeon-Pennell, K. Y., and Lindsey, M. L. (2019). Neutrophil proteome shifts over the myocardial infarction time continuum. Basic Res. Cardiol. 114, 37. doi:10.1007/S00395-019-0746-X
Di Lullo, G. A., Sweeney, S. M., Körkkö, J., Ala-Kokko, L., and San Antonio, J. D. (2002). Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. J. Biol. Chem. 277, 4223–4231. doi:10.1074/JBC.M110709200
Diegelmann, R. F., and Evans, M. C. (2004). Wound healing: An overview of acute, fibrotic and delayed healing. Front. Biosci. 9, 283–289. doi:10.2741/1184
Díez, J. (2004). Profibrotic effects of angiotensin II in the heart: A matter of mediators. Hypertension 43, 1164–1165. doi:10.1161/01.HYP.0000128620.57061.67
Dokainish, H., Teo, K., Zhu, J., Roy, A., AlHabib, K. F., ElSayed, A., et al. (2017). Global mortality variations in patients with heart failure: Results from the international congestive heart failure (INTER-CHF) prospective cohort study. Lancet. Glob. Health 5, e665–e672. doi:10.1016/S2214-109X(17)30196-1
Domanski, M. J., Krause-Steinrauf, H., Massie, B. M., Deedwania, P., Follmann, D., Kovar, D., et al. (2003). A comparative analysis of the results from 4 trials of β-blocker therapy for heart failure: BEST, CIBIS-II, MERIT-HF, and COPERNICUS. J. Card. Fail. 9, 354–363. doi:10.1054/S1071-9164(03)00133-7
Duncan, S. E., Gao, S., Sarhene, M., Coffie, J. W., Linhua, D., Bao, X., et al. (2020). Macrophage activities in myocardial infarction and heart failure. Cardiol. Res. Pract. 2020, 4375127. doi:10.1155/2020/4375127
Engel, M. E., McDonnell, M. A., Law, B. K., and Moses, H. L. (1999). Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J. Biol. Chem. 274, 37413–37420. doi:10.1074/JBC.274.52.37413
Euler-Taimor, G., and Heger, J. (2006). The complex pattern of SMAD signaling in the cardiovascular system. Cardiovasc. Res. 69, 15–25. doi:10.1016/J.CARDIORES.2005.07.007/2/69-1-15-FIG4
Faita, F., Vecoli, C., Foffa, M., and Andreassi, G. (2012). Next generation sequencing in cardiovascular diseases. World J. Cardiol. 4, 288–295. doi:10.4330/WJC.V4.I10.288
Fava, M., Barallobre-Barreiro, J., Mayr, U., Lu, R., Didangelos, A., Baig, F., et al. (2018). Role of ADAMTS-5 in aortic dilatation and extracellular matrix remodeling. Arterioscler. Thromb. Vasc. Biol. 38, 1537–1548. doi:10.1161/ATVBAHA.117.310562
Fedarko, N. S. (2014). Osteoblast/osteoclast development and function in osteogenesis imperfecta. Osteogenes. Imperfecta A Transl. Approach Brittle Bone Dis., 45–56. doi:10.1016/B978-0-12-397165-4.00005-8
Fernández-Real, J. M., Bulló, M., Moreno-Navarrete, J. M., Ricart, W., Ros, E., Estruch, R., et al. (2012). A Mediterranean diet enriched with olive oil is associated with higher serum total osteocalcin levels in elderly men at high cardiovascular risk. J. Clin. Endocrinol. Metab. 97, 3792–3798. doi:10.1210/JC.2012-2221
Flevari, P., Theodorakis, G., Leftheriotis, D., Kroupis, C., Kolokathis, F., Dima, K., et al. (2012). Serum markers of deranged myocardial collagen turnover: Their relation to malignant ventricular arrhythmias in cardioverter-defibrillator recipients with heart failure. Am. Heart J. 164, 530–537. doi:10.1016/J.AHJ.2012.07.006
Foronjy, R. F., Sun, J., Lemaitre, V., and d’Armiento, J. M. (2008). Transgenic expression of matrix metalloproteinase-1 inhibits myocardial fibrosis and prevents the transition to heart failure in a pressure overload mouse model. Hypertens. Res. 31 (4 31), 725–735. doi:10.1291/hypres.31.725
Foulquier, S., Daskalopoulos, E. P., Lluri, G., Hermans, K. C. M., Deb, A., and Blankesteijn, W. M. (2018). WNT signaling in cardiac and vascular disease. Pharmacol. Rev. 70, 68–141. doi:10.1124/PR.117.013896
Francki, A., Bradshaw, A. D., Bassuk, J. A., Howe, C. C., Couser, W. G., and Sage, E. H. (1999). SPARC regulates the expression of collagen type I and transforming growth factor-beta1 in mesangial cells. J. Biol. Chem. 274, 32145–32152. doi:10.1074/jbc.274.45.32145
Frangogiannis, N. G. (2019). Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 65, 70–99. doi:10.1016/J.MAM.2018.07.001
Frangogiannis, N. G., Ren, G., Dewald, O., Zymek, P., Haudek, S., Koerting, A., et al. (2005). Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation 111, 2935–2942. doi:10.1161/CIRCULATIONAHA.104.510354
Frangogiannis, N. G. (2017). The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 127, 1600–1612. doi:10.1172/JCI87491
Freue, G. V. C., and Borchers, C. H. (2012). Multiple reaction monitoring (MRM): Principles and application to coronary artery disease. Circ. Cardiovasc. Genet. 5, 378. doi:10.1161/CIRCGENETICS.111.959528
Frevert, C. W., and Wight, T. N. (2006). “Extracellular matrix: Matrix proteoglycans,” in Encyclopedia of respiratory medicine, four-volume set (Elsevier), 184–188. doi:10.1016/B0-12-370879-6/00150-2
Frieler, R. A., and Mortensen, R. M. (2015). Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation 131, 1019–1030. doi:10.1161/CIRCULATIONAHA.114.008788
Gagliano, N., Pelillo, F., Chiriva-Internati, M., Picciolini, O., Costa, F., Schutt, R. C., et al. (2009). Expression profiling of genes involved in collagen turnover in tendons from cerebral palsy patients. doi:10.1080/03008200802613630
Garcia-Puig, A., Mosquera, J. L., Jiménez-Delgado, S., García-Pastor, C., Jorba, I., Navajas, D., et al. (2019). Proteomics analysis of extracellular matrix remodeling during zebrafish heart regeneration. Mol. Cell. Proteomics 18, 1745–1755. doi:10.1074/MCP.RA118.001193
Gonzalez-Avila, G., Sommer, B., Mendoza-Posada, D. A., Ramos, C., Garcia-Hernandez, A. A., and Falfan-Valencia, R. (2019). Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 137, 57–83. doi:10.1016/J.CRITREVONC.2019.02.010
Gonzalez-Quesada, C., Cavalera, M., Biernacka, A., Kong, P., Lee, D. W., Saxena, A., et al. (2013). Thrombospondin-1 induction in the diabetic myocardium stabilizes the cardiac matrix in addition to promoting vascular rarefaction through angiopoietin-2 upregulation. Circ. Res. 113, 1331–1344. doi:10.1161/CIRCRESAHA.113.302593
González-Santamaría, J., Villalba, M., Busnadiego, O., López-Olañeta, M. M., Sandoval, P., Snabel, J., et al. (2016). Matrix cross-linking lysyl oxidases are induced in response to myocardial infarction and promote cardiac dysfunction. Cardiovasc. Res. 109, 67–78. doi:10.1093/CVR/CVV214
Granger, C. B., McMurray, J. J. V., Yusuf, S., Held, P., Michelson, E. L., Olofsson, B., et al. (2003). Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: The CHARM-alternative trial. Lancet (London, Engl. 362, 772–776. doi:10.1016/S0140-6736(03)14284-5
Greenberg, C. S., Birckbichler, P. J., and Rice, R. H. (1991). Transglutaminases: Multifunctional cross-linking enzymes that stabilize tissues. FASEB J. official Publ. Fed. Am. Soc. Exp. Biol. 5, 3071–3077. doi:10.1096/FASEBJ.5.15.1683845
Guazzi, M., Vicenzi, M., Arena, R., and Guazzi, M. D. (2011). PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: Results of a 1-year, prospective, randomized, placebo-controlled study. Circ. Heart Fail. 4, 8–17. doi:10.1161/CIRCHEARTFAILURE.110.944694
Haudek, S. B., Trial, J. A., Xia, Y., Gupta, D., Pilling, D., and Entman, M. L. (2008). Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proc. Natl. Acad. Sci. U. S. A. 105, 10179–10184. doi:10.1073/PNAS.0804910105/ASSET/A52E9537-2050-45D5-BE45-90F01246B997/
Hay, E. D. (1995). An overview of epithelio-mesenchymal transformation. Acta Anat. 154, 8–20. doi:10.1159/000147748
Hedtke, T., Schräder, C. U., Heinz, A., Hoehenwarter, W., Brinckmann, J., Groth, T., et al. (2019). A comprehensive map of human elastin cross-linking during elastogenesis. FEBS J. 286, 3594–3610. doi:10.1111/FEBS.14929
Hennet, T. (2019). Collagen glycosylation. Curr. Opin. Struct. Biol. 56, 131–138. doi:10.1016/J.SBI.2019.01.015
Hildebrand, A., Romaris, M., Rasmussen, M., Heinegard, D., Twardzik, D. R., Border, W. A., et al. (1994). Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem. J. 302, 527–534. doi:10.1042/bj3020527
Hinderer, S., and Schenke-Layland, K. (2019). Cardiac fibrosis – a short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 146, 77–82. doi:10.1016/J.ADDR.2019.05.011
Homan, E. P., Lietman, C., Grafe, I., Lennington, J., Morello, R., Napierala, D., et al. (2014). Differential effects of collagen prolyl 3-hydroxylation on skeletal tissues. PLoS Genet. 10, e1004121. doi:10.1371/JOURNAL.PGEN.1004121
Hudson, D. M., Joeng, K. S., Werther, R., Rajagopal, A., Weis, M., Lee, B. H., et al. (2015). Post-translationally abnormal collagens of prolyl 3-hydroxylase-2 null mice offer a pathobiological mechanism for the high myopia linked to human LEPREL1 mutations. J. Biol. Chem. 290, 8613–8622. doi:10.1074/JBC.M114.634915
Humphrey, S. J., Karayel, O., James, D. E., and Mann, M. (2018). High-throughput and high-sensitivity phosphoproteomics with the EasyPhos platform. Nat. Protoc. 13 (9 13), 1897–1916. doi:10.1038/s41596-018-0014-9
ICC NHFR Report (2022). The current situation. Available at: https://www.iccnhfr.org/the-current-situation#heartFailure (Accessed August 27, 2022).
Iozzo, R. V., and Murdoch, A. D. (1996). Proteoglycans of the extracellular environment: Clues from the gene and protein side offer novel perspectives in molecular diversity and function. FASEB J. 10, 598–614. doi:10.1096/fasebj.10.5.8621059
Iozzo, R. V., Zoeller, J. J., and Nyström, A. (2009). Basement membrane proteoglycans: Modulators Par Excellence of cancer growth and angiogenesis. Mol. Cells 27, 503–513. doi:10.1007/s10059-009-0069-0
Iruela-Arispe, M. L., Liska, D. A. J., Sage, E. H., and Bornstein, P. (1993). Differential expression of thrombospondin 1, 2, and 3 during murine development. Dev. Dyn. 197, 40–56. doi:10.1002/aja.1001970105
Jane-Lise, S., Corda, S., Chassagne, C., and Rappaport, L. (2000). The extracellular matrix and the cytoskeleton in heart hypertrophy and failure. Heart fail. Rev. 5, 239–250. doi:10.1023/A:1009857403356
Johnson, K., Farley, D., Hu, S. I., and Terkeltaub, R. (2003). One of two chondrocyte-expressed isoforms of cartilage intermediate-layer protein functions as an insulin-like growth factor 1 antagonist. Arthritis Rheum. 48, 1302–1314. doi:10.1002/ART.10927
Jones, F. S., and Jones, P. L. (2000). The tenascin family of ECM glycoproteins: Structure, function, and regulation during embryonic development and tissue remodeling. Dev. Dyn. official Publ. Am. Assoc. Anatomists, 218. 2-G, 235–259. doi:10.1002/(SICI)1097-0177
Judd, R. M., Atalay, M. K., Rottman, G. A., and Zerhouni, E. A. (1995). Effects of myocardial water exchange on T1 enhancement during bolus administration of MR contrast agents. Magn. Reson. Med. 33, 215–223. doi:10.1002/MRM.1910330211
Jürgensen, H. J., Madsen, D. H., Ingvarsen, S., Melander, M. C., Gårdsvoll, H., Patthy, L., et al. (2011). A novel functional role of collagen glycosylation: Interaction with the endocytic collagen receptor uPARAP/endo180. J. Biol. Chem. 286, 32736–32748. doi:10.1074/JBC.M111.266692
Kamato, D., Do, B. H., Osman, N., Ross, B. P., Mohamed, R., Xu, S., et al. (2020). Smad linker region phosphorylation is a signalling pathway in its own right and not only a modulator of canonical TGF-β signalling. Cell. Mol. Life Sci. 77, 243–251. doi:10.1007/s00018-019-03266-3
Kandalam, V., Basu, R., Moore, L., Fan, D., Wang, X., Jaworski, D. M., et al. (2011). Lack of tissue inhibitor of metalloproteinases 2 leads to exacerbated left ventricular dysfunction and adverse extracellular matrix remodeling in response to biomechanical stress. Circulation 124, 2094–2105. doi:10.1161/CIRCULATIONAHA.111.030338
Kassiri, Z., Defamie, V., Hariri, M., Oudit, G. Y., Anthwal, S., Dawood, F., et al. (2009). Simultaneous transforming growth factor β-tumor necrosis factor activation and cross-talk cause aberrant remodeling response and myocardial fibrosis in Timp3-deficient heart. J. Biol. Chem. 284, 29893–29904. doi:10.1074/jbc.M109.028449
Katare, P. B., Nizami, H. L., Paramesha, B., Dinda, A. K., and Banerjee, S. K. (2020). Activation of toll like receptor 4 (TLR4) promotes cardiomyocyte apoptosis through SIRT2 dependent p53 deacetylation. Sci. Rep. 10 (1 10), 19232–19246. doi:10.1038/s41598-020-75301-4
Kawano, H., Do, Y. S., Kawano, Y., Starnes, V., Barr, M., Law, R. E., et al. (2000). Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation 101, 1130–1137. doi:10.1161/01.CIR.101.10.1130
Kawashima, H., Hirose, M., Hirose, J., Nagakubo, D., Plaas, A. H. K., and Miyasaka, M. (2000). Binding of a large chondroitin sulfate/dermatan sulfate proteoglycan, versican, to L-selectin, P-selectin, and CD44. J. Biol. Chem. 275, 35448–35456. doi:10.1074/jbc.M003387200
Kim, R. J., Chen, E. L., Lima, J. A. C., and Judd, R. M. (1996). Myocardial Gd-DTPA kinetics determine MRI contrast enhancement and reflect the extent and severity of myocardial injury after acute reperfused infarction. Circulation 94, 3318–3326. doi:10.1161/01.CIR.94.12.3318
Kim, Y., Nurakhayev, S., Nurkesh, A., Zharkinbekov, Z., and Saparov, A. (2021). Macrophage polarization in cardiac tissue repair following myocardial infarction. Int. J. Mol. Sci. 22, 2715–15. doi:10.3390/IJMS22052715
Klappacher, G., Franzen, P., Haab, D., Mehrabi, M., Binder, M., Plesch, K., et al. (1995). Measuring extracellular matrix turnover in the serum of patients with idiopathic or ischemic dilated cardiomyopathy and impact on diagnosis and prognosis. Am. J. Cardiol. 75, 913–918. doi:10.1016/S0002-9149(99)80686-9
Klotz, S., Danser, A. H. J., Foronjy, R. F., Oz, M. C., Wang, J., Mancini, D., et al. (2007). The impact of angiotensin-converting enzyme inhibitor therapy on the extracellular collagen matrix during left ventricular assist device support in patients with end-stage heart failure. J. Am. Coll. Cardiol. 49, 1166–1174. doi:10.1016/J.JACC.2006.10.071
Komatsubara, I., Murakami, T., Kusachi, S., Nakamura, K., Hirohata, S., Hayashi, J., et al. (2003). Spatially and temporally different expression of osteonectin and osteopontin in the infarct zone of experimentally induced myocardial infarction in rats. Cardiovasc. Pathol. 12, 186–194. doi:10.1016/S1054-8807(03)00042-5
Kong, P., Christia, P., and Frangogiannis, N. G. (2014a). The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 71, 549–574. doi:10.1007/S00018-013-1349-6
Kong, P., Christia, P., and Frangogiannis, N. G. (2014b). The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 71, 549–574. doi:10.1007/s00018-013-1349-6
Konishi, H., Katoh, Y., Takaya, N., Kashiwakura, Y., Itoh, S., Ra, C., et al. (2002). Platelets activated by collagen through immunoreceptor tyrosine-based activation motif play pivotal role in initiation and generation of neointimal hyperplasia after vascular injury. Circulation 105, 912–916. doi:10.1161/HC0802.105256
Konstandin, M. H., Völkers, M., Collins, B., Quijada, P., Quintana, M., De La Torre, A., et al. (2013). Fibronectin contributes to pathological cardiac hypertrophy but not physiological growth. Basic Res. Cardiol. 108, 375. doi:10.1007/S00395-013-0375-8
Kostin, S. (2000). “The structural correlate of reduced cardiac function in failing human hearts BT - the hypertrophied heart,” in, eds. N. Takeda, M. Nagano, and N. S. Dhalla (Boston, MA: Springer US), 423–439. doi: doi:10.1007/978-1-4615-4423-4_35
Kuhota, T., McTiernan, C. F., Frye, C. S., Slawson, S. E., Lemster, B. H., Koretsky, A. P., et al. (1997). Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ. Res. 81, 627–635. doi:10.1161/01.RES.81.4.627
Kukreja, R. C., Yin, C., and Salloum, F. N. (2011). MicroRNAs: New players in cardiac injury and protection. Mol. Pharmacol. 80, 558–564. doi:10.1124/MOL.111.073528
Kupfahl, C., Pink, D., Friedrich, K., Zurbrügg, H. R., Neuss, M., Warnecke, C., et al. (2000). Angiotensin II directly increases transforming growth factor beta1 and osteopontin and indirectly affects collagen mRNA expression in the human heart. Cardiovasc. Res. 46, 463–475. doi:10.1016/s0008-6363(00)00037-7
Kuzmanov, U., Wang, E. Y., Vanderlaan, R., Kim, D. H., Lee, S. H., Hadipour-Lakmehsari, S., et al. (2020a2020). Mapping signalling perturbations in myocardial fibrosis via the integrative phosphoproteomic profiling of tissue from diverse sources. Nat. Biomed. Eng. 4 (9 4), 889–900. doi:10.1038/s41551-020-0585-y
Kuzmanov, U., Wang, E. Y., Vanderlaan, R., Kim, D. H., Lee, S. H., Hadipour-Lakmehsari, S., et al. (2020b2020). Mapping signalling perturbations in myocardial fibrosis via the integrative phosphoproteomic profiling of tissue from diverse sources. Nat. Biomed. Eng. 4 (9 4), 889–900. doi:10.1038/s41551-020-0585-y
Ladage, D., Yaniz-Galende, E., Rapti, K., Ishikawa, K., Tilemann, L., Shapiro, S., et al. (2013). Stimulating myocardial regeneration with periostin peptide in large mammals improves function post-myocardial infarction but increases myocardial fibrosis. PLoS ONE 8, e59656. doi:10.1371/journal.pone.0059656
Lee, A. A., Dillmann, W. H., McCulloch, A. D., and Villarreal, F. J. (1995). Angiotensin II stimulates the autocrine production of transforming growth factor-beta 1 in adult rat cardiac fibroblasts. J. Mol. Cell. Cardiol. 27, 2347–2357. doi:10.1016/s0022-2828(95)91983-x
Levick, S. P., and Goldspink, P. H. (2014). Could interferon-gamma be a therapeutic target for treating heart failure? Heart fail. Rev. 19, 227–236. doi:10.1007/S10741-013-9393-8
Lindsey, M. L., Jung, M., Hall, M. E., and DeLeon-Pennell, K. Y. (2018). Proteomic analysis of the cardiac extracellular matrix: Clinical research applications. Expert Rev. Proteomics 15, 105–112. doi:10.1080/14789450.2018.1421947
Liu, Y., Wu, M., Zhong, C., Xu, B., and Kang, L. (2022). M2-like macrophages transplantation protects against the doxorubicin-induced heart failure via mitochondrial transfer. Biomater. Res. 26, 14. doi:10.1186/S40824-022-00260-Y
Liu, Y., Zeng, R., Wang, R., Weng, Y., Wang, R., Zou, P., et al. (2021). Spatiotemporally resolved subcellular phosphoproteomics. Proc. Natl. Acad. Sci. U. S. A. 118, e2025299118. doi:10.1073/PNAS.2025299118/SUPPL_FILE/PNAS.2025299118.SD06
López, B., Querejeta, R., González, A., Larman, M., and Díez, J. (2012). Collagen cross-linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure: Potential role of lysyl oxidase. Hypertension 60, 677–683. doi:10.1161/HYPERTENSIONAHA.112.196113
Lorenzo, P., Bayliss, M. T., and Heinegård, D. (2004). Altered patterns and synthesis of extracellular matrix macromolecules in early osteoarthritis. Matrix Biol. 23, 381–391. doi:10.1016/J.MATBIO.2004.07.007
Lorenzo, P., Bayliss, M. T., and Heinegårdt, D. (1998a). A novel cartilage protein (CILP) present in the mid-zone of human articular cartilage increases with age. J. Biol. Chem. 273, 23463–23468. doi:10.1074/JBC.273.36.23463
Lorenzo, P., Neame, P., Sommarin, Y., and Heinegård, D. (1998b). Cloning and deduced amino acid sequence of a novel cartilage protein (CILP) identifies a proform including a nucleotide pyrophosphohydrolase. J. Biol. Chem. 273, 23469–23475. doi:10.1074/JBC.273.36.23469
Lu, M., Qin, Q., Yao, J., Sun, L., and Qin, X. (2019). Induction of LOX by TGF-β1/Smad/AP-1 signaling aggravates rat myocardial fibrosis and heart failure. IUBMB life 71, 1729–1739. doi:10.1002/IUB.2112
Lundby, A., Andersen, M. N., Steffensen, A. B., Horn, H., Kelstrup, C. D., Francavilla, C., et al. (2013). In vivo phosphoproteomics analysis reveals the cardiac targets of β-adrenergic receptor signaling. Sci. Signal. 6. doi:10.1126/SCISIGNAL.2003506/SUPPL_FILE/TABLES5
Lundgren, E., Gullberg, D., Rubin, K., Borg, T. K., Terracio, M. J., and Terracio, L. (1988). In vitro studies on adult cardiac myocytes: Attachment and biosynthesis of collagen type IV and laminin. J. Cell. Physiol. 136, 43–53. doi:10.1002/jcp.1041360106
Ma, F., Li, Y., Jia, L., Han, Y., Cheng, J., Li, H., et al. (2012). Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PloS one 7, e35144. doi:10.1371/JOURNAL.PONE.0035144
Ma, Y., Halade, G. V., Zhang, J., Ramirez, T. A., Levin, D., Voorhees, A., et al. (2013). Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ. Res. 112, 675–688. doi:10.1161/CIRCRESAHA.111.300502
Martos, R., Baugh, J., Ledwidge, M., O’Loughlin, C., Murphy, N. F., Conlon, C., et al. (2009). Diagnosis of heart failure with preserved ejection fraction: Improved accuracy with the use of markers of collagen turnover. Eur. J. Heart Fail. 11, 191–197. doi:10.1093/EURJHF/HFN036
Massagué, J. (2000). How cells read TGF-beta signals. Nat. Rev. Mol. Cell. Biol. 1, 169–178. doi:10.1038/35043051
Massoullié, G., Sapin, V., Ploux, S., Rossignol, P., Mulliez, A., Jean, F., et al. (2019). Low fibrosis biomarker levels predict cardiac resynchronization therapy response. Sci. Rep. 9 (1 9), 6103. doi:10.1038/s41598-019-42468-4
Masuda, I., Iyama, K. I., Halligan, B. D., Barbieri, J. T., Haas, A. L., Mccarty, D. J., et al. (2001). Variations in site and levels of expression of chondrocyte nucleotide pyrophosphohydrolase with aging. J. Bone Min. Res. 16, 868–875. doi:10.1359/JBMR.2001.16.5.868
Matsui, Y., Ikesue, M., Danzaki, K., Morimoto, J., Sato, M., Tanaka, S., et al. (2011). Syndecan-4 prevents cardiac rupture and dysfunction after myocardial infarction. Circ. Res. 108, 1328–1339. doi:10.1161/CIRCRESAHA.110.235689
Matsumoto, K., Saga, Y., Ikemura, T., Sakakura, T., and Chiquet-Ehrismann, R. (1994). The distribution of tenascin-X is distinct and often reciprocal to that of tenascin-C. J. Cell. Biol. 125, 483–493. doi:10.1083/jcb.125.2.483
McCabe, M. C., Saviola, A. J., and Hansen, K. C. (2022). A mass spectrometry-based atlas of extracellular matrix proteins across 25 mouse organs. bioRxiv 2022, 03.04.482898. doi:10.1101/2022.03.04.482898
McCabe, M. C., Schmitt, L. R., Hill, R. C., Dzieciatkowska, M., Maslanka, M., Daamen, W. F., et al. (2021). Evaluation and refinement of sample preparation methods for extracellular matrix proteome coverage. Mol. Cell. Proteomics. 20, 100079. doi:10.1016/J.MCPRO.2021.100079
McLellan, M. A., Skelly, D. A., Dona, M. S. I., Squiers, G. T., Farrugia, G. E., Gaynor, T. L., et al. (2020). High-resolution transcriptomic profiling of the heart during chronic stress reveals cellular drivers of cardiac fibrosis and hypertrophy. Circulation 142, 1448–1463. doi:10.1161/CIRCULATIONAHA.119.045115
Medeiros, N. I., Gomes, J. A. S., Fiuza, J. A., Sousa, G. R., Almeida, E. F., Novaes, R. O., et al. (2019). MMP-2 and MMP-9 plasma levels are potential biomarkers for indeterminate and cardiac clinical forms progression in chronic Chagas disease. Sci. Rep. 9 (1 9), 14170–14179. doi:10.1038/s41598-019-50791-z
Meier, F., Köhler, N. D., Brunner, A. D., Wanka, J. M. H., Voytik, E., Strauss, M. T., et al. (2021). Deep learning the collisional cross sections of the peptide universe from a million experimental values. Nat. Commun. 12, 1185. doi:10.1038/S41467-021-21352-8
Merl-Pham, J., Basak, T., Knüppel, L., Ramanujam, D., Athanason, M., Behr, J., et al. (2019). Quantitative proteomic profiling of extracellular matrix and site-specific collagen post-translational modifications in an in vitro model of lung fibrosis. Matrix Biol. Plus 1, 100005. doi:10.1016/j.mbplus.2019.04.002
Merline, R., Schaefer, R. M., and Schaefer, L. (2009). The matricellular functions of small leucine-rich proteoglycans (SLRPs). J. Cell. Commun. Signal. 3, 323–335. doi:10.1007/s12079-009-0066-2
Messroghli, D. R., Radjenovic, A., Kozerke, S., Higgins, D. M., Sivananthan, M. U., and Ridgway, J. P. (2004). Modified Look-Locker inversion recovery (MOLLI) for high-resolution T1 mapping of the heart. Magn. Reson. Med. 52, 141–146. doi:10.1002/MRM.20110
Moll, S., Desmoulière, A., Moeller, M. J., Pache, J. C., Badi, L., Arcadu, F., et al. (2019). DDR1 role in fibrosis and its pharmacological targeting. Biochim. Biophys. Acta. Mol. Cell. Res. 1866, 118474. doi:10.1016/J.BBAMCR.2019.04.004
Molnar, J., Fong, K. S. K., He, Q. P., Hayashi, K., Kim, Y., Fong, S. F. T., et al. (2003). Structural and functional diversity of lysyl oxidase and the LOX-like proteins. Biochim. Biophys. Acta 1647, 220–224. doi:10.1016/S1570-9639(03)00053-0
Mongiat, M., Sweeney, S. M., San Antonio, J. D., Fu, J., and Iozzo, R. V. (2003). Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J. Biol. Chem. 278, 4238–4249. doi:10.1074/JBC.M210445200
Moreira, L. M., Takawale, A., Hulsurkar, M., Menassa, D. A., Antanaviciute, A., Lahiri, S. K., et al. (2020). Paracrine signalling by cardiac calcitonin controls atrial fibrogenesis and arrhythmia. Nature 587, 460–465. doi:10.1038/S41586-020-2890-8
Mori, M., Nakajima, M., Mikami, Y., Seki, S., Takigawa, M., Kubo, T., et al. (2006). Transcriptional regulation of the cartilage intermediate layer protein (CILP) gene. Biochem. Biophys. Res. Commun. 341, 121–127. doi:10.1016/J.BBRC.2005.12.159
Motamed, K., Funk, S. E., Koyama, H., Ross, R., Raines, E. W., and Sage, E. H. (2002). Inhibition of PDGF-stimulated and matrix-mediated proliferation of human vascular smooth muscle cells by SPARC is independent of changes in cell shape or cyclin-dependent kinase inhibitors. J. Cell. Biochem. 84, 759–771. doi:10.1002/jcb.10095
Murphy-Ullrich, J. E. (2001). The de-adhesive activity of matricellular proteins: Is intermediate cell adhesion an adaptive state? J. Clin. Investig. 107, 785–790. doi:10.1172/JCI12609
Murtha, L. A., Schuliga, M. J., Mabotuwana, N. S., Hardy, S. A., Waters, D. W., Burgess, J. K., et al. (2017). The processes and mechanisms of cardiac and pulmonary fibrosis. Front. Physiol. 8, 777. doi:10.3389/fphys.2017.00777
Myllyharju, J. (2005). Intracellular post-translational modifications of collagens. Top. Curr. Chem. (Cham). 247, 115–147. doi:10.1007/B103821
Myllyharju, J. (2008). Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann. Med. 40, 402–417. doi:10.1080/07853890801986594
Nandi, S. S., Katsurada, K., Sharma, N. M., Anderson, D. R., Mahata, S. K., and Patel, K. P. (2020). MMP9 inhibition increases autophagic flux in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 319, H1414-H1437–H1437. doi:10.1152/AJPHEART.00032.2020
Nauroy, P., Hughes, S., Naba, A., and Ruggiero, F. (2018a). The in-silico zebrafish matrisome: A new tool to study extracellular matrix gene and protein functions. Matrix Biol. 65, 5–13. doi:10.1016/J.MATBIO.2017.07.001
Nauroy, P., Hughes, S., Naba, A., and Ruggiero, F. (2018b). The in-silico zebrafish matrisome: A new tool to study extracellular matrix gene and protein functions. Matrix Biol. 65, 5–13. doi:10.1016/J.MATBIO.2017.07.001
Nevers, T., Salvador, A. M., Grodecki-Pena, A., Knapp, A., Velázquez, F., Aronovitz, M., et al. (2015). Left ventricular T-cell recruitment contributes to the pathogenesis of heart failure. Circ. Heart Fail. 8, 776–787. doi:10.1161/CIRCHEARTFAILURE.115.002225
Nevers, T., Salvador, A. M., Velazquez, F., Ngwenyama, N., Carrillo-Salinas, F. J., Aronovitz, M., et al. (2017). Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J. Exp. Med. 214, 3311–3329. doi:10.1084/JEM.20161791
Ohmura, H., Yasukawa, H., Minami, T., Sugi, Y., Oba, T., Nagata, T., et al. (2012). Cardiomyocyte-specific transgenic expression of lysyl oxidase-like protein-1 induces cardiac hypertrophy in mice. Hypertens. Res. 35, 1063–1068. doi:10.1038/HR.2012.92
Ohnishi, H., Oka, T., Kusachi, S., Nakanishi, T., Takeda, K., Nakahama, M., et al. (1998). Increased expression of connective tissue growth factor in the infarct zone of experimentally induced myocardial infarction in rats. J. Mol. Cell. Cardiol. 30, 2411–2422. doi:10.1006/jmcc.1998.0799
Oliviéro, P., Chassagne, C., SalichoNN., , Corbier, A., Hamon, G., MarotteF., , et al. (2000). Expression of laminin alpha2 chain during normal and pathological growth of myocardium in rat and human. Cardiovasc. Res. 46, 346–355. doi:10.1016/S0008-6363(00)00034-1
Pitt, B., Zannad, F., Remme, W. J., Cody, R., Castaigne, A., Perez, A., et al. (1999). The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 341, 709–717. doi:10.1056/NEJM199909023411001
Plavelil, N., Goldstein, R., Klein, M., Ho, V. B., Haigney, M. C., and Hood, M. N. (2020). Experimental biology 2020 meeting abstracts. FASEB J. 34, 1. doi:10.1096/fsb2.v34.S1
Pokidysheva, E., Boudko, S., Vranka, J., Zientek, K., Maddox, K., Moser, M., et al. (2014). Biological role of prolyl 3-hydroxylation in type IV collagen. Proc. Natl. Acad. Sci. U. S. A. 111, 161–166. doi:10.1073/pnas.1307597111
Prabhu, S. D., and Frangogiannis, N. G. (2016). The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ. Res. 119, 91–112. doi:10.1161/CIRCRESAHA.116.303577
Prockop, D. J., and Kivirikko, K. I. (1995). Collagens: Molecular biology, diseases, and potentials for therapy. Annu. Rev. Biochem. 64, 403–434. doi:10.1146/ANNUREV.BI.64.070195.002155
Querejeta, R., López, B., González, A., Sánchez, E., Larman, M., Martínez Ubago, J. L., et al. (2004). Increased collagen type I synthesis in patients with heart failure of hypertensive origin: Relation to myocardial fibrosis. Circulation 110, 1263–1268. doi:10.1161/01.CIR.0000140973.60992.9A
Rappu, P., Salo, A. M., Myllyharju, J., and Heino, J. (2019). Role of prolyl hydroxylation in the molecular interactions of collagens. Essays Biochem. 63, 325–335. doi:10.1042/EBC20180053
Riley, N. M., and Coon, J. J. (2016). Phosphoproteomics in the age of rapid and deep proteome profiling. Anal. Chem. 88, 74–94. doi:10.1021/ACS.ANALCHEM.5B04123/ASSET/IMAGES/LARGE/AC-2015-04123X_0009.JPEG
{kind=link}
Rostam, M. A., Kamato, D., Piva, T. J., Zheng, W., Little, P. J., and Osman, N. (2016). The role of specific Smad linker region phosphorylation in TGF-β mediated expression of glycosaminoglycan synthesizing enzymes in vascular smooth muscle. Cell. Signal. 28, 956–966. doi:10.1016/J.CELLSIG.2016.05.002
Rouf, R., MacFarlane, E. G., Takimoto, E., Chaudhary, R., Nagpal, V., Rainer, P. P., et al. (2017). Nonmyocyte ERK1/2 signaling contributes to load-induced cardiomyopathy in Marfan mice. JCI insight 2, 91588. doi:10.1172/JCI.INSIGHT.91588
Russo, I., and Frangogiannis, N. G. (2016). Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 90, 84–93. doi:10.1016/J.YJMCC.2015.12.011
Salo, A. M., and Myllyharju, J. (2021). Prolyl and lysyl hydroxylases in collagen synthesis. Exp. Dermatol. 30, 38–49. doi:10.1111/EXD.14197
Sánchez-Iranzo, H., Galardi-Castilla, M., Sanz-Morejón, A., González-Rosa, J. M., Costa, R., Ernst, A., et al. (2018). Transient fibrosis resolves via fibroblast inactivation in the regenerating zebrafish heart. Proc. Natl. Acad. Sci. U. S. A. 115, 4188–4193. doi:10.1073/PNAS.1716713115
Sane, D. C., Kontos, J. L., and Greenberg, C. S. (2007). Roles of transglutaminases in cardiac and vascular diseases. Front. Biosci. 12, 2530–2545. doi:10.2741/2253
Sarohi, V., Srivastava, S., and Basak, T. (2022a). A comprehensive outlook on dilated cardiomyopathy (DCM): State-Of-The-Art developments with special emphasis on OMICS-based approaches. J. Cardiovasc. Dev. Dis. 9, 174. doi:10.3390/JCDD9060174
Sarohi, V., Srivastava, S., and Basak, T. (2022b). Comprehensive mapping and dynamics of site-specific prolyl-hydroxylation, lysyl-hydroxylation and lysyl O-glycosylation of collagens deposited in ECM during zebrafish heart regeneration. Front. Mol. Biosci. 9, 892763. doi:10.3389/FMOLB.2022.892763
Sasse, P., Malan, D., Fleischmann, M., Roell, W., Gustafsson, E., Bostani, T., et al. (2008). Perlecan is critical for heart stability. Cardiovasc. Res. 80, 435–444. doi:10.1093/CVR/CVN225
Savage, S. R., and Zhang, B. (2020). Using phosphoproteomics data to understand cellular signaling: A comprehensive guide to bioinformatics resources. Clin. Proteomics 17, 27–18. doi:10.1186/s12014-020-09290-x
Savarese, G., and Lund, L. H. (2017). Global public health burden of heart failure. Card. Fail. Rev. 3, 7–11. doi:10.15420/CFR.2016:25:2
Schelbert, E. B., Piehler, K. M., Zareba, K. M., Moon, J. C., Ugander, M., Messroghli, D. R., et al. (2015). Myocardial fibrosis quantified by extracellular volume is associated with subsequent hospitalization for heart failure, death, or both across the spectrum of ejection fraction and heart failure stage. J. Am. Heart Assoc. 4, e002613. doi:10.1161/JAHA.115.002613
Schellings, M. W. M., Vanhoutte, D., Swinnen, M., Cleutjens, J. P., Debets, J., Van Leeuwen, R. E. W., et al. (2009). Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J. Exp. Med. 206, 113–123. doi:10.1084/jem.20081244
Schellings, M. W. M., Vanhoutte, D., Van Almen, G. C., Swinnen, M., Leenders, J. J. G., Kubben, N., et al. (2010). Syndecan-1 amplifies angiotensin II-induced cardiac fibrosis. Hypertension 55, 249–256. doi:10.1161/HYPERTENSIONAHA.109.137885
Schroen, B., Heymans, S., Sharma, U., Blankesteijn, W. M., Pokharel, S., Cleutjens, J. P. M., et al. (2004). Thrombospondin-2 is essential for myocardial matrix integrity: Increased expression identifies failure-prone cardiac hypertrophy. Circ. Res. 95, 515–522. doi:10.1161/01.RES.0000141019.20332.3e
Schroer, A. K., and Merryman, W. D. (2015). Mechanobiology of myofibroblast adhesion in fibrotic cardiac disease. J. Cell. Sci. 128, 1865–1875. LP – 1875. doi:10.1242/jcs.162891
Schwarzbauer, J. E. (1991). Fibronectin: From gene to protein. Curr. Opin. Cell. Biol. 3, 786–791. doi:10.1016/0955-0674(91)90051-y
Segura, A. M., Frazier, O. H., and Buja, L. M. (2014). Fibrosis and heart failure. Heart fail. Rev. 19, 173–185. doi:10.1007/s10741-012-9365-4
Seki, S., Kawaguchi, Y., Chiba, K., Mikami, Y., Kizawa, H., Oya, T., et al. (2005). A functional SNP in CILP, encoding cartilage intermediate layer protein, is associated with susceptibility to lumbar disc disease. Nat. Genet. 37, 607–612. doi:10.1038/NG1557
Sen, S., Chen, S., Feng, B., Iglarz, M., and Chakrabarti, S. (2012). Renal, retinal and cardiac changes in type 2 diabetes are attenuated by macitentan, a dual endothelin receptor antagonist. Life Sci. 91, 658–668. doi:10.1016/J.LFS.2012.03.032
Shapiro, S. D. (1998). Matrix metalloproteinase degradation of extracellular matrix: Biological consequences. Curr. Opin. Cell. Biol. 10, 602–608. doi:10.1016/S0955-0674(98)80035-5
Sharma, K., D’Souza, R. C. J., Tyanova, S., Schaab, C., Wiśniewski, J. R., Cox, J., et al. (2014). Ultradeep human phosphoproteome reveals a distinct regulatory nature of tyr and ser/thr-based signaling. Cell. Rep. 8, 1583–1594. doi:10.1016/j.celrep.2014.07.036
Shimizu, T., and Liao, J. K. (2016). Rho kinases and cardiac remodeling. Circ. J. 80, 1491–1498. doi:10.1253/CIRCJ.CJ-16-0433
Shiraishi, M., Shintani, Y., Shintani, Y., Ishida, H., Saba, R., Yamaguchi, A., et al. (2016). Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 126, 2151–2166. doi:10.1172/JCI85782
Simões, F. C., Cahill, T. J., Kenyon, A., Gavriouchkina, D., Vieira, J. M., Sun, X., et al. (2020). Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 11 (1 11), 600–617. doi:10.1038/s41467-019-14263-2
Sims, J. R., Faça, V. M., Pereira, C., Ascenção, C., Comstock, W., Badar, J., et al. (2022). Phosphoproteomics of ATR signaling in mouse testes. eLife 11, e68648. doi:10.7554/ELIFE.68648
Sipila, K. H., Drushinin, K., Rappu, P., Jokinen, J., Salminen, T. A., Salo, A. M., et al. (2018). Proline hydroxylation in collagen supports integrin binding by two distinct mechanisms. J. Biol. Chem. 293, 7645–7658. doi:10.1074/JBC.RA118.002200
Smith-Mungo, L. I., and Kagan, H. M. (1998). Lysyl oxidase: Properties, regulation and multiple functions in biology. Matrix Biol. 16, 387–398. doi:10.1016/S0945-053X(98)90012-9
Soni, R. K. (2022). High-throughput plasma proteomic profiling. Methods Mol. Biol. 2546, 411–420. doi:10.1007/978-1-0716-2565-1_36
Stawikowski, M. J., Aukszi, B., Stawikowska, R., Cudic, M., and Fields, G. B. (2014). Glycosylation modulates melanoma cell α2β1 and α3β1 integrin interactions with type IV collagen. J. Biol. Chem. 289, 21591–21604. doi:10.1074/JBC.M114.572073
Steffensen, L. B., and Rasmussen, L. M. (2018). A role for collagen type IV in cardiovascular disease? Am. J. Physiol. Heart Circ. Physiol. 315, H610-H625–H625. doi:10.1152/AJPHEART.00070.2018
Steger, M., Tonelli, F., Ito, G., Davies, P., Trost, M., Vetter, M., et al. (2016). Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 5, e12813. doi:10.7554/eLife.12813
St. John Sutton, M., Ghio, S., Plappert, T., Tavazzi, L., Scelsi, L., Daubert, C., et al. (2009). Cardiac resynchronization induces major structural and functional reverse remodeling in patients with New York Heart Association class I/II heart failure. Circulation 120, 1858–1865. doi:10.1161/CIRCULATIONAHA.108.818724
Suna, G., Wojakowski, W., Lynch, M., Barallobre-Barreiro, J., Yin, X., Mayr, U., et al. (2018). Extracellular matrix proteomics reveals interplay of aggrecan and aggrecanases in vascular remodeling of stented coronary arteries. Circulation 137, 166–183. doi:10.1161/CIRCULATIONAHA.116.023381
Swinnen, M., Vanhoutte, D., Van Almen, G. C., Hamdani, N., Schellings, M. W. M., D’Hooge, J., et al. (2009). Absence of thrombospondin-2 causes age-related dilated cardiomyopathy. Circulation 120, 1585–1597. doi:10.1161/CIRCULATIONAHA.109.863266
Taha, I. N., and Naba, A. (2019). Exploring the extracellular matrix in health and disease using proteomics. Essays Biochem. 63, 417–432. doi:10.1042/EBC20190001
Takahashi, K., Ghatei, M. A., Lam, H. C., O’Halloran, D. J., and Bloom, S. R. (1990). Elevated plasma endothelin in patients with diabetes mellitus. Diabetologia 33, 306–310. doi:10.1007/BF00403325
Takaluoma, K., Hyry, M., Lantto, J., Sormunen, R., Bank, R. A., Kivirikko, K. I., et al. (2007). Tissue-specific changes in the hydroxylysine content and cross-links of collagens and alterations in fibril morphology in lysyl hydroxylase 1 knock-out mice. J. Biol. Chem. 282, 6588–6596. doi:10.1074/JBC.M608830200
Takawale, A., Zhang, P., Patel, V. B., Wang, X., Oudit, G., and Kassiri, Z. (2017). Tissue inhibitor of matrix metalloproteinase-1 promotes myocardial fibrosis by mediating CD63-integrin β1 interaction. Hypertension 69, 1092–1103. doi:10.1161/HYPERTENSIONAHA.117.09045
Tan, H., Yang, K., Li, Y., Shaw, T. I., Wang, Y., Blanco, D. B., et al. (2017). Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity 46, 488–503. doi:10.1016/J.IMMUNI.2017.02.010
Terajima, M., Perdivara, I., Sricholpech, M., Deguchi, Y., Pleshko, N., Tomer, K. B., et al. (2014). Glycosylation and cross-linking in bone type I collagen. J. Biol. Chem. 289, 22636–22647. doi:10.1074/JBC.M113.528513
Termine, J. D., Kleinman, H. K., Whitson, S. W., Conn, K. M., McGarvey, M. L., and Martin, G. R. (1981). Osteonectin, a bone-specific protein linking mineral to collagen. Cell. 26, 99–105. doi:10.1016/0092-8674(81)90037-4
Testa, M., Yeh, M., Lee, P., Fanelli, R., Loperfido, F., Berman, J. W., et al. (1996). Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J. Am. Coll. Cardiol. 28, 964–971. doi:10.1016/S0735-1097(96)00268-9
Thiery, J. P. (2002). Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2, 442–454. doi:10.1038/nrc822
Tian, J., An, X., and Niu, L. (2017). Myocardial fibrosis in congenital and pediatric heart disease. Exp. Ther. Med. 13, 1660–1664. doi:10.3892/etm.2017.4224
Troilo, H., Steer, R., Collins, R. F., Kielty, C. M., and Baldock, C. (2016). Independent multimerization of Latent TGFβ Binding Protein-1 stabilized by cross-linking and enhanced by heparan sulfate. Sci. Rep. 6, 34347. doi:10.1038/SREP34347
Trueblood, N. A., Xie, Z., Communal, C., Sam, F., Ngoy, S., Liaw, L., et al. (2001). Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ. Res. 88, 1080–1087. doi:10.1161/hh1001.090842
Tryggvason, K. (1993). The laminin family. Curr. Opin. Cell. Biol. 5, 877–882. doi:10.1016/0955-0674(93)90038-r
Valencik, M. L., Zhang, D., Punske, B., Hu, P., McDonald, J. A., and Litwin, S. E. (2006). Integrin activation in the heart: A link between electrical and contractile dysfunction? Circ. Res. 99, 1403–1410. doi:10.1161/01.RES.0000252291.88540.ac
Valente, A. J., Yoshida, T., Gardner, J. D., Somanna, N., Delafontaine, P., and Chandrasekar, B. (2012). Interleukin-17A stimulates cardiac fibroblast proliferation and migration via negative regulation of the dual-specificity phosphatase MKP-1/DUSP-1. Cell. Signal. 24, 560–568. doi:10.1016/J.CELLSIG.2011.10.010
Valiente-Alandi, I., Potter, S. J., Salvador, A. M., Schafer, A. E., Schips, T., Carrillo-Salinas, F., et al. (2018). Inhibiting fibronectin attenuates fibrosis and improves cardiac function in a model of heart failure. Circulation 138, 1236–1252. doi:10.1161/CIRCULATIONAHA.118.034609
van den Akker, G. G., van Beuningen, H. M., Vitters, E. L., Koenders, M. I., van de Loo, F. A., van Lent, P. L., et al. (2017). Interleukin 1 β-induced SMAD2/3 linker modifications are TAK1 dependent and delay TGFβ signaling in primary human mesenchymal stem cells. Cell. Signal. 40, 190–199. doi:10.1016/J.CELLSIG.2017.09.010
Van Nieuwenhoven, F. A., Munts, C., Op’T Veld, R. C., González, A., Diéz, J., Heymans, S., et al. (2017). Cartilage intermediate layer protein 1 (CILP1): A novel mediator of cardiac extracellular matrix remodelling. Sci. Rep. 7, 16042. doi:10.1038/S41598-017-16201-Y
Vanhoutte, D., Schellings, M. W. M., Götte, M., Swinnen, M., Herias, V., Wild, M. K., et al. (2007). Increased expression of syndecan-1 protects against cardiac dilatation and dysfunction after myocardial infarction. Circulation 115, 475–482. doi:10.1161/CIRCULATIONAHA.106.644609
Veerappan, A., O’Connor, N. J., Brazin, J., Reid, A. C., Jung, A., McGee, D., et al. (2013). Mast cells: A pivotal role in pulmonary fibrosis. DNA Cell. Biol. 32, 206–218. doi:10.1089/DNA.2013.2005
Villarreal, F., Epperson, S. A., Ramirez-Sanchez, I., Yamazaki, K. G., and Brunton, L. L. (2009). Regulation of cardiac fibroblast collagen synthesis by adenosine: Roles for epac and PI3K. Am. J. Physiol. Cell. Physiol. 296, C1178–C1184. doi:10.1152/AJPCELL.00291.2008
Villarreal, F. J., and Dillmann, W. H. (1992). Cardiac hypertrophy-induced changes in mRNA levels for TGF-beta 1, fibronectin, and collagen. Am. J. Physiol. 262, H1861–H1866. doi:10.1152/ajpheart.1992.262.6.H1861
Viola, A., Munari, F., Sánchez-Rodríguez, R., Scolaro, T., and Castegna, A. (2019). The metabolic signature of macrophage responses. Front. Immunol. 10, 1462. doi:10.3389/FIMMU.2019.01462
Virani, S. S., Alonso, A., Benjamin, E. J., Bittencourt, M. S., Callaway, C. W., Carson, A. P., et al. (2020). Heart disease and stroke statistics-2020 update: A report from the American heart association. Circulation 141, E139–E596. doi:10.1161/CIR.0000000000000757
Voll, R. E., Herrmann, M., Roth, E. A., Stach, C., Kalden, J. R., and Girkontaite, I. (1997). Immunosuppressive effects of apoptotic cells. Nature 3906658 390, 350–351. doi:10.1038/37022
Vranka, J. A., Sakai, L. Y., and Bächinger, H. P. (2004). Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 279, 23615–23621. doi:10.1074/jbc.M312807200
Waller, A. H., Sanchez-Ross, M., Kaluski, E., and Klapholz, M. (2010). Osteopontin in cardiovascular disease: A potential therapeutic target. Cardiol. Rev. 18, 125–131. doi:10.1097/CRD.0b013e3181cfb646
Wang, J., Hoshijima, M., Lam, J., Zhou, Z., Jokiel, A., Dalton, N. D., et al. (2006). Cardiomyopathy associated with microcirculation dysfunction in laminin alpha4 chain-deficient mice. J. Biol. Chem. 281, 213–220. doi:10.1074/jbc.M505061200
Wang, K. X., and Denhardt, D. T. (2008). Osteopontin: Role in immune regulation and stress responses. Cytokine Growth Factor Rev. 19, 333–345. doi:10.1016/j.cytogfr.2008.08.001
Wang, W., Zhou, G., Hu, M. C. T., Yao, Z., and Tan, T. H. (1997). Activation of the hematopoietic progenitor kinase-1 (HPK1)-dependent, stress-activated c-Jun N-terminal kinase (JNK) pathway by transforming growth factor beta (TGF-beta)-activated kinase (TAK1), a kinase mediator of TGF beta signal transduction. J. Biol. Chem. 272, 22771–22775. doi:10.1074/JBC.272.36.22771
Wang, X., Chen, W., Zhang, J., Khan, A., Li, L., Huang, F., et al. (2017). Critical role of ADAMTS2 (A disintegrin and metalloproteinase with thrombospondin motifs 2) in cardiac hypertrophy induced by pressure overload. Hypertension 69, 1060–1069. doi:10.1161/HYPERTENSIONAHA.116.08581
Wang, Z., Stuckey, D. J., Murdoch, C. E., Camelliti, P., Lip, G. Y. H., and Griffin, M. (2018). Cardiac fibrosis can be attenuated by blocking the activity of transglutaminase 2 using a selective small-molecule inhibitor. Cell. Death Dis. 9, 613. doi:10.1038/S41419-018-0573-2
Weber, K. T., and Brilla, C. G. (1991). Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 83, 1849–1865. doi:10.1161/01.cir.83.6.1849
Weber, K. T., Sun, Y., Tyagi, S. C., and Cleutjens, J. P. (1994a). Collagen network of the myocardium: Function, structural remodeling and regulatory mechanisms. J. Mol. Cell. Cardiol. 26, 279–292. doi:10.1006/jmcc.1994.1036
Weber, K. T., Sun, Y., Tyagi, S. C., and Cleutjens, J. P. M. (1994b). Collagen network of the myocardium: Function, structural remodeling and regulatory mechanisms. J. Mol. Cell. Cardiol. 26, 279–292. doi:10.1006/JMCC.1994.1036
White, E. S., and Mantovani, A. R. (2013). Inflammation, wound repair, and fibrosis: Reassessing the spectrum of tissue injury and resolution. J. Pathol. 229, 141–144. doi:10.1002/PATH.4126
Widyantoro, B., Emoto, N., Nakayama, K., Anggrahini, D. W., Adiarto, S., Iwasa, N., et al. (2010). Endothelial cell–derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 121, 2407–2418. doi:10.1161/CIRCULATIONAHA.110.938217
Wu, M., Feng, G., Zhang, B., Xu, K., Wang, Z., Cheng, S., et al. (2020). Phosphoproteomics reveals novel targets and phosphoprotein networks in cell cycle mediated by Dsk1 kinase. ACS Appl. Mater. Interfaces 19, 1776–1787. doi:10.1021/ACS.JPROTEOME.0C00027/ASSET/IMAGES/LARGE/PR0C00027_0007
Wu, Y. J., La Pierre, D. P., Wu, J., Yee, A. J., and Yang, B. B. (2005). The interaction of versican with its binding partners. Cell. Res. 15, 483–494. doi:10.1038/sj.cr.7290318
Xing, S. S., Bi, X. P., Tan, H. W., Zhang, Y., Xing, Q. C., and Zhang, W. (2010). Overexpression of interleukin-18 aggravates cardiac fibrosis and diastolic dysfunction in fructose-fed rats. Mol. Med. 16, 465–470. doi:10.2119/MOLMED.2010.00028
Yamamoto, K., Dang, Q. N., Kennedy, S. P., Osathanondh, R., Kelly, R. A., and Lee, R. T. (1999). Induction of tenascin-C in cardiac myocytes by mechanical deformation. Role of reactive oxygen species. J. Biol. Chem. 274, 21840–21846. doi:10.1074/jbc.274.31.21840
Yang, J., Liu, Q., Yu, B., Han, B., and Yang, B. (2022). 4D-quantitative proteomics signature of asthenozoospermia and identification of extracellular matrix protein 1 as a novel biomarker for sperm motility. Mol. Omics 18, 83–91. doi:10.1039/D1MO00257K
Yang, J., Savvatis, K., Kang, J. S., Fan, P., Zhong, H., Schwartz, K., et al. (2016). Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat. Commun. 7, 13710. doi:10.1038/NCOMMS13710
Yao, X. Q., Liu, Z. Y., Chen, J. Y., Huang, Z. C., Liu, J. H., Sun, B. H., et al. (2021). Proteomics and bioinformatics reveal insights into neuroinflammation in the acute to subacute phases in rat models of spinal cord contusion injury. FASEB J. 35. doi:10.1096/FJ.202100081RR
Yarbrough, W. M., Baicu, C., Mukherjee, R., Van Laer, A., Rivers, W. T., McKinney, R. A., et al. (2014). Cardiac-restricted overexpression or deletion of tissue inhibitor of matrix metalloproteinase-4: Differential effects on left ventricular structure and function following pressure overload-induced hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 307, H752–H761. doi:10.1152/ajpheart.00063.2014
Yokota, T., McCourt, J., Ma, F., Ren, S., Li, S., Kim, T. H., et al. (2020). Type V collagen in scar tissue regulates the size of scar after heart injury. Cell. 182, 545–562. e23. doi:10.1016/J.CELL.2020.06.030
Yoon, P. O., Lee, M. A., Cha, H., Jeong, M. H., Kim, J., Jang, S. P., et al. (2010). The opposing effects of CCN2 and CCN5 on the development of cardiac hypertrophy and fibrosis. J. Mol. Cell. Cardiol. 49, 294–303. doi:10.1016/j.yjmcc.2010.04.010
Zeisberg, E. M., Tarnavski, O., Zeisberg, M., Dorfman, A. L., McMullen, J. R., Gustafsson, E., et al. (2007). Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13, 952–961. doi:10.1038/nm1613
Zhang, C. L., Zhao, Q., Liang, H., Qiao, X., Wang, J. Y., Wu, D., et al. (2018). Cartilage intermediate layer protein-1 alleviates pressure overload-induced cardiac fibrosis via interfering TGF-β1 signaling. J. Mol. Cell. Cardiol. 116, 135–144. doi:10.1016/J.YJMCC.2018.02.006
Zhang, J., Yang, R., Liu, Z., Hou, C., Zong, W., Zhang, A., et al. (2015). Loss of lysyl oxidase-like 3 causes cleft palate and spinal deformity in mice. Hum. Mol. Genet. 24, 6174–6185. doi:10.1093/HMG/DDV333
Zhang, Q. J., He, Y., Li, Y., Shen, H., Lin, L., Zhu, M., et al. (2021). Matricellular protein Cilp1 promotes myocardial fibrosis in response to myocardial infarction. Circ. Res. 129, 1021–1035. doi:10.1161/CIRCRESAHA.121.319482
Keywords: extracellular matrix, cardiac fibrosis, heart failure, proteomics, mass-spectrometry
Citation: Sarohi V, Chakraborty S and Basak T (2022) Exploring the cardiac ECM during fibrosis: A new era with next-gen proteomics. Front. Mol. Biosci. 9:1030226. doi: 10.3389/fmolb.2022.1030226
Received: 28 August 2022; Accepted: 31 October 2022;
Published: 22 November 2022.
Edited by:
Georgina Gonzalez-Avila, Instituto Nacional de Enfermedades Respiratorias-México (INER), MexicoReviewed by:
Criselda Mendozo-Milla, Instituto Nacional de Enfermedades Respiratorias-México (INER), MexicoPrachi Umbarkar, University of Alabama at Birmingham, United States
Copyright © 2022 Sarohi, Chakraborty and Basak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Trayambak Basak, dHJheWFtYmFrQGlpdG1hbmRpLmFjLmlu