 Cassandra Kennedy
Cassandra Kennedy Katherine McPhie
Katherine McPhie Katrin Rittinger
Katrin Rittinger- Molecular Structure of Cell Signalling Laboratory, The Francis Crick Institute, London, United Kingdom
The ubiquitin system contains a wealth of potential drug targets for many diseases and conditions, including neurodegenerative, immune, metabolic and developmental diseases, as well as multiple cancers. Despite years of research, relatively few clinical inhibitors or specific chemical probes for proteins within the ubiquitin system exist, with many interesting target proteins yet to be explored. Fragment-based drug discovery (FBDD) offers efficient and broad coverage of chemical space with small libraries, using covalent and non-covalent approaches. Coupled with advances in structural biology and proteomics, FBDD now provides a thorough screening platform for inhibitor discovery within the ubiquitin system. In this mini review, we summarise the current scope of FBDD and how it has been applied to ubiquitin-activating (E1), ubiquitin-conjugating (E2), ubiquitin ligase (E3) and deubiquitinating (DUB) enzymes. We also discuss the newest frontiers of FBDD and how they could be applied to enable inhibitor and novel chemical probe discovery and provide functional insight into the ubiquitin system.
Introduction
Ubiquitination is a post-translational modification (PTM) which regulates the majority of cellular processes, from protein degradation and homeostasis to cell cycle control and immune signalling (Komander and Rape, 2012). Proteins are marked with single molecules of ubiquitin (Ub) or poly-ubiquitin chains, via surface lysine residues. Ubiquitination is mediated by an ATP-dependant enzymatic cascade of E1-activating, E2-conjugating and E3 ligase enzymes. Proteins containing ubiquitin-binding domains (UBDs) recognise the modification; and proteases called deubiquitinating enzymes (DUBs) cleave ubiquitin chains. Together, this cellular machinery comprises the ubiquitin system (Figure 1A).
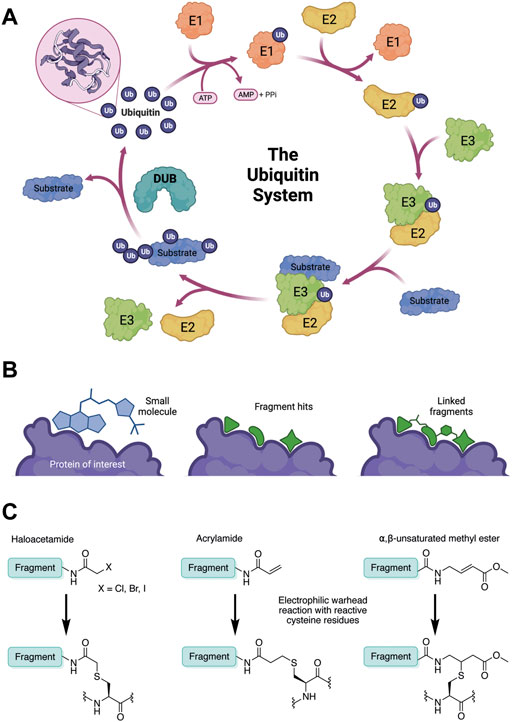
FIGURE 1. (A) The ubiquitin system involves a cascade of E1 (orange), E2 (yellow) and E3 (green) enzymes to add ubiquitin (purple) to the substrate protein (blue). DUB enzymes (teal) can remove ubiquitin from the substrate. (B) FBDD can better identify binding hot spots in pockets than traditional small molecule screens. Fragments can then be linked together to create molecules with higher binding affinities. (C) Common cysteine-reactive warheads for covalent fragment screens include halo-acetamides, acrylamides and α,β-unsaturated methyl esters.
The diversity and complexity of the ubiquitin code is vast. Poly-ubiquitin chains have multiple conformations: chains can be homotypic, mixed or branched, and each promote different cellular outcomes. For example, Lys48-linked chains mark proteins for proteasomal degradation, whereas Lys63-linked chains activate immune signalling pathways (Swatek and Komander, 2016). Furthermore, recent studies have shown that ubiquitination is not limited to lysine residues and can even occur on other biomolecules (Otten et al., 2021; Squair and Virdee, 2022). Specificity of chain type and substrate recognition comes from the specific E2/E3 combination used. Dysregulation of the ubiquitin system is a driver for many different diseases, and so targeting the ubiquitination machinery offers attractive therapeutic opportunities, as well as scope for tool compound development to better understand cellular function and disease development (Wertz and Wang, 2019).
With over 600 E3 ligases, around 100 DUBs (Harrigan et al., 2018), 40 E2 enzymes, and only two E1 enzymes known in humans (Jin et al., 2007), each class of enzyme offers both advantages and disadvantages as targets for tool compound development and therapeutic intervention. Several small molecule inhibitors of the proteasome, E1s and DUBs are progressing through clinical trials (Wang et al., 2016; Hyer et al., 2018), however few exist as approved drugs (Manasanch and Orlowski, 2017; Wertz and Wang, 2019). Achieving specific responses to inhibition of the proteasome and E1 enzymes is difficult since both have such broad reactivities and form so many interactions. For inhibiting specific cellular pathways, the E3 and DUB families are the most favourable drug targets within the ubiquitin system, however few E3 ligases or DUBs have modulators in the clinic or are liganded (Wu et al., 2020).
Fragment-based drug discovery
Traditional high throughput screening (HTS) methods for drug discovery involve screening libraries of hundreds of thousands of small molecules against a protein of interest or a phenotype (MacArron et al., 2011). While this approach has successfully identified many lead compounds for many different targets, there are key limitations. The relatively large and complex “drug-like” molecules used in HTS often form sub-optimal interactions with the target protein due to steric hinderance and conformational inflexibility. This means that other, stronger interactions may be masked, resulting in an inferior starting point for hit-to-lead optimisation (Figure 1B). Furthermore, due to the structural complexity of these “drug-like” molecules, very large libraries are required to attain sufficient hit rates. Fragment-based drug discovery (FBDD) has emerged as an alternative hit-finding method in the last two decades (Erlanson et al., 2016). In FBDD, much smaller molecules (“fragments”) are screened, with molecular recognition between optimal pharmacophores and small protein pockets more likely to be identified. The decreased molecular size enables greater coverage of chemical space with a much smaller library of compounds (Kirsch et al., 2019), therefore screening is faster and cost effective. Although this decreased molecular size limits the strength and specificity of target-fragment interactions, this can be regained through fragment elaboration and targeted medicinal chemistry campaigns after hit identification (Figure 1B). Furthermore, fragment hits typically have a much higher ligand efficiency than traditional HTS “drug-like” hits, so desirable physicochemical properties can be maintained during hit-lead optimisation (Kirsch et al., 2019). More recently, covalent fragment screening has developed as an additional modality, where the weaker target-fragment interactions are stabilised by covalent bond formation (Keeley et al., 2020).
Fragments physiochemical properties are typically guided by the “rule of 3”: molecular weight <300 Da, logP ≤3, and fewer than 3 hydrogen-bond donors, hydrogen-bond acceptors and rotatable bonds (Congreve et al., 2003; Jhoti et al., 2013). Non-covalent and covalent fragments require different considerations when developing fragment campaigns against a certain target or phenotype, particularly in library design and detection method. Traditional HTS detection methods, such as in vitro activity assays (enzymatic inhibition, or ELISA), are less applicable to fragment campaigns since higher compound concentrations are required to compensate for the lower affinities of fragments compared to small molecules, and this can lead to assay interference (Inglese et al., 2007).
Non-covalent fragments
The key advantage of non-covalent fragments is that target proteins are not required to have a readily accessible reactive residue. Unless some inherent structural library bias is desired, for example if the target protein favours a particular chemical motif, the library should be designed such that the broadest coverage of chemical space is achieved. Some libraries, such as the Diamond-SGC-iNEXT Poised (DSi-Poised) library, contain fragments which can be easily conjugated post-screening (Cox et al., 2016), or can be grown in multiple vectors (Bancet et al., 2020; Consortium et al., 2021). In addition, libraries can be simplified and streamlined by removing enantiomeric compounds.
Techniques for non-covalent fragment hit identification can be protein-based or ligand-based. In the case of protein-based detection, identification of hits relies on the biophysical properties of the protein target to change upon fragment binding; alternatively, NMR can be used for ligand-based detection. The most common biophysical detection techniques include differential scanning fluorimetry (DSF), nuclear magnetic resonance (NMR) and surface plasmon resonance (SPR) (Kirsch et al., 2019; Gao et al., 2020). More recently, combining hit identification directly with structural insight by X-ray crystallography has become popular, as demonstrated by the development of the XChem platform at Diamond Light Source (DLS) (Douangamath et al., 2021). Fragments are particularly amenable for crystallographic screening: smaller library size ensures practical aspects such as crystal mounting (which is yet to be automated) are manageable, and high fragment solubility ensures the required concentrations can be soaked into the crystal.
Once fragment hits have been identified, typically, a follow-up screen is performed to investigate structure-activity relationships (SAR). Lead fragments are then grown, merged or linked into larger molecules which retain the optimal binding efficiency of the original fragments, but with increased specificity and affinity for the desired protein target. Access to structures of fragment-protein complexes is often necessary to aid the synthetic elaboration of fragments by informing on suitable vectors to optimise target-fragment interactions. Crystallographic fragment screening platforms such as XChem provide this structural insight in a high throughput manner, and have greatly expedited the FBDD process.
Covalent fragments
In contrast to non-covalent fragments, covalent fragments contain a reactive functional group (often referred to as a “warhead”) in addition to the molecular pharmacophore. As such, covalent fragment engagement is first driven by molecular recognition between pharmacophore and target protein, followed by covalent bond formation between the electrophilic “warhead” and a nearby nucleophilic residue. This additional functionality increases the complexity of the library design, as variations in the electrophile also need to be considered (Lu et al., 2021). Cysteines are the most commonly targeted nucleophilic residues within the proteome; they are the most reactive at physiological pH, and are often key to enzyme catalytic activity, for example in cysteine proteases and some DUBs and E3 ligases. For cysteine-targeting, commonly used electrophiles include α,β-unsaturated methyl esters (Kathman et al., 2015; Johansson et al., 2019), chloroacetamides and acrylamides (Resnick et al., 2019) (Figure 1C). A balance between reactivity and specificity is required, and acrylamides and chloroacetamides are often chosen for this reason. More recently, novel electrophiles have been developed for targeting residues beyond cysteine, including lysines, tyrosines and histidines (Hacker et al., 2017; Ward et al., 2017; Abbasov et al., 2021; Zanon et al., 2021). The main advantage of covalent fragment screening over non-covalent screening is the increased simplicity of hit detection. Hit fragments form a covalent bond with their target residue, resulting in a change in molecular weight which can be easily detected by liquid chromatography coupled mass spectrometry (LC-MS) for purified proteins, and with LC-MS/MS for cell lysates.
FBDD has become increasingly reliable as a drug discovery method, with multiple examples of FDA approved compounds derived from initial fragment hits (Erlanson et al., 2016), including BRAF inhibitor, Vemurafenib (Bollag et al., 2012), and BCL-2 inhibitor, Venetoclax (Souers et al., 2013). In addition, there are several general efforts to identify probes for the entire human proteome (Arrowsmith et al., 2015; Drewes and Knapp, 2018; Carter et al., 2019), towards which FBDD will undoubtedly contribute. Developing high-quality tool compounds to better understand molecular systems and provide insight into protein pathways is becoming commonplace before starting expensive drug campaigns, and FBDD could help to expand the number of probes available. Overall, FBDD is proving to be an incredibly powerful technique for drug and probe discovery, and its application to the ubiquitin system is only just beginning. A summary of the fragment campaigns discussed in the following sections is given in Table 1.
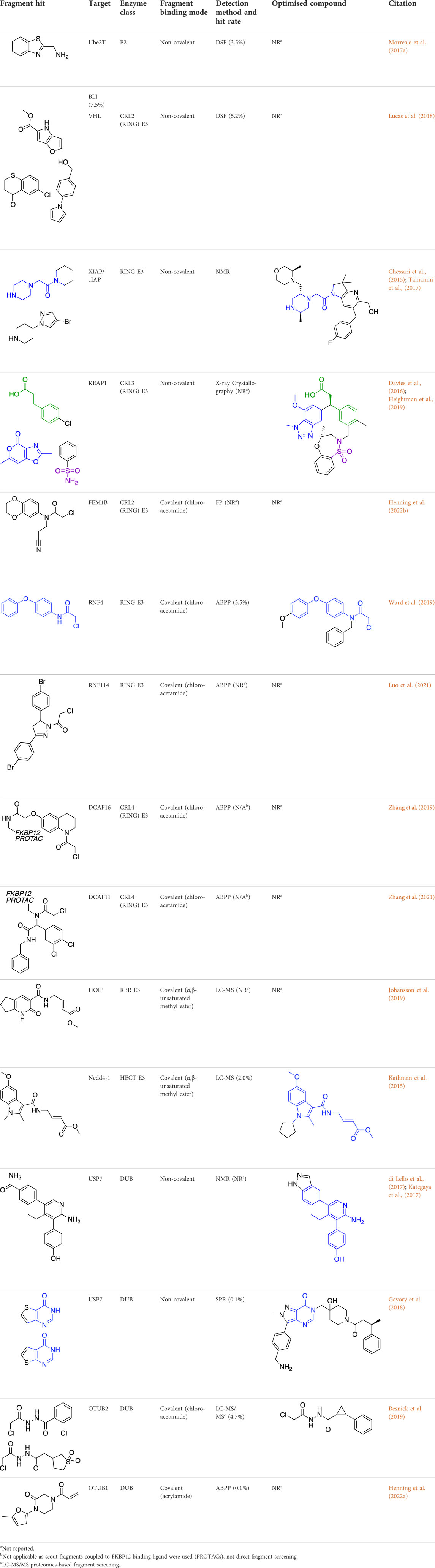
TABLE 1. Summary of fragment hits and fragment-derived modulators of ubiquitin system proteins. Pharmacophores from original fragment hits which were retained in optimised compounds are highlighted in blue/green/purple. Potency/binding affinity of optimised compounds was defined individually in each report (see Citation).
Ubiquitin-activating (E1) and ubiquitin-conjugating (E2) enzyme fragment-based drug discovery
While E1-activating enzymes are the least specific component of the ubiquitin system, a handful of E1 enzyme inhibitors have been published. E1 inhibitors can be targeted to the ATP binding site, and therefore show exquisite selectivity over other ubiquitin system proteins which are ATP-independent and typically do not have existing small molecule binding sites. One example, small molecule UBE1 inhibitor, TAK-243, showed promise as an anti-tumour agent in vivo (Hyer et al., 2018), suggesting that despite risks of high toxicity, E1 proteins could be interesting targets. No fragment campaigns have yet been reported against E1 enzymes, even though any new tool compounds would enable researchers to better understand the role of E1 enzymes in the ubiquitin system and related diseases.
Due to their limited specificity, not many E2 inhibitors currently exist. However, whilst using FBDD to screen the E2 enzyme Ube2T, a novel allosteric binding site was identified (Morreale et al., 2017a). Following a 1200-fragment screening campaign using both DSF and biolayer interferometry (BLI) to identify hits, NMR spectroscopy and X-ray crystallography were used for further validation. This allosteric site provided opportunities for novel inhibitor development and insights into allosteric modulation mechanisms for Ube2T. However, following conflicting SAR results, further biophysical and structural analysis ultimately identified that binding from a zinc impurity was responsible for the inhibition observed (Morreale et al., 2017b).
Ubiquitin ligase (E3) fragment-based drug discovery
E3 ligases determine substrate specificity as well as chain linkage specificity (either directly, or through E2 selection) and are therefore highly valuable drug targets. As the largest family of enzymes within the ubiquitin system, new tool compounds and probes targeting E3 ligases are needed to better understand their biology and physiological roles. Binders for E3 ligases can be distinguished between inhibitors, which inactivate the enzymes, and recruiters, which are commonly used for targeted protein degradation (TPD), including modalities such as proteolysis-targeting chimeras (PROTACs) (Sakamoto et al., 2001).
The E3 ligase family was originally subdivided into three main classes: Homologous with E6-associated protein C-terminus (HECT), Really Interesting New Gene (RING) and Ring-between-Ring (RBR) (Komander and Rape, 2012; Morreale and Walden, 2016) but more recently, further classes of E3 ligases including RING-Cys-Relay (RCR) enzymes have been identified (Pao et al., 2018; Otten et al., 2021). Of the 600 E3s known, the majority belong to the RING class; only 28 are HECT E3 ligases and 14 belong to the RBR class (Chaugule and Walden, 2016; Morreale and Walden, 2016; de Cesare et al., 2018). These classes have two distinct mechanisms of action. HECT and RBR E3 ligases facilitate transfer of ubiquitin to the target by first accepting a molecule of ubiquitin from the E2-Ub conjugate, via a transthiolation reaction forming an E3-Ub thioester bond, before final transfer of ubiquitin to the target. This E3-Ub conjugation occurs on the catalytic cysteine residue in the HECT, RBR, or RCR E3 active site (Walden and Rittinger, 2018). Conversely, RING E3 ligases do not possess this active site cysteine. Instead, they mediate ubiquitin transfer by acting as a scaffold, binding both the E2-Ub conjugate and the target substrate, bringing them into close proximity. The E3 stabilises the active E2-Ub conformation, thus priming the complex for final ubiquitin transfer onto the target substrate (Berndsen and Wolberger, 2014).
Non-covalent and covalent fragment campaigns against RING, HECT, RBR and newly-discovered E3 ligases could yield starting motifs for both inhibition and TPD recruitment purposes, depending on the location of the binding site. However, for the development of inhibitors, the active site cysteine of HECT, RBR and RCR E3 ligases offers an excellent target, thus cysteine-reactive fragment campaigns may be advantageous. Recent work to understand the “ligandable cysteinome” through chemoproteomics suggested that 51 E3 ligases would be susceptible to specific probe design. Interestingly, not all the “ligandable” residues identified were catalytic cysteines, suggesting that covalent fragment screening approaches should not be disregarded for RING E3 ligases (Belcher et al., 2021).
RING ubiquitin ligases
Within the RING E3 ligases, the largest subclass are the Cullin-RING ubiquitin ligases (CRL) (Nguyen et al., 2017). Differentiation and substrate specificity within this subclass relies on adaptor proteins and substrate receptors, for example Von Hippel Lindau (VHL), cereblon (CRBN) and Kelch Like ECH Associated Protein 1 (KEAP1). It is these appended adaptors which have been the targets of most RING E3 ligase ligand efforts (Rothweiler et al., 2022).
The most studied of the liganded E3 ligases is VHL, a CRL2 E3 ligase, which has a robust and well-used small molecule ligand targeting the VHL:HIFα protein-protein interaction (PPI) interface (Galdeano et al., 2014). Fragment campaigns using FBDD and in silico screening approaches have identified several novel VHL binding pockets (Lucas et al., 2018). After a 1200-fragment screen by DSF and NMR, 82 hits were validated by NMR and crystallography, which revealed 18 fragments as true hits. Displacement assays using an HIFα peptide were carried out, highlighting allosteric binding for 17 of the fragments, which was further confirmed by crystallography, and may now form a basis for further tool development.
The CRL3 E3 ligase, KEAP1, was subjected to a screen of 330 non-covalent fragments by crystallography. Key protein-fragment interactions were identified at the KEAP1 interface with its substrate, NRF2 (Davies et al., 2016). Significant fragment elaboration and medicinal chemistry efforts (Heightman et al., 2019) led to a potent compound, KI-696, which showed cellular activity and in vivo efficacy, and is now a high-quality chemical probe for the KEAP1-NRF2 PPI interface.
The inducer of apoptosis protein family (IAP) has proved one of the most successfully targeted RING E3 ligase families by FBDD. Previous inhibitors of this family had been peptidomimetic (Sharma et al., 2006) but fragment campaigns identified novel small molecule dual inhibitors of cIAP2 and XIAP (Chessari et al., 2015). 1151 fragments were screened by 1D NMR against XIAP-Bir3, and hits were characterised further using NMR and crystallography. Two overlapping hits demonstrated promising inhibition in fluorescence polarisation (FP) assays and, following significant medicinal chemistry efforts, achieved nanomolar potency and efficacy in mouse models. The elaborated molecule, ASTX660, is now in Phase II trials for the treatment of advanced solid tumours and lymphomas (Chessari et al., 2015; Tamanini et al., 2017).
More recently, covalent FBDD has driven novel ligand discovery for RING E3 ligases. Uncharacteristically, FEM1B, a CRL2 E3 ligase was screened against a covalent cysteine-reactive fragment library using an FP-assay, as opposed to LC-MS. The lead fragment utilised a chloroacetamide electrophile which reacted with a key cysteine residue responsible for substrate recognition (Henning et al., 2022b). The fragment was further developed into a FEM1B recruiter for novel PROTACs, and further SAR and medicinal chemistry is ongoing to develop the lead fragment into a stable and effective tool compound.
RNF4 and RNF114 have also been screened against covalent fragment libraries. Using gel-based activity-based protein profiling (ABPP) with a fluorescent iodoacetamide probe, an RNF4 hit fragment was identified. This fragment bound to two key zinc-coordinating cysteine residues without inhibiting RNF4 auto-ubiquitination activity, making it an ideal ligand for development into a TPD recruiter motif. Following further SAR optimisation, the fragment was developed into a PROTAC for BRD4 degradation but was found to be nonspecific for RNF4 (Ward et al., 2019). Similarly, RNF114 was subjected to a covalent fragment screen using gel-based and proteomics ABPP, and the lead fragment developed into PROTACs for BRD4 and BCL-ABL degradation (Luo et al., 2021).
A novel method, using broadly reactive electrophilic scout fragments (Backus et al., 2016; Bar-Peled et al., 2017) as E3 ligase recruiters in PROTAC molecules, identified two novel CRL4 E3 ligases, DCAF16 and DCAF11, for TPD purposes. Following the identification of a PROTAC which degraded FKBP12, affinity enrichment, proteomics and genomic sequencing were used to validate DCAF16 as the E3 enzyme responsible for the degradation (Zhang et al., 2019). This method identified degraders of specific targets, and then retrospectively deconvoluted the responsible E3 ligase. This contrasts with previously described methods, where fragment screens were used to identify recruiter ligands for specific E3 ligases.
HECT and RBR ubiquitin ligases
HECT, RBR, and RCR E3 ligases rely on a catalytic cysteine for their activity and are therefore obvious targets for covalent fragment screening to identify novel inhibitors. As these E3 classes are less numerous than RING E3 ligases, there are fewer examples of fragment campaigns against them.
The first example of a covalent fragment screening campaign against an E3 ligase with a catalytic cysteine was the HECT E3 ligase, Nedd4-1. The HECT domain of Nedd4-1 was screened against an α,β-unsaturated methyl ester fragment library by LC-MS (Kathman et al., 2015). Hit fragments were found to bind to a non-catalytic cysteine at the ubiquitin-binding interface and validated by X-ray crystallography. However, at endogenous levels, ubiquitin outcompeted fragment binding. Following SAR optimisation, an N-cyclopentyl substituted fragment derivative was found to be the most potent, whilst retaining specificity. Interestingly, this tool compound was then used to show that Nedd4-1 switches from elongating ubiquitin chains through a processive mechanism to a distributive mechanism when bound to the compound. Previously, it was thought that all HECT E3 ligases were processive, and so this finding provided new insights into the mechanism of HECT E3 ligases, and highlights how FBDD can be used to develop excellent tool compounds for the ubiquitin system.
HOIP RBR E3 ligase is one of the three subunits which comprise the linear ubiquitin chain assembly complex (LUBAC). A fragment library of pooled α,β-unsaturated methyl esters was screened against HOIP using LC-MS to identify hit fragments, which were confirmed to bind to the active site cysteine. Despite structure-based fragment optimisation, efficacy could not be improved significantly, however, inhibition of HOIP and impressive selectivity over other ubiquitin system proteins was demonstrated in cells, suggesting that further medicinal chemistry efforts could yield a potent and specific tool compound (Johansson et al., 2019).
Whilst other HECT and RBR E3 ligases have been liganded through small molecule screening (Watt et al., 2018; Tian et al., 2019), many E3 ligases do not have high-quality chemical probes yet and would be worthy targets for future FBDD campaigns.
Deubiquitinating (DUB) enzyme fragment-based drug discovery
DUBs have been implicated in many different disease pathways, including cancers, neurodegenerative diseases, and immunity and infection (Harrigan et al., 2018). A number of DUB inhibitors are in the drug development pipeline (Rowinsky et al., 2020; Schauer et al., 2020), some of which have been discovered through FBDD.
In 2017, USP7 was the first DUB targeted with a non-covalent fragment screen, using NMR as a hit detection method (Kategaya et al., 2017). Lead compounds were assessed using ABPP, and prioritised based on their specificity, biophysical properties and toxicity in multiple cell lines. These hits were subjected to in silico development and medicinal chemistry, resulting in small molecule inhibitors of USP7 (di Lello et al., 2017). USP7 was also subjected to a non-covalent fragment screen using SPR (Gavory et al., 2018). Hit fragments were compared to known inhibitors of USP7, and a stereocentre discovered to be key to activity of the fragments. Crystallography was then used to understand and optimise fragment-protein interactions, before biochemical and biophysical analysis showed excellent selectivity of the final compound for USP7 against other related DUBs. The final compound showed efficacy in cells, resulting in stabilisation of p53 and decreased levels of MDM2.
More recently, electrophilic covalent fragment screening against recombinant proteins with mass spectrometry was combined with high-throughput crystallography (Resnick et al., 2019). A panel of clinically relevant proteins, including DUBs OTUB2 and USP8 were screened, and hits were identified by LC-MS. 47 fragment hits were identified for OTUB2, which were further developed into a lead series, guided by crystallography. Hits were validated for selectivity using cell and lysate gel-based and proteomics ABPP experiments in HEK293T cells, highlighting the power of combining mass spectrometry with high throughput crystallography for FBDD.
Chemical biology has seen a huge increase in PROTAC development, with the recruitment of a range of novel E3 ligases demonstrated, such as for RNF4 (Ward et al., 2019) and FEM1B (Henning et al., 2022b). Excitingly, the first deubiquitinase-targeting chimera (DUBTAC) was recently reported (Henning et al., 2022a), where DUBs are recruited to new targets for targeted protein stabilisation. A cysteine-reactive fragment campaign identified specific recruiters for DUB proteins through gel-based ABPP. A fragment hit for OTUB1 was further developed and coupled with a small molecule binder for a mutant CFTR chloride channel to create the first DUBTAC, highlighting how FBDD can be used to make novel tools.
Future Outlooks
FBDD has enabled the development of novel tool compounds for a range of E1, E2, E3, and DUB proteins. However, many ubiquitin system proteins remain unliganded, and novel technologies are emerging to expand the scope of FBDD.
So far, covalent fragment-based campaigns have been focused on targeting cysteine residues. However, other nucleophilic residues beyond cysteine are more prevalent, and more likely to be post-translationally modified in situ. Profiling these residues with fragments could provide interesting insights into biology and highlight new drug targets. Despite their lower intrinsic reactivity, advances have been made in targeting lysine residues with sulfotetrafluorophenyl ester electrophiles (Hacker et al., 2017; Abbasov et al., 2021), and serine, threonine, tyrosine and histidine residues with N-hydroxysuccinimide esters (Ward et al., 2017; Zanon et al., 2021). In addition, profiling of cysteines, lysines, tyrosines and histidines has been characterised with sulfonyl fluorides (Gilbert et al., 2022), and tyrosines with sulfur-triazole exchange chemistry (Brulet et al., 2020). These exciting advances in non-cysteine residue profiling have yet to be fully translated into FBDD but are likely to be at the forefront of the field in the future.
Photoaffinity labelling (PAL), a technique initially developed for target identification (Smith and Collins, 2015), has also been utilised for FBDD. A library of highly reactive carbene intermediates was formed in situ using UV irradiation of diazirine precursors. The carbene inserts into proximal bonds within 15 Å, forming a covalent bond, even when there is no nucleophilic residue available. This has so far been reported with recombinant proteins (Grant et al., 2020) and directly in cells (Bar-Peled et al., 2017). Despite challenges with non-specific crosslinking and low crosslinking yields, the technology remains a powerful method to identify novel tool compounds for the ubiquitin system.
Recently, metallofragments have been highlighted as a promising new avenue for FBDD. Using a metal ion to coordinate fragments adds 3D character and increases shape diversity in library design. This could be particularly useful for target systems where metallo-compounds have already shown efficacy, such as antibacterial compounds (Hess, 2022). A novel metallofragment library has already shown activity and specificity against a few protein targets (Morrison et al., 2020), but has yet to be screened against ubiquitin system proteins.
We believe that the newest, cutting-edge discoveries in FBDD will be seen through the development and use of fragment campaigns in collaboration with phenotypic screening. Advances in proteomics methods for reactive residue profiling have been reported recently, for example by the Gygi and Cravatt groups (Roberts et al., 2017; Maurais and Weerapana, 2019; Kuljanin et al., 2021). By coupling reactive residue profiling with covalent fragment screening in relevant cellular models for diseases of interest, researchers will be able to identify fragment and residue pairs on a proteome-wide level and identify whether they affect the disease phenotype. If this method is applied to the ubiquitin system, we could rapidly see a rise in specific and potent chemical tools and therapeutic molecules entering drug development, enabling researchers to better understand this complex and dynamic signalling pathway.
Author contributions
CK, KM, and KR wrote and revised the manuscript, and approved the submitted version.
Funding
This work was supported by the Francis Crick Institute which receives its core funding from Cancer Research United Kingdom (CC 2075), the United Kingdom Medical Research Council (CC 2075), and the Wellcome Trust (CC 2075), and by the Biotechnology and Biological Research Council, BB/T014547/1 to KR. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission. Figures 1A,B were created in BioRender.com under the institutional license belonging to the Francis Crick Institute.
Acknowledgments
We thank Dr. William McCarthy for critical reading of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abbasov, M. E., Kavanagh, M. E., Ichu, T. A., Lazear, M. R., Tao, Y., Crowley, V. M., et al. (2021). A proteome-wide atlas of lysine-reactive chemistry. Nat. Chem. 13, 1081–1092. doi:10.1038/s41557-021-00765-4
Arrowsmith, C. H., Audia, J. E., Austin, C., Baell, J., Bennett, J., Blagg, J., et al. (2015). The promise and peril of chemical probes. Nat. Chem. Biol. 11, 536–541. doi:10.1038/nchembio.1867
Backus, K. M., Correia, B. E., Lum, K. M., Forli, S., Horning, B. D., González-Páez, G. E., et al. (2016). Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. doi:10.1038/nature18002
Bancet, A., Raingeval, C., Lomberget, T., le Borgne, M., Guichou, J. F., and Krimm, I. (2020). Fragment linking strategies for structure-based drug design. J. Med. Chem. 63, 11420–11435. doi:10.1021/acs.jmedchem.0c00242
Bar-Peled, L., Kemper, E. K., Suciu, R. M., Vinogradova, E. v., Backus, K. M., Horning, B. D., et al. (2017). Chemical proteomics identifies druggable vulnerabilities in a genetically defined cancer. Cell 171, 696–709. doi:10.1016/j.cell.2017.08.051
Belcher, B. P., Ward, C. C., and Nomura, D. K. (2021). Ligandability of E3 ligases for targeted protein degradation applications. Biochemistry 2021, 1c00464. doi:10.1021/acs.biochem.1c00464
Berndsen, C. E., and Wolberger, C. (2014). New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 21, 301–307. doi:10.1038/nsmb.2780
Bollag, G., Tsai, J., Zhang, J., Zhang, C., Ibrahim, P., Nolop, K., et al. (2012). Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 11, 873–886. doi:10.1038/nrd3847
Brulet, J. W., Borne, A. L., Yuan, K., Libby, A. H., and Hsu, K. L. (2020). Liganding functional tyrosine sites on proteins using sulfur-triazole exchange chemistry. J. Am. Chem. Soc. 142, 8270–8280. doi:10.1021/jacs.0c00648
Carter, A. J., Kraemer, O., Zwick, M., Mueller-Fahrnow, A., Arrowsmith, C. H., and Edwards, A. M. (2019). Target 2035: Probing the human proteome. Drug Discov. Today 24, 2111–2115. doi:10.1016/j.drudis.2019.06.020
Chaugule, V. K., and Walden, H. (2016). Specificity and disease in the ubiquitin system. Biochem. Soc. Trans. 44, 212–227. doi:10.1042/BST20150209
Chessari, G., Buck, I. M., Day, J. E. H., Day, P. J., Iqbal, A., Johnson, C. N., et al. (2015). Fragment-based drug discovery targeting inhibitor of apoptosis proteins: Discovery of a non-alanine lead series with dual activity against cIAP1 and XIAP. J. Med. Chem. 58, 6574–6588. doi:10.1021/acs.jmedchem.5b00706
Congreve, M., Carr, R., Murray, C., and Jhoti, H. (2003). A ‘Rule of Three’ for fragment-based lead discovery? Drug Discov. Today 8, 876–877. doi:10.1016/S1359-6446(03)02831-9
Consortium, T. C. M., Chodera, J., Lee, A., London, N., and Delft, F., 2021. Open science discovery of oral non-covalent SARS-CoV-2 main protease inhibitors. CHEMRXIV. doi: doi:10.26434/CHEMRXIV-2021-585KS-V2
Cox, O. B., Krojer, T., Collins, P., Monteiro, O., Talon, R., Bradley, A., et al. (2016). A poised fragment library enables rapid synthetic expansion yielding the first reported inhibitors of PHIP(2), an atypical bromodomain. Chem. Sci. 7, 2322–2330. doi:10.1039/C5SC03115J
Davies, T. G., Wixted, W. E., Coyle, J. E., Griffiths-Jones, C., Hearn, K., McMenamin, R., et al. (2016). Monoacidic inhibitors of the kelch-like ECH-associated protein 1: Nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) protein-protein interaction with high cell potency identified by fragment-based discovery. J. Med. Chem. 59, 3991–4006. doi:10.1021/acs.jmedchem.6b00228
de Cesare, V., Johnson, C., Barlow, V., Hastie, J., Knebel, A., and Trost, M. (2018). The MALDI-TOF E2/E3 ligase assay as universal tool for drug discovery in the ubiquitin pathway. Cell Chem. Biol. 25, 1117–1127. doi:10.1016/j.chembiol.2018.06.004
di Lello, P., Pastor, R., Murray, J. M., Blake, R. A., Cohen, F., Crawford, T. D., et al. (2017). Discovery of small-molecule inhibitors of ubiquitin specific protease 7 (USP7) using integrated NMR and in silico techniques. J. Med. Chem. 60, 10056–10070. doi:10.1021/acs.jmedchem.7b01293
Douangamath, A., Powell, A., Fearon, D., Collins, P. M., Talon, R., Krojer, T., et al. (2021). Achieving efficient fragment screening at XChem facility at Diamond Light Source. J. Vis. Exp. 1, 62414. doi:10.3791/62414
Drewes, G., and Knapp, S. (2018). Chemoproteomics and chemical probes for target discovery. Trends Biotechnol. 36, 1275–1286. doi:10.1016/j.tibtech.2018.06.008
Erlanson, D. A., Fesik, S. W., Hubbard, R. E., Jahnke, W., and Jhoti, H. (2016). Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 15 (9), 605–619. doi:10.1038/nrd.2016.109
Galdeano, C., Gadd, M. S., Soares, P., Scaffidi, S., van Molle, I., Birced, I., et al. (2014). Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von hippel-lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 57, 8657–8663. doi:10.1021/jm5011258
Gao, K., Oerlemans, R., and Groves, M. R. (2020). Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys. Rev. 12, 85–104. doi:10.1007/s12551-020-00619-2
Gavory, G., O’Dowd, C. R., Helm, M. D., Flasz, J., Arkoudis, E., Dossang, A., et al. (2018). Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat. Chem. Biol. 14, 118–125. doi:10.1038/nchembio.2528
Gilbert, K., Vuorinen, A., Aatkar, A., Pogany, P., Pettinger, J., Grant, E., et al. (2022). Profiling sulfur(VI) fluorides as reactive functionalities for chemical biology tools and expansion of the ligandable proteome. CHEMRXIV. doi: doi:10.26434/CHEMRXIV-2022-J8B8D-V2
Grant, E. K., Fallon, D. J., Hann, M. M., Fantom, K. G. M., Quinn, C., Zappacosta, F., et al. (2020). A photoaffinity-based fragment-screening platform for efficient identification of protein ligands. Angew. Chem. Int. Ed. Engl. 59, 21096–21105. doi:10.1002/anie.202008361
Hacker, S. M., Backus, K. M., Lazear, M. R., Forli, S., Correia, B. E., and Cravatt, B. F. (2017). Global profiling of lysine reactivity and ligandability in the human proteome. Nat. Chem. 9 (12), 1181–1190. doi:10.1038/nchem.2826
Harrigan, J. A., Jacq, X., Martin, N. M., and Jackson, S. P. (2018). Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 17, 57–78. doi:10.1038/nrd.2017.152
Heightman, T. D., Callahan, J. F., Chiarparin, E., Coyle, J. E., Griffiths-Jones, C., Lakdawala, A. S., et al. (2019). Structure-activity and structure-conformation relationships of aryl propionic acid inhibitors of the kelch-like ECH-associated protein 1/nuclear factor erythroid 2-related factor 2 (KEAP1/NRF2) protein-protein interaction. J. Med. Chem. 62, 4683–4702. doi:10.1021/acs.jmedchem.9b00279
Henning, N. J., Boike, L., Spradlin, J. N., Ward, C. C., Liu, G., Zhang, E., et al. (2022a2022). Deubiquitinase-targeting chimeras for targeted protein stabilization. Nat. Chem. Biol. 18 (4), 412–421. doi:10.1038/s41589-022-00971-2
Henning, N. J., Manford, A. G., Spradlin, J. N., Brittain, S. M., Zhang, E., McKenna, J. M., et al. (2022b). Discovery of a covalent FEM1B recruiter for targeted protein degradation applications. J. Am. Chem. Soc. 144, 701–708. doi:10.1021/jacs.1c03980
Hess, J. (2022). Rational approaches towards inorganic and organometallic antibacterials. Biol. Chem. 403, 363–375. doi:10.1515/hsz-2021-0253
Hyer, M. L., Milhollen, M. A., Ciavarri, J., Fleming, P., Traore, T., Sappal, D., et al. (2018). A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med. 24, 186–193. doi:10.1038/nm.4474
Inglese, J., Johnson, R. L., Simeonov, A., Xia, M., Zheng, W., Austin, C. P., et al. (2007). High-throughput screening assays for the identification of chemical probes. Nat. Chem. Biol. 3, 466–479. doi:10.1038/nchembio.2007.17
Jhoti, H., Williams, G., Rees, D. C., and Murray, C. W. (2013). The “rule of three” for fragment-based drug discovery: Where are we now? Nat. Rev. Drug Discov. 12 (8 12), 644–645. doi:10.1038/nrd3926-c1
Jin, J., Li, X., Gygi, S. P., and Harper, J. W. (2007). Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 447, 1135–1138. doi:10.1038/nature05902
Johansson, H., Tsai, Y.-C. I., Fantom, K., Chung, C.-W., Kü, S., Martino, L., et al. (2019). Fragment-based covalent ligand screening enables rapid discovery of inhibitors for the RBR E3 ubiquitin ligase HOIP. J. Am. Chem. Soc. 141, 2703–2712. doi:10.1021/jacs.8b13193
Kategaya, L., di Lello, P., Rougé, L., Pastor, R., Clark, K. R., Drummond, J., et al. (2017). USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 550, 534–538. doi:10.1038/nature24006
Kathman, S. G., Span, I., Smith, A. T., Xu, Z., Zhan, J., Rosenzweig, A. C., et al. (2015). A small molecule that switches a ubiquitin ligase from a processive to a distributive enzymatic mechanism. J. Am. Chem. Soc. 137, 12442–12445. doi:10.1021/jacs.5b06839
Keeley, A., Petri, L., Ábrányi-Balogh, P., and Keserű, G. M. (2020). Covalent fragment libraries in drug discovery. Drug Discov. Today 25, 983–996. doi:10.1016/j.drudis.2020.03.016
Kirsch, P., Hartman, A. M., Hirsch, A. K. H., and Empting, M. (2019). Concepts and core principles of fragment-based drug design. Molecules 24, E4309. doi:10.3390/molecules24234309
Komander, D., and Rape, M. (2012). The ubiquitin code. Annu. Rev. Biochem. 81, 203–229. doi:10.1146/annurev-biochem-060310-170328
Kuljanin, M., Mitchell, D. C., Schweppe, D. K., Gikandi, A. S., Nusinow, D. P., Bulloch, N. J., et al. (2021). Reimagining high-throughput profiling of reactive cysteines for cell-based screening of large electrophile libraries. Nat. Biotechnol. 39, 630–641. doi:10.1038/s41587-020-00778-3
Lu, W., Kostic, M., Zhang, T., Che, J., Patricelli, M. P., Jones, L. H., et al. (2021). Fragment-based covalent ligand discovery. RSC Chem. Biol. 2, 354–367. doi:10.1039/D0CB00222D
Lucas, X., van Molle, I., and Ciulli, A. (2018). Surface probing by fragment-based screening and computational methods identifies ligandable pockets on the von Hippel-lindau (VHL) E3 ubiquitin ligase. J. Med. Chem. 61, 7387–7393. doi:10.1021/acs.jmedchem.8b00842
Luo, M., Spradlin, J. N., Boike, L., Tong, B., Brittain, S. M., McKenna, J. M., et al. (2021). Chemoproteomics-enabled discovery of covalent RNF114-based degraders that mimic natural product function. Cell Chem. Biol. 28, 559–566.e15. doi:10.1016/j.chembiol.2021.01.005
MacArron, R., Banks, M. N., Bojanic, D., Burns, D. J., Cirovic, D. A., Garyantes, T., et al. (2011). Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 10, 188–195. doi:10.1038/nrd3368
Manasanch, E. E., and Orlowski, R. Z. (2017). Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 14, 417–433. doi:10.1038/nrclinonc.2016.206
Maurais, A. J., and Weerapana, E. (2019). Reactive-cysteine profiling for drug discovery. Curr. Opin. Chem. Biol. 50, 29–36. doi:10.1016/j.cbpa.2019.02.010
Morreale, F. E., Bortoluzzi, A., Chaugule, V. K., Arkinson, C., Walden, H., and Ciulli, A. (2017a). Allosteric targeting of the fanconi anemia ubiquitin-conjugating enzyme Ube2T by fragment screening. J. Med. Chem. 60, 4093–4098. doi:10.1021/acs.jmedchem.7b00147
Morreale, F. E., Testa, A., Chaugule, V. K., Bortoluzzi, A., Ciulli, A., and Walden, H. (2017b). Mind the metal: A fragment library-derived zinc impurity binds the E2 ubiquitin-conjugating enzyme Ube2T and induces structural rearrangements. J. Med. Chem. 60, 8183–8191. doi:10.1021/ACS.JMEDCHEM.7B01071/SUPPL_FILE/JM7B01071_SI_001.CSV
Morreale, F. E., and Walden, H. (2016). Types of ubiquitin ligases. Cell 165, 248. doi:10.1016/j.cell.2016.03.003
Morrison, C. N., Prosser, K. E., Stokes, R. W., Cordes, A., Metzler-Nolte, N., and Cohen, S. M. (2020). Expanding medicinal chemistry into 3D space: Metallofragments as 3D scaffolds for fragment-based drug discovery. Chem. Sci. 11, 1216–1225. doi:10.1039/c9sc05586j
Nguyen, H. C., Wang, W., and Xiong, Y. (2017). Cullin-RING E3 ubiquitin ligases: Bridges to destruction. Sub-Cellular Biochem. 83, 323–347. doi:10.1007/978-3-319-46503-6_12
Otten, E. G., Werner, E., Casado, A. C., Boyle, K. B., Dharamdasani, V., Pathe, C., et al. (2021). Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature 594, 111–116. doi:10.1038/s41586-021-03566-4
Pao, K. C., Wood, N. T., Knebel, A., Rafie, K., Stanley, M., Mabbitt, P. D., et al. (2018). Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature 556, 381–385. doi:10.1038/s41586-018-0026-1
Resnick, E., Bradley, A., Gan, Jinrui, Douangamath, A., Krojer, T., Sethi, R., et al. (2019). Rapid covalent-probe discovery by electrophile-fragment screening. J. Am. Chem. Soc. 141, 8951–8968. doi:10.1021/jacs.9b02822
Roberts, A. M., Ward, C. C., and Nomura, D. K. (2017). Activity-based protein profiling for mapping and pharmacologically interrogating proteome-wide ligandable hotspots. Curr. Opin. Biotechnol. 43, 25–33. doi:10.1016/j.copbio.2016.08.003
Rothweiler, E. M., Brennan, P. E., and Huber, K. V. M. (2022). Covalent fragment-based ligand screening approaches for identification of novel ubiquitin proteasome system modulators. Biol. Chem. 403, 391–402. doi:10.1515/hsz-2021-0396
Rowinsky, E. K., Paner, A., Berdeja, J. G., Paba-Prada, C., Venugopal, P., Porkka, K., et al. (2020). Phase 1 study of the protein deubiquitinase inhibitor VLX1570 in patients with relapsed and/or refractory multiple myeloma. Invest. New Drugs 38, 1448–1453. doi:10.1007/s10637-020-00915-4
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., and Deshaies, R. J. (2001). Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 98, 8554–8559. doi:10.1073/pnas.141230798
Schauer, N. J., Magin, R. S., Liu, X., Doherty, L. M., and Buhrlage, S. J. (2020). Advances in discovering deubiquitinating enzyme (DUB) inhibitors. J. Med. Chem. 63, 2731–2750. doi:10.1021/acs.jmedchem.9b01138
Sharma, S. K., Straub, C., and Zawel, L. (2006). Development of peptidomimetics targeting IAPs. Int. J. Pept. Res. Ther. 12, 21–32. doi:10.1007/s10989-005-9003-2
Smith, E., and Collins, I. (2015). Photoaffinity labeling in target- and binding-site identification. Future Med. Chem. 7, 159–183. doi:10.4155/fmc.14.152
Souers, A. J., Leverson, J. D., Boghaert, E. R., Ackler, S. L., Catron, N. D., Chen, J., et al. (2013). ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208. doi:10.1038/nm.3048
Squair, D. R., and Virdee, S. (2022). A new dawn beyond lysine ubiquitination. Nat. Chem. Biol. 18, 802–811. doi:10.1038/s41589-022-01088-2
Swatek, K. N., and Komander, D. (2016). Ubiquitin modifications. Cell Res. 26, 399–422. doi:10.1038/cr.2016.39
Tamanini, E., Buck, I. M., Chessari, G., Chiarparin, E., Day, J. E. H., Frederickson, M., et al. (2017). Discovery of a potent nonpeptidomimetic, small-molecule antagonist of cellular inhibitor of apoptosis protein 1 (cIAP1) and X-linked inhibitor of apoptosis protein (XIAP). J. Med. Chem. 60, 4611–4625. doi:10.1021/acs.jmedchem.6b01877
Tian, M., Zeng, T., Liu, M., Han, S., Lin, Huayue, Lin, Q., et al. (2019). A cell-based high-throughput screening method based on a ubiquitin-reference technique for identifying modulators of E3 ligases. J. Biol. Chem. 294, 2880–2891. doi:10.1074/jbc.RA118.003822
Walden, H., and Rittinger, K. (2018). RBR ligase-mediated ubiquitin transfer: A tale with many twists and turns. Nat. Struct. Mol. Biol. 25, 440–445. doi:10.1038/s41594-018-0063-3
Wang, X., Mazurkiewicz, M., Hillert, E. K., Olofsson, M. H., Pierrou, S., Hillertz, P., et al. (2016). The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci. Rep. 6, 26979. doi:10.1038/srep26979
Ward, C. C., Kleinman, J. I., Brittain, S. M., Lee, P. S., Chung, C. Y. S., Kim, K., et al. (2019). Covalent ligand screening uncovers a RNF4 E3 ligase recruiter for targeted protein degradation applications. ACS Chem. Biol. 14, 2430–2440. doi:10.1021/acschembio.8b01083
Ward, C. C., Kleinman, J. I., and Nomura, D. K. (2017). NHS-esters as versatile reactivity-based probes for mapping proteome-wide ligandable hotspots. ACS Chem. Biol. 12, 1478–1483. doi:10.1021/acschembio.7b00125
Watt, J. E., Hughes, G. R., Walpole, S., Monaco, S., Stephenson, G. R., Bulman Page, P. C., et al. (2018). Discovery of small molecule WWP2 ubiquitin ligase inhibitors. Chemistry 24, 17677–17680. doi:10.1002/chem.201804169
Wertz, I. E., and Wang, X. (2019). From discovery to bedside: Targeting the ubiquitin system. Cell Chem. Biol. 26, 156–177. doi:10.1016/j.chembiol.2018.10.022
Wu, H. Q., Baker, D., and Ovaa, H. (2020). Small molecules that target the ubiquitin system. Biochem. Soc. Trans. 48, 479–497. doi:10.1042/BST20190535
Zanon, P. R. A., Yu, F., Musacchio, P., Lewald, L., Zollo, M., Krauskopf, K., et al. (2021). Profiling the proteome-wide selectivity of diverse electrophiles. CHEMRXIV. doi: doi:10.26434/CHEMRXIV-2021-W7RSS-V2
Zhang, X., Crowley, V. M., Wucherpfennig, T. G., Dix, M. M., and Cravatt, B. F. (2019). Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 15, 737–746. doi:10.1038/s41589-019-0279-5
Keywords: ubiquitin, drug discovery, fragment-based drug discovery, chemical tools, ubiquitination
Citation: Kennedy C, McPhie K and Rittinger K (2022) Targeting the ubiquitin system by fragment-based drug discovery. Front. Mol. Biosci. 9:1019636. doi: 10.3389/fmolb.2022.1019636
Received: 15 August 2022; Accepted: 05 September 2022;
Published: 27 September 2022.
Edited by:
Peter Mabbitt, New Zealand Forest Research Institute Limited (Scion), New ZealandReviewed by:
Satpal Virdee, University of Dundee, United KingdomCopyright © 2022 Kennedy, McPhie and Rittinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katrin Rittinger, a2F0cmluLnJpdHRpbmdlckBjcmljay5hYy51aw==
†These authors have contributed equally to this work