 Roman Zadorozhnyi1,2
Roman Zadorozhnyi1,2 Sucharita Sarkar1,2Caitlin M. Quinn1Kaneil K. Zadrozny3Barbie K. Ganser-Pornillos3
Sucharita Sarkar1,2Caitlin M. Quinn1Kaneil K. Zadrozny3Barbie K. Ganser-Pornillos3 Owen Pornillos3Angela M. Gronenborn2,4*
Owen Pornillos3Angela M. Gronenborn2,4* Tatyana Polenova1,2*
Tatyana Polenova1,2*- 1Department of Chemistry and Biochemistry, University of Delaware, Newark, DE, United States
- 2Pittsburgh Center for HIV Protein Interactions, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 3Department of Molecular Physiology and Biological Physics, University of Virginia School of Medicine, Charlottesville, VA, United States
- 4Department of Structural Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
Histidine residues play important structural and functional roles in proteins, such as serving as metal-binding ligands, mediating enzyme catalysis, and modulating proton channel activity. Many of these activities are modulated by the ionization state of the imidazole ring. Here we present a fast MAS NMR approach for the determination of protonation and tautomeric states of His at frequencies of 40–62 kHz. The experiments combine 1H detection with selective magnetization inversion techniques and transferred echo double resonance (TEDOR)–based filters, in 2D heteronuclear correlation experiments. We illustrate this approach using microcrystalline assemblies of HIV-1 CACTD-SP1 protein.
Introduction
Histidines (His) play important structural and functional roles in proteins such as metal binding (Stryer et al., 1964; Perutz and Mathews, 1966; Adams et al., 1969; Liljas et al., 1972), proton transfer (Hoffee et al., 1967; Blow et al., 1969; Campbell et al., 1974), and stability (Perutz et al., 1969; Lewis et al., 1976; Loewenthal et al., 1992). These functions are often correlated with the ionization state of the histidine sidechain (Figure 1A) (Bachovchin and Roberts, 1978; Kossiakoff and Spencer, 1981; Lewis et al., 1981). While the pKa of the imidazole ring for free histidine is 6.5 (Blomberg et al., 1977), in proteins the pKa values vary widely, from 3 to 9, depending on the interactions with neighboring residues and degree of burial (Zhou et al., 1993; Plesniak et al., 1996). At pH values above the pKa, anionic τ and π tautomers with hydrogens at either Nε2 or Nδ1 are present, while below the pKa the protonated imidazole ring possesses hydrogens at both Nε2 and Nδ1. For a protein at intermediate pH values, it is possible that a fraction of His residues is protonated and the remaining fraction unprotonated (French and Hammes, 1965; Edwards and Sykes, 1980; Hass et al., 2008).
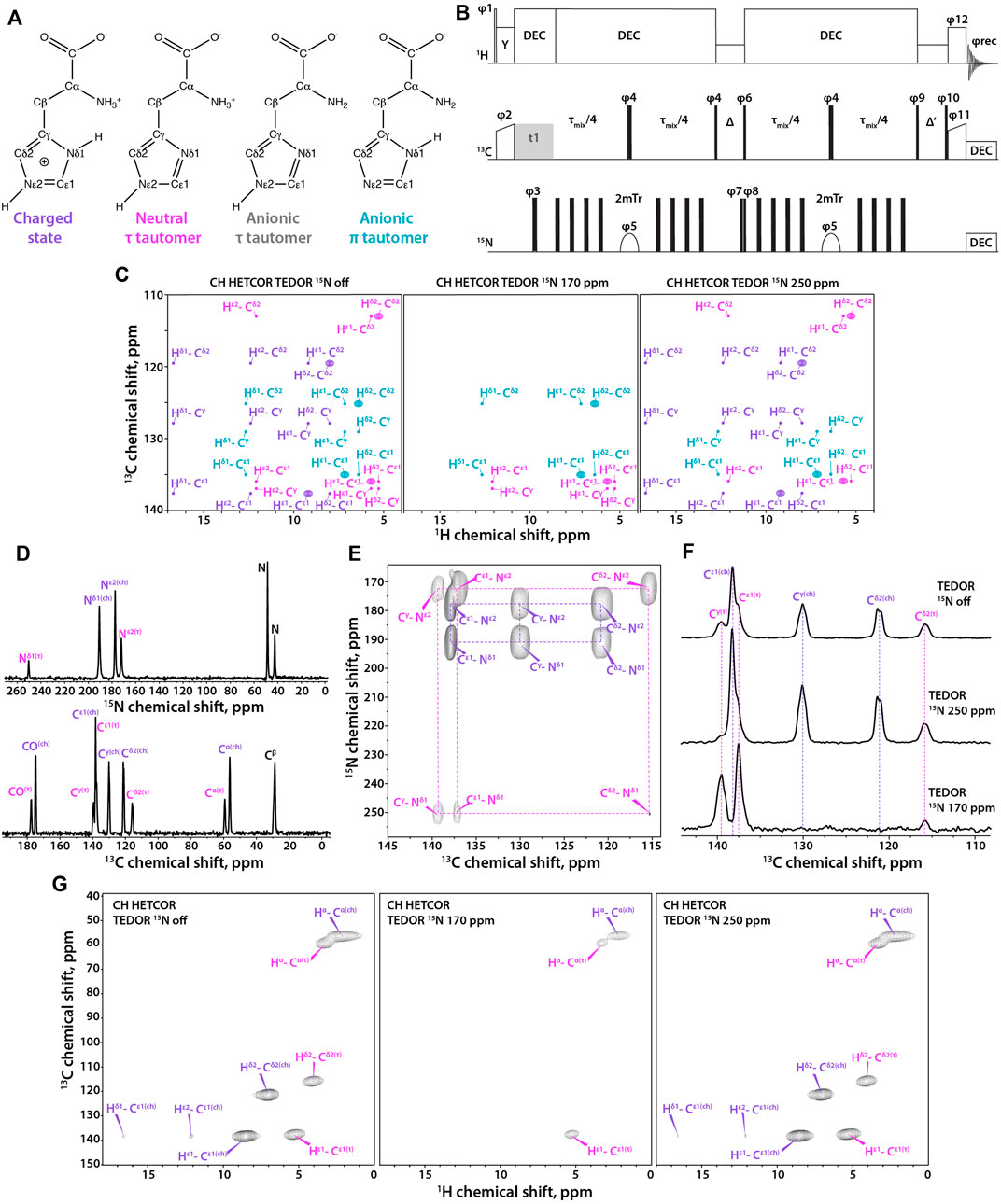
FIGURE 1. (A) Four states of histidine: left to right, charged state, neutral τ tautomer, anionic τ tautomer, and anionic π tautomer. (B) Pulse sequence for the 1H-detected TEDOR-based 15N selective filtered experiment. Tr is the MAS rotor period, τmix is the total TEDOR mixing time. The phase on the individual pulses are: φ1 = 16 × (0) 16 × (2), φ2 = 1, φ3 = 0,φ4 = 0, φ5 = 0213 2031, φ6 = 2, φ7 = 0, φ8 = 02, φ9 = 1133, φ10 = 4 × (0) 4 × (1) 4 × (2) 4 × (3), φ11 = 4 × (1) 4 × (0), φ12 = 4 × (1) 4 × (0) 4 × (1) 4 × (0) 4 × (3) 4 × (2) 4 × (3) 4 × (2), φrec = 3113 0220 1331 2002, where 0 = x, 1 = y, 2 = –x, and 3 = –y. Δ is set to one rotor period during which 1H rf field of ωr amplitude is applied for effective Z-filtering. MISSISSIPPI water suppression sequence is applied during Δ′ time period. (C) Synthetic 1H-detected TEDOR-based 15N selective filtered CH HETCOR spectra showing cross peaks expected for each tautomer. Left to right, soft pulse turned off, soft pulse at 170 ppm, soft pulse at 250 ppm. The filtering patterns for neutral and anionic τ tautomers are identical. (D) 15N (top) and 13C (bottom) CPMAS NMR spectra of crystalline histidine. (E) Aromatic region expansion of 2D NCA spectrum of crystalline histidine. (F) 1D 13C spectra using TEDOR-based 15N selective filtering in the aromatic region. Top to bottom, soft pulse turned off; soft pulse at 250 ppm; soft pulse at 170 ppm. (G) Three complementary 1H-detected TEDOR-based 15N selective filtered CH HETCOR spectra. Left to right, soft pulse turned off, soft pulse at 170 ppm, soft pulse at 250 ppm. The MAS frequency was 60 kHz in all experiments. Signals of charged state are shown in purple, neutral τ tautomer – in magenta, anionic τ tautomer - in grey, and anionic π tautomer – in teal.
Methods to determine His ionization states in proteins are solution NMR (Kilmartin et al., 1973; Markley, 1975; Bachovchin and Roberts, 1978; Perutz et al., 1985; Pelton et al., 1993; Shimba et al., 1998; Hass et al., 2008; Hansen and Kay, 2014) or neutron diffraction (Kossiakoff and Spencer, 1980; Maeda et al., 2004; Kovalevsky et al., 2010), with the latter limited to very large single crystals and requiring a neutron source, both difficult conditions to meet routinely. Therefore, solid-state magic angle spinning (MAS) NMR constitutes a viable alternative (Wei et al., 1999). Similar to solution NMR, the tautomeric state of histidines can be unambiguously determined from a unique combination of 15N sidechain chemical shifts (Munowitz et al., 1982; Wei et al., 1999; Miao et al., 2014) and the corresponding N-H distances can be estimated, allowing for hydrogen bonding studies (Shenderovich et al., 2015). Protonation states for the crystalline histidine amino acid have been determined by MAS NMR for different pH values (Li and Hong, 2011) and crystalline short peptides (Platzer et al., 2014). Using 15N selective filtered, 13C-detected experiments with the inversion pulses at frequencies of the different tautomers (Miao et al., 2014) permits their identification. For proteins containing several histidine residues, the above experiments are challenging due to low sensitivity and spectral overlap. Therefore, only a handful of such studies have been reported to date (Hu et al., 2006; Hu et al., 2010; Miao et al., 2015; Kwon et al., 2019; Maciejko et al., 2019; Vasa et al., 2019; Movellan et al., 2020). In order to increase resolution, the original pulse sequence can be reconfigured as a 2D experiment by introducing a 13C-13C mixing period based on proton-driven spin diffusion (PDSD) (Bloembergen, 1949) and extending the second Z-filter (Miao et al., 2014). 2D and 3D proton-based experiments were also introduced with 1H chemical shifts either recorded in the indirect dimension (Miao et al., 2015) or detected directly (Shenderovich et al., 2015; Vasa et al., 2019; Movellan et al., 2020).
Herein, we present an alternative MAS experiment that uses 1H detected transferred-echo double resonance (TEDOR)-based 15N selectively filtered 2D correlations at fast MAS frequencies of 40–60 kHz. The advantages of the 1H-detected fast-MAS experiments presented here are: i) improved sensitivity due to 1H detection, and ii) improved resolution via the second dimension and selective recoupling of aromatic resonances directly attached to 15N atoms. Microcrystalline assemblies of U-13C,15N- and fractionally deuterated (FD) (Mance et al., 2015) 13C,15N-HIV-1 CACTD-SP1 protein samples, possessing solely a single His residue, His-226, are ideally suited for pulse sequence optimization and therefore were selected for illustrating our current approach. Extension to ultrafast MAS frequencies (up to 110 kHz), should yield even higher sensitivity and resolution for proteins with multiple histidines.
Materials and Methods
Sample Preparation
U-13C,15N-L-histidine was purchased from Cambridge Isotope Laboratories, recrystallized from an aqueous solution at pH 6.0, adjusted by mixing HCl and NaOH. The sample was packed into a 1.3 mm MAS rotor. Microcrystalline assemblies of U-13C,15N- and FD-13C,15N-HIV-1 CACTD-SP1 were prepared in the presence of the assembly cofactor inositol hexakisphosphate (IP6) as described previously (Wagner et al., 2016) except for growing Escherichia coli in M9 medium containing 13C glucose, 15N NH4Cl, isotopically labeled precursors, and (for the deuterated sample) D2O. Proteins were assembled with 1.6 mM IP6 (Sigma-Aldrich), for a final reaction volume of 1 ml at pH 8.0. Assemblies were incubated overnight at 20°C and packed into 3.2 mm (U-13C,15N), 1.9 mm (FD-13C,15N), or 1.3 mm MAS rotors (U-13C,15N).
MAS NMR Spectroscopy
MAS NMR experiments on U-13C,15N-CACTD-SP1 and FD-13C,15N-CACTD-SP1 microcrystalline assemblies were performed on a 20.0 T Bruker AVIII spectrometer outfitted with 3.2 mm E-Free HCN and 1.9 HCN probes, respectively. The MAS frequency was 14 and 40 kHz, respectively, controlled to within ± 10 Hz by a Bruker MAS controller. The actual sample temperature was maintained at 4 ± 1°C throughout the experiments using the Bruker temperature controller.
The Larmor frequencies were 850.4 MHz (1H), 213.9 MHz (13C) and 86.2 MHz (15N). The typical 90° pulse lengths were 2.6–3.0 μs for 1H, 4.3–4.5 μs for 13C, and 4.2–4.7 μs for 15N. The 1H-13C and 1H-15N cross-polarization employed a linear amplitude ramp of 90–110% on 1H, and the center of the ramp was matched to a Hartmann–Hahn condition at the first spinning sideband; contact times of 0.7–1.5 ms and 1.0–1.7 ms were used, respectively. 50 ms CORD (Hou et al., 2013) mixing time was applied to facilitate 13C-13C mixing.
MAS NMR experiments on U-13C,15N-L-histidine and FD-13C,15N-CACTD-SP1 microcrystalline assemblies were performed on a 14.1 T Bruker AVIII spectrometer outfitted with 1.3 mm HCN probe. Larmor frequencies were 599.8 MHz (1H), 150.8 MHz (13C), and 60.7 MHz (15N). The MAS frequency was 60 kHz, controlled to within ± 10 Hz by a Bruker MAS controller. The actual sample temperature was maintained at 40 ± 1°C throughout the experiments using the Bruker temperature controller. The typical 90° pulse lengths were 1.4–1.6 μs for 1H, 2.7–3.0 μs for 13C, and 3.3–3.6 μs for 15N. The 1H-13C and 1H-15N cross-polarization employed a linear amplitude ramp of 90–110% on 1H, center of the ramp was matched to a Hartmann–Hahn condition at the first spinning sideband, with contact times of 1.0–5.0 ms and 1.3–5.0 ms, respectively. Band-selective 15N-13C SPECIFIC-CP contact time was 5.0–6.0 ms. SWFTPPM (Vinod Chandran et al., 2008) decoupling (15 kHz) was used during the TEDOR block and acquisition periods. The selective 15N 180° r-SNOB (Kupce et al., 1995) pulse length in the Z-filtered TEDOR experiments was 500 µs and the bandwidth — 2 kHz; the rf power was 4 kHz. During the Z-filter time period Δ, 60 kHz CW decoupling was applied for τr on 1H channel, while during the time period Δ’, MISSISSIPPI (Zhou and Rienstra, 2008) water suppression was applied. The TEDOR block duration was 1–3 ms.
Data Processing
All MAS NMR data were processed using NMRPipe (Delaglio et al., 1995). The 13C and 15N chemical shifts were referenced with respect to the external standards adamantane (Morcombe and Zilm, 2003) and ammonium chloride (Bertani et al., 2014), respectively. The 2D and 3D data sets were processed by applying 30, 45, 60, and 90° shifted sine bell apodization followed by a Lorentzian-to-Gaussian transformation in both dimensions. Forward linear prediction to twice the number of the original data points was used in the indirect dimension followed by zero filling. The processed spectra were analyzed in NMRFAM-Sparky (Goddard and Kneller, 2004; Lee et al., 2015) and CCPN (Stevens et al., 2011).
Results
Here, we report on a 2D 1H-detected TEDOR-based Z-filtered experiment, which incorporates 15N selective filters for the determination of histidine tautomeric states. The pulse sequence is shown in Figure 1B. The experiment is well suited for fast MAS frequencies of 40 kHz and above. The tautomeric states of His residues are unambiguously determined using a combination of three CH HETCOR experiments comprising: i) 15N selective TEDOR filter, containing 13C resonances of all protonation and tautomeric states present; ii) 15N selective TEDOR filter with a soft pulse at 170 ppm, removing resonances of the protonated state while Cε1 and Cδ2 atoms of π tautomer and Cε1 and Cγ atoms of τ tautomer remain; and iii) 15N selective TEDOR filter with a soft pulse at 250 ppm, retaining all signals of the charged state, Cε1 and Cγ of the π tautomer as well as Cε1 and Cδ2 atoms of the τ tautomer. Cε1 of anionic tautomers is always present in TEDOR filtered spectra, but has reduced peak intensity when 15N selective pulse is applied as C-N dipolar interaction with the non-selectively irradiated nitrogen atom is recoupled. The sequence was first tested on a crystalline L-histidine sample prepared at pH 6.0. The 13C and 15N 1D CPMAS and 2D NCA spectra are shown in Figures 1D, E, respectively. The spectra clearly indicate the presence of two forms of L-histidine, the charged monohydrate and the τ tautomer, in approximately 2:1 ratio. As shown in Figure 1F, conventional 13C-detected TEDOR-based experiments are well suited for the determination of protonation states in this sample. To test the 1H-detected sequences proposed herein, three complementary experiments were performed. As shown in Figure 1G, 15N selective TEDOR-filtered CH HETCOR without or with a soft pulse at 250 ppm (left and right panels, respectively) yield the sidechain signals of both protonation states, while 15N selective TEDOR-filtered CH HETCOR with soft pulse at 170 ppm retains only Cε1 resonance of the τ tautomer (chemical shifts provided in Supplementary Table S1). Water suppression was incorporated into the second Z-filter, allowing to record spectra on hydrated samples.
HIV-1 CACTD-SP1 (Figure 2A) contains a single His residue, His-226. The outstanding high spectral resolution in the microcrystalline FD-13C,15N-CACTD-SP1 sample allows for the determination of histidine protonation and tautomeric states even in the 13C-detected mode (Figure 2B). The Cε1 and Cγ resonances are present in 1D experiments, while the Cδ2 resonance is absent in the 15N selective TEDOR-filtered 13C CPMAS experiment with the soft pulse at 170 ppm since its magnetization does not build up during the TEDOR block due to the very weak dipolar coupling to Nδ1 (chemical shifts provided in Supplementary Table S2). The 2D 13C-13C CORD spectrum clearly shows a single set of resonances, indicating the presence of only one histidine species (Figure 2C), although the protonation and tautomeric state cannot be determined without additional experiments. The three complementary 1H-detected TEDOR-based 15N selective CH HETCOR spectra (Figure 2D) also indicate the presence of a single species, which is unambiguously assigned as τ tautomer. These 1H-detected 2D spectra contain no resonances of aromatic residues other than His (shown in black in the CH HETCOR spectrum) and Trp (these are weak or absent in the spectra of the deuterated sample), as only carbons attached to nitrogens are selected, making assignment of histidine resonances straightforward.
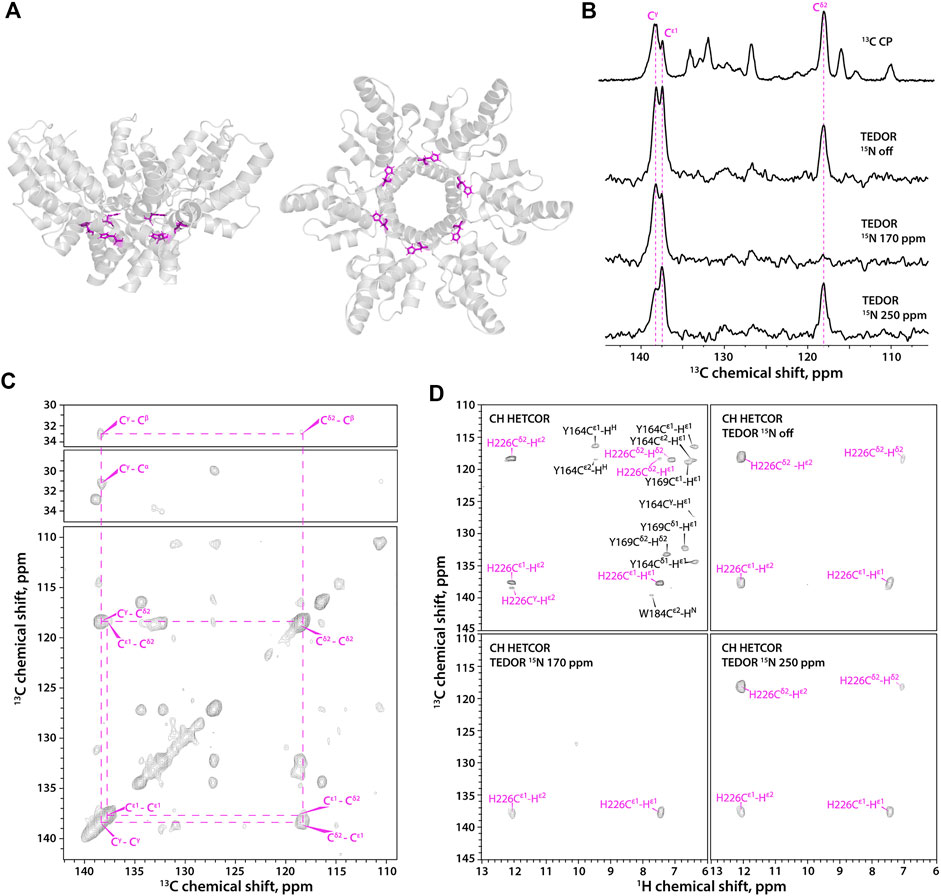
FIGURE 2. (A) A hexameric unit of HIV-1 CACTD-SP1 in the microcrystalline assembly (PDB 5I4T) shown as side view (left) and top view (right). (B) 1D 13C MAS NMR spectra of FD-13C,15N-CACTD-SP1 with TEDOR-based 15N selective filtering in the aromatic region. Top to bottom: CPMAS spectrum; TEDOR-based 15N selectively filtered spectra with soft pulse turned off, soft pulse at 170 ppm, and soft pulse at 250 ppm. (C) 2D CORD spectrum of FD-13C,15N-CACTD-SP1 (MAS frequency 14 kHz). (D) Aromatic regions of 1H-detected TEDOR-based 15N selective filtered CH HETCOR spectra in FD-13C,15N-CACTD-SP1: TEDOR filter and soft pulse turned off (top left), soft pulse turned off (top right), soft pulse at 170 ppm (bottom left), soft pulse at 250 ppm (bottom right). The MAS frequency was 40 kHz in all experiments, unless indicated otherwise. Signals of τ tautomer are shown in magenta.
In contrast to the FD-13C,15N-CACTD-SP1, the His-226 protonation state in U-13C,15N-CACTD-SP1 assemblies cannot be easily determined using the 1D 13C-detected version of TEDOR-based 15N selective filtered experiments due to low resolution and spectral overlap (Figure 3A). In contrast, the 2D 1H-detected TEDOR-based 15N selective filtered spectra (Figure 3B) suggest the presence of a small fraction of π tautomer along with the predominant τ tautomer in this sample (chemical shifts provided in Supplementary Table S2).
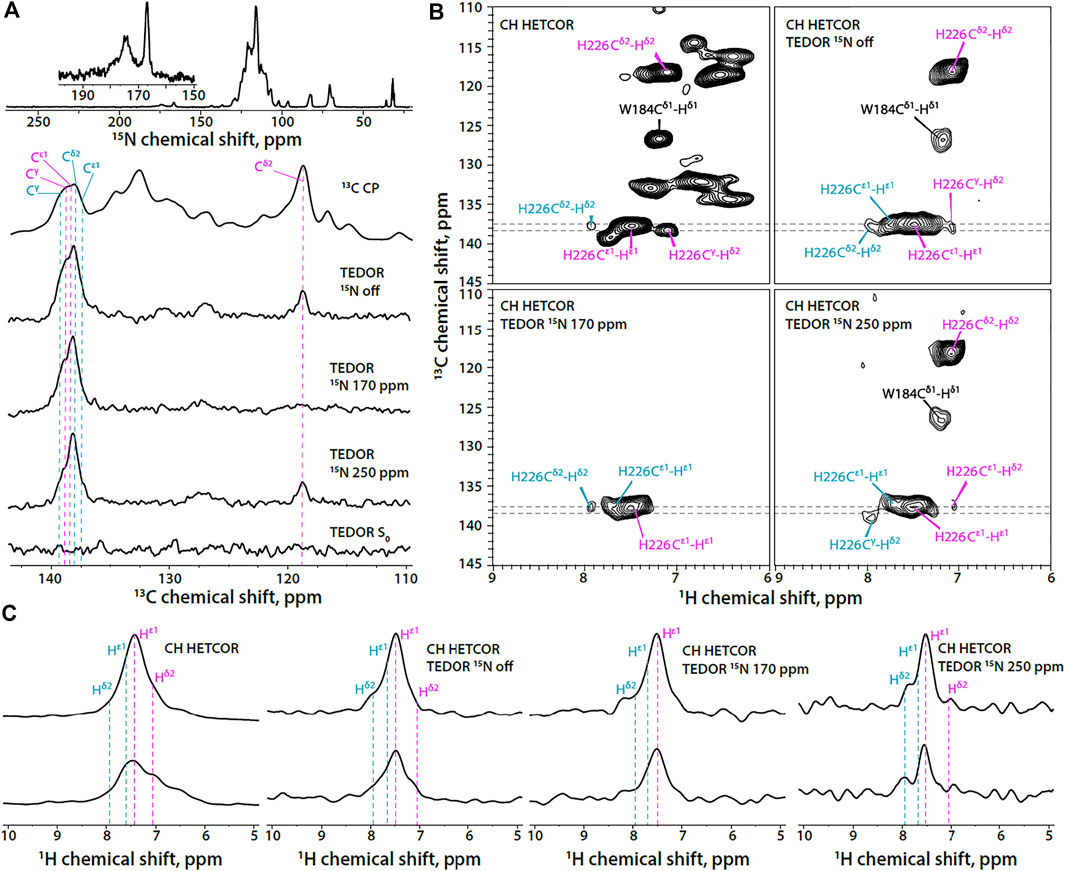
FIGURE 3. (A) 1D 15N CPMAS and 13C MAS NMR spectra of U-13C,15N-CACTD-SP1 with TEDOR-based 15N selective filtering in the aromatic region (MAS frequency 14 kHz). Top to bottom: 15N CPMAS spectrum; 13C CPMAS spectrum; TEDOR-based 15N selectively filtered spectra with soft pulse turned off, soft pulse at 170 ppm, soft pulse at 250 ppm, and a reference (S0) experiment. (B) Aromatic regions of 1H-detected TEDOR-based 15N selective filtered CH HETCOR spectra in U-13C,15N-CACTD-SP1: TEDOR filter and soft pulse turned off (top left), soft pulse turned off (top right), soft pulse at 170 ppm (bottom left), soft pulse at 250 ppm (bottom right). The first contour was set at 5× the noise rmsd. (C) 1D 1H slices of 1H-detected TEDOR-based 15N selective filtered CH HETCOR spectra in U-13C,15N-CACTD-SP1, extracted at 13C shifts shown as gray dashed lines in panel (B). Left to right: TEDOR filter and soft pulse turned off, soft pulse turned off, soft pulse at 170 ppm, soft pulse at 250 ppm. The MAS frequency was 60 kHz. Signals of τ tautomer and π tautomer are shown in magenta and teal, respectively.
In addition to the His signals, the indole ring signals of the Trp184 residue are also present in the 1H-detected TEDOR-based experiments when the soft pulse is either turned off or centered at 250 ppm. This is expected due to the nitrogen atom Nε1 in the indole ring, which allows for magnetization build up on adjacent carbon atoms (Cδ1 and Cε2) during TEDOR transfer. Tryptophan sidechain resonances appear much stronger in non-deuterated protein assemblies compared to the FD-13C,15N -CACTD-SP1 and can be distinguished from those corresponding to the histidine based on chemical shift.
Conclusion
We demonstrated that 1H-detected 2D Z-filtered TEDOR experiments incorporating 15N selective filters permit unambiguous assignment of histidine protonation and tautomeric states in microcrystalline proteins and protein assemblies. This approach combines all the advantages of fast MAS and proton detection. Extending the experiments to MAS frequencies of 110 kHz and above can further improve the quality of data sets and allow unambiguous assignment of His protonation and tautomeric states in larger proteins and protein assemblies.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
TP and AMG conceived the project and guided the work. RZ performed NMR experiments and analyzed the experimental data. CMQ assisted with the NMR experiments and data analysis. SS assigned the CACTD-SP1 chemical shifts. KKZ, BKG-P, and OP prepared samples of microcrystalline CACTD-SP1 assemblies. RZ and TP took the lead in writing the manuscript. All authors discussed the results and contributed to the manuscript preparation.
Funding
This work was supported by the National Institutes of Health (NIH Grants P50AI1504817 and R01AI129678). We acknowledge the support of the National Science Foundation (NSF Grant CHE0959496) for the acquisition of the 850 MHz NMR spectrometer and of the National Institutes of Health (NIH Grant P30GM110758) for the support of core instrumentation infrastructure at the University of Delaware; and of the National Institutes of Health (NIH Grant S10OD012213) for the acquisition of the 750 MHz NMR spectrometer at the University of Pittsburgh.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.767040/full#supplementary-material
References
Adams, M. J., Blundell, T. L., Dodson, E. J., Dodson, G. G., Vijayan, M., Baker, E. N., et al. (1969). Structure of Rhombohedral 2 Zinc Insulin Crystals. Nature 224, 491–495. doi:10.1038/224491a0
Bachovchin, W. W., and Roberts, J. D. (1978). Nitrogen-15 Nuclear Magnetic Resonance Spectroscopy. The State of Histidine in the Catalytic Triad of Alpha-Lytic Protease. Implications for the Charge-Relay Mechanism of Peptide-Bond Cleavage by Serine Proteases. J. Am. Chem. Soc. 100, 8041–8047. doi:10.1021/ja00494a001
Bertani, P., Raya, J., and Bechinger, B. (2014). 15N Chemical Shift Referencing in Solid State NMR. Solid State. Nucl. Magn. Reson. 61-62, 15–18. doi:10.1016/j.ssnmr.2014.03.003
Bloembergen, N. (1949). On the Interaction of Nuclear Spins in a Crystalline Lattice. Physica 15, 386–426. doi:10.1016/0031-8914(49)90114-7
Blomberg, F., Maurer, W., and Rueterjans, H. (1977). Nuclear Magnetic Resonance Investigation of Nitrogen-15-Labeled Histidine in Aqueous Solution. J. Am. Chem. Soc. 99, 8149–8159. doi:10.1021/ja00467a005
Blow, D. M., Birktoft, J. J., and Hartley, B. S. (1969). Role of a Buried Acid Group in the Mechanism of Action of Chymotrypsin. Nature 221, 337–340. doi:10.1038/221337a0
Campbell, I. D., Lindskog, S., and White, A. I. (1974). A Study of the Histidine Residues of Human Carbonic Anhydrase B Using 270 MHz Proton Magnetic Resonance. J. Mol. Biol. 90, 469–489. doi:10.1016/0022-2836(74)90229-0
Delaglio, F., Grzesiek, S., Vuister, G., Zhu, G., Pfeifer, J., and Bax, A. (1995). NMRPipe: a Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR 6, 277–293. doi:10.1007/bf00197809
Edwards, B. F. P., and Sykes, B. D. (1980). Nuclear Magnetic Resonance Evidence for the Coexistence of Several Conformational States of Rabbit Cardiac and Skeletal Tropomyosins. Biochemistry 19, 2577–2583. doi:10.1021/bi00553a007
French, T. C., and Hammes, G. G. (1965). Relaxation Spectra of Ribonuclease. II. Isomerization of Ribonuclease at Neutral pH Values. J. Am. Chem. Soc. 87, 4669–4673. doi:10.1021/ja00949a002
Hansen, A. L., and Kay, L. E. (2014). Measurement of Histidine pKa Values and Tautomer Populations in Invisible Protein States. Proc. Natl. Acad. Sci. 111, E1705–E1712. doi:10.1073/pnas.1400577111
Hass, M. A. S., Hansen, D. F., Christensen, H. E. M., Led, J. J., and Kay, L. E. (2008). Characterization of Conformational Exchange of a Histidine Side Chain: Protonation, Rotamerization, and Tautomerization of His61 in Plastocyanin from Anabaena Variabilis. J. Am. Chem. Soc. 130, 8460–8470. doi:10.1021/ja801330h
Hoffee, P., Lai, C. Y., Pugh, E. L., and Horecker, B. L. (1967). The Function of Histidine Residues in Rabbit Muscle Aldolase. Proc. Natl. Acad. Sci. 57, 107–113. doi:10.1073/pnas.57.1.107
Hou, G., Yan, S., Trébosc, J., Amoureux, J.-P., and Polenova, T. (2013). Broadband Homonuclear Correlation Spectroscopy Driven by Combined R2nv Sequences Under Fast Magic Angle Spinning for NMR Structural Analysis of Organic and Biological Solids. J. Magn. Reson. 232, 18–30. doi:10.1016/j.jmr.2013.04.009
Hu, J., Fu, R., Nishimura, K., Zhang, L., Zhou, H.-X., Busath, D. D., et al. (2006). Histidines, Heart of the Hydrogen Ion Channel from Influenza A Virus: toward an Understanding of Conductance and Proton Selectivity. Proc. Natl. Acad. Sci. 103, 6865–6870. doi:10.1073/pnas.0601944103
Hu, F., Luo, W., and Hong, M. (2010). Mechanisms of Proton Conduction and Gating in Influenza M2 Proton Channels from Solid-State NMR. Science 330, 505–508. doi:10.1126/science.1191714
Kilmartin, J. V., Breen, J. J., Roberts, G. C. K., and Ho, C. (1973). Direct Measurement of the pK Values of an Alkaline Bohr Group in Human Hemoglobin. Proc. Natl. Acad. Sci. 70, 1246–1249. doi:10.1073/pnas.70.4.1246
Kossiakoff, A. A., and Spencer, S. A. (1980). Neutron Diffraction Identifies His 57 as the Catalytic Base in Trypsin. Nature 288, 414–416. doi:10.1038/288414a0
Kossiakoff, A. A., and Spencer, S. A. (1981). Direct Determination of the Protonation States of Aspartic Acid-102 and Histidine-57 in the Tetrahedral Intermediate of the Serine Proteases: Neutron Structure of Trypsin. Biochemistry 20, 6462–6474. doi:10.1021/bi00525a027
Kovalevsky, A. Y., Chatake, T., Shibayama, N., Park, S.-Y., Ishikawa, T., Mustyakimov, M., et al. (2010). Direct Determination of Protonation States of Histidine Residues in a 2 Å Neutron Structure of Deoxy-Human Normal Adult Hemoglobin and Implications for the Bohr Effect. J. Mol. Biol. 398, 276–291. doi:10.1016/j.jmb.2010.03.016
Kupce, E., Boyd, J., and Campbell, I. D. (1995). Short Selective Pulses for Biochemical Applications. J. Magn. Reson. Ser. B 106, 300–303. doi:10.1006/jmrb.1995.1049
Kwon, B., Roos, M., Mandala, V. S., Shcherbakov, A. A., and Hong, M. (2019). Elucidating Relayed Proton Transfer through a His-Trp-His Triad of a Transmembrane Proton Channel by Solid-State NMR. J. Mol. Biol. 431, 2554–2566. doi:10.1016/j.jmb.2019.05.009
Lee, W., Tonelli, M., and Markley, J. L. (2015). NMRFAM-SPARKY: Enhanced Software for Biomolecular NMR Spectroscopy. Bioinformatics 31, 1325–1327. doi:10.1093/bioinformatics/btu830
Lewis, S. D., Johnson, F. A., and Shafer, J. A. (1976). Potentiometric Determination of Ionizations at the Active Site of Papain. Biochemistry 15, 5009–5017. doi:10.1021/bi00668a010
Lewis, S. D., Johnson, F. A., and Shafer, J. A. (1981). Effect of Cysteine-25 on the Ionization of Histidine-159 in Papain as Determined by Proton Nuclear Magnetic Resonance Spectroscopy. Evidence for a Histidine-159-Cysteine-25 Ion Pair and its Possible Role in Catalysis. Biochemistry 20, 48–51. doi:10.1021/bi00504a009
Li, S., and Hong, M. (2011). Protonation, Tautomerization, and Rotameric Structure of Histidine: a Comprehensive Study by Magic-Angle-Spinning Solid-State NMR. J. Am. Chem. Soc. 133, 1534–1544. doi:10.1021/ja108943n
Liljas, A., Kannan, K. K., Bergstén, P.-C., Waara, I., Fridborg, K., Strandberg, B., et al. (1972). Crystal Structure of Human Carbonic Anhydrase C. Nat. New Biol. 235, 131–137. doi:10.1038/newbio235131a0
Loewenthal, R., Sancho, J., and Fersht, A. R. (1992). Histidine-aromatic Interactions in Barnase. J. Mol. Biol. 224, 759–770. doi:10.1016/0022-2836(92)90560-7
Maciejko, J., Kaur, J., Becker-Baldus, J., and Glaubitz, C. (2019). Photocycle-dependent Conformational Changes in the Proteorhodopsin Cross-Protomer Asp-His-Trp Triad Revealed by DNP-Enhanced MAS-NMR. Proc. Natl. Acad. Sci. USA 116, 8342–8349. doi:10.1073/pnas.1817665116
Maeda, M., Chatake, T., Tanaka, I., Ostermann, A., and Niimura, N. (2004). Crystallization of a Large Single crystal of Cubic Insulin for Neutron Protein Crystallography. J. Synchrotron Radiat. 11, 41–44. doi:10.1107/s0909049503023859
Mance, D., Sinnige, T., Kaplan, M., Narasimhan, S., Daniëls, M., Houben, K., et al. (2015). An Efficient Labelling Approach to Harness Backbone and Side‐Chain Protons in 1H‐Detected Solid‐State NMR Spectroscopy. Angew. Chem. Int. Ed. 54, 15799–15803. doi:10.1002/anie.201509170
Markley, J. L. (1975). Observation of Histidine Residues in Proteins by Nuclear Magnetic Resonance Spectroscopy. Acc. Chem. Res. 8, 70–80. doi:10.1021/ar50086a004
Miao, Y., Cross, T. A., and Fu, R. (2014). Differentiation of Histidine Tautomeric States Using 15N Selectively Filtered 13C Solid-State NMR Spectroscopy. J. Magn. Reson. 245, 105–109. doi:10.1016/j.jmr.2014.06.005
Miao, Y., Fu, R., Zhou, H.-X., and Cross, T. A. (2015). Dynamic Short Hydrogen Bonds in Histidine Tetrad of Full-Length M2 Proton Channel Reveal Tetrameric Structural Heterogeneity and Functional Mechanism. Structure 23, 2300–2308. doi:10.1016/j.str.2015.09.011
Morcombe, C. R., and Zilm, K. W. (2003). Chemical Shift Referencing in MAS Solid State NMR. J. Magn. Reson. 162, 479–486. doi:10.1016/S1090-7807(03)00082-X
Movellan, K. T., Wegstroth, M., Overkamp, K., Leonov, A., Becker, S., and Andreas, L. B. (2020). Imidazole-Imidazole Hydrogen Bonding in the pH-Sensing Histidine Side Chains of Influenza A M2. J. Am. Chem. Soc. 142, 2704–2708. doi:10.1021/jacs.9b10984
Munowitz, M., Bachovchin, W. W., Herzfeld, J., Dobson, C. M., and Griffin, R. G. (1982). Acid-base and Tautomeric Equilibriums in the Solid State: Nitrogen-15 NMR Spectroscopy of Histidine and Imidazole. J. Am. Chem. Soc. 104, 1192–1196. doi:10.1021/ja00369a007
Pelton, J. G., Torchia, D. A., Meadow, N. D., and Roseman, S. (1993). Tautomeric States of the Active-Site Histidines of Phosphorylated and Unphosphorylated IIIGlc, a Signal-Transducing Protein from Escherichia coli, Using Two-Dimensional Heteronuclear NMR Techniques. Protein Sci. 2, 543–558. doi:10.1002/pro.5560020406
Perutz, M. F., and Mathews, F. S. (1966). An X-ray Study of Azide Methaemoglobin. J. Mol. Biol. 21, 199–202. doi:10.1016/0022-2836(66)90088-x
Perutz, M. F., Muirhead, H., Mazzarella, L., Crowther, R. A., Greer, J., and Kilmartin, J. V. (1969). Identification of Residues Responsible for the Alkaline Bohr Effect in Haemoglobin. Nature 222, 1240–1243. doi:10.1038/2221240a0
Perutz, M. F., Gronenborn, A. M., Clore, G. M., Fogg, J. H., and Shih, D. T.-b. (1985). The pKa Values of Two Histidine Residues in Human Haemoglobin, the Bohr Effect, and the Dipole Moments of α-helices. J. Mol. Biol. 183, 491–498. doi:10.1016/0022-2836(85)90016-6
Platzer, G., Okon, M., and Mcintosh, L. P. (2014). pH-Dependent Random Coil 1H, 13C, and 15N Chemical Shifts of the Ionizable Amino Acids: a Guide for Protein pK a Measurements. J. Biomol. NMR 60, 109–129. doi:10.1007/s10858-014-9862-y
Plesniak, L. A., Connelly, G. P., Mcintosh, L. P., and Wakarchuk, W. W. (1996). Characterization of a Buried Neutral Histidine Residue in Bacillus Circulansxylanase: NMR Assignments, pH Titration, and Hydrogen Exchange. Protein Sci. 5, 2319–2328. doi:10.1002/pro.5560051118
Shenderovich, I. G., Lesnichin, S. B., Tu, C., Silverman, D. N., Tolstoy, P. M., Denisov, G. S., et al. (2015). NMR Studies of Active-Site Properties of Human Carbonic Anhydrase II by Using 15N-Labeled 4-Methylimidazole as a Local Probe and Histidine Hydrogen-Bond Correlations. Chem. Eur. J. 21, 2915–2929. doi:10.1002/chem.201404083
Shimba, N., Takahashi, H., Sakakura, M., Fujii, I., and Shimada, I. (1998). Determination of Protonation and Deprotonation Forms and Tautomeric States of Histidine Residues in Large Proteins Using Nitrogen−Carbon J Couplings in Imidazole Ring. J. Am. Chem. Soc. 120, 10988–10989. doi:10.1021/ja982153g
Stevens, T. J., Fogh, R. H., Boucher, W., Higman, V. A., Eisenmenger, F., Bardiaux, B., et al. (2011). A Software Framework for Analysing Solid-State MAS NMR Data. J. Biomol. NMR 51, 437–447. doi:10.1007/s10858-011-9569-2
Stryer, L., Kendrew, J. C., and Watson, H. C. (1964). The Mode of Attachment of the Azide Ion to Sperm Whale Metmyoglobin. J. Mol. Biol. 8, 96–IN10. doi:10.1016/s0022-2836(64)80152-2
Vasa, S. K., Singh, H., Grohe, K., and Linser, R. (2019). Assessment of a Large Enzyme-Drug Complex by Proton‐Detected Solid‐State NMR Spectroscopy without Deuteration. Angew. Chem. Int. Ed. 58, 5758–5762. doi:10.1002/anie.201811714
Vinod Chandran, C., Madhu, P. K., Kurur, N. D., and Bräuniger, T. (2008). Swept-frequency Two-Pulse Phase Modulation (SWf-TPPM) Sequences with Linear Sweep Profile for Heteronuclear Decoupling in Solid-State NMR. Magn. Reson. Chem. 46, 943–947. doi:10.1002/mrc.2285
Wagner, J. M., Zadrozny, K. K., Chrustowicz, J., Purdy, M. D., Yeager, M., Ganser-Pornillos, B. K., et al. (2016). Crystal Structure of an HIV Assembly and Maturation Switch. eLife 5, e17063. doi:10.7554/eLife.17063
Wei, Y., De Dios, A. C., and McDermott, A. E. (1999). Solid-State 15N NMR Chemical Shift Anisotropy of Histidines: Experimental and Theoretical Studies of Hydrogen Bonding. J. Am. Chem. Soc. 121, 10389–10394. doi:10.1021/ja9919074
Zhou, D. H., and Rienstra, C. M. (2008). High-performance Solvent Suppression for Proton Detected Solid-State NMR. J. Magn. Reson. 192, 167–172. doi:10.1016/j.jmr.2008.01.012
Keywords: Magic angle spinning (MAS), nuclear magnetic resonance (NMR) spectroscopy, histidine protonation state, transferred echo double resonance (TEDOR), Fast MAS NMR, solid-state NMR
Citation: Zadorozhnyi R, Sarkar S, Quinn CM, Zadrozny KK, Ganser-Pornillos BK, Pornillos O, Gronenborn AM and Polenova T (2021) Determination of Histidine Protonation States in Proteins by Fast Magic Angle Spinning NMR. Front. Mol. Biosci. 8:767040. doi: 10.3389/fmolb.2021.767040
Received: 30 August 2021; Accepted: 15 October 2021;
Published: 10 December 2021.
Edited by:
Amir Goldbourt, Tel Aviv University, IsraelReviewed by:
Loren B. Andreas, Max Planck Institute for Biophysical Chemistry, GermanyHans-Heinrich Limbach, Freie Universität Berlin, Germany
Copyright © 2021 Zadorozhnyi, Sarkar, Quinn, Zadrozny, Ganser-Pornillos, Pornillos, Gronenborn and Polenova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tatyana Polenova, dHBvbGVub3ZAdWRlbC5lZHU=; Angela M. Gronenborn, YW1nMTAwQHBpdHQuZWR1