
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 30 November 2021
Sec. Cellular Biochemistry
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.753283
This article is part of the Research Topic Venom Peptides: A Rich Combinatorial Library for Drug Development View all 14 articles
 Igor E. Kasheverov1†
Igor E. Kasheverov1† Alexey I. Kuzmenkov1†
Alexey I. Kuzmenkov1† Denis S. Kudryavtsev1
Denis S. Kudryavtsev1 Ivan S. Chudetskiy1Irina V. Shelukhina1
Ivan S. Chudetskiy1Irina V. Shelukhina1 Evgeny P. Barykin2
Evgeny P. Barykin2 Igor A. Ivanov1
Igor A. Ivanov1 Andrei E. Siniavin1Rustam H. Ziganshin1
Andrei E. Siniavin1Rustam H. Ziganshin1 Mikhail S. Baranov1Victor I. Tsetlin1
Mikhail S. Baranov1Victor I. Tsetlin1 Alexander A. Vassilevski1,3
Alexander A. Vassilevski1,3 Yuri N. Utkin1*
Yuri N. Utkin1*Fluorescence can be exploited to monitor intermolecular interactions in real time and at a resolution up to a single molecule. It is a method of choice to study ligand-receptor interactions. However, at least one of the interacting molecules should possess good fluorescence characteristics, which can be achieved by the introduction of a fluorescent label. Gene constructs with green fluorescent protein (GFP) are widely used to follow the expression of the respective fusion proteins and monitor their function. Recently, a small synthetic analogue of GFP chromophore (p-HOBDI-BF2) was successfully used for tagging DNA molecules, so we decided to test its applicability as a potential fluorescent label for proteins and peptides. This was done on α-cobratoxin (α-CbTx), a three-finger protein used as a molecular marker of muscle-type, neuronal α7 and α9/α10 nicotinic acetylcholine receptors (nAChRs), as well as on azemiopsin, a linear peptide neurotoxin selectively inhibiting muscle-type nAChRs. An activated N-hydroxysuccinimide ester of p-HOBDI-BF2 was prepared and utilized for toxin labeling. For comparison we used a recombinant α-CbTx fused with a full-length GFP prepared by expression of a chimeric gene. The structure of modified toxins was confirmed by mass spectrometry and their activity was characterized by competition with iodinated α-bungarotoxin in radioligand assay with respective receptor preparations, as well as by thermophoresis. With the tested protein and peptide neurotoxins, introduction of the synthetic GFP chromophore induced considerably lower decrease in their affinity for the receptors as compared with full-length GFP attachment. The obtained fluorescent derivatives were used for nAChR visualization in tissue slices and cell cultures.
Three-finger toxins (TFTs) are common widespread non-enzymatic components of elapid snake venoms. TFTs comprise ∼60–80 amino acid residues folded into three β-hairpin loops extending from a hydrophobic core (Kini and Doley, 2010; Kini and Koh, 2020). The name “three-finger toxins” originates from these three loops resembling three fingers of the human hand. Usually, these toxins are stabilized by four or five disulfide bridges, four of which are conserved (Kessler et al., 2017). Among these polypeptides there are well-known ligands of several ion channels including α-neurotoxins (nicotinic acetylcholine receptors, nAChRs) (Barber et al., 2013), Tx7335 (voltage-gated potassium channels) (Rivera-Torres et al., 2016), calliotoxin (sodium channels) (Yang et al., 2016), calciseptine (calcium channels) (de Weille et al., 1991), mambalgins (acid-sensing ion channels) (Diochot et al., 2012), and micrurotoxins (receptors of γ-aminobutyric acid) (Rosso et al., 2015). TFTs represent one of the best studied natural combinatorial libraries with immense potential for drug discovery.
The history of TFTs began with the discovery of α-bungarotoxin (α-BgTx) in the venom of the many-banded krait Bungarus multicinctus about 60 years ago (Chang and Lee, 1963). Dozens of TFTs, such as α-cobratoxin (α-CbTx) from the monocled cobra Naja kaouthia (Karlsson et al., 1972), neurotoxin NTII from the Caspian cobra Naja oxiana, and LsIII from the black-banded sea krait Laticauda semifasciata, were further purified and characterized (Utkin, 2019; Tsetlin et al., 2021). Those toxins served as high-affinity and selective molecular probes per se, and their labeled derivatives are traditionally utilized as robust molecular reporter tools in channel research (Anderson and Cohen, 1974; Stiles, 1993; Kuzmenkov and Vassilevski, 2018; Kudryavtsev et al., 2020). Pioneer works, where tritiated and iodinated α-BgTx was used, introduced the methodology of nAChR purification, identification of binding sites, and determination of the receptors distribution in the brain (O’Brien et al., 1972; Silver and Billiar, 1976; Lukasiewicz and Bennett, 1978). TFTs with isotopic moieties are still widely used in radioligand binding assays for the detection of novel compounds and establishing their kinetic properties (Martin et al., 2007; Anderson, 2008). An alternative approach to develop toxin-based molecular markers is the utilization of fluorescent dyes. Fluorescent derivatives of α-BgTx were used for visualizing the distribution of nAChR in vertebrate skeletal muscle fibers, expression profiling and description of the clusterization process of diffusely distributed receptors (Ravdin and Axelrod, 1977; Stya and Axelrod, 1983; Anderson, 2008; Shelukhina et al., 2009). Simultaneously, fluorescently-modified NTII and α-CbTx were also successfully produced and applied for binding studies and mapping of nAChR (Tsetlin et al., 1979, 1982; Kang and Maelicke, 1980).
Here, we extend the techniques of toxin labeling by the design and production of novel fluorescent probes for nAChR imaging. For these purposes we obtained three derivatives of well-studied toxins. The first marker is the recombinant protein eGFP–α-CbTx, which is a fusion of α-CbTx and the enhanced green fluorescent protein (eGFP). α-CbTx binds with high affinity and practically irreversibly to nAChRs; therefore, even after some decrease in affinity as a result of modification this toxin retains the capacity to bind to its receptor. Earlier, it was shown that attachment of a fluorescein label at Lys23 in the central loop of α-CbTx resulted in a fluorescently labeled derivative retaining nanomolar affinity to Torpedo nAChR (Johnson and Taylor, 1982). For quite a long time we have been using α-CbTx and its derivatives for the study of different receptors (Utkin et al., 1998; Kudryavtsev et al., 2015) and gained extensive experience in work with just this toxin. Moreover, we possess sufficient quantities of native α-CbTx for comparison. All of this contributed to our decision to choose α-CbTx for preparation of chimera with eGFP. We have already successfully utilized conjugation with eGFP to produce and characterize selective probes to voltage-gated potassium channels (Kuzmenkov et al., 2016a). Such method of labeling is aimed at obtaining mono-tagged derivatives and claims to dramatically decrease the costs of the final products (Kuzmenkov and Vassilevski, 2018). The second fluorescent probe is presented by the same neurotoxin covalently linked to eGFP chromophore-mimicking moiety (p-HOBDI-BF2) (Baranov et al., 2012). This tag was previously applied only for labeling DNA molecules (Stakheev et al., 2018) and used in the preparation of an inhibitor for irreversible interactions with the active-site cysteine of human cathepsins (Frizler et al., 2013). Moreover, to design specific muscle-type nAChR reporter we utilized p-HOBDI-BF2 moiety for the preparation of a mono-labeled fluorescent derivative of azemiopsin (Aze), a linear neurotoxic peptide earlier isolated from the Azemiops feae viper venom (Utkin et al., 2012).
α-CbTx was purified from N. kaouthia venom as described (Osipov et al., 2008). Aze was synthesized as in (Shelukhina et al., 2018). Radiolabeled 125I-α-BgTx with specific radioactivity of 500 Ci/mmol was prepared as in (Kryukova et al., 2018). Alexa Fluor 555 and 647 conjugates of α-BgTx (AF555–α-BgTx and AF647–α-BgTx) were purchased from Thermo Fisher Scientific.
Chymotrypsin was from Sigma and Lys-C protease was from BDH Chemicals (Sweden). Recombinant human enteropeptidase light chain was a gift from Dr. Marine Gasparian (Gasparian et al., 2003). Acetonitrile was from PanReac-AppliChem (ITW Reagents, Spain). Commercially available reagents were used for activated p-HOBDI-BF2 synthesis without additional purification. All other reagents were of analytical or higher grade.
To construct an expression vector bearing the eGFP–α-CbTx gene, we used pET-28a (Novagen). The gene of α-CbTx including a stop codon was synthesized by Evrogen (Russia).
Murine neuroblastoma Neuro-2a cells were obtained from ATCC-LGC (France) and maintained as described previously (Shelukhina et al., 2017). THP-1 (ATCC TIB-202), Raji (ATCC CCL-86), and RPMI 1788 (ATCC CCL-156) cells were maintained in RPMI 1640 medium, supplemented with 10% fetal bovine serum, 1× GlutaMAX and 1× penicillin-streptomycin solution (all from Gibco, Thermo Fisher Scientific) at 37°C with 5% CO2.
At the first step the pET-28a-eGFP vector was obtained by cloning the eGFP gene alone, which was amplified with eGFP-f and eGFP-r primers from the plasmid pUC-eGFP, as we described previously (Kuzmenkov et al., 2016a) (Table 1). NdeI and BamHI sites were used for cloning. On the second step, a sequence encoding a short linker was synthesized from primers (Link-f and Link-r) and added at BamHI and EcoRI restriction sites. Finally, the gene of α-CbTx with a stop codon was amplified with CbTx-f and CbTx-r oligonucleotides and then subcloned in pET-28a-eGFP using EcoRI and SalI restriction sites. Additionally, α-CbTx gene was produced with modified human enteropeptidase site (DDDDR) at the N-terminus by changing CbTx-f to CbTx-f-EK primer. All oligonucleotides from which the full genes encoding eGFP–α-CbTx and eGFP–EK-α-CbTx were generated by PCR amplification are listed in Table 1. Correct cloning of the genes was confirmed by sequencing.

TABLE 1. List of oligonucleotides used for eGFP–α-CbTx and eGFP–EK-α-CbTx gene construction.
Escherichia coli BL21 (DE3) cells transformed with pET-28a-eGFP–α-CbTx and pET-28a-eGFP–EK-α-CbTx were cultured at 37°C in LB medium in the presence of 50 ng/ml kanamycin to the mid-log phase. Expression of the fluorescent chimera gene was induced by 0.5 mM IPTG and the culture was further incubated at 25°C for 20 h. Cells were harvested by centrifugation, disrupted by sonication, and the chimeric proteins were purified from the soluble fraction by affinity chromatography on a TALON Superflow resin (Clontech) following the manufacturer’s protocol. Further purification was performed by size-exclusion chromatography on a TSK 2000SW column (7.5 × 600 mm, 12.5 nm pore size, 10 μm particle size; Toyo Soda Manufacturing) in phosphate-buffered saline (PBS), pH 7.4, at a flow rate of 0.5 ml/min. Expression and purification of the target proteins was monitored by SDS-PAGE. Concentration of the final eGFP–α-CbTx and eGFP–EK-α-CbTx preparations was measured by absorption spectroscopy using ε (489 nm) = 55,000 M−1cm−1.
eGFP–EK-α-CbTx chimera produced in bacteria was subjected to treatment by enteropeptidase. The recombinant protein was dissolved in 50 mM Tris-HCl (pH 8.0) to a concentration of 1 mg/ml. Protein cleavage with human enteropeptidase light chain (1 U of enzyme per 1 mg of substrate) was performed overnight (16 h) at 37°C. The mixture was separated by reversed-phase HPLC on a Jupiter C5 column (4.6 × 250 mm; Phenomenex) in a linear gradient of acetonitrile concentration (0–60% in 60 min) in the presence of 0.1% trifluoroacetic acid, at a flow rate of 1 ml/min. Purified recombinant α-CbTx was assessed by MALDI mass-spectrometry.
Ultraflex TOF-TOF (Bruker Daltonik, Germany) spectrometer was used for MALDI mass spectrometry as described previously (Kuzmenkov et al., 2016b). 2,5-Dihydroxybenzoic acid (Sigma-Aldrich) was used as a matrix. Measurements were performed in the linear mode. Mass spectra were analyzed with the Data Analysis 4.3 and Data Analysis Viewer 4.3 software (Bruker).
High-resolution mass spectra of activated p-HOBDI-BF2 were recorded on a TripleTOF 5600+ System (SCIEX) using electrospray ionization (ESI). The measurements were done in a positive ion mode (interface capillary voltage of 5500 V); mass range from m/z 50 to m/z 3,000; external or internal calibration was done with the ESI Tuning Mix (Agilent). A syringe injection was used for solutions in acetonitrile, methanol, or water (flow rate of 20 μl/min). Nitrogen was applied as a dry gas; interface temperature was set at 180°C.
ESI mass spectra of Aze fragments were recorded on an LTQ Orbitrap XL instrument (Thermo Fisher Scientific) equipped with HESI-II ion source. The acquisition was performed using full-scan FTMS mode at 30K resolution in a positive ions detection mode within the 700–2000 mass range.
Kieselgel 60 (Merck, Germany) was used for column chromatography. Thin-layer chromatography was performed on silica gel 60 F254 glass-backed plates (Merck). Visualization was performed using UV light (254 or 312 nm) and staining with KMnO4. NMR spectra were recorded on a 700 MHz Bruker Avance III instrument at 303 K. Chemical shifts are reported relative to residual peaks of DMSO-d6 (2.51 ppm for 1H and 39.5 ppm for 13C). Melting points were measured on a Stuart SMP30 apparatus (Cole-Parmer) without correction. IUPAC compound names were generated using ChemDraw software (PerkinElmer).
1.0 mmol of (Z)-4-(4-(2-(difluoroboryl)-4-hydroxybenzylidene)-2-methyl-5-oxo-4,5-dihydro-1H-imidazol-1-yl)butanoic acid (Frizler et al., 2013) was dissolved in dry THF (30 ml), and N,N,N′,N′-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate (330 mg, 1.1 mmol) and triethylamine (120 mg, 1.2 mmol) were added. The mixture was stirred at room temperature for 1 h. Hydrofluoric acid (water solution, 48%, 0.5 ml) was added and the mixture was stirred for additional 10 min. The mixture was dissolved by EtOAc (150 ml), washed by brine (3 × 30 ml), dried over Na2SO4, and evaporated. The product (N-hydroxysuccinimide ester of p-HOBDI-BF2, p-HOBDI-BF2-OSu) was purified by flash chromatography (using EtOAc:THF = 2:1 mixture as an eluent): yellow solid, 370 mg (85%), m. p. = 163–165°С.
1H NMR (DMSO-d6) δ 10.19 (s, 1H), 7.55 (s, 1H), 7.49 (d, J = 8.1Hz, 1H), 7.00 (d, J = 2.5Hz, 1H), 6.74 (dd, J1 = 8.2Hz, J2 = 2.5Hz, 1H), 3.80 (t, J = 7.27Hz, 2H), 2.85 (t, J = 7.48Hz, 2H), 2.82 (br.s, 4H), 2.74 (s, 3H), 1.99 (p, J = 7.48Hz, 2H).
13C NMR (DMSO-d6) δ 12.65 (CH3), 22.90 (CH2), 25.40 (2×CH2), 27.52, 39.88, 115.12 (CH), 118.48 (CH), 124.00, 124.87, 129.25 (CH), 134.27 (CH), 161.41, 162.93, 164.18, 168.51, 170.11.
High resolution mass spectrometry (ESI) [M + H]+ m/z: 434.1341 found; 434.1335 calculated for C19H19BF2N3O6.
α-CbTx was modified with p-HOBDI-BF2-OSu and the derivatives were purified exactly under conditions used earlier for the preparation of α-CbTx mono-labeled derivatives (Utkin et al., 1998). To obtain Aze derivatives, 5.3 mg of Aze were dissolved in 1 ml of 0.2 M sodium phosphate buffer (pH 8.2) and 1.1 mg of p-HOBDI-BF2-OSu in 100 µl of dioxane was added to the toxin solution. The mixture was incubated overnight at room temperature and desalted on a Sephadex G-10 column (1 × 90 cm) equilibrated in 0.1 M acetic acid. After freeze-drying the mixture was separated by reversed-phase HPLC on a Jupiter C18 column (10 × 250 mm; Phenomenex) in a linear gradient of acetonitrile (15–30% in 75 min) in the presence of 0.1% trifluoroacetic acid, at a flow rate of 2 ml/min. After freeze-drying, the obtained derivatives (HOBDI-BF2–Aze) were used for further studies.
Purified HOBDI-BF2–Aze conjugate was dissolved in 50 mM ammonium bicarbonate solution (pH 9.0) to a final concentration of 1 mg/ml, then subjected to digestion by chymotrypsin or Lys-C protease (1 : 25, enzyme: substrate, w/w) at 37°C for 24 h, followed by formic acid quenching (1% by vol). The digested samples were analyzed by LC-MS on a YMC-Triart C18 column (2.1 × 150 mm; YMC) in a linear gradient of acetonitrile (5–55%) at flow rate of 0.3 ml/min. The modified peptide fragments were detected by specific dye absorption at 415 nm.
In competition experiments with 125I-α-BgTx (500 Ci/mmol), all tested compounds (α-CbTx, HOBDI-BF2–α-CbTx, eGFP–α-CbTx, eGFP, Aze, and HOBDI-BF2–Aze derivatives) at different concentrations were pre-incubated for 2.5 h at room temperature with the great pond snail Lymnaea stagnalis acetylcholine binding protein (AChBP) (at the final concentration of 2.5 nM), or with GH4C1 cells (final concentration of 0.4 nM of toxin-binding sites), or Torpedo californica electric organ membranes (final concentration of 0.5 nM of toxin-binding sites) in 50 μL of binding buffer (20 mM Tris-HCl, 1 mg/ml bovine serum albumin (BSA), pH 7.5). After that 125I-α-BgTx was added to AChBP, GH4C1 cells or membranes to a final concentration of 0.5 nM, and the mixtures were additionally incubated for 5 min. Binding was stopped by rapid filtration on double DE-81 filters (Whatman) pre-soaked in the binding buffer (for AChBP) or on GF/C filters (Whatman) pre-soaked in 0.25% polyethylenimine (for GH4C1 cells or membranes), unbound radioactivity being removed from the filters by washout (3 × 3 ml) with the binding buffer. Non-specific binding was determined in all cases using 2.5 h pre-incubation with 10 μM α-CbTx. The binding results were analyzed using OriginPro 2015 program (OriginLab Corporation) fitting to a one-site dose-response curve with the following equation:
For the microscale thermophoresis (MST) experiments, both the fluorescent ligands (HOBDI-BF2–α-CbTx or eGFP–α-CbTx) and AChBP were diluted in PBS, pH 7.2, containing 0.05% Tween. Then, glass capillaries (NanoTemper Technologies, Germany) were filled with different concentrations of AChBP and with the same concentration of the fluorescent ligand. The MST experiments were performed with Monolith NT.115 (NanoTemper Technologies, Germany) according to the manufacturer’s recommendations. Before each experiment, a pretest was carried out to check the fluorophore’s stability and exclude the sorption of the fluorescent molecule on the capillary or aggregation during thermophoresis. Notably, no aggregation or adsorption was observed for either HOBDI-BF2–α-CbTx or eGFP–α-CbTx. The MST experiment was carried out at 40% MST power using a green light emitting diode for excitation. MST data were analyzed and binding parameters were calculated using MO. Affinity Analysis 2 Software (NanoTemper Technologies, Germany). Data points were fitted to a Kd model equation according to built-in software recommendations.
One day before the transfection, Neuro-2a cells were applied to coverslips and placed inside 35-mm cell culture dishes with 2 ml of Eagle’s medium. Transfection medium contained cDNA of human α7 nAChR (3 mg/ml) and the Lipofectamine 2000 transfection reagent (Invitrogen, Thermo Fisher Scientific). The cDNA-containing solution was replaced by cell culture medium 6–8 h after the transfection.
Cryostat 10 μm-thick sections of rat tongue were fixed with isopropanol for 10 min at 4°C, rinsed with PBS and distilled water, air dried for 1 h (all further procedures were carried out at room temperature) and incubated for 1 h with PBS, pH 7.4, containing 10 mg/ml BSA and 5 ml/L Tween 20 to block unspecific binding of toxins. The sections were pre-incubated for 1–2 h in buffer A (1 mg/ml BSA and 150 mM NaCl in PBS). Controls were run simultaneously by adding 300-fold excess of unlabeled α-CbTx (unless specified otherwise). Then, AF555–α-BgTx was added to the slides to reach 50 nM final concentration and they were incubated for 1–15 h. Sections were subsequently washed with PBS, fixed with 40 mg/ml paraformaldehyde for 10 min, rinsed with PBS again and coverslipped in carbonate-buffered glycerol at pH 8.6. The slides were analyzed using CellA Imaging Software (Olympus Soft Imaging Solutions, Germany) coupled to epifluorescent microscope with cooled CCD CAM-XM10 (Olympus, Japan).
For differentiation of THP-1 into macrophages (THP-1 Mϕ), cells were seeded in 24-well plates and treated with 100 nM phorbol 12-myristate 13-acetate (Sigma-Aldrich) for 48 h. Сells were stained as previously described (Siniavin et al., 2020). Briefly, THP-1 Mϕ, Raji, or RPMI 1788 cells were incubated with 100 nM HOBDI-BF2–α-CbTx, eGFP–α-CbTx, eGFP, or AF647–α-BgTx (1 h, at 4°C). Cells were then washed two times with PBS and assayed using BD FACSCalibur or MACSQuant Analyzer 10 flow cytometer (Miltenyi Biotec, Germany) and analyzed using FlowJo software 10.0.8. To determine non-specific binding, cells were pre-incubated with 10 µM α-CbTx before fluorescent toxin staining.
A model of eGFP–α-CbTx was constructed using UCSF Chimera 1.10.1 software (Pettersen et al., 2004) for Modeller 9.14 (Webb and Sali, 2016). We utilized eGFP (PDB ID: 2Y0G) and α-CbTx (PDB ID: 1CTX) spatial structures as templates. The N-terminal site of chimeric protein and the (G4S)3-linker between the eGFP and α-CbTx modules were represented in a disordered conformation. A model of eGFP–α-CbTx was visualized by the PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC.
We designed an expression cassette encoding a three-finger toxin fused with a fluorescent protein via a flexible linker (Figure 1A). The well-studied α-CbTx (UniProt ID: P01391) from the venom of N. kaouthia (Karlsson et al., 1972) was selected because this protein is highly active towards several targets including muscle-type, neuronal α7 and α9/α10 nAChRs (Chandna et al., 2019), as well as AChBP from L. stagnalis (Hansen et al., 2002); α-CbTx binds to these targets with sub-nanomolar affinities. Additionally, it was shown that α-CbTx is able to inhibit ionotropic γ-aminobutyric acid receptors (GABAA) at sub-micromolar concentrations (Kudryavtsev et al., 2015). As a fluorescent module, we selected one of the best-studied fluorescent proteins eGFP (FPbase ID: R9NL8) (Cormack et al., 1996) which is characterized by high brightness and weak dimerization properties.
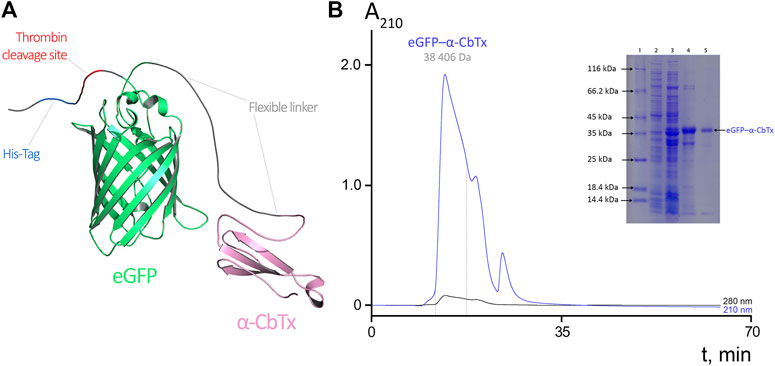
FIGURE 1. (A) Molecular model of eGFP–α-CbTx chimeric protein. eGFP and α-CbTx modules are connected by a short flexible linker. The His-Tag and adjacent thrombin cleavage site are located at the N-terminus of the chimera. (B) Size-exclusion chromatography of eGFP–α-CbTx. The dotted lines mark the area corresponding to the collected fraction that contained the chimeric protein. Detection was performed at 210 and 280 nm. The inset shows electrophoretic separation of fractions obtained during the purification of eGFP–α-CbTx in 12% polyacrylamide gel. 1, standards (Pierce Unstained Protein MW Marker, #26610, Thermo Fisher Scientific) for protein electrophoresis; 2, soluble fraction of total cellular protein before induction of expression; 3, after induction by IPTG; 4, chimeric protein after metal-chelate affinity chromatography; 5, chimeric protein after size-exclusion chromatography.
The eGFP–α-CbTx chimera was produced recombinantly, isolated from E. coli lysate by affinity chromatography, and purified by size-exclusion chromatography (Figure 1B). Fluorescence excitation and emission spectra of this hybrid were found identical to those of native eGFP protein (excitation and emission maxima were at 488 and 507 nm, respectively). This fact indicates that the spectral properties of the fluorescent protein did not change when it was fused with α-CbTx. The yield of recombinant eGFP–α-CbTx was ∼10 mg per 1 L of bacterial culture; the purity of the final product was not less than 95%.
To obtain p-HOBDI-BF2-labeled toxins, they were reacted with p-HOBDI-BF2-OSu (Figure 2) using conditions elaborated earlier for α-CbTx modification by activated esters of photoactivatable labels. The modified α-CbTx was purified using high-performance ion-exchange and reversed-phase chromatography as described earlier (Utkin et al., 1998), and similarly one predominant derivative (HOBDI-BF2–α-CbTx) was obtained. This derivative was analyzed by high-resolution mass spectrometry, which showed that it possessed a molecular mass of 8,112.76 Da. This value was 19.92 Da lower than the theoretical mass; the difference is explained by the loss of HF from fluorine-containing label under mass spectroscopy conditions, this phenomenon was earlier described for the BODIPY label (Qi et al., 2015). Peptide mass fingerprinting using trypsin digestion revealed the labeled fragment 13–33 (data not shown), which indicates that α-CbTx derivative contains the label at Lys23.
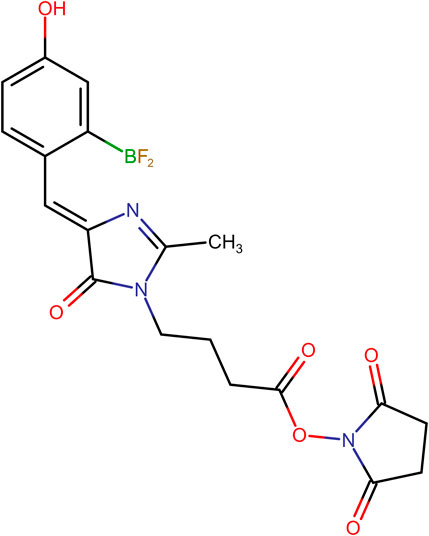
FIGURE 2. Chemical structure of (Z)-2,5-dioxopyrrolidin-1-yl 4-(4-(2-(difluoroboryl)-4-hydroxybenzylidene)-2-methyl-5-oxo-4,5-dihydro-1H-imidazol-1-yl)butanoate (p-HOBDI-BF2-OSu).
A similar technique was applied to introduce the p-HOBDI-BF2 label into the molecule of amidated Aze (see structure in Figure 3A). The Aze derivatives were purified by reversed-phase HPLC (Figure 3A) and analyzed by mass spectrometry. It was found that fraction 2 with the molecular mass of 2,539 Da represents unmodified toxin. Fraction 3 contains a di-labeled derivative (3,132 Da) contaminated with unmodified toxin. Fractions 4–6 contain different mono-modified products (HOBDI-BF2–Aze) with molecular masses of 2,836 Da, fraction 6 being contaminated with a di-labeled product. Interestingly, the mass corresponding to the loss of HF was also observed. As there are 3 amino groups (N-terminal and ε-amino groups of Lys6 and Lys20) in Aze, our results show that three corresponding mono-labeled derivatives were obtained.
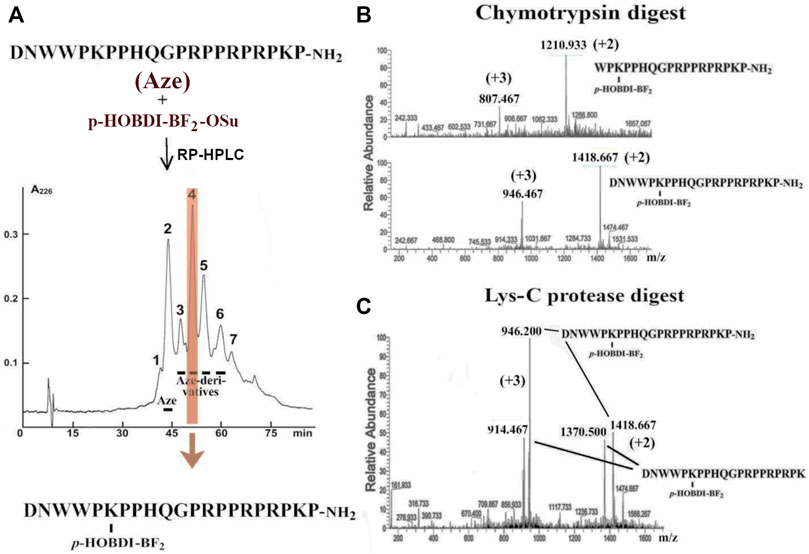
FIGURE 3. (A) Preparation of Aze derivatives. Aze was reacted with p-HOBDI-BF2-OSu and the obtained derivatives were separated by reversed-phase HPLC on a Jupiter C18 column (10 × 250 mm) in a linear gradient of acetonitrile (15–30% in 75 min). Peak 4 (shaded in orange) was found as the best purified individual mono-derivative (HOBDI-BF2–Aze) with localization of the photolabel at the ε-amino group of Lys6, as established by chromato-mass spectrometry of its fragments after digestion with chymotrypsin or Lys-C proteinase. (B) Chromato-mass spectrometry analysis of peak 4 digested with chymotrypsin revealed the fragments with the measured m/z values 1,418.667 (+2) and 1,210.933 (+2) or 946.467 (+3) and 807.467 (+3) Da. They correspond to the masses of the non-digested HOBDI-BF2–Aze and the fragment Aze (4–21) with p-HOBDI-BF2 label. (C) Chromato-mass spectrometry analysis of peak 4 digested with Lys-C protease. Identified m/z values 1,418.667 (+2) and 1,370.5 (+2), or 946.2 (+3) and 914.487 (+3) Da, correspond to the non-digested HOBDI-BF2–Aze and the reaction product Aze(1–20) with p-HOBDI-BF2 label.
According to chromato-mass spectrometry analysis, among all these fractions only peak 4 was an individual compound (data not shown). To localize the fluorophore in this mono-derivative, it was digested with two proteases, and the obtained fragments were analyzed by mass spectrometry. The digestion of this derivative with chymotrypsin resulted in a fragment with molecular mass of 2,420 Da containing the fluorescent label (Figure 3B). This product corresponds to the C-terminal fragment 4–21 of HOBDI-BF2–Aze, which indicates that the N-terminal amino group is not modified. To find out which of the two lysine residues (Lys6 or Lys20) is modified, the derivative was digested with Lys-C endoproteinase, and the obtained products were analyzed by mass spectrometry. A peptide with molecular mass of 2,740 Da was found (Figure 3C). It corresponds to the N-terminal fragment 1–20 containing the fluorescent label, suggesting that the fluorophore is attached to Lys6 (see structure in Figure 3A).
We evaluated the affinity of the prepared fluorescent α-CbTx derivatives to the muscle-type nAChR from T. californica ray electric organ and neuronal human α7 nAChR expressed in GH4C1 cells, as well as to AChBP from L. stagnalis, which is a homolog of the ligand-binding domain of all nAChRs. This was done by measuring their ability to compete with 125I-α-BgTx used as a radioligand. When testing fluorescent α-CbTx (eGFP–α-CbTx and HOBDI-BF2–α-CbTx), α-CbTx and eGFP were also used in parallel experiments. The respective inhibition curves on muscle-type and α7 nAChRs, and AChBP are shown in Figures 4A–C and corresponding IC50 values and slopes (Hill coefficients) are presented in Table 2.
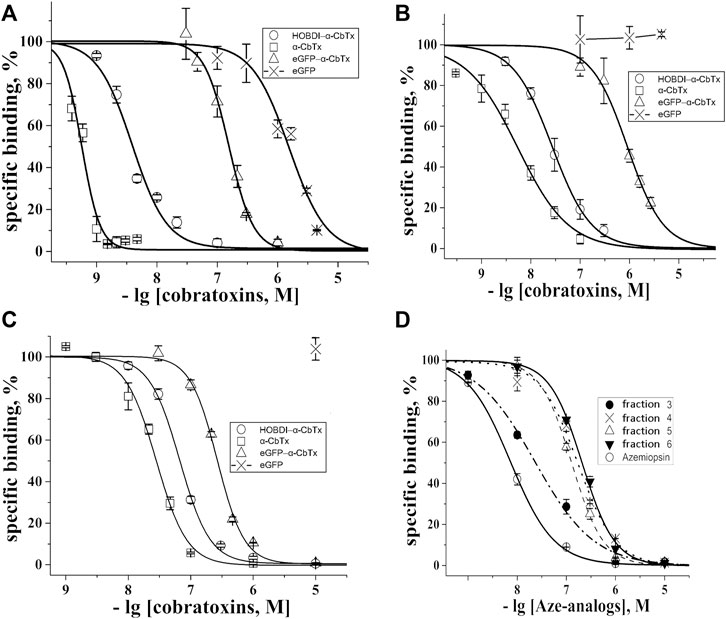
FIGURE 4. Inhibition of the initial rate for 125I-α-BgTx binding to (A) T. californica nAChR, (B) human α7 nAChR and (C) L. stagnalis AChBP with α-CbTx and its fluorescent analogs (eGFP–α-CbTx, HOBDI-BF2–α-CbTx) as well as eGFP. (D) Inhibition of initial rate for 125I-α-BgTx binding to T. californica nAChR with Aze and its p-HOBDI-BF2 derivatives from fractions 3–6 (see Figure 3A). Each point is a mean ± S.E.M. value of three measurements for each concentration. The curves were calculated from the means ± S.E.M. using OriginPro 2015, and the respective IC50 values in nM are presented in Table 2.
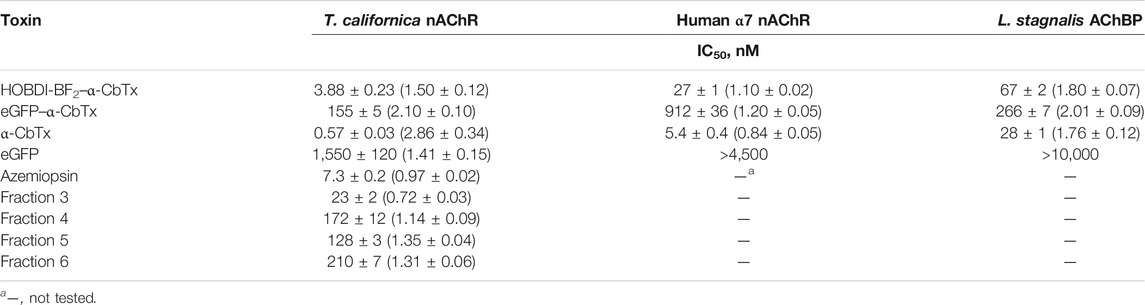
TABLE 2. Affinity of the fluorescent toxins tested in competition with125I-α-BgTx for binding to muscle-type nAChR from T. californica, human neuronal α7 nAChR, and AChBP from L. stagnalis calculated from the respective inhibition curves in Figure 4 as the IC50 values and Hill coefficients (nH) using OriginPro 2015.
It was found that modification of α-CbTx with eGFP leads to a decrease in affinity to all three targets, whereas the incorporation of the small fluorophore HOBDI-BF2 has a significantly weaker effect. When p-HOBDI-BF2 label was inserted, the affinity of the derivative towards muscle-type and neuronal α7 nAChRs, and AChBP was 3.88 ± 0.23, 27 ± 1 and 67 ± 2 nM, respectively, i.e., it decreased 6.8, 5 and 2.4 times (Figures 4A–C; Table 2). The corresponding IC50 values for the chimeric eGFP–α-CbTx were substantially higher and reached 155 ± 5, 912 ± 36, and 266 ± 7 nM (Table 2) demonstrating the decrease in affinity 272, 169 and 9.5 times for muscle-type, neuronal α7 nAChRs, and AChBP, respectively. Interestingly, the eGFP itself showed the ability to compete with 125I-α-BgTx for binding to the nAChR from T. californica with IC50 = 1,550 ± 120 nM (Figure 4A), but when the N-terminal His-tag was removed from eGFP, this inhibitory activity disappeared (data not shown).
In order to figure out the reason for such a noticeable drop in affinity of eGFP–α-CbTx, α-CbTx was cleaved from the chimeric fusion protein eGFP–EK-α-CbTx and purified by reversed-phase HPLC. The affinity of purified recombinant α-CbTx towards muscle-type nAChR was markedly lower than that of the native α-CbTx (IC50 value was >10 nM vs 0.57 ± 0.03 nM, respectively). This result suggests that recombinant α-CbTx in the chimeric eGFP–α-CbTx protein does not fold properly.
A competitive radioligand analysis was also used to evaluate the affinity of the obtained fluorescent Aze derivatives from fractions 3–6 (Figure 3A) towards the muscle-type T. californica nAChR. The inhibition curves and the corresponding IC50 values are shown in Figure 4D and Table 2. The affinity of the derivatives from fractions 4–6 decreased after modification by more than an order of magnitude (IC50 of 170 ± 50 nM compared to IC50 = 7.3 ± 0.2 nM for unmodified Aze). Fraction 3 showed a markedly higher affinity (IC50 = 23 ± 2 nM), most likely due to the presence of an unmodified toxin detected by mass spectrometric analysis (see above). The small differences in affinity of mono-labeled derivatives from fractions 4–6 probably reflect the similar importance of modified amino groups for binding to the receptor.
Interaction of eGFP–α-CbTx and HOBDI-BF2–α-CbTx with L. stagnalis AChBP was further characterized using MST. To generate a binding curve, we used 70 nM eGFP–α-CbTx and 100 nM HOBDI-BF2–α-CbTx, whereas the concentration of AChBP varied from 60 pM to 1 µM. First, the parameters of MST were adjusted to achieve a substantial initial fluorescence. It was found that eGFP–α-CbTx fluorescence is roughly an order of magnitude brighter than HOBDI-BF2–α-CbTx, so the excitation power values of 20% for eGFP–α-CbTx and 80% for HOBDI-BF2–α-CbTx were used in the subsequent experiments. Pre-MST scan of the capillaries revealed that HOBDI-BF2–α-CbTx fluorescence changes significantly upon binding to AChBP (Figure 5A), which allowed a pre-MST estimation of Kd ≈ 200 nM. Such changes in initial fluorescence were not observed for eGFP–α-CbTx. Then, the MST experiment was performed and the resulting data points (Figure 5B) were fitted with a Kd model to generate binding curves for eGFP–α-CbTx and HOBDI-BF2–α-CbTx (Figure 5D). Dissociation constants were calculated: 7 ± 6 nM for eGFP–α-CbTx and 194 ± 89 nM for HOBDI-BF2–α-CbTx. Thus, the MST experiment demonstrated a stronger binding of eGFP–α-CbTx to AChBP compared to HOBDI-BF2–α-CbTx.
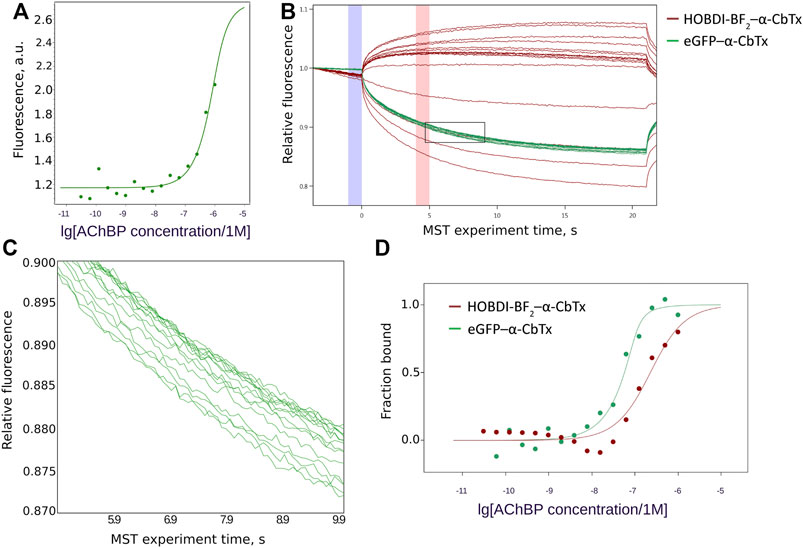
FIGURE 5. HOBDI-BF2–α-CbTx and eGFP–α-CbTx binding to L. stagnalis AChBP. (A) Changes in initial fluorescence intensity of HOBDI-BF2–α-CbTx dependent on AChBP concentration. (B) MST traces showing the change of fluorescence over time during the MST experiment. For both HOBDI-BF2–α-CbTx (red) and eGFP–α-CbTx (green) a set of MST curves was obtained, each representing a single MST measurement for every concentration of L. stagnalis AChBP. Binding curves were calculated from fluorescence values at 5 s (red column on the graph) corrected for fluorescence at 0 time point (blue column on the graph). The region of eGFP–α-CbTx curves enclosed by a rectangle is shown enlarged in the next panel. (C) Enlarged region of eGFP–α-CbTx MST traces showing the signal dependence on AChBP concentration. (D) Binding curves of eGFP–α-CbTx (70 nM) and HOBDI-BF2–α-CbTx (100 nM) to AChBP (60 pM–1 µM). The bound fraction of fluorescent target dependent on ligand (AChBP) concentration is presented.
Fluorescence microscopy of Neuro-2a cells transfected with human α7 nAChR, and rat tongue preparation containing muscle fiber end plates with muscle α1β1εδ nAChR, revealed a difference in applicability of the HOBDI-BF2 and eGFP derivatives of α-CbTx. Control commercial AF555–α-BgTx (50 nM) shows bright staining of Neuro-2a cells and rat tongue end plates (Figure 6A). Both eGFP–α-CbTx chimeric protein (500 nM) and HOBDI-BF2–α-CbTx (100 nM) bind to Neuro-2a cells transfected with human α7 nAChR. This staining is abolished by 1 h preincubation with a 30–50 molar excess of α-CbTx, confirming the specificity of binding (Figure 6A). eGFP alone does not stain cells (Figure 6A), thus confirming that the staining could not be attributed to a non-specific uptake of the fluorescent protein.
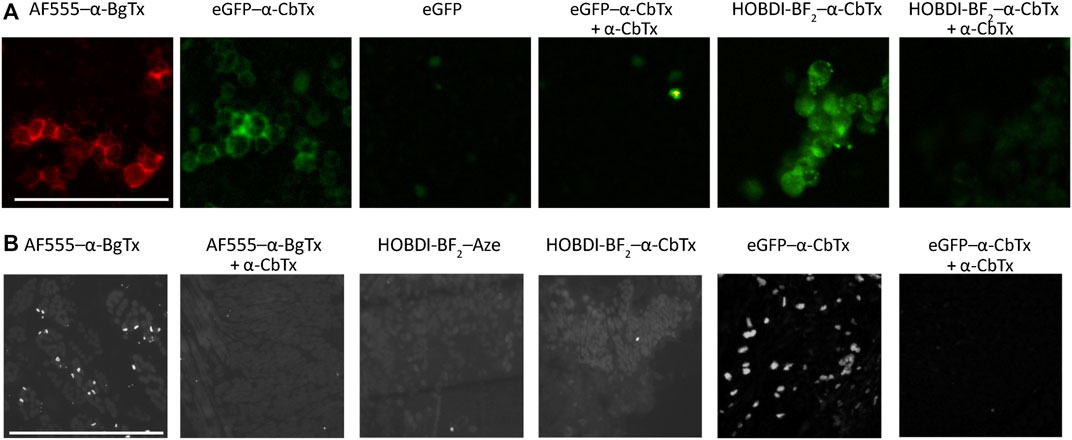
FIGURE 6. Fluorescence microscopy of Neuro-2a cells transfected with human α7 nAChR, and rat tongue preparation containing muscle fiber end plates with muscle α1β1εδ nAChR. (A) Control staining of Neuro-2a cells with 50 nM AF555–α-BgTx shows peripheral staining of the cells, which is reproduced by the eGFP–α-CbTx chimeric protein (500 nM). eGFP alone does not stain the cells. Complementary to that, specific eGFP–α-CbTx staining is abolished by 1 h preincubation with 15 µM α-CbTx. HOBDI-BF2–α-CbTx (100 nM) also stains Neuro-2a cells, and it is prevented by 1 h pre-treatment using 5 µM α-CbTx. Scale bar in the first panel, 50 μm. (B) AF555–α-BgTx (100 nM) specifically stains end plates in rat tongue cross-sections, and no staining is observed in cross-sections pre-treated with 5 µM α-CbTx. Neither HOBDI-BF2–α-CbTx, nor HOBDI-BF2–Aze produce end plate staining, whereas 500 nM eGFP–α-CbTx chimeric protein (12 h of incubation) stained cross-sections brightly. End-plate staining by eGFP–α-CbTx chimeric protein is prevented by 1 h pre-treatment with 15 µM α-CbTx. Scale bar in the first panel, 50 μm.
eGFP–α-CbTx stained rat tongue end plates brightly and specifically (Figure 6B). Surprisingly, we failed to achieve end-plate staining with HOBDI-BF2–α-CbTx. We also tried to stain end plates with HOBDI-BF2–Aze, however, no end-plate staining was detected in this case either (Figure 6B). It is worth noting that rat tongue preparations are made using denaturating conditions (isopropyl alcohol). The absence of specific staining of rat tongue cross-sections by the HOBDI-BF2 derivatives of neurotoxins could be explained by the intrinsic properties of this fluorophore preventing it from binding to partially denaturated muscle nAChR. On the other hand, the absence of staining by HOBDI-BF2–α-CbTx or HOBDI-BF2–Aze could be attributed to their fast wash-out kinetics, which might be due to the modification of lysine side chains crucial to the binding.
In flow cytometry experiments, we visualized the binding of eGFP–α-CbTx and HOBDI-BF2–α-CbTx to different transformed immune cells, such as Raji, RPMI 1788, and THP-1 Mϕ, expressing α7 nAChR (Figure 7). It was revealed that 100 nM eGFP–α-CbTx stained THP-1 Mϕ cells (Figure 7A). However, the average fraction of stained cells was only 9% (Figure 7C). Pre-incubation of the cells with an excess of free α-CbTx led to a decrease in the percentage of eGFP positive cells (eGFP+) (∼10.2 vs 6.4%; Figure 7A, central panels). Staining of cells with 500 nM eGFP–α-CbTx gave similar results (data not shown). Free eGFP also stained cells (∼3.9% eGFP+; Figure 7A, left panel, bottom row) indicating partial nonspecific binding. In contrast, HOBDI-BF2–α-CbTx did not stain the cells, and the level of HOBDI-BF2+ cells did not differ from autofluorescence (auto; 0.26 vs 0.29%; Figure 7A, left and right panels, top row). AF647–α-BgTx, which was used as a control, stained THP-1 Mϕ cells ∼7 times better than eGFP–α-CbTx (∼63.1% positive cells; Figure 7A, right panel, bottom row, and Figure 7C).
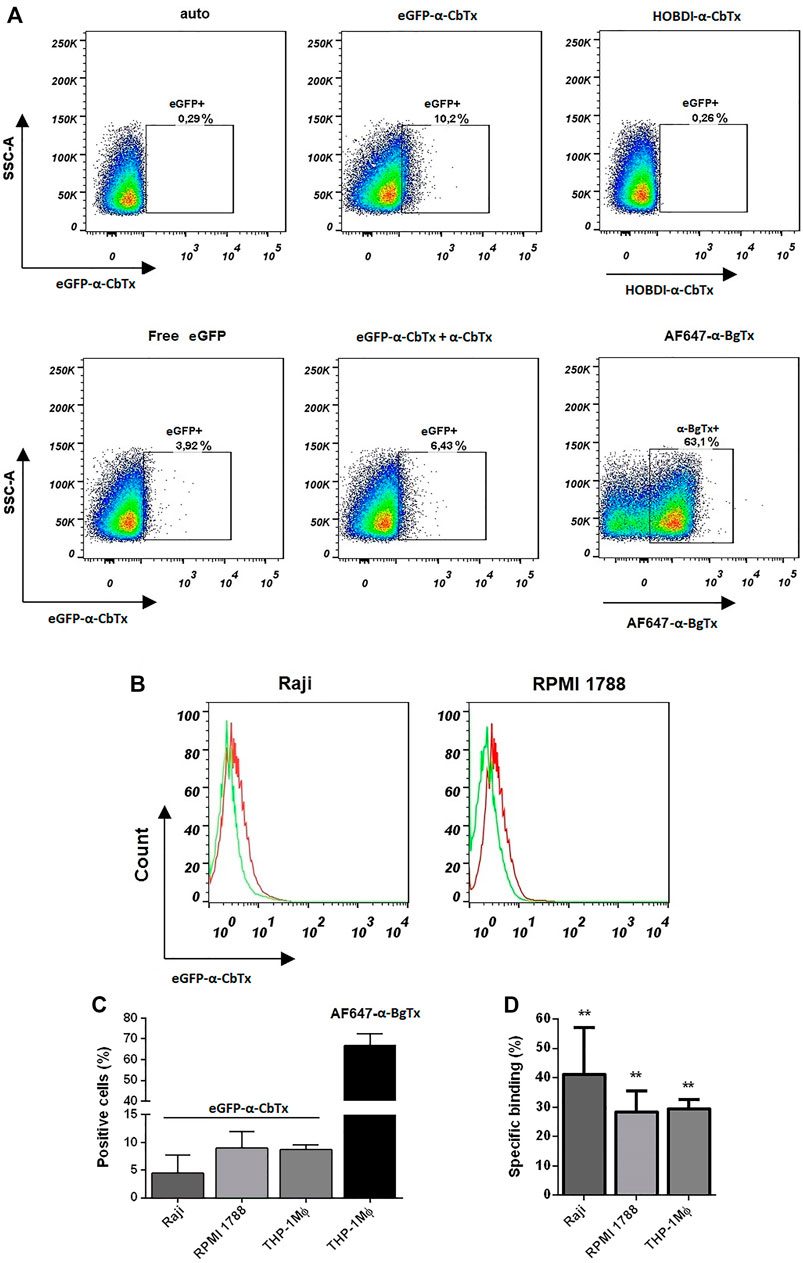
FIGURE 7. Investigation of cell binding and staining using fluorescent α-CbTx analogs by flow cytometry. (A) Representative dot plots of stained THP-1 Mϕ cells using two fluorescent toxins, i.e. eGFP–α-CbTx and HOBDI-BF2–α-CbTx. In the top row the autofluorescence, cells stained with eGFP–α-CbTx and HOBDI-BF2–α-CbTx, are shown. In the bottom row cells stained with free eGFP, eGFP–α-CbTx with excess of α-CbTx and cells stained with AF647–α-BgTx as a control, are shown. Regions in dot plots represent the percentage of stained cells. (B) Representative histograms of Raji or RPMI 1788 cells stained with eGFP–α-CbTx. Red histogram, cells stained with eGFP–α-CbTx; green histogram, cells pre-incubated with an excess of free α-CbTx and then stained with eGFP–α-CbTx. A decrease in the fluorescence of cells due to the binding of free α-CbTx is revealed. (C) Diagrams showing the number of positive cells (Raji, RPMI 1788, and THP-1 Mϕ) when stained with eGFP–α-CbTx or AF647–α-BgTx. (D) Diagrams of specific binding (%) of eGFP–α-CbTx on different cells from three independent experiments (mean ± S.E.M.)). One-way ANOVA tests with Tukey post hoc test (**p < 0.01) were used.
Next, we performed binding assays using eGFP–α-CbTx to measure the surface expression of α7 nAChR levels on Raji and RPMI 1788 cells. Figure 7B shows that eGFP–α-CbTx can stain both cell lines (red histograms), and this binding is specific because it decreased after pre-incubation with an excess of free α-CbTx (green histograms). However, again the calculated average values for Raji and RPMI 1788 cells labeled with fluorescent toxin (positive cells) were only ∼5 and 9%, respectively (Figure 7C). The average values of specific binding of eGFP–α-CbTx for Raji, RPMI 1788, and THP-1 Mϕ cells were 41, 28, and 30%, respectively (Figure 7D). Flow cytometry measurements showed that eGFP–α-CbTx specifically stains cell lines that express α7 nAChR (Figure 7C). On the other hand, HOBDI-BF2–α-CbTx was found unsuitable for this purpose.
Studies of ligand-receptor interactions require adequate molecular instruments and methods. Radioligand analysis was the method of choice for such studies for a long time. However, this method has limitations related to safety and ecological aspects. Fluorescence-based methods became more convenient and popular due to the development of new approaches including super-resolution techniques such as Photoactivated Localization Microscopy (PALM) and Stochastic Optical Resolution Microscopy (STORM) and highly fluorescent labels (e.g., quantum dots) (see reviews (Baddeley and Bewersdorf, 2018; Himmelstoß and Hirsch, 2019)). Among the developed methodologies is the application of fluorescent proteins (GFP and analogs) for the labeling. Modern fluorescent proteins cover a wide range of excitation and emission wavelengths and possess high quantum yields (Chudakov et al., 2010). Recently, we proposed a novel approach for toxin labeling, which is based on the production of chimeric proteins consisting of two modules: a fluorescent protein and a polypeptide toxin specific for certain channels where the ligand part serves for selective channel recognition, and the fluorescent part is used for effective detection. Chimeric fusion proteins comprising GFP (or analogs) and the protein ligand under the study can be easily produced recombinantly in high quantities. Relying on this approach we produced a chimera (eGFP-OSK1) for potassium channel recognition (Kuzmenkov et al., 2016a). Further, this approach was also applied for KV1.3-specific chimera production due to clinical importance of this isoform (Nekrasova et al., 2020).
The preparation of fluorescently labeled proteins and peptides by chemical modification is another frequently used method for which appropriate reagents have long been developed. N-Hydroxysuccinimide esters of respective fluorophores are most often used, resulting in modification of amino groups (the N-terminal α-amino group or ε-amino groups of lysine residues). This method allows introducing labels into different parts of the molecule, which is not always possible with fluorescent proteins. However, sometimes the introduction of large fluorophores into peptides or small proteins can lead to a loss of activity. Nevertheless, fluorescently labeled toxins were successfully used to study toxin-receptor interactions. We were among the first in application of fluorescently labeled TFTs for the investigation of nAChRs (Tsetlin et al., 1979, 1982; Surin et al., 1983) and we continue to successfully use them for this purpose until now (Shelukhina et al., 2009; Pivovarov et al., 2020). The first studies showed the preferential modification of the lysine in the central loop of TFTs, which did not lead to a significant change in the affinity to the receptor, and allowed to characterize the toxin site involved in the interaction with nAChR.
When introducing bulk moieties into small peptides, a noticeable decrease in affinity, a change in specificity or an increase in non-specific interactions is more often observed, as we demonstrated earlier with the example of some photoactivated derivatives of α-conotoxins MI and GI (Kasheverov et al., 2001, 2006). However, fluorescent derivatives of some α-conotoxins were prepared, which became selective markers of different nAChR subtypes (Hone et al., 2010; Surin et al., 2012; Barloscio et al., 2017). For instance, using a fluorescent derivative of α-conotoxin MII, a selective α6β2 nAChR antagonist, α6-containing nAChRs were localized in the retinal tissue and found to be predominantly localized to the ganglion cell layer (Barloscio et al., 2017).
The introduction of a bulky fluorescent label can result in a decrease of affinity and change of selectivity of toxins. However cryo-EM structure of Torpedo acetylcholine receptor in complex with α-bungarotoxin (Rahman et al., 2020) reveals that both N- and C-termini of TFTs are less important for the interaction with the receptor and can tolerate the attachment of a sizeable substituent. Keeping this in mind we prepared a chimeric protein, in which eGFP was fused with α-CbTx N-terminus through a linker of 19 amino acid residues (Figure 1). In radioligand assays, the chimeric protein with a full-sized GFP (eGFP–α-CbTx) showed a significantly diminished affinity towards all tested targets as compared to α-CbTx. The decrease in affinity for muscle-type T. californica, human α7 nAChR, and L. stagnalis AChBP was ∼272, 169 and 9.5 times (Figures 4A–C; Table 2). To find out the reasons for this discrepancy, the toxin was split from eGFP and purified. The obtained recombinant α-CbTx was substantially less active than the native toxin, leading us to the assumption that an essential part of the recombinant toxin was incorrectly folded.
As mentioned in the Introduction, the synthetic analog of GFP chromophore p-HOBDI-BF2 was prepared earlier (Baranov et al., 2012). This label is similar in many of its spectral characteristics to the well-known dye fluorescein. It has a similar spectrum, close extinction coefficient, and quantum yield of fluorescence. However, compared to fluorescein, HOBDI-BF2 is minute, its molecular mass is 4 times smaller. It is also assumed as more biomimetic since it is based on the GFP chromophore. Thus, the use of this label should affect the properties and behavior of the stained biological objects to a significantly lower degree. To utilize this chromophore for specific amino group labeling in proteins, an activated N-hydroxysuccinimide ester was synthesized. We introduced p-HOBDI-BF2 into α-CbTx, and one predominant fluorescent derivative HOBDI-BF2–α-CbTx was obtained. Compared to native α-CbTx, this derivative showed a reduced affinity to muscle-type T. californica, human α7 nAChR, and L. stagnalis AChBP by ∼6.8, 5.0 and 2.4 times, respectively (Figures 4A–C; Table 2), still retaining a high affinity to all tested targets. Since α-CbTx binds to both muscle-type and some neuronal nAChRs, we also prepared fluorescent derivatives of Aze, a selective antagonist of muscle-type nAChR. p-HOBDI-BF2 introduction into Aze led to the formation of multiple reaction products (mono- and di-derivatives with possible label localization at the α-amino group of Asp1 and ε-amino groups of Lys6 or Lys20; Figure 3A). To identify the exact localization of the label for the best purified derivative (fraction 4), a chromato-mass spectrometrу analysis of its chymotrypsin (Figure 3B) and Lys-C fragments (Figure 3C) was performed. Interestingly, the introduction of a p-HOBDI-BF2 chromophore into a small peptide Aze did not dramatically decrease its affinity to muscle-type nAChR (Figure 4D).
The evaluation of the binding of two fluorescent toxins (eGFP–α-CbTx and HOBDI-BF2–α-CbTx) to AChBP using MST (Figure 5) showed that the affinity of the photoprobes lies in the same concentration range, i.e. 10–200 nM. This result suggests that both methods of obtaining fluorescent products (recombinant and chemical modification) used by us lead to sufficiently effective tools for detecting the respective receptor targets.
Optical microscopy is one of the major fields for the application of fluorescent derivatives of neurotoxins. Both HOBDI-BF2–α-CbTx and eGFP–α-CbTx were applicable for the detection of nAChR in live cell imaging (Figure 6A). eGFP–α-CbTx is able to detect muscle nAChR in a tissue preparation and represents a cheap analog of the chemically modified α-BgTx conventionally utilized for this task (Figure 6B). However, neither HOBDI-BF2–α-CbTx nor HOBDI-BF2–Aze were able to stain histological cross-sections of rat tongue containing muscle nAChR. Their inability to bind to nAChRs that are present in rat tongue preparations could be explained by partial denaturation of the receptor due to isopropyl alcohol treatment. As a result of such treatment some fluorescent toxins retain the ability to bind to muscle nAChR (AF555–α-BgTx, eGFP–α-CbTx), but others lose their affinity. These features of new fluorescent derivatives should be taken into account in experimental design.
In flow cytometry experiments (Figure 7), it was revealed that a very small proportion (<10%) of the three studied cell types (Raji, RPMI 1788, and THP-1 Mϕ) expressing α7 nAChR (Siniavin et al., 2020), is specifically stained by eGFP–α-CbTx. The use of HOBDI-BF2–α-CbTx produced results at the level of autofluorescence (Figure 7A). This is in particular contrast to the commercially available AF647–α-BgTx (Figures 7A,C), which stained ∼60% of cells. Thus, there are applications where our novel derivatives are efficient, but there are numerous cases where traditional readioactive and fluorescent labels retain their role.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Institutional Policy on the Use of Laboratory Animals of the Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry Russian Academy of Sciences (Protocol Number 327/2021).
IK, AK, DK, AV, and YU designed research; IK, AK, DK, AV, and YU analyzed data; IK, AK, DK, AV, and YU performed research; IK, AK, DK, AV, and YU wrote the paper; IC performed gene engineering and chimera production; IS, DK, performed cyto and histochemistry; IK synthesized radioligand and performed radioligand analysis; EB obtained thermophoresis spectra; II synthesized peptides and localized the fluorescent label, AS performed flow-cytometry, RZ measured molecular masses, MB performed fluorescent label synthesis, VT, AV, and YU supervised research.
This study was funded by RFBR, project number 20-54-00033. Fluorescent chimera design and production were supported by RFBR, project number 20-34-70031.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Eli Lilly (United Kingdom) for GH4C1 cells transfected with human α7 nAChR and Prof. Ferdinand Hucho (Free University of Berlin, Berlin, Germany) for T. californica electric organ preparation as well as Prof. Sulan Luo (Hainan University, Haikou, People’s Republic of China) for L. stagnalis AChBP.
Anderson, D. J. (2008). Radioligand Binding Characterization of Neuronal Nicotinic Acetylcholine Receptors. Curr. Protoc. Pharmacol. 43Chapter 1, Unit1.8. doi:10.1002/0471141755.ph0108s43
Anderson, M. J., and Cohen, M. W. (1974). Fluorescent Staining of Acetylcholine Receptors in Vertebrate Skeletal Muscle. J. Physiol. 237, 385–400. doi:10.1113/jphysiol.1974.sp010487
Baddeley, D., and Bewersdorf, J. (2018). Biological Insight From Super-Resolution Microscopy: What We Can Learn From Localization-Based Images. Annu. Rev. Biochem. 87, 965–989. doi:10.1146/annurev-biochem-060815-014801
Baranov, M. S., Lukyanov, K. A., Borissova, A. O., Shamir, J., Kosenkov, D., Slipchenko, L. V., et al. (2012). Conformationally Locked Chromophores as Models of Excited-State Proton Transfer in Fluorescent Proteins. J. Am. Chem. Soc. 134, 6025–6032. doi:10.1021/ja3010144
Barber, C. M., Isbister, G. K., and Hodgson, W. C. (2013). Alpha Neurotoxins. Toxicon. 66, 47–58. doi:10.1016/j.toxicon.2013.01.019
Barloscio, D., Cerri, E., Domenici, L., Longhi, R., Dallanoce, C., Moretti, M., et al. (2017). In Vivo Study of the Role of α6‐Containing Nicotinic Acetylcholine Receptor in Retinal Function Using Subtype‐Specific RDP‐MII(E11R) Toxin. FASEB j. 31, 192–202. doi:10.1096/fj.201600855R
Chandna, R., Tae, H.-S., Seymour, V. A. L., Chathrath, S., Adams, D. J., and Kini, R. M. (2019). Drysdalin, an Antagonist of Nicotinic Acetylcholine Receptors Highlights the Importance of Functional rather Than Structural Conservation of Amino Acid Residues. FASEB bioAdvances. 1, 115–131. doi:10.1096/FBA.1027
Chang, C. C., and Lee, C. Y. (1963). Isolation of Neurotoxins from the Venom of bungarus Multicinctus and Their Modes of Neuromuscular Blocking Action. Arch. Int. Pharmacodyn Ther. 144, 241–257.
Chudakov, D. M., Matz, M. V., Lukyanov, S., and Lukyanov, K. A. (2010). Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues. Physiol. Rev. 90, 1103–1163. doi:10.1152/physrev.00038.2009
Cormack, B. P., Valdivia, R. H., and Falkow, S. (1996). FACS-Optimized Mutants of the Green Fluorescent Protein (GFP). Gene. 173, 33–38. doi:10.1016/0378-1119(95)00685-0
de Weille, J. R., Schweitz, H., Maes, P., Tartar, A., and Lazdunski, M. (1991). Calciseptine, a Peptide Isolated From Black Mamba Venom, Is a Specific Blocker of the L-Type Calcium Channel. Proc. Natl. Acad. Sci. 88, 2437–2440. doi:10.1073/pnas.88.6.2437
Diochot, S., Baron, A., Salinas, M., Douguet, D., Scarzello, S., Dabert-Gay, A.-S., et al. (2012). Black Mamba Venom Peptides Target Acid-Sensing Ion Channels to Abolish Pain. Nature. 490, 552–555. doi:10.1038/nature11494
Frizler, M., Yampolsky, I. V., Baranov, M. S., Stirnberg, M., and Gütschow, M. (2013). Chemical Introduction of the Green Fluorescence: Imaging of Cysteine Cathepsins by an Irreversibly Locked GFP Fluorophore. Org. Biomol. Chem. 11, 5913. doi:10.1039/c3ob41341a
Gasparian, M. E., Ostapchenko, V. G., Schulga, A. A., Dolgikh, D. A., and Kirpichnikov, M. P. (2003). Expression, Purification, and Characterization of Human Enteropeptidase Catalytic Subunit in Escherichia coli. Protein Expr. Purif. 31, 133–139. doi:10.1016/S1046-5928(03)00159-1
Hansen, S. B., Radić, Z., Talley, T. T., Molles, B. E., Deerinck, T., Tsigelny, I., et al. (2002). Tryptophan Fluorescence Reveals Conformational Changes in the Acetylcholine Binding Protein. J. Biol. Chem. 277, 41299–41302. doi:10.1074/JBC.C200462200
Himmelstoß, S. F., and Hirsch, T. (2019). A Critical Comparison of Lanthanide Based Upconversion Nanoparticles to Fluorescent Proteins, Semiconductor Quantum Dots, and Carbon Dots for Use in Optical Sensing and Imaging. Methods Appl. Fluoresc. 7, 022002. doi:10.1088/2050-6120/ab0bfa
Hone, A. J., Whiteaker, P., Mohn, J. L., Jacob, M. H., and Michael McIntosh, J. (2010). Alexa Fluor 546-ArIB[V11L;V16A] Is a Potent Ligand for Selectively Labeling α7 Nicotinic Acetylcholine Receptors. J. Neurochem. 114, no. doi:10.1111/j.1471-4159.2010.06819.x
Johnson, D. A., and Taylor, P. (1982). Site-Specific Fluorescein-Labeled Cobra Alpha-Toxin. Biochemical and Spectroscopic Characterization. J. Biol. Chem. 257, 5632–5636. doi:10.1016/s0021-9258(19)83824-3
Kang, S., and Maelicke, A. (1980). Fluorescein Isothiocyanate-Labeled Alpha-Cobratoxin. Biochemical Characterization and Interaction with Acetylcholine Receptor From Electrophorus Electricus. J. Biol. Chem. 255, 7326–7332. doi:10.1016/s0021-9258(20)79707-3
Karlsson, E., Eaker, D., and Ponterius, G. (1972). Modification of Amino Groups in Naja Naja Neurotoxins and the Preparation of Radioactive Derivatives. Biochim. Biophys. Acta (Bba) - Protein Struct. 257, 235–248. doi:10.1016/0005-2795(72)90275-9
Kasheverov, I. E., Chiara, D. C., Zhmak, M. N., Maslennikov, I. V., Pashkov, V. S., Arseniev, A. S., et al. (2006). Alpha-Conotoxin GI Benzoylphenylalanine Derivatives. 1H-NMR Structures and Photoaffinity Labeling of the Torpedo californica Nicotinic Acetylcholine Receptor. FEBS J. 273, 1373–1388. doi:10.1111/J.1742-4658.2006.05161.X
Kasheverov, I., Rozhkova, A., Zhmak, M., Utkin, Y., Ivanov, V., and Tsetlin, V. I. (2001). Photoactivatable α-Conotoxins Reveal Contacts With All Subunits as Well as Antagonist-Induced Rearrangements in the Torpedo Californicaacetylcholine Receptor. Eur. J. Biochem. 268, 3664–3673. doi:10.1046/J.1432-1327.2001.02272.X
Kessler, P., Marchot, P., Silva, M., and Servent, D. (2017). The Three-Finger Toxin Fold: a Multifunctional Structural Scaffold Able to Modulate Cholinergic Functions. J. Neurochem. 142 (Suppl. 2), 7–18. doi:10.1111/jnc.13975
Kini, R. M., and Doley, R. (2010). Structure, Function and Evolution of Three-Finger Toxins: Mini Proteins With Multiple Targets. Toxicon. 56, 855–867. doi:10.1016/j.toxicon.2010.07.010
Kini, R. M., and Koh, C. Y. (2020). Snake Venom Three-Finger Toxins and Their Potential in Drug Development Targeting Cardiovascular Diseases. Biochem. Pharmacol. 181, 114105. doi:10.1016/j.bcp.2020.114105
Kryukova, E., Ivanov, I., Lebedev, D., Spirova, E., Egorova, N., Zouridakis, M., et al. (2018). Orthosteric And/or Allosteric Binding of α-Conotoxins to Nicotinic Acetylcholine Receptors and Their Models. Mar. Drugs. 16, 460. doi:10.3390/md16120460
Kudryavtsev, D. S., Shelukhina, I. V., Son, L. V., Ojomoko, L. O., Kryukova, E. V., Lyukmanova, E. N., et al. (2015). Neurotoxins From Snake Venoms and α-Conotoxin ImI Inhibit Functionally Active Ionotropic γ-Aminobutyric Acid (GABA) Receptors. J. Biol. Chem. 290, 22747–22758. doi:10.1074/jbc.M115.648824
Kudryavtsev, D. S., Tabakmakher, V. М., Budylin, G. S., Egorova, N. S., Efremov, R. G., Ivanov, I. A., et al. (2020). Complex Approach for Analysis of Snake Venom α-Neurotoxins Binding to HAP, the High-Affinity Peptide. Sci. Rep. 10, 3861. doi:10.1038/s41598-020-60768-y
Kuzmenkov, A. I., Nekrasova, O. V., Kudryashova, K. S., Peigneur, S., Tytgat, J., Stepanov, A. V., et al. (2016a). Fluorescent Protein-Scorpion Toxin Chimera Is a Convenient Molecular Tool for Studies of Potassium Channels. Sci. Rep. 6, 33314. doi:10.1038/srep33314
Kuzmenkov, A. I., Sachkova, M. Y., Kovalchuk, S. I., Grishin, E. V., and Vassilevski, A. A. (2016b). Lachesana Tarabaevi, an Expert in Membrane-Active Toxins. Biochem. J. 473, 2495–2506. doi:10.1042/bcj20160436
Kuzmenkov, A. I., and Vassilevski, A. A. (2018). Labelled Animal Toxins as Selective Molecular Markers of Ion Channels: Applications in Neurobiology and beyond. Neurosci. Lett. 679, 15–23. doi:10.1016/j.neulet.2017.10.050
Lukasiewicz, R. J., and Bennett, E. L. (1978). α-Bungarotoxin Binding Properties of a Central Nervous System Nicotinic Acetylcholine Receptor. Biochim. Biophys. Acta (Bba) - Gen. Subjects. 544, 294–308. doi:10.1016/0304-4165(78)90098-3
Martin, I. L., Davies, M., and Dunn, S. M. J. (2007). “Characterization of Receptors by Radiolabelled Ligand Binding Techniques,” in Handbook of Neurochemistry and Molecular Neurobiology (Boston, MA: Springer US), 257–274. doi:10.1007/978-0-387-30401-4_10
Nekrasova, O. V., Primak, A. L., Ignatova, A. A., Novoseletsky, V. N., Geras’kina, O. V., Kudryashova, K. S., et al. (2020). N-Terminal Tagging With GFP Enhances Selectivity of Agitoxin 2 to Kv1.3-Channel Binding Site. Toxins. 12, 802. doi:10.3390/toxins12120802
O’Brien, R. D., Eldefrawi, M. E., and Eldefrawi, A. T. (1972). Isolation of Acetylcholine Receptors. Annu. Rev. Pharmacol. 12, 19–34. doi:10.1146/annurev.pa.12.040172.000315
Osipov, A. V., Kasheverov, I. E., Makarova, Y. V., Starkov, V. G., Vorontsova, O. V., Ziganshin, R. K., et al. (2008). Naturally Occurring Disulfide-Bound Dimers of Three-Fingered Toxins. J. Biol. Chem. 283, 14571–14580. doi:10.1074/JBC.M802085200
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera?A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 25, 1605–1612. doi:10.1002/JCC.20084
Pivovarov, A. S., Palikhova, T. A., Nikolaev, G. M., Velikanov, A. N., Vasilieva, N. A., Kasheverov, I. E., et al. (2020). Atypical Acetylcholine Receptors on the Neurons of the Turkish Snail. Dokl. Biochem. Biophys. 491, 81–84. doi:10.1134/S1607672920020118
Qi, Y., Geib, T., Huynh, A.-M., Jung, G., and Volmer, D. A. (2015). Fragmentation Patterns of Boron-Dipyrromethene (BODIPY) Dyes by Electrospray Ionization High-Resolution Tandem Mass Spectrometry. Rapid Commun. Mass. Spectrom. 29, 885–890. doi:10.1002/rcm.7179
Rahman, M. M., Teng, J., Worrell, B. T., Noviello, C. M., and Lee, M. (2020). Structure of the Native Muscle-type Nicotinic Receptor and Inhibition by Snake Venom Toxins. Neuron. 106, 952–962. e5. doi:10.1016/J.NEURON.2020.03.012
Ravdin, P., and Axelrod, D. (1977). Fluorescent Tetramethyl Rhodamine Derivatives of Alpha-Bungarotoxin: Preparation, Separation, and Characterization. Anal. Biochem. 80, 585–592. doi:10.1016/0003-2697(77)90682-0
Rivera-Torres, I. O., Jin, T. B., Cadene, M., Chait, B. T., and Poget, S. F. (2016). Discovery and Characterisation of a Novel Toxin From Dendroaspis Angusticeps, Named Tx7335, That Activates the Potassium Channel KcsA. Sci. Rep. 6, 23904. doi:10.1038/srep23904
Rosso, J.-P., Schwarz, J. R., Diaz-Bustamante, M., Céard, B., Gutiérrez, J. M., Kneussel, M., et al. (2015). MmTX1 and MmTX2 From Coral Snake Venom Potently Modulate GABAA Receptor Activity. Proc. Natl. Acad. Sci. U. S. A. 112, E891–E900. doi:10.1073/pnas.1415488112
Shelukhina, I., Spirova, E., Kudryavtsev, D., Ojomoko, L., Werner, M., Methfessel, C., et al. (2017). Calcium Imaging With Genetically Encoded Sensor Case12: Facile Analysis of α7/α9 nAChR Mutants. PLoS One. 12, e0181936. doi:10.1371/journal.pone.0181936
Shelukhina, I. V., Kryukova, E. V., Lips, K. S., Tsetlin, V. I., and Kummer, W. (2009). Presence of α7 Nicotinic Acetylcholine Receptors on Dorsal Root Ganglion Neurons Proved Using Knockout Mice and Selective α-neurotoxins in Histochemistry. J. Neurochem. 109, 1087–1095. doi:10.1111/j.1471-4159.2009.06033.x
Shelukhina, I., Zhmak, M., Lobanov, A., Ivanov, I., Garifulina, A., Kravchenko, I., et al. (2018). Azemiopsin, a Selective Peptide Antagonist of Muscle Nicotinic Acetylcholine Receptor: Preclinical Evaluation as a Local Muscle Relaxant. Toxins (Basel). 10, 34. doi:10.3390/toxins10010034
Silver, J., and Billiar, R. B. (1976). An Autoradiographic Analysis of [3H]alpha-Bungarotoxin Distribution in the Rat Brain After Intraventricular Injection. J. Cell Biol. 71, 956–963. doi:10.1083/jcb.71.3.956
Siniavin, A. E., Streltsova, M. A., Kudryavtsev, D. S., Shelukhina, I. V., Utkin, Y. N., and Tsetlin, V. I. (2020). Activation of α7 Nicotinic Acetylcholine Receptor Upregulates HLA-DR and Macrophage Receptors: Potential Role in Adaptive Immunity and in Preventing Immunosuppression. Biomolecules. 10, 507. doi:10.3390/biom10040507
Stakheev, A. A., Ryazantsev, D. Y., Zvezdina, Y. K., Baranov, M. S., and Zavriev, S. K. (2018). A Novel Fluorescent GFP Chromophore Analog-Based Dye for Quantitative PCR. Biochem. 83, 855–860. doi:10.1134/S000629791807009X
Stiles, B. G. (1993). Acetylcholine Receptor Binding Characteristics of Snake and Cone Snail Venom Postsynaptic Neurotoxins: Further Studies With a Non-Radioactive Assay. Toxicon. 31, 825–834. doi:10.1016/0041-0101(93)90217-7
Stya, M., and Axelrod, D. (1983). Diffusely Distributed Acetylcholine Receptors Can Participate in Cluster Formation on Cultured Rat Myotubes. Proc. Natl. Acad. Sci. U. S. A. 80, 449–453. doi:10.1073/pnas.80.2.449
Surin, A., Kriukova, E., Strukov, A., Zhmak, M., Talka, R., Tuominen, R., et al. (2012). Effect of Alpha-Conotoxin MII and its N-Terminal Derivatives on Ca2+ and Na+ Signals Induced by Nicotine in Neuroblastoma Cell Line SH-Sy5y. Bioorg. Khim. 38, 214–222. doi:10.1134/S1068162012020112
Surin, A., Pluzhnikov, K., Utkin, Y., Karlsson, E., and Tsetlin, V. I. (1983). Study of Long-Chain Neurotoxins From Naja naja Siamensis and Naja naja Oxiana Venoms by Circular Dichroism and Fluorescence. Bioorg. Khim 9, 756–767. Available at: http://www.rjbc.ru/arc/9/6/0756-0767.pdf
Tsetlin, V. I., Karlsson, E., Arseniev, A. S., Utkin, Y. N., Surin, A. M., Pashkov, V. S., et al. (1979). EPR and Fluorescence Study of Interaction of Naja naja Oxiana Neurotoxin II and its Derivatives With Acetylcholine Receptor Protein From Torpedo marmorata. FEBS Lett. 106, 47–52. doi:10.1016/0014-5793(79)80692-4
Tsetlin, V. I., Karlsson, E., YuN, U., Pluzhnikov, K. A., Arseniev, A. S., Surin, A. M., et al. (1982). Interaction Surfaces of Neurotoxins and Acetylcholine Receptor. Toxicon. 20, 83–93. doi:10.1016/0041-0101(82)90171-4
Tsetlin, V. I., Kasheverov, I. E., and Utkin, Y. N. (2021). Three-Finger Proteins From Snakes and Humans Acting on Nicotinic Receptors: Old and New. J. Neurochem. 158, 1223–1235. doi:10.1111/jnc.15123
Utkin, Y. N., Krivoshein, A. V., Davydov, V. L., Kasheverov, I. E., Franke, P., Maslennikov, I. V., et al. (1998). Labeling of Torpedo californica Nicotinic Acetylcholine Receptor Subunits by Cobratoxin Derivatives With Photoactivatable Groups of Different Chemical Nature at Lys23. Eur. J. Biochem. 253, 229–235. doi:10.1046/j.1432-1327.1998.2530229.x
Utkin, Y. N. (2019). Last Decade Update for Three-Finger Toxins: Newly Emerging Structures and Biological Activities. World J. Biol. Chem. 10, 17–27. doi:10.4331/wjbc.v10.i1.17
Utkin, Y. N., Weise, C., Kasheverov, I. E., Andreeva, T. V., Kryukova, E. V., Zhmak, M. N., et al. (2012). Azemiopsin From Azemiops Feae Viper Venom, a Novel Polypeptide Ligand of Nicotinic Acetylcholine Receptor. J. Biol. Chem. 287, 27079–27086. doi:10.1074/jbc.M112.363051
Webb, B., and Sali, A. (2016). Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinforma. 54, 1–5. doi:10.1002/CPBI.3
Yang, D. C., Deuis, J. R., Dashevsky, D., Dobson, J., Jackson, T. N. W., Brust, A., et al. (2016). The Snake With the Scorpion’s Sting: Novel Three-Finger Toxin Sodium Channel Activators From the Venom of the Long-Glanded Blue Coral Snake (Calliophis Bivirgatus). Toxins (Basel). 8, 303. doi:10.3390/toxins8100303
Keywords: nicotinic acetylcholine receptor, acetylcholine-binding protein, α-cobratoxin, azemiopsin, fluorescent protein, fluorescent toxin, ion channel, peptide toxin
Citation: Kasheverov IE, Kuzmenkov AI, Kudryavtsev DS, Chudetskiy IS, Shelukhina IV, Barykin EP, Ivanov IA, Siniavin AE, Ziganshin RH, Baranov MS, Tsetlin VI, Vassilevski AA and Utkin YN (2021) Snake Toxins Labeled by Green Fluorescent Protein or Its Synthetic Chromophore are New Probes for Nicotinic acetylcholine Receptors. Front. Mol. Biosci. 8:753283. doi: 10.3389/fmolb.2021.753283
Received: 04 August 2021; Accepted: 10 November 2021;
Published: 30 November 2021.
Edited by:
Maria Elena de Lima, Grupo Santa Casa BH, BrazilReviewed by:
Steve Peigneur, KU Leuven, BelgiumCopyright © 2021 Kasheverov, Kuzmenkov, Kudryavtsev, Chudetskiy, Shelukhina, Barykin, Ivanov, Siniavin, Ziganshin, Baranov, Tsetlin, Vassilevski and Utkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuri N. Utkin, dXRraW5AaWJjaC5ydQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.