 Anshupriya Si
Anshupriya Si Steven J. Sucheck
Steven J. Sucheck- Department of Chemistry and Biochemistry, University of Toledo, Toledo, OH, United States
Pseudomonas aeruginosa is a highly prevalent gram-negative bacterium that is becoming more difficult to treat because of increasing antibiotic resistance. As chemotherapeutic treatment options diminish, there is an increased need for vaccines. However, the creation of an effective P. aeruginosa vaccine has been elusive despite intensive efforts. Thus, new paradigms for vaccine antigens should be explored to develop effective vaccines. In these studies, we have focused on the synthesis of two L-rhamnose–bearing epitopes common to glycoforms I and II of the outer core domain of Pseudomonas aeruginosa lipopolysaccharide, α-L-Rha-(1→6)-α-D-Glc-(1→4)-α-D-GalN-(Ala)-α-aminooxy (3) and α-L-Rha-(1→3)-β-D-Glc-(1→3)-α-D-GalN-(Ala)-α-aminooxy (4), respectively. The target trisaccharides were both prepared starting from a suitably protected galactosamine glycoside, followed by successive deprotection and glycosylation with suitably protected D-glucose and L-rhamnose thioglycosides. Global deprotection resulted in the formation of targets 3 and 4 in 22 and 35% yield each. Care was required to modify basic reaction conditions to avoid early deprotection of the N-oxysuccinamido group. In summary, trisaccharides related to the L-rhamnose–bearing epitopes common to glycoforms I and II of the outer core domain of Pseudomonas aeruginosa lipopolysaccharide have been prepared as their aminooxy glycosides. The latter are expected to be useful in chemoselective oxime-based bioconjugation reactions to form Pseudomonas aeruginosa vaccines.
Introduction
Pseudomonas aeruginosa is a widely distributed, encapsulated, gram-negative bacterium. In the early 1900s, it was recognized as a bacterial pathogen, and in the past 50 years, it has become one of the most concerning pathogens. It is reported that P. aeruginosa is the cause of 1 in 10 nosocomial infections associated with serious illness such as ventilator-associated pneumonia and various sepsis syndromes, and it has the highest mortality rate (37%) (Klevens et al., 2008; Lister et al., 2009). Most P. aeruginosa is resistant to at least one of the classes of antibiotics, and some P. aeruginosa is resistant to all the available antibiotics (Talbot et al., 2006; Pier, 2007). Therefore, vaccines are potential solutions to overcome the antimicrobial resistance (AMR) developed by P. aeruginosa. Lipopolysaccharide (LPS) is a complex glycolipid present on the outer layer of gram-negative bacteria. It plays a vital role as an essential virulence factor in the pathogenicity of P. aeruginosa strains and, hence, is a potential antigen target for a prophylactic vaccine (Pier, 2007). In a recent study, Liu et al. published that this bacterium could have one of twenty different O-polysaccharides as a part of its LPS (Liu et al., 1983; Liu and Wang, 1990). This is a proposed reason for inconsistent efficiency of some of the vaccines that were formulated based on isolated LPS as target antigens. Therefore, we have shifted our focus to synthetic outer core domains of the LPS that have been shown to react with protective monoclonal antibodies that promoted macrophage-mediated opsonophagocytosis against several serotypes and are also believed to target the rhamnose moiety and neighboring saccharides (Yokota et al., 1989; Terashima et al., 1991). It is noted that LPS is made up of O-polysaccharides linked to Lipid A via an intervening core oligosaccharide. Outer-membrane LPS consists of both smooth-type (S) LPS and rough-type (R) LPS based on the presence and absence of O-polysaccharides, respectively (Rocchetta et al., 1999; Raetz and Whitfield, 2002). Both the (S)-type laboratory strains and (R)-type clinical strains of P. aeruginosa share the presence of structurally similar outer-core glycoform I (1) and glycoform II (2) (Figure 1) consisting of one D-galactosamine (GalN) residue, three D-glucose residues, and one L-rhamnose residue. The GalN residue is acylated with an L-alanyl group in all LPSs (Sadovskaya et al., 1998; Sadovskaya et al., 2000; Knirel et al., 2001; Bystrova et al., 2002; Bystrova et al., 2003; Bystrova et al., 2004; Bystrova et al., 2006).
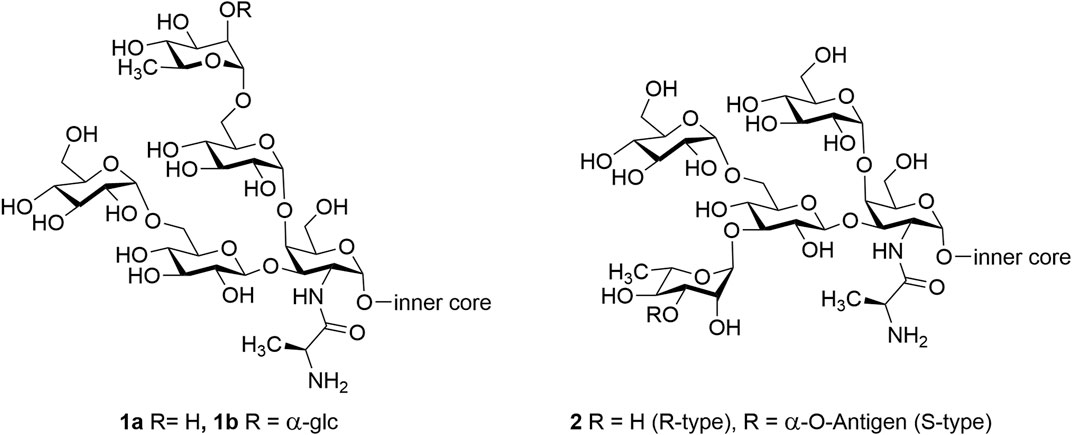
FIGURE 1. P. aeruginosa LPS outer core structure of glycoform I (1a,b) and glycoform II (2).
Glycan epitopes common to both S-type and R-type LPS are potentially ideal antigen targets as they are less serotype dependent. However, isolation of these minimal epitopes from the biological source is not possible. Therefore, it is essential to develop synthetic strategies to obtain these materials for study. In this context, synthesis of two common trisaccharide fragments found within the outer core domain of P. aeruginosa related to glycoform I and glycoform II, noted as α-L-Rha-(1→6)-α-d-Glc-(1→4)-α-D-GalN-(Ala)-α-aminooxy (3) and α-L-Rha-(1→3)-β-D-Glc-(1→3)-α-D-GalN-(Ala)-α-aminooxy (4) (Figure 2), respectively, is reported herein. We have selected the succinimidyl group as the aminooxy precursor and introduced it at the preliminary stage of the synthetic strategies based on the previous reports (Marcaurelle et al., 2001; De Silva et al., 2009) for developing biologically stable oxime bonds for glycoconjugate vaccines. The use of N-hydroxysuccinimide (NHS) was essential for introducing the aminooxy (-ONH2) functionality which has already proved very useful in several reports (Bourgault et al., 2014; Ghosh and Andreana, 2014; Ghosh et al., 2016; Ghosh et al., 2020; Kleski et al., 2020). The presence of an aminooxy group at the reducing terminal of the trisaccharides provides a readily available form for conjugation to an appropriate carbonyl-modified carrier protein (Agten et al., 2016) without destroying the cyclic structure of the reducing end saccharide. The resulting glycoconjugates can then be evaluated in immunological experiments.
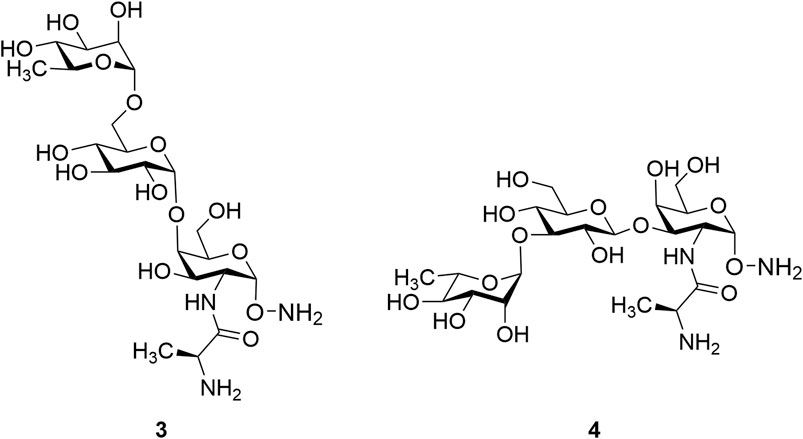
FIGURE 2. Two target trisaccharide fragments common to glycoform I and glycoform II, respectively.
Results and Discussion
Earlier syntheses of pentasaccharide and trisaccharide fragments of outer core domains corresponding to glycoforms I and II of P. aeruginosa containing a methoxy group and tert-butyldiphenylsilyl (TBDPS)-protected hydroquinone (TPH) as a multifunctional reducing-end capping group have been reported (Komarova et al., 2006; Komarova et al., 2008, 2012; Vartak et al., 2018). In these studies, we sought a route to an aminooxy glycoside that could be used in an oxime-based conjugation which would leave the unique L-Ala–modified galactosamine in its native state.
The target trisaccharide fragments of glycoforms I and II were synthesized as their α-aminooxy glycosides from suitably functionalized monosaccharides using stereoselective sequential glycosylations and functional group manipulations. A set of suitably functionalized donor thioglycoside building blocks, A (Tam and Lowary, 2010), B (Rajput and Mukhopadhyay, 2008), C (He et al., 2019), and D (Mukhopadhyay et al., 2004) (Figure 3), were prepared from the naturally available reducing sugars applying a number of reaction conditions reported earlier. D-(+)-glucose–based thioglycoside donors A and C were obtained from commercially available D-(+)-glucose. L-rhamnose–based glycosyl donors B and D were synthesized starting from L-rhamnose according to reported literature (Mukhopadhyay et al., 2004; Rajput and Mukhopadhyay, 2008; Tam and Lowary, 2010; He et al., 2019).
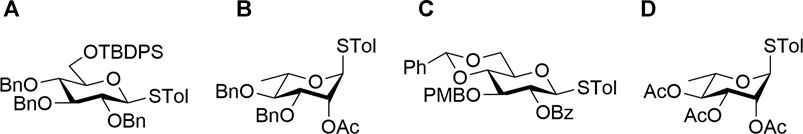
FIGURE 3. Thioglycoside donor building blocks (A–D).
Syntheses of the two trisaccharides, 3 and 4, proceeded from common intermediate p-methylphenyl 2-azido-4,6-O-benzylidene-2-deoxy-1-thio-α/β-D-galactopyranoside (5α/β) (Scheme 1) which was prepared from tri-O-acetyl-D-galactal over six steps according to the reported procedure (Lemieux and Ratcliffe, 1979; Santra et al., 2012).
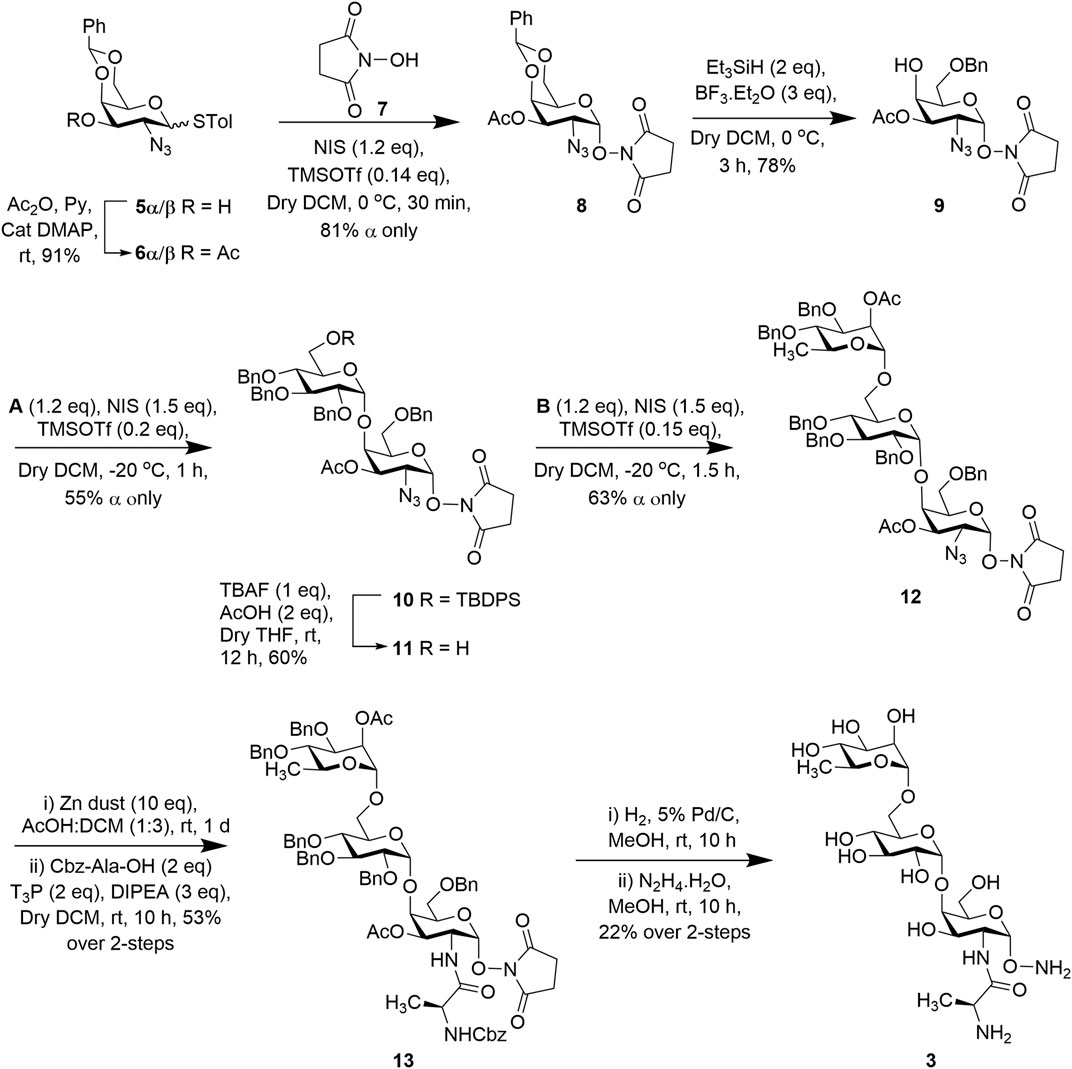
SCHEME 1. Synthesis of trisaccharide fragment target 3.
To access the target trisaccharide 3 of P. aeruginosa LPS, the 3-OH positions of both 5α/β were acetylated using acetic anhydride and pyridine to obtain p-methylphenyl 2-azido-3-O-acetyl-4,6-O-benzylidene-2-deoxy-1-thio-α/β-D-galactopyranoside (6α/β) in 91% yield (Santra et al., 2012). Both the isomers were used as donors for stereoselective glycosylation with N-hydroxy succinimide (NHS) (7) in the presence of a combination of N-iodosuccinimide (NIS) and trimethylsilyl trifluoromethanesulfonate (TMSOTf) (Nicolaou et al., 1983; Konradsson et al., 1990; Veeneman et al., 1990) at 0°C to furnish compound 8 (exclusively α product) in 81% yield. NMR spectral analysis of this compound supported its formation; signals at [δ = 5.60 ppm (d, J = 3.7 Hz, 1H, H-1) in the 1H NMR and at 102.80 (C-1) in the 13C NMR spectra]. Compound 8 was subjected to regioselective reductive cleavage by treatment with borontrifluoride diethyletherate and triethylsilane (Et3SiH) (Debenham and Toone, 2000) at 0°C to selectively open the benzylidene ring to afford acceptor 9 containing a free C-4 hydroxyl in 78% yield (Scheme 1). Acceptor 9 was then coupled with thioglucoside donor A in the presence of a combination of NIS and TMSOTf (as before) in a mixed solvent (CH2Cl2-Et2O; 1:3) at −20°C for 40 min to furnish disaccharide 10 in 55% yield (α only) (Scheme 1). The newly formed glycosyl linkage in the compound was confirmed by its NMR spectral analysis: signals at δ = 5.60 (d, J = 3.8 Hz, 1H, H-1) and 4.91 ppm (brs, 1H, H-1′) in the 1H NMR and at 101.82 (C-1) and 99.94 ppm (C-1′) in the 13C NMR spectra. The TBDPS group was deprotected using 1 M tetra-n-butyl ammonium fluoride (TBAF) solution in THF and in the presence of a catalytic amount of acetic acid (AcOH) to afford disaccharide acceptor 11 in 60% yield. The compound was subsequently glycosylated with rhamnose-based thioglycoside donor B in the presence of a combination of NIS and TMSOTf (as before) at −20°C for 30 min to afford trisaccharide 12 in 63% yield (α only), which was confirmed from its spectral analysis: signals at δ = 5.64 (d, J = 3.8 Hz, 1H, H-1), 4.84 (brs, 1H, H-1′), and 4.67 ppm (brs, 1H, H-1″) in the 1H NMR and at 101.78 (C-1), 99.52 (C-1′), and 97.86 ppm (C-1″) in the 13C NMR spectra. The Cbz-alanine residue was then introduced by reducing the azide group of the trisaccharide 12 with Zn/AcOH, followed by coupling with Cbz-Ala-OH using propylphosphonic anhydride (T3P) as the coupling reagent and DIPEA to afford trisaccharide derivative 13 in 53% overall yield. Finally, the benzyl groups were removed by hydrogenation using 5% Pd/C followed by treatment with hydrazine hydrate for deprotection of the succinimide group and the remaining acetate groups to furnish the final product 3 (Scheme 1) in 22% yield. Spectral analysis of compound 3 unambiguously supported its formation [signals at δ 4.93 (brs, 1H, H-1″), 4.88 (brs, 1H, H-1), and 4.85 ppm (brs, 1H, H-1′) in the 1H NMR and at 100.37 (C-1′) and 100.22 ppm (C-1, C-1″) in the 13C NMR spectra].
To access the second trisaccharide common outer core fragment of P. aeruginosa LPS, we selected the chloroacetyl group (ClAc) for 3-OH protection inspired by previous uses (Jiaang et al., 2000; Bourgault et al., 2014). Treatment of both 5α/β with chloroacetyl chloride and pyridine afforded p-methylphenyl 2-azido-4,6-O-benzylidene-3-O-chloroacetyl-2-deoxy-1-thiol-α/β-D-galactopyranoside (14α/β) in 85% yield. Both the compounds were then used for stereoselective 1,2-cis glycosylation with N-hydroxy succinimide (7) in the presence of NIS and TMSOTf (as before) at 0°C to furnish compound succinimidyl 2-azido-4,6-O-benzylidene-2-deoxy-α-D-galactopyranoside (15) in 70% yield. NMR spectral analysis of compound 15 supported its formation: signals at δ = 5.56 ppm (d, J = 3.7 Hz, 1H, H-1) in the 1H NMR and at 103.01 (C-1) in the 13C NMR spectra and it matches with the previously reported data (Bourgault et al., 2014). The chloroacetyl group was selectively removed (Masaki et al., 1968) by the treatment with a nonbasic nucleophile, thiourea, to afford the first monosaccharide acceptor 16 in 65% yield without affecting the base labile NHS group (Scheme 2).
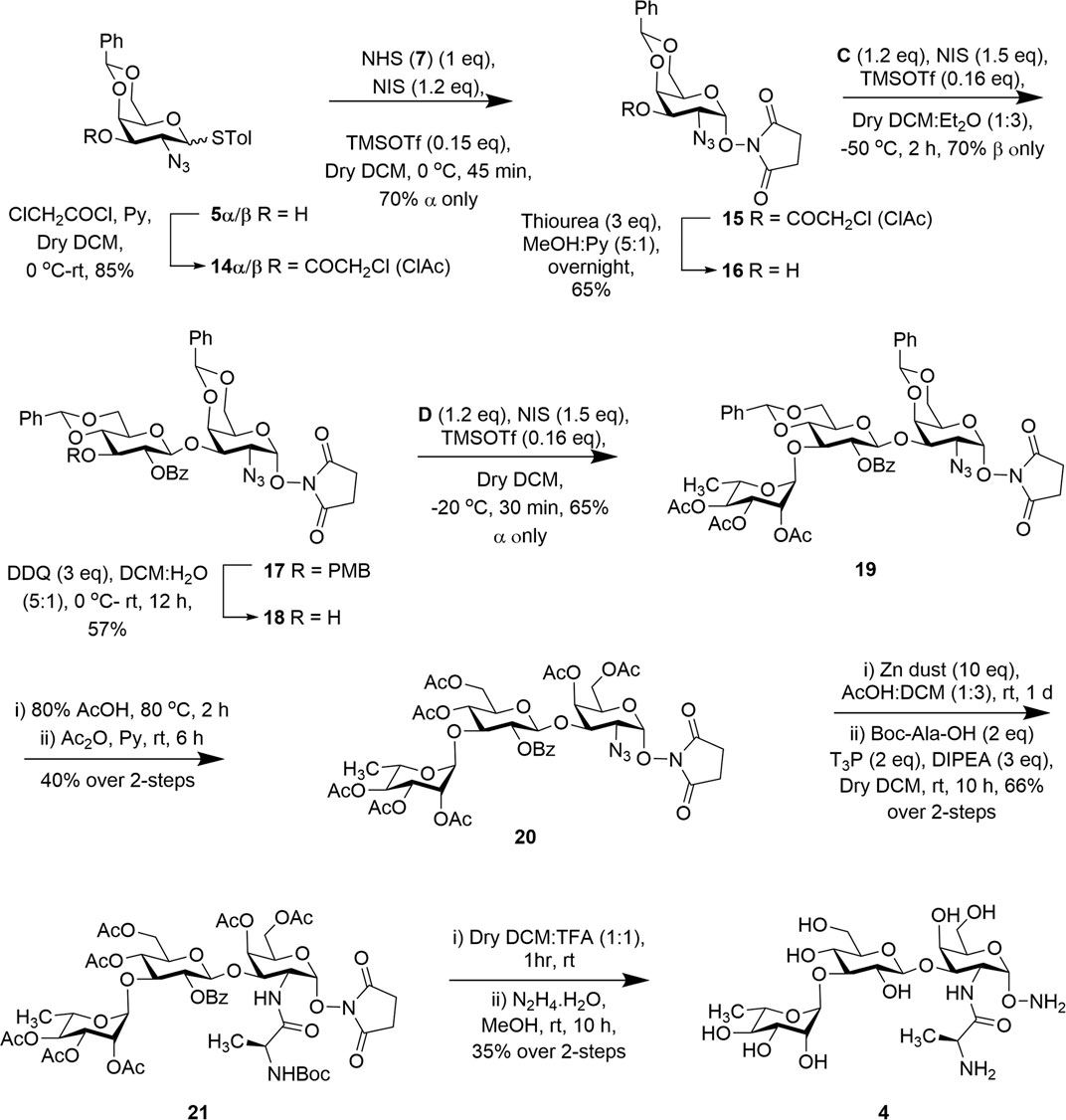
SCHEME 2. Synthesis of trisaccharide fragment target 4.
With a reasonable amount of acceptor in hand, iodonium ion–mediated stereoselective 1,2-trans glycosylation was achieved with thioglucoside donor C in the presence of a combination of NIS and TMSOTf (as before) at −50°C to afford β (1 → 3) disaccharide 17 in 70% yield. Exclusive formation of compound 17 was confirmed from its spectral analysis: signals at δ = 5.53 (d, J = 3.7 Hz, 1H, H-1) and 4.93 ppm (d, J = 7.7 Hz, 1H, H-1′) in the 1H NMR and at 103.04 (C-1) and 102.83 ppm (C-1′) in the 13C NMR spectra. To build the target trisaccharide, oxidative removal of the p-methoxybenzyl (PMB) (Oikawa et al., 1982) group from compound 17 was achieved by the treatment with 2,3-dicholoro-5,6-dicyano-1,4-benzoquinone (DDQ) to give disaccharide acceptor 18 in 57% yield. Acceptor 18 and rhamnoside donor D were then coupled using the NIS:TMSOTf (as before) promoter system at −20°C to furnish the product 19 in 65% yield. From spectral analysis it was determined that the product was exclusively the desired trisaccharide without producing unwanted orthoester. The stereochemistry at the glycosidic linkages in compound 19 was confirmed from its spectral analysis: signals at δ = 5.52 (d, J = 3.7 Hz, 1H, H-1), 5.06 (d, J = 7.3 Hz, 1H, H-1′), and 4.83 ppm (brs, 1H, H-1″) in the 1H NMR and at 102.96 (C-1), 102.27 (C-1′), and 97.78 ppm (C-1″) in the 13C NMR spectra. The global deprotection was achieved by treatment of the compound 19 with 80% acetic acid at 80°C to remove the benzylidene acetal groups. This step was followed by acetylation with acetic anhydride and pyridine to afford trisaccharide 20 in 40% yield over two steps. The azido group was reduced to an amine by the treatment with Zn/AcOH and coupled with Boc-Ala-OH in the presence of T3P in one pot to obtain trisaccharide derivative 21 in 66% yield in two steps. Finally, trisaccharide was subjected to deprotection reactions including (a) removal of the Boc-group using trifluoroacetic acid (TFA) and (b) removal of the remaining acetyl groups and the replacement of the succinimidyl protecting group as the α-aminooxy group by the treatment with hydrazine monohydrate to afford the target trisaccharide 4 in 35% overall yield (Scheme 2). The formation of compound 4 was confirmed by spectroscopic analysis: signal δ 5.00 (brs, 1H, H-1″), 4.86 (d, J = 4.0 Hz, 1H, H-1), and 4.43 ppm (d, J = 8.0 Hz, 1H, H-1′) in the 1H NMR and at 103.50 (C-1′), 100.91 (C-1″), and 100.44 ppm (C-1) in the 13C NMR spectra.
Difficult-to-remove side products were generated during the removal of the succinimide group due to the use of excess hydrazine monohydrate (Renaudet and Dumy, 2004; Ghosh and Andreana, 2014). Furthermore, it was challenging to purify both deprotected compounds using only a C18 silica gel column. The problem was solved by passing the compounds through size exclusion chromatography (Bio-gel P-2) using water as the eluent. Fractions containing aminooxy sugars 3 and 4 (identified by TLC staining) were collected, frozen, and lyophilized. The resulting white solid was characterized by NMR and HRMS.
Conclusion
In conclusion, oligosaccharide fragments corresponding to the outer core domain of P. aeruginosa LPS were synthesized in good yield using a sequential glycosylation approach. During the synthesis of the target molecules, similar reaction conditions were used in each of the intermediate glycosylation reaction steps to obtain excellent stereochemical outcomes. This report again presents the importance of the NHS group for the synthesis of aminooxy glycosides where the base-sensitive NHS group remains stable through multiple glycosylations, protecting group modifications, and deprotections. However, care was required to avoid basic conditions to maintain NHS stability. For example, the modification of TBAF with 2 equivalents of acetic acid was essential for NHS stability. Furthermore, the route to compound 3 was tolerant of carefully controlled hydrogenolysis to avoid reduction of the N-O bond. Formation of the aminooxy linkage at the reducing end is expected to afford a convenient handle for highly chemoselective oxime conjugation with an appropriately modified carrier protein.
Experimental
General Methods
All chemicals and solvents were purchased from Fisher Scientific, Acros Organics, Alfa Aesar, or Sigma-Aldrich. Reactions are carried out under an atmosphere of nitrogen using a nitrogen balloon. Solvents were dried using a solvent purification system by passing them through activated alumina and copper catalyst columns. Reactions were monitored by TLC (silica gel, f254) under UV light or by charring (5% H2SO4-MeOH), and the purification was performed by column chromatography on silica gel (230–400 mesh), C-18, and P-2 biogel using the solvent system specified; solvents were used without purification for chromatography. 1H NMR was recorded on a Bruker Avance III 600 MHz spectrometer using CDCl3 and D2O as an internal reference. 13C were recorded on a Bruker Avance III 600 MHz spectrometer using CDCl3 and D2O as the internal reference. High-resolution mass spectrometry was recorded on a Thermo LTQ XL Orbitrap instrument from the Ohio State University Mass Spectrometry Center.
p-methylphenyl 2-azido-3-O-acetyl-4,6-O-benzylidene-2-deoxy-1-thio-α/β-D-galactopyranoside (6α/β)
Acetic anhydride (1.07 ml, 11.3 mmol) was added to p-methylphenyl 2-azido-4,6-O-benzylidene-2-deoxy-1-thio-α/β-D-galactopyranoside (5α/β) (1.50 g, 3.76 mmol) in pyridine (1.50 ml, 18.6 mmol) and the mixture was stirred at room temperature for 1 h. After completion of the reaction, the solvents were removed under reduced pressure and co-evaporated with toluene three times (3 × 10 ml). The reaction mixture was diluted with ethyl acetate and successively washed with satd. CuSO4 (2 × 100 ml), 1 N HCl (2 × 50 ml), and brine (2 × 100 ml) solution. The organic layer was collected, dried over anhydrous Na2SO4, and concentrated. The residue was purified by silica gel flash column chromatography to isolate the compound as white solid 6α/β: yield 91% (1.50 g); silica gel TLC Rf = 0.65 (50% ethyl acetate: hexane). All the spectral data match with reported data (Santra et al., 2012).
Succinimidyl 2-azido-3-O-acetate-4,6-O-benzylidene-2-deoxy-α-D-galactopyranoside (8)
Both 6α/β compound (1.20 g, 2.72 mmol) and N-hydroxy succinimide (7) (0.31 g, 2.70 mmol) were mixed in a flame-dried round bottom flask and left under high vacuum overnight. To the dried reagents were added flame-dried 4-Å molecular sieves and 20 ml of anhydrous DCM. This mixture was then stirred at room temperature for 15 min. NIS (0.73 g, 3.26 mmol) was added to it, and the reaction mixture was cooled to 0°C. Subsequently, (70 μL, 0.38 mmol) TMSOTf was added. After 30 min, the reaction was diluted with DCM and filtered. The filtrate was washed with aq. sodium thiosulfate (2 × 50 ml), aq. sodium bicarbonate (2 × 50 ml), and brine (2 × 100 ml) solution. The organic layer was collected, dried over anhydrous Na2SO4, and concentrated. The residue was purified by silica gel flash column chromatography to isolate the compound as white solid 8: yield 81% (0.95 g); silica gel TLC Rf = 0.20 (50% ethyl acetate: hexane). 1H NMR (600 MHz, CDCl3) δ 7.58–7.33 (Ar-H, 5H), 5.60 (d, J = 3.7 Hz, 1H, H-1), 5.56 (s, 1H, PhCH), 5.42 (dd, J = 11.4, 3.3 Hz, 1H, H-3), 4.72 (m, 1H, H-5), 4.56 (dd, J = 3.4, 1.2 Hz, 1H, H-4), 4.23–4.07 (m, 3H, H-2, H-6a, H-6b), 2.75 (s, 4H), 2.18 (s, 3H, COCH3). 13C NMR (151 MHz, CDCl3) δ 170.86 (2C), 170.29, 137.40–126.12 (Ar-C), 102.80, 100.78, 73.13, 68.88, 68.87, 64.65, 56.33, 25.47 (2C), 20.97. mass spectrum (HRMS), m/z = 455.1168 (M+Na)+, C19H20N4O8 requires 455.1173.
Succinimidyl 2-azido-3-O-acetate-6-O-benzyl-2-deoxy-α-D-galactopyranoside (9)
To the solution of compound 8 (0.93 g, 2.14 mmol) in anhydrous DCM (25 ml) at 0°C, BF3.OEt2 (0.80 ml, 6.45 mmol) and triethylsilane (Et3SiH) (0.70 ml, 4.30 mmol) were added successively using a syringe. The reaction was stirred under N2 and monitored by TLC. After 3 h, the reaction was quenched with aq. sodium bicarbonate (2 × 50 ml) solution. The compound was extracted with DCM (2 × 50 ml). The organic layer was collected, dried over anhydrous Na2SO4, and purified by silica gel flash column chromatography to obtain the compound as white solid 9: yield 78% (725 mg); silica gel TLC Rf = 0.15 (50% ethyl acetate: hexane). mass spectrum 1H NMR (600 MHz, CDCl3) δ 7.46–7.22 (Ar-H, 5H), 5.58 (d, J = 3.8 Hz, 1H, H-1), 5.35 (dd, J = 11.2, 2.8 Hz, 1H, H-3), 4.86 (t, J = 3.5 Hz, 1H, H-5), 4.64 (d, J = 11.7 Hz, 1H, PhCH), 4.52 (d, J = 11.7 Hz, 1H, PhCH), 4.46–4.39 (m, 1H, H-4), 4.21 (dd, J = 11.1, 3.9 Hz, 1H, H-2), 3.90–3.76 (m, 2H, H-6a, H-6b), 2.76 (s, 4H), 2.22 (s, 3H, COCH3). 13C NMR (151 MHz, CDCl3) δ 170.70 (2C), 170.07, 136.80–127.87 (Ar-C), 102.63, 74.08, 71.06, 70.48, 70.04, 69.19, 56.23, 25.47 (2C), 20.99 (HRMS), m/z = 457.1327 (M+Na)+, C19H22N4O8 requires 457.1330.
Succinimidyl [2,3,4-tri-O-benzyl-6-tertbutyldiphenylsillyl-α-D-glucopyranosyl]-(1→4)-2-azido-3-O-acetate-6-O-benzyl-2-deoxy-α-D-galactopyranoside (10)
The acceptor 9 (0.70 g, 1.56 mmol) and thioglycoside donor A (1.50 g, 1.88 mmol) were dissolved in dry DCM (5 ml) and dry diethyl ether (15 ml) and stirred using 4-Å molecular sieves for 30 min. NIS (0.51 g, 2.26 mmol) and TMSOTf (50 μl, 0.27 mmol) were added successively to the reaction mixture at −20°C. The solution was then allowed to warm to room temperature. The reaction was stirred under N2 and observed to be completed after 1 h. The reaction was diluted with DCM (50 ml) and filtered. The filtrate was washed with aq. sodium thiosulfate (2 × 50 ml), aq. sodium bicarbonate (2 × 50 ml), and brine (2 × 50 ml) solution. The organic layer was collected and dried over anhydrous Na2SO4, and the residue was subjected to silica gel flash column chromatography to afford the compound as white solid 10: yield 55% (0.98 g α only); silica gel TLC Rf = 0.50 (30% ethyl acetate: hexane). 1H NMR (600 MHz, CDCl3) δ 7.78–7.63 (Ar-H, 4H), 7.49–7.22 (Ar-H, 26H), 5.60 (d, J = 3.8 Hz, 1H, H-1), 5.35 (dd, J = 11.5, 2.7 Hz, 1H, H-3), 5.00 (d, J = 11.6 Hz, 1H, PhCH), 4.96 (d, J = 11.6 Hz, 1H, PhCH), 4.94–4.88 (m, 2H, H-1′, H-5), 4.80 (dd, J = 13.6, 11.1 Hz, 2H, 2 PhCH), 4.68 (d, J = 11.6 Hz, 1H, PhCH), 4.40 (d, J = 11.9 Hz, 1H, PhCH), 4.32 (d, J = 11.9 Hz, 1H, PhCH), 4.28–4.24 (m, 1H, H-4), 4.12 (dd, J = 11.4, 2.3 Hz, 1H, H-6b’), 4.08–3.95 (m, 2H, H-4′, H-5′), (dd, J = 11.5, 3.8 Hz, 1H, H-2), 3.93–3.81 (m, 3H, H-2′, H-6b, H-6a), 3.55 (ddd, J = 9.8, 4.9, 2.8 Hz, 2H, H-3′, H-6a’), 2.74–2.59 (m, 4H), 1.70 (s, 3H, COCH3), 1.10 (s, 9H, t-Bu-H). 13C NMR (151 MHz, CDCl3) δ 170.55 (2C), 170.01, 138.55–127.58 (Ar-C), 101.82, 99.94, 81.65, 80.91, 76.85, 75.79, 75.65, 75.47, 74.11, 72.83, 72.44, 71.28, 69.65, 67.53, 62.34, 56.88, 26.92, 25.43 (2C), 20.70, 19.45. mass spectrum (HRMS), m/z = 1127.4434 (M+Na)+, C62H68N4O13Si requires 1127.4552.
Succinimidyl [2,3,4-tri-O-benzyl-α-D-glucopyranosyl]-(1→4)-2-azido-3-O-acetate-6-O-benzyl-2-deoxy α-D-galactopyranoside (11)
To the disaccharide 10 (0.97 g, 0.88 mmol) in THF (20 ml) was added acetic acid (0.10 ml, 1.75 mmol), followed by tetrabutylammonium fluoride (TBAF) (1.08 ml, 0.87 mmol, 1.0 M in THF). After stirring at room temperature for 12 h, the solution was washed with aq. sodium bicarbonate (2 × 50 ml) and brine (2 × 50 ml) solution and extracted with DCM (2 × 50 ml). The organic layer was collected and dried over anhydrous Na2SO4, and purification of the resulting residue was done by flash chromatography, yielding the disaccharide acceptor as white solid 11: yield 60% (0.46 g); silica gel TLC Rf = 0.19 (30% ethyl acetate: hexane). 1H NMR (600 MHz, CDCl3) δ 7.29 (Ar-H, 20H), 5.62 (d, J = 3.9 Hz, 1H, H-1), 5.41 (dd, J = 11.6, 2.8 Hz, 1H, H-3), 5.03–4.85 (m, 5H, 3 PhCH, H-1′, H-5), 4.75 (d, J = 12.6 Hz, 1H, PhCH), 4.69 (d, J = 12.6 Hz, 1H, PhCH), 4.63 (d, J = 12.6 Hz, 1H, PhCH), 4.43 (d, J = 11.9 Hz, 1H, PhCH), 4.38–4.29 (m, 2H, PhCH, H-4), 4.08–3.95 (m, 3H, H-2, H-3′, H-5′), 3.88–3.73 (m, 3H, H-6a, H-6b, H-6b’), 3.59 (dd, J = 10.0, 9.0 Hz, 1H, H-4′), 3.54–3.46 (m, 2H, H-6a’, H-2′), 2.73–2.63 (m, 4H), 2.16 (s, 3H, COCH3). 13C NMR (151 MHz, CDCl3) δ 170.60 (2C), 170.13, 138.49–127.55 (Ar-C), 101.85, 99.38, 81.48, 80.27, 77.62, 75.57, 75.36, 75.17, 74.06, 72.89, 71.70, 71.00, 69.76, 67.23, 61.82, 56.97, 25.44 (2C), 21.10. mass spectrum (HRMS), m/z = 889.3259 (M+Na)+, C46H50N4O13 requires 889.3374.
Succinimidyl [2-O-acetyl-3,4-di-O-benzyl-α-L-rhamnopyranosyl]-(1→6)-[2,3,4-tri-O-benzyl-α-D-glucopyranosyl]-(1→4)-2-azido-3-O-acetate-6-O-benzyl-2-deoxy-α-D-galactopyranoside (12)
Disaccharide acceptor 11 (0.45 g, 0.52 mmol) and rhamnose thioglycoside donor B (0.30 g, 0.62 mmol) were dried together in high vacuum overnight. The compounds were dissolved in dry DCM (20 ml), followed by the addition of 4-Å molecular sieves, and stirred for 30 min. The solution temperature was lowered to −20°C, and NIS (0.17 g, 0.78 mmol) and TMSOTf (15 μL, 0.08 mmol) were added. The reaction was monitored by TLC and appeared complete after 1.5 h. The reaction temperature was raised to 0°C. After completion, the reaction was diluted with DCM (50 ml), filtered, and washed with aq. sodium thiosulfate (2 × 30 ml), aq. sodium bicarbonate (2 × 30 ml), and brine (2 × 50 ml) solution. The organic layer was collected, dried over anhydrous Na2SO4, and concentrated. The residue was purified by silica gel flash column chromatography to isolate the compound as white solid 12: yield 63% (0.40 g, α only); silica gel TLC Rf = 0.45 (30% ethyl acetate: hexane). 1H NMR (600 MHz, CDCl3) δ 7.44–7.17 (Ar-H, 30H), 5.64 (d, J = 3.8 Hz, 1H, H-1), 5.42 (ddd, J = 11.6, 2.7, 1.5 Hz, 1H, H-3), 5.27–5.24 (m, 1H, H-2″), 5.01–4.87 (m, 5H, 4 PhCH, H-5), 4.84 (brs, 1H, H-1′), 4.73 (dt, J = 11.7, 1.8 Hz, 2H, 2 PhCH), 4.67 (brs, 1H, H-1″), 4.65–4.56 (m, 4H, 4 PhCH), 4.38–4.31 (m, 3H, 2 PhCH, H-4), 4.04–3.95 (m, 3H, H-5″, H-4′, H-2), 3.93 (ddd, J = 9.4, 3.5, 1.5 Hz, 1H, H-3″), 3.83–3.76 (m, 3H, H-3′, H-6a, H-6b), 3.69–3.64 (m, 1H, H-6a’), 3.63–3.55 (m, 2H, H-5′, H-6b’), 3.48–3.42 (m, 2H, H-2′, H-4″), 2.76–2.60 (s, 4H), 2.14 (s, 6H, 2 COCH3), 1.30 (d, J = 6.2 Hz, 3H, Rha-CH3). 13C NMR (151 MHz, CDCl3) δ 170.56 (2C), 170.29, 169.99, 138.51–127.52 (Ar-C), 101.78, 99.52, 97.87, 81.50, 80.53, 80.06, 77.63, 77.47, 75.70, 75.65, 75.60, 75.42, 73.95, 72.77, 71.97, 71.25, 70.80, 69.74, 69.21, 67.75, 67.69, 66.00, 56.99, 25.43 (2C), 21.16, 21.10, 17.93. mass spectrum (HRMS), m/z = 1257.4881 (M+Na)+, C68H74N4O18 requires 1257.4998.
Succinimidyl [2-O-acetyl-3,4-di-O-benzyl-α-L-rhamnopyranosyl]-(1→6)-[2,3,4-tri-O-benzyl-α-D-glucopyranosyl]-(1→4)-2-N-benzyloxycarbonylalanine-3-O-acetate-6-O-benzyl-2-deoxy-α-D-galactopyranoside (13)
To the solution of trisaccharide 12 (0.38 g, 0.30 mmol) in dry DCM:AcOH (3:1, 12 ml), zinc dust (100 mg, 1.53 mmol) was added, and the reaction was stirred under N2 at room temperature. After 1 day, the reaction was observed to be complete by TLC. The reaction was diluted with DCM (25 ml) and washed with aq. sodium bicarbonate (2 × 30 ml) and brine (2 × 30 ml). The organic layer was separated, dried over anhydrous Na2SO4, and evaporated. The residue was dried and used for the next reaction without further purification. To the solution of the residue in dry DCM, Cbz-Ala-OH (0.14 g, 0.61 mmol), T3P (0.18 ml, 0.61 mmol), and DIPEA (0.16 ml, 0.92 mmol) were added successively at 0°C. The solution was stirred under a N2 atmosphere and allowed to warm to room temperature. The reaction appeared complete after 12 h. The reaction was diluted with DCM (30 ml) and washed with 1N HCl (2 × 15 ml), followed by aq. sodium bicarbonate (2 × 30 ml). The organic layer was collected and purified by silica gel flash column chromatography to obtain the compound as fluffy white solid 13: yield 53% over two steps (0.23 g); silica gel TLC Rf = 0.20 (50% ethyl acetate: hexane). 1H NMR (600 MHz, CDCl3) δ 7.47–7.12 (m, 36H), 6.66 (d, J = 9.5 Hz, 1H, NH), 5.38 (ddd, J = 11.6, 2.7, 1.5 Hz, 1H, H-3), 5.35 (d, J = 3.8 Hz, 1H, H-1), 5.26 (m, 1H, H-2″), 5.14 (brs, 2H, PhCH2), 5.02–4.84 (m, 6H, 4 PhCH, H-5, H-1″), 4.81–4.67 (m, 3H, 2 PhCH, H-2), 4.67–4.53 (m, 7H, 6 PhCH, H-1′), 4.45 (d, J = 12.0 Hz, 1H, PhCH), 4.41–4.29 (m, 2H, PhCH, CH), 4.30–4.19 (m, 2H, H-4, H-5′), 4.10 (t, J = 9.5 Hz, 1H, H-3′), 3.99–3.86 (m, 2H, H-3″, H-6a), 3.86–3.76 (m, 2H, H-5″, H-6a’), 3.70 (dd, J = 11.1, 3.6 Hz, 1H, H-6b’), 3.59 (t, J = 9.6 Hz, 1H, H-4′), 3.57–3.37 (m, 3H, H-6b, H-4″, H-2′), 2.67 (s, 4H), 2.10 (2 s, 6H, 2 COCH3), 1.45 (d, J = 7.1 Hz, 3H, Ala-CH3), 1.30 (d, J = 6.2 Hz, 3H, Rha-CH3). 13C NMR (151 MHz, CDCl3) δ 172.61 (2C), 170.73, 170.26, 138.72–127.50, 103.92, 98.85, 97.77, 81.66, 80.50, 80.14, 77.75, 77.71, 75.69, 75.60, 75.07, 74.13, 73.90, 72.82, 71.90, 71.36, 70.57, 69.13, 68.48, 67.60, 67.13, 66.08, 50.94, 47.94, 25.40 (2C), 21.14, 21.07, 18.17, 17.95. mass spectrum (HRMS), m/z = 1436.5712 (M+Na)+, C79H87N3O21 requires 1436.5832.
Aminooxy [α-L-rhamnopyranosyl]-(1→6)-[α-D-glucopyranosyl]-(1→4)-2-N-alanine-2-deoxy-α-D-galactopyranoside (3)
To a solution of compound 13 (0.21 g, 0.15 mmol) in dry MeOH (10 ml) was added 25 mg of Pd/C (5 wt%). The reaction was stirred for 6 h under 1 atm of H2. The reaction was observed to be complete by TLC and ESI-MS. The reaction was diluted with MeOH (15 ml) and filtered through Celite 545. The filtrate was evaporated and dried and used for the next reaction without further purification. The residue (0.06 g, 0.08 mmol) was dissolved in methanol (3 ml), and then hydrazine hydrate (0.08 ml, 1.60 mmol) was added and the reaction was stirred for 10 h. The reaction mixture was then concentrated to dryness. This residue was dissolved in a minimal amount of water and purified using P-2 biogel with water as the eluent (collecting ∼0.5 ml fractions), to provide white solid 3: yield 22% (10 mg); silica gel TLC Rf = 0.20 (50% methanol: DCM). 1H NMR (600 MHz, D2O) δ 1H NMR (600 MHz, D2O) δ 4.93 (brs, 1H, H-1″), 4.88 (brs, 1H, H-1), 4.85 (brs, 1H, H-1′), 4.10–4.02 (m, 2H, H-3, H-5), 3.97–3.93 (m, 3H, H-2, H-5″, H-3″), 3.86–3.82 (m, 2H, H-5′, H-4′), 3.70–3.66 (m, 5H, H-4″, CH, H-2′, H-3′, H-2″), 3.50–3.46 (m, 2H, H-6a, H-6b), 3.37–3.30 (m, 3H, H-4, H-6a’, H-6b’), 1.45 (d, J = 7.1 Hz, 3H, Ala-CH3), 1.17 (d, J = 6.3 Hz, 3H, Rha-CH3). 13C NMR (151 MHz, D2O) δ 171.89, 100.37, 100.22 (2C), 72.50, 71.92, 71.60, 71.24, 70.99, 70.12, 69.88, 69.23, 68.50, 67.12, 65.95, 60.29 (2C), 49.21 (2C), 16.50 (2 C). Data for. mass spectrum (HRMS), m/z = 574.2451 (M + H)+, C21H39N3O15 requires 574.2381.
p-Methylphenyl 2-azido-4,6-O-benzylidene-3-O-chloroacetyl-2-deoxy-1-thiol-α/β-D-galactopyranoside (14α/β)
A solution of the compound p-methylphenyl 2-azido-4,6-O-benzylidene-2-deoxy-1-thio-α/β-D-galactopyranoside (5α/β) (Santra et al., 2012) (1.50 g, 3.75 mmol) in dry DCM (20 ml) and pyridine (0.30 ml, 3.75 mmol) was cooled to 0°C. To the cooled reaction mixture was added chloroacetyl chloride (0.36 ml, 4.52 mmol) using a syringe, and the reaction mixture was stirred for another 2 h at room temperature. After completion, the reaction mixture was diluted with DCM (50 ml). The organic layer was washed successively with satd. sodium bicarbonate (2 × 100 ml) and brine (2 × 100 ml) solution. The organic layer was collected, dried over anhydrous Na2SO4, and concentrated. The residue was purified by silica gel flash column chromatography to isolate the compound as white solid 14α/β: yield 85% (1.51 g); silica gel TLC Rf = 0.60 (50% ethyl acetate: hexane). All the spectral data match with reported data (Zhang et al., 1999; Sarkar et al., 2003).
Succinimidyl 2-azido-4,6-O-benzylidene-3-O-chloroacetyl-2-deoxy-α-D-galactopyranoside (15)
Both the 14α/β compound (1.40 g, 2.94 mmol) and N-hydroxy succinimide (7) (0.34 g, 2.91 mmol) were mixed in a flame-dried round bottom flask and left under high vacuum overnight. To the dried reagents were added flame-dried 4-Å molecular sieves and 20 ml of anhydrous DCM. This mixture was then stirred at room temperature for 15 min. NIS (0.78 g, 3.50 mmol) was added to it, and the reaction mixture was cooled to 0°C. Subsequently, (80 μL, 0.44 mmol) TMSOTf was added. After 45 min, the reaction was diluted with DCM (50 ml), filtered, and washed with aq. sodium thiosulfate (2 × 50 ml), aq. sodium bicarbonate (2 × 50 ml), and brine (2 × 100 ml) solution. The organic layer was collected, dried over anhydrous Na2SO4, and concentrated. The residue was purified by silica gel flash column chromatography to isolate the compound as white solid 15: yield 70% (0.96 g); silica gel TLC Rf = 0.30 (50% ethyl acetate: hexane). 1H NMR (600 MHz, CDCl3) δ 7.49–7.40 (Ar-H, 5H), 5.64 (t, J = 2.7 Hz, 1H, H-1), 5.58 (s, 1H, PhCH), 5.52–5.42 (m, 1H, H-5), 4.75 (t, J = 1.7, 1.7 Hz, 1H, H-3), 4.62 (dt, J = 3.3, 1.6 Hz, 1H, H-4), 4.27–4.17 (m, 4H, H-2, H-6a, COCH2), 4.11 (dt, J = 12.8, 1.9 Hz, 1H, H-6b), 2.80 (s, 4H). 13C NMR (151 MHz, CDCl3) δ 170.72, 166.76, 137.18, 129.30, 128.33, 126.08, 102.71, 100.82, 72.77, 70.78, 68.82, 64.54, 56.30, 40.65, 25.48. mass spectrum (HRMS), m/z = 498.0857 (M+Na)+, C22H22ClN3O5S requires 498.0784.
Succinimidyl 2-azido-4,6-O-benzylidene-2-deoxy-α-D-galactopyranoside (16)
To a solution of compound 15 (0.93 g, 1.99 mmol) in MeOH:pyridine (90 ml, 5:1) was added thiourea (0.45 g, 5.98 mmol), and this reaction mixture was stirred overnight at room temperature. After completion of the reaction as shown in TLC, the reaction mixture was concentrated and co-evaporated with toluene two times. The residue was diluted with DCM (50 ml) and successively washed with aq. sodium bicarbonate (2 × 100 ml) and brine (2 × 100 ml) solution. The organic layer was collected, dried over anhydrous Na2SO4, and concentrated. The residue was purified by silica gel flash column chromatography to isolate the compound as white solid 16: yield 65% (0.51 g); silica gel TLC Rf = 0.25 (50% ethyl acetate: hexane). 1H NMR (600 MHz, CDCl3) δ 7.54–7.46 (Ar-H, 2H), 7.46–7.37 (Ar-H, 4H), 5.61 (s, 1H, PhCH), 5.57 (d, J = 3.7 Hz, 1H, H-1), 4.69 (s, 1H, H-5), 4.39 (d, J = 3.7 Hz, 1H, H-4), 4.28 (d, J = 3.6 Hz, 1H, H-3), 4.21 (dd, J = 12.9, 1.3 Hz, 1H, H-6a), 4.10 (dd, J = 12.9, 1.6 Hz, 1H, H-6b), 3.88 (dd, J = 11.0, 3.5 Hz, 1H, H-2), 2.76 (s, 4H). 13C NMR (151 MHz, CDCl3) δ 170.89, 137.20, 129.49, 128.42, 126.22, 103.01, 101.29, 77.28, 77.07, 76.85, 75.07, 68.96, 67.28, 64.85, 59.76, 25.48. All the spectral data match with reported data (Bourgault et al., 2014).
Succinimidyl [2-O-benzoyl-4,6-O-benzylidene-3-O-(p-methoxy)benzyl-β-D-glucopyranosyl]-(1→3)-2-azido-4,6-O-benzylidene-2-deoxy-α-D-galactopyranoside (17)
The acceptor 16 (0.50 g, 1.28 mmol) and thioglycoside donor C (0.92 g, 1.53 mmol) were dissolved in dry DCM (15 ml) and stirred using 4-Å molecular sieves for 10 min. NIS (0.42 g, 1.88 mmol) and TMSOTf (40 μL, 0.21 mmol) were added to the reaction at −50°C. The solution was then allowed to warm to −30°C. The reaction was stirred under N2 and observed to be complete after 2 h. The reaction was diluted with DCM (30 ml) and filtered. The filtrate was washed with aq. sodium thiosulfate (2 × 30 ml), aq. sodium bicarbonate (2 × 30 ml), and brine (2 × 50 ml) solution. The organic layer was collected and dried over anhydrous Na2SO4, and the resulting residue was subjected to silica gel flash column chromatography to afford the compound as white solid 17: yield 70% (0.78 g β only); silica gel TLC Rf = 0.50 (50% ethyl acetate: hexane). 1H NMR (600 MHz, CDCl3) δ 8.03–7.97 (ArH, 2H), 7.57–7.28 (ArH, 22H), 7.06–6.99 (ArH, 2H), 6.60–6.55 (ArH, 2H), 5.56 (s, 1H, PhCH), 5.57 (s, 1H, PhCH), 5.53 (d, J = 3.7 Hz, 1H, H-1), 5.33 (t, J = 8.2, 8.2 Hz, 1H, H-2′), 4.93 (d, J = 7.7 Hz, 1H, H-1′), 4.72 (d, J = 11.8 Hz, 1H, PhCH), 4.62–4.54 (m, 2H, PhCH, H-5), 4.45–4.37 (m, 2H, H-4, H-5′), 4.19–4.12 (m, 2H, H-6a, H-3), 4.03 (ddd, J = 15.0, 11.9, 2.7 Hz, 2H, H-6b, H-2), 3.89–3.84 (m, 3H, H-6a’, H-4′, H-3′), 3.67 (s, 3H), 3.57–3.52 (m, 1H, H-6b’), 2.73 (s, 4H). 13C NMR (151 MHz, CDCl3) δ 170.94 (2C), 165.06, 159.04, 137.35–126.05 (Ar-C) 113.52, 103.04, 102.83, 101.29, 100.71, 81.18, 75.54, 74.36, 73.55, 73.09, 68.79, 68.63, 66.40, 65.19, 57.87, 55.12, 25.46 (2C). mass spectrum (HRMS), m/z = 887.2742 (M+Na)+, C45H44N4O14 requires 887.2746.
Succinimidyl [2-O-benzoyl-4,6-O-benzylidene-β-D-glucopyranosyl]-(1→3)-2-azido-4,6-O-benzylidene-2-deoxy-α-D-galactopyranoside (18)
The solution of disaccharide 17 (0.76 g, 0.88 mmol) in DCM–H2O (40 ml, 5:1) was cooled to 0°C. To the cooled reaction mixture was added 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (0.60 g, 2.63 mmol). The mixture was heated to room temperature and was stirred for 12 h. Then it was quenched with saturated aq. sodium bicarbonate (2 × 50 ml) and brine (2 × 50 ml), extracted with DCM, and dried over anhydrous Na2SO4, and the solvent was concentrated in vacuum. The residue was subjected to silica gel flash column chromatography to afford the compound as white solid 18: yield 57% (0.37 g); silica gel TLC Rf = 0.40 (50% ethyl acetate: hexane). 1H NMR (600 MHz, CDCl3) δ 8.18–8.10 (Ar-H, 2H), 7.59–7.36 (Ar-H, 18H), 5.61–5.54 (m, 3H, 2 PhCH, H-1), 5.31 (dd, J = 8.9, 7.7 Hz, 1H, H-2′), 5.07 (d, J = 7.7 Hz, 1H, H-1′), 4.62 (brs, J = 1.3 Hz, 1H, H-5), 4.48–4.40 (m, 2H, H-4, H-6a’), 4.24 (dd, J = 11.1, 3.1 Hz, 1H, H-3), (4.22–4.07 (m, 4H, H-6a, H-6b, H-2, H-3′), 3.87 (t, J = 10.3 Hz, 1H, H-6b’), 3.77 (t, J = 9.4 Hz, 1H, H-4′), 3.61 (d, J = 4.9 Hz, 1H, H-5′), 2.76 (s, 4H). 13C NMR (151 MHz, CDCl3) δ 170.88 (2C), 166.15, 137.39, 136.80, 133.41, 129.93, 129.02, 128.48, 128.40, 128.21, 126.32, 126.12, 102.99, 102.53, 101.99, 100.69, 80.61, 77.26, 77.05, 76.84, 75.60, 74.81, 74.29, 72.70, 68.78, 68.55, 66.28, 65.23, 60.44, 57.99, 25.47 (2C). mass spectrum (HRMS), m/z = 767.2166 (M+Na)+, C37H36N4O13 requires 767.2171.
Succinimidyl [2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl]-(1→3)-[2-O-benzoyl-4,6-O-benzylidene-β-D-glucopyranosyl]-(1→3)-2-azido-4,6-O-benzylidene-2-deoxy-α-D-galactopyranoside (19)
Disaccharide acceptor 18 (0.35 g, 0.47 mmol) and donor D (0.22 g, 0.56 mmol) were dried together in high vacuum overnight. The compounds were dissolved in dry DCM (10 ml), followed by the addition of 4-Å molecular sieves, and stirred for 30 min. The solution temperature was lowered to −20°C, and NIS (0.15 g, 0.67 mmol) and TMSOTf (12 μL, 0.06 mmol) were added. The reaction was monitored by TLC and appeared complete after 30 min. The reaction temperature was raised to 0°C. After completion, the reaction was diluted with DCM (50 ml), filtered, and washed with aq. sodium thiosulfate (2 × 30 ml), aq. sodium bicarbonate (2 × 30 ml), and brine (2 × 30 ml) solution. The organic layer was collected, dried over anhydrous Na2SO4, and concentrated. The residue was purified by silica gel flash column chromatography to isolate the compound as white solid 19: yield 65% (0.31 g); silica gel TLC Rf = 0.30 (50% ethyl acetate: hexane). 1H NMR (600 MHz, CDCl3) δ 8.13–7.96 (Ar-H, 2H), 7.46 (Ar-H, 10H), 5.59 (s, 1H, PhCH), 5.52 (d, J = 3.7 Hz, 1H, H-1), 5.50 (s, 1H, PhCH), 5.44 (dd, J = 8.2, 7.4 Hz, 1H, H-2′), 5.27 (dd, J = 10.1, 3.6 Hz, 1H, H-3″), 5.08 (dd, J = 3.6, 1.7 Hz, 1H, H-2″), 5.06 (d, J = 7.3 Hz, 1H, H-1′), 4.87 (t, J = 10.0 Hz, 1H, H-4″), 4.83 (brs, 1H, H-1″), 4.62–4.60 (m, 1H, H-5), 4.47 (dd, J = 3.3, 1.2 Hz, 1H, H-4), 4.44 (dd, J = 10.5, 4.9 Hz, 1H, H-6a’), 4.22 (dd, J = 11.0, 3.2 Hz, 1H, H-3), 4.19–4.05 (m, 5H, H-5″, H-3′, H-2, H-6a, H-6b), 3.92 (t, J = 9.4 Hz, 1H, H-4′), 3.85 (t, J = 10.3 Hz, 1H, H-6b’), 3.66 (td, J = 9.8, 5.0 Hz, 1H, H-5′), 2.74 (s, 4H), 1.95 (2s, 6H, 2COCH3), 1.82 (s, 3H, COCH3), 0.75 (d, J = 6.2 Hz, 3H, Rha-CH3). 13C NMR (151 MHz, CDCl3) δ 170.90 (2C), 169.91, 169.86, 169.07, 164.69, 137.37-126.11 (Ar-C), 102.96, 102.27, 101.81, 100.58, 97.78, 78.43, 77.29, 77.08, 76.87, 76.22, 75.45, 74.43, 74.16, 71.04, 69.36, 68.81, 68.73, 68.65, 66.61, 66.36, 65.15, 57.97, 25.45 (2C), 20.75, 20.68, 20.47, 17.30. mass spectrum (HRMS), m/z = 1039.3059 (M+Na)+, C49H52N4O20 requires 1039.3067.
Succinimidyl [2,3,4-tri-O-acetyl-α-L-rhamno-pyranosyl]-(1→3)-[4,6-O-acetyl-2-O-benzoyl-β-D-glucopyranosyl]-(1→3)-2-azido-4,6-O-acetyl-2-deoxy-α-D-galactopyranoside (20)
The solution of compound 19 (0.30 g, 0.30 mmol) in 80% acetic acid (50 ml) was stirred at 80°C for 2 h, and the solvents were evaporated and co-evaporated with toluene (2 × 10 ml). To the solution of the crude compound in pyridine (2 ml) was added acetic anhydride (1 ml), and the reaction mixture was stirred at room temperature for 6 h. The solvents were removed under reduced pressure to yield a residue which was purified by silica gel flash column chromatography to isolate pure trisaccharide as white solid 20: yield 40% over two steps (0.12 g); silica gel TLC Rf = 0.15 (50% ethyl acetate: hexane). 1H NMR (600 MHz, CDCl3) δ 8.12–7.97 (Ar-H, 2H), 7.61–7.43 (Ar-H, 3H), 5.62 (dd, J = 3.4, 1.3 Hz, 1H, H-4), 5.45 (d, J = 3.9 Hz, 1H, H-1), 5.30 (dd, J = 9.5, 7.8 Hz, 1H, H-2′), 5.18 (t, J = 9.6 Hz, 1H, H-4′), 5.14–5.09 (m, 1H, H-2″), 4.92–4.84 (m, 5H, H-3, H-5, H-3″, H-1′, H-1″), 4.32 (dd, J = 11.8, 4.6 Hz, 1H, H-6a), 4.23–4.14 (m, 3H, H-5″, H-6a’, H-6b’), 4.04 (t, J = 9.3 Hz, 1H, H-3′), 3.83 (ddd, J = 12.0, 8.8, 5.4 Hz, 3H, H-2, H-6b, H-4″), 3.67 (ddd, J = 10.0, 4.8, 2.8 Hz, 1H, H-5′), 2.74 (s, 4H), 2.17 (s, 3H, COCH3), 2.14 (s, 3H, COCH3), 2.11 (s, 3H, COCH3), 2.07 (s, 3H, COCH3), 2.01 (s, 3H, COCH3),1.88 (s, 3H COCH3), 1.81 (s, 3H, COCH3), 1.16 (d, J = 6.3 Hz, 3H, Rha-CH3). 13C NMR (151 MHz, CDCl3) δ 170.89 (2C), 170.52, 170.46, 170.05, 169.49, 169.44, 169.28, 169.12, 164.52, 133.24, 129.97, 129.08, 128.31 (Ar-C), 102.21, 101.18, 99.14, 79.95, 73.39, 72.87, 72.26, 70.86, 69.84, 69.78, 69.24, 69.07, 68.18, 67.36, 62.34, 61.87, 58.53, 25.41 (2C), 20.95 (2C), 20.83, 20.78, 20.64, 20.59, 20.45, 17.32. mass spectrum (HRMS), m/z = 1031.2860 (M+Na)+, C43H52N4O24 requires 1031.2864.
Succinimidyl [2,3,4-tri-O-acetyl-α-L-rham-nopyranosyl]-(1→3)-[4,6-O-acetyl-2-O-benzoyl-β-D-glucopyranosyl]-(1→3)-2-N-tertbutyloxycarbonylalanine-4,6-O-acetyl-2-deoxy-α-D-galactopyranoside (21)
To a solution of trisaccharide 20 (0.10 g, 0.10 mmol) in dry DCM:AcOH (3:1, 8 ml), zinc dust (0.07 g, 1.01 mmol) was added, and the reaction was stirred under N2 at room temperature. After 1 day, the reaction was observed to be complete by TLC. The reaction was diluted with DCM (15 ml) and washed with aq. sodium bicarbonate (2 × 30 ml) and brine (2 × 30 ml) solution. The organic layer was separated, dried over anhydrous Na2SO4, and evaporated. The residue was dried and used for the next reaction without further purification. The residue was dissolved in dry DCM (3 ml), and Boc-Ala-OH (0.04 g, 0.20 mmol), T3P (0.06 ml, 0.20 mmol), and DIPEA (0.05 ml, 0.30 mmol) were successively added at 0°C. The solution was stirred under a N2 atmosphere and allowed to warm to room temperature. The reaction appeared complete after 12 h. The reaction was diluted with DCM (20 ml) and washed with 1N HCl (2 × 15 ml), followed by aq. sodium bicarbonate (2 × 20 ml) solution. The organic layer was collected and subjected to silica gel flash column chromatography to afford the compound as fluffy white solid 21: yield 66% over two steps (75.0 mg); silica gel TLC Rf = 0.50 (100% ethyl acetate). 1H NMR (600 MHz, CDCl3) δ 8.13–8.02 (Ar-H, 2H), 7.55–7.39 (Ar-H, 3H), 6.74 (d, J = 9.3 Hz, 1H, NH), 5.58 (dd, J = 3.2, 1.3 Hz, 1H, H-4), 5.32–5.29 (m, 1H, CH), 5.27–5.14 (m, 3H, H-4′, H-2′, H-1), 5.10 (dd, J = 10.0, 2.8 Hz, 1H, H-2″), 4.92–4.84 (m, 4H, H-1′, H-3″, H-5, H-1″), 4.58 (ddd, J = 11.2, 9.2, 3.9 Hz, 1H, H-2), 4.33 (dd, J = 11.7, 4.7 Hz, 1H, H-6a), 4.20 (d, J = 4.3 Hz, 2H, H-6a’, H-6b’), 4.11–4.03 (m, 2H, H-3, H-3′), 4.00–3.91 (m, 1H, H-5″), 3.88–3.83 (m, 2H, H-6b, H-4″), 3.68–3.65 (m, 1H, H-5′), 2.72 (s, 4H), 2.17 (s, 3H, COCH3), 2.11 (s, 3H, COCH3), 2.10 (s, 3H, COCH3), 2.09 (s, 3H, COCH3), 2.00 (s, 3H, COCH3), 1.88 (s, 3H, COCH3), 1.79 (s, 3H, COCH3), 1.44 (s, 12H, Ala-CH3, tBu-H), 1.15 (d, J = 6.3 Hz, 3H, Rha-CH3). 13C NMR (151 MHz, CDCl3) δ 172.33, 170.96 (2C), 170.69, 170.48, 170.08, 169.94, 169.33, 169.23, 169.04, 164.39, 133.03, 130.15, 129.31, 128.19, 104.34, 100.20, 99.09, 79.92, 79.83, 72.93, 72.04, 71.66, 70.92, 69.78, 69.76, 69.08, 68.75, 68.22, 67.28, 62.42, 61.65, 53.46, 48.31, 28.38 (3C), 25.35 (2C), 20.94 (2C), 20.87, 20.79, 20.60, 20.58, 20.42, 17.33 (2C). mass spectrum (HRMS), m/z = 1176.3854 (M+Na)+, C51H67N3O27 requires 1176.3854.
Aminooxy [α-L-rhamnopyranosyl]-(1→3)-[β-D-glucopyranosyl]-(1→3)-2-N-alanine-2-deoxy-α-D-galactopyranoside (4)
Compound 21 (60.0 mg, 0.05 mmol) was dissolved in trifluoroacetic acid (TFA)/DCM (3 ml, 1:1) and stirred for 1 h. The mixture was diluted with DCM (10 ml) and washed with sodium bicarbonate solution (2 × 20 ml); the organic layer was separated, dried over Na2SO4, and filtered. The filtrate was evaporated, dried, and used for the next reaction without further purification. The residue (40.0 mg, 0.03 mmol) was dissolved in methanol (3 ml), and then hydrazine hydrate (0.03 ml, 0.62 mmol) was added and the reaction was stirred for 10 h. The reaction mixture was then concentrated to dryness. This residue was dissolved in a minimal amount of water and purified using P-2 biogel with water as the eluent (collecting ∼0.2 ml fractions), to provide as a white solid 4: yield 35% over two steps (10 mg); silica gel TLC Rf = 0.20 (50% methanol: DCM). 1H NMR (600 MHz, D2O) δ 5.00 (brs, 1H, H-1″), 4.86 (d, J = 4.0 Hz, 1H, H-1), 4.43 (d, J = 8.0 Hz, 1H, H-1′), 4.29 (dd, J = 11.3, 4.1 Hz, 1H, H-2), 4.17 (dd, J = 3.1, 1.2 Hz, 1H, H-4), 3.99–3.82 (m, 4H, H-5, H-3, H-2″, CH), 3.76 (dd, J = 12.4, 2.3 Hz, 1H, H-6a), 3.72–3.59 (m, 5H, H-3″, H-5″, H-6b, H-6a’, H-6b’), 3.44 (t, J = 9.1 Hz, 1H, H-3′), 3.40–3.23 (m, 4H, H-2′, H-4′, H-4″, H-5′), 1.41 (d, J = 7.1 Hz, 3H, Ala-CH3), 1.13 (d, J = 6.3 Hz, 3H, Rha-CH3). 13C NMR (151 MHz, D2O) δ 171.64, 103.50, 100.91, 100.44, 82.06, 76.69, 75.61, 73.53, 71.86, 70.60, 70.25, 70.11, 68.75, 68.41, 67.64, 61.01, 60.30, 49.32, 48.03, 16.77, 16.38. mass spectrum (HRMS), m/z = 574.2444 (M + H)+, C21H39N3O15 requires 574.2453.
Data Availability Statement
The original contributions presented in the study are included in the Supplementary Material; further inquiries can be directed to the corresponding author.
Author Contributions
The experiments were performed in the laboratory of SS at the University of Toledo. AS and SS designed the experiments, wrote the manuscript, and critically revised the manuscript; AS performed and analyzed the experiments.
Funding
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases (NIAID) (AI148570) to SS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.750502/full#supplementary-material
References
Agten, S. M., Dawson, P. E., and Hackeng, T. M. (2016). Oxime Conjugation in Protein Chemistry: from Carbonyl Incorporation to Nucleophilic Catalysis. J. Pept. Sci. 22 (5), 271–279. doi:10.1002/psc.2874
Bourgault, J. P., Trabbic, K. R., Shi, M., and Andreana, P. R. (2014). Synthesis of the Tumor Associative α-aminooxy Disaccharide of the TF Antigen and its Conjugation to a Polysaccharide Immune Stimulant. Org. Biomol. Chem. 12 (11), 1699–1702. doi:10.1039/C4OB00128A
Bystrova, O. V., Knirel, Y. A., Lindner, B., Kocharova, N. A., Kondakova, A. N., Zähringer, U., et al. (2006). Structures of the Core Oligosaccharide and O-Units in the R- and SR-type Lipopolysaccharides of Reference Strains of Pseudomonas aeruginosa O-Serogroups. FEMS Microbiol. Immunol. 46 (1), 85–99. doi:10.1111/j.1574-695x.2005.00004.x
Bystrova, O. V., Lindner, B., Moll, H., Kocharova, N. A., Knirel, Y. A., Zähringer, U., et al. (2004). Full Structure of the Lipopolysaccharide of Pseudomonas aeruginosa Immunotype 5. Biochemistry (Moscow) 69 (2), 170–175. doi:10.1023/b:biry.0000018947.60328.8d
Bystrova, O. V., Lindner, B., Moll, H., Kocharova, N. A., Knirel, Y. A., Zähringer, U., et al. (2003). Structure of the Lipopolysaccharide of Pseudomonas aeruginosa O-12 with a Randomly O-Acetylated Core Region. Carbohydr. Res. 338 (18), 1895–1905. doi:10.1016/s0008-6215(03)00290-8
Bystrova, O. V., Shashkov, A. S., Kocharova, N. A., Knirel, Y. A., Lindner, B., Zähringer, U., et al. (2002). Structural Studies on the Core and the O-Polysaccharide Repeating Unit of Pseudomonas aeruginosa Immunotype 1 Lipopolysaccharide. Eur. J. Biochem. 269 (8), 2194–2203. doi:10.1046/j.1432-1033.2002.02875.x
De Silva, R. A., Wang, Q., Chidley, T., Appulage, D. K., and Andreana, P. R. (2009). Immunological Response from an Entirely Carbohydrate Antigen: Design of Synthetic Vaccines Based on Tn−PS A1 Conjugates. J. Am. Chem. Soc. 131 (28), 9622–9623. doi:10.1021/ja902607a
Debenham, S. D., and Toone, E. J. (2000). Regioselective Reduction of 4,6-O-Benzylidenes Using Triethylsilane and BF3·Et2O. Tetrahedron: Asymmetry 11 (2), 385–387. doi:10.1016/S0957-4166(99)00584-4
Ghosh, S., and Andreana, P. R. (2014). Synthesis of an Aminooxy Derivative of the Trisaccharide Globotriose Gb3. J. Carbohydr. Chem. 33 (7-8), 381–394. doi:10.1080/07328303.2014.925913
Ghosh, S., Nishat, S., and Andreana, P. R. (2016). Synthesis of an Aminooxy Derivative of the Tetrasaccharide Repeating Unit of Streptococcus Dysgalactiae 2023 Polysaccharide for a PS A1 Conjugate Vaccine. J. Org. Chem. 81 (11), 4475–4484. doi:10.1021/acs.joc.6b00195
Ghosh, S., Trabbic, K. R., Shi, M., Nishat, S., Eradi, P., Kleski, K. A., et al. (2020). Chemical Synthesis and Immunological Evaluation of Entirely Carbohydrate Conjugate Globo H-PS A1. Chem. Sci. 11 (48), 13052–13059. doi:10.1039/D0SC04595K
He, H., Chen, D., Li, X., Li, C., Zhao, J.-H., and Qin, H.-B. (2019). Synthesis of Trisaccharide Repeating Unit of Fucosylated Chondroitin Sulfate. Org. Biomol. Chem. 17 (11), 2877–2882. doi:10.1039/C9OB00057G
Jiaang, W.-T., Chang, M.-Y., Tseng, P.-H., and Chen, S.-T. (2000). A Concise Synthesis of the O-Glycosylated Amino Acid Building Block; Using Phenyl Selenoglycoside as a Glycosyl Donor. Tetrahedron Lett. 41 (17), 3127–3130. doi:10.1016/S0040-4039(00)00367-1
Kleski, K. A., Shi, M., Lohman, M., Hymel, G. T., Gattoji, V. K., and Andreana, P. R. (2020). Synthesis of an Aminooxy Derivative of the GM3 Antigen and its Application in Oxime Ligation. J. Org. Chem. 85 (24), 16207–16217. doi:10.1021/acs.joc.0c00320
Klevens, R. M., Edwards, J. R., Gaynes, R. P., and System, N. N. I. S. (2008). The Impact of Antimicrobial‐Resistant, Health Care-Associated Infections on Mortality in the United States. Clin. Infect. Dis. 47 (7), 927–930. doi:10.1086/591698
Knirel, Y. A., Bystrova, O. g. V., Shashkov, A. S., Lindner, B., Kocharova, N. A., Senchenkova, S. y. N., et al. (2001). Structural Analysis of the Lipopolysaccharide Core of a Rough, Cystic Fibrosis Isolate of Pseudomonas aeruginosa. Eur. J. Biochem. 268 (17), 4708–4719. doi:10.1046/j.1432-1327.2001.02396.x
Komarova, B. S., Tsvetkov, Y. E., Knirel, Y. A., Zähringer, U., Pier, G. B., and Nifantiev, N. E. (2006). Synthesis of a Common Trisaccharide Fragment of Glycoforms of the Outer Core Region of the Pseudomonas aeruginosa Lipopolysaccharide. Tetrahedron Lett. 47 (21), 3583–3587. doi:10.1016/j.tetlet.2006.03.045
Komarova, B. S., Tsvetkov, Y. E., Pier, G. B., and Nifantiev, N. E. (2008). First Synthesis of Pentasaccharide Glycoform I of the Outer Core Region of the Pseudomonas aeruginosa Lipopolysaccharide. J. Org. Chem. 73 (21), 8411–8421. doi:10.1021/jo801561p
Komarova, B. S., Tsvetkov, Y. E., Pier, G. B., and Nifantiev, N. E. (2012). Synthesis of Pentasaccharides Corresponding to the Glycoform II of the Outer Core Region of the Pseudomonas aeruginosa Lipopolysaccharide. Carbohydr. Res. 360, 56–68. doi:10.1016/j.carres.2012.07.019
Konradsson, P., Udodong, U. E., and Fraser-Reid, B. (1990). Iodonium Promoted Reactions of Disarmed Thioglycosides. Tetrahedron Lett. 31 (30), 4313–4316. doi:10.1016/S0040-4039(00)97609-3
Lemieux, R. U., and Ratcliffe, R. M. (1979). The Azidonitration of Tri-O-acetyl-D-galactal. Can. J. Chem. 57 (10), 1244–1251. doi:10.1139/v79-203
Lister, P. D., Wolter, D. J., and Hanson, N. D. (2009). Antibacterial-Resistant Pseudomonas aeruginosa : Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms. Clin. Microbiol. Rev. 22 (4), 582–610. doi:10.1128/cmr.00040-09
Liu, P. V., Matsumoto, H., Kusama, H., and Bergan, T. (1983). Survey of Heat-Stable, Major Somatic Antigens of Pseudomonas aeruginosa. Int. J. Syst. Bacteriol. 33 (2), 256–264. doi:10.1099/00207713-33-2-256
Liu, P. V., and Wang, S. (1990). Three New Major Somatic Antigens of Pseudomonas aeruginosa. J. Clin. Microbiol. 28 (5), 922–925. doi:10.1128/jcm.28.5.922-925.1990
Marcaurelle, L. A., Shin, Y., Goon, S., and Bertozzi, C. R. (2001). Synthesis of Oxime-Linked Mucin Mimics Containing the Tumor-Related Tn and Sialyl Tn Antigens. Org. Lett. 3 (23), 3691–3694. doi:10.1021/ol0166247
Masaki, M., Kitahara, T., Kurita, H., and Ohta, M. (1968). A New Method for the Removal of Chloroacetyl Groups. J. Am. Chem. Soc. 90 (16), 4508–4509. doi:10.1021/ja01018a088
Mukhopadhyay, B., Kartha, K. P. R., Russell, D. A., and Field, R. A. (2004). Streamlined Synthesis of Per-O-Acetylated Sugars, Glycosyl Iodides, or Thioglycosides from Unprotected Reducing Sugars1. J. Org. Chem. 69 (22), 7758–7760. doi:10.1021/jo048890e
Nicolaou, K. C., Seitz, S. P., and Papahatjis, D. P. (1983). A Mild and General Method for the Synthesis of O-glycosides. J. Am. Chem. Soc. 105 (8), 2430–2434. doi:10.1021/ja00346a053
Oikawa, Y., Yoshioka, T., and Yonemitsu, O. (1982). Specific Removal of O-Methoxybenzyl protection by DDQ Oxidation. Tetrahedron Lett. 23 (8), 885–888. doi:10.1016/S0040-4039(00)86974-9
Pier, G. (2007). Pseudomonas aeruginosa Lipopolysaccharide: a Major Virulence Factor, Initiator of Inflammation and Target for Effective Immunity. Int. J. Med. Microbiol. 297 (5), 277–295. doi:10.1016/j.ijmm.2007.03.012
Raetz, C. R. H., and Whitfield, C. (2002). Lipopolysaccharide Endotoxins. Annu. Rev. Biochem. 71 (1), 635–700. doi:10.1146/annurev.biochem.71.110601.135414
Rajput, V. K., and Mukhopadhyay, B. (2008). Concise Synthesis of a Pentasaccharide Related to the Anti-leishmanial Triterpenoid Saponin Isolated from Maesa Balansae. J. Org. Chem. 73 (17), 6924–6927. doi:10.1021/jo801171f
Renaudet, O., and Dumy, P. (2004). Chemoselectively Template-Assembled Glycopeptide Presenting Clustered Cancer Related T-Antigen. Tetrahedron Lett. 45 (1), 65–68. doi:10.1016/j.tetlet.2003.10.126
Rocchetta, H. L., Burrows, L. L., and Lam, J. S. (1999). Genetics of O-Antigen Biosynthesis in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 63 (3), 523–553. doi:10.1128/mmbr.63.3.523-553.1999
Sadovskaya, I., Brisson, J.-R., Lam, J. S., Richards, J. C., and Altman, E. (1998). Structural Elucidation of the Lipopolysaccharide Core Regions of the Wild-type Strain PAO1 and O-Chain-Deficient Mutant Strains AK1401 and AK1012 from Pseudomonas aeruginosa Serotype O5. Eur. J. Biochem. 255 (3), 673–684. doi:10.1046/j.1432-1327.1998.2550673.x
Sadovskaya, I., Brisson, J.-R., Thibault, P., Richards, J. C., Lam, J. S., and Altman, E. (2000). Structural Characterization of the Outer Core and the O-Chain Linkage Region of Lipopolysaccharide from Pseudomonas aeruginosa Serotype O5. Eur. J. Biochem. 267(6), 1640–1650. doi:10.1046/j.1432-1327.2000.01156.x
Santra, A., Ghosh, T., and Misra, A. K. (2012). Expedient Synthesis of Two Structurally Close Tetrasaccharides Corresponding to the O-Antigens of Escherichia coli O127 and Salmonella enterica O13. Tetrahedron: Asymmetry 23 (18-19), 1385–1392. doi:10.1016/j.tetasy.2012.08.006
Sarkar, K., Mukherjee, I., and Roy, N. (2003). Synthesis of the Trisaccharide Repeating Unit of the O‐Antigen Related to the Enterohemorrhagic Escherichia coli Type O26:H. J. Carbohydr. Chem. 22 (2), 95–107. doi:10.1081/car-120020480
Talbot, G. H., Bradley, J., Edwards, J. E., Gilbert, D., Scheld, M., and Bartlett, J. G. (2006). Bad Bugs Need Drugs: an Update on the Development Pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin. Infect. Dis. 42 (5), 657–668. doi:10.1086/499819
Tam, P. H., and Lowary, T. L. (2010). Epimeric and Amino Disaccharide Analogs as Probes of an α-(1→6)-mannosyltransferase Involved in Mycobacterial Lipoarabinomannanbiosynthesis. Org. Biomol. Chem. 8 (1), 181–192. doi:10.1039/b916580k
Terashima, M., Uezumi, I., Tomio, T., Kato, M., Irie, K., Okuda, T., et al. (1991). A Protective Human Monoclonal Antibody Directed to the Outer Core Region of Pseudomonas aeruginosa Lipopolysaccharide. Infect. Immun. 59 (1), 1–6. doi:10.1128/iai.59.1.1-6.1991
Vartak, A., Hefny, F. M., and Sucheck, S. J. (2018). Synthesis of Oligosaccharide Components of the Outer Core Domain of P. aeruginosa Lipopolysaccharide Using a Multifunctional Hydroquinone-Derived Reducing-End Capping Group. Org. Lett. 20 (2), 353–356. doi:10.1021/acs.orglett.7b03590
Veeneman, G. H., Van Leeuwen, S. H., and Van Boom, J. H. (1990). Iodonium Ion Promoted Reactions at the Anomeric centre. II an Efficient Thioglycoside Mediated Approach toward the Formation of 1,2-trans Linked Glycosides and Glycosidic Esters. Tetrahedron Lett. 31 (9), 1331–1334. doi:10.1016/S0040-4039(00)88799-7
Yokota, S., Ochi, H., Ohtsuka, H., Kato, M., and Noguchi, H. (1989). Heterogeneity of the L-Rhamnose Residue in the Outer Core of Pseudomonas aeruginosa Lipopolysaccharide, Characterized by Using Human Monoclonal Antibodies. Infect. Immun. 57 (6), 1691–1696. doi:10.1128/Iai.57.6.1691-1696.1989
Keywords: thioglycosides, vaccine, lipopolysaccharide, Pseudomonas aeruginosa, outer core, aminooxy glycosides
Citation: Si A and Sucheck SJ (2021) Synthesis of Aminooxy Glycoside Derivatives of the Outer Core Domain of Pseudomonas aeruginosa Lipopolysaccharide. Front. Mol. Biosci. 8:750502. doi: 10.3389/fmolb.2021.750502
Received: 30 July 2021; Accepted: 09 September 2021;
Published: 08 November 2021.
Edited by:
Lei Li, Georgia State University, United StatesReviewed by:
Hai Yu, University of California, Davis, United StatesJoseph John Barchi, National Cancer Institute (NCI), United States
Copyright © 2021 Si and Sucheck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven J. Sucheck, c3RldmUuc3VjaGVja0B1dG9sZWRvLmVkdQ==