
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 10 December 2021
Sec. Molecular Diagnostics and Therapeutics
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.749842
 Songchang Chen1,2,3,4†Hongjun Fei2,3†Junyun Zhang2,3Yiyao Chen2,3Hefeng Huang1,2,3Daru Lu4
Songchang Chen1,2,3,4†Hongjun Fei2,3†Junyun Zhang2,3Yiyao Chen2,3Hefeng Huang1,2,3Daru Lu4 Chenming Xu1,2,3*
Chenming Xu1,2,3*Background: The lifespan of Marfan Syndrome (MFS) patients is shortened, especially in patients without early diagnostics, preventive treatment, and elective surgery. Clinically, MFS diagnosis is mainly dependent on phenotypes, but for children, sporadic cases, or suspicious MFS patients, molecular genetic testing, and mainly FBN1 mutation screening, plays a significant role in the diagnosis of MFS. PGT-M gives couples that had a family history of monogenic disorders the opportunity to avoid the occurrence of MFS.
Methods: In this study, 11 families with MFS were recruited and complete clinical features were collected. Variants were classified and interpreted through pedigree analysis according to guidelines. Two families chose to undergo PGT-M; 16 blastocysts were biopsied and amplified. Haplotype analysis was performed to deduce the embryo’s genotype by using single nucleotide polymorphisms (SNPs) identified in each sample.
Results: We identified 11 potential disease-causing FBN1 variants, six of which are novel. All variants were assessed with prediction tools to assess mutation pathogenicity, population databases to evaluate population allele frequency, literature databases to identify whether the variant had been reported in MFS patients, and multiple sequence alignment to carry out conservative analysis. Finally, nine variants were classified as likely pathogenic/pathogenic variants. Among 11 variants, eight variants were missense, and seven of them were located in the Ca-binding EGF-like motifs, moreover, half of them substituted conserved Cysteine residues. We also identified a splice site variant, a frameshift variant, and a synonymous variant. There are two variants that are de novo variants. PGT-M helped two MFS families give birth to a healthy baby not carrying the FBN1 mutation.
Conclusions: In the present study, the FBN1 mutation spectrum was enriched, and may help further elucidate the pathogenesis, benefiting clinical diagnosis and management of MFS. We make use of a reliable PGT-M method for the successful birth of healthy babies to two MFS families.
Marfan syndrome (MFS, OMIM 154700) is a connective tissue disease that is caused by mutations in fibrillin-1; it mainly affects several systems including cardiovascular, ocular, and musculoskeletal systems. MFS is inherited in an autosomal dominant pattern with an estimated prevalence of 1–5/10,000 (Ramirez and Dietz, 2007; Radke and Baumgartner, 2014). Clinically, cardiovascular manifestations (aortic dilatation and dissection) are the most serious complications that cause the death of MFS patients, although ocular and skeletal involvement may also pose a great burden on MFS patients (Ammash et al., 2008). Multiple studies confirmed that the lifespan of MFS patients is shortened, especially in patients without diagnostics and surgical treatment (Krause, 2000; Yetman et al., 2003; Groth et al., 2018).
FBN1 gene (OMIM 134797) contained 66 exons and encoded fibrillin-1 protein; its heterozygous mutations are detected in most patients with MFS (Holcomb, 2000). Compound heterozygous or homozygous FBN1 variants in MFS patients are rare (Arnaud et al., 2017). There are 2890 FBN1 mutations that had been described in the professional Human Gene Mutation database. However, more pathogenic genes or atypical mutations in specific populations remain to be identified and interpreted (Arslan-Kirchner et al., 2008). Clinically, MFS diagnosis is mainly dependent on manifestations or phenotypes (Loeys et al., 2010), but for children or suspicious MFS patients, molecular genetic testing, and mainly FBN1 mutation screening, plays a significant role in the diagnosis of MFS (Cañadas et al., 2010; Groth et al., 2017). There is a specific guideline for the interpretation of sequenced variants in the FBN1 Gene for Marfan Syndrome besides the American College of Medical Genetics (ACMG) guideline to help us identify novel mutations in FBN1 likely to cause MFS (Muiño-Mosquera et al., 2018). With the identification of FBN1 as the genetic basis of MFS (Stark et al., 2020), the hope for MFS patients’ early diagnosis, preventive treatment, and elective surgery is feasible.
Pre-implantation genetic testing for monogenic diseases (PGT-M) is a part of the in vitro fertilization (IVF) process, which genetically profiles oocytes or embryos before implantation, and is generally available for any monogenic condition in which the causative variant is known (Besser et al., 2019; Brown, 2020; Doroftei et al., 2020). PGT-M gives couples that had a family history of monogenic disorders the opportunity to avoid the occurrence of such diseases (Hamid and Loyd, 2012; Arian et al., 2020). There are two crucial problems for couples who choose to do IVF with PGT-M: determining the disease-causing variant and effective biopsy and subsequent genetic analysis which prevent any damage to embryo viability (Priner et al., 2019).
In this study, we investigated the clinical manifestations and molecular basis of 11 unrelated suspected MFS families, to screen and identify disease-causing mutations, and two MFS families want to block the transmission of the disease by PGT-M. We reported a successful application of targeted capture sequencing and haplotype analysis-based PGT in MFS families, coupled with prenatal testing for fetal aneuploidy and large chromosomal imbalance arrangement, to help give birth to a healthy baby.
Eleven nonconsanguineous families with MFS were recruited from International Peace Maternity and Child Health Hospital in this study. The ethics committee of International Peace Maternity and Child Health Hospital approved the project and investigators followed the principles of the Declaration of Helsinki. Informed consent was obtained from each patient and their related families before genetic testing. MFS was diagnosed according to Ghent criteria by cardiologists, ophthalmologists, internists, and geneticists.
Clinical data were retrospectively collected based on patients’ medical records kept at our hospital. Case inclusion criteria and clinical data inclusion scope are (Radke and Baumgartner, 2014): diagnosed patients or family history of MFS (Ramirez and Dietz, 2007); Cardiovascular system phenotype: aortic dilatation, aortic dissection, or mitral valve prolapse (Ammash et al., 2008); Ocular system phenotype: high myopia >6.0D or ectopia lentis (Yetman et al., 2003); Skeletal system phenotype: arachnodactyly, scoliosis, pectus excavatum, or flatfeet.
Genomic DNA was isolated from peripheral blood or using the MagNA Pure LC DNA Isolation Kit (Roche Diagnostics, GmbH, Mannheim, Germany). Whole-exome sequencing library construction and sequencing were performed using the Illumina platform by Beijing Genomics Institute (BGI) according to the manufacturer’s protocols. The detection covers exons (over 180,000) and 10bp flanking sequences of 22,000 genes. Exome sequencing was performed on the HiSeq2000 sequencing platform (Illumina). In-solution whole-exome capture and massively parallel sequencing was performed using the Agilent SureSelectXT All Exon Kit 51 Mb. Sequenced reads were collected, filtered for quality, and aligned to the human reference sequence (UCSC Genome Browser hg19) with the Burrows-Wheeler Aligner. On average, over 95% of exons were covered at >20×. Sequence variants including single-nucleotide variants (SNVs) and small insertions or deletions (indels) were annotated by ANNOVAR software. Common variants (defined as 10% frequency in 1,000 Genomes) were excluded if they were present in the dbSNP (v.142) database, the 1,000 Genomes Project, or the Exome Aggregation Consortium (ExAC) Browser. The detected variants were annotated and filtered with Annovar based on public databases [such as Mendelian Inheritance in Man (OMIM), Exome Aggregation Consortium (ExAC) Browser and MutationTaster2] in accordance with the criteria set by the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines. We focus on screen mutations of the FBN1 (NM_000138.5) gene with bioinformatics analysis of FASTQ files. Each mutation we found will be confirmed by bidirectional Sanger sequencing.
The sequences of screened variations sites in FBN1 (NM_000138.5) were obtained from UCSC Human Genome Browser. Pedigree analysis was performed to identify the disease-causing mutation. The variants were detected in probands and his/her family members by polymerase chain reaction (PCR). All variants were classified according to the FBN1-Specific Guideline for the Interpretation of Sequenced Variants in the FBN1 gene. The involved databases and criteria are as follows:
1) Prediction tools which are used to assess mutation pathogenicity contained SIFT (http://sift.jcvi.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), Rare Exome Variant Ensemble Learner (REVEL) (https://sites.google.com/site/revelgenomics/), ClinPred (https://sites.google.com/site/clinpred/), Human Splicing Finder (http://www.umd.be/HSF3/index.html/), and NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html).
2) Population databases which are used to evaluate population allele frequency information contained gnomAD (https://gnomad.broadinstitute.org) and integrated online website VarCards (http://varcards.biols.ac.cn/).
3) Literature Databases which are used to identify whether the variant had been reported in MFS patients contained Human Gene Mutation database (HGMD) (http://www.hgmd.cf.ac.uk/ac/index.php), PubMed (https://pubmed.ncbi.nlm.nih.gov), and Mastermind Genomic Search Engine (https://mastermind.genomenon.com/).
4) Some helpful databases which are used to distinguish whether the variants are located in functional domain such as Cysteine substitutions in the cb-EGF domains of fibrillin-1 are considered variants that happen in functional domains. The database contained VarSome (https://varsome.com/), subRVIS (http://subrvis.org/), and InterVar (http://wintervar.wglab.org/).
Proband in family five is a female (III-2). She was a 26-year-old woman who was diagnosed with Marfan syndrome (MFS). Her mother and grandfather are all MFS patients and are treated for to aortic dilatation and mitral valve prolapse. She and her mother are very tall. All three MFS patients in this family suffered cardiac anomalies and skeletal dysplasia. Her father, husband, and aunt were apparently healthy. The variant is classified as uncertain significance with a pathogenic possibility of 67.5%–81.2. The patient had a strong desire to block the disease and to have an unaffected child via PGT-M. After expert consultation, informed consent from the patient and her husband, and approval from the ethics committee, the couples were determined to be pregnant through IVF and PGT-M, and signed an informed consent form for the PGT-M cycle.
Proband in family eight is a Male (I-2). He was a 36-year-old man who was diagnosed with Marfan syndrome (MFS). He had a c.5498G > T [p(Cys1833Phe)] mutation in FBN1 gene. He was a typical MFS patient with disease phenotype in the Cardiovascular system, Ocular system, and Skeletal system. The variant is classified as pathogenic which is a pathogenic possibility over 99.7%. The MFS patient and his wife decided to undergo an IVF cycle associated with PGT-M and signed their informed consent.
Family five and family eight obtained 18 and 10 embryos respectively; embryo biopsy was performed on Day 3 (cleavage stage) and blastocysts biopsy was performed in embryos of grade 3 or higher according to Gardner’s grading scale on day 6. The multiple displacement amplification (MDA) products and gDNA libraries were prepared and captured using a 1.5 Mb customized probe covering 350 kb upstream to 350 kb downstream of the FBN1 gene. The SNPs identified in the couple and their parents were used for haplotype construction. Embryos diagnosed as unaffected were selected for transfer. Prenatal molecular diagnosis was performed through amniocentesis at the 13th-20th gestational week. The fetal genotype was confirmed by Sanger sequencing.
Clinical information of MFS probands and patients was summarized in Table 1. Affected patients from these families exhibited similar clinical symptoms of MFS. All the healthy family members had no features of MFS. We recruited 11 probands with MFS and recorded the phenotype of all MFS patients. The phenotype and family history of affected patients with FBN1 mutation coincided with MFS except in Family 4 and Family 6, the clinical manifestations of the two families are mainly ocular defects including ectopia lentis and high myopia >6.0D. The clinical feature of the proband in Family 11 is missing since the proband died before birth.
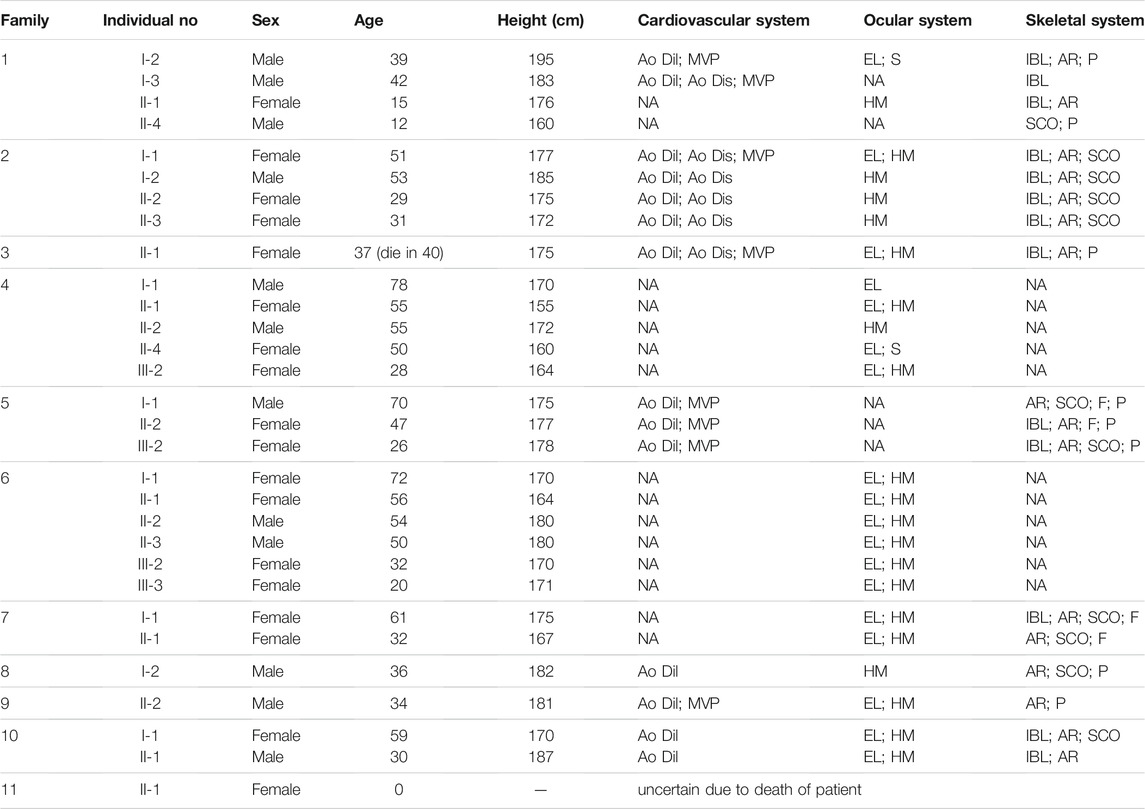
TABLE 1. Clinical features of fibrillin-1 (FBN1) mutation patients in 11 Marfan families.
In this study, we analyzed the genomic DNA of 11 probands with MFS. The quality and reliability of targeted NGS data were evaluated based on the percentage of readable bases and the coverage depth in the targeted region, to ensure complete sequencing coverage of all coding regions in candidate genes. Altogether, 11 potentially disease-causal FBN1 gene variants were screened out in 11 probands, 6/11 (54.5%) variants had not been reported before and 5/11 (45.5%) variants had been reported in MFS patients. We showed all mutations and their location in the protein domain of FBN1 (Figure 1). The hot spot mutations were considered to happen frequently in exon 1–16 and secondarily exon 32–40. Among 11 variants, six variants marked in red were first reported through this study. The location and basic information of all mutations are summarized in Table 2. Except for one splicing variant, nine variants are in Ca-binding EGF-like domains and one is not in a Ca-binding EGF-like domain. There are eight missense variants, and four of eight (50%) missense variants affected conserved Cysteine residues of fibrillin 1 protein.
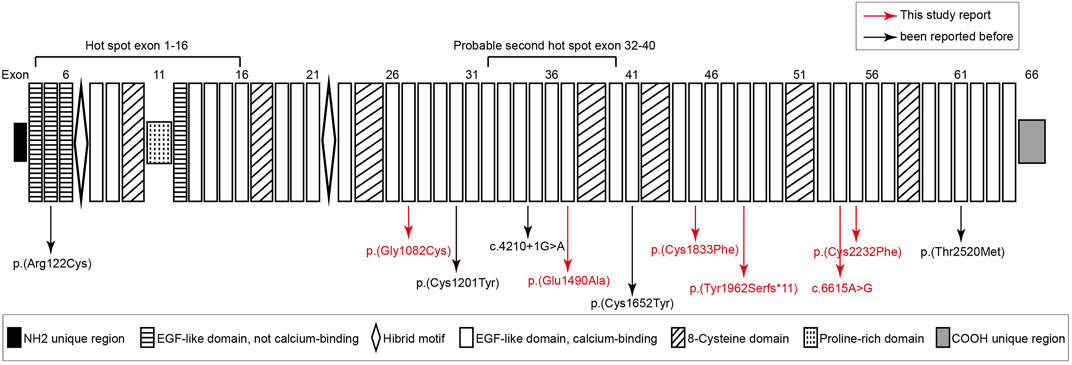
FIGURE 1. Structure diagram of FBN1 sequence, and distribution of FBN1 mutations identified from Marfan syndrome patients. six variants marked in red are novel variants never reported before. Except for a splicing variant (c.4210+1G > A) and c.364C > T [p(Arg122Cys)] that occurred in the not calcium binding EGF-like domain, other variants all occurred in the calcium binding EGF-like domain and four mutations happened in the cysteine residues of FBN1.
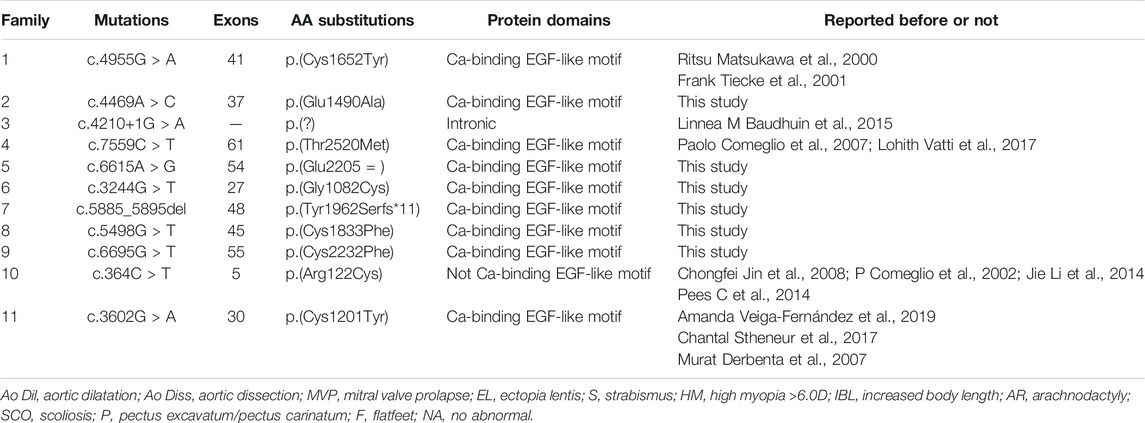
TABLE 2. FBN1 variants identified for affected individuals in 11 Marfan families.
Pedigree analyses were performed to obtain familial segregation data and determine whether the mutation is a de novo mutation. Each variant, considered as a causative candidate and pathogenic mutation, was further validated using the Sanger sequencing method in other family members. Figure 2 is the pedigree chart of 11 MFS families. FBN1 mutations in Family 3 and Family 11 were do novo mutations. Figure 3 showed the validation result of FBN1 mutations in MFS families by sanger sequencing.
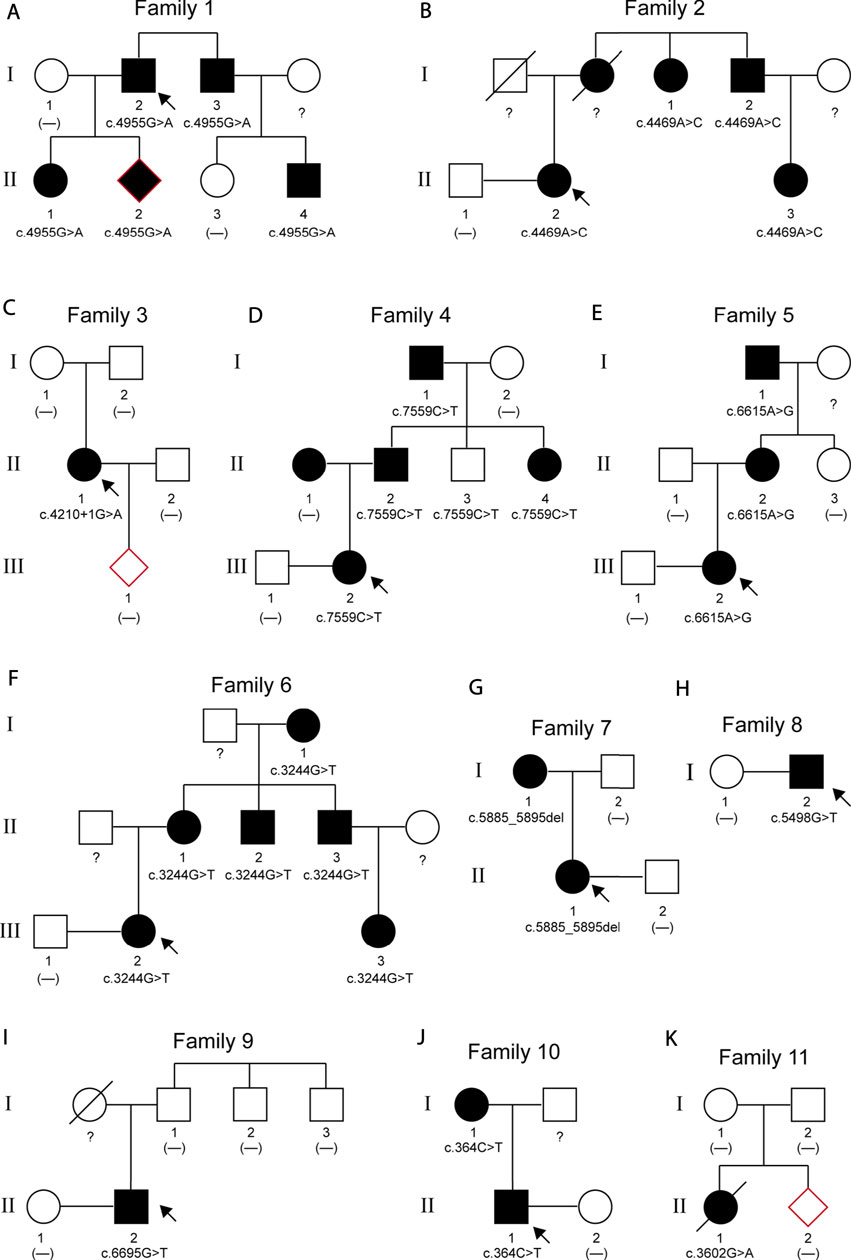
FIGURE 2. Pedigrees of 11 families with MFS. The arrows indicate the proband of each family. Squares represent males; circles represent females; solid symbols indicate affected patients; open symbols indicate unaffected subjects; a slash through the symbol means deceased.
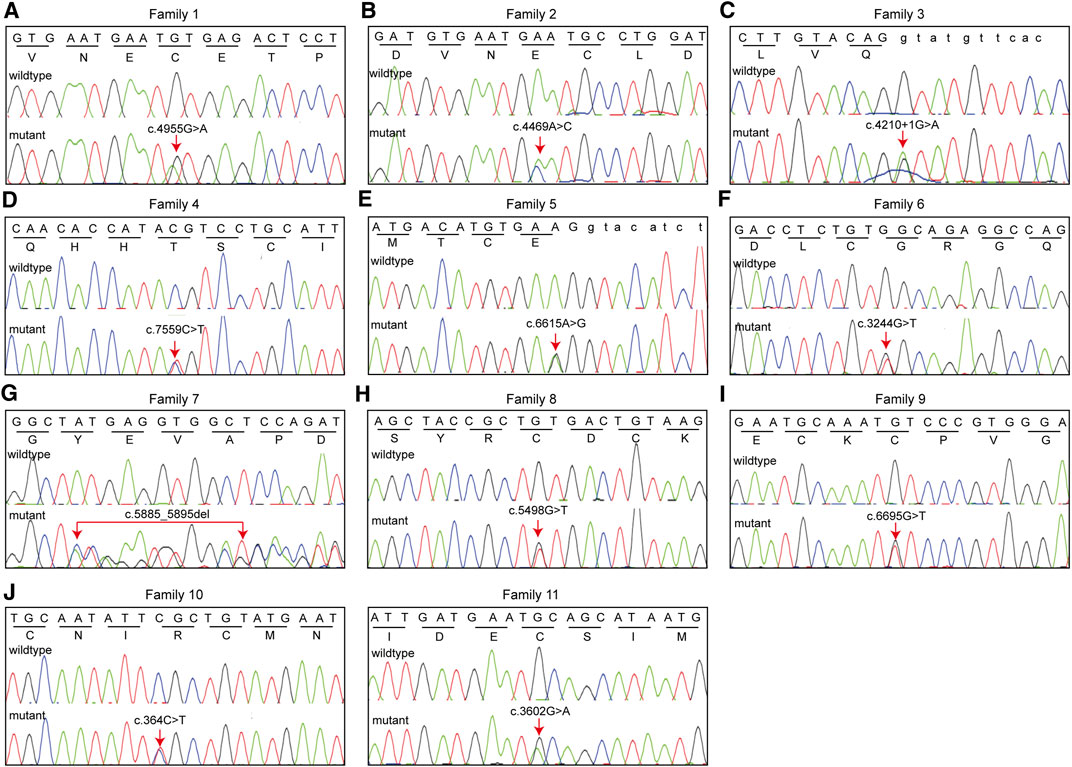
FIGURE 3. Eleven FBN1 mutations identified in 11 families with MFS were validated using Sanger sequencing. We presented nucleotide sequence and corresponding amino acid sequence.
To identify disease-causing mutations for MFS families, stringent criteria according to ACMG guidelines were performed. We carefully examined all available literature and mutation-related database for sequence variant interpretation. In Table 3, allele frequencies of variants detected in populations, pieces of computational evidence of pathogenicity prediction, the clinical significance of variants in Clinvar, and whether the variant is a de novo variant are listed. We explored the classification evidence for each variant and did sequence variant interpretation according to the ACMG criterion. In Figure 4, conservation analysis of the related homologous proteins in eight FBN1 missense mutation sites was conducted by referring to the UniProt database. Orthologous protein sequence alignment for eight missense mutations showed the eight mutation sites happened in a highly conserved region of FBN1 among different species. Finally, we confirmed 11 causative candidate and pathogenic heterozygous mutations including five known mutations of the FBN1 gene in the patients, including c.4955G > A [p(Cys1652Tyr)], c.4210+1G > A [p(?)], c.7559C > T [p(Thr2520Met)], c.364C > T [p(Arg122Cys)], and c.3602G > A [p(Cys1201Tyr)], as well as six novel mutations never reported before, including c.4469A > C [p(Glu1490Ala)], c.6615A > G [p(Glu2205 = )], c.3244G > T [p(Gly1082Cys)], c.5885_5895del [p(Tyr1962Serfs*11)], c.5498G > T [p(Cys1833Phe)], and c.6695G > T [p(Cys2232Phe)].
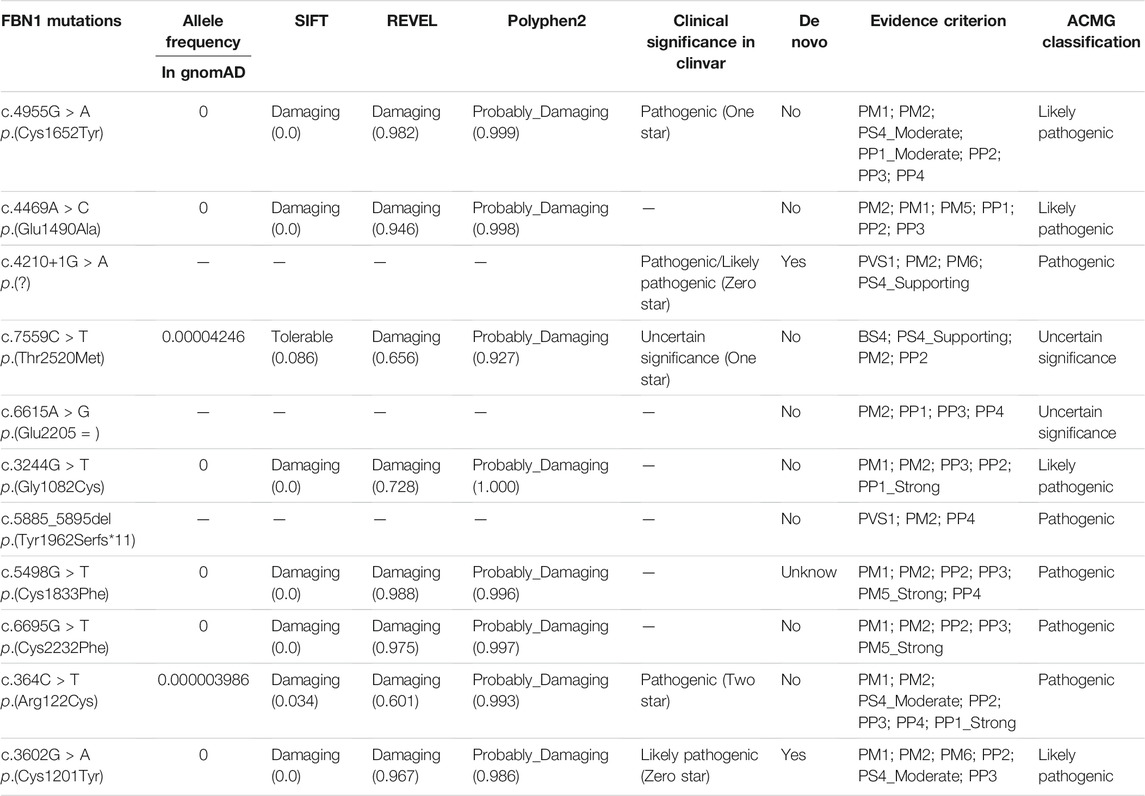
TABLE 3. Classification of pathogenicity of FBN1 mutations identified from 11 Marfan families.

FIGURE 4. Orthologous protein sequence alignment of FBN1 sequence. Conservation analysis among nine species showed all 11 mutations sites happened in a highly conserved region of FBN1 among different species.
For the FBN1 gene, missense variants are a common mechanism of MFS disease (PP2). c.4955G > A [p(Cys1652Tyr)] is a missense variant (PP2), it had been reported by several groups in patients with MFS (PS4_Moderate). The variant is not present in GnomAD and ExAC (PM2). The variant results in a Cysteine substitution in the critical function domain of the calcium-binding EGF-like domain of FBN1 protein (PM1). In Family 1, the mutation was segregated with the phenotype in five affected patients and was absent in unaffected individuals (eLOD = 1.20, PP1_Moderate). Phenotype and family history of this family and reported patients were consistent with FBN1-induced MFS (PP4). Cys1652 is conserved between species and predicted to be disease-causing according to in silico prediction (PP3). In summary, this variation is classified as “likely pathogenic variation”. In August 2016, the patient and his wife became pregnant naturally, and the fetus (II-2) was diagnosed with FBN1 c.4955G > A mutation after prenatal diagnosis.
c.4210+1G > A [p(?)] is in intron 34 and affects the canonical +1 donor splice site most likely leading to abnormal splicing (PVS1). It has been reported previously in association with MFS (PS4_Supporting). The mutation was not detected in the samples of proband’s parent (PM6) and was not present in GnomAD (PM2). In summary, this variation is classified as “pathogenic variation”. In December 2019, the patient and his wife became pregnant naturally, and the fetus (III-1) did not carry this mutation after prenatal diagnosis.
c.7559C > T [p(Thr2520Met)] is a missense variant (PP2), and is found at a very low frequencies in GnomAD (0.00004246) (PM2). Although the variant had been reported in an MFS patient (PS4_Supporting), in our study, the mutation was not segregated with the phenotype in five affected patients and was detected in unaffected individuals (BS4). In summary, this variation is classified as “Uncertain significance variation”.
c.364C > T [p(Arg122Cys)] is a missense variant (PP2) and located in a critical function domain of the not calcium-binding EGF-like domain of FBN1 protein (PM1). The variant is found at a very low frequency in GnomAD (0.000003986) (PM2) and many pieces of prediction software predict this variant to be damaging (PolyPhen2, SIFT, REVEL) (PP3). The variant had been reported in MFS patients before (PS4_Moderate) and was segregated with the phenotype in eight affected patients (eLOD = 2.00, PP1_Strong). In Family 10 and the previous reports, phenotypes and family histories of patients were consistent with MFS (PP4). In summary, this variation is classified as “pathogenic variation”.
c.3602G > A [p(Cys1201Tyr)] is a missense variant (PP2) and results in a Cysteine substitution in the critical function domain of the calcium-binding EGF-like domain of FBN1 protein (PM1). The mutation was not present in GnomAD (PM2) and was predicted to be disease-causing by in silico analysis (PP3). In Family 11, the mutation is a de novo mutation (PM6), and the mutation had been reported in the literature (PS4_Moderate). In summary, this variation is classified as “likely pathogenic variation”. In May 2019, the parents of the probands conceived naturally, and the fetus (II-2) did not carry FBN1 c.3602G > A mutation after prenatal diagnosis.
c.4469A > C [p(Glu1490Ala)] is a novel missense variant (PP2) and located in a critical function domain of the calcium-binding EGF-like domain of FBN1 protein (PM1). The mutation was not present in GnomAD (PM2) and was predicted to be disease-causing by in silico analysis (PP3). In Family 2, the mutation was segregated with the phenotype in four affected patients and was absent in unaffected individuals (eLOD = 0.90, PP1). Moreover, mutation at Glu1490 locus [p(Glu1490Lys)] had been classified as likely pathogenic (PM5). In summary, this variation is classified as “likely pathogenic variation".
c.6615A > G [p(Glu2205 = )] is a novel synonymous variant that had not been reported before, all three MFS patients had c.6615A > G mutation in FBN1 but the other healthy members in her family did not carry this mutation (eLOD = 0.90, PP1). The variant is absent from all population databases (PM2), and multiple lines of computational evidence support a deleterious effect on the gene product through disturb splicing (PP3). The phenotype and family history of the proband in this family are highly consistent with MFS characteristics (PP4). All three MFS patients in this family suffered cardiac anomalies and skeletal dysplasia. In Figure 5, we can see three affected patients have arachnodactyly, flatfeet, pectus excavatum, or pectus carinatum. In summary, this variation is classified as “Uncertain significance variation”. The variant is classified as uncertain significance with a pathogenic possibility of 67.5%–81.2. The patient had a strong desire to block the disease and to have an unaffected child via PGT-M. After expert consultation, informed consent from the patient and her husband, and approval from the ethics committee, the couples were determined to be pregnant through IVF and PGT-M, and signed an informed consent form for the PGT-M cycle.

FIGURE 5. Clinical features of MFS patients in Family five who chose to do IVF with PGT-M. All three patients posted cardiac surgery. They all had a clinical manifestation of arachnodactyly and pectus excavatum/pectus carinatum. I-1 and III-2 had scoliosis. I-1 and II-2 have flatfeet.
c.3244G > T [p(Gly1082Cys)] is a novel missense variant (PP2) and located in a critical function domain of the calcium-binding EGF-like domain of FBN1 protein (PM1). The mutation was not present in GnomAD (PM2) and was predicted to be disease-causing by in silico analysis (PP3). In Family 6, the mutation was segregated with the phenotype in six affected patients and was absent in unaffected individuals (eLOD = 1.50, PP1_Strong). In summary, this variation is classified as “likely pathogenic variation”.
c.5885_5895del [p(Tyr1962Serfs*11)] is a novel frameshift variant that likely leads to a truncated protein (PVS1). The variant is absent from all population databases (PM2), and the phenotype of affected patients is consistent with MFS (PP4). In summary, this variation is classified as “pathogenic variation”.
c.5498G > T [p(Cys1833Phe)] and c.6695G > T [p(Cys2232Phe)] are all novel missense variants (PP2) and result in a Cysteine substitution in the critical function domain of the calcium-binding EGF-like domain of FBN1 protein (PM1). The mutations were not all present in GnomAD (PM2) and were predicted to be disease-causing by in silico analysis (PP3). In Family eight and Family 9, the phenotype of affected patients is consistent with MFS (PP4). Moreover, mutation at Cys1833 locus [p(Cys1833Arg), p(Cys1833Ser)], and mutation at Cys2232 locus [p(Cys2232Tyr), p(Cys2232Arg)] had been reported to be pathogenic (PM5_Strong). In summary, the two variations are classified as “pathogenic variation".
In Family 5, we defined the haplotype linked to c.6615A > G and associated with MFS as F0, and the haplotype linked to wildtype allele as F1. In Family 8, we defined the haplotype linked to c.5498G > T and associated with MFS as M0, while the haplotype linked to wildtype allele was defined as M1. The genotypes of 16 embryos were all successfully determined using the Hidden Markov model approach (Figure 6; Table 4). In Family 5, five embryos were free of maternal FBN1 c.6615A > G variant; embryos 2, 7, and 8 were carriers of c.6615A > G variant. In Family 8, five embryos were free of paternal FBN1 c.5498G > T variant; embryos 1, 6, and 8 were carriers of c.5498G > T variant.
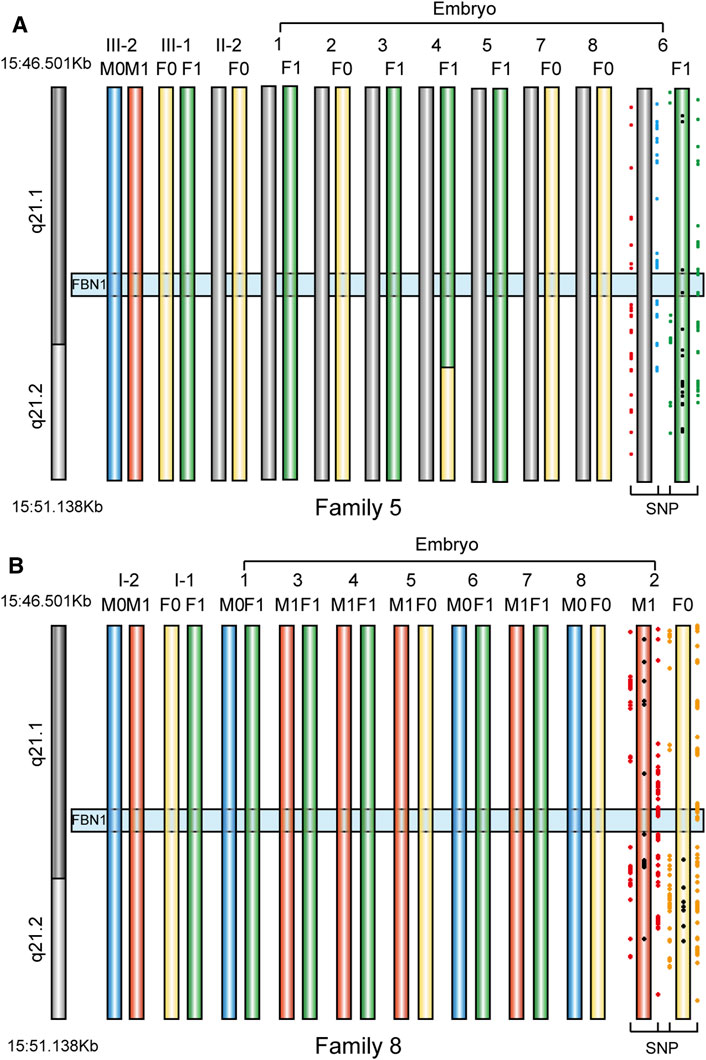
FIGURE 6. The haplotype in FBN1 gene in eight embryos from two families. We showed informative SNPs that supported the haplotype of only one embryo used for implantation (A) PGT haplotype analysis in embryos 1 to eight in Family 5. F0 means Female disease-causing chromosome, F1 means Female normal chromosome, M0 and M1 means Male normal chromosome. Embryo 4 had a recombination event in the maternal allele (length 869.56 Kb), but it did not influence the genotype deduction of the FBN1 gene, because the recombination loci were outside the gene region (B) PGT haplotype analysis in embryos 1 to eight in Family 8. M0 means Male disease-causing chromosome, M1 means Male normal chromosome, F0 and F1 mean Female normal chromosome.
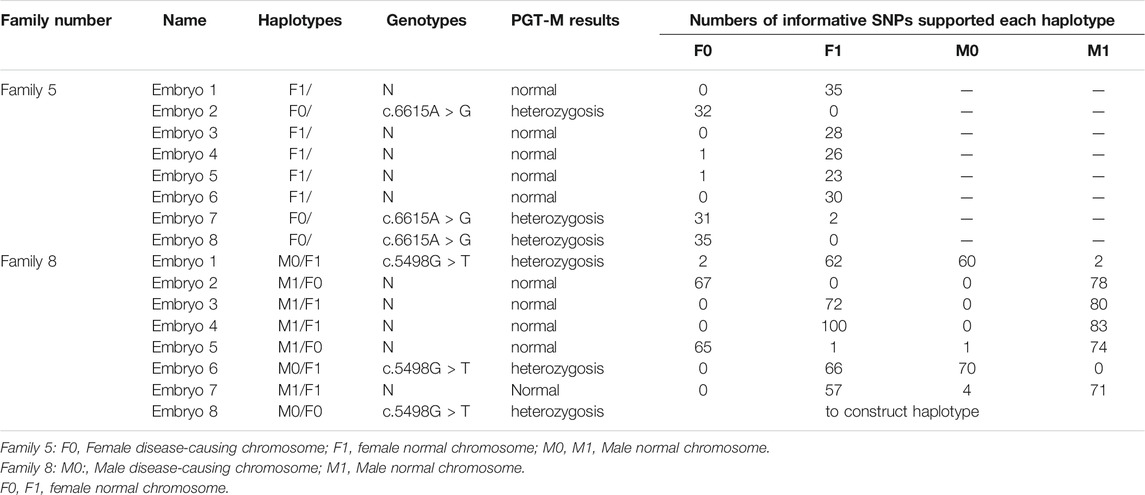
TABLE 4. Details of haplotype in FBN1 gene in embryos from two Marfan families who chose to do IVF with PGT-M.
Embryo 6 in Family 5 and embryo 2 in Family 8 were selected for transfer and a successful pregnancy was confirmed by human chorionic gonadotropin (hCG) and ultrasound examination. Sanger sequencing results showed that the fetus did not carry mutations. This confirmed the accuracy of PGT-M. The chromosome imbalance anomaly results showed that no copy number variant (CNV) larger than 100 kb was identified in the fetus.
In this study, we collected clinical features of all MFS patients in 11 families with the monogenetic disease. We found that the clinical phenotypes of MFS patients in the same family are broadly similar and remain different. It is consistent with the reports in the literatures (Ramirez, 1996; Seo et al., 2018). In Family 1, two children (12 and 15 years old) did not have cardiac phenotypes such as aortic dilatation, aortic dissection, and mitral valve prolapse but adults had; it reminded us that molecular genetic testing for children or atypical MFS patients is important and to warn patients to take medical prophylaxis to prevent cardiovascular pathologies causing the highest mortality in MFS (Comeglio et al., 2007; Franken et al., 2017; Manchola-Linero et al., 2018).
Here, we identified 11 potential disease-causing FBN1 variants in patients with Marfan syndrome, and six of them are novel. Among 11 variants, eight variants were missense, and seven of them were located in the Ca-binding EGF-like motifs, moreover half of them substituted conserved Cysteine residues. According to previous reports, Cysteine residues are highly conserved in fibrillin 1 (Corson et al., 2004), mutations affecting Cysteine residues may disrupt one of the three disulfide bonds and critical functional domain (Schrijver et al., 1999; Faivre et al., 2007). Our results proved that Cysteine substitutions in Ca-binding EGF-like domains of fibrillin 1 may be the most common and critical causal for FBN1 induced MFS.
Besides eight missense variants, we also identified a splice site variant, a frameshift variant, and a synonymous variant. C.6615A > G [p(Glu2205 = )] variant had never been reported before, the affected patient who carries this variant is a typical patient, her mother and grandfather all had typical MFS phenotype (pieces of variant classification evidence including: PM2, PP1, PP3 and PP4). All of them underwent cardiac surgery. The proband and her husband hope to give birth to a healthy baby. Many pieces of prediction software predicted the variant will affect mRNA splicing, however, more functional evidence is needed.
According to statistics, over 25% of MFS patients are sporadic cases, and these patients always have the severe neonatal phenotype (Collod-Béroud and Boileau, 2002; Tan et al., 2017; Weerakkody et al., 2018). In our study, II-1in Family 3 and II-1 in Family 11 are sporadic cases, their parents are normal, they had no family history, and the variants are de novo. Obviously, they had a severe disease phenotype, II-1in Family 3 died when she was 40 years old and II-1 in Family 11 died after birth. The clinical criteria in the revised Ghent nosology are not always suitable for children, particularly those sporadic cases. It further proved that molecular diagnosis of MFS is useful and necessary (Toudjarska et al., 2001; Ades and Group, 2007). Further, making the pathogenesis clear can help patients block the transmission of the pathogenic FBN1 mutation by PGT and produce healthy babies.
Due to phenotypic variability, a high rate of sporadic cases, and lack of a genetic or biochemical test for MFS, the actual incidence of MFS may be considerably higher than the reported 1–5/10,000 (Chiu et al., 2014). And clinical demand for PGT has increased as research and cases on MFS increase. Over the past few decades, PGT always used STR as a genetic marker, but it is time-consuming to select appropriate markers. The capture sequencing and linkage analysis of SNPs located nearby the gene of interest provides a convenient and efficient way for PGT-M experiment design (Brezina et al., 2016). In two families that underwent PGT-M, informative SNPs were distributed from upstream to downstream of the FBN1 gene, ensuring any recombination will be identified. We determined each embryo successfully. Embryo 4 in Family five had a recombination event in the maternal allele (869.56 Kb), but it did not influence the genotype deduction of the FBN1 gene, because the recombination loci were outside the gene region. Of course, we did invasive prenatal diagnosis for two families who underwent PGT-M to avoid misdiagnosis (Chen et al., 2018).
We identified six novel variants and five known variants in the FBN1 gene from 11 Chinese families with MFS. We performed detailed classification and interpretation for 11 variants. In the present study, the FBN1 mutation spectrum was enriched and may help further elucidate the pathogenesis, benefiting clinical diagnosis and management of MFS. We make use of a reliable PGT-M method to enable the successful birth of healthy babies for two MFS families.
ACMG, American College of Medical Genetics and Genomics; IVF, in vitro fertilization; MDA, multiple displacement amplification; MFS, Marfan syndrome; PGT, Pre-implantation genetic testing; PGT-M, Pre-implantation genetic testing for monogenic diseases
The data presented in the study are deposited in the Sequence Read Archive (SRA) repository, accession number PRJNA778868.
The studies involving human participants were reviewed and approved by The International Peace Maternity and Child Health Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
SC and CX conceived of the study and HF drafted the manuscript. HF collected clinical data of MFS patients and family pedigrees. HF and SC performed acquisition of data and conducted interpretation of sequenced variants. SC performed PGT-M test and analyzed the biopsy results. CX supervised all the work and revised the manuscript. All the authors have read and approved the manuscript.
This work was supported by the research grant from the National Key R&D Program of China (2018YFC1004900), the National Natural Science Foundation of China (Nos.81971344, 81771638 and 81901495), the Shanghai Municipal Commission of Science and Technology Program (No.21Y21901002) Shanghai Municipal Health Commission (GW-10.1-XK07), the Shanghai “Rising Stars of Medical Talent” Youth Development Program Clinical Laboratory Practitioners Program (201972), Shanghai Municipal Commission of Health and family planning (202140110, 20215Y0216), Shanghai Sailing Program (19YF1452200, 18YF1424800), clinical research special projects from Shanghai Municipal Health Commission (No.20204Y0230).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ACMG, American College of Medical Genetics and Genomics; IVF, in vitro fertilization; MDA, multiple displacement amplification; MFS, Marfan syndrome; PGT, Pre-implantation genetic testing; PGT-M, Pre-implantation genetic testing for monogenic diseases.
Ades, L., and Group, C. C. G. W. (2007). Guidelines for the Diagnosis and Management of Marfan Syndrome. Heart Lung Circ. 16 (1), 28–30. doi:10.1016/j.hlc.2006.10.022
Ammash, N. M., Sundt, T. M., and Connolly, H. M. (2008). Marfan Syndrome-Diagnosis and Management. Curr. Probl. Cardiol. 33 (1), 7–39. doi:10.1016/j.cpcardiol.2007.10.001
Arian, S. E., Erfani, H., Westerfield, L. E., Buffie, A., Nassef, S., Gibbons, W. E., et al. (2020). Prenatal Testing in Pregnancies Conceived by In Vitro Fertilization with Pre‐implantation Genetic Testing. Prenatal Diagn. 40 (7), 846–851. doi:10.1002/pd.5711
Arnaud, P., Hanna, N., Aubart, M., Leheup, B., Dupuis-Girod, S., Naudion, S., et al. (2017). Homozygous and Compound Heterozygous Mutations in theFBN1gene: Unexpected Findings in Molecular Diagnosis of Marfan Syndrome. J. Med. Genet. 54 (2), 100–103. doi:10.1136/jmedgenet-2016-103996
Arslan-Kirchner, M., Kodolitsch, Y. v., and Schmidtke, J. (2008). The Importance of Genetic Testing in the Clinical Management of Patients with Marfan Syndrome and Related Disorders. Deutsches Arzteblatt Int. 105 (27), 483–491. doi:10.3238/arztebl.2008.0483
Besser, A. G., Blakemore, J. K., Grifo, J. A., and Mounts, E. L. (2019). Transfer of Embryos with Positive Results Following Preimplantation Genetic Testing for Monogenic Disorders (PGT-M): Experience of Two High-Volume Fertility Clinics. J. Assist. Reprod. Genet. 36 (9), 1949–1955. doi:10.1007/s10815-019-01538-2
Brezina, P. R., Anchan, R., and Kearns, W. G. (2016). Preimplantation Genetic Testing for Aneuploidy: what Technology Should You Use and what Are the Differences? J. Assist. Reprod. Genet. 33 (7), 823–832. doi:10.1007/s10815-016-0740-2
Brown, S. (2020). Identity-by-state Analysis: a New Method for PGT-M. Hum. Reprod. 35 (3), 485–487. doi:10.1093/humrep/deaa011
Cañadas, V., Vilacosta, I., Bruna, I., and Fuster, V. (2010). Marfan Syndrome. Part 1: Pathophysiology and Diagnosis. Nat. Rev. Cardiol. 7 (5), 256–265. doi:10.1038/nrcardio.2010.30
Chen, H.-F., Chen, S.-U., Ma, G.-C., Hsieh, S.-T., Tsai, H.-D., Yang, Y.-S., et al. (2018). Preimplantation Genetic Diagnosis and Screening: Current Status and Future Challenges. J. Formos. Med. Assoc. 117 (2), 94–100. doi:10.1016/j.jfma.2017.08.006
Chiu, H.-H., Wu, M.-H., Chen, H.-C., Kao, F.-Y., and Huang, S.-K. (2014). Epidemiological Profile of Marfan Syndrome in a General Population: a National Database Study. Mayo Clinic Proc. 89 (1), 34–42. doi:10.1016/j.mayocp.2013.08.022
Collod-Béroud, G., and Boileau, C. (2002). Marfan Syndrome in the Third Millennium. Eur. J. Hum. Genet. 10 (11), 673–681. doi:10.1038/sj.ejhg.5200876
Comeglio, P., Johnson, P., Arno, G., Brice, G., Evans, A., Aragon-Martin, J., et al. (2007). The Importance of Mutation Detection in Marfan Syndrome and Marfan-Related Disorders: Report of 193FBN1 Mutations. Hum. Mutat. 28 (9), 928. doi:10.1002/humu.9505
Corson, G. M., Charbonneau, N. L., Keene, D. R., and Sakai, L. Y. (2004). Differential Expression of Fibrillin-3 Adds to Microfibril Variety in Human and Avian, but Not Rodent, Connective Tissues. Genomics 83 (3), 461–472. doi:10.1016/j.ygeno.2003.08.023
Doroftei, B., Nemtanu, L., Ilie, O.-D., Simionescu, G., Ivanov, I., Anton, E., et al. (2020). In Vitro Fertilisation (IVF) Associated with Preimplantation Genetic Testing for Monogenic Diseases (PGT-M) in a Romanian Carrier Couple for Congenital Disorder of Glycosylation Type Ia (CDG-Ia): A Case Report. Genes 11 (6), 697. doi:10.3390/genes11060697
Faivre, L., Collod-Beroud, G., Loeys, B. L., Child, A., Binquet, C., Gautier, E., et al. (2007). Effect of Mutation Type and Location on Clinical Outcome in 1,013 Probands with Marfan Syndrome or Related Phenotypes and FBN1 Mutations: an International Study. Am. J. Hum. Genet. 81 (3), 454–466. doi:10.1086/520125
Franken, R., Teixido-Tura, G., Brion, M., Forteza, A., Rodriguez-Palomares, J., Gutierrez, L., et al. (2017). Relationship between Fibrillin-1 Genotype and Severity of Cardiovascular Involvement in Marfan Syndrome. Heart 103 (22), 1795–1799. doi:10.1136/heartjnl-2016-310631
Groth, K. A., Stochholm, K., Hove, H., Andersen, N. H., and Gravholt, C. H. (2018). Causes of Mortality in the Marfan Syndrome(from a Nationwide Register Study). Am. J. Cardiol. 122 (7), 1231–1235. doi:10.1016/j.amjcard.2018.06.034
Groth, K. A., Von Kodolitsch, Y., Kutsche, K., Gaustadnes, M., Thorsen, K., Andersen, N. H., et al. (2017). Evaluating the Quality of Marfan Genotype-Phenotype Correlations in Existing FBN1 Databases. Genet. Med. 19 (7), 772–777. doi:10.1038/gim.2016.181
Hamid, R., and Loyd, J. (2012). Pre-implantation Genetic Testing for Hereditary Pulmonary Arterial Hypertension: Promise and Caution. Eur. Respir. J. 39 (6), 1292–1293. doi:10.1183/09031936.00218911
Holcomb, S. S. (2000). Marfan Syndrome. Dimensions Crit. Care nursingDCCN 19 (4), 22–23. doi:10.1097/00003465-200019040-00006
Krause, K. J. (2000). Marfan Syndrome: Literature Review of Mortality Studies. J. Insur Med. 32 (2), 79–88. Available at: https://www.crossref.org/guestquery
Loeys, B. L., Dietz, H. C., Braverman, A. C., Callewaert, B. L., De Backer, J., Devereux, R. B., et al. (2010). The Revised Ghent Nosology for the Marfan Syndrome. J. Med. Genet. 47 (7), 476–485. doi:10.1136/jmg.2009.072785
Manchola-Linero, A., Gran Ipiña, F., Teixidó-Tura, G., López Grondona, F., Rosés Noguer, F., and Sabaté-Rotés, A. (2018). Marfan Syndrome and Loeys-Dietz Syndrome in Children: A Multidisciplinary Team Experience. Revista Española de Cardiología (English Edition) 71 (7), 585–587. doi:10.1016/j.rec.2017.03.031
Muiño-Mosquera, L., Steijns, F., Audenaert, T., Meerschaut, I., De Paepe, A., Steyaert, W., et al. (2018). Tailoring the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Guidelines for the Interpretation of Sequenced Variants in the FBN1 Gene for Marfan Syndrome. Circ. Genomic Precision Med. 11 (6), e002039. doi:10.1161/CIRCGEN.117.002039
Priner, S., Altarescu, G., Schonberger, O., Holzer, H., Rubinstein, E., Dekel, N., et al. (2019). The Effect of Repeated Biopsy on Pre-implantation Genetic Testing for Monogenic Diseases (PGT-M) Treatment Outcome. J. Assist. Reprod. Genet. 36 (1), 159–164. doi:10.1007/s10815-018-1359-2
Radke, R. M., and Baumgartner, H. (2014). Diagnosis and Treatment of Marfan Syndrome: an Update. Heart 100 (17), 1382–1391. doi:10.1136/heartjnl-2013-304709
Ramirez, F. (1996). Fibrillin Mutations in Marfan Syndrome and Related Phenotypes. Curr. Opin. Genet. Dev. 6 (3), 309–315. doi:10.1016/s0959-437x(96)80007-4
Ramirez, F., and Dietz, H. C. (2007). Marfan Syndrome: from Molecular Pathogenesis to Clinical Treatment. Curr. Opin. Genet. Dev. 17 (3), 252–258. doi:10.1016/j.gde.2007.04.006
Schrijver, I., Liu, W., Brenn, T., Furthmayr, H., and Francke, U. (1999). Cysteine Substitutions in Epidermal Growth Factor-like Domains of Fibrillin-1: Distinct Effects on Biochemical and Clinical Phenotypes. Am. J. Hum. Genet. 65 (4), 1007–1020. doi:10.1086/302582
Seo, G. H., Kim, Y.-M., Kang, E., Kim, G.-H., Seo, E.-J., Lee, B. H., et al. (2018). The Phenotypic Heterogeneity of Patients with Marfan-Related Disorders and Their Variant Spectrums. Medicine 97 (20), e10767. doi:10.1097/MD.0000000000010767
Stark, V. C., Hensen, F., Kutsche, K., Kortüm, F., Olfe, J., Wiegand, P., et al. (2020). Genotype-Phenotype Correlation in Children: The Impact of FBN1 Variants on Pediatric Marfan Care. Genes 11 (7), 799. doi:10.3390/genes11070799
Tan, L., Li, Z., Zhou, C., Cao, Y., Zhang, L., Li, X., et al. (2017). FBN1 Mutations Largely Contribute to Sporadic Non-syndromic Aortic Dissection. Hum. Mol. Genet. 26 (24), 4814–4822. doi:10.1093/hmg/ddx360
Toudjarska, I., Kilpatrick, M. W., Lembessis, P., Carra, S., Harton, G. L., Sisson, M. E., et al. (2001). Novel Approach to the Molecular Diagnosis of Marfan Syndrome: Application to Sporadic Cases and in Prenatal Diagnosis. Am. J. Med. Genet. 99 (4), 294–302. doi:10.1002/ajmg.1174
Weerakkody, R., Ross, D., Ross, D., Parry, D. A., Ziganshin, B., Vandrovcova, J., et al. (2018). Targeted Genetic Analysis in a Large Cohort of Familial and Sporadic Cases of Aneurysm or Dissection of the Thoracic Aorta. Genet. Med. 20 (11), 1414–1422. doi:10.1038/gim.2018.27
Keywords: haplotype analysis, interpretation of sequenced variants, Marfan syndrome, pre-implantation genetic testing, molecular genetic testing, FBN1
Citation: Chen S, Fei H, Zhang J, Chen Y, Huang H, Lu D and Xu C (2021) Classification and Interpretation for 11 FBN1 Variants Responsible for Marfan Syndrome and Pre-implantation Genetic Testing (PGT) for Two Families Successfully Blocked Transmission of the Pathogenic Mutations. Front. Mol. Biosci. 8:749842. doi: 10.3389/fmolb.2021.749842
Received: 30 July 2021; Accepted: 10 November 2021;
Published: 10 December 2021.
Edited by:
Aleksandra Jezela-Stanek, National Institute of Tuberculosis and Lung Diseases, PolandReviewed by:
Karolina Chwialkowska, Medical University of Bialystok, PolandCopyright © 2021 Chen, Fei, Zhang, Chen, Huang, Lu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chenming Xu, Y2hlbm1pbmdfeHUyMDA2QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.