 Taylor R. Cole
Taylor R. Cole Tatyana I. Igumenova
Tatyana I. Igumenova- Department of Biochemistry and Biophysics, Texas A&M University, College Station, TX, United States
Conserved homology 1 (C1) domains are peripheral zinc finger domains that are responsible for recruiting their host signaling proteins, including Protein Kinase C (PKC) isoenzymes, to diacylglycerol-containing lipid membranes. In this work, we investigated the reactivity of the C1 structural zinc sites, using the cysteine-rich C1B regulatory region of the PKCα isoform as a paradigm. The choice of Cd2+ as a probe was prompted by previous findings that xenobiotic metal ions modulate PKC activity. Using solution NMR and UV-vis spectroscopy, we found that Cd2+ spontaneously replaced Zn2+ in both structural sites of the C1B domain, with the formation of all-Cd and mixed Zn/Cd protein species. The Cd2+ substitution for Zn2+ preserved the C1B fold and function, as probed by its ability to interact with a potent tumor-promoting agent. Both Cys3His metal-ion sites of C1B have higher affinity to Cd2+ than Zn2+, but are thermodynamically and kinetically inequivalent with respect to the metal ion replacement, despite the identical coordination spheres. We find that even in the presence of the oxygen-rich sites presented by the neighboring peripheral membrane-binding C2 domain, the thiol-rich sites can successfully compete for the available Cd2+. Our results indicate that Cd2+ can target the entire membrane-binding regulatory region of PKCs, and that the competition between the thiol- and oxygen-rich sites will likely determine the activation pattern of PKCs.
Introduction
Approximately ∼10% of the human proteome uses Zn2+ as a cofactor (Andreini et al., 2006). While Zn2+ is not redox active, it plays a critical role in many vital cellular processes. Functional annotation of Zn proteome predicts a wide range of biological and enzymatic activities (Bertini et al., 2010), with over 40% of the assigned sequences involved in the regulation of gene expression. One of the key signaling enzymes that require Zn2+ is the family of Protein Kinase C isoenzymes (PKCs). By serving as the key node in the phosphoinositide signaling pathway, PKCs regulate cell growth and differentiation (Dempsey et al., 2000; Newton, 2010). Aberrant PKC activity has been implicated in many human diseases including cancer progression (Antal et al., 2015; Rahimova et al., 2020), diabetes (Koya and King, 1998; Mishra and Dey, 2021), as well as neurological (Khan et al., 2009) and cardiovascular dysfunctions (Johnson et al., 1995; Budas et al., 2007; Churchill et al., 2008; Drosatos et al., 2011). Exposure to divalent xenobiotic metal ions, such as Pb2+ (Markovac and Goldstein, 1988; Tomsig and Suszkiw, 1995; Sun et al., 1999; Morales et al., 2011) and Cd2+ (Beyersmann et al., 1994; Morales et al., 2013b) modulates PKC activity. Specifically, Cd2+ can exert both activating and inhibitory effects on PKCs (Block et al., 1992; Beyersmann et al., 1994; Long, 1997) (Saijoh et al., 1988; Speizer et al., 1989). Cadmium(II) is a known carcinogen (Waalkes and Rehm, 1992; Jarup et al., 1998; Waalkes, 2003; Faroon et al., 2012) with elevated levels in the environment due to human activity. The deleterious effects of cadmium are compounded by its relatively long half-life in the human body (Faroon et al., 2012). The molecular mechanism of how Cd2+ modulates PKC activity remains unresolved.
The regulatory domain of conventional (i.e., Ca2+-dependent) PKC isoforms consists of three peripheral membrane binding modules: the tandem C1A and C1B domains that penetrate the membrane in response to binding a signaling lipid, diacylglycerol, and the C2 domain that binds to anionic phospholipids in a Ca2+ dependent manner (Figure 1A). The membrane recruitment step, mediated by both C1 and C2, removes the autoinhibition of the enzyme and enables it to phosphorylate its targets. C1 and C2 make use of two metal-ion cofactors: Zn2+ and Ca2+, respectively. The Zn2+ ions, 2 per C1 domain, are coordinated by the Cys3His motifs each in a tetrahedral geometry (Hubbard et al., 1991; Hommel et al., 1994; Zhang et al., 1995) and are essential for the 3D fold of C1 domains. Ca2+ ions are required for the membrane-binding function of C2 but not for its fold (Verdaguer et al., 1999; Morales et al., 2011). Up to three Ca2+ ions can bind to the all-oxygen coordination site harbored by the apical loops of C2.
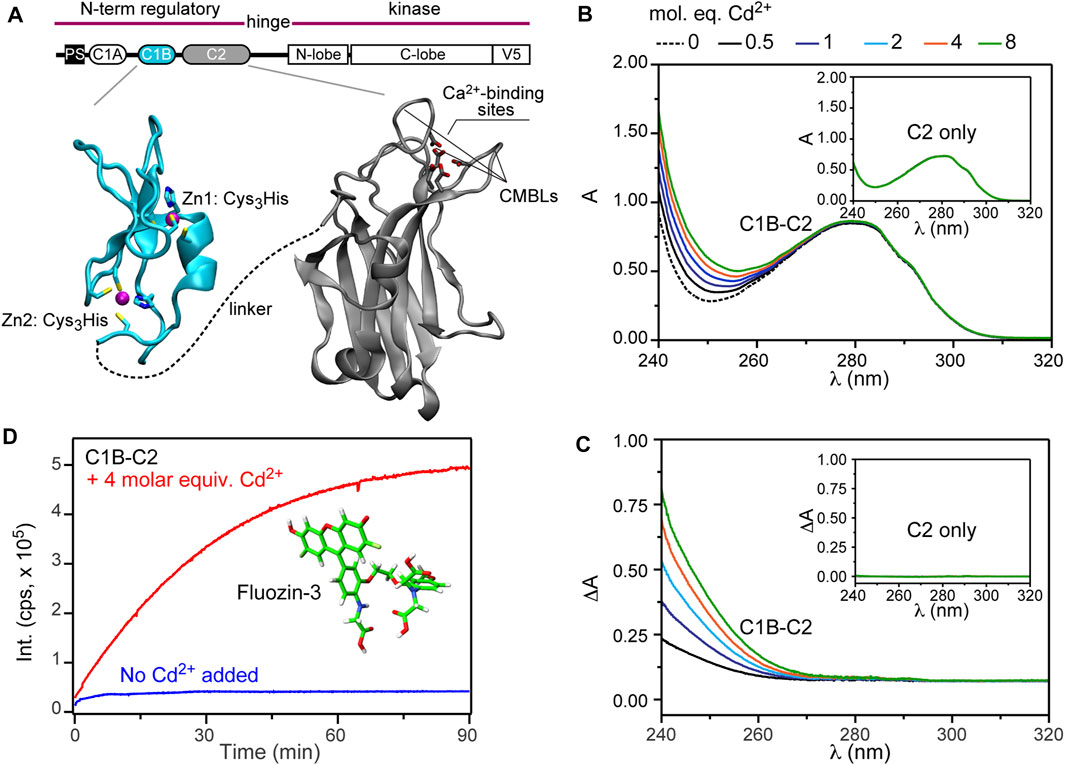
FIGURE 1. Cd2+ replaces Zn2+ in the C1B domain. (A) Ribbon diagrams of C1B (2ELI) and C2 (4L1L) highlighting the metal-ion ligands. CMBLs stand for Ca2+- and membrane-binding loop loops. (B) UV-vis absorption spectra for the Cd2+ titration of 25 μM C1B-C2. Inset: UV-vis absorption spectra for the Cd2+ titration of 25 μM isolated C2 domain. The spectrum of free Cd2+ served as the reference and was subtracted from each spectrum. (C) Difference UV-vis absorption spectra between C1B-C2 and C2 obtained at increasing molar equivalents of Cd2+. The position of the absorption shoulder is consistent with the formation of the Cd2+-thiolate bonds. (D) Cd2+-stimulated Zn2+ release from the C1B-C2 domain monitored using fluorescence intensity of FluoZin-3 (Pubchem CID 101165894) at λ = 516 nm. The no-Cd2+ control is shown in blue.
In this work, we applied solution NMR spectroscopy to probe Cd2+ interactions with the regulatory region from PKCα, with the primary objective to evaluate the reactivity of the thiol-rich Zn2+-coordinating sites towards Cd2+ substitution. For our experiments, we chose the two-domain unit from PKCα (denoted C1B-C2) that comprises C1B and the neighboring C2 connected by the native linker region (Figure 1A). C1B-C2 represents the minimum membrane-binding unit of PKCα capable of coincidence detection of four signaling molecules: diacylglycerol (C1B) and Ca2+/phosphatidylserine/phosphatidylinositol-4,5-bisphosphate (C2). We found that Cd2+ readily displaces Zn2+ at both structural sites in C1B, and that this process successfully competes with the Cd2+ interactions with the oxygen-rich C2 sites. Furthermore, despite the identical coordination spheres, the two Zn2+ sites show different thermodynamics and kinetics of Cd2+ binding. C1 and C2 domains are the basic building blocks of more than 100 proteins involved in signal transduction. Hence, the knowledge gained from this study will be applicable to other C1- and C2-containing proteins (Lemmon, 2008), leading to a more complete understanding of how xenobiotic metal ions interfere with the mechanisms of signal transduction and elicit a toxic response.
Results
Cd2+ Coordinates Thiol Groups and Ejects Zn2+ From C1B
The first step was to determine how Cd2+ interacts with the C1B-C2 domain using UV-vis absorption spectroscopy. It is well established that thiolate-Cd2+ charge transfer bands have characteristic wavelengths at around ∼240 nm (Busenlehner et al., 2001; Habjanič et al., 2020). The C1B domain has six cysteine residues, all of which are involved in coordinating the structural Zn2+ ions (Figure 1A). C2 is cysteine-free, but can bind Cd2+ with high affinity through the vacant oxygen-rich sites formed by the aspartate carboxyl groups and the carbonyl oxygens of W247 and M186 (Morales et al., 2013a). Thus, the presence of thiolate-Cd2+ charge transfer bands upon C1B-C2 treatment with Cd2+ can only originate from Cd2+ coordinating Cys residues of C1B.
Addition of increasing amounts of Cd2+ to C1B-C2 resulted in significant spectral changes (Figure 1B). Based on the C2-only control experiment with Cd2+ (inset of Figure 1B), these changes can only be attributed to the C1B-Cd2+ interactions. The difference UV-Vis spectra, where the protein contribution to the absorbance is subtracted out, clearly shows the build-up of a shoulder near λ = 270 nm (Figure 1C). The wavelength range is consistent with the position of thiolate-Cd2+ charge transfer bands observed in other studies (Busenlehner et al., 2001; Habjanič et al., 2020). Based on this information and previous work on the Zn2+-containing proteins with Cys-rich sites (Wang et al., 2005; Chakraborty et al., 2011; Malgieri et al., 2011), we conclude that Cd2+ forms coordination bonds with the cysteine residues of C1B, even in the presence of Cd2+-sequestering C2.
Two scenarios are possible: Cd2+ can either eject and substitute for Zn2+, or Cd2+ can peripherally coordinate cysteines without displacing Zn2+, forming a binuclear metal cluster similar to that observed in the GAL4 transcription factor (Pan and Coleman, 1990). To distinguish between these two scenarios, we used a highly selective Zn2+ fluorophore, FluoZin-3. Four molar equivalents of Cd2+ were added to the C1B-C2 domain in the presence of FluoZin-3, and the time-dependent fluorescence intensity was monitored at 516 nm. We observed a steady increase in the fluorescence intensity, indicating that Zn2+ is being displaced from the protein as a result of Cd2+ treatment (Figure 1D, red trace). There was no time-dependent increase in fluorescence for an identical experiment conducted in the absence of externally added Cd2+ (Figure 1D, blue trace), indicating that Fluozin-3 alone cannot strip Zn2+ off C1B. Collectively, these experiments show that Cd2+ successfully ejects Zn2+ from C1B and forms coordination bonds with cysteines.
Cd2+ Binds to Both Cys3His Sites With the Formation of All-Cd and Cd/Zn Mixed C1B Species
While the UV-vis data show that Cd2+ is displacing Zn2+ from C1B they do not contain any site-specific information. We used solution NMR spectroscopy to gain insight into how Cd2+ interacts with sites 1 and 2 of C1B (see Figure 1 for site definitions). The site-specific information was obtained by collecting 2D [15N, 1H] HSQC spectra of [U-15N] enriched C1BZn in the absence and presence of Cd2+. Each N-H group in C1BZn gives rise to a cross-peak in the 2D NMR spectra that we assigned in our previous work (Figure 2A, red spectrum) (Stewart et al., 2011). Upon addition of Cd2+, we observed an appearance of a new subset of well-dispersed C1B cross-peaks (Figure 2A, black spectrum). We were able to assign this subset to specific Cd/Zn C1B states based on their relative peak intensities and the chemical shifts of the refolded C1BCd (vide infra). From the spectral overlay, it is evident that the N-H resonances of many C1B residues, particularly those coordinating Zn1 and Zn2, experience large chemical shift perturbations upon C1B binding Cd2+.
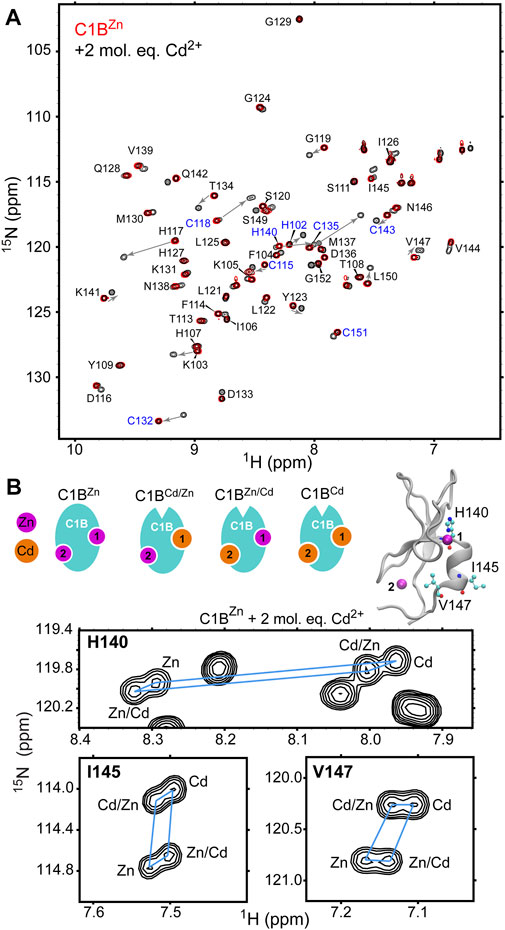
FIGURE 2. Cd2+ treatment results in the formation of fully Cd-bound and Zn/Cd mixed C1B species. (A) [15N- 1H] HSQC of 0.1 mM [U-15N] C1BZn by itself (red) and in the presence of 2 molar-equivalents of Cd2+ (black). Addition of Cd2+ results in an appearance of a new subset of cross-peaks. Arrows indicate the residue-specific changes in chemical shifts associated with Cd2+ binding to C1B. Zn2+-coordinating residues are highlighted in blue. (B) Expansions of the [15N-1H] HSQC spectra for three residues, His140, Ile145, and Val147 that show four distinct cross-peaks upon treatment of C1BZn with Cd2+. His140 is a Zn2+-coordinating residue; Ile145 and Val147 reside on the C-terminal α helix. The four Zn/Cd C1B species are shown in cartoon representation.
In addition to native C1BZn, there are three other possible Cd/Zn protein states: C1BCd, C1BZn/Cd, and C1BCd/Zn that can co-exist in solution. The N-H groups of three residues in C1B: His140, Ile145, and Val147 show four cross-peaks each (Figure 2B) and serve as direct evidence for the formation of the all-Cd and Zn/Cd mixed C1B species. Moreover, the distinct chemical shifts of the four cross-peaks enable the calculation of the relative affinities of Cd2+ to each metal ion coordination site, using the procedures described in the Materials and Methods section. The relative affinity data presented in Table 1 show that: (i) Cd2+ has a ∼2-fold and ∼1.6-fold higher affinities than Zn2+ for the C1B sites 1 and 2, respectively; and (ii) relative Cd2+ affinity for either site does not depend on the chemical identity of the metal ion, Cd2+ or Zn2+, that occupies the other site (i.e. for a given site the χ and μ values are essentially identical). We conclude that both thiol-rich coordination sites in C1B are reactive with respect to Cd2+ substituting for the native Zn2+ ion.
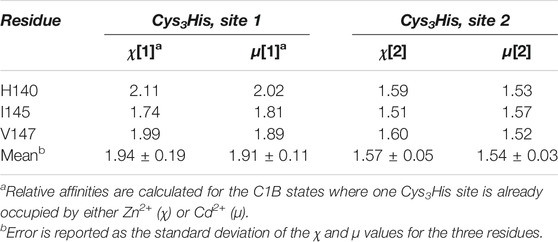
TABLE 1. Relative affinities of Cd2+ to the C1B Cys3His metal ion coordination sites.
In the PKCα regulatory region, C1B is adjacent to the C2 domain. C2 is metal-ion free in the inactive state of the kinase, but binds Ca2+ that is released as a result of the signaling events preceding PKCα activation. The Ca2+ binding site is formed by the Ca2+- and membrane-binding loops or CMBLs (Figure 1A). To determine the effect of the C2 domain on the C1B-Cd2+ interactions, we compared the [15N,1H] HSQC spectra of C1B-C2 in the absence and presence of 2 molar equivalents of Cd2+. We observed the same signatures of Zn2+ replacement as in the isolated C1B domain, including the presence of four cross peaks for Ile145 and Val147 (Figure 3A). Overall, there is an excellent correlation between the chemical shift perturbations due to Cd2+ binding for isolated C1B and C1B in the context of its neighboring C2 (Figure 3B, inset). The full chemical shift perturbation (CSP) plot shows that not only C1B resonances are affected by interactions with Cd2+, but also the CMBLs of C2 (Figure 3B). We previously demonstrated that the isolated C2 domain can bind Cd2+ with high affinity (Kd < 1 μM) through the loop regions (Morales et al., 2013a). Collectively, these data indicate that Cd2+ binds simultaneously to both C1B and C2 domains and that the thiol-rich C1B Cys3His sites can effectively compete for Cd2+ with the C2 oxygen-rich sites.
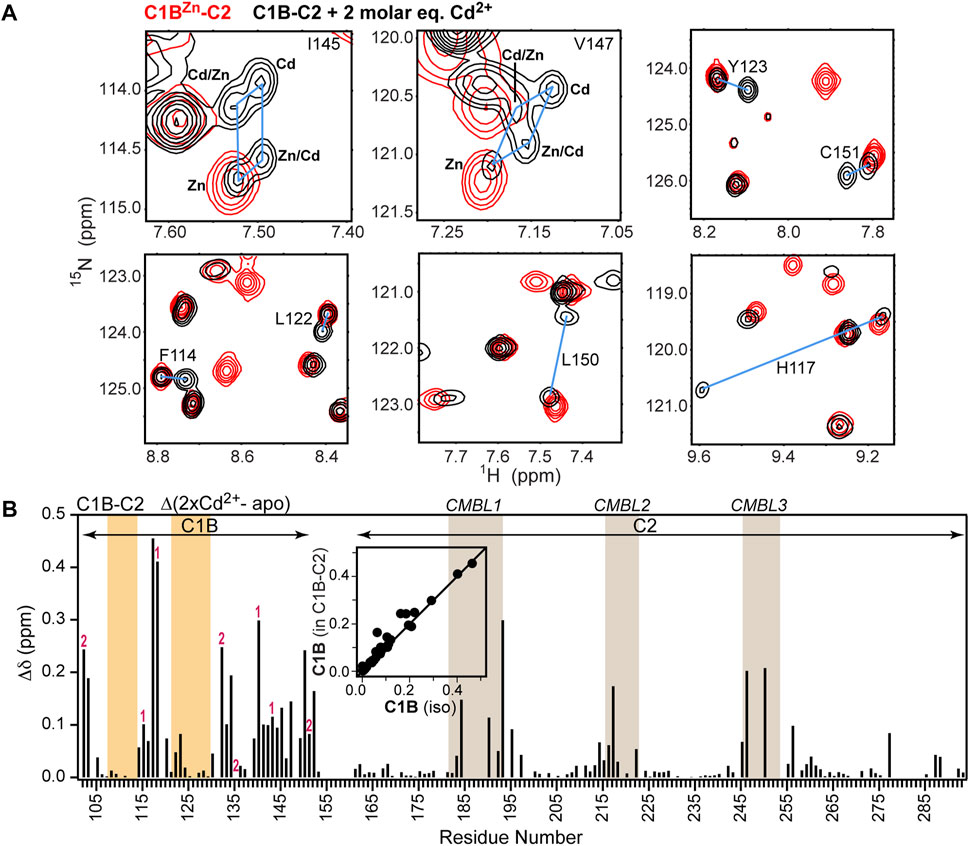
FIGURE 3. Cd2+ simultaneously populates thiol- and oxygen-rich sites in C1B-C2. (A) Overlays of the expansions of the [15N-1H] HSQC spectra of C1BZn-C2 (red) and Cd2+-treated C1B-C2 (black). The N-H cross-peaks of Cd2+ and Zn2+ containing species are connected with blue lines. (B) Chemical shift perturbations (CSPs) for backbone N-H groups as a function of C1B-C2 primary structure. The CSP values were calculated between the C1BZn-C2 and Cd2+-treated C1B-C2. Cys and His residues that coordinate Zn2+ in sites 1 and 2 are labeled accordingly. The C1B and C2 membrane-binding loops are highlighted in orange and tan, respectively. Inset: Correlation of C1B CSPs in the presence of 2 molar equivalents Cd 2+ in the isolated domain and in C1B-C2.
C1B Function Is Preserved Upon Zn2+ Replacement With Cd2+
It is evident from the chemical shift dispersion in the 2D spectra that C1B remains folded upon incorporating Cd2+ (Figure 2 and Figure 3A). To test if C1BCd retains its function, we conducted NMR-detected binding experiments between C1BCd and a tumor-promoting agent, phorbol-12,13-dibutyrate (PDBu, Figure 4A). PDBu is an extremely potent exogenous agonist of PKC that binds specifically to C1 domains and drives their membrane insertion as part of the PKC activation sequence. These properties have made PDBu the most commonly used agonist (Katti and Igumenova, 2021) in the PKC field to assess the C1 domain functional competency. To generate C1BCd as the dominant species in solution, C1BZn was denatured and refolded in the presence of Cd2+. The 2D [15N,1H] HSQC spectrum of the refolded C1BCd showed distinct chemical shifts compared to those of C1BZn (Figure 4B), but superimposed exactly onto the spectrum of the Cd2+-bound species that were formed as a result of C1BZn treatment with Cd2+ (Figure 2A).
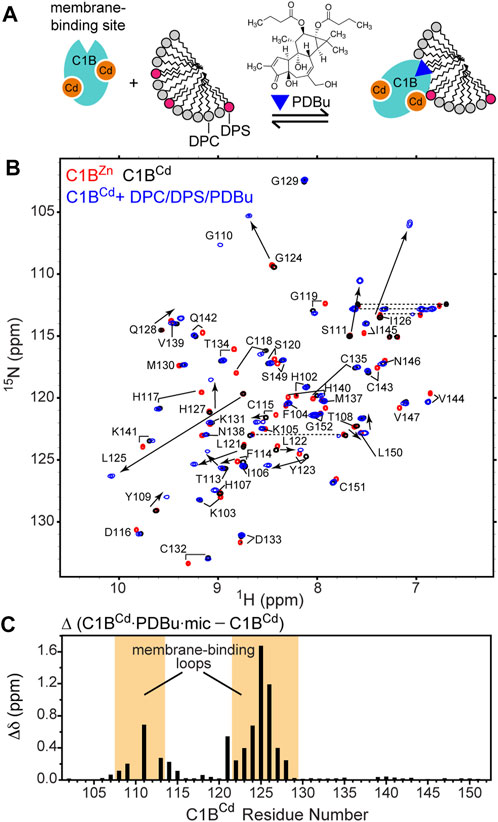
FIGURE 4. Cd2+ supports the PKC agonist-binding function of C1B. (A) Schematic representation of the experimental setup that involves C1BCd, mixed micelles, and the PKC agonist PDBu. (B) [15N-1H] HSQC spectra of isolated native C1BZn (red), C1BCd (black), and C1BCd complexed to PDBu and mixed micelles (blue). (C) Chemical shift perturbations (CSPs) upon micelle/PDBu binding for the backbone N-H groups as a function of C1B primary structure. The membrane binding loops of C1B are highlighted in orange.
PDBu is an extremely hydrophobic ligand that requires a membrane-mimicking environment to form a soluble complex with C1 domains. To provide such an environment, we used the DPC/DPS mixed micelle system that supports the C1 ligand-binding function (Stewart et al., 2011; Stewart et al., 2014) and faithfully reproduces the outcomes of in-cell experiments. Upon addition of PDBu and mixed micelles to C1BCd, we observed dramatic changes in the NMR spectrum (Figure 4B). Several residues, such as Ser111, Gly124, Leu125, and Ile126 experienced significant chemical shift perturbations upon the formation of the ternary C1BCd-PDBu-micelle complex. The CSP plot comparing the complex with the apo state showed that the changes are localized to the C1B membrane-binding loop regions, which is responsible for capturing the ligand in the membrane environment (Figure 4C). This CSP pattern is essentially identical to that observed for the native C1BZn protein upon PDBu binding in micelles (Stewart et al., 2011). Because NMR chemical shifts are exquisitely sensitive to the electronic environment of the reporting nuclei, we conclude that C1BCd interacts with PDBu and partitions into micelles in a manner identical to that of the native C1BZn.
Kinetics of Cd2+ Binding Reports on the Inequivalency of the Cys3His Structural Sites
To investigate the site-specific kinetics of Zn2+ replacement with Cd2+, we used SOFAST HMQC experiments to monitor the build-up of the Cd2+-bound C1B species. The population in % was calculated as the ratio of the N-H cross-peak intensities of the Cd2+-bound C1B, ICd, and the combined peak intensities I0 = ICd + IZn. The data were plotted as the mean of the ICd/I0 values for a subset of residues (listed in the Methods section) that report on Cd2+ binding to either site 1 or site 2. The kinetics data shown in Figure 5A revealed that sites 1 and 2 differ with respect to their kinetic behavior.
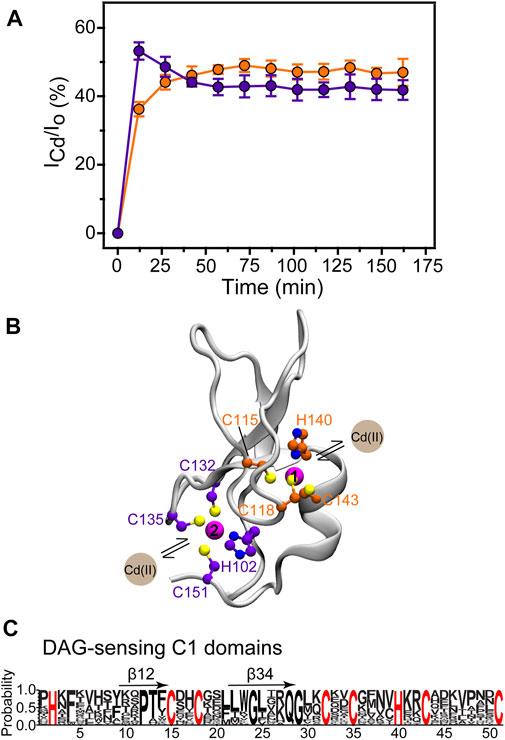
FIGURE 5. Site-resolved kinetics of Cd2+ binding to C1B. (A) The build-up of the Cd2+-bound C1B is plotted for the Cys3His sites 1 (orange) and 2 (purple). The error bars represent the standard deviations of the ICd/I0 values within a given residue subset. (B) The residues that form sites 1 and 2 are highlighted on the ribbon diagram of C1B (2ELI). (C) WebLogo representations of the sequence alignment of 31 DAG-sensitive C1 domains found in DAG effector proteins (R. norvegicus). The sequence homology values are all between 52 and 92%. Cys3His motifs are strictly conserved. β12 and β34 denote the membrane-binding loops.
Site 2 is more reactive towards Cd2+, reaching the Cd2+-bound population of 53% within the first 15 min of the experiment. This exceeds the equilibrium value by ∼10%, and the site 1 population by 17%. As shown on the 3D structure of the C1B domain in Figure 5B, Zn2+ at site 2 brings the termini of C1B together by coordinating His102 at the N-terminus and Cys151 at the C-terminus. This part of the protein has a relatively high degree of solvent exposure and is therefore readily accessible to Cd2+. Another distinct feature of site 2 is the presence of a reactive Cys residue, Cys151, which serves as the entry point for the reactive oxygen species that activate PKCα in a process involving Zn2+ release. The structural dynamics of site 2, associated with the loss of Cys151 coordination bond with Zn2+ (Stewart and Igumenova, 2012), is likely to be another factor that makes site 2 susceptible to Cd2+ interactions. Under the conditions of our experiment, the system reached equilibrium within 1 hour. At equilibrium, the Cd2+ population of site 1 is higher than that of site 2, fully consistent with the pattern of relative Cd2+ affinities (Table 1). Together, the data of Figure 1D, 5(A), and Table 1 show that Cd2+ binding accompanied by Zn2+ ejection is a slow process, and that sites 1 and 2 are non-equivalent kinetically and thermodynamically.
Discussion
Due to the similarities in charge and ligand preferences, xenobiotic Cd2+ ions target proteins that rely on Ca2+ and Zn2+ for their function (Choong et al., 2014; Petering, 2017; Duan et al., 2018; Ben Mimouna et al., 2019). Cd2+ has high affinity for thiol groups (Krizek et al., 1993) and, just like Zn2+, prefers tetrahedral geometry when coordinated by sulfur and nitrogen ligands. Cd2+ interactions with thiol groups in proteins were proposed to drive aggregation of nascent proteins through inhibition of folding in vitro (Sharma et al., 2008) and in vivo (Jacobson et al., 2017), whereas treatment with Zn2+ was shown to have a protective effect. Cd2+ can also target protein oxygen-rich sites and engage in either specific interactions in lieu of Ca2+ (Morales et al., 2013a; Katti et al., 2017) or opportunistic interactions that result in the formation of well-defined protein aggregates (Cole et al., 2019).
Here, we used Cd2+ to probe the reactivity of the structural Zn2+ sites in the regulatory membrane-binding region of the Ca2+-activated Protein Kinase Cα. Previous work on Zn2+ replacement by Cd2+ at protein structural sites suggests that generally this process can have varying consequences for the protein structure and function (Pan et al., 1990; Huang et al., 2004; Michalek et al., 2012). While in some cases Cd2+ was demonstrated to support the protein fold and function (Malgieri et al., 2014), global structural rearrangements due to Cd2+ replacing Zn2+ and loss of or change in function were also reported (Huang et al., 2004; Malgieri et al., 2011; Michalek et al., 2012). The use of Cd2+ in folding the C1 peptides derived from PKCα, β, and γ isoforms revealed isoform-specific differences in the functional behavior (Irie et al., 1998) and highlighted the need to investigate the Cd2+ response in the context of the fully folded PKC regulatory region that harbors all potential metal-ion binding sites. To that end, we used the C1B-C2 membrane-binding regulatory region to evaluate the site-specific response and reactivity of the structural thiol-rich Zn2+ sites towards Cd2+ substitution in the context of the neighboring Ca2+-sensing C2 domain. Despite the presence of competing oxygen-rich C2 metal ion binding sites, Cd2+ was able to partially eject Zn2+ from C1B-C2 (Figure 1D) with the formation of the all-Cd and Zn/Cd mixed metal ion protein species (Figure 3A). The solution NMR approach was critical here, as it enabled us to follow the Cd2+-binding process in the site-specific manner, starting with the fully folded domains and a native Zn2+ ion populating the C1B structural sites.
By specifically focusing on the isolated C1B domain, we were able to identify the spectroscopic signatures of Zn2+ replacement with Cd2+ (Figure 2) and use them to obtain thermodynamic and kinetic properties of the two Cys3His sites. The Cd2+ replacement occurs spontaneously, due to the ∼1.6- (site 2) and ∼2-fold (site 1) higher affinity to Cd2+ relative to Zn2+ (Table 1). The relative affinities can be explained by Cd2+ being a softer Lewis acid (larger ionic radius and polarizability) than Zn2+ and therefore forming stronger interactions with thiolate ligands (Puljung and Zagotta, 2011). This property confers thermodynamic advantages onto Cd2+ interactions with protein sites that are thiol-rich, such as Cys3His and Cys4 (Kluska et al., 2018). With respect to the Cd2+/Zn2+ replacement kinetics (Figure 1D and Figure 5), the reaction is slow to reach full equilibrium, likely due to the small Zn2+ koff rate constants that are typical for the high-affinity structural sites. Despite the coordination spheres being identical, site 2 is more reactive with respect to Cd2+ binding. This is evidenced by the sharp increase in the respective population of Cd2+-bound C1B species that get equilibrated within an hour to form all four possible metal-ion bound states (Figure 5A). We attribute the reactivity of site 2 to Cd2+ to its higher solvent exposure and the presence of the reactive Cys residue, Cys 151, in the coordination sphere. We previously demonstrated that in addition to being susceptible to oxidation and alkylation, Cys151 undergoes a dynamic process that slightly opens up site 2 of the C1B structure (Stewart and Igumenova, 2012). Given that this cysteine residue is proposed to be the PKC entry point of reactive oxygen species, we speculate that Cd2+ could have a protective effect by forming a stronger bond with the Cys151 residue. The diacylglycerol-sensitive C1 domains share significant sequence homology (Figure 5C), and the two Zn2+-coordinating Cys3His motifs are strictly conserved. This strongly suggests that our findings on the reactivity of the Cys3His sites in C1B from PKCα are broadly applicable to the other C1 domains. It remains to be established if the other C1s show a similar pattern of relative site reactivity, with site 2 being more reactive than site 1.
Our results for the regulatory region of PKC suggest a possible explanation of how Cd2+ can modulate PKC activity. Cd2+ spontaneously incorporates itself into the C1B structural sites without compromising the fold and PDBu-binding (Figure 4). It is therefore likely that Cd2+-substituted C1 domains will retain at least part of their diacylglycerol-binding function. The membrane-binding function of Ca2+-responsive C2 domains, however, is inhibited by Cd2+—despite its relatively high-affinity to the oxygen-rich sites of the C2 membrane-binding loops (Morales et al., 2013a; Katti et al., 2017). Since the membrane association of both domains is necessary for PKC activation, the inhibitory effect of C2 might be predominant at high Cd2+ concentrations. These findings may also have implications for the mechanisms of Cd2+ toxicity in the cell, where the identity and occupancy of target protein sites will depend on the concentration of bioavailable Cd2+.
Materials and Methods
Buffers and Metal Ion Stock Solutions
The Cd2+ stock solutions were prepared by dissolving Cd(NO3)2·4H2O (>99% purity, Sigma-Aldrich) in the appropriate buffer. Unless indicated otherwise, the experiments were conducted in the “MES buffer” comprising 10 mM 2-(N-morpholino)ethanesulfonic acid (MES) at pH 6.0 in HPLC-grade water (Avantor), 150 mM KCl, and 1 mM tris(2-carboxyethyl) phosphine (TCEP). The buffers were passed through the Chelex® 100 (Sigma-Aldrich) column to remove residual divalent metal ions.
Protein Expression
The DNA sequences encoding C1B-C2 (residues 100–293), isolated C1B (residues 100–152) or C2 (residues 155–293) of PKCα (M. musculus for C1B-C2 and C1B; R. Norvegicus for C2) were amplified by PCR using the cDNA clone of PKCα (Open Biosystems) as a template and cloned into the pET-SUMO vector (Invitrogen). Isolated C1B, C2, and C1B-C2 were expressed and purified as described previously (Morales et al., 2011; Stewart et al., 2011; Cole et al., 2019). [U-15N, 75%-2H]-enriched C1B-C2 and [U-15N]- or [U-13C, 15N]-enriched C1B were used for the NMR experiments.
UV-Vis Spectroscopy
UV-vis spectra were collected on a Beckman DU 640 spectrophotometer. 25 μM protein (C1B-C2, C2, or C1B) solution or MES buffer (for metal ion-only reference experiments) were placed in the sample cuvette; the reference cuvette always contained metal ion-free MES buffer. Cd2+ was added stepwise from the corresponding stock solutions to the sample cuvette. The samples were incubated for 1 h prior to the start of the measurements. The post-acquisition processing included the subtraction of the free Cd2+ spectra from the spectra of protein-containing samples. To eliminate contribution of protein-only absorption bands, the difference spectra were generated by subtracting the spectrum of the apo protein from the spectra of the metal-ion-containing protein. All spectra were corrected for dilution prior to subtraction.
C1B Refolding
[U-15N]-enriched C1B was dissolved in 6 M guanidine hydrochloride (Acros Organics) and the “refolding buffer” comprising 20 mM MES at pH 6.0 and 1 mM TCEP. The final protein concentration was between 15 and 35 μM during the denaturation step. The refolding was conducted in three dialysis steps, all of them carried out in the refolding buffer: (1) against 8 M urea at room temperature, for 8 h; (2) against 1.5 M urea and 100 μM Cd(II) nitrate at 4°C, overnight; and (3) against urea-free buffer at 4°C, for 3 days to ensure complete removal of urea. The refolded protein was concentrated in a Vivaspin® spin concentrator with a 3 kDa cut-off and subsequently exchanged into an “NMR buffer” (10 mM MES at pH 6.0, 150 mM KCl, 1 mM TCEP, 0.02% NaN3, and 8% (v/v) D2O using a Midi-Trap G25 desalting column (GE Healthcare).
NMR Spectroscopy
All proteins were concentrated and buffer exchanged using 10 kDa (C1B-C2), 3 kDa (C1B) and 5 kDa (C2) cut-off Vivaspin® 15R concentrators into an NMR buffer. The experiments were carried out on Avance III HD NMR spectrometers (Bruker Biospin), operating at the 1H Larmor frequencies of 800 MHz (18.8 Tesla) and 600 MHz (14.1 Tesla) equipped with cryogenically cooled probes, and 500 MHz (11.7 Tesla) equipped with a room temperature probe. The temperature was calibrated using deuterated (D4, 98%) methanol for cryogenically cooled probes and protonated methanol for the room temperature probe. Spectra were processed using NMRPipe (Delaglio et al., 1995). The cross-peak intensities were obtained using Sparky (Si et al., 2015). Sequence-specific assignments of the 1HN and 15N resonances for apo C1B-C2 were obtained using 2H-decoupled 3D HN(CA)CB, HNCA(CB), HN(COCA)CB, and HN(CO)CA (Yamazaki et al., 1994) experiments on a [U-13C,15N; 55%-2H] C1B-C2 sample. Resonance assignments for Cd2+-substituted C1B (C1BCd) were transferred from those for the native Zn2+-containing protein (C1BZn) and subsequently verified using 3D CBCA(CO)NH and HNCACB (Muhandiram and Kay, 1994) spectra collected at 14.1 Tesla. Resonance assignments for Cd2+-bound C1B-C2 were transferred from those for the isolated C1BCd and the Cd2+-complexed C2 (Morales et al., 2013b) domains. Chemical shift perturbations Δ were calculated between Cd2+-free and Cd2+-containing C1B-C2 as well as micelle/PDBu bound C1BCd and apo C1BCd according to the following equation:
where ΔδH and ΔδN are residue-specific 1HN and 15N chemical shift differences. For the NMR-detected binding experiments, the C1B ligand, phorbol-12,13-dibutyrate (PDBu, Sigma-Aldrich) was dissolved in [2H6] DMSO (Cambridge Isotopes) and added to the sample containing 94 μM of [U-15N] enriched C1BCd in the presence of 10 mM mixed micelles. Mixed micelles comprising [2H38] dodecylphosphocholine, (DPC, Cambridge Isotopes) and 2-dihexanoyl-sn-glycero-3-[phospho-l-serine] (DPS, Avanti Polar Lipids) at a molar ratio of seven to three were prepared as previously described (Stewart et al., 2011). The final concentration of PDBu in the NMR sample was 100 μM.
Determination of Relative Cd2+ and Zn2+ Affinities to C1B
The four possible Zn/Cd metallated protein states are identified using the following nomenclature: C1BZn (native C1B with Zn2+ at both structural sites), C1BCd (Cd2+ at both structural sites), C1BZn/Cd (Zn2+ at site 1 and Cd2+ at site 2), and C1BCd/Zn (Cd2+ at site 1 and Zn2+ at site 2). The fractional populations of those protein species can be defined as:
where I is the intensity of the corresponding N-H cross peaks in the 15N-1H HSQC spectra for H140, I145, and V147. The concentrations of free Cd2+ ([Cd2+]) and Zn2+ ([Zn2+]) can be calculated from the following mass balance equations:
where P0, [Cd2+]0, and [Zn2+]0 = 2×P0 are the total concentrations of protein, Cd2+, and Zn2+, respectively. It is convenient to define the affinities of metal ions to C1B in terms of individual sites. For the single metal-ion bound species, we use the M [n] notation, where M = Zn or Cd, and n = 1 or 2. For example, C1BZn[2] defines C1B with site 2 populated by Zn2+ and a vacant site 1, and KaZn[1] defines the association constant for the binding of Zn2+ to site 1 when site 2 is already populated by Zn2+. The following equilibria describe the binding processes and the associated Ka values:
The relative affinity of Cd2+ and Zn2+ to sites 1 and 2 can then be defined as the ratio of the association constants:
The χ[n] (n = 1 or 2) values report on the relative affinities of Cd2+ and Zn2+ to a given site C1B site when Zn2+ populates the other. A similar set of equilibria can be constructed to obtain the relative Cd2+ and Zn2+ affinities when Cd2+ populates the other site:
χ[n] and μ[n] for sites 1 and 2 (Table 1) were calculated using the NMR cross-peak intensities and the total concentrations of Cd2+, C1B, and Zn2+ in the system (see Eqs. 1–3). The NMR cross-peaks intensities were determined using the [15N-1H] HSQC spectrum of 0.1 mM [U-15N] C1BZn, equilibrated overnight in the presence of 0.1 mM Cd2+.
Site-specific Kinetics of Cd2+ Binding to C1B
To monitor the kinetics of Cd2+ binding to C1B, 2-fold molar excess of Cd2+ was added to 200 μM [U-13C,15N; ∼75%-2H] C1BZn in 10 mM HEPES buffer at pH 7.2, 75 mM KCl, and 1 mM TCEP. The process of Zn2+ replacement with Cd2+ was monitored using SOFAST-HMQC NMR experiments that were conducted on a 500 MHz instrument (11.7 Tesla) equipped with a room temperature probe. The first time point started 12 min post Cd2+ addition, and each SOFAST HMQC experiment took 15 min. Because the inter-conversion between Zn2+- and Cd2+-complexed states is in the slow exchange regime, at any time point their fractional population can be determined from the intensities of the corresponding amide cross-peaks in the SOFAST-HMQC spectra. We used the N-H resonances of His102, Cys132, Thr134, Cys135, and Leu150 as the reporters of Cd2+ binding to site 1; and Phe114, Cys115, His117, Cys118, Gly119, Ser120, Tyr123, Lys141, and Cys143 as the reporters of Cd2+ binding to site 2.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
TI conceived the study and administered the research project. TC and TI designed the study and wrote the article. TC conducted all experimental work and analyzed the data.
Funding
This work was supported by the National Science Foundation grant CHE-1905116. TC was supported in part by the National Institutes of Health grant R01 GM108998.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Sachin Katti for generating the sequence alignment shown in Figure 5C and for the critical reading of the article.
References
Andreini, C., Banci, L., Bertini, I., and Rosato, A. (2006). Zinc Through the Three Domains of Life. J. Proteome Res. 5 (11), 3173–3178. doi:10.1021/pr0603699
Antal, C. E., Hudson, A. M., Kang, E., Zanca, C., Wirth, C., Stephenson, N. L., et al. (2015). Cancer-Associated Protein Kinase C Mutations Reveal Kinase's Role as Tumor Suppressor. Cell. 160 (3), 489–502. doi:10.1016/j.cell.2015.01.001
Ben Mimouna, S., Le Charpentier, T., Lebon, S., Steenwinckel, J., Messaoudi, I., and Gressens, P. (2019). Involvement of the Synapse‐specific Zinc Transporter ZnT3 in Cadmium‐induced Hippocampal Neurotoxicity. J. Cel Physiol. 234, 15872–15884. doi:10.1002/jcp.28245
Bertini, I., Decaria, L., and Rosato, A. (2010). The Annotation of Full Zinc Proteomes. J. Biol. Inorg. Chem. 15 (7), 1071–1078. doi:10.1007/s00775-010-0666-6
Beyersmann, D., Block, C., and Malviya, A. N. (1994). Effects of Cadmium on Nuclear Protein Kinase C. Environ. Health Perspect. 102 (Suppl. 3), 177–180. doi:10.2307/3431783
Block, C., Freyermuth, S., Beyersmann, D., and Malviya, A. N. (1992). Role of Cadmium in Activating Nuclear Protein Kinase C and the Enzyme Binding to Nuclear Protein. J. Biol. Chem. 267 (28), 19824–19828. doi:10.1016/s0021-9258(19)88628-3
Budas, G., Churchill, E., and Mochlyrosen, D. (2007). Cardioprotective Mechanisms of PKC Isozyme-Selective Activators and Inhibitors in the Treatment of Ischemia-Reperfusion Injury. Pharmacol. Res. 55 (6), 523–536. doi:10.1016/j.phrs.2007.04.005
Busenlehner, L. S., Cosper, N. J., Scott, R. A., Rosen, B. P., Wong, M. D., and Giedroc, D. P. (2001). Spectroscopic Properties of the Metalloregulatory Cd(II) and Pb(II) Sites ofS. AureuspI258 CadC. Biochemistry. 40 (14), 4426–4436. doi:10.1021/bi010006g
Chakraborty, S., Yudenfreund Kravitz, J., Thulstrup, P. W., Hemmingsen, L., DeGrado, W. F., and Pecoraro, V. L. (2011). Design of a Three-Helix Bundle Capable of Binding Heavy Metals in a Triscysteine Environment. Angew. Chem. Int. Ed. 50 (9), 2049–2053. doi:10.1002/anie.201006413
Choong, G., Liu, Y., and Templeton, D. M. (2014). Interplay of Calcium and Cadmium in Mediating Cadmium Toxicity. Chemico-Biological Interactions. 211, 54–65. doi:10.1016/j.cbi.2014.01.007
Churchill, E., Budas, G., Vallentin, A., Koyanagi, T., and Mochly-Rosen, D. (2008). PKC Isozymes in Chronic Cardiac Disease: Possible Therapeutic Targets?. Annu. Rev. Pharmacol. Toxicol. 48, 569–599. doi:10.1146/annurev.pharmtox.48.121806.154902
Cole, T. R., Erickson, S. G., Morales, K. A., Sung, M., Holzenburg, A., and Igumenova, T. I. (2019). Cd(II)- and Pb(II)-Induced Self-Assembly of Peripheral Membrane Domains From Protein Kinase C. Biochemistry. 58 (6), 509–513. doi:10.1021/acs.biochem.8b01235
Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995). NMRPipe: a Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR. 6 (3), 277–293. doi:10.1007/bf00197809
Dempsey, E. C., Newton, A. C., Mochly-Rosen, D., Fields, A. P., Reyland, M. E., Insel, P. A., et al. (2000). Protein Kinase C Isozymes and the Regulation of Diverse Cell Responses. Am. J. Physiology-Lung Cell Mol. Physiol. 279 (3), L429–L438. doi:10.1152/ajplung.2000.279.3.L429
Drosatos, K., Bharadwaj, K. G., Lymperopoulos, A., Ikeda, S., Khan, R., Hu, Y., et al. (2011). Cardiomyocyte Lipids Impair β-adrenergic Receptor Function via PKC Activation. Am. J. Physiology-Endocrinology Metab. 300 (3), E489–E499. doi:10.1152/ajpendo.00569.2010
Duan, L., Kong, J.-J., Wang, T.-Q., and Sun, Y. (2018). Binding of Cd(II), Pb(II), and Zn(II) to a Type 1 Metallothionein From maize (Zea mays). Biometals. 31 (4), 539–550. doi:10.1007/s10534-018-0100-z
Faroon, O., Ashizawa, A., Wright, S., Tucker, P., Jenkins, K., Ingerman, L., et al. (2012). Toxicological Profile for Cadmium. Atlanta (GA): Agency for Toxic Substances and Disease Registry.
Habjanič, J., Chesnov, S., Zerbe, O., and Freisinger, E. (2020). Impact of Naturally Occurring Serine/cysteine Variations on the Structure and Function of Pseudomonas Metallothioneins. Metallomics. 12 (1), 23–33. doi:10.1039/C9MT00213H
Hommel, U., Zurini, M., and Luyten, M. (1994). Solution Structure of a Cysteine Rich Domain of Rat Protein Kinase C. Nat. Struct. Mol. Biol. 1 (6), 383–387. doi:10.1038/nsb0694-383
Huang, M., Krepkiy, D., Hu, W., and Petering, D. H. (2004). Zn-, Cd-, and Pb-Transcription Factor IIIA: Properties, DNA Binding, and Comparison With TFIIIA-finger 3 Metal Complexes. J. Inorg. Biochem. 98 (5), 775–785. doi:10.1016/j.jinorgbio.2004.01.014
Hubbard, S. R., Bishop, W., Kirschmeier, P., George, S., Cramer, S., and Hendrickson, W. (1991). Identification and Characterization of Zinc Binding Sites in Protein Kinase C. Science. 254 (5039), 1776–1779. doi:10.1126/science.1763327
Irie, K., Oie, K., Nakahara, A., Yanai, Y., Ohigashi, H., Wender, P. A., et al. (1998). Molecular Basis for Protein Kinase C Isozyme-Selective Binding: The Synthesis, Folding, and Phorbol Ester Binding of the Cysteine-Rich Domains of All Protein Kinase C Isozymes. J. Am. Chem. Soc. 120 (36), 9159–9167. doi:10.1021/ja981087f
Jacobson, T., Priya, S., Sharma, S. K., Andersson, S., Jakobsson, S., Tanghe, R., et al. (2017). Cadmium Causes Misfolding and Aggregation of Cytosolic Proteins in Yeast. Mol. Cel Biol. 37 (17). doi:10.1128/MCB.00490-16
Järup, L., Berglund, M., Elinder, C. G., Nordberg, G., and Vahter, M. (1998). Health Effects of Cadmium Exposure-a Review of the Literature and a Risk Estimate. Scand. J. Work Environ. Health. 24 (Suppl. 1), 1–51. https://www.sjweh.fi/show_abstract.php?abstract_id=281
Johnson, J. A., Adak, S., and Mochly-Rosen, D. (1995). Prolonged Phorbol Ester Treatment Down-Regulates Protein Kinase C Isozymes and Increases Contraction Rate in Neonatal Cardiac Myocytes. Life Sci. 57 (11), 1027–1038. doi:10.1016/0024-3205(95)02048-n
Katti, S., and Igumenova, T. I. (2021). Structural Insights Into C1-Ligand Interactions: Filling the Gaps by In Silico Methods. Adv. Biol. Regul. 79, 100784. doi:10.1016/j.jbior.2020.100784
Katti, S., Nyenhuis, S. B., Her, B., Srivastava, A. K., Taylor, A. B., Hart, P. J., et al. (2017). Non-Native Metal Ion Reveals the Role of Electrostatics in Synaptotagmin 1-Membrane Interactions. Biochemistry. 56 (25), 3283–3295. doi:10.1021/acs.biochem.7b00188
Khan, T. K., Nelson, T. J., Verma, V. A., Wender, P. A., and Alkon, D. L. (2009). A Cellular Model of Alzheimer's Disease Therapeutic Efficacy: PKC Activation Reverses Aβ-Induced Biomarker Abnormality on Cultured Fibroblasts. Neurobiol. Dis. 34 (2), 332–339. doi:10.1016/j.nbd.2009.02.003
Kluska, K., Adamczyk, J., and Krężel, A. (2018). Metal Binding Properties, Stability and Reactivity of Zinc Fingers. Coord. Chem. Rev. 367, 18–64. doi:10.1016/j.ccr.2018.04.009
Koya, D., and King, G. L. (1998). Protein Kinase C Activation and the Development of Diabetic Complications. Diabetes. 47 (6), 859–866. doi:10.2337/diabetes.47.6.859
Krizek, B. A., Merkle, D. L., and Berg, J. M. (1993). Ligand Variation and Metal Ion Binding Specificity in Zinc finger Peptides. Inorg. Chem. 32(6), 937–940. doi:10.1021/ic00058a030
Lemmon, M. A. (2008). Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9 (2), 99–111. doi:10.1038/nrm2328
Long, G. J. (1997). The Effect of Cadmium on Cytosolic Free Calcium, Protein Kinase C, and Collagen Synthesis in Rat Osteosarcoma (ROS 17/2.8) Cells. Toxicol. Appl. Pharmacol. 143 (1), 189–195. doi:10.1006/taap.1996.8060
Malgieri, G., Palmieri, M., Esposito, S., Maione, V., Russo, L., Baglivo, I., et al. (2014). Zinc to Cadmium Replacement in the Prokaryotic Zinc-finger Domain. Metallomics. 6 (1), 96–104. doi:10.1039/c3mt00208j
Malgieri, G., Zaccaro, L., Leone, M., Bucci, E., Esposito, S., Baglivo, I., et al. (2011). Zinc to Cadmium Replacement in the A. Thaliana SUPERMAN Cys2His2 Zinc finger Induces Structural Rearrangements of Typical DNA Base Determinant Positions. Biopolymers. 95 (11), a–n. doi:10.1002/bip.21680
Markovac, J., and Goldstein, G. W. (1988). Picomolar Concentrations of lead Stimulate Brain Protein Kinase C. Nature 334 (6177), 71–73. doi:10.1038/334071a0
Michalek, J. L., Lee, S. J., and Michel, S. L. J. (2012). Cadmium Coordination to the Zinc Binding Domains of the Non-Classical Zinc finger Protein Tristetraprolin Affects RNA Binding Selectivity. J. Inorg. Biochem. 112, 32–38. doi:10.1016/j.jinorgbio.2012.02.023
Mishra, D., and Dey, C. S. (2021). PKCα: Prospects in Regulating Insulin Resistance and AD. Trends Endocrinol. Metab. 32 (6), 341–350. doi:10.1016/j.tem.2021.03.006
Morales, K. A., Lasagna, M., Gribenko, A. V., Yoon, Y., Reinhart, G. D., Lee, J. C., et al. (2011). Pb2+as Modulator of Protein-Membrane Interactions. J. Am. Chem. Soc. 133 (27), 10599–10611. doi:10.1021/ja2032772
Morales, K. A., Yang, Y., Long, Z., Li, P., Taylor, A. B., Hart, P. J., et al. (2013a). Cd2+ as a Ca2+ Surrogate in Protein-Membrane Interactions: Isostructural but Not Isofunctional. J. Am. Chem. Soc. 135 (35), 12980–12983. doi:10.1021/ja406958k
Morales, K. A., Yang, Y., Long, Z., Li, P., Taylor, A. B., Hart, P. J., et al. (2013b). Cd2+ as a Ca2+ Surrogate in Protein-Membrane Interactions: Isostructural but Not Isofunctional. J. Am. Chem. Soc. 135 (35), 12980–12983. doi:10.1021/ja406958k
Muhandiram, D. R., and Kay, L. E. (1994). Gradient-Enhanced Triple-Resonance Three-Dimensional NMR Experiments With Improved Sensitivity. J. Magn. Reson. Ser. B. 103(3), 203–216. doi:10.1006/jmrb.1994.1032
Newton, A. C. (2010). Protein Kinase C: Poised to Signal. Am. J. Physiology-Endocrinology Metab. 298 (3), E395–E402. doi:10.1152/ajpendo.00477.2009
Pan, T., and Coleman, J. E. (1990). GAL4 Transcription Factor Is Not a "zinc finger" but Forms a Zn(II)2Cys6 Binuclear Cluster. Proc. Natl. Acad. Sci. 87 (6), 2077–2081. doi:10.1073/pnas.87.6.2077
Pan, T., Freedman, L. P., and Coleman, J. E. (1990). Cadmium-113 NMR Studies of the DNA Binding Domain of the Mammalian Glucocorticoid Receptor. Biochemistry. 29 (39), 9218–9225. doi:10.1021/bi00491a016
Petering, D. H. (2017). Reactions of the Zn Proteome with Cd2+and Other Xenobiotics: Trafficking and Toxicity. Chem. Res. Toxicol. 30 (1), 189–202. doi:10.1021/acs.chemrestox.6b00328
Puljung, M. C., and Zagotta, W. N. (2011). Labeling of Specific Cysteines in Proteins Using Reversible Metal Protection. Biophysical J. 100 (10), 2513–2521. doi:10.1016/j.bpj.2011.03.063
Rahimova, N., Cooke, M., Zhang, S., Baker, M. J., and Kazanietz, M. G. (2020). The PKC Universe Keeps Expanding: From Cancer Initiation to Metastasis. Adv. Biol. Regul. 78, 100755. doi:10.1016/j.jbior.2020.100755
Saijoh, K., Inoue, Y., Katsuyama, H., and Sumino, K. (1988). The Interaction of Cations With Activity of Soluble Protein Kinase C from Mouse Brain. Pharmacol. Toxicol. 63 (4), 221–224. doi:10.1111/j.1600-0773.1988.tb00944.x
Sharma, S. K., Goloubinoff, P., and Christen, P. (2008). Heavy Metal Ions Are Potent Inhibitors of Protein Folding. Biochem. Biophysical Res. Commun. 372 (2), 341–345. doi:10.1016/j.bbrc.2008.05.052
Si, Y.-X., Lee, J., Cai, Y., Yin, S.-J., Yang, J.-M., Park, Y.-D., et al. (2015). Molecular Dynamics Simulations Integrating Kinetics for Pb2+-Induced Arginine Kinase Inactivation and Aggregation. Process Biochem. 50 (5), 729–737. doi:10.1016/j.procbio.2015.02.008
Speizer, L. A., Watson, M. J., Kanter, J. R., and Brunton, L. L. (1989). Inhibition of Phorbol Ester Binding and Protein Kinase C Activity by Heavy Metals. J. Biol. Chem. 264 (10), 5581–5585. doi:10.1016/s0021-9258(18)83586-4
Stewart, M. D., Cole, T. R., and Igumenova, T. I. (2014). Interfacial Partitioning of a Loop Hinge Residue Contributes to Diacylglycerol Affinity of Conserved Region 1 Domains. J. Biol. Chem. 289 (40), 27653–27664. doi:10.1074/jbc.M114.585570
Stewart, M. D., and Igumenova, T. I. (2012). Reactive Cysteine in the Structural Zn2+ Site of the C1B Domain from PKCα. Biochemistry. 51 (37), 7263–7277. doi:10.1021/bi300750w
Stewart, M. D., Morgan, B., Massi, F., and Igumenova, T. I. (2011). Probing the Determinants of Diacylglycerol Binding Affinity in the C1B Domain of Protein Kinase Cα. J. Mol. Biol. 408 (5), 949–970. doi:10.1016/j.jmb.2011.03.020
Sun, X., Tian, X., Tomsig, J. L., and Suszkiw, J. B. (1999). Analysis of Differential Effects of Pb2+on Protein Kinase C Isozymes. Toxicol. Appl. Pharmacol. 156 (1), 40–45. doi:10.1006/taap.1999.8622
Tomsig, J. L., and Suszkiw, J. B. (1995). Multisite Interactions Between Pb2+ and Protein Kinase C and its Role in Norepinephrine Release from Bovine Adrenal Chromaffin Cells. J. Neurochem. 64 (6), 2667–2673. doi:10.1046/j.1471-4159.1995.64062667.x
Verdaguer, N., Corbalan-Garcia, S., Ochoa, W. F., Fita, I., and Gómez-Fernández, J. C. (1999). Ca2+ Bridges the C2 Membrane-Binding Domain of Protein Kinase Cα Directly to Phosphatidylserine. EMBO J. 18 (22), 6329–6338. doi:10.1093/emboj/18.22.6329
Waalkes, M. P. (2003). Cadmium Carcinogenesis. Mutat. Res. 533 (1-2), 107–120. doi:10.1016/j.mrfmmm.2003.07.011
Waalkes, M. P., and Rehm, S. (1992). Carcinogenicity of Oral Cadmium in the Male Wistar (WF/NCr) Rat: Effect of Chronic Dietary Zinc Deficiency. Toxicol. Sci. 19 (4), 512–520. doi:10.1093/toxsci/19.4.512
Wang, Y., Hemmingsen, L., and Giedroc, D. P. (2005). Structural and Functional Characterization of Mycobacterium tuberculosis CmtR, a PbII/CdII-Sensing SmtB/ArsR Metalloregulatory Repressor. Biochemistry. 44 (25), 8976–8988. doi:10.1021/bi050094v
Yamazaki, T., Lee, W., Arrowsmith, C. H., Muhandiram, D. R., and Kay, L. E. (1994). A Suite of Triple Resonance NMR Experiments for the Backbone Assignment of 15N, 13C, 2H Labeled Proteins with High Sensitivity. J. Am. Chem. Soc. 116 (26), 11655–11666. doi:10.1021/ja00105a005
Keywords: protein kinase C, C1 domain, zinc finger, cadmium, thiol-rich sites, cysteine reactivity, NMR spectroscopy, metal ion toxicity
Citation: Cole TR and Igumenova TI (2021) Reactivity of Thiol-Rich Zn Sites in Diacylglycerol-Sensing PKC C1 Domain Probed by NMR Spectroscopy. Front. Mol. Biosci. 8:728711. doi: 10.3389/fmolb.2021.728711
Received: 21 June 2021; Accepted: 27 July 2021;
Published: 10 August 2021.
Edited by:
Vincenzo Venditti, Iowa State University, United StatesReviewed by:
Francesca Massi, University of Massachusetts Medical School, United StatesJustin Lorieau, University of Illinois at Chicago, United States
Copyright © 2021 Cole and Igumenova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tatyana I. Igumenova, dGlndW1lbm92YUB0YW11LmVkdQ==