
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci., 26 October 2021
Sec. Molecular Diagnostics and Therapeutics
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.716735
This article is part of the Research TopicBreast Cancer Resistance, Biomarkers and Therapeutics Development in the Era of Artificial IntelligenceView all 12 articles
 Iqra Muneer1†
Iqra Muneer1† Sajjad Ahmad2†
Sajjad Ahmad2† Anam Naz3
Anam Naz3 Sumra Wajid Abbasi4Adel Alblihy5Abdulaziz A. Aloliqi6
Sumra Wajid Abbasi4Adel Alblihy5Abdulaziz A. Aloliqi6 Faris F. Aba Alkhayl7
Faris F. Aba Alkhayl7 Faris Alrumaihi7*
Faris Alrumaihi7* Sarfraz Ahmad8
Sarfraz Ahmad8 Youness El Bakri9
Youness El Bakri9 Muhammad Tahir Ul Qamar10*
Muhammad Tahir Ul Qamar10*V-domain Ig suppressor of T cell activation (VISTA) is an immune checkpoint and is a type I transmembrane protein. VISTA is linked to immunotherapy resistance, and it is a potential immune therapeutic target, especially for triple-negative breast cancer. It expresses at a high concentration in regulatory T cells and myeloid-derived suppressor cells, and its functional blockade is found to delay tumor growth. A useful medicinal plant database for drug designing (MPD3), which is a collection of phytochemicals from diverse plant families, was employed in virtual screening against VISTA to prioritize natural inhibitors against VISTA. Three compounds, Paratocarpin K (PubChem ID: 14187087), 3-(1H-Indol-3-yl)-2-(trimethylazaniumyl)propanoate (PubChem ID: 3861164), and 2-[(5-Benzyl-4-ethyl-1,2,4-triazol-3-yl)sulfanylmethyl]-5-methyl-1,3,4-oxadiazole (PubChem ID: 6494266), having binding energies stronger than −6 kcal/mol were found to have two common hydrogen bond interactions with VISTA active site residues: Arg54 and Arg127. The dynamics of the compound–VISTA complexes were further explored to infer binding stability of the systems. Results revealed that the compound 14187087 and 6494266 systems are highly stable with an average RMSD of 1.31 Å. Further affirmation on the results was achieved by running MM-GBSA on the MD simulation trajectories, which re-ranked 14187087 as the top-binder with a net binding energy value of −33.33 kcal/mol. In conclusion, the present study successfully predicted natural compounds that have the potential to block the function of VISTA and therefore can be utilized further in experimental studies to validate their real anti-VISTA activity.
Immunotherapy has turned into an important pillar of cancer treatment due to the successful blocking of the programmed cell death protein 1 (PD-1) and its ligand-programmed death-ligand 1 (PD-L1) immune checkpoints. Immune checkpoint receptors control the duration and intensity of immune response by inhibiting T cell activation (Tang et al., 2018). Several immune checkpoint proteins have been discovered, such as PD-1/PD-L1, TIGIT, VISTA, cytotoxic T lymphocyte antigen-4 (CTLA-4), TIM3, BTLA, and LAG3. PD-1 inhibitors such as nivolumab, pembrolizumab, and cemiplimab and the human IgG1 k anti-CTLA-4 monoclonal antibody ipilimumab have been approved by the Food and Drug Administration (FDA). These approved drugs have become successful cancer therapies. However, the relatively low response rate of current immunotherapy drugs (less than 30%) is still a serious challenge, and efforts are needed to identify and overcome other immunosuppressive pathways (Ventola, 2017).
In the ever-expanding list of immune checkpoints, VISTA (V-domain immunoglobulin suppressor of T cell activation) is considered to be an important regulator of the immune system. VISTA immune checkpoint protein is a type 1 transmembrane protein that is encoded by the C10orf54 gene (Wang et al., 2011). VISTA is part of the B7 family consisting of a single extracellular N-terminal Ig-V domain, a stalk with approximately 30 amino acids, a transmembrane domain, and a cytoplasmic domain (Flies et al., 2011). The closest homolog of VISTA in the B7 family is PD-L1, sharing 23% sequence identity. VISTA is highly expressed in tumor-infiltrating lymphocytes. VISTA is also expressed in CD4+ and CD8+ cells, where it negatively regulates T cell responses (Borggrewe et al., 2018; ElTanbouly et al., 2019). It has also been observed that VISTA is highly expressed in breast cancer as compared to other cancer types, indicating that targeting VISTA may benefit breast cancer immunotherapy (Xie et al., 2020). Interestingly, expression of VISTA has also been observed in different cancer types such as breast invasive carcinoma (BRCA), invasive ductal carcinoma (IDC), bladder urothelial carcinoma (BLCA), colon adenocarcinoma (COAD), kidney chromophobe (KICH), lung squamous cell carcinoma (LUSC), uterine carcinosarcoma (UCS), and skin cutaneous melanoma (SKCM). Recently, it has been reported that VISTA is the acidic pH selective ligand of PSGL-1, which means that it may engender resistance to antitumor immune response (Johnston et al., 2019). Research on a variety of clinical samples, autoimmune disease models, and tumor models has shown that VISTA has a key regulatory effect on the immune system and has the potential to be used as a therapeutic or combined drug target. These findings indicated that the high expression of VISTA on tumor cells in about 20% of NSCLC specimens can prove the feasibility of targeting VISTA for cancer therapy (Cuzick et al., 2015). Clinical studies have shown that the expression of VISTA is upregulated in oral squamous cell carcinoma and gastric cancer (Böger et al., 2017; Wu et al., 2017). After ipilimumab therapy, the VISTA immune checkpoint has also increased in patients with prostate cancer (Gao et al., 2017; Kakavand et al., 2017). In addition, previous studies have shown that VISTA is highly expressed in the immune cell subsets of human pancreatic cancer patients (Xie et al., 2018b).
Currently, compound CA-170 is undergoing phase I clinical trial for advanced tumors and lymphoma. CA-170 exhibits powerful activity to stop the lymphocyte proliferation and effector functions inhibited by VISTA proteins. CA-170 also exhibits selectivity for other immune checkpoints such as CTLA4, BTLA, and LAG3. These nonclinical data provide a strong basis for the clinical development of CA-170 (Sasikumar and Ramachandra, 2018; Wang et al., 2018; Blevins et al., 2019). In this study, we performed molecular docking to select natural drugable molecules from medicinal plants which may act as antagonists against VISTA. Molecular dynamics (MD) studies were carried out to further verify the binding of natural leads with VISTA protein.
The MPD3 database’s (https://www.bioinformation.info/) (Mumtaz et al., 2017) diverse and ready-to-dock library of phytochemicals was retrieved and filtered for lead-like molecules. Lead molecules may serve as the starting point for further structural optimization and have the best chance to become good drug candidates. The lead molecule filtration was accomplished through the online FAF-Drugs4 server (Lagorce et al., 2017). The different physicochemical parameters applied during filtration include the following: molecular weight (150–400 kDa), logP (−3 to 4), hydrogen bond donor number (≤4), hydrogen bond acceptor number (≤7), TPSA (≤160), rotatable bonds (≤9), rigid bonds (≤30), rings (≤4), maximum ring size of system (≤18), number of heteroatoms (1–15), carbon number (3–35), charges (≤4), ratio of H/C (0.1–1.1), total charge (−4 to 4), and stero centers (≤2). These parameters were applied in accordance with Lipinski’s (Lipinski et al., 1997), Veber’s (Veber et al., 2002), and Egan’s (Egan et al., 2000) rules, to screen out the most promising hits for downward analysis.
The human VISTA extracellular domain crystal structure present in the RCSB PDB database with the PDB ID: 6OIL was retrieved and processed in UCSF Chimera (Pettersen et al., 2004) for the molecular docking process. The structure was prepared first by removing co-crystalized water molecules and the NAG ligand, and then missing hydrogen atoms were added and minimized for energy via two algorithms, conjugate gradient and steepest descent, keeping the step size of 0.02 Å. Autodock Vina (Trott and Olson, 2010) was used to dock the ligand library against VISTA. We set the number of binding modes to 20 and exhaustiveness to 20. The grid dimensions were 40 × 40 × 40 (x, y, z), focused on the binding site of the VISTA native ligand along the XYZ dimension of 28.474 × 31.645 × 34.012 Å. Each docked pose was ranked using the Vina empirical scoring function where the most negative binding energy implies the most stable complex. The top 20 docked ligands with the lowest docking energy were considered for further analyses. Protein–ligand interactions were visualized using Pymol (DeLano, 2002).
The FF14SB force field of the AMBER 18 (Case et al., 2012) molecular dynamics (MD) simulation package was used for preparation of protein parameters, while its GAFF force field was used for generating ligand parameters (Wang et al., 2004). The whole system was solvated in the water box (TIP3P), considering the padding distance of 12 Å (Jorgensen et al., 1983). Particle mesh Ewald (PME) was employed for processing long-range electrostatic interactions (Darden et al., 1993), and for the nonbonded interactions, the distance cutoff was allowed to be 10 Å. The SHAKE algorithm was used to constrain the bonds involving hydrogen (Ryckaert et al., 1977). All the systems were subjected to energy minimization by running 1,000 steps of the steepest descent and conjugate gradient algorithms. Temperature of each system was equilibrated to 300 K using NVT for a time period of 20 picoseconds (ps), gradually. Afterward, the system equilibration was achieved using NPT ensemble. Finally, a production run of 50 ns was performed, and each trajectory was saved after every 2 fs. Root mean square deviation (RMSD) and root mean square fluctuation (RMSF) analysis of all trajectories was performed to check the system stability by using module CPPTRAJ (Roe and Cheatham, 2013).
The MM-GBSA method in AMBER 18 was used to estimate free energies binding for complexes (Miller et al., 2012). 100 snapshots separated at equal intervals were collected from MD trajectories to carry out the binding free energy calculations. In MM-GBSA, estimation of the net binding free energy (
In Equation 1,
In Equation 2,
The selected compounds were also subjected to different predictions such as drug-likeness, lead-likeness, pharmacokinetics, medicinal chemistry, and toxicity to guide synthetic chemists in optimizing the structure to be successful in clinical studies. Computational predictions of the compound parameters as discussed above were done using the SWISSADME server (Daina et al., 2017).
The proposed research involves virtual screening of the MPD3 database against VISTA protein, followed by MD simulations and MM-GBSA methods. MPD3 is a collection of uniquely retrieved phytochemical compounds with reported therapeutic potential. The natural compounds were preferred because they are safer, possess better pharmacokinetics, and are easy to test in further experimental studies (Riaz et al., 2017). The lead-like compounds from MPD3 were considered to be therapeutically useful in the drug discovery process, as such compounds have improved selectivity, potency, and medicinal chemistry parameters (Hughes et al., 2011). Additionally, such compounds’ structures can be easily optimized to get the desired biological activity. Previously, only Li et al. (2020) and Gabr and Gambhir (2020) reported small-molecule inhibitors against VISTA. Thus, lead-like natural compounds from MPD3 were retrieved (Figure 1). In total, 1,634 molecules were able to fulfill the criteria of lead-like compounds. Theses 1,634 compounds were used for subsequent docking studies.
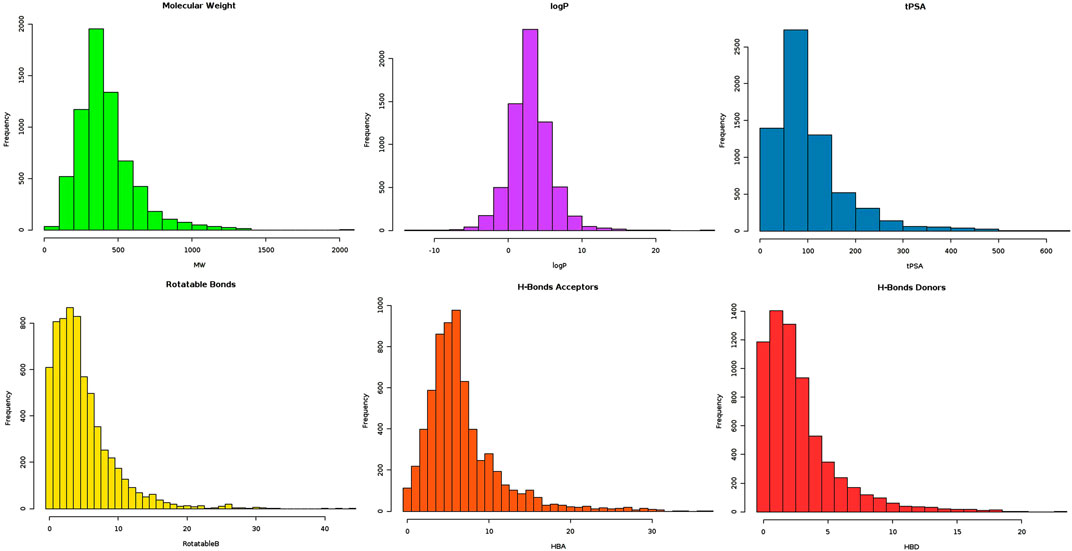
FIGURE 1. Graphical illustration of FAF-Drugs4 server output, highlighting distribution of lead-like compounds on different parameters (bars of different colors are associated with different parameter results).
The CA-170 small molecule has been reported as a dual inhibitor of PDL1/L2 and VISTA in order to treat advanced solid tumors and lymphomas. CA-170 is under phase II clinical trials for head and neck/oral cavity cancer, MSI-H positive cancers, lung cancer, and Hodgkin lymphoma in India. Its exact chemical structure has not been disclosed; however, some studies suggested that CA-170 is a peptidomimetic compound, composed of D-asparagine, L-serine, and L-threonine (Sasikumar et al., 2018; Musielak et al., 2019). Recently, the X-ray structure of the human VISTA extracellular domain has been solved at a resolution of 1.85 Å (PDB ID: 6OIL). VISTA is implicated in different cancers, including breast (Zong et al., 2020), skin (melanoma) (Kakavand et al., 2017), prostate (Gao et al., 2017), colon (Xie et al., 2018a), pancreatic (Xie et al., 2018b), ovarian (Mulati et al., 2019), and lung cancer (Villarroel-Espindola et al., 2018). A single-point mutation study to find the essential residues involved in the interaction of anti-VISTA antibody VSTB showed that three residues, Arg54, Phe62, and Gln63, are essential for the binding of VISTA to VSTB. The latter suggested that targeting these residues would be a valuable approach to inhibiting the VISTA immune checkpoint. In order to predict the binding pocket of CA-170 within the VISTA immune checkpoint, a flexible structure-based docking of CA-170 (PubChem ID: 123843830) using Autodock vina software was performed, following the same protocol as mentioned in the methodology. The grid box which represents the docking search area was centered to cover three key residues (Arg54, Phe62, and Gln63). Interestingly, the top pose of CA-170 with the lowest binding energy was forming hydrogen bonds with the Tyr41, Tyr37, Cys51, Ser52, and Arg127, including two crucial residues, Arg54 and Gln63 (Figure 2). Previous study indicated that a single-point mutation of Arg54 into Ala led to the abolition of the binding of anti-VISTA antibody VSTB to VISTA (Mehta et al., 2019). These results validated the docking protocol being applied in this study.
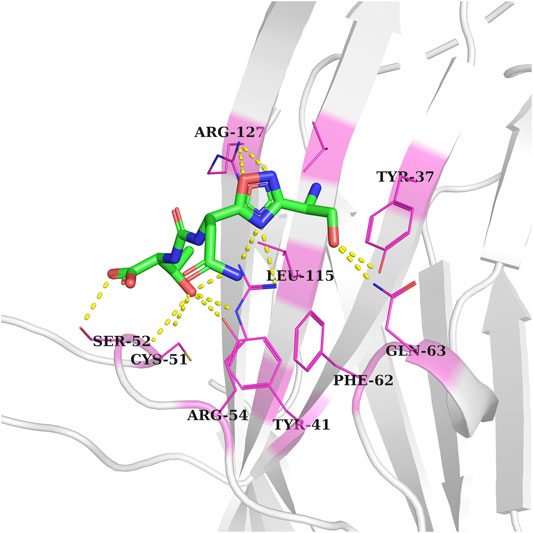
FIGURE 2. Docked conformation of the CA-170 inhibitor in the VISTA active pocket (gray color). Interacting residue of VISTA is shown as pink-colored lines-cartoons and labeled. Meanwhile, hydrogen bonding between residues is represented by yellow-colored dotted lines.
The molecular docking approach, one of the reliable approaches in the drug discovery process, was used to determine the natural inhibitors of VISTA protein. Docked ligands were graded based on their binding energy scores. The pose with the lowest score compared to CA-170 was regarded as the stable binding mode of the ligand. The top 20 compounds were visually analyzed using PyMol; out of those 20, three compounds were selected based on the binding conformation and interactions with the active site key residues. These top selected natural ligands were successfully docked in the target active site. The ligand binding poses are depicted in Figure 3.
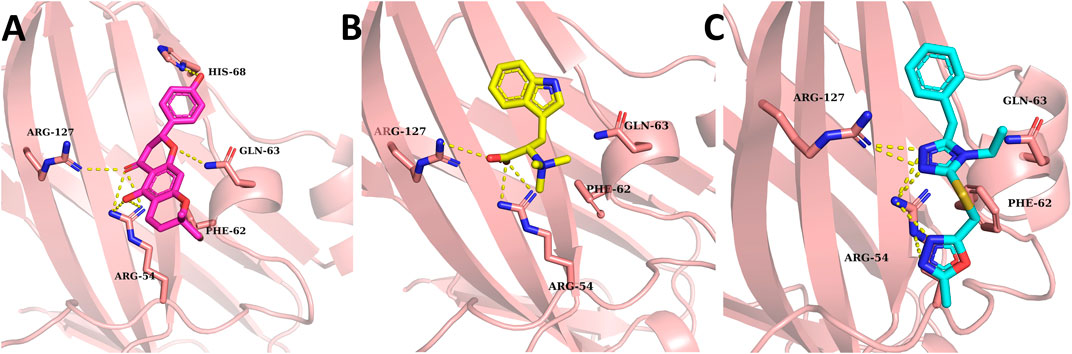
FIGURE 3. Binding conformation and hydrogen binding residues of the top three compounds at the active site of VISTA (light pink). (A) 14187087 (dark pink), (B) 3861164 (yellow), and (C) 6494266 (cyan). Meanwhile, hydrogen bonding between residues is represented by yellow-colored dotted lines.
All the compounds had at least one hydrogen bond with the critical active site residues. Among all three ligands’ compounds, 14187087 has a greater number of hydrogen bonds with an energy value of −6.3 kcal/mol. It formed hydrogen bonds with Arg54, Gln63, His68, and Arg127 residues, out of which two residues (Arg54 and Gln63) are important active site residues. Compounds 3861164 and 6494266 formed two hydrogen bonds with Arg54 and Arg127 with the binding scores −6.8 kcal/mol and −6.7 kcal/mol, respectively (Table 1). All the ligands have two common interactions with Arg54 and Arg127. As the compounds revealed favorable docking scores and good atomic-level chemical interactions, including hydrogen bonding with the VISTA, dynamics supported by binding free estimation were undertaken to further investigate the applicability of these compounds as effective VISTA inhibitors.

TABLE 1. Filtered best affinity binders of VISTA. The binding affinity score, total number of hydrogen bonds, and VISTA residues involved in hydrogen bonding with the compound.
All atom MD simulation was conducted using the AMBER package to assess the validity of the docking data and results by analyzing the dynamics behavior of protein atoms and the stability of the compounds at the binding site. For a time scale of 50 ns, the systems were analyzed for structure stability using RMSD and RMSF. The CPPTRAJ module of AMBER 18 was used to calculate the RMSD values to determine the convergence of the trajectories. RMSF values were calculated to determine the structure flexibilities of protein. Even though docking studies have been used effectively for calculating the ligand binding pose for several proteins, they failed to assess the ligand binding affinity (Cheng et al., 2012). During docking, proteins are treated as rigid molecules which do not consider the conformational changes that occur due to the ligand binding (Heitz and Van Mau, 2002). These conformational changes can be studied using molecular dynamics simulations. MD simulations have been extensively used to study the conformational changes in the protein–ligand interactions (Li et al., 2011).
Compound 6494266 fluctuated up to 3.5 Å during the first 5 ns, but later, after 15 ns, it reached equilibrium. Among all the complexes, the 14187087 compound was the most stable complex throughout the simulation with an average RMSD of 1.31 Å. However, the 3861164 complex kept oscillating throughout the simulation, indicating that this complex might be unstable among all the complexes. Thereafter, the 14187087 and 6494266 complexes were stabilized and showed steady state dynamic behavior, as shown in Figure 4.
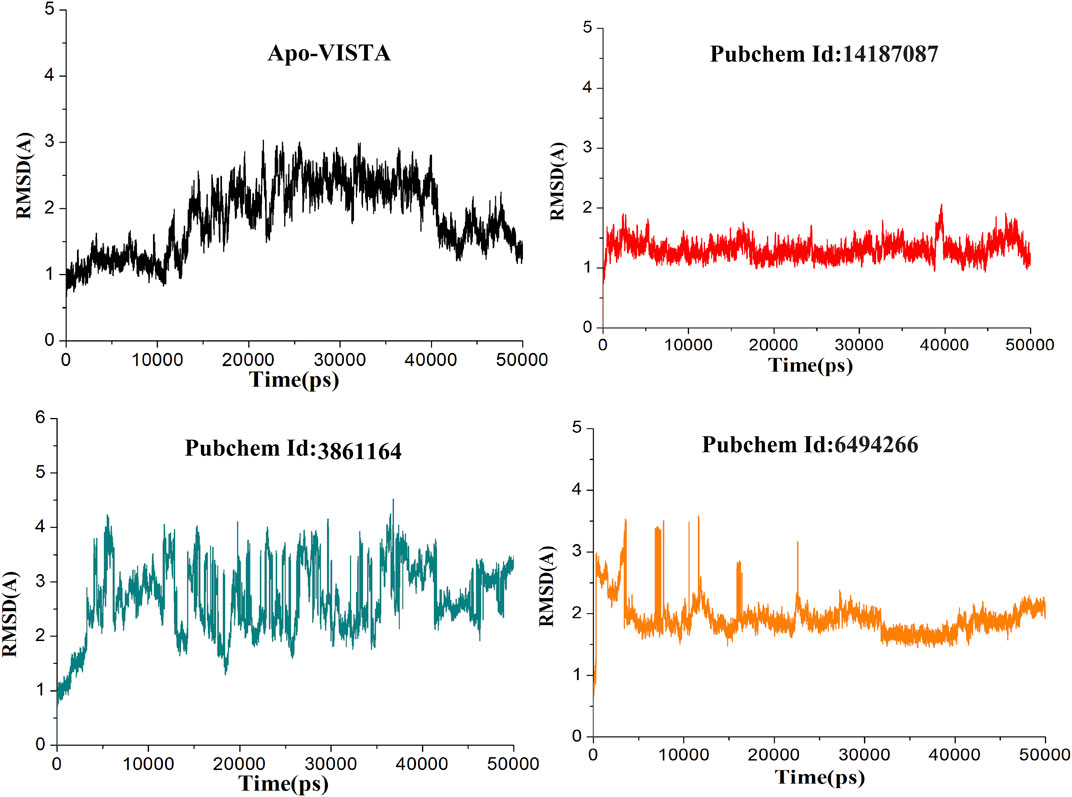
FIGURE 4. RMSD of all simulated systems as a function of time (Apo-VISAT: black, PubChem ID: 14187087-VISTA: red, PubChem ID: 3861164-VISTA: green, and PubChem ID: 6494266-VISTA: orange).
The variability in the conformation of trajectories can be monitored by calculating RMSF for individual atoms. In order to investigate and explore the conformational variability of each trajectory, RMSF of residues was plotted with respect to the residue number to show the local conformational changes for all the simulated complexes (Figure 5). Among all the docked complexes, compound 3861164 and compound 6494266 showed high fluctuations as compared to other systems, which is also consistent with the RMSD results. It can be concluded that the apo-VISTA structure, despite one large peak (40–52 amino acids), is highly stable compared to the VISTA–compound complexes. Conformational rearrangement of the loops than the rest of the protein in the presence of compounds has been previously reported and is linked to greater flexibility (Streaker and Beckett, 1999; Danielson and Lill, 2012). As VISTA protein has higher loop percentage and has a small size, upon ligand binding it is highly likely that loops may behave more dynamically. However, these fluctuations did not disturb the ligand binding conformation and the chemical interaction network, which are key to the stable binding of the compounds throughout the simulation time.
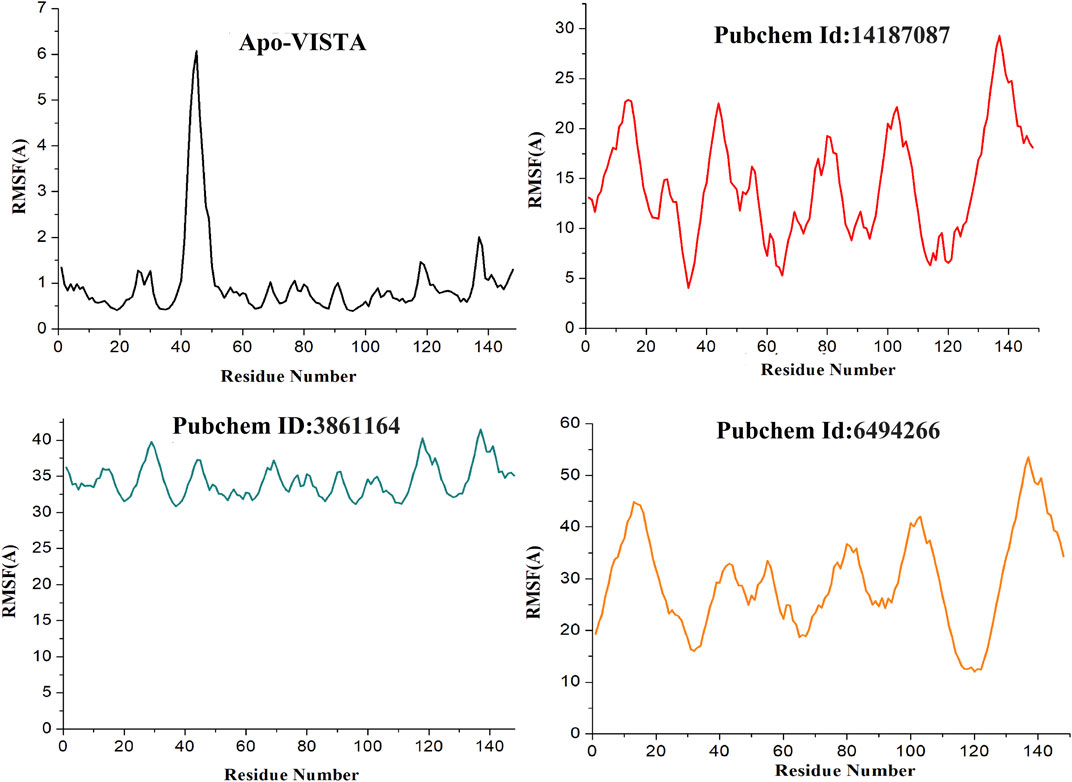
FIGURE 5. RMSF of simulated systems. The protein residues (on the x-axis) are plotted versus their flexibility (on the y-axis) from the mean position in simulation time (Apo-VISAT: black, PubChem ID: 14187087-VISTA: red, PubChem ID: 3861164-VISTA: green, and PubChem ID: 6494266-VISTA: orange).
MM-GBSA binding free energies of the complexes were estimated to validate the docking and simulation results. Such MM-GBSA binding free energy is now regularly applied in drug-designing protocols as they are more reliable than conventional docking techniques and less computationally expensive (Alamri et al., 2020a; Alamri et al., 2020b). The binding energies of complexes are presented in Table 2. It was observed that van der Waals energy and electrostatic energy dominated chemical interactions between the compounds and VISTA protein and contributed majorly to the total energy. In the interaction of 14187087, the van der Waals and electrostatic energy values were −32.2723 and −49.3294 kcal/mol, respectively, suggesting that electrostatic interactions were the major forces in the binding of VISTA and compound-1. In the case of 3861164, the contribution of van der Waals energy was −21.4642 kcal/mol and that of electrostatic energy was −16.8891 kcal/mol. In the case of complex 6494266, van der Waals and electrostatic energy was −27.7207 and −37.0189 kcal/mol, respectively. Among all three complexes, 14187087 had the minimum binding energy, indicating it as an effective inhibitor.
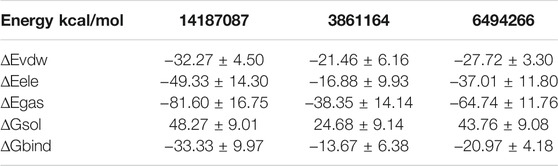
TABLE 2. Binding free energy calculations of all three complexes. ∆Egas, molecular mechanics energy in the gas phase; ∆Eele, electrostatic energy; ∆Evdw, van der Waals potential energy; ∆Gsol, solvation free energy; ∆Gbind, binding energy.
SwissADME is an online server for calculating different physical and chemical indicators and predicting drug-like properties, ADME parameters, pharmacochemical friendliness, and pharmacokinetic properties to help drug discovery. Detailed results of all the compounds are listed in Table 3. The oral bioavailability radar of the compounds is shown in Figure 6.
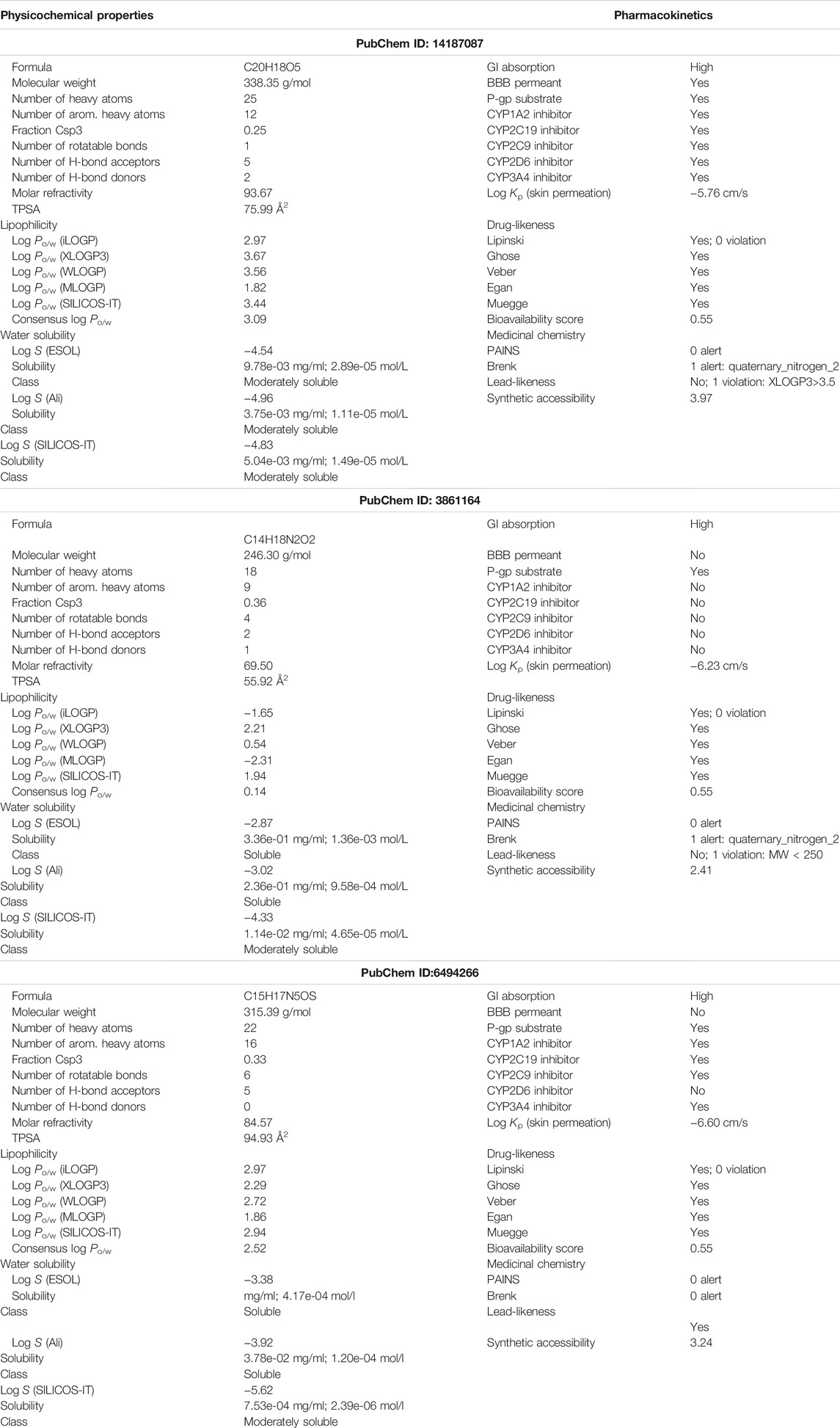
TABLE 3. Overview of different physicochemical properties, pharmacokinetics, medicinal chemistry, and drug-likeness of the compounds.
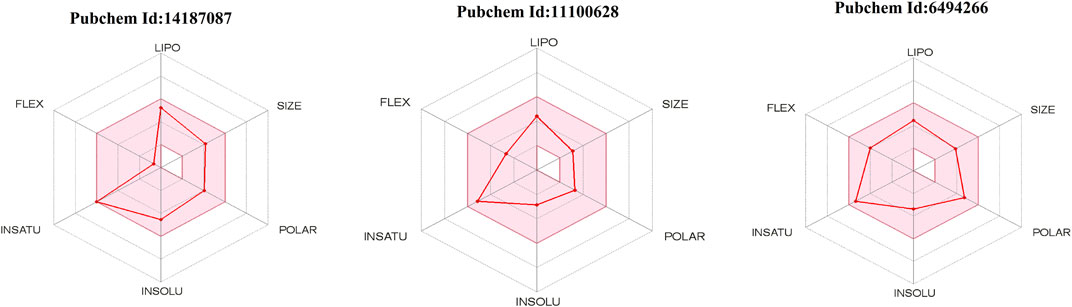
FIGURE 6. Radar of oral bioavailability (shown by red line). The pink-colored zone presents the physicochemical space allowed for orally bioavailable drugs. INSATU (instauration), INSOLU (insolubility), LIPO (lipophility), POLAR (polarity), FLEX (flexibility), and SIZE (molecular weight).
The physicochemical properties of the compounds are within the scope of drug-likeness and do not violate any Lipinski rule parameter. In addition, the compounds have good lipophilicity, so they can be transported to the maximum extent and reach the target site (Arnott and Planey, 2012). The compounds were also demonstrated to fulfill all requirements of the prominent Lipinski (Lipinski, 2004), Egan (Egan et al., 2000), Muegge (Muegge et al., 2001), and Veber (Veber et al., 2002) drug-ability rules. The compounds were predicted to be soluble and thus can be good candidates for oral administration. All the compounds were also predicted to not contain pan-assay interference compounds (PAINS) alerts and will not interact nonspecifically with multiple biological targets. This analysis revealed that the screened hits are VISTA-specific and will not have off-target effects. The compounds also have good gastrointestinal absorption, thus indicating that the good concentration of the drugs can reach the target site for performing the required action. Also, the compounds have good synthetic accessibility scores; therefore, they are easy to synthesize for experimental studies.
The study short-listed Paratocarpin K (PubChem ID: 14187087), 3-(1H-Indol-3-yl)-2-(trimethylazaniumyl)propanoate (PubChem ID: 3861164), and 2-[(5-Benzyl-4-ethyl-1,2,4-triazol-3-yl)sulfanylmethyl]-5-methyl-1,3,4-oxadiazole (PubChem ID: 6494266) from the MPD3 database as effective natural lead inhibitory molecules against VISTA protein, which is an immune checkpoint protein and is considered as a potential therapeutic target, especially for treating triple-negative breast cancer. These molecules unveiled good binding affinity as predicted by the docking technique and showed stable binding modes at the active pocket of VISTA protein. The compounds’ docked conformation dynamics study validated their stable binding nature and compounds remained intact at the active site by both hydrophobic and hydrophilic interactions with key active residues of the protein. Additionally, confirmation on the binding stability of the compounds was accomplished through the binding free energy approach, which also revealed consistent results with the docking and MD simulation outcomes. The study employed a comprehensive computational framework to identify anticancer molecules by targeting VISTA protein. Although each step is thoroughly validated and the results are investigated for accuracy via follow-up computational approaches, the study suffers from lack of experimental validation. Altogether, the study findings are promising and could be subjected to further experimental evaluation to disclose their anti-VISTA/cancer potency.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
IM and SaA: conceptualization, data curation, software, methodology, investigation, and writing—original draft preparation. AN, SuA, AdA, AbA, FFA, SFA, and YE: visualization, validation, and writing—reviewing and editing. FA and MT: resources, supervision, project administration, funding acquisition, and writing—reviewing and editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Researchers would like to thank the Deanship of Scientific Research, Qassim University, for funding publication of this project.
Alamri, M. A., Tahir Ul Qamar, M., Mirza, M. U., Bhadane, R., Alqahtani, S. M., Muneer, I., et al. (2020a). Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn., 1–13. doi:10.1080/07391102.2020.1782768
Alamri, M. A., Tahir ul Qamar, M., Afzal, O., Alabbas, A. B., Riadi, Y., and Alqahtani, S. M. (2021). Discovery of anti-MERS-CoV small covalent inhibitors through pharmacophore modeling, covalent docking and molecular dynamics simulation. J. Mol. Liquids 330, 115699. doi:10.1016/j.molliq.2021.115699
Alamri, M. A., Tahir ul Qamar, M., Mirza, M. U., Alqahtani, S. M., Froeyen, M., and Chen, L.-L. (2020b). Discovery of human coronaviruses pan-papain-like protease inhibitors using computational approaches. J. Pharm. Anal. 10, 546–559. doi:10.1016/j.jpha.2020.08.012
Arnott, J. A., and Planey, S. L. (2012). The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 7, 863–875. doi:10.1517/17460441.2012.714363
Blevins, D. J., Hanley, R., Bolduc, T., Powell, D. A., Gignac, M., Walker, K., et al. (2019). In vitro assessment of putative PD-1/PD-L1 Inhibitors: suggestions of an alternative mode of action. ACS Med. Chem. Lett. 10, 1187–1192. doi:10.1021/acsmedchemlett.9b00221
Böger, C., Behrens, H.-M., Krüger, S., and Röcken, C. (2017). The novel negative checkpoint regulator VISTA is expressed in gastric carcinoma and associated with PD-L1/PD-1: A future perspective for a combined gastric cancer therapy. Oncoimmunology 6, e1293215. doi:10.1080/2162402x.2017.1293215
Borggrewe, M., Grit, C., Den Dunnen, W. F. A., Burm, S. M., Bajramovic, J. J., Noelle, R. J., et al. (2018). VISTA expression by microglia decreases during inflammation and is differentially regulated in CNS diseases. Glia 66, 2645–2658. doi:10.1002/glia.23517
Case, D., Darden, T., Cheatham, T., Simmerling, C., Wang, J., Duke, R., et al. (2012). AMBER 12 OR. San Francisco, CA, USA: University of California.
Cheng, T., Li, Q., Zhou, Z., and Bryant, S. H. (2012). Structure-Based Virtual Screening for Drug Discovery: a Problem-Centric Review. Aaps J. 14, 133–141. doi:10.1208/s12248-012-9322-0
Cuzick, J., Thorat, M. A., Bosetti, C., Brown, P. H., Burn, J., Cook, N. R., et al. (2015). Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann. Oncol. 26, 47–57. doi:10.1093/annonc/mdu225
Daina, A., Michielin, O., and Zoete, V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717–42813. doi:10.1038/srep42717
Danielson, M. L., and Lill, M. A. (2012). Predicting flexible loop regions that interact with ligands: the challenge of accurate scoring. Proteins 80, 246–260. doi:10.1002/prot.23199
Darden, T., York, D., and Pedersen, L. (1993). Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. doi:10.1063/1.464397
Delano, W. L. (2002). Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 40, 82–92.
Egan, W. J., Merz, K. M., and Baldwin, J. J. (2000). Prediction of drug absorption using multivariate statistics. J. Med. Chem. 43, 3867–3877. doi:10.1021/jm000292e
Eltanbouly, M. A., Croteau, W., Noelle, R. J., and Lines, J. L. (2019). )VISTA: a novel immunotherapy target for normalizing innate and adaptive immunity. Semin. Immunol. Elsevier, 101308.
Flies, D. B., Wang, S., Xu, H., and Chen, L. (2011). Cutting edge: A monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J.I. 187, 1537–1541. doi:10.4049/jimmunol.1100660
Gabr, M. T., and Gambhir, S. S. (2020). Discovery and Optimization of Small-Molecule Ligands for V-Domain Ig Suppressor of T-Cell Activation (VISTA). J. Am. Chem. Soc. 142, 16194–16198. doi:10.1021/jacs.0c07276
Gao, J., Ward, J. F., Pettaway, C. A., Shi, L. Z., Subudhi, S. K., Vence, L. M., et al. (2017). VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 23, 551–555. doi:10.1038/nm.4308
Gohlke, H., Kuhn, L. A., and Case, D. A. (2004). Change in protein flexibility upon complex formation: Analysis of Ras-Raf using molecular dynamics and a molecular framework approach. Proteins 56, 322–337. doi:10.1002/prot.20116
Heitz, F., and Van Mau, N. (2002). Protein structural changes induced by their uptake at interfaces. Biochim. Biophys. Acta (Bba) - Protein Struct. Mol. Enzymol. 1597, 1–11. doi:10.1016/s0167-4838(02)00273-x
Hou, T., Wang, J., Li, Y., and Wang, W. (2011). Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 51, 69–82. doi:10.1021/ci100275a
Hughes, J., Rees, S., Kalindjian, S., and Philpott, K. (2011). Principles of early drug discovery. Br. J. Pharmacol. 162, 1239–1249. doi:10.1111/j.1476-5381.2010.01127.x
Johnston, R. J., Su, L. J., Pinckney, J., Critton, D., Boyer, E., Krishnakumar, A., et al. (2019). VISTA is an acidic pH-selective ligand for PSGL-1. Nature 574, 565–570. doi:10.1038/s41586-019-1674-5
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. doi:10.1063/1.445869
JyrkkäRinne, J., KüBlbeck, J., Pulkkinen, J., Honkakoski, P., Laatikainen, R., Poso, A., et al. (2012). Molecular dynamics simulations for human CAR inverse agonists. J. Chem. Inf. Model. 52, 457–464.
Kakavand, H., Jackett, L. A., Menzies, A. M., Gide, T. N., Carlino, M. S., Saw, R. P. M., et al. (2017). Negative immune checkpoint regulation by VISTA: a mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod. Pathol. 30, 1666–1676. doi:10.1038/modpathol.2017.89
Lagorce, D., Bouslama, L., Becot, J., Miteva, M. A., and Villoutreix, B. O. (2017). FAF-Drugs4: free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 33, 3658–3660. doi:10.1093/bioinformatics/btx491
Li, T.-T., Jiang, J.-W., Qie, C., Xuan, C.-X., Hu, X.-L., Liu, W.-M., et al. (2020). Identification of Active Small-Molecule Modulators Targeting the Novel Immune Checkpoint VISTA. BMC Immunol 22, 55. doi:10.1186/s12865-021-00446-4
Li, W., Shen, J., Liu, G., Tang, Y., and Hoshino, T. (2011). Exploring coumarin egress channels in human cytochrome P450 2A6 by random acceleration and steered molecular dynamics simulations. Proteins 79, 271–281. doi:10.1002/prot.22880
Lipinski, C. A. (2004). Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Tech. 1, 337–341. doi:10.1016/j.ddtec.2004.11.007
Lipinski, C. A., Lombardo, F., Dominy, B. W., and Feeney, P. J. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23, 3–25. doi:10.1016/s0169-409x(96)00423-1
Liu, J., Xie, X., Xuan, C., Li, T., Wang, L., Teng, L., et al. (2018b). High-density infiltration of V-domain immunoglobulin suppressor of T-cell activation up-regulated immune cells in human pancreatic cancer. Pancreas 47, 725–731. doi:10.1097/MPA.0000000000001059
Mehta, N., Maddineni, S., Mathews, I. I., Andres Parra Sperberg, R., Huang, P.-S., and Cochran, J. R. (2019). Structure and functional binding epitope of V-domain Ig suppressor of T cell activation. Cel Rep. 28, 2509–2516. e2505. doi:10.1016/j.celrep.2019.07.073
Miller, B. R., Mcgee, T. D., Swails, J. M., Homeyer, N., Gohlke, H., and Roitberg, A. E. (2012). MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theor. Comput. 8, 3314–3321. doi:10.1021/ct300418h
Muegge, I., Heald, S. L., and Brittelli, D. (2001). Simple selection criteria for drug-like chemical matter. J. Med. Chem. 44, 1841–1846. doi:10.1021/jm015507e
Mulati, K., Hamanishi, J., Matsumura, N., Chamoto, K., Mise, N., Abiko, K., et al. (2019). VISTA expressed in tumour cells regulates T cell function. Br. J. Cancer 120, 115–127. doi:10.1038/s41416-018-0313-5
Mumtaz, A., Ashfaq, U. A., Ul Qamar, M. T., Anwar, F., Gulzar, F., Ali, M. A., et al. (2017). MPD3: a useful medicinal plants database for drug designing. Nat. Prod. Res. 31, 1228–1236. doi:10.1080/14786419.2016.1233409
Musielak, B., Kocik, J., Skalniak, L., Magiera-Mularz, K., Sala, D., Czub, M., et al. (2019). CA-170 - A Potent Small-Molecule PD-L1 Inhibitor or Not. Molecules 24, 2804. doi:10.3390/molecules24152804
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi:10.1002/jcc.20084
Riaz, M., Ashfaq, U. A., Qasim, M., Yasmeen, E., Ul Qamar, M. T., and Anwar, F. (2017). Screening of medicinal plant phytochemicals as natural antagonists of p53-MDM2 interaction to reactivate p53 functioning. Anti-cancer drugs 28, 1032–1038. doi:10.1097/cad.0000000000000548
Roe, D. R., and Cheatham, T. E. (2013). PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theor. Comput. 9, 3084–3095. doi:10.1021/ct400341p
Ryckaert, J.-P., Ciccotti, G., and Berendsen, H. J. C. (1977). Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. doi:10.1016/0021-9991(77)90098-5
Sasikumar, P. G. N., Ramachandra, M., and Naremaddepalli, S. S. S. (2018). 1, 3, 4-Oxadiazole and 1, 3, 4-Thiadiazole Derivatives as Immunomodulators. Washington, DC: U.S. Patent and Trademark Office.
Sasikumar, P. G., and Ramachandra, M. (2018). Small-molecule immune checkpoint inhibitors targeting PD-1/PD-L1 and other emerging checkpoint pathways. BioDrugs 32, 481–497. doi:10.1007/s40259-018-0303-4
Streaker, E. D., and Beckett, D. (1999). Ligand-linked structural changes in the Escherichia coli biotin repressor: the significance of surface loops for binding and allostery. J. Mol. Biol. 292, 619–632. doi:10.1006/jmbi.1999.3086
Tahir ul Qamar, M., Ahmad, S., Fatima, I., Ahmad, F., Shahid, F., Naz, A., et al. (2021). Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 132, 104389. doi:10.1016/j.compbiomed.2021.104389
Tang, J., Yu, J. X., Hubbard-Lucey, V. M., Neftelinov, S. T., Hodge, J. P., and Lin, Y. (2018). The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat. Rev. Drug Discov. 17, 854–855. doi:10.1038/nrd.2018.210
Trott, O., and Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. doi:10.1002/jcc.21334
Veber, D. F., Johnson, S. R., Cheng, H.-Y., Smith, B. R., Ward, K. W., and Kopple, K. D. (2002). Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623. doi:10.1021/jm020017n
Villarroel-Espindola, F., Yu, X., Datar, I., Mani, N., Sanmamed, M., Velcheti, V., et al. (2018). Spatially Resolved and Quantitative Analysis of VISTA/PD-1H as a Novel Immunotherapy Target in Human Non-small Cell Lung Cancer. Clin. Cancer Res. 24, 1562–1573. doi:10.1158/1078-0432.ccr-17-2542
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., and Case, D. A. (2004). Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174. doi:10.1002/jcc.20035
Wang, L., Rubinstein, R., Lines, J. L., Wasiuk, A., Ahonen, C., Guo, Y., et al. (2011). VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 208, 577–592. doi:10.1084/jem.20100619
Wang, T., Wu, X., Guo, C., Zhang, K., Xu, J., Li, Z., et al. (2018). Development of inhibitors of the programmed cell death-1/programmed cell death-ligand 1 signaling pathway. J. Med. Chem. 62, 1715–1730. doi:10.1021/acs.jmedchem.8b00990
Wu, L., Deng, W.-W., Huang, C.-F., Bu, L.-L., Yu, G.-T., Mao, L., et al. (2017). Expression of VISTA correlated with immunosuppression and synergized with CD8 to predict survival in human oral squamous cell carcinoma. Cancer Immunol. Immunother. 66, 627–636. doi:10.1007/s00262-017-1968-0
Xie, S., Huang, J., Qiao, Q., Zang, W., Hong, S., Tan, H., et al. (2018a). Expression of the inhibitory B7 family molecule VISTA in human colorectal carcinoma tumors. Cancer Immunol. Immunother. 67, 1685–1694. doi:10.1007/s00262-018-2227-8
Xie, X., Zhang, J., Shi, Z., Liu, W., Hu, X., Qie, C., et al. (2020). The Expression Pattern and Clinical Significance of the Immune Checkpoint Regulator VISTA in Human Breast Cancer. Front. Immunol. 11, 563044. doi:10.3389/fimmu.2020.563044
Keywords: VISTA, breast cancer, medicinal plant, phytochemical, MD simulation
Citation: Muneer I, Ahmad S, Naz A, Abbasi SW, Alblihy A, Aloliqi AA, Aba Alkhayl FF, Alrumaihi F, Ahmad S, El Bakri Y and Tahir Ul Qamar M (2021) Discovery of Novel Inhibitors From Medicinal Plants for V-Domain Ig Suppressor of T-Cell Activation. Front. Mol. Biosci. 8:716735. doi: 10.3389/fmolb.2021.716735
Received: 29 May 2021; Accepted: 20 September 2021;
Published: 26 October 2021.
Edited by:
Dongqing Wei, Shanghai Jiao Tong University, ChinaReviewed by:
Sanjay Rathod, University of Pittsburgh, United StatesCopyright © 2021 Muneer, Ahmad, Naz, Abbasi, Alblihy, Aloliqi, Aba Alkhayl, Alrumaihi, Ahmad, El Bakri and Tahir Ul Qamar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Faris Alrumaihi, Zl9hbHJ1bWFpaGlAcXUuZWR1LnNh; Muhammad Tahir Ul Qamar, bS50YWhpcnVscWFtYXJAaG90bWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.