 Mahmudul Hasan
Mahmudul Hasan Ipsita Banerjee
Ipsita Banerjee Inna Rozman Grinberg2
Inna Rozman Grinberg2 Britt-Marie Sjöberg
Britt-Marie Sjöberg Derek T. Logan
Derek T. Logan- 1Biochemistry and Structural Biology, Dept. of Chemistry, Lund University, Lund, Sweden
- 2Dept. of Biochemistry and Biophysics, Stockholm University, Stockholm, Sweden
The essential enzyme ribonucleotide reductase (RNR) is highly regulated both at the level of overall activity and substrate specificity. Studies of class I, aerobic RNRs have shown that overall activity is downregulated by the binding of dATP to a small domain known as the ATP-cone often found at the N-terminus of RNR subunits, causing oligomerization that prevents formation of a necessary α2β2 complex between the catalytic (α2) and radical generating (β2) subunits. In some relatively rare organisms with RNRs of the subclass NrdAi, the ATP-cone is found at the N-terminus of the β subunit rather than more commonly the α subunit. Binding of dATP to the ATP-cone in β results in formation of an unusual β4 tetramer. However, the structural basis for how the formation of the active complex is hindered by such oligomerization has not been studied. Here we analyse the low-resolution three-dimensional structures of the separate subunits of an RNR from subclass NrdAi, as well as the α4β4 octamer that forms in the presence of dATP. The results reveal a type of oligomer not previously seen for any class of RNR and suggest a mechanism for how binding of dATP to the ATP-cone switches off catalysis by sterically preventing formation of the asymmetrical α2β2 complex.
Introduction
The enzyme ribonucleotide reductase (RNR) catalyzes the reduction of ribonucleotides to deoxyribonucleotides. Being the only source of dNTPs for DNA synthesis and repair, RNR is essential for all but a very few living organisms. Since its discovery in the 1950s, RNR has continuously delivered surprises in terms of radical chemistry, allosteric regulation and molecular organisation, which have all been extensively reviewed previously (Hofer et al., 2012; Ahmad and Dealwis, 2013; Torrents, 2014; Lundin et al., 2015; Mathews, 2018; Greene et al., 2020; Högbom et al., 2020). The very large family of RNR enzymes can be divided into three major functional classes based on their radical generation mechanisms (Högbom et al., 2020). All RNRs have a catalytic subunit with a 10-stranded α-β barrel fold, in class I called NrdA or α. Class I RNRs are activated by a radical generating subunit from the ferritin superfamily of all-α-helical proteins (NrdB or β). Class II RNRs, which can function both aerobically and anaerobically, have only a catalytic subunit and generate a 5′-deoxyadenosyl radical directly in the active site by homolytic cleavage of the C-Co bond in adenosylcobalamin. Class III, anaerobic RNRs also generate a 5′-deoxyadenosyl radical but do so by homolytic cleavage of S-adenosylmethionine by an accessory activase. This radical in turn generates a glycyl radical in the active site of the catalytic subunit. All three classes are unified by the transfer of these radicals to a cysteine next to the substrate in the active site.
RNR proteins are regulated at two levels: overall activity and substrate specificity. RNRs are active in the presence of ATP and downregulated by dATP through binding of these nucleotides to a small, approximately 100-residue domain known as the ATP-cone (Aravind et al., 2000), most often found at the N-terminus of the catalytic α subunit. Over the past decade it has become evident that the ATP-cone, in the presence of dATP, directs the formation of a variety of catalytically incompetent oligomers (Figure 1), all of which have in common that they break the sensitive chain of residues necessary for the proton-coupled electron transfer (PCET) that must occur upon each cycle of catalysis. In Escherichia coli, two dimers of NrdA and two dimers of NrdB form a ring-shaped α4β4 octamer in which the β2 dimers are sandwiched between the α2 dimers in such a manner that the PCET chain is broken (Ando et al., 2011). In eukaryotic RNR, three dimers of NrdA form a ring-shaped α6 hexamer (Fairman et al., 2011; Brignole et al., 2018) to which one β2 dimer can attach (Rofougaran et al., 2006; Fairman et al., 2011; Ando et al., 2016), though the interactions with NrdB and how they break the PCET chain are not fully structurally characterized for this system. More recently, higher order oligomers such as helices have also been proposed both for class Ib RNRs with partial ATP cones (Thomas et al., 2019) and class Id RNRs lacking ATP cones (Rose et al., 2019).
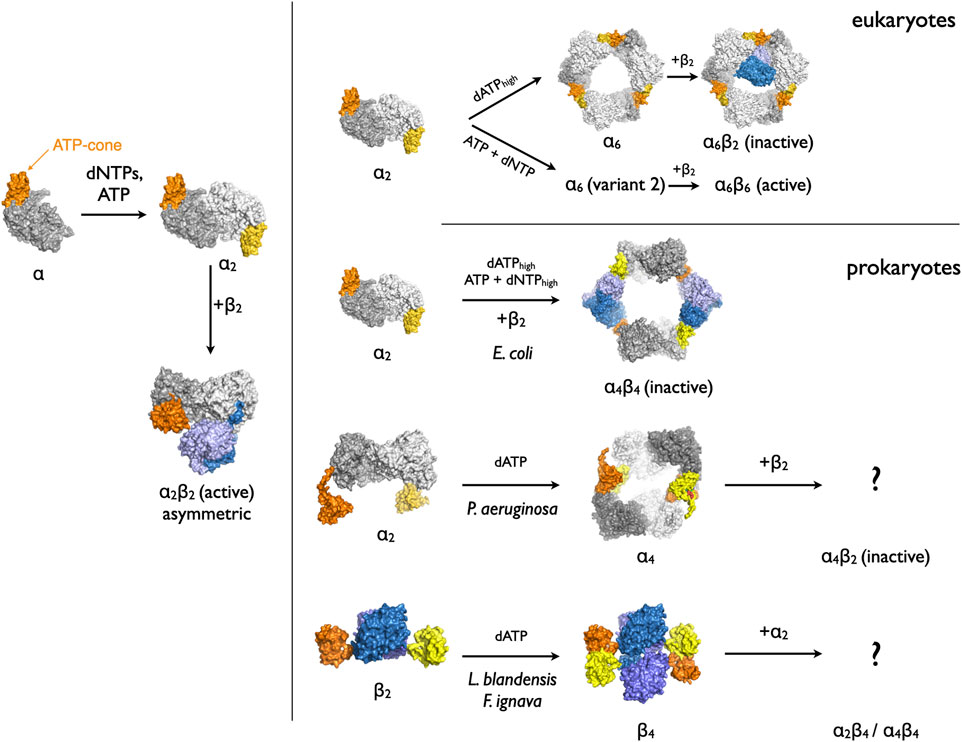
FIGURE 1. The variety of oligomers generated by binding of dATP to the ATP-cone in RNRs from different organisms. Low concentrations of dATP or other specificity-regulating dNTPs shift the monomer-dimer equilibrium towards dimers. Higher dATP concentrations induce the formation of inactive complexes of differing stoichiometries. The core domains of each NrdA subunit are shown in two shades of grey. The two ATP-cone domains in each dimer are shown in orange and yellow. The NrdB proteins are coloured in two shades of blue.
Another type of ATP-cone was identified through bioinformatics, biochemical and structural studies of the class I RNR from Pseudomonas aeruginosa (PaNrdA), which belongs to the subfamily NrdAz having multiple ATP-cones (Johansson et al., 2016). Remarkably, in P. aeruginosa the ATP-cone domain was found to bind two molecules of dATP rather than the one molecule previously observed for the subfamily NrdAg (Jonna et al., 2015). Binding of dATP induces the formation of a ring-shaped α4 tetramer consisting of two PaNrdA dimers. Furthermore, the contacts between ATP-cone domains that lead to tetramer formation involve a different area of the surface of the ATP-cone to those in subfamily NrdAg.
In some organisms, the ATP-cone is not found at the N-terminus of the catalytic subunit NrdA but rather at the N-terminus of the radical generating subunit NrdB (Rozman Grinberg et al., 2018a). In these organisms the NrdA subunit lacks an ATP-cone. Biochemical and structural studies of the class I RNR from the marine bacterium Leeuwenhoekiella blandensis have shown that the ATP-cone responds to the binding of dATP by inducing formation of an unusual β4 tetramer of two NrdB dimers, held together only by the interaction of the ATP-cones (Rozman Grinberg et al., 2018a). The details of these interactions are remarkably similar to those seen in PaNrdA despite the fact that the ATP-cone is fused to a subunit completely different in size and fold. The ATP-cone thus appears to be a genetically transposable “molecular adhesive” that can induce protein-protein interactions somewhat independently of the subunit it is attached to. The L. blandensis NrdB (LbNrdB) was also found to belong to a family of NrdBs with a novel dimanganese metal center that does not use a tyrosyl radical. A high-valent Mn2(III,IV) radical is instead generated directly at the metal center, as shown both for LbNrdB the closely related Facklamia ignava NrdB (FiNrdB) (Rozman Grinberg et al., 2018a; Rozman Grinberg et al., 2019).
When an ATP-cone-containing β is co-incubated with α, inactive oligomers with stoichiometries α2β4 and α4β4 can be isolated using size exclusion chromatography in the presence of dATP, for both the L. blandensis system (Rozman Grinberg et al., 2018a) and the one from F. ignava (Rozman Grinberg et al., 2018b). These complexes are inactive, since activity assays demonstrated a Ki of 20 μM for dATP, and at ∼90 μM dATP the enzyme is fully inactive.
One important unresolved question is how the dATP-induced tetramerization of LbNrdB and NrdBs with similar ATP-cone fusions inhibits enzymatic activity, since in the dATP-induced LbNrdB tetramer, the face of NrdB that should interact with NrdA in the active α2β2 NrdAB complex is left exposed (Rozman Grinberg et al., 2018a). How then do the α2 dimers bind to the tetramer in such a way that the resulting complexes are inactive? In 1994, Uhlin and Eklund proposed a symmetrical docking model for the active α2β2 complex in E. coli, based on the assumption that the symmetry axes of the dimers would coincide and that there would be maximal alignment of residues in the long-range PCET pathway (Uhlin and Eklund, 1994). The LbNrdB tetramer would superficially seem to present no hindrance to formation of such an active complex. However, a recent structure of the active E. coli RNR holoenzyme (EcNrdAB) determined using cryo-electron microscopy showed that the active complex was in fact highly asymmetrical (Kang et al., 2020). One EcNrdA monomer makes extensive interactions with one EcNrdB monomer, in which large stretches of the C-termini of EcNrdA and EcNrdB become ordered in the interface, while the other pair of monomers make almost no interactions (Figure 1, lower left). One hypothesis as to how the tetramerization of LbNrdB via the ATP-cone inhibits enzymatic activity is by sterically blocking the formation of such an asymmetric complex. Thus one important question is the structural organisation of the inactive complexes of this class of RNRs and how they differ from those of active complexes.
Here we present a study of the low-resolution three-dimensional structures of the two components of L. blandensis RNR in the presence of the inhibitory nucleotide dATP, namely LbNrdA alone, LbNrdB alone, and the dATP-induced inactive RNR complex LbNrdAB with stoichiometry α4β4, using small-angle X-ray scattering and cryo-electron microscopy. The results reveal a quasi-symmetrical complex in which the dimer axes of the LbNrdA and LbNrdB dimers are almost aligned and are consistent with a model in which the ATP cone sterically prevents the formation of a more asymmetrical active complex.
Materials and Methods
LbNrdA Homology Model Preparation
A homology model for residues 40–596 of LbNrdA was made using the SwissModel server (Waterhouse et al., 2018) and the sequence of LbNrdA from UniProt (accession no. A3XHF8, gene MED217_17,130). The best template found was the 1.76 Å resolution crystal structure of the class Id NrdA from Actinobacillus ureae (PDB ID 6DQX) (Rose et al., 2019), which has 59% sequence identity to LbNrdA over 92% of the length of LbNrdA. In addition, models were generated using the Phyre2 (Kelley et al., 2015) and I-TASSER (Yang et al., 2015) servers. Geometry statistics for all models were evaluated using the SwissModel server, including analysis using MolProbity (Chen et al., 2010) and are presented in Supplementary Table 1, along with RMS deviations in Cα positions between the models.
LbNrdA/LbNrdB Complex Model Preparation
The LbNrdA homology model obtained from SwissModel was placed on both sides of the LbNrdB tetramer complex (PDB ID 5OLK) in orientations compatible with the docking model for active class Ia RNR from E. coli proposed in 1994 (Uhlin and Eklund, 1994) by superimposing one of the dimers of 5OLK with the dimer of E. coli NrdB in the docking model, which was a gift from Ulla Uhlin and Hans Eklund.
Expression and Purification of LbNrdA and LbNrdB
Both LbNrdA and LbNrdB were purified as described previously (Rozman Grinberg et al., 2018a), except that the 6-His tags on the N-termini of both proteins were not removed. Protein concentrations were determined by A280 measurements using a Nanodrop spectrophotometer (Thermo Scientific) using molar extinction coefficients of 91,135 and 46,870 M−1 cm−1 for LbNrdA and LbNrdB respectively, as calculated from ProtParam (Gasteiger et al., 2005). Further preparation of the protein samples for SAXS experiments is described below.
SAXS Data Collection
SAXS experiments were carried out at beamline BM29 of the ESRF, Grenoble, France (Pernot et al., 2013) at a wavelength of 0.9919 Å. Data for LbNrdA with dATP were collected in batch mode. Dilution series of 5, 2.5, 1.25, and 0.625 mg/ml were prepared in buffer containing 50 mM Tris-HCl pH 7.6, 300 mM NaCl, 2 mM DTT, 5% glycerol, 2 mM magnesium acetate and 1 mM dATP, incubated for 5 min and centrifuged at high speed. The same buffer was used for buffer subtraction. Samples were loaded using the ESRF/EMBL SAXS sample changer (Round et al., 2015). Software implemented at the beamline was used for data collection, radial averaging of the images, background subtraction and conversion to 1D scattering profiles (Supplementary Figure 1). The protein solutions were flowed slowly through the quartz SAXS measurement capillary (diameter 1.5 mm) to avoid radiation damage and the temperature was kept constant at 10 °C. The scattered intensities were recorded on a pixel detector, Pilatus 1 M (Dectris), at a sample-detector distance of 2.867 m, allowing a range of momentum transfer q from 0.025 to 5.0 nm−1 (q = 4π sin Θ/λ, where 2q is the scattering angle and λ is the X-ray wavelength). Twenty frames of 45 ms each were recorded and compared to each other to check for radiation damage before merging. Further details are provided in Table 1.
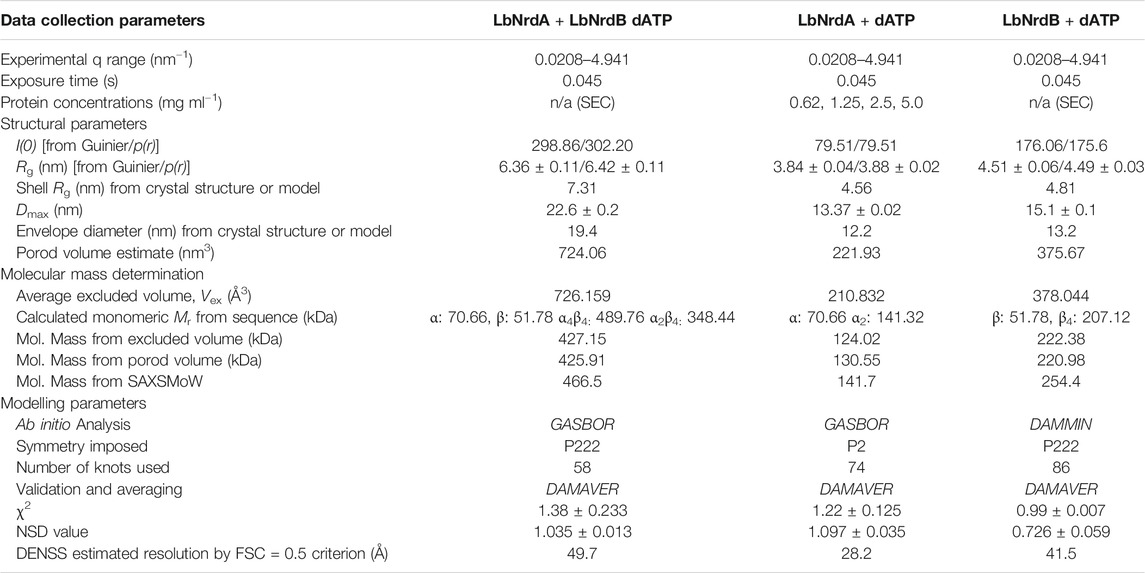
TABLE 1. SAXS data collection and derived parameters. Ab initio models were calculated in expert mode where the number of knots used from the scattering curve was adjusted to make better agreement between the data and model. Structural parameters from crystal structures or homology model were calculated using CRYSOL.
For the complex of LbNrdA and LbNrdB, as well as LbNrdB with dATP, in-line size exclusion chromatography with SAXS (SEC-SAXS) was employed, where purified protein sample was loaded onto a HiLoad 10/300 GL Superdex S200 (GE Healthcare) column. For the complex, NrdA and NrdB were mixed to reach a final concentration of 85 μM each, in buffer containing 50 mM Tris-HCl pH 7.6, 300 mM NaCl, 10% glycerol, 2 mM DTT, 1 mM dATP and 2 mM magnesium acetate. After 5 min incubation, the mixture was centrifuged at high speed and 350 μL were injected onto the column. The proteins were eluted at a flow rate of 0.5 ml min−1 and passed through the capillary cell. Frames were collected every 1 s with a total of 3,000 frames. The buffer for SEC-SAXS was 50 mM Tris-HCl pH 7.6, 300 mM NaCl, 2 mM DTT, 1 mM Mg acetate, 0.4 mM dATP and 5% glycerol. LbNrdB with dATP was prepared in the same buffer as the LbNrdA-LbNrdB complex, 350 μL of 11 mg/ml protein were loaded into the column and the same flow rate and SEC buffer were used in the SEC-SAXS data collection. Total scattering profiles and Rg plots for the elutions are shown in Supplementary Figures 2, 3.
SAXS Data Processing
The datasets collected in batch mode at different protein concentrations were compared to look for concentration-dependent aggregation effects. Having found no such artefacts, the datasets were then processed and scaled using Primus (Konarev et al., 2003). The radii of gyration (Rg) of the proteins in solution were determined from the lowest q values of the SAXS data, using the Guinier approximation. The value I(0)/c, the scattering intensity at zero angle normalized to the protein concentration of the sample, is proportional to the molecular mass M of the protein, estimated after calibration of the intensity using reference samples. The distance distribution function, representing the distribution of distances between any pair of volume elements within the particle, together with the structural parameters derived from p(r), i.e. the maximum dimension of the particle (Dmax), and the radius of gyration, were evaluated using the indirect transform method as implemented in the program GNOM (Guinier, 1939; Petoukhov et al., 2012). SEC-SAXS profiles were processed by CHROMIXS (Panjkovich and Svergun, 2018) and best fractions were chosen according to their expected elution volumes based on their molecular weight. The figures of envelopes from SAXS data and their corresponding molecular models were made using the programs PyMOL (www.pymol.org), Situs (Wriggers, 2012) and Chimera (Pettersen et al., 2004).
Ab initio SAXS Modelling
The model calculations were performed using GASBOR (Svergun et al., 2001) or DAMMIN (Franke and Svergun, 2009). The scattering profiles up to qmax = 1.26 to 4.2 nm−1 were used and 20 independent runs of modelling were carried out with each method. The 20 models were averaged and filtered using DAMAVER (Volkov and Svergun, 2003). As an additional check on the oligomeric state of LbNrdA and LbNrdB and their complexes by GASBOR or DAMMIN, several ab-initio dummy residue reconstructions were performed assuming monomeric as well as other oligomeric organizations. In all cases the models were generated assuming that either symmetry (P2 or P222) or no symmetry was present, and the best results were selected.
Electron Density Map Generation Using DENSS
As an additional check of the correctness of the ab initio structures obtained using GASBOR and DAMMIN, electron density maps were generated using the phase retrieval method implemented in the program DENSS (Grant, 2018). The same output pair distribution functions and Dmax values from GNOM as used for DAMMIN and GASBOR were used. Twenty independent models were generated for each dataset. When appropriate, 2-fold symmetry was applied to the simulation (P222 is not possible in DENSS). The best principal axis of the ellipsoid describing the molecular dimensions to which to apply the symmetry was determined by applying it to each axis in turn in independent simulations.
Cryo-Electron Microscopy
Sample Preparation
For making the LbNrdAB complex, 230 μL of LbNrdB were taken from an initial stock solution of 6 mg/ml, 2 mM dATP was added followed by 170 μL of LbNrdA from an initial stock solution of 10 mg/ml. The mixture was incubated for 10 min on ice. This was followed by centrifugation of the sample at 5,000 rpm for 6 min. Protein was initially present in buffer containing 50 mM Tris (pH 7.6), 50 mM NaCl, 2% glycerol, 2 mM MgCl2, 2 mM TCEP and 0.4 mM dATP. A Superdex 200 column was used for the purification of the complex. Gentle glutaraldehyde crosslinking was used, as initial trials showed that the complex fell apart almost completely on cryoEM grids. The original buffer was changed from 50 mM Tris to 50 mM HEPES as the Tris would react with the glutaraldehyde. Buffer exchange was done on the complex using an Amicon Ultra 4 ml centrifugal filter membrane. All other buffer components remained the same. Following wash of the Superdex 200 column using HEPES buffer, 4 ml of 0.05% glutaraldehyde were injected into the column. A further 2 ml of HEPES buffer (without glutaraldehyde) were then injected, followed by injection of the protein complex and elution. Fractions of 200 μL were collected. The formation of α4β4 complexes (theoretical molecular weight 490 kDa) was verified by the presence of a band at > 460 kDa on a Coomassie-stained reducing SDS-PAGE gel (not shown).
Single particle cryoEM grids were prepared using Quantifoil R 1.2/1.3 grids (without continuous carbon coating) using sample concentration of 2 mg/ml. The Vitrobot settings were T = 4°C, humidity 100%, blot time 5 s, blot force -5, wait and drain time 1 and 0 s respectively. The grids had a good particle distribution in thin ice.
Data Collection
Data were collected from the grids using a Titan Krios microscope (ThermoFisher) equipped with a Gatan K2 detector. A total of 493 movies were collected in counting mode using an accelerating voltage of 300 kV. The data acquisition parameters were as follows: objective aperture 100, energy filter slit 20 eV, illuminated area 1.02 μm, spot size 6 nm, spherical aberration 2.7, defocus range −1.5 to −1.3 μm, pixel size 0.82 Å2, dose 11.77 e−Å−2s−1 with an exposure time of 4 s, making a total dose of 47.1 e−Å−2. The total number of dose fractions was 40.
Data Processing
Data processing was done using cryoSPARC v2.15 (Punjani et al., 2017; Punjani, 2020). A total of 493 movies were imported then patch motion correction (multi) and patch CTF estimation (multi) were carried out using default parameters. A volume of a hypothetical octamer was made with one pair of LbNrdA dimers from the homology model and one LbNrdB tetramer from the crystal structure, based on the docking model of active complex suggested by Uhlin and Eklund, and was imported into cryoSPARC. From this volume, a set of 50 equally spaced templates was created for template-based particle picking from the CTF-corrected micrographs. The particle diameter was 210 Å and the minimum separation distance was 0.5 particle diameters. This template, low-pass filtered at 20 Å, gave 200,948 particles from the 493 micrographs. Particles with an NCC score <0.300 and power threshold <961 and >4,971 were then discarded, leaving 149,794 particles, which were then extracted with a box size of 448 pixels. All particles were used for 2D classification with 80 classes and default parameters. Fifteen 2D classes were selected. The 2D classes showed the tetramer of LbNrdB and one dimer of LbNrdA distinctly while the other dimer of LbNrdA seemed less well ordered. These 2D classes had 35,473 particles, which were used for a second round of 2D classification into 50 classes. From here, 27 2D classes having 29,902 particles were selected and used in two separate runs to make a single ab-initio model and two ab-initio models. The particles from the single model were then refined against the two models using heterogeneous refinement. One of the resulting models (model A) had 19,630 particles and a gold standard FSC resolution of 9.9 Å while the other had 10,272 particles with FSC resolution 10.8 Å. Finally, the 19,630 particles were refined against model A using homogeneous refinement, which produced a refined model with FSC resolution 8.2 Å.
Alignment of Crystal Structures and Homology Models to Ab Initio Envelopes From SAXS
The bead models from DAMMIN and GASBOR, which were obtained from independent 20 averaged and filtered models, were converted to volumes using SITUS. The crystal structures and homology models were aligned to the resulting envelopes using a combination of rigid body fitting in Chimera and manual adjustment. Molecular structures were aligned to the DENSS volumes using the script denss.align.py and some manual adjustment.
Results
LbNrdA (α2) Homology Model
The LbNrdA construct used consists of 600 amino acids. No crystal structure of LbNrdA has been determined to date. A reliable homology model for residues 40–596 was prepared using the SwissModel server with the dimeric crystal structure of Actinobacillus ureae (PDB ID 6DQX) (Rose et al., 2019) as template. This NrdA has 59% sequence identity to LbNrdA over 92% of the length of the latter. The QMEAN score for the homology model was −1.20. Two further models were constructed using the Phyre2 and I-TASSER servers. All three models were evaluated using the same criteria on the SwissModel server. Comparative statistics are shown in Supplementary Table 1. The Phyre2 and I-TASSER models had poorer overall quality but predicted a structure for the N-terminal 39 residues based on weak homology to three helices in the “connector domain” (Parker et al., 2018; Rose et al., 2019) that falls between the ATP-cone and the core in many class Ia RNRs. However, this three-helix bundle packed against the core of the protein such that its hydrophobic core enclosed three charged residues from the surface of the core: Glu69, Arg72, and Asp76. Furthermore, the predicted 3-helix bundle clearly protrudes from the cryoEM reconstruction (see below). No homology to known structures could be found for the first 39 residues of LbNrdA using a BLAST search (Altschul et al., 1990). Taken together, the evidence points towards the N-terminal 39 residues of LbNrdA being disordered.
As expected, the homology model strongly resembles an NrdA dimer lacking an ATP-cone. The SwissModel coordinates were used for further interpretation of the interactions between LbNrdA and LbNrdB. The crystal structure with PDB ID 5OLK was used for NrdB (Rozman Grinberg et al., 2018a).
Small-Angle X-Ray Scattering
SAXS data were collected for three samples: LbNrdA in the presence of dATP, LbNrdB in the presence of dATP, and finally the inactive complex of LbNrdA and LbNrdB in the presence of dATP (LbNrdAB). To ensure sample homogeneity, the full-length LbNrdB and LbNrdAB preparations were recorded using inline size exclusion chromatography. Key structural parameters derived from the SAXS data are presented in Table 1.
LbNrdA Forms a Dimer in the Presence of dATP
The solution scattering data from dATP-bound LbNrdA indicate a globular protein with an Rg of 3.92 nm and Dmax of 13.9 nm (Table 1). The Rg value compares well with the value of 3.66 nm from the homology model, which lacks a total of 43 residues at the termini of each monomer, consistent with its lower Rg. The distance distribution function is shown in Figure 2. The form of the p(r) function, with a maximum at less than Dmax/2, suggests that dATP-bound LbNrdA NrdA forms a species that is wider in one axis and the long tail on the right is consistent with the presence of unstructured terminal residues.
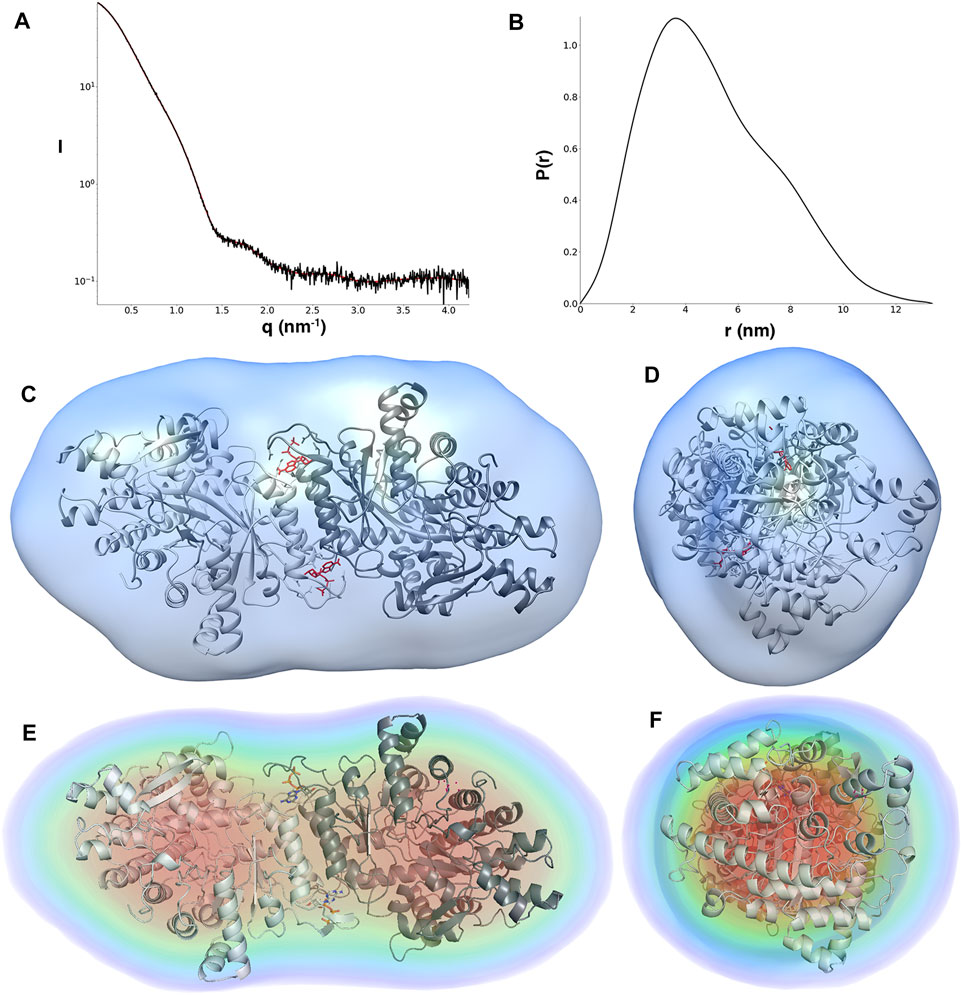
FIGURE 2. (A) Scattering curve (black line) and fitting (red line) to generate pair distance distribution function of dATP-bound LbNrdA. (B) Pair distance distribution function. The angular range used for fitting to generate p(r) is from 0.13 to 4.22 nm. Ab initio models of dATP bound LbNrdA superimposed on the homology model. (C,D) Two views of the envelope from GASBOR processed to a volume using SITUS, related by a 90° rotation around the vertical axis. (E,F) Similar views of the DENSS density reconstruction at approximately 42 Å resolution.
Furthermore, models for dATP-bound LbNrdA obtained by GASBOR ab initio reconstruction and DENSS phase recovery show a good fit to the dimeric homology model (Figure 2). The resolution of the DENSS reconstruction is estimated to be 28 Å by the FSC 0.5 criterion (Supplementary Figure 4). The correlation score of a 15 Å resolution map calculated from the homology model and the DENSS reconstruction is 0.889, as calculated in DENSS.
LbNrdB in Complex With dATP Shows a Tetrameric Arrangement in Solution Consistent With the Crystal Structure
SAXS data for LbNrdB in complex with dATP are shown in Figure 3. The p(r) function obtained from dATP-bound LbNrdB (Figure 3B) shows tailing at high values of r consistent with small separate domains connected to the ends of the core domain, i.e. the four ATP-cones of the tetramer. The distribution of p(r) peaks with a maximum at less than Dmax/2 also confirms the elongated form of dATP-bound LbNrdB. The Rg values of 4.49 nm obtained from the Guinier approximation and from the p(r) function are identical. These and the Dmax value of 15.1 nm confirm the tetrameric arrangement in the known crystal structure (5OLK). The maximum distance between ordered atoms in the crystal structure is about 12.5 nm, thus Dmax is consistent with the presence of disordered residues at the N- and C-termini (see below). The molecular mass calculated from the Porod volume is 221 kDa and the one calculated from the excluded volume is 222 kDa, both of which are close to the calculated one of 250 kDa, confirming the tetrameric arrangement of LbNrdB in solution.
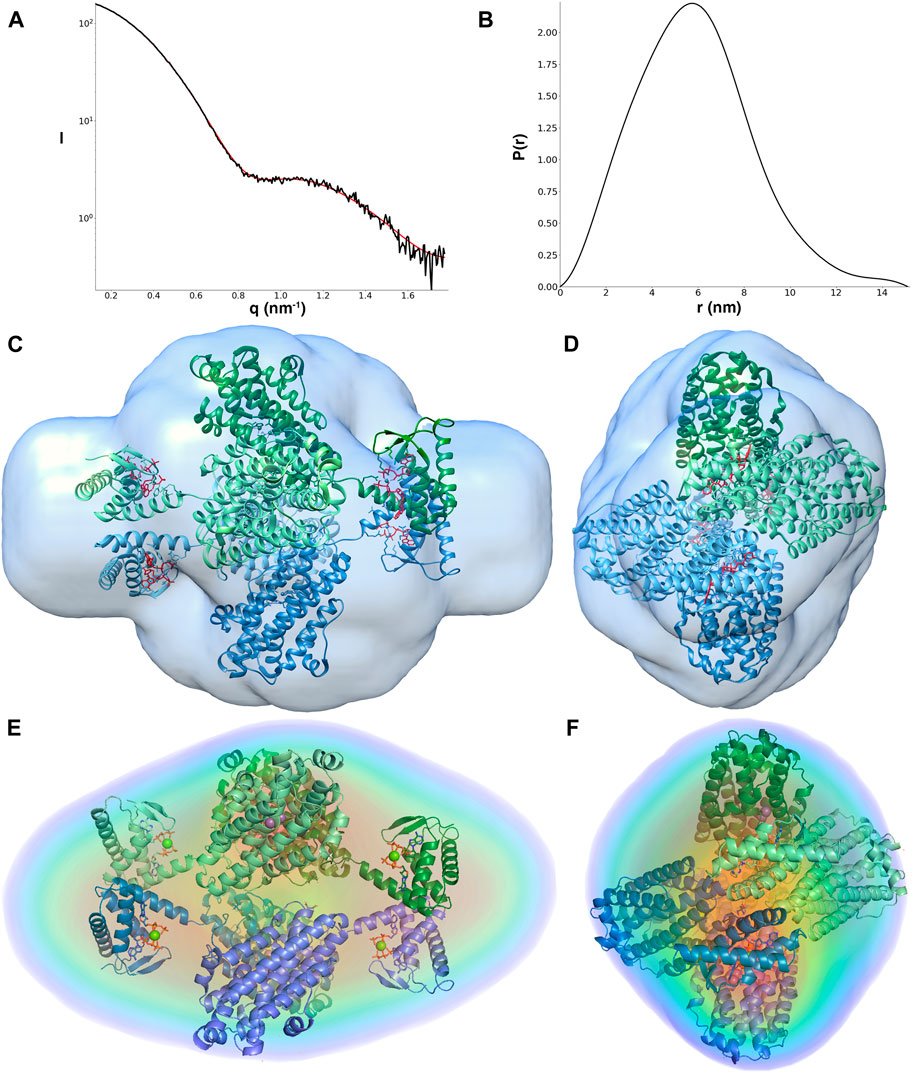
FIGURE 3. (A) Fit of the GASBOR model (red line) to the experimental scattering curve (black line); (B) pair distance distribution function of LbNrdB in the presence of dATP. The angular range used for fitting to generate p(r) was from 0.13 to 1.77 nm−1. (C–F) The tetrameric arrangement of LbNrdB as in PDB structure 5OLK fits well with the ab initio models derived from solution scattering data. (C,D) show the crystal structure fitted to the reconstruction from DAMMIN processed to a volume in SITUS. (E,F) show the fit of the crystal structure to the electron density reconstructed by DENSS at approximately 28 Å resolution. Views in (D,F) are rotated by 90° around the vertical axis relative to (C,E) respectively. The cartoon representation of LbNrdB is coloured by chain. The 8 dATP molecules are shown as sticks and the 4 Mg2+ ions as spheres.
The overall ab initio models obtained from the SAXS data also show good agreement with the crystal structure. The resolution of the DENSS reconstruction is estimated to be 42 Å (Supplementary Figure 4). The correlation score between the DENSS reconstruction and a 15 Å map calculated from the crystal structure is 0.740. The excluded volume obtained from the model is also consistent with a homotetrameric oligomeric state. The crystal structure of LbNrdB lacks between 24 and 29 residues at the N-terminus and 28–29 residues at the C-terminus of each chain, including the N-terminal His-tag and its cleavage site, which are not visible in the electron density due to flexibility. These regions may account for the extra volume close to the N-terminus (near the ATP-cone) and C-terminus.
Modelling of the Missing N- and C-Terminal Residues in the LbNrdB Tetramer
The residues missing in the crystal structure at the N- and C-termini of each chain were modelled as dummy atoms using the program CORAL, which uses a fixed protein conformation with flexible ends and/or linkers. Thus, CORAL modelling gives good structural reconstructions for a more rigid and less flexible protein that does not possess a large structural heterogeneity. In contrast, CORAL yields poor reconstructions for systems that are better represented by a broader structural ensemble. In addition to the termini, the connecting neck between the N-terminal ATP-cone domain and C terminal domain consisting of 16 residues (from residue 103–118) was also deleted and rebuilt with CORAL.
CORAL was used to generate five representative models where χ2 values varied from 1.23 to 1.32 (Figure 4A). Through the CORAL modelling the χ2 value was reduced from on average 3.27 for the crystal structure alone to 1.27. This good fit (Figure 4B) indicates that the overall structure, including the relative orientations of the ATP-cone domains relative to the catalytic core domains, is very similar to the crystal structure and that the tetramer as a whole is not very flexible. In the CORAL model, in all four monomers the N-terminal residues project directly outwards from their respective ATP-cones. In contrast, the missing C-terminal residues show more variability in that they either lie close to the structured domains or project outwards in opposite directions. The modelled termini correspond well to the regions of density not occupied by the crystal structure in the DAMMIN and DENSS reconstructions.
FIGURE 4. (A) Models of the LbNrdB tetramer with built missing N- and C-termini, each with 24–29 residues modelled using CORAL. The neck region between the core and ATP cone domains consisting of 16 residues was also deleted and rebuilt. Five possible models obtained from independent runs of CORAL are shown. (C) The scattering curve of LbNrdB (black) and its fit with the CORAL model (red).
LbNrdA and LbNrdB Form a Hetero-Octamer With Stoichiometry α4β4 in Solution in the Presence of dATP
The Porod volume of the LbNrdAB species induced by dATP is 724.1 nm3, which indicates a molecular mass of 425.9 kDa. The calculated molecular mass of an α4β4 octamer that consists of the LbNrdB tetramer with two LbNrdA dimers attached is 489.8 kDa, while the molecular mass of an α2β4 hexamer is only 348.4 kDa. Thus the most plausible stoichiometry for this species in solution is α4β4, though we cannot exclude the presence of a small amount of α2β4. The Rg of the LbNrdAB complex is 6.26 nm and Dmax is 22.6 nm, both of which are significantly larger than either of the β4 or α2 species described above. During ab-initio modelling with GASBOR, several possible symmetries were imposed and only P222 symmetry gave a reconstruction that fitted well with the data. This strongly supports an arrangement where two dimers of NrdA are arranged on opposite sides of the dATP-induced NrdB tetramer observed in the crystal structure and confirmed by SAXS (Figure 5). Furthermore, this arrangement is compatible with the concentration-dependent formation of α4β4 species observed in previous gas phase electrophoretic mobility analysis GEMMA and analytical SEC experiments (Rozman Grinberg et al., 2018a).
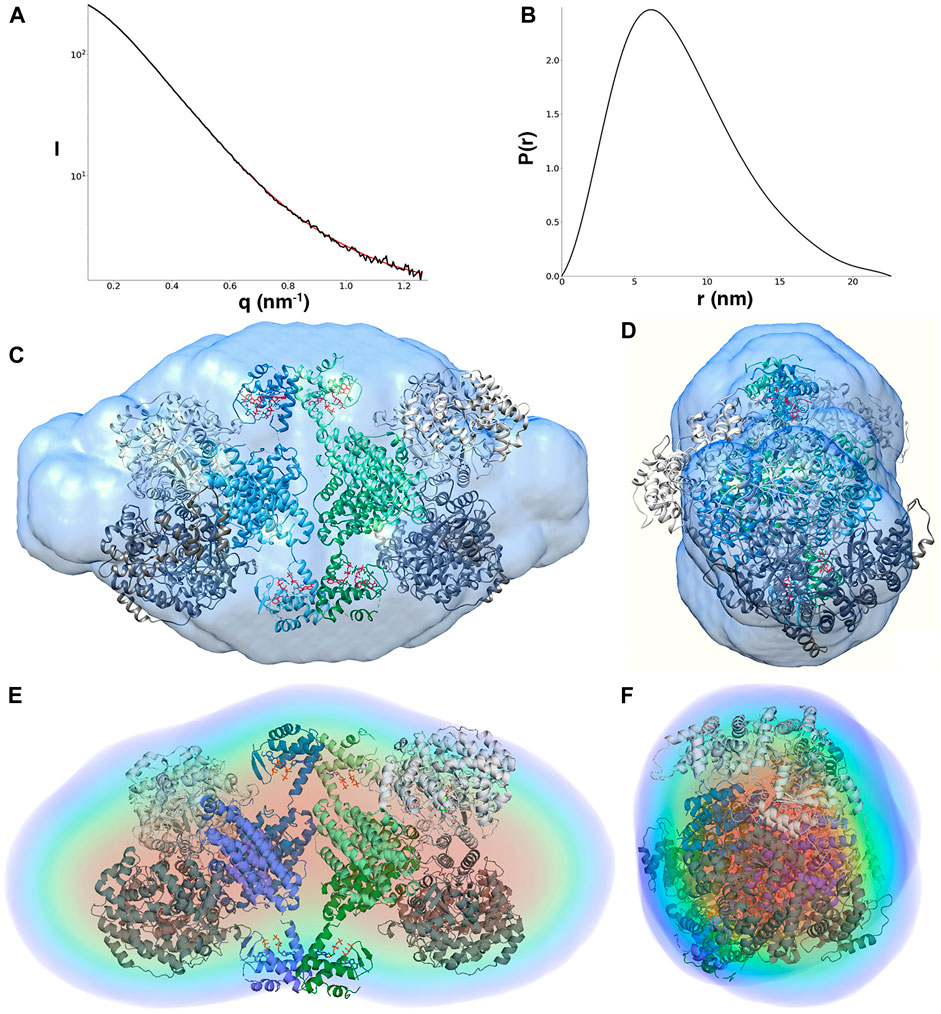
FIGURE 5. (A) Scattering curve of the LbNrdA/NrdB complex in the presence of dATP. (B)p(r) function. The angular range used to generate p(r) is from 0.11 to 1.26 nm. (C,D) Approximately orthogonal views of a fit of the docking model of the complex to the envelope from GASBOR. (E,F) Approximately orthogonal views of the fit of the same model to the DENNS reconstruction.
To test the compatibility of a symmetrical model for the inhibited α4β4 complex with the SAXS data, one copy of the homology model for the dimeric LbNrdA was placed on each side of the LbNrdB tetramer from the crystal structure in a way compatible with the docking model of the active complex of E. coli (Figure 5). This model fits reasonably well with the ab initio models, both from GASBOR and DENSS. The resolution of the DENSS model was estimated to be 50 Å (Supplementary Figure 4). The correlation score of a 15 Å map of the docking model with the DENSS volume was 0.768 as calculated by DENSS. Once again, the extra density present at both ends is most likely explained by the flexible N-terminus.
CryoEM of the dATP-Inactivated LbNrdAB Complex
Analytical SEC had previously demonstrated the existence of species with molecular weights consistent with both α4β4 and α2β4 stoichiometries, with the stoichiometry being concentration-dependent (Rozman Grinberg et al., 2018a). Initial attempts were made to prepare cryoEM grids of the LbNrdAB complex by isolating fractions from a SEC run where LbNrdA had been mixed with excess LbNrdB. However, despite taking fractions from the centre of the peak in a well-defined elution profile that should correspond to the α4β4 complex, 2D classes generated from micrographs taken from these grids showed near-complete dissociation of the complex. We thus applied gentle glutaraldehyde cross-linking to the complex during the SEC run according to a published protocol previously applied to a GPCR/arrestin complex (Shukla et al., 2014). Here, the protein travels through a front of glutaraldehyde as it traverses the SEC column. Furthermore, the sample was applied to carbon-coated grids to maximize the amount of intact complex.
Two-dimensional class averages from a small set of 29,902 particles (Figure 6A) suggest an organization similar to that revealed by SAXS, with one α2 dimer symmetrically bound to each side of a β4 tetramer. However, the sample is most likely a mixture of α4β4 and α2β4 species, as one of the α2 dimers is less well-ordered than the other, as already evident from the 2D classes (Figure 6A). The grids also contained a population of non-complexed β4 tetramers (not shown). A 3D reconstruction was made from these particles with a resolution of 8.2 Å according to a gold standard Fourier shell correlation value of 0.143 (Figure 6B and Supplementary Figure 5). No symmetry was imposed. The 3D volume confirms that one of the α2 dimers is significantly less well ordered or less occupied than the other, as the density is discontinuous and appears at a lower contour level than for the more ordered dimer (right hand side of Figure 6B). The dimeric homology model from the present work was fitted as a rigid body into the volume for α2 using Chimera and the crystal structure of the β4 tetramer was fitted into the corresponding volume as a single rigid body. Considering the more well-ordered of the two α2 dimers, it is arranged on top of one of the dimers of the β4 tetramer such that the active sites of both α2 monomers face the β subunits. However, the 2-fold axes of α2 and β2 are not exactly superimposed and the α2 dimer is tilted such that one of the monomers approaches one of the ATP-cones more closely. The shortest distance between two Cα atoms on one side is ∼10 Å, between Lys560 on α and Thr16 in β, while in the other pair of monomers the same atoms are ∼20 Å apart. Interestingly, neither of the ATP-cone domains apparently makes direct contact with the α2 dimers through hydrogen bonds or hydrophobic interactions. The molecular model fitted to the cryoEM map also fits at least as well as the docking model to the SAXS reconstruction from DENSS (Figures 6D,E).
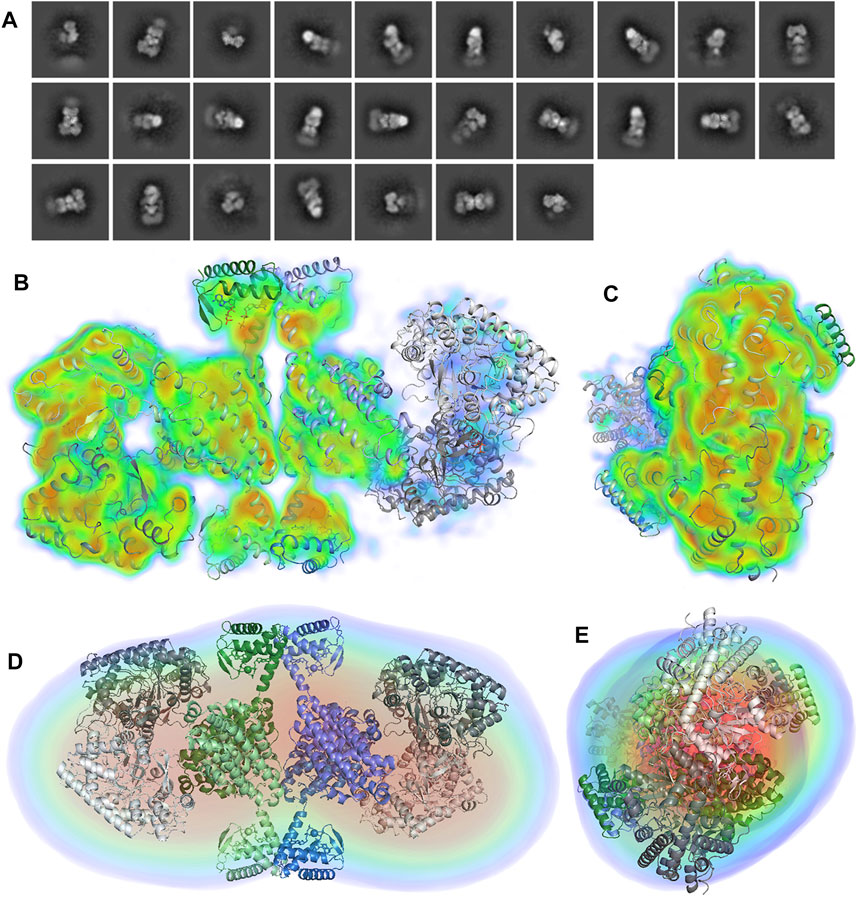
FIGURE 6. (A) Initial 2D classification of 29,902 particles of the cross-linked LbNrdAB complex in cryoSPARC. The first 27 classes are shown. (B,C) Approximately orthogonal views of the cryoEM volume of the α4β4 complex at 8.2 Å α2 dimers are shown in two shades of grey while the two dimers of the β4 tetramer are shown in in shades of green and blue respectively. The volume is coloured according to electrostatic potential level, from transparent blue at 2 σ through cyan at 6 σ, green at 8 σ, yellow at 10 σ to red at 15 σ, where σ is the number of standard deviations of the potential above the mean for the reconstruction box. Note the strong density for several helices. The lack of potential at higher levels for the right-hand dimer indicates lower occupancy and/or higher disorder. (D,E) Approximately orthogonal views of the fit of the cryoEM model to the SAXS envelope from DENSS (the GASBOR envelope is not shown for brevity).
The distance between the last ordered amino acid in the PCET chain in α (the hydroxyl group of Tyr579; Figure 7) and the last ordered residue of the chain in β (the Cβ atom of Trp133) varies between 26 and 29 Å in the four pairs of interacting chains and there is no density in the 8.2 Å map that clearly suggests ordered structure at the interface between any α/β pair. In contrast, in the active complex from E. coli, the corresponding distance between the hydroxyl group of Tyr731 and the CB atom of Trp48 is only 19 Å and the intervening space is filled by the ordered C-terminus of the β subunit, including Tyr356, which is critical for the PCET pathway (Kang et al., 2020).
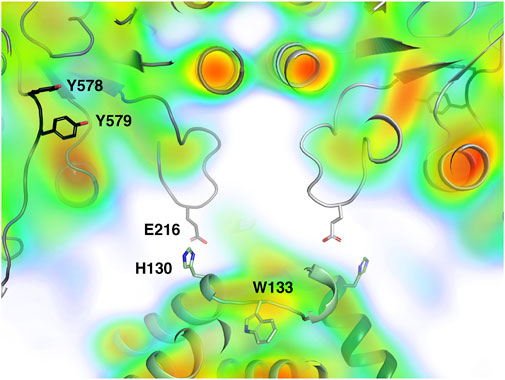
FIGURE 7. Closeup view of the central part of the interaction area between LbNrdA and LbNrdB in the dATP-inhibited complex, closest to the misaligned 2-fold symmetry axes. The homology model of LbNrdA and the crystal structure of LbNrdB are superposed on the 8.2 Å cryoEM reconstruction coloured as in Figure 6. Note the lack of electrostatic potential in the interface between the two proteins. The potential area of closest contact involves the loop containing Glu216 in the α subunit.
The closest approach between the α and β subunits occurs between the tip of the loop 213–222 between β-strands C and D of the α subunit and helix 118–131 on the β subunit (Figure 7). The distance between the Cα atom of Asp α216 and His β130 is around 10 Å. This interaction area is close to Trp133, one of the last residues in the PCET pathway on β. The side chains of α216 and β130 may be projected towards each other, but this interaction would be insufficient to stabilize the formation of a complex. It should be borne in mind that the α subunit in the current analysis is a homology model and that the interaction between subunits may involve structural elements not resolved at the current resolution. There is no well-defined density for this loop in the cryoEM reconstruction. Work is ongoing to deconvolute possible heterogeneity in the particle population and to obtain a larger dataset for higher resolution.
Discussion
Class I ribonucleotide reductases are dependent for their activity on the formation of a productive complex between the α and β subunits to enable long-range proton-coupled electron transfer from β to α and back again on each catalytic cycle. The RNRs from L. blandensis and F. ignava are both inactivated by the formation of a tetramer of β subunits in the presence of dATP that is catalytically incompetent, most likely by holding the subunits in a relative orientation that breaks the PCET chain, as has been observed previously in various forms for other class I RNRs (Ando et al., 2011; Fairman et al., 2011). Here we have studied the oligomeric organization of the individual α and β components from L. blandensis, as well as the inactive complex, all in the presence of the inhibitory nucleotide dATP. The SAXS-derived structures agree well with a homology model for the α2 dimer and the previously published crystal structure of the β4 tetramer. Furthermore, the SAXS studies suggest that the ATP-cones of the β4 tetramer are not highly flexible in solution with respect to the core domains, and good agreement to the experimental scattering profile can be obtained simply by modelling the additional disordered residues at the N- and C-termini. Thus, the crystal structure is a faithful representation of the conformation in solution.
Biochemical and biophysical studies of the dATP-inactivated complex indicated the concentration-dependent formation of oligomers with stoichiometry α2β4 and α4β4 when α2 dimers were added to β4 tetramers (Rozman Grinberg et al., 2018a). However, the structures of these complexes, and thus the molecular mechanisms of inactivation of class II RNRs, in which the ATP-cone is found in the β subunit, have not been elucidated previously. Here we studied these complexes with both SEC-SAXS and cryoEM. The SAXS studies are consistent with a predominantly α4β4 stoichiometry and a hypothetical structure in which two α2 dimers attach symmetrically or pseudo-symmetrically to each side of a β4 tetramer. Such an arrangement would be inconsistent with an intact PCET pathway, as cryoEM studies on the class I RNR from E. coli have indicated that such a complex is in fact highly asymmetrical (Figure 1). However, the SAXS envelopes are of too low resolution to dissect the structure of the complex in detail.
To confirm and extend the solution results, we also studied the inactivated complexes by single particle cryoEM. The complexes were isolated using size exclusion chromatography in the presence of dATP using the same protocol as for SEC-SAXS. In addition, gentle on-column crosslinking using glutaraldehyde was applied. The latter was necessary because the forces exerted at the air-water interface in the cryoEM freezing entirely disrupted the complexes without crosslinking, though the SEC-SAXS results show that the same complexes are stable in solution. A reconstruction was obtained at 8.2 Å resolution that also reveals binding of two α2 dimers, one on either side of the β4 tetramer, with both active sites in the α2 dimer facing towards the active site of one β2 dimer, in a near-productive conformation. Despite cross-linking to prevent disruption of the complex during grid preparation, one of the dimers is less well-occupied than the other, but the SAXS results are most consistent with an α4β4, stoichiometry in solution. Most surprisingly, the cryoEM volume shows the subunits at some distance from each other and in a slightly asymmetrical orientation such that one ATP-cone on β4 approaches an α subunit more closely than does the other from the same dimer of the tetramer.
Tetramerization May Inhibit L. blandensis RNR by Blocking Formation of an Asymmetric Active Complex
The observation that the dATP-inhibited α4β4 LbNrdAB complex is quasi-symmetrical in combination with the recent discovery that the active complex of the E. coli enzyme is highly asymmetrical led us to hypothesize about how the inhibition might occur for L. blandensis, F. ignava and similar organisms. We used the homology model of LbNrdA in combination with the crystal structure of the dATP-inhibited LbNrdB tetramer and superimposed these on the cryoEM structure of the active E. coli α2β2 complex (EcNrdAB, PDB ID 6W4X (Kang et al., 2020)) to check for possible steric hindrances to the formation of an active complex (Figure 8). More specifically, we superposed the “active” monomer of the NrdB component of EcNrdAB (light green) on one chain of the LbNrdB tetramer. Then we superposed the homology model of LbNrdA (light grey) on the “active” monomer of EcNrdA from EcNrdAB. This provides a current best model of the interactions between LbNrdA and LbNrdB in the putative active complex. It can clearly be seen that in this conformation the ATP-cone of the “inactive” LbNrdB monomer (dark green in Figure 8) would clash with the “inactive” LbNrdA monomer (dark grey). Since the LbNrdA dimer is relatively rigid, this clash would also prevent the interaction of the two “active” chains.
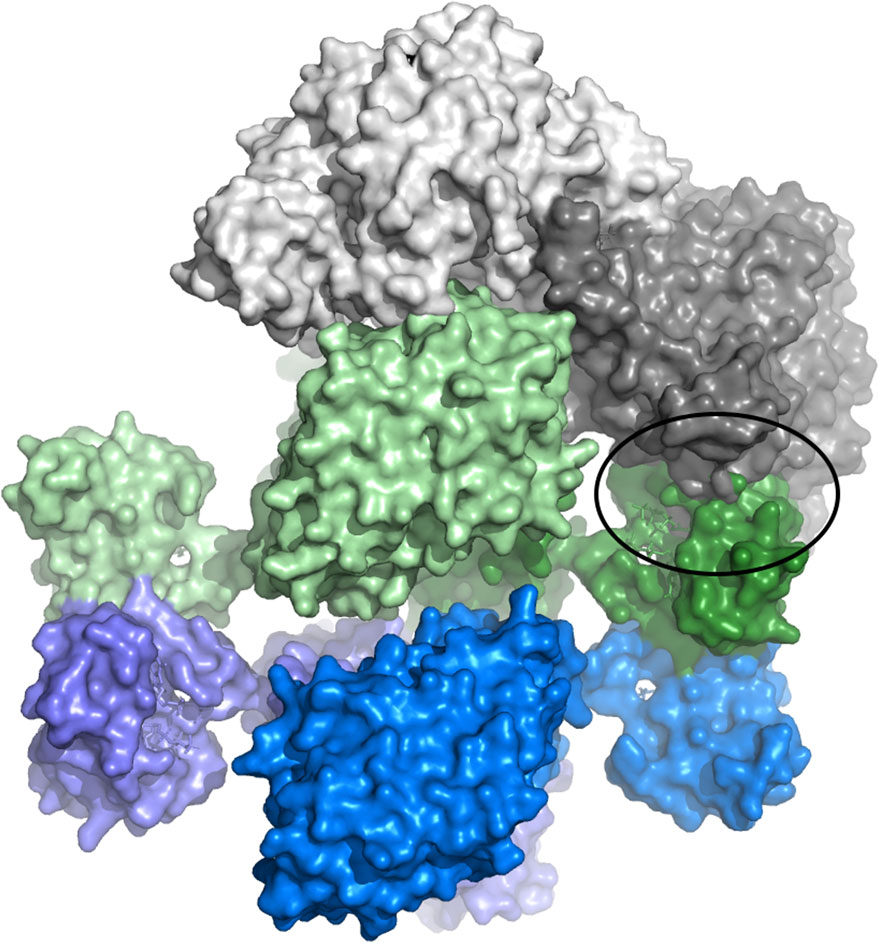
FIGURE 8. Model of the hypothetical steric clash that may prevent formation of an active complex of L. blandensis RNR when LbNrdB has tetramerized in the presence of dATP. Dimeric LbNrdA is shown in two shades of grey. The two dimers of LbNrdB that form the tetramer are shown in light and dark shades of green and blue respectively. The model has been generated to recreate the interactions that would occur in the active complex, based on the structure of E. coli NrdAB, between the light grey LbNrdA and pale green LbNrdB subunits. This orientation is prevented by a severe steric clash of the ATP-cone of the dark green LbNrdB subunit with the light grey LbNrdA subunit (circled).
Thus, despite subtle differences, our SAXS and cryoEM results are both compatible with the hypothesis that the ATP-cone functions as a steric block to formation of the highly asymmetrical complex that would be necessary for an intact PCET chain. These results begin to illuminate how the ATP-cone functions as a transposable functional module that, through oligomerization and steric hindrance, can inhibit RNR activity even when transposed from its usual place in the catalytic subunit to a new one in the radical generating subunit.
Data Availability Statement
The cryoEM data presented in this study can be found in the Electron Microscopy Database (EMDB) https://www.ebi.ac.uk/pdbe/emdb/, with accession number EMD-12985.
Author Contributions
MH collected and analysed SAXS data. IB analysed SAXS data, prepared cryoEM samples, collected and analysed the cryoEM data. IRG prepared the LbNrdA and LbNrdB proteins used in this study, prepared samples for SAXS and collected the SAXS data. B-MS contributed to the discussion. DL conceived and supervised the experiments and analysis. All authors contributed to the manuscript.
Funding
This work was funded by grant 2016-04855 from the Swedish Research Council to DTL and 2019-01400 to BMS, as well as grant 2018/820 from the Swedish Cancer Foundation and a grant from the Wenner-Gren Foundations to BMS. IB is the recipient of a scholarship from the Lawski Foundation. Molecular graphics and analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Petra Pernot and other staff at beamline BM29 of the ESRF for help with SAXS data collection and Michael Hall for help with cryoEM grid preparation and data collection. The cryoEM data were collected at the Umeå Core Facility for Electron Microscopy, a node of the Cryo-EM Swedish National Facility, funded by the Knut and Alice Wallenberg, Family Erling Persson and Kempe Foundations, SciLifeLab, Stockholm University and Umeå University.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.713608/full#supplementary-material
References
Ahmad, M. F., and Dealwis, C. G. (2013). The Structural Basis for the Allosteric Regulation of Ribonucleotide Reductase. Prog. Mol. Biol. Transl Sci. 117, 389–410. doi:10.1016/B978-0-12-386931-9.00014-3
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic Local Alignment Search Tool. J. Mol. Biol. 215, 403–410. doi:10.1016/S0022-2836(05)80360-2S0022-2836(05)80360-2
Ando, N., Brignole, E. J., Zimanyi, C. M., Funk, M. A., Yokoyama, K., Asturias, F. J., et al. (2011). Structural Interconversions Modulate Activity of Escherichia coli Ribonucleotide Reductase. Proc. Natl. Acad. Sci. 108, 21046–21051. doi:10.1073/pnas.1112715108
Ando, N., Li, H., Brignole, E. J., Thompson, S., McLaughlin, M. I., Page, J. E., et al. (2016). Allosteric Inhibition of Human Ribonucleotide Reductase by dATP Entails the Stabilization of a Hexamer. Biochemistry 55, 373–381. doi:10.1021/acs.biochem.5b01207
Aravind, L., Wolf, Y. I., and Koonin, E. V. (2000). The ATP-Cone: an Evolutionarily mobile, ATP-Binding Regulatory Domain. J. Mol. Microbiol. Biotechnol. 2, 191–194. Available at: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10939243.
Brignole, E. J., Tsai, K.-L., Chittuluru, J., Li, H., Aye, Y., Penczek, P. A., et al. (2018). 3.3-Å Resolution Cryo-EM Structure of Human Ribonucleotide Reductase with Substrate and Allosteric Regulators Bound. Elife 7, e31502. doi:10.7554/elife.31502.023Available at: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=29460780&retmode=ref&cmd=prlinks
Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., et al. (2010). MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. D Biol. Cryst. 66, 12–21. doi:10.1107/S0907444909042073
Fairman, J. W., Wijerathna, S. R., Ahmad, M. F., Xu, H., Nakano, R., Jha, S., et al. (2011). Structural Basis for Allosteric Regulation of Human Ribonucleotide Reductase by Nucleotide-Induced Oligomerization. Nat. Struct. Mol. Biol. 18, 316–322. doi:10.1038/nsmb.2007
Franke, D., and Svergun, D. I. (2009). DAMMIF, a Program for Rapidab-Initioshape Determination in Small-Angle Scattering. J. Appl. Cryst. 42, 342–346. doi:10.1107/s0021889809000338Available at: http://scripts.iucr.org/cgi-bin/paper?S0021889809000338
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S. e., Wilkins, M. R., Appel, R. D., et al. (2005). “Protein Identification and Analysis Tools on the ExPASy Server,” in The Proteomics Protocols Handbook, Totowa, NJ: Humana Press. 571–607. doi:10.1385/1-59259-890-0:571
Grant, T. D. (2018). Ab Initio electron Density Determination Directly from Solution Scattering Data. Nat. Methods 15, 191–193. doi:10.1038/nmeth.4581
Greene, B. L., Kang, G., Cui, C., Bennati, M., Nocera, D. G., Drennan, C. L., et al. (2020). Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu. Rev. Biochem. 89, 45–75. doi:10.1146/annurev-biochem-013118-111843
Guinier, A. (1939). La diffraction des rayons X aux très petits angles : application à l'étude de phénomènes ultramicroscopiques. Ann. Phys. 11, 161–237. doi:10.1051/anphys/193911120161
Hofer, A., Crona, M., Logan, D. T., and Sjöberg, B.-M. (2012). DNA Building Blocks: Keeping Control of Manufacture. Crit. Rev. Biochem. Mol. Biol. 47, 50–63. doi:10.3109/10409238.2011.630372
Högbom, M., Sjöberg, B. M., and Berggren, G. (2020). Radical Enzymes. eLS 1, 375–393. doi:10.1002/9780470015902.a0029205
Johansson, R., Jonna, V. R., Kumar, R., Nayeri, N., Lundin, D., Sjöberg, B.-M., et al. (2016). Structural Mechanism of Allosteric Activity Regulation in a Ribonucleotide Reductase with Double ATP Cones. Structure 24, 906–917. doi:10.1016/j.str.2016.03.025
Jonna, V. R., Crona, M., Rofougaran, R., Lundin, D., Johansson, S., Brännström, K., et al. (2015). Diversity in Overall Activity Regulation of Ribonucleotide Reductase. J. Biol. Chem. 290, 17339–17348. doi:10.1074/jbc.M115.649624
Kang, G., Taguchi, A. T., Stubbe, J., and Drennan, C. L. (2020). Structure of a Trapped Radical Transfer Pathway within a Ribonucleotide Reductase Holocomplex. Science 368, 424–427. doi:10.1126/science.aba6794
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 Web portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 10, 845–858. doi:10.1038/nprot.2015.053
Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., and Svergun, D. I. (2003). PRIMUS: a Windows PC-Based System for Small-Angle Scattering Data Analysis. J. Appl. Cryst. 36, 1277–1282. doi:10.1107/S0021889803012779
Lundin, D., Berggren, G., Logan, D., and Sjöberg, B.-M. (2015). The Origin and Evolution of Ribonucleotide Reduction. Life 5, 604–636. doi:10.3390/life5010604
Mathews, C. K. (2018). Still the Most Interesting Enzyme in the World. FASEB j. 32, 4067–4069. doi:10.1096/fj.201800790rAvailable at: https://www.fasebj.org/doi/10.1096/fj.201800790R
Panjkovich, A., and Svergun, D. I. (2018). CHROMIXS: Automatic and Interactive Analysis of Chromatography-Coupled Small-Angle X-ray Scattering Data. Bioinformatics 34, 1944–1946. doi:10.1093/bioinformatics/btx846
Parker, M. J., Maggiolo, A. O., Thomas, W. C., Kim, A., Meisburger, S. P., Ando, N., et al. (2018). An Endogenous dAMP Ligand in Bacillus Subtilis Class Ib RNR Promotes Assembly of a Noncanonical Dimer for Regulation by dATP. Proc. Natl. Acad. Sci. USA 115, E4594–E4603. doi:10.1073/pnas.1800356115Available at: https://www.pnas.org/content/115/20/E4594.abstract
Pernot, P., Round, A., Barrett, R., De Maria Antolinos, A., Gobbo, A., Gordon, E., et al. (2013). Upgraded ESRF BM29 Beamline for SAXS on Macromolecules in Solution. J. Synchrotron Radiat. 20, 660–664. doi:10.1107/S0909049513010431
Petoukhov, M. V., Franke, D., Shkumatov, A. V., Tria, G., Kikhney, A. G., Gajda, M., et al. (2012). New Developments in theATSASprogram Package for Small-Angle Scattering Data Analysis. J. Appl. Cryst. 45, 342–350. doi:10.1107/S0021889812007662
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera?A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 25, 1605–1612. doi:10.1002/jcc.20084
Punjani, A. (2020). Algorithmic Advances in Single Particle Cryo-EM Data Processing Using CryoSPARC. Microsc. Microanal. 26, 2322–2323. doi:10.1017/s1431927620021194
Punjani, A., Rubinstein, J. L., Fleet, D. J., and Brubaker, M. A. (2017). cryoSPARC: Algorithms for Rapid Unsupervised Cryo-EM Structure Determination. Nat. Methods 14, 290–296. doi:10.1038/nmeth.4169Available at: http://www.nature.com/articles/nmeth.4169
Rofougaran, R., Vodnala, M., and Hofer, A. (2006). Enzymatically Active Mammalian Ribonucleotide Reductase Exists Primarily as an α6β2 Octamer. J. Biol. Chem. 281, 27705–27711. doi:10.1074/jbc.m605573200Available at: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16861739
Rose, H. R., Maggiolo, A. O., McBride, M. J., Palowitch, G. M., Pandelia, M.-E., Davis, K. M., et al. (2019). Structures of Class Id Ribonucleotide Reductase Catalytic Subunits Reveal a Minimal Architecture for Deoxynucleotide Biosynthesis. Biochemistry 58, 1845–1860. doi:10.1021/acs.biochem.8b01252
Rozman Grinberg, I., Berglund, S., Hasan, M., Lundin, D., Ho, F. M., Magnuson, A., et al. (2019). Class Id Ribonucleotide Reductase Utilizes a Mn2(IV,III) Cofactor and Undergoes Large Conformational Changes on Metal Loading. J. Biol. Inorg. Chem. 24, 863–877. doi:10.1007/s00775-019-01697-8
Rozman Grinberg, I., Lundin, D., Hasan, M., Crona, M., Jonna, V. R., Loderer, C., et al. (2018a). Novel ATP-Cone-Driven Allosteric Regulation of Ribonucleotide Reductase via the Radical-Generating Subunit. Elife 7, 1–26. doi:10.7554/eLife.31529
Rozman Grinberg, I., Lundin, D., Sahlin, M., Crona, M., Berggren, G., Hofer, A., et al. (2018b). A Glutaredoxin Domain Fused to the Radical-Generating Subunit of Ribonucleotide Reductase (RNR) Functions as an Efficient RNR Reductant. J. Biol. Chem. 293, 15889–15900. doi:10.1074/jbc.ra118.004991Available at: http://www.jbc.org/lookup/doi/10.1074/jbc.RA118.004991
Shukla, A. K., Westfield, G. H., Xiao, K., Reis, R. I., Huang, L.-Y., Tripathi-Shukla, P., et al. (2014). Visualization of Arrestin Recruitment by a G-Protein-Coupled Receptor. Nature 512, 218–222. doi:10.1038/nature13430
Svergun, D. I., Petoukhov, M. V., and Koch, M. H. J. (2001). Determination of Domain Structure of Proteins from X-ray Solution Scattering. Biophysical J. 80, 2946–2953. doi:10.1016/s0006-3495(01)76260-1Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1301478/pdf/11371467.pdf
Thomas, W. C., Brooks, F. P., Burnim, A. A., Bacik, J.-P., Stubbe, J., Kaelber, J. T., et al. (2019). Convergent Allostery in Ribonucleotide Reductase. Nat. Commun. 10, 1–13. doi:10.1038/s41467-019-10568-4
Torrents, E. (2014). Ribonucleotide Reductases: Essential Enzymes for Bacterial Life. Front. Cel. Infect. Microbiol. 4, 52. doi:10.3389/fcimb.2014.00052
Uhlin, U., and Eklund, H. (1994). Structure of Ribonucleotide Reductase Protein R1. Nature 370, 533–539. doi:10.1038/370533a0
Volkov, V. V., and Svergun, D. I. (2003). Uniqueness Ofab Initioshape Determination in Small-Angle Scattering. J. Appl. Cryst. 36, 860–864. doi:10.1107/S0021889803000268
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 46, W296–W303. doi:10.1093/nar/gky427
Wriggers, W. (2012). Conventions and Workflows for Using Situs. Acta Crystallogr. D Biol. Cryst. 68, 344–351. doi:10.1107/S0907444911049791
Keywords: ribonucleotide reductase, allosteric regulation, oligomerization, nucleotide binding, small-angle X-ray scattering, single particle cryo-EM
Citation: Hasan M, Banerjee I, Rozman Grinberg I, Sjöberg B-M and Logan DT (2021) Solution Structure of the dATP-Inactivated Class I Ribonucleotide Reductase From Leeuwenhoekiella blandensis by SAXS and Cryo-Electron Microscopy. Front. Mol. Biosci. 8:713608. doi: 10.3389/fmolb.2021.713608
Received: 23 May 2021; Accepted: 21 June 2021;
Published: 26 July 2021.
Edited by:
Giovanni Gotte, University of Verona, ItalyReviewed by:
Chris Dealwis, Case Western Reserve University, United StatesGiovanna Di Nardo, University of Turin, Italy
Copyright © 2021 Hasan, Banerjee, Rozman Grinberg, Sjöberg and Logan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Derek T. Logan, ZGVyZWsubG9nYW5AYmlvY2hlbWlzdHJ5Lmx1LnNl
†These authors have contributed equally to this work and share first authorship