 Louis Bleuzé
Louis Bleuzé- 1University de Rennes 1, Rennes, France
- 2Humanitas Clinical and Research Center-IRCCS, Rozzano (Mi), Italy
- 3Institute of Biochemistry and Cell Biology, National Research Council (CNR-IBBC), International Campus A. Buzzati Traverso, Monterotondo, Italy
- 4Institute of Neuroscience-National Research Council (CNR-IN), Milan, Italy
Fragile X mental retardation protein (FMRP) is an RNA binding protein (RBP) whose absence is essentially associated to Fragile X Syndrome (FXS). As an RNA Binding Protein (RBP), FMRP is able to bind and recognize different RNA structures and the control of specific mRNAs is important for neuronal synaptic plasticity. Perturbations of this pathway have been associated with the autistic spectrum. One of the FMRP partners is the APP mRNA, the main protagonist of Alzheimer’s disease (AD), thereby regulating its protein level and metabolism. Therefore FMRP is associated to two neurodevelopmental and age-related degenerative conditions, respectively FXS and AD. Although these pathologies are characterized by different features, they have been reported to share a number of common molecular and cellular players. The aim of this review is to describe the double-edged sword of FMRP in autism and AD, possibly allowing the elucidation of key shared underlying mechanisms and neuronal circuits. As an RBP, FMRP is able to regulate APP expression promoting the production of amyloid β fragments. Indeed, FXS patients show an increase of amyloid β load, typical of other neurological disorders, such as AD, Down syndrome, Parkinson’s Disease, etc. Beyond APP dysmetabolism, the two neurodegenerative conditions share molecular targets, brain circuits and related cognitive deficits. In this review, we will point out the potential common neuropathological pattern which needs to be addressed and we will hopefully contribute to clarifying the complex phenotype of these two neurorological disorders, in order to pave the way for a novel, common disease-modifying therapy.
FMRP as RNA Binding Protein: The Double Role in FXS and AD
Fragile X mental retardation protein (FMRP) is an RNA binding protein (RBP) encoded by the fmr1 gene located on the long arm of the X chromosome. The mutation in the 5′UTR and consequent elongation of the triplet cause hypermethylation, FMRP silencing, and strong elongation of the long chromosome arm, which becomes thinner and fragile. The absence of FMRP is the main cause of a common intellectual disability, known as Fragile X Syndrome (FXS). In more detail, the main symptoms of FXS patients include cognitive disability, hyperactivity, and autism-related disorders (Santoro et al., 2012; Sethna et al., 2014). Furthermore, morphological analysis of the brain of FXS individuals reveals an increase in spine density, which is also present in the fmr1KO mouse model (Grossman et al., 2010). This abnormal phenotype reflects what happens locally at synapses in the absence of FMRP. Notably, FMRP has different mRNA partners relevant for synaptic plasticity. The functional role of FMRP at synapses as a regulator of the translation of these specific mRNAs is actively controlled by its phosphorylation status (Bartley et al., 2014; Bartley et al., 2016). Independently, by the increased triplet repetition in the 5′UTR of the gene, other mutations in the coding region of the fmr1 gene have been identified. These mutations are mainly localized in the functional FMRP domain required for the RNA binding (Okray et al., 2015; Maddirevula et al., 2020) or affect its subcellular distribution (Fu et al., 2020).
Based on this primary role, most of the studies focused their attention on the local function of FMRP at synapses. However, the first report of the missense mutation c.148G>A mutation in the fmr1 gene suggests that a subset of patients with FXS preserved the post-synaptic functions of FMRP at the KH helix alpha A domain.
The data herein demonstrate the prominent role of FMRP in RNA metabolism. Of note, several mutations have been found in the KH domain of FMRP that affect its RNA-binding site (Di Marino et al., 2014; Golden et al., 2019). Most fmr1 mutations are associated with the overproduction of triplets in the 5′UTR of the gene. Few missense mutations in the fmr1 gene have been well characterized and identified as responsible for the FXS phenotype (De Boulle et al., 1993, Prieto et al., 2021; Myrick et al., 2015; Sitzman et al., 2018).
For the entire list of mutations found in the fmr1 gene, see the Table 1.
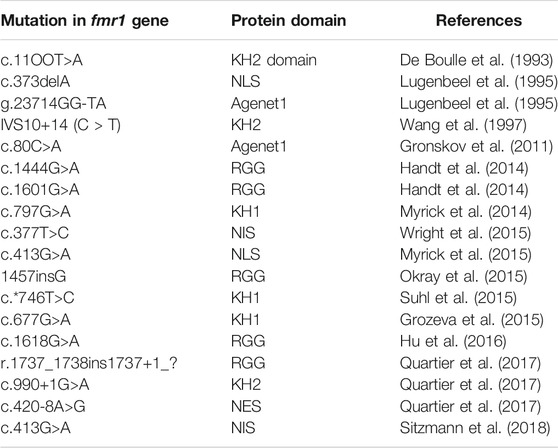
TABLE 1. List of missense mutation in fmr1 gene.
The main role of FMRP is related to mRNA metabolism: transport, stability and expression. In particular, FMRP is known as the RNA binding protein which is able to bind a plethora of mRNAs that have special roles in neuronal plasticity (Table 2, Figure 1). FMRP is able to bind to different mRNAs, microRNAs and long non-coding RNA (lncRNA) thanks to its peculiar structure (D’Annessa et al., 2019), including the APP mRNA (Westmark and Malter, 2007).
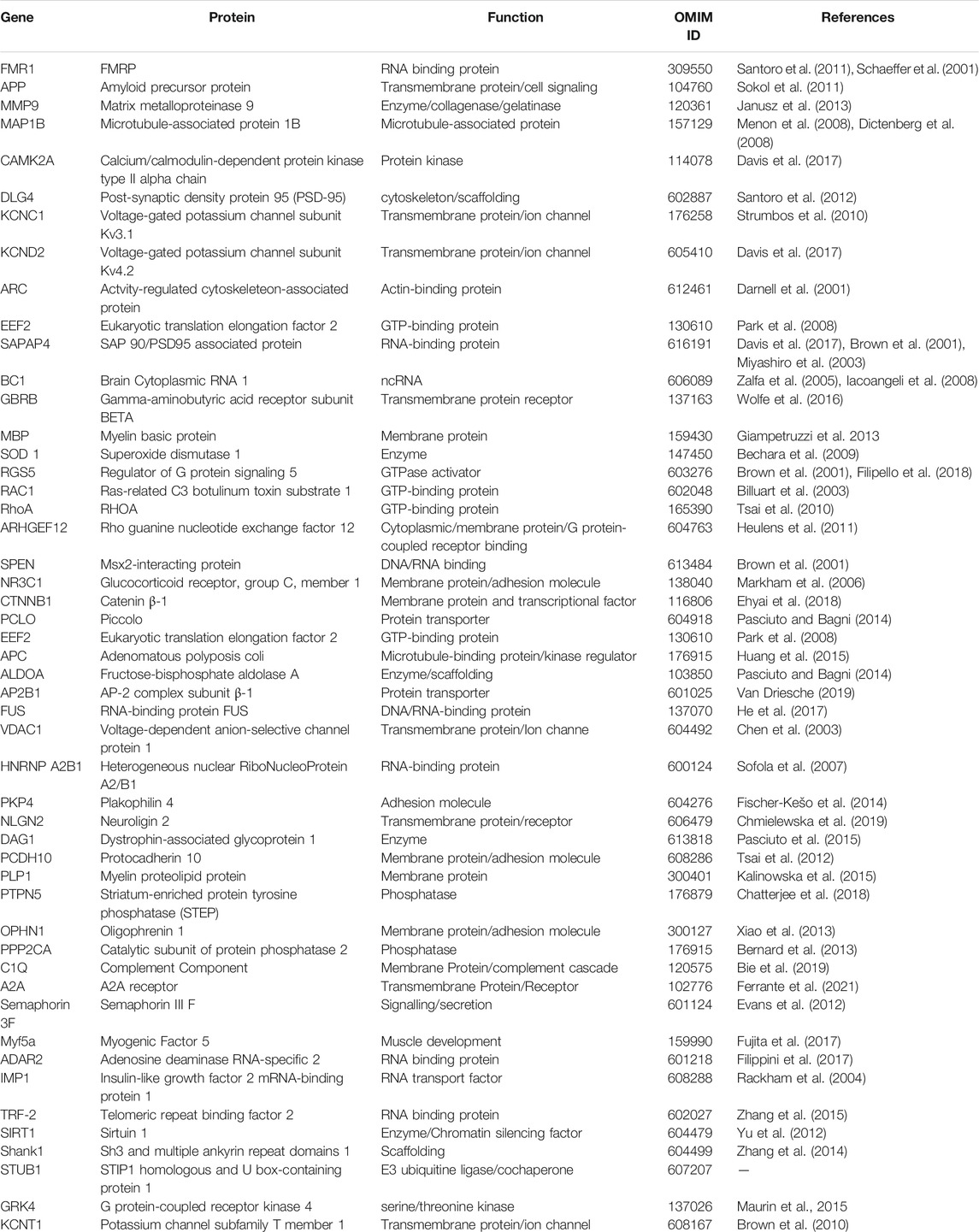
TABLE 2. FMRP mRNAs interactors.
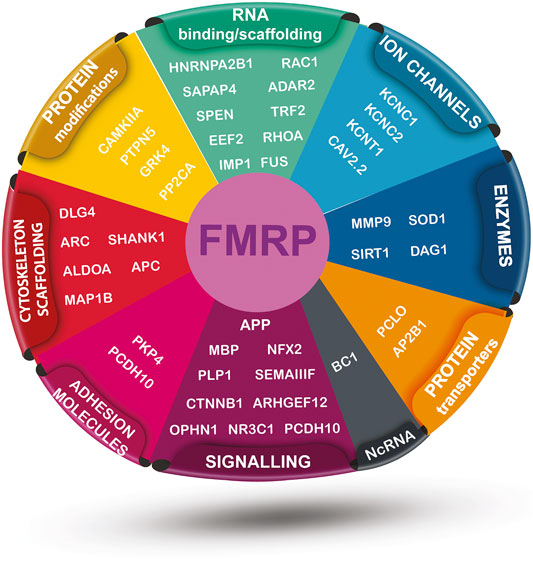
FIGURE 1. FMRP-interacting targets mRNAs sorted by their general function. The illustration includes vectors modified from “Vecteezy” and it is distributed under the Creative Commons Attribution 4.0 license (CC BY 4.0).
Theoretically, FMRP is structured in N-terminal, C-terminal and central domains. Apart its RNA binding domain, the structure of FMRP presents a nuclear export signal and a nuclear localization signal, able to transport ribonuclear complexes outside the nucleus toward synapses (Eberhart et al., 1996).
The nuclear localization signal suggests the presence of the protein in the nucleus and a related, still unknown nuclear function.
FMRP is characterized by different RNA binding motifs: namely, the KH and RGG box domains. Only a few mutations have been identified in the coding region of the fmr1 gene (De Boulle et al.1993; Myrick et al., 2014) in FXS patients and all of them are localized in these two motifs, pinpointing their key role in protein functions (Di Marino et al., 2014). A newly reported R138Q mutation results in impaired hippocampal long-term potentiation and socio-cognitive deficits in mice (Prieto et al., 2021).
The RNA binding takes place directly or via non-coding RNAs, such as BC1 (Lacoux et al., 2012) or miRNAs (Ifrim et al., 2015). Interestingly, the BC1 non coding RNA is able to regulate the expression of the APP mRNA upon FMRP binding. Inhibition of BC1 or of BC1-FMRP interaction represses APP translation and hence rescues the amyloid beta level, as well as spatial learning, and memory impairments in AD mice with dysregulated RNA production (Zhang et al., 2018).
As an RBP, FMRP regulates the expression of specific mRNA locally at synapses under specific conditions and in response to external stimuli (Dolen et al., 2007). The absence of the protein causes an abnormal translational expression of its RNA partners, causing an alteration of normal synaptic plasticity. In more detail, Bear and collaborators speculated that mGluR5 serves as a sensor of ongoing synaptic excitation in the brain and, among other actions, stimulates local mRNA translation, so that the supply of rapidly turned-over proteins keeps up with demand. As with all biochemical pathways, this process is balanced by negative regulators. In the case of mGluR5-dependent protein synthesis, FMRP might be one of these negative regulators. In the absence of FMRP, one might expect that the protein synthesis-dependent effect of mGluR5 activation would be exaggerated, and indeed this is the case (Huber et al., 2002; Chuang et al., 2005).
Taken together, the findings that 1) mGluR5 stimulates protein synthesis and that 2) FMRP negatively regulates protein synthesis led to the “mGluR theory of fragile X,” which posits that the neurological and psychiatric symptoms of FXS are generated as a consequence of abnormal responses to mGluR activation (Bear et al., 2004). If this theory is valid, then it might be possible to correct some aspects of fragile X by specifically reducing mGluR5 signaling.
Under mGluR stimulation FMRP is phosphorylated and releases all partner mRNAs, thus favoring their translation. APP expression is downstream from the FMRP cascade, and patients with FXS are characterized by increase in APP level and concomitant accumulation of amyloid plaques (Westmark and Malter, 2007). In line with this, drugs aimed at inhibiting mGluR5 activation are also able to reduce Aβ accumulation in FXS patients (Westmark et al., 2009; Thomas et al., 2012; Gandhi et al., 2014). Different antagonists of mGluR5 have been shown to downregulate APP protein expression and interfere with the accumulation of amyloid-β fragments (Kumar et al., 2016).
FMRP binds the APP mRNA through the RGG box domain which recognizes a G quartet region that is extremely conserved in all species and is also present in coding region and 3′UTR of APP mRNA. The G-quartet region is also present in PSD95 mRNA, another well-known FMRP partner (Zalfa et al., 2007).
The control by FMRP of APP mRNA is confirmed by another RNA binding protein: hnRNPC (Rajagopalan et al., 1998). While FMRP acts as a repressor of APP mRNA, hnRNPC is able to stabilize it, thus facilitating APP expression (Lee et al., 2010). Notably, an imbalance has been demonstrated in both FMRP (Renoux et al., 2014a; Borreca et al., 2016) and hnRNPC expression levels in AD mouse models (Borreca et al., 2016). This alteration has been reported in the early phase of pathology, highlighting the important role of APP mRNA level upstream of any APP cleavage dysregulation. This may explain the different FMRP outcomes reported in the two studies (Renoux et al., 2014b; Borreca et al., 2016), together with the use of two different AD mouse models.
These results support a possible double role of FMRP in FXS and in AD, although further studies are required. How FMRP mutations can affect two apparently different pathologies is still unknow and the role of neuroscientists is fundamental to disclose the shared underlying mechanism.
FMRP and Protein Synthesis in FXS and AD
The role of FMRP in protein synthesis has been clearly described. FMRP acts as translational repressor of specific target mRNAs and its absence or dysfunction affects the protein synthesis machinery.
In more detail, FMRP acts as negative regulator of local translation at synapses. By acting on RNA metabolism, FMRP is able to bind specific RNA, transfer them locally at synapses, and release them upon specific stimuli, allowing their translation when necessary.
In FXS patients, basal translation of FMRP mRNA partners is unstable and constantly active. Among them, ARC mRNA is overexpressed in FXS patients inducing AMPA internalization and unstable spine function (Park et al., 2008).
A local protein synthesis at synapses plays a relevant role in the synaptic formation and storage of the information necessary for performing specific memory tasks (Holt et al., 2019; Perrone-Capano et al., 2021).
Recent data demonstrate how protein synthesis levels are increased in individuals with FXS (Jacquemont et al., 2018; Utami et al., 2020). FXS individuals have an increase in the synthesis of de novo proteins such as ERK1/2 and Akt, involved in the mTOR pathway, which is known for regulating the expression of key proteins involved in neurodevelopmental and neurodegenerative diseases, such as FXS, ASD, T2D, and AD. This pinpoints its relevance for the identification of new common therapeutic strategies.
The activation of the metabotropic glutamate receptor 5 (mGluR5) in FXS triggers a cascade of reactions involving PI3K/Akt survival signal, the activation of mTOR, and its subsequent interaction with mTORC1 (Sharma et al., 2010; Casingal et al., 2020).
Following mTOR pathway activation, there is a regulation of eIF4E by 4E-BP such as CYFIP1, with increase of the protein synthesis in normal conditions (Napoli et al., 2008; De Rubeis et al., 2013). While in FXS the lack or absence of FMRP leads to an upstream dysregulation of the mTOR pathway that results in an increased protein synthesis.
Since microglia, the brain immune cells, are also involved in synaptic pruning and one of the main features of FXS individuals is excess of synapses, a general protein synthesis is fundamental for the functional role of synapses. Accordingly, alteration of protein synthesis machinery in microglia induces an autistic-like behavior (Xu et al., 2020).
Furthermore, it has been largely demonstrated the presence of FMRP in polysomes and mRNA granules in neuronal cells, suggesting its translational control in dendrites. In particular, granules are normally composed of a repertoire of mRNAs transported in a translationally silent state into dendrites for subsequent site-specific utilization at synapses undergoing protein synthesis-dependent changes (Krichevsky et al., 2001; Keene et al., 2002; Antic et al., 1998). FMRP is associated with translating polysomes and with stalled polysomes (Khandjian et al., 2004; Stefani et al., 2004; Ceman et al., 2003), confirming the idea that it gates translation after the initiation step. This means that specific mRNAs is stalled on ribosomes by FMRP, suggesting a prominent role in the first step of translational initiation.
On the other hand, AD is characterized by an accumulation of amyloid β plaques in synapses and the hyperphosphorylation of tau proteins. Several lines of evidence pinpoint that brain neurodegeneration in AD shares some common pathological patterns with FXS.
In line, FXS patients show an increase of Aβ production due to the FMRP negative control on APP mRNA. The absence of FMRP induces the release of APP mRNA which is locally translated at synapses, leading to an increase of amyloidogenic products. Amyloid β, a product of the amyloidogenic APP processing pathway, is able to bind mGluR5 and promote a positive feedback loop controlling amyloid β within normal levels in healthy conditions (Westmark, 2013). Given the key role of FMRP on controlling mGluR5 signalling and protein synthesis at synapses, an involvement of FMRP in AD typical events cannot be excluded.
Interestingly, protein synthesis alteration has been found also in AD brains (Hernandez-Ortega et al., 2016; Cefaliello et al., 2020; Ghosh et al., 2020). Polysomal profiles from wild-type (wt) and the Tg2576 AD mouse model (Borreca et al., 2020) reveals the shift of APP mRNA in the polysomal fraction, with APP more prone to be translated. Furthermore, a translational initiation factor, EIF2α is involved in AD cognitive deficit (Costa Mattioli et al., 2006). In particular, EIF2α is upregulated upon phosphorylation and the protein synthesis is blocked in late phase of AD pathology. Surprisingly, in the early phase of AD, the phosphorylation status of EIF2α is reduced, suggesting an increase of protein synthesis machinery and as a consequence a compensatory mechanism taking place in the late phase of the disease with high phosphorylation status of EIF2α and block of protein synthesis. Therefore, the block of protein synthesis will occur later in the disease, possibly as a feedback control (Borreca et al., 2020). Several protein synthesis molecules are involved in other neurodegenerative disorders such as EIF4G in Parkinson disease, thus suggesting a fundamental role of protein synthesis in the functional role of synapses and the cognitive deficit observed (Dhungel et al., 2015).
Overall, it can be hypothesized that FMRP control of mGluR5 signaling, APP expression and synaptic proteins represents a common dysregulated pathway contributing to the increase in protein expression apparently occurring in both FXS and prodromal AD.
THE miRNA-Based Pathway in FXS and AD
Non-coding RNA plays a pivotal role in different cellular processes and are implicated in epigenetic mechanisms, translational and transcriptional processes, of interest for understanding more complicated cellular pathways.
Noteworthy, FMRP is known to be implicated in the RNA interference (RNAi) mechanism and epigenetic silencing. The process of RNAi is a very intriguing mechanism that regulates post transcriptional gene expression within the cells. The silencing of the gene is regulated by the RISC complex. In particular, AGO2 protein combines with the RISC complex before binding to the functional strand RNA, inducing silencing of gene. In this scenario, the fmr1 gene containing full mutation CGG triplets in the 5′UTR is recognized by the RNAi complex and has a key role in the epigenetic silencing of the fmr1 gene.
Moreover, FMRP is able to bind specifically to some miRNAs, via its RNA binding domain, and in particular, to two miRNAs known to be also involved in AD pathologies: miR-132 and miR-125b (Edbauer et al., 2010). The miR132 has a protective effect on neuronal cells and its expression is reduced in AD disease progression (Zhu et al., 2016). In contrast, miR-125b is extremely deleterious for neuronal cells, with an increase in the hippocampus of AD mice (Lukiw, 2007; Ma et al., 2017). Furthermore, there is ongoing debate over whether FMRP is able to bind mRNA through long non-coding RNA (lncRNA) BC1 (Zhong et al., 2010). In AD mouse models, once linked to BC1 FMRP releases the APP mRNA with full protein overexpression (Zhang et al., 2018). This idea is still controversial and understanding the molecular mechanism of APP expression is still open. BC1 is found overexpressed in AD mice models and human AD samples (Mus et al., 2007). Furthermore, inhibition of BC1 blocks APP expression in AD mouse models and rescues cognitive defects in those mice (Zhang et al., 2017). APP expression is also regulated post-transcriptionally by different miRNAs. Most of them are in the regulatory region of APP mRNA (Vilardo et al., 2010; Long et al., 2019).
A growing body of evidence indicates that miRNAs regulate synaptic homeostasis and plasticity processes, suggesting that they may be involved in early synaptic dysfunction during AD. Based on this, it is thought that miRNA may be a potential biomarker in the prodromal phase of pathology (Siedlecki-Wullich et al., 2021).
Identifying the molecular mechanisms of miRNA-driven gene expression will be helpful in understanding the complex pathologies of AD and FXS.
Neuroinflammation Studies in FXS and AD
The brain is composed of neuronal cells, and also non-neuronal cells, such as microglia and astrocytes. The concomitant work of all brain cell is fundamental for synapses to function. The role of FMRP has been well described in neurons and synapses. The role of this protein in other brain cells has only recently been questioned. It has been demonstrated that astrocytic glutamate transport 1 (GLT1), a major glutamate transporter of glia, is reduced in fmr1KO astrocytes compared to wt (Higashimori et al., 2013; Higashimori et al., 2016). It has also been demonstrated that fmr1KO astrocytes are more active compared to wt (Cerdeno et al., 2018) and that there is a reduced number of microglia cells in fmr1KO mice compared to wt (Lee et al., 2019). This demonstrates the important role of different brain cell types and shows how they contribute to the physiological state of neuronal cells. In particular, astrocytes and microglia contribute to synapse refinement, and it is possible, alike that their alteration would contribute to the excessive and immature synapses observed in FXS mice models. This idea is also supported by the alteration of different cytokines in fmr1KO mice (Hodges et al., 2017). Furthermore, it has been demonstrated that FMRP is associated to C1q mRNA, which is fundamental for complement cascade and for the recognition of synapses to eliminate (Bie et al., 2019). In particular, artificial activation of mGluR1 signaling promotes dephosphorylation of FMRP and facilitates the local translation machinery of synaptic C1q mRNA, thus mimicking the C1q-mediated microglial phagocytosis of hippocampal glutamatergic synapses and cognitive deficiency in rodent models (Bie et al., 2019). C1q is part of the complement system and also contributes to synaptic pruning by tagging weak or damaged synapses, resulting in their subsequent phagocytosis by microglia (Schafer et al., 2012).
C1q immunoreactivity is not visualized in synapses, which indicates that C1q expression from other brain cells (microglia) might contribute to the pathological processes. In particular, this mechanism suggests a fundamental communication between neurons and microglia in physiological conditions. Alteration of C1q-mediated synaptic elimination is observed in the pathogenesis of several neurological disorders (Wang et al., 2020a; Wang et al., 2020b).
An alteration of the neuroinflammatory system is also a common feature of AD patients and it has been clearly demonstrated that the brain of AD samples are “inflamed.”
AD is a complex pathology and the involvement of non-neuronal cell types make this disease difficult to understand. The Amyloid β production and accumulation of neurofibrillary tangles extracellularly activate microglia and astrocytes (Fakhoury et al., 2018). Furthermore, it has been demonstrated how mutations in TREM2 microglia receptors are associated with Alzheimer’s disease (Ulland et al., 2018). This further suggests a prominent role of immune cells in the pathogenesis of AD. Trem2 alteration are also observed in human ASD patients (Filipello et al., 2018) suggesting a further common feature in AD and Autism.
Amelioration of FXS Cognition and Behavior Upon Rescue of Basal Forebrain Cholinergic Neurons Deficits by AD Drugs
Neurodegeneration of cholinergic neurons of the basal forebrain (BF) and of its cortical and hippocampal targets is one of the most prominent and prototypical sign of AD pathology (Fernandez-Cabello et al., 2020; Schmitz et al., 2016). Interestingly, increasing evidence on BF abnormalities were reported in several syndromes, including Down syndrome and Rett syndrome, as well as in developmental conditions like autism spectrum disorders (ASD), in particular FXS.
Functionally, the basal forebrain cholinergic system (BFCS) has been referred to as an “action system,” allowing correct focusing of the individual on the environment and providing a coherent behavioral response to external stimuli. The BFCS subserves sensory processing, working memory, attention, learning, and memory by activating ACh receptors in the hippocampus, entorhinal, and frontal cortices (Li et al., 2018; Ballinger et al., 2016). Thus, BFCS disruption may contribute to abnormal development of neuronal circuits in both FXS animal models and patients (Perry et al., 2001; Kesler et al., 2009). FXS is the most common heritable cause of developmental disability, and it is associated with IQ, memory, visuospatial processing deficits, and a profile of cognitive-behavioral deficits predominantly related to executive function, social behavior, communication, and cognitive skills (Reiss and Dant, 2003).
The fmr1 gene is prominently transcribed during early brain development in the basal forebrain and the hippocampus, two regions critical to memory encoding and attention (Abitbol et al., 1993). More specifically, FMR1-mRNA is expressed with the highest level in BFCN of the Nucleus basalis Magnocellularis (NbM) and in hippocampal pyramidal neurons (Reiss and Dant, 2003; Greicius 2004). The early transcription of fmr1 gene and brain distribution of FMR1-mRNAs in human fetuses suggest that alterations of fmr1 gene expression in BFCS may be responsible for FXS typical executive and cognitive dysfunctions (Sarter et al., 2003; Hoeft 2010), and intellectual disability (Abitbol 1993). Furthermore, fmr1 KO animal models show aberrant cholinergic function in the subiculum and the CA1 region of the hippocampus (D’Antuono et al., 2003), with concurrent behavioral deficits resembling those observed in human FXS patients.
Neuroimaging studies have repeatedly demonstrated abnormalities in prefrontal cortex of individuals with FXS and have shown that lower FMRP is correlated with impaired performance and abnormal prefrontal brain activation during executive tasks (Menon et al., 2004). Males with FXS are at risk for significant cognitive and behavioral deficits, particularly those involving executive prefrontal systems. Reduced grey matter volumes in low functioning autistic children were found in the nucleus accumbens, a BF area critical for adaptive response to aversive stimuli and reward (Riva et al., 2011).
Also, auditory hyper-reactivity is a common sensory-perceptual abnormality in ASD and increases the risk of maladaptive behavior in ASD children. Importantly, BFCS has been implicated in these reflex responses and cholinesterase inhibitors have been proposed to ameliorate auditory sensory response in rodent models (Hohnadel et al., 2007; Jin et al., 2019).
These findings are consistent with a hypothesis of functional disruption of cholinergic systems as a direct consequence of reduced FMRP expression level, resulting in turn in delayed neuronal development and permanent changes in cortical cytoarchitecture, finally leading to cognitive dysfunctions.
Since the etiological causes of ASD remain unknown, the current treatments of this disturbing behavioral profile are largely symptomatic (Mukaetova-Ladinska and Perry, 2015). The use of acetylcholinesterase inhibitors to sustain ach levels, including donepezil, galantamine and rivastigmine, is reported to improve cholinergic synaptic plasticity in rodent models, and executive functions in open label human trials (Ginestet et al., 2007).
Several studies on ASD animal models have shown the involvement of nAChRs in modulating social and repetitive behaviors (Wang et al., 2015). Moreover, CHRNA7 gene mutations have been correlated to autistic-like phenotypes (Yasui et al., 2011). Of note, behavioral abnormalities induced by maternal brain inflammation were prevented by gestational choline supplementation in the offspring of a rodent model (Wu et al., 2015). In line, BFCN have been shown to downregulate brain inflammation via stimulation of microglial alpha7 nAChAR (Wang and El-Deiry, 2003; Shytle et al., 2004).
Neuropathological studies conducted on human post-mortem brain tissue of autistic adults confirmed the clinical relevance of both muscarinic and nicotinic acetylcholine receptors in regulating memory and behavioral flexibility deficits in autism. ASD associated changes in α7 nAChAR, as well as muscarinic 1–2 (M1–M2) receptors, have been reported by several groups (Martin-Ruiz et al., 2004; Palma et al., 2012). Further, selective nicotinic and M1–M4 muscarinic agonists lead to enhancement of memory and cognitive performance (Deutsch et al., 2010; Ragozzino et al., 2012; Thomson et al., 2017).
Overall, findings of loss of muscarinic receptors in the brain tissue in autism and FXS and the observed beneficial action of cholinomimetics and Ache inhibitors represent a relevant step forward the development of novel targeted pharmacological treatments for AD, FXS and ASD related disorders.
FMRP Alterations in Autism and Alzheimer: A Shared Neuropathology?
It is interesting to note that on one hand APP metabolites are considered as potential strategies for FXS, and in turn mGluR5 blocking drugs have been used for AD therapies (Westmark and App, 2019). Two diseases that are characteristic of opposite ends of the human lifespan converge into the same molecular neuropathology. An increasing number of findings highlight the common features between neurodevelopmental issues, on one side, and age-related neurodegeneration, on the other (Triaca and Calissano, 2016; Lahiri et al., 2021), although conclusive evidence is needed.
FMRP is mainly involved in RNA regulation and its absence induces APP overexpression in FXS (Westmark and Malter, 2007; Westmark et al., 2009), as well as in AD (Borreca et al., 2016). Furthermore, and related to FMRP metabolic control at the synapses, the protein synthesis machinery is another common feature found to be altered in both diseases.
These features are only small part of the long story where FMRP plays a key role in two completely different pathologies.
Moreover, a further relevant role for FMRP has also been demonstrated in AD. In fact, fragile X-associated tremor/ataxia syndrome (FXTAS) patients are characterized by an FMRP mutation in 5′UTR of the gene with CGG repetition comprising between 55 and 200, showing a neurocognitive deficit associated with AD (Aydin et al., 2020). Also, other neurodegenerative disorders are associated to FMR1 premutation, such as Parkinson disease or multiple sclerosis (Sacino et al., 2019; Dijkstra et al., 2021), pinpointing a crucial effect of FMRP mutations in common neuronal function and synaptic dysfunction.
Furthermore, FXS and AD share more than molecular substrates, based on compelling evidence on the role of BFCS attention impairment in AD and FXS typical behavioral deficits.
Understanding the molecular link among pathologies affecting brain lifespan from development to ageing is of foremost importance for drug discovery to vanquish pathologies, like FXS, AD, PD, and autism with a deep medical and financial impact on the society.
Common Future Perspectives
We have highlighted the common features of neurodevelopmental and neurodegenerative disorders. The key molecule, FMRP, is associated to both pathologies and contributes to their phenotypes. How it may contribute to neuronal loss and altered synaptic plasticity in AD is still unknown. This review has described the role of the protein in both pathologies and may offer insight into its double role in neurodevelopmental and neurodegenerative diseases. In more detail, we also described how FMRP has some common features with AD pathology and how this help us to uncover converging mechanisms shared by FXS and other brain disorders. This contributes to the development of a broad-spectrum therapeutic agent.
Author Contributions
This review article was conceived by AB with contributions from all authors. VT supported AB on the preparation of the review with a main focus on the cholinergic system. LB prepared the table and read the manuscript. The figure was conceived by AB and illustrated by VT and LB.
Funding
AB is grateful for the support of the FRAXA Research Foundation with a project titled “Characterization of microglia transcriptional profile in fmr1ko mice model”.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abitbol, M., Menini, C., Delezoide, A.-L., Rhyner, T., Vekemans, M., and Mallet, J. (1993). Nucleus Basalis Magnocellularis and hippocampus Are the Major Sites of FMR-1 Expression in the Human Fetal Brain. Nat. Genet. 4 (2), 147–153. doi:10.1038/ng0693-147
Antic, D., and Keene, J. D. Messenger Ribonucleoprotein Complexes Containing Human ELAV Proteins: Interactions with Cytoskeleton and Translational Apparatus. J. Cel Sci. (1998); 111:183, 197. doi: doi:10.1242/jcs.111.2.183
Aydin, H., Bucak, İ. H., and Bağiş, H. (2020). Bir Sendrom, Farklı Fenotipler; Fragile X Sendromu. Turkish J. Pediatr. Dis., 1–5. doi:10.12956/tchd.613271
Ballinger, E. C., Ananth, M., Talmage, D. A., and Role, L. W. (2016). Basal Forebrain Cholinergic Circuits and Signaling in Cognition and Cognitive Decline. Neuron 91, 1199–1218. doi:10.1016/j.neuron.2016.09.006
Bartley, C. M., O’Keefe, R. A., Blice-Baum, A., Mihailescu, M.-R., Gong, X., Miyares, L., et al. Mammalian FMRP S499 Is Phosphorylated by CK2 and Promotes Secondary Phosphorylation of FMRP, eNeuro (2016), 3(6), 0092–116. doi:10.1523/ENEURO.0092-16.2016
Bartley, C. M., O’Keefe, R. A., and Bordey, A. (2014). FMRP S499 Is Phosphorylated Independent of mTORC1-S6k1 Activity. PLOS ONE 9, e96956. doi:10.1371/journal.pone.0096956
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR Theory of Fragile X Mental Retardation. Trends Neurosciences 27, 370–377. doi:10.1016/j.tins.2004.04.009
Bechara, E. G., Didiot, M. C., Melko, M., Davidovic, L., Bensaid, M., Martin, P., et al. (2009). A Novel Function for Fragile X Mental Retardation Protein in Translational Activation. Plos Biol. 7 (1), e1000016. doi:10.1371/journal.pbio.1000016
Bernard, P. B., Castano, A. M., O'Leary, H., Simpson, K., Browning, M. D., and Benke, T. A. (2013). Phosphorylation of FMRP and Alterations of FMRP Complex Underlie Enhanced mLTD in Adult Rats Triggered by Early Life Seizures. Neurobiol. Dis. 59, 1–17. doi:10.1016/j.nbd.2013.06.013
Bie, B., Wu, J., Foss, J. F., and Naguib, M. (2019). Activation of mGluR1 Mediates C1q-dependent Microglial Phagocytosis of Glutamatergic Synapses in Alzheimer's Rodent Models. Mol. Neurobiol. 56 (8), 5568–5585. doi:10.1007/s12035-019-1467-8
Billuart, P., and Chelly, J. (2003). From Fragile X Mental Retardation Protein to Rac1 GTPase. Neuron 38 (6), 843–845. doi:10.1016/s0896-6273(03)00362-3
Borreca, A., Gironi, K., Amadoro, G., and Ammassari-Teule, M. (2016). Opposite Dysregulation of Fragile-X Mental Retardation Protein and Heteronuclear Ribonucleoprotein C Protein Associates with Enhanced APP Translation in Alzheimer Disease. Mol. Neurobiol. 53, 3227–3234. doi:10.1007/s12035-015-9229-8
Borreca, A., Valeri, F., De Luca, M., Ernst, L., Russo, A., Nobili, A., et al. (2020). Transient Upregulation of Translational Efficiency in Prodromal and Early Symptomatic Tg2576 Mice Contributes to Aβ Pathology. Neurobiol. Dis. 139, 104787. doi:10.1016/j.nbd.2020.104787
Brown, M. R., Kronengold, J., Gazula, V.-R., Chen, Y., Strumbos, J. G., Sigworth, F. J., et al. (2010). Fragile X Mental Retardation Protein Controls Gating of the Sodium-Activated Potassium Channel Slack. Nat. Neurosci. 13, 819–821. doi:10.1038/nn.2563
Brown, V., Jin, P., Ceman, S., Darnell, J. C., O'Donnell, W. T., Tenenbaum, S. A., et al. (2001). Microarray Identification of FMRP-Associated Brain mRNAs and Altered mRNA Translational Profiles in Fragile X Syndrome. Cell 107 (4), 477–487. doi:10.1016/s0092-8674(01)00568-2
Casingal, C. R., Kikkawa, T., Inada, H., Sasaki, Y., and Osumi, N. (2020). Identification of FMRP Target mRNAs in the Developmental Brain: FMRP Might Coordinate Ras/MAPK, Wnt/β-Catenin, and mTOR Signaling during Corticogenesis. Mol. Brain 13 (1), 167. doi:10.1186/s13041-020-00706-1
Cefaliello, C., Penna, E., Barbato, C., Di Ruberto, G., Mollica, M. P., Trinchese, G., et al. (2020). Deregulated Local Protein Synthesis in the Brain Synaptosomes of a Mouse Model for Alzheimer's Disease. Mol. Neurobiol. 57, 1529–1541. doi:10.1007/s12035-019-01835-y
Ceman, S., O'Donnell, W. T., Reed, M., Patton, S., Pohl, J., and Warren, S. T. (2003). Phosphorylation Influences the Translation State of FMRP-Associated Polyribosomes. Hum. Mol. Genet. 12, 3295–3305. doi:10.1093/hmg/ddg350
Chatterjee, M., Kurup, P. K., Lundbye, C. J., Hugger Toft, A. K., Kwon, J., Benedict, J., et al. (2018). STEP Inhibition Reverses Behavioral, Electrophysiologic, and Synaptic Abnormalities in Fmr1 KO Mice. Neuropharmacology 128, 43–53. doi:10.1016/j.neuropharm.2017.09.026
Chen, L., Yun, S.-W., Seto, J., Liu, W., and Toth, M. (2003). The Fragile X Mental Retardation Protein Binds and Regulates a Novel Class of mRNAs Containing U Rich Target Sequences. Neuroscience 120 (4), 1005–1017. doi:10.1016/S0306-4522(03)00406-8
Chmielewska, J. J., Kuzniewska, B., Milek, J., Urbanska, K., and Dziembowska, M. (2019). Neuroligin 1, 2, and 3 Regulation at the Synapse: FMRP-dependent Translation and Activity-Induced Proteolytic Cleavage. Mol. Neurobiol. 56 (4), 2741–2759. doi:10.1007/s12035-018-1243-1
Chuang, S.-C., Zhao, W., Bauchwitz, R., Yan, Q., Bianchi, R., and Wong, R. K. (2005). Prolonged Epileptiform Discharges Induced by Altered Group I Metabotropic Glutamate Receptor-Mediated Synaptic Responses in Hippocampal Slices of a Fragile X Mouse Model. J. Neurosci. 25, 8048–8055. doi:10.1523/JNEUROSCI.1777-05.2005
D'Annessa, I., Cicconardi, F., and Di Marino, D. (2019). Handling FMRP and its Molecular Partners: Structural Insights into Fragile X Syndrome. Prog. Biophys. Mol. Biol. 141, 3–14. doi:10.1016/j.pbiomolbio.2018.07.001
D'Antuono, M., Merlo, D., and Avoli, M. (2003). Involvement of Cholinergic and Gabaergic Systems in the Fragile X Knockout Mice. Neuroscience 119 (1), 9–13. doi:10.1016/s0306-4522(03)00103-9
Darnell, J. C., Jensen, K. B., Jin, P., Brown, V., Warren, S. T., and Darnell, R. B. (2001). Fragile X Mental Retardation Protein Targets G Quartet mRNAs Important for Neuronal Function. Cell 107 (4), 489–499. doi:10.1016/s0092-8674(01)00566-9
Davis, J. K., and Broadie, K. (2017). Multifarious Functions of the Fragile X Mental Retardation Protein. Trends Genet. 33 (10), 703–714. doi:10.1016/j.tig.2017.07.008
De Boulle, K., Verkerk, A. J. M. H., Reyniers, E., Vits, L., Hendrickx, J., Van Roy, B., et al. (1993). A point Mutation in the FMR-1 Gene Associated with Fragile X Mental Retardation. Nat. Genet. 3, 31–35. doi:10.1038/ng0193-31
De Rubeis, S., Pasciuto, E., Li, K. W., Fernández, E., Di Marino, D., Buzzi, A., et al. (2013). CYFIP1 Coordinates mRNA Translation and Cytoskeleton Remodeling to Ensure Proper Dendritic Spine Formation. Neuron 79 (6), 1169–1182. doi:10.1016/j.neuron.2013.06.039
Deutsch, S. I., Urbano, M. R., Neumann, S. A., Burket, J. A., and Katz, E. (2010). Cholinergic Abnormalities in Autism. Clin. Neuropharmacol 33, 114–120. doi:10.1097/WNF.0b013e3181d6f7ad
Dhungel, N., Eleuteri, S., Li, L.-b., Kramer, N. J., Chartron, J. W., Spencer, B., et al. (2015). Parkinson's Disease Genes VPS35 and EIF4G1 Interact Genetically and Converge on α-Synuclein. Neuron 85 (1), 76–87. doi:10.1016/j.neuron.2014.11.027
Di Marino, D., Achsel, T., Lacoux, C., Falconi, M., and Bagni, C. (2014). Molecular Dynamics Simulations Show How the FMRP Ile304Asn Mutation Destabilizes the KH2 Domain Structure and Affects its Function. J. Biomol. Struct. Dyn. 32, 337–350. doi:10.1080/07391102.2013.768552
Dictenberg, J. B., Swanger, S. A., Antar, L. N., Singer, R. H., and Bassell, G. J. (2008). A Direct Role for FMRP in Activity-dependent Dendritic mRNA Transport Links Filopodial-Spine Morphogenesis to Fragile X Syndrome. Develop. Cel 14 (6), 926–939. Jun. doi:10.1016/j.devcel.2008.04.003
Dijkstra, A. A., Haify, S. N., Verwey, N. A., Prins, N. D., van der Toorn, E. C., Rozemuller, A. J. M., et al. (2021). Neuropathology of FMR1-Premutation Carriers Presenting with Dementia and Neuropsychiatric Symptoms. Brain Commun. 3 (1), fcab007. doi:10.1093/braincomms/fcab007
Dölen, G., Osterweil, E., Rao, B. S. S., Smith, G. B., Auerbach, B. D., Chattarji, S., et al. (2007). Correction of Fragile X Syndrome in Mice. Neuron 56, 955–962. doi:10.1016/j.neuron.2007.12.001
Eberhart, D., Malter, H. E., Feng, Y., and Warren, S. T. (1996). The Fragile X Mental Retardation Protein Is a Ribonucleoprotein Containing Both Nuclear Localization and Nuclear export Signals. Hum. Mol. Genet. 5, 1083–1091. doi:10.1093/hmg/5.8.1083
Edbauer, D., Neilson, J. R., Foster, K. A., Wang, C.-F., Seeburg, D. P., Batterton, M. N., et al. (2010). Regulation of Synaptic Structure and Function by FMRP-Associated microRNAs miR-125b and miR-132. Neuron 65 (3), 373–384. doi:10.1016/j.neuron.2010.01.005
Ehyai, S., Miyake, T., Williams, D., Vinayak, J., Bayfield, M. A., and McDermott, J. C. (2018). FMRP Recruitment of β‐catenin to the Translation Pre‐initiation Complex Represses Translation. EMBO Rep. 19 (12), e45536. doi:10.15252/embr.201745536
Evans, T. L., Blice-Baum, A. C., and Mihailescu, M.-R. (2012). Analysis of the Fragile X Mental Retardation Protein Isoforms 1, 2 and 3 Interactions with the G-Quadruplex Forming Semaphorin 3F mRNA. Mol. Biosyst. 8 (2), 642–649. doi:10.1039/c1mb05322a
Fakhoury, M. (2018). Microglia and Astrocytes in Alzheimer's Disease: Implications for Therapy. Cn 16, 508–518. doi:10.2174/1570159X15666170720095240
Fernández-Cabello, S., Kronbichler, M., Van Dijk, K. R. A., Goodman, J. A., Spreng, R. N., and Schmitz, T. W. (2020). Basal Forebrain Volume Reliably Predicts the Cortical Spread of Alzheimer's Degeneration. Brain 143 (3), 993–1009. 1;. doi:10.1093/brain/awaa012
Ferrante, A., Boussadia, Z., Borreca, A., Mallozzi, C., Pedini, G., Pacini, L., et al. (2021). Adenosine A2A Receptor Inhibition Reduces Synaptic and Cognitive Hippocampal Alterations in Fmr1 KO Mice. Transl Psychiatry 11, 112. doi:10.1038/s41398-021-01238-5
Filipello, F., Morini, R., Corradini, I., Zerbi, V., Canzi, A., Michalski, B., et al. (2018). The Microglial Innate Immune Receptor TREM2 is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 48 (5), 979–991.e8. doi:10.1016/j.immuni.2018.04.016
Filippini, A., Bonini, D., Lacoux, C., Pacini, L., Zingariello, M., Sancillo, L., et al. (2017). Absence of the Fragile X Mental Retardation Protein Results in Defects of RNA Editing of Neuronal mRNAs in Mouse. RNA Biol. 14, 1580–1591. doi:10.1080/15476286.2017.1338232
Fischer-Kešo, R., Breuninger, S., Hofmann, S., Henn, M., Röhrig, T., Ströbel, P., et al. (2014). Plakophilins 1 and 3 Bind to FXR1 and Thereby Influence the mRNA Stability of Desmosomal Proteins. Mol. Cel Biol. 34 (34), 4244–4256. doi:10.1128/MCB.00766-14
Fu, X.-g., Yan, A.-z., Xu, Y.-j., Liao, J., Guo, X.-y., Zhang, D., et al. Splicing of Exon 9a in FMR1 Transcripts Results in a Truncated FMRP with Altered Subcellular Distribution. Gene, 731. (2020). 144359. doi: doi:10.1016/j.gene.2020.144359
Fujita, R., Zismanov, V., Jacob, J.-M., Jamet, S., Asiev, K., and Crist, C. (2017). Fragile X Mental Retardation Protein Regulates Skeletal Muscle Stem Cell Activity by Regulating the Stability of Myf5 mRNA. Skeletal Muscle 7, 18. doi:10.1186/s13395-017-0136-8
Gandhi, R. n. M., Kogan, C. S., and Messier, C. (2014). 2-Methyl-6-(phenylethynyl) Pyridine (MPEP) Reverses Maze Learning and PSD-95 Deficits in Fmr1 Knock-Out Mice. Front. Cel. Neurosci. 8, 8. doi:10.3389/fncel.2014.00070
Ghosh, A., Mizuno, K., Tiwari, S. S., Proitsi, P., Gomez Perez-Nievas, B., Glennon, E., et al. (2020). Alzheimer's Disease-Related Dysregulation of mRNA Translation Causes Key Pathological Features with Ageing. Transl Psychiatry 10, 192. doi:10.1038/s41398-020-00882-7
Giampetruzzi, A., Carson, J. H., and Barbarese, E. (2013). FMRP and Myelin Protein Expression in Oligodendrocytes. Mol. Cell Neurosci. 56, 333–341. doi:10.1016/j.mcn.2013.07.009
Ginestet, L., Ferrario, J. E., Raisman-Vozari, R., Hirsch, E. C., and Debeir, T. (2007). Donepezil Induces a Cholinergic Sprouting in Basocortical Degeneration. J. Neurochem. 102, 434–440. doi:10.1111/j.1471-4159.2007.04497.x
Golden, C. E. M., Breen, M. S., Koro, L., Sonar, S., Niblo, K., Browne, A., et al. (2019). Deletion of the KH1 Domain ofFmr1Leads to Transcriptional Alterations and Attentional Deficits in Rats. Cereb. Cortex 29 (5), 2228–2244. doi:10.1093/cercor/bhz029
Greicius, M. D., Boyett-Anderson, J. M., Menon, V., and Reiss, A. L. (2004). Reduced Basal Forebrain and Hippocampal Activation during Memory Encoding in Girls with Fragile X Syndrome. Neuroreport 15 (10), 1579–1583. doi:10.1097/01.wnr.0000134472.44362.be
Grønskov, K., Brøndum-Nielsen, K., Dedic, A., Hjalgrim, H., and Hjalgrim, H. (2011). A Nonsense Mutation in FMR1 Causing Fragile X Syndrome. Eur. J. Hum. Genet. 19, 489–491. doi:10.1038/ejhg.2010.223
Grossman, A. W., Aldridge, G. M., Lee, K. J., Zeman, M. K., Jun, C. S., Azam, H. S., et al. (2010). Developmental Characteristics of Dendritic Spines in the Dentate Gyrus of Fmr1 Knockout Mice. Brain Res. 1355, 221–227. doi:10.1016/j.brainres.2010.07.090
Grozeva, D., Carss, K., Spasic‐Boskovic, O., Tejada, M. I., Gecz, J., Shaw, M., et al. (2015). Targeted Next‐Generation Sequencing Analysis of 1,000 Individuals with Intellectual Disability. Hum. Mutat. 36, 1197–1204. doi:10.1002/humu.22901
Handt, M., Epplen, A., Hoffjan, S., Mese, K., Epplen, J. T., and Dekomien, G. (2014). Point Mutation Frequency in the FMR1 Gene as Revealed by Fragile X Syndrome Screening. Mol. Cell Probes 28, 279–283. doi:10.1016/j.mcp.2014.08.003
He, Q., and Ge, W. (2017). The Tandem Agenet Domain of Fragile X Mental Retardation Protein Interacts with FUS. Sci. Rep. 7 (1), 962. doi:10.1038/s41598-017-01175-8
Hernández-Ortega, K., Garcia-Esparcia, P., Gil, L., Lucas, J. J., and Ferrer, I. Altered Machinery of Protein Synthesis in Alzheimer's: From the Nucleolus to the Ribosome, Brain Pathol. (2016), 26(5) Zurich, Switzerland:, 593–605. doi:10.1111/bpa.12335
Heulens, I., and Kooy, F. (2011). Fragile X Syndrome: from Gene Discovery to Therapy. Front. Biosci. 16, 1211–1232. doi:10.2741/3785
Higashimori, H., Morel, L., Huth, J., Lindemann, L., Dulla, C., Taylor, A., et al. (2013). Astroglial FMRP-dependent Translational Down-Regulation of mGluR5 Underlies Glutamate Transporter GLT1 Dysregulation in the Fragile X Mouse. Hum. Mol. Genet. 22 (10), 2041–2054. doi:10.1093/hmg/ddt055
Higashimori, H., Schin, C. S., Chiang, M. S. R., Morel, L., Shoneye, T. A., Nelson, D. L., et al. (2016). Selective Deletion of Astroglial FMRP Dysregulates Glutamate Transporter GLT1 and Contributes to Fragile X Syndrome Phenotypes In Vivo. J. Neurosci. 36, 7079–7094. doi:10.1523/JNEUROSCI.1069-16.2016
Hodges, S. L., Nolan, S. O., Taube, J. H., and Lugo, J. N. (2017). Adult Fmr1 Knockout Mice Present with Deficiencies in Hippocampal Interleukin-6 and Tumor Necrosis Factor-α Expression. Neuroreport 28, 1246–1249. doi:10.1097/WNR.0000000000000905
Hoeft, F., Carter, J. C., Lightbody, A. A., Cody Hazlett, H., Piven, J., and Reiss, A. L. (2010). Region-specific Alterations in Brain Development in One- to Three-Year-Old Boys with Fragile X Syndrome. Proc. Natl. Acad. Sci. 107 (20), 9335–9339. doi:10.1073/pnas.1002762107
Hohnadel, E., Bouchard, K., and Terry, A. V. (2007). Galantamine and Donepezil Attenuate Pharmacologically Induced Deficits in Prepulse Inhibition in Rats. Neuropharmacology 52 (2), 542–551. doi:10.1016/j.neuropharm.2006.08.025
Holt, C. E., Martin, K. C., and Schuman, E. M. (2019). Local Translation in Neurons: Visualization and Function. Nat. Struct. Mol. Biol. 26 (7), 557–566. doi:10.1038/s41594-019-0263-5
Hu, H., Haas, S. A., Chelly, J., Van Esch, H., Raynaud, M., de Brouwer, A. P. M., et al. (2016). X-exome Sequencing of 405 Unresolved Families Identifies Seven Novel Intellectual Disability Genes. Mol. Psychiatry 21, 133–148. doi:10.1038/mp.2014.193
Hu, Y., Chen, Z., Fu, Y., He, Q., Jiang, L., Zheng, J., et al. (2015). The Amino-Terminal Structure of Human Fragile X Mental Retardation Protein Obtained Using Precipitant-Immobilized Imprinted Polymers. Nat. Commun. 6, 6634. doi:10.1038/ncomms7634
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered Synaptic Plasticity in a Mouse Model of Fragile X Mental Retardation. Proc. Natl. Acad. Sci. 99, 7746–7750. doi:10.1073/pnas.122205699
Iacoangeli, A., Rozhdestvensky, T. S., Dolzhanskaya, N., Tournier, B., Schütt, J., Brosius, J., et al. (2008). On BC1 RNA and the Fragile X Mental Retardation Protein. Proc. Natl. Acad. Sci. 105 (2), 734–739. doi:10.1073/pnas.0710991105
Ifrim, M. F., Williams, K. R., and Bassell, G. J. (2015). Single-Molecule Imaging of PSD-95 mRNA Translation in Dendrites and its Dysregulation in a Mouse Model of Fragile X Syndrome. J. Neurosci. 35, 7116–7130. doi:10.1523/JNEUROSCI.2802-14.2015
Jacquemont, S., Pacini, L., Jønch, A. E., Cencelli, G., Rozenberg, I., He, Y., et al. (2018). Protein Synthesis Levels Are Increased in a Subset of Individuals with Fragile X Syndrome. Hum. Mol. Genet. 27, 2039–2051. doi:10.1093/hmg/ddy099
Janusz, A., Milek, J., Perycz, M., Pacini, L., Bagni, C., Kaczmarek, L., et al. (2013). The Fragile X Mental Retardation Protein Regulates Matrix Metalloproteinase 9 mRNA at Synapses. J. Neurosci. 33 (46), 18234–18241. doi:10.1523/JNEUROSCI.2207-13.2013
Jin, J., Cheng, J., Lee, K.-W., Amreen, B., McCabe, K. A., Pitcher, C., et al. (2019). Cholinergic Neurons of the Medial Septum Are Crucial for Sensorimotor Gating. J. Neurosci. 39 (26), 5234–5242. doi:10.1523/JNEUROSCI.0950-18.2019
Kalinowska, M., Chávez, A. E., Lutzu, S., Castillo, P. E., Bukauskas, F. F., and Francesconi, A. Actinin-4 Governs Dendritic Spine Dynamics and Promotes Their Remodeling by Metabotropic Glutamate Receptors. J. Biol. Chem. 2015 Jun 26;290(26):15909–15920. doi: doi:10.1074/jbc.M115.640136
Keene, J. D., and Tenenbaum, S. A. (2002). Eukaryotic mRNPs May Represent Posttranscriptional Operons. Mol. Cel. 9(6):1161-7. doi:10.1016/S1097-2765(02)00559-2
Kesler, S. R., Lightbody, A. A., and Reiss, A. L. (2009). Cholinergic Dysfunction in Fragile X Syndrome and Potential Intervention: A preliminary1H MRS Study. Am. J. Med. Genet. 149A (3), 403–407. doi:10.1002/ajmg.a.32697
Khandjian, E. W., Huot, M.-E., Tremblay, S., Davidovic, L., Mazroui, R., and Bardoni, B. (2004). Biochemical Evidence for the Association of Fragile X Mental Retardation Protein with Brain Polyribosomal Ribonucleoparticles. Proc. Natl. Acad. Sci. 101, 13357–13362. doi:10.1073/pnas.0405398101
Krichevsky, A. M., and Kosik, K. S. (2001). Neuronal RNA Granules: a Link between RNA Localization and Stimulation-dependent Translation. Neuron 32, 683–696. doi:10.1016/s0896-6273(01)00508-6
Kumar, A., Roy, S., Tripathi, S., and Sharma, A. (2016). Molecular Docking Based Virtual Screening of Natural Compounds as Potential BACE1 Inhibitors: 3D QSAR Pharmacophore Mapping and Molecular Dynamics Analysis. J. Biomol. Struct. Dyn. 34 (2), 239–249. doi:10.1080/07391102.2015.1022603
Lacoux, C., Di Marino, D., Pilo Boyl, P., Zalfa, F., Yan, B., Ciotti, M. T., et al. (2012). BC1-FMRP Interaction is Modulated by 2′-O-Methylation: RNA-Binding Activity of the Tudor Domain and Translational Regulation at Synapses. Nucleic Acids Res. 40 (9), 4086–4096. doi:10.1093/nar/gkr1254
Lahiri, D. K., Maloney, B., Wang, R., Sokol, D. K., Rogers, J. T., and Westmark, C. J. (2021). How Autism and Alzheimer's Disease are TrAPPed. Mol. Psychiatry 26, 26–29. doi:10.1038/s41380-020-00928-8
Lee, E. K., Kim, H. H., Kuwano, Y., Abdelmohsen, K., Srikantan, S., Subaran, S. S., et al. (2010). hnRNP C Promotes APP Translation by Competing with FMRP for APP mRNA Recruitment to P Bodies. Nat. Struct. Mol. Biol. 17 (6), 732–739. doi:10.1038/nsmb.1815
Lee, F. H. F., Lai, T. K. Y., Su, P., and Liu, F. (2019). Altered Cortical Cytoarchitecture in the Fmr1 Knockout Mouse. Mol. Brain 12, 56. doi:10.1186/s13041-019-0478-8
Li, X., Yu, B., Sun, Q., Zhang, Y., Ren, M., Zhang, X., et al. (2018). Generation of a Whole-Brain Atlas for the Cholinergic System and Mesoscopic Projectome Analysis of Basal Forebrain Cholinergic Neurons. Proc. Natl. Acad. Sci. USA 115 (2), 415–420. doi:10.1073/pnas.1703601115
Long, J. M., Maloney, B., Rogers, J. T., and Lahiri, D. K. (2019). Novel Upregulation of Amyloid-β Precursor Protein (APP) by microRNA-346 via Targeting of APP mRNA 5′-untranslated Region: Implications in Alzheimer's Disease. Mol. Psychiatry 24, 345–363. doi:10.1038/s41380-018-0266-3
Lugenbeel, K. A., Peier, A. M., Carson, N. L., Chudley, A. E., and Nelson, D. L. (1995). Intragenic Loss of Function Mutations Demonstrate the Primary Role of FMR1 in Fragile X Syndrome. Nat. Genet. 10, 483–485. doi:10.1038/ng0895-483
Lukiw, W. J. (2007). Micro-RNA Speciation in Fetal, Adult and Alzheimer's Disease hippocampus. Neuroreport 18 (3), 297–300. doi:10.1097/WNR.0b013e3280148e8b
Ma, X., Liu, L., and Meng, J. (2017). MicroRNA-125b Promotes Neurons Cell Apoptosis and Tau Phosphorylation in Alzheimer's Disease. Neurosci. Lett. 661, 57–62. doi:10.1016/j.neulet.2017.09.043
Maddirevula, S., Alsaif, H. S., Ibrahim, N., and Alkuraya, F. S. (2020). A De Novo Mutation in FMR1 in a Patient with Intellectual Disability. Eur. J. Med. Genet. 63 (3), 103763. doi:10.1016/j.ejmg.2019.103763
Markham, J. A., Beckel-Mitchener, A. C., Estrada, C. M., and Greenough, W. T. (2006). Corticosterone Response to Acute Stress in a Mouse Model of Fragile X Syndrome. Psychoneuroendocrinology 31 (6), 781–785. doi:10.1016/j.psyneuen.2006.02.008
Martin-Ruiz, C. M., Lee, M., Perry, R. H., Baumann, M., Court, J. A., and Perry, E. K. (2004). Molecular Analysis of Nicotinic Receptor Expression in Autism. Mol. Brain Res. 123, 81–90. doi:10.1016/j.molbrainres.2004.01.003
Martínez Cerdeño, V., Hong, T., Amina, S., Lechpammer, M., Ariza, J., Tassone, F., et al. (2018). Microglial Cell Activation and Senescence Are Characteristic of the Pathology FXTAS. Mov Disord. 33, 12. doi:10.1002/mds.27553
Maurin, T., Melko, M., Abekhoukh, S., Khalfallah, O., Davidovic, L., Jarjat, M., et al. (2015). The FMRP/GRK4mRNA Interaction Uncovers a New Mode of Binding of the Fragile X Mental Retardation Protein in Cerebellum. Nucleic Acids Res. 43, 8540–8550. doi:10.1093/nar/gkv801
Menon, L., Mader, S. A., and Mihailescu, M.-R. (2008). Fragile X Mental Retardation Protein Interactions with the Microtubule Associated Protein 1B RNA. RNA 14 (8), 1644–1655. doi:10.1261/rna.1100708
Menon, V., Leroux, J., White, C. D., and Reiss, A. L. (2004). Frontostriatal Deficits in Fragile X Syndrome: Relation to FMR1 Gene Expression. Proc. Natl. Acad. Sci. 101 (10), 3615–3620. doi:10.1073/pnas.0304544101
Miyashiro, K. Y., Beckel-Mitchener, A., Purk, T. P., Becker, K. G., Barret, T., Liu, L., et al. (2003). RNA Cargoes Associating with FMRP Reveal Deficits in Cellular Functioning in Fmr1 Null Mice. Neuron 37 (3), 417–431. doi:10.1016/s0896-6273(03)00034-5
Mukaetova-Ladinska, E. B., and Perry, E. K. (2015). “Molecular Basis of Cholinergic Changes in Autism Spectrum Disorders and Relevance for Treatment Interventions,” in The Molecular Basis of Autism. Contemporary Clinical Neuroscience. Editor S Fatemi (New York, USA: Springer), 307–335. doi:10.1007/978-1-4939-2190-4_15
Mus, E., Hof, P. R., and Tiedge, H. (2007). Dendritic BC200 RNA in Aging and in Alzheimer's Disease. Proc. Natl. Acad. Sci. 104, 10679–10684. doi:10.1073/pnas.0701532104
Myrick, L. K., Deng, P.-Y., Hashimoto, H., Oh, Y. M., Cho, Y., Poidevin, M. J., et al. (2015). Independent Role for Presynaptic FMRP Revealed by an FMR1 Missense Mutation Associated with Intellectual Disability and Seizures. Proc. Natl. Acad. Sci. USA 112 (4), 949–956. doi:10.1073/pnas.1423094112
Myrick, L. K., Nakamoto-Kinoshita, M., Lindor, N. M., Kirmani, S., Cheng, X., and Warren, S. T. (2014). Fragile X Syndrome Due to a Missense Mutation. Eur. J. Hum. Genet. 22, 1185–1189. doi:10.1038/ejhg.2013.311
Napoli, I., Mercaldo, V., Boyl, P. P., Eleuteri, B., Zalfa, F., De Rubeis, S., et al. (2008). The Fragile X Syndrome Protein Represses Activity-dependent Translation through CYFIP1, a New 4E-BP. Cell 134 (6), 1042–1054. doi:10.1016/j.cell.2008.07.031
Okray, Z., Esch, C. E., Van Esch, H., Devriendt, K., Claeys, A., Yan, J., et al. (2015). A Novel Fragile X Syndrome Mutation Reveals a Conserved Role for the Carboxy‐terminus in FMRP Localization and Function. EMBO Mol. Med. 7, 423–437. doi:10.15252/emmm.201404576
Palma, E., Conti, L., Roseti, C., and Limatola, C. (2012). Novel Approaches to Study the Involvement of α7-nAChR in Human Diseases. Cdt 13, 579–586. doi:10.2174/138945012800398838
Park, S., Park, J. M., Kim, S., Kim, J.-A., Shepherd, J. D., Smith-Hicks, C. L., et al. (2008). Elongation Factor 2 and Fragile X Mental Retardation Protein Control the Dynamic Translation of Arc/Arg3.1 Essential for mGluR-LTD. Neuron 59 (1), 70–83. doi:10.1016/j.neuron.2008.05.023
Pasciuto, E., Ahmed, T., Wahle, T., Gardoni, F., D’Andrea, L., Pacini, L., et al. (2015). Dysregulated ADAM10-Mediated Processing of APP during a Critical Time Window Leads to Synaptic Deficits in Fragile X Syndrome. Neuron 87 (2), 382–398. doi:10.1016/j.neuron.2015.06.032
Pasciuto, E., and Bagni, C. (2014). SnapShot: FMRP mRNA Targets and Diseases. Cell 158 (6), 1446. e1. doi:10.1016/j.cell.2014.08.035
Perrone-Capano, C., Volpicelli, F., Penna, E., Chun, J. T., and Crispino, M. (2021). Presynaptic Protein Synthesis and Brain Plasticity: From Physiology to Neuropathology. Prog. Neurobiol. 202, 102051. doi:10.1016/j.pneurobio.2021.102051
Perry, E. K., Lee, M. L. W., Martin-Ruiz, C. M., Court, J. A., Volsen, S. G., Merrit, J., et al. (2001). Cholinergic Activity in Autism: Abnormalities in the Cerebral Cortex and Basal Forebrain. Ajp 158 (7), 1058–1066. doi:10.1176/appi.ajp.158.7.1058
Prieto, M., Folci, A., Poupon, G., Schiavi, S., Buzzelli, V., Pronot, M., et al. (2021). Missense Mutation of Fmr1 Results in Impaired AMPAR-Mediated Plasticity and Socio-Cognitive Deficits in Mice. Nat. Commun. 12, 1557. doi:10.1038/s41467-021-21820-1
Quartier, A., Poquet, H., Gilbert-Dussardier, B., Rossi, M., Casteleyn, A.-S., Portes, V. d., et al. (2017). Intragenic FMR1 Disease-Causing Variants: A Significant Mutational Mechanism Leading to Fragile-X Syndrome. Eur. J. Hum. Genet. 25, 423–431. doi:10.1038/ejhg.2016.204
Rackham, O., and Brown, C. M. (2004). Visualization of RNA-Protein Interactions in Living Cells: FMRP and IMP1 Interact on mRNAs. EMBO J. 23, 3346–3355. doi:10.1038/sj.emboj.7600341
Ragozzino, M. E., Artis, S., Singh, A., Twose, T. M., Beck, J. E., and Messer, W. S. (2012). The Selective M1 Muscarinic Cholinergic Agonist CDD-0102A Enhances Working Memory and Cognitive Flexibility. J. Pharmacol. Exp. Ther. 340, 588–594. doi:10.1124/jpet.111.187625
Rajagopalan, L. E., Westmark, C. J., Jarzembowski, J. A., and Malter, J. S. (1998). hnRNP C Increases Amyloid Precursor Protein (APP) Production by Stabilizing APP mRNA. Nucleic Acids Res. 26, 3418–3423. doi:10.1093/nar/26.14.3418
Reiss, A. L., and Dant, C. C. (2003). The Behavioral Neurogenetics of Fragile X Syndrome: Analyzing Gene-Brain-Behavior Relationships in Child Developmental Psychopathologies. Dev. Psychopathol 15 (4), 927–968. doi:10.1017/s0954579403000464
Renoux, A. J., Carducci, N. M., Ahmady, A. A., and Todd, P. K. (2014a). Fragile X Mental Retardation Protein Expression in Alzheimerâ€s Disease. Front. Genet. 5, 360. doi:10.3389/fgene.2014.00360
Renoux, A. J., Sala-Hamrick, K. J., Carducci, N. M., Frazer, M., Halsey, K. E., Sutton, M. A., et al. (2014b). Impaired Sensorimotor Gating in Fmr1 Knock Out and Fragile X Premutation Model Mice. Behav. Brain Res. 267 (267), 42–45. doi:10.1016/j.bbr.2014.03.013
Riva, D., Bulgheroni, S., Aquino, D., Di Salle, F., Savoiardo, M., and Erbetta, A. (2011). Basal Forebrain Involvement in Low-Functioning Autistic Children: a Voxel-Based Morphometry Study. AJNR Am. J. Neuroradiol 32 (8), 1430–1435. doi:10.3174/ajnr.A2527
Sacino, A. N., Prokop, S., Walsh, M. A., Adamson, J., Subramony, S. H., Krans, A., et al. (2019). Fragile X-Associated Tremor Ataxia Syndrome with Co-occurrent Progressive Supranuclear Palsy-like Neuropathology. Acta Neuropathol. Commun. 7, 158. doi:10.1186/s40478-019-0818-z
Santoro, M. R., Bray, S. M., and Warren, S. T. (2012). Molecular Mechanisms of Fragile X Syndrome: a Twenty-Year Perspective. Annu. Rev. Pathol. Mech. Dis. 7, 219–245. doi:10.1146/annurev-pathol-011811-132457
Sarter, M., Bruno, J. P., and Givens, B. (2003). Attentional Functions of Cortical Cholinergic Inputs: what Does it Mean for Learning and Memory? Neurobiol. Learn. Mem. 80 (3), 245–256. doi:10.1016/s1074-7427(03)00070-4
Schaeffer, C., Bardoni, B., Mandel, J. L., Ehresmann, B., Ehresmann, C., and Moine, H. (2001). The Fragile X Mental Retardation Protein Binds Specifically to its mRNA via a Purine Quartet Motif. Embo J. 20 (17), 4803–4813. doi:10.1093/emboj/20.17.4803
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-dependent Manner. Neuron 74, 691–705. doi:10.1016/j.neuron.2012.03.026
Schmitz, T. W., Nathan Spreng, R., and Nathan Spreng, R. (2016). Basal Forebrain Degeneration Precedes and Predicts the Cortical Spread of Alzheimer's Pathology. Nat. Commun. 7, 13249. doi:10.1038/ncomms13249
Sethna, F., Moon, C., and Wang, H. (2014). From FMRP Function to Potential Therapies for Fragile X Syndrome. Neurochem. Res. 39, 1016–1031. doi:10.1007/s11064-013-1229-3
Sharma, A., Hoeffer, C. A., Takayasu, Y., Miyawaki, T., McBride, S. M., Klann, E., et al. (2010). Dysregulation of mTOR Signaling in Fragile X Syndrome. J. Neurosci., 30(2), 694–702. doi:10.1523/JNEUROSCI.3696-09.2010
Shytle, R. D., Mori, T., Townsend, K., Vendrame, M., Sun, N., Zeng, J., et al. (2004). Cholinergic Modulation of Microglial Activation by α7 Nicotinic Receptors. J. Neurochem. 89 (2), 337–343. Apr. doi:10.1046/j.1471-4159.2004.02347.x
Siedlecki-Wullich, D., Miñano-Molina, A. J., and Rodríguez-Álvarez, J. (2021). microRNAs as Early Biomarkers of Alzheimer's Disease: A Synaptic Perspective. Cells 10, 113. doi:10.3390/cells10010113
Sitzmann, A. F., Hagelstrom, R. T., Tassone, F., Hagerman, R. J., and Butler, M. G. (2018). Rare FMR1 Gene Mutations Causing Fragile X Syndrome: A Review. Am. J. Med. Genetpart A. 176, 11–18. doi:10.1002/ajmg.a.38504
Sofola, O. A., Jin, P., Qin, Y., Duan, R., Liu, H., de Haro, M., et al. (2007). RNA-binding Proteins hnRNP A2/B1 and CUGBP1 Suppress Fragile X CGG Premutation Repeat-Induced Neurodegeneration in a Drosophila Model of FXTAS. Neuron 55 (4), 565–571. doi:10.1016/j.neuron.2007.07.021
Sokol, D. K., Maloney, B., Long, J. M., Ray, B., and Lahiri, D. K. (2011). Autism, Alzheimer Disease, and Fragile X: APP, FMRP, and mGluR5 Are Molecular Links. Neurology 76 (15), 1344–1352. doi:10.1212/WNL.0b013e3182166dc7
Sonenberg, N., and Costa-Mattioli, M. (2006). Translational Control of Long-Term Synaptic Plasticity and Memory Storage by eIF2α. Crit. Rev. Neurobiol. 18 (1-2), 187–195. doi:10.1615/critrevneurobiol.v18.i1-2.190
Stefani, G., Fraser, C. E., Darnell, J. C., and Darnell, R. B. (2004). Fragile X Mental Retardation Protein Is Associated with Translating Polyribosomes in Neuronal Cells. J. Neurosci. 24, 7272–7276. doi:10.1523/JNEUROSCI.2306-04.2004
Strumbos, J. G., Brown, M. R., Kronengold, J., Polley, D. B., and Kaczmarek, L. K. (2010). Fragile X Mental Retardation Protein Is Required for Rapid Experience-dependent Regulation of the Potassium Channel Kv3.1b. J. Neurosci. 30 (31), 10263–10271. doi:10.1523/JNEUROSCI.1125-10.2010
Suhl, J. A., and Warren, S. T. Single-nucleotide Mutations in FMR1 Reveal Novel Functions and Regulatory Mechanisms of the Fragile X Syndrome Protein FMRP. J. Exp. Neurosci. (2015) 9s2, JEN.S25524–41 doi: doi:10.4137/JEN.S25524
Thomas, A. M., Bui, N., Perkins, J. R., Yuva-Paylor, L. A., and Paylor, R. (2012). Group I Metabotropic Glutamate Receptor Antagonists Alter Select Behaviors in a Mouse Model for Fragile X Syndrome. Psychopharmacology 219 (1), 47–58. doi:10.1007/s00213-011-2375-4
Thomson, S. R., Seo, S. S., Barnes, S. A., Louros, S. R., Muscas, M., Dando, O., et al. (2017). Cell-Type-Specific Translation Profiling Reveals a Novel Strategy for Treating Fragile X Syndrome. Neuron 95, 550–563. doi:10.1016/j.neuron.2017.07.013
Triaca, V., and Calissano, P. (2016). Impairment of the Nerve Growth Factor Pathway Driving Amyloid Accumulation in Cholinergic Neurons: The Incipit of the Alzheimer’s Disease story?. Neural Regen. Res. 11 (10), 1553–1556. doi:10.4103/1673-5374.193224
Tsai, N.-P., and Wei, L.-N. (2010). RhoA/ROCK1 Signaling Regulates Stress Granule Formation and Apoptosis. Cell Signal. 22 (4), 668–675. doi:10.1016/j.cellsig.2009.12.001
Tsai, N.-P., Wilkerson, J. R., Guo, W., Maksimova, M. A., DeMartino, G. N., Cowan, C. W., et al. (2012). Multiple Autism-Linked Genes Mediate Synapse Elimination via Proteasomal Degradation of a Synaptic Scaffold PSD-95. Cell 151 (7), 1581–1594. doi:10.1016/j.cell.2012.11.040
Ulland, T. K., and Colonna, M. (2018). TREM2 - a Key Player in Microglial Biology and Alzheimer Disease. Nat. Rev. Neurol. 14, 667–675. doi:10.1038/s41582-018-0072-1
Utami, K. H., Yusof, N. A. B. M., Kwa, J. E., Peteri, U.-K., Castrén, M. L., and Pouladi, M. A. (2020). Elevated De Novo Protein Synthesis in FMRP-Deficient Human Neurons and its Correction by Metformin Treatment. Mol. Autism 11 (1), 41. doi:10.1186/s13229-020-00350-5
Van Driesche, S. J., Sawicka, K., Zhang, C., Hung, S. K. Y., Park, C. Y., Fak, J. J., et al. FMRP Binding to a Ranked Subset of Long Genes Is Revealed by Coupled CLIP and TRAP in Specific Neuronal Cell Types. bioRxiv,(2019) 762500; doi:10.1101/762500
Vilardo, E., Barbato, C., Ciotti, M., Cogoni, C., and Ruberti, F. (2010). MicroRNA-101 Regulates Amyloid Precursor Protein Expression in Hippocampal Neurons. J. Biol. Chem. 285 (24), 18344–18351. doi:10.1074/jbc.M110.112664
Wang, D.-D., Hou, X.-H., Li, H.-Q., Xu, W., Ma, Y.-H., Dong, Q., et al. (2020b). Association of Serum Complement C1q Concentration with Severity of Neurological Impairment and Infarct Size in Patients with Acute Ischemic Stroke. J. Stroke Cerebrovasc. Dis. 29, 105363. doi:10.1016/j.jstrokecerebrovasdis.2020.105363
Wang, H., Liu, Q., and Zhang, X. (2020a). C1q/tumor Necrosis Factor-Related Protein-1 Attenuates Microglia Autophagy and Inflammatory Response by Regulating the Akt/mTOR Pathway. Life Sci. 256, 117992. doi:10.1016/j.lfs.2020.117992
Wang, L., Almeida, L. E. F., Spornick, N. A., Kenyon, N., Kamimura, S., Khaibullina, A., et al. (2015). Modulation of Social Deficits and Repetitive Behaviors in a Mouse Model of Autism: The Role of the Nicotinic Cholinergic System. Psychopharmacology 232, 4303–4316. doi:10.1007/s00213-015-4058-z
Wang, S., and El-Deiry, W. S. (2003). TRAIL and Apoptosis Induction by TNF-Family Death Receptors. Oncogene 22, 8628–8633. doi:10.1038/sj.onc.1207232
Wang, Y. C., Lin, M. L., Lin, S. J., Li, Y. C., and Li, S. Y. (1997). Novel point Mutation within Intron 10 of FMR-1 Gene Causing Fragile X Syndrome. Hum. Mutat. 10, 393–399. doi:10.1002/(SICI)1098-1004(1997)10:5<393:AID-HUMU10>3.0.CO;2-V
Westmark, C. J., Westmark, P. R., and Malter, J. S. (2009). MPEP Reduces Seizure Severity in Fmr-1 KO Mice over Expressing Human Abeta. Int. J. Clin. Exp. Pathol. 3, 56–68. www.ijcep.com/IJCEP907006.
Westmark, C. J., and App, (2019). Fragile X and APP: a Decade in Review, a Vision for the Future. Mol. Neurobiol. 56, 3904–3921. doi:10.1007/s12035-018-1344-x
Westmark, C. J., and Malter, J. S. (2007). FMRP Mediates mGluR5-dependent Translation of Amyloid Precursor Protein. Plos Biol. 5, e52. doi:10.1371/journal.pbio.0050052
Westmark, C. J. (2013). What's hAPPening at Synapses? the Role of Amyloid β-protein Precursor and β-amyloid in Neurological Disorders. Mol. Psychiatry 18, 425–434. doi:10.1038/mp.2012.122
Wolfe, S. A., Workman, E. R., Heaney, C. F., Niere, F., Namjoshi, S., Cacheaux, L. P., et al. (2016). FMRP Regulates an Ethanol-dependent Shift in GABABR Function and Expression with Rapid Antidepressant Properties. Nat. Commun. 7, 12867. doi:10.1038/ncomms12867
Wright, C. F., Fitzgerald, T. W., Jones, W. D., Clayton, S., McRae, J. F., Van Kogelenberg, M., et al. (2015). Genetic Diagnosis of Developmental Disorders in the DDD Study: A Scalable Analysis of Genome-wide Research Data. The Lancet 385, 1305–1314. doi:10.1016/s0140-6736(14)61705-0
Wu, W.-L., Adams, C. E., Stevens, K. E., Chow, K.-H., Freedman, R., and Patterson, P. H. (2015). The Interaction between Maternal Immune Activation and Alpha 7 Nicotinic Acetylcholine Receptor in Regulating Behaviors in the Offspring. Brain Behav. Immun. 46, 192–202. doi:10.1016/j.bbi.2015.02.005
Xiao, J., Neylon, C. B., Hunne, B., and Furness, J. B. Oligophrenin-1, a Rho GTPase-Activating Protein (RhoGAP) Involved in X-Linked Mental Retardation, Is Expressed in the Enteric Nervous System. Anat. Rec. (2003). 273A:671– 676. doi: doi:10.1002/ar.a.10072
Xu, Z.-X., Kim, G. H., Tan, J.-W., Riso, A. E., Sun, Y., Xu, E. Y., et al. (2020). Elevated Protein Synthesis in Microglia Causes Autism-like Synaptic and Behavioral Aberrations. Nat. Commun. 11, 1. doi:10.1038/s41467-020-15530-3
Yasui, D. H., Scoles, H. A., Horike, S.-i., Meguro-Horike, M., Dunaway, K. W., Schroeder, D. I., et al. (2011). 15q11.2-13.3 Chromatin Analysis Reveals Epigenetic Regulation of CHRNA7 with Deficiencies in Rett and Autism Brain. Hum. Mol. Genet. 20, 4311–4323. doi:10.1093/hmg/ddr357
Yu, Z., Fan, D., Gui, B., Shi, L., Xuan, C., Shan, L., et al. (2012). Neurodegeneration-associated TDP-43 Interacts with Fragile X Mental Retardation Protein (FMRP)/Staufen (STAU1) and Regulates SIRT1 Expression in Neuronal Cells. J. Biol. Chem. 287, 22560–22572. doi:10.1074/jbc.M112.357582
Zalfa, F., Adinolfi, S., Napoli, I., Kühn-Hölsken, E., Urlaub, H., Achsel, T., et al. (2005). Fragile X Mental Retardation Protein (FMRP) Binds Specifically to the Brain Cytoplasmic RNAs BC1/BC200 via a Novel RNA-Binding Motif. J. Biol. Chem. 280 (39), 33403–33410. doi:10.1074/jbc.M504286200
Zalfa, F., Eleuteri, B., Dickson, K. S., Mercaldo, V., De Rubeis, S., di Penta, A., et al. (2007). A New Function for the Fragile X Mental Retardation Protein in Regulation of PSD-95 mRNA Stability. Nat. Neurosci. 10, 578–587. doi:10.1038/nn1893
Zhang, M., Chen, D., Xia, J., Han, W., Cui, X., Neuenkirchen, N., et al. (2017). Post-transcriptional Regulation of Mouse Neurogenesis by Pumilio Proteins. Genes Dev. 31 (13), 1354–1369. doi:10.1101/gad.298752.117
Zhang, P., Abdelmohsen, K., Liu, Y., Tominaga-Yamanaka, K., Yoon, J.-H., Ioannis, G., et al. (2015). Novel RNA- and FMRP-Binding Protein TRF2-S Regulates Axonal mRNA Transport and Presynaptic Plasticity. Nat. Commun. 6, 8888. doi:10.1038/ncomms9888
Zhang, T., Pang, P., Fang, Z., Guo, Y., Li, H., Li, X., et al. (2018). Expression of BC1 Impairs Spatial Learning and Memory in Alzheimer's Disease via APP Translation. Mol. Neurobiol. 55, 6007–6020. doi:10.1007/s12035-017-0820-z
Zhang, Y., Gaetano, C. M., Williams, K. R., Bassell, G. J., and Mihailescu, M. R. (2014). FMRP Interacts with G-Quadruplex Structures in the 3'-UTR of its Dendritic Target Shank1 mRNA. RNA Biol. 11, 1364–1374. doi:10.1080/15476286.2014.996464
Zhong, J., Chuang, S.-C., Bianchi, R., Zhao, W., Paul, G., Thakkar, P., et al. (2010). Regulatory BC1 RNA and the Fragile X Mental Retardation Protein: Convergent Functionality in Brain. PloS one 5, e15509. doi:10.1371/journal.pone.0015509
Keywords: FMRP, RNA binding protein, RNA metabolism, APP mRNA, Alzheimer disease, fragile X syndrome
Citation: Bleuzé L, Triaca V and Borreca A (2021) FMRP-Driven Neuropathology in Autistic Spectrum Disorder and Alzheimer's disease: A Losing Game. Front. Mol. Biosci. 8:699613. doi: 10.3389/fmolb.2021.699613
Received: 23 April 2021; Accepted: 24 August 2021;
Published: 25 October 2021.
Edited by:
Rossella Di Giaimo, University of Naples Federico II, ItalyReviewed by:
Maria Giuseppina Miano, Institute of Genetics and Biophysics (CNR), ItalyCarolina Cefaliello, University of Massachusetts Medical School, United States
Copyright © 2021 Bleuzé, Triaca and Borreca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viviana Triaca, Viviana.triaca@cnr.it; Antonella Borreca, antonella.borreca@gmail.com, antonella.borreca@cnr.it