 Liena E. O. Elsayed
Liena E. O. Elsayed Isra Zuhair Eltazi
Isra Zuhair Eltazi Ammar E. Ahmed2
Ammar E. Ahmed2 Giovanni Stevanin
Giovanni Stevanin- 1Department of Basic Sciences, College of Medicine, Princess Nourah bint Abdulrahman University [PNU], Riyadh, Saudi Arabia
- 2Faculty of Medicine, University of Khartoum, Khartoum, Sudan
- 3Institut du Cerveau – Paris Brain Institute - ICM, Sorbonne Université, INSERM, CNRS, APHP, Paris, France
- 4CNRS, INCIA, Université de Bordeaux, Bordeaux, France
- 5Ecole Pratique des Hautes Etudes, EPHE, PSL Research University, Paris, France
Hereditary spastic paraplegias (HSP) are a heterogeneous group of motor neurodegenerative disorders that have the core clinical presentation of pyramidal syndrome which starts typically in the lower limbs. They can present as pure or complex forms with all classical modes of monogenic inheritance reported. To date, there are more than 100 loci/88 spastic paraplegia genes (SPG) involved in the pathogenesis of HSP. New patterns of inheritance are being increasingly identified in this era of huge advances in genetic and functional studies. A wide range of clinical symptoms and signs are now reported to complicate HSP with increasing overall complexity of the clinical presentations considered as HSP. This is especially true with the emergence of multiple HSP phenotypes that are situated in the borderline zone with other neurogenetic disorders. The genetic diagnostic approaches and the utilized techniques leave a diagnostic gap of 25% in the best studies. In this review, we summarize the known types of HSP with special focus on those in which spasticity is the principal clinical phenotype (“SPGn” designation). We discuss their modes of inheritance, clinical phenotypes, underlying genetics, and molecular pathways, providing some observations about therapeutic opportunities gained from animal models and functional studies. This review may pave the way for more analytic approaches that take into consideration the overall picture of HSP. It will shed light on subtle associations that can explain the occurrence of the disease and allow a better understanding of its observed variations. This should help in the identification of future biomarkers, predictors of disease onset and progression, and treatments for both better functional outcomes and quality of life.
Introduction
Hereditary spastic paraplegias (HSP) are a group of heterogeneous neurodegenerative disorders which are characterized by an insidiously progressive weakness and spasticity of the lower extremities as their core defining clinical features. They constitute the second most frequent motor neuron disease (MND) with an estimated prevalence of 3–10/100,000 in most populations (Noreau et al., 2014). HSPs are perfect models for understanding current challenges in inherited neurodegenerative disorders, as they overlap genetically with different neurogenetic diseases, although they were previously considered as completely separate entities (Harding, 1983).
Classification of HSP
Classical classification of HSP was historically based on the clinical phenotype and the mode of inheritance (Harding, 1983). Recently, there has been an increasing tendency to involve the underlying molecular pathogenetic mechanisms in the categorization of HSP with multiple trials to establish classifications and groupings of identified HSP forms/genes based on the affected function (Lo Giudice et al., 2014; Noreau et al., 2014; Vallat et al., 2016; de Souza et al., 2017; Blackstone, 2018; Boutry et al., 2019a; Darios et al., 2020; Darios and Stevanin, 2020). More recently, machine-learning approaches were also used in their clinico-genetic stratification (Vavouraki et al., 2021). This group of diseases suffers from the fact that it gathers diseases in which spasticity/pyramidal signs are sometimes only part of the clinical phenotype such as some neurodevelopmental disorders due to AP4n genes mutations, and, at the other end of the spectrum, disorders in which the spasticity/pyramidal signs are the core and sometimes the only symptoms in patients, such as most SPG4 and SPG8 patients. In addition, spasticity and pyramidal signs are often observed in almost all neurogenetic conditions at some point of the disease evolution. A nosology restricted to HSP disorders will then be inefficient and a new nosology of all neurogenetic conditions is then necessary to take into account these now common observations in almost all neurogenetic conditions (Cassis et al., 2015; Synofzik and Schüle, 2017; Elsayed et al., 2019b).
All current classifications are therefore far from optimum, and are just used to give a clue about the disease and its clinical presentation and molecular basis.
The Landscape of HSP Genetics
HSP shows remarkable genetic and clinical heterogeneity and is by far the most heterogeneous condition together with peripheral neuropathies. It is transmitted via all classical modes of monogenic inheritance which include autosomal dominant (AD), autosomal recessive (AR), recessive or dominant X-Linked (X-L), and mitochondrial/maternal transmission. The prevalence of AD HSP varies between 0.5 and 5.5/100,000 whereas that of the AR HSP ranges from 0.3 to 5.3/100,000 in hospital-based studies and 0.6 to 2.6/100,000 in multisource studies (Ruano et al., 2014). X-linked and mitochondrial inheritance are rare and mainly affect congenital cases; this is particularly the case with X-linked inheritance.
While AD HSP is more frequent in northern Europe, AR HSP predominates in highly consanguineous populations with one of the highest values reported in Tunisia, where it is probably attributed to the high rates of consanguinity in Northern Africa (Ben Hamida et al., 1986; Boukhris et al., 2008, 2009; Ruano et al., 2014).
To date, more than 100 loci/88 genes are known to be implicated in the pathogenesis of HSP. Of those, 83 clinical-genetic forms have the designation spastic paraplegia gene “n” or SPGn. However, there are more than 25 genes that have been reported to cause HSP but not assigned an SPG designation including the four mitochondrial genes (MTND4, MTTI, MTCO3, MTATP6) that were associated with HSP/HSP-like phenotypes. In 12 of the 83 SPG forms, the underlying gene/protein is not yet identified (Klebe et al., 2015; Tesson et al., 2015; de Souza et al., 2017; Boutry et al., 2019a).
Consanguinity has been of great help in identifying HSP causative genes since only 17 HSP subtypes show AD inheritance whereas the number of identified AR HSP subtypes is progressively increasing mounting to 53 HSP forms. X-L inheritance is relatively rare being reported in five forms only, all with recessive inheritance. SPG4 is the commonest AD HSP form in many populations, particularly in Caucasians. It accounts for 17–79% of AD HSP cases with monoallelic forms of KIF1A variant carriers, SPG3A and SPG31 coming next (Schüle et al., 2016; Burguez et al., 2017; Dong et al., 2018; Boutry et al., 2019a; Méreaux et al., 2021b). The collective frequency of SPG4 together with SPG3A amounted to 90% of the AD HSP in some series (Dong et al., 2018). On the other hand, SPG11 and SPG7 are the most frequent AR HSP subtypes followed by SPG15 and SPG5 (Ruano et al., 2014; Tesson et al., 2015; Boutry et al., 2019b; Coarelli et al., 2019; Du, 2021).
HSP can also be caused by mutations in “non-SPG” genes. The majority of the “non-SPG” forms have an original phenotype and HSP stands as an allelic presentation, frequently reported in a single family (Table 1). Although the non-SPG forms will be included in multiple sections of this article, the focus will be on the 83 SPG forms.
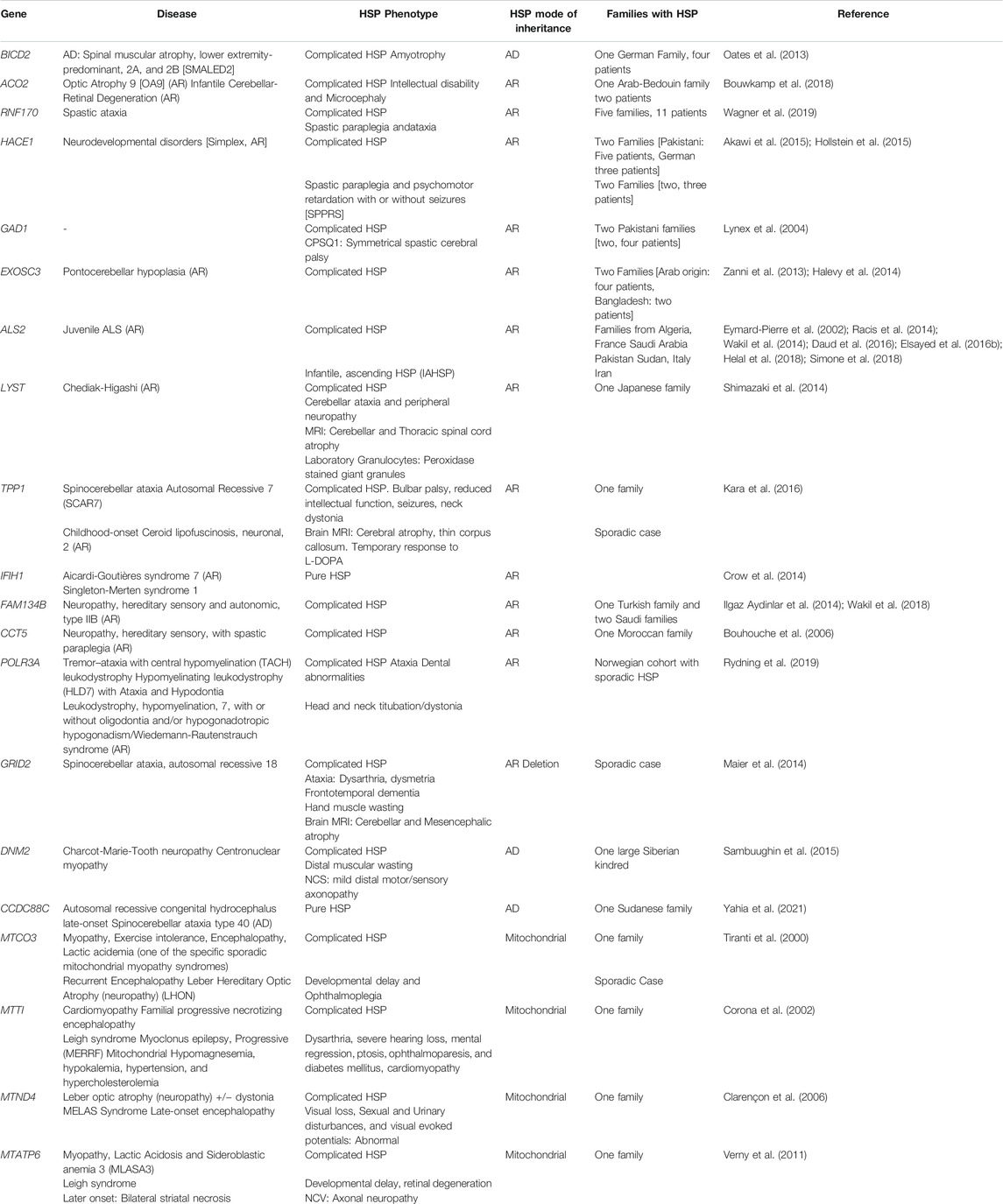
TABLE 1. Table summarizing HSP forms associated with non-SPG genes and the main allelic phenotypes associated with causative variants in these genes.
Over the last decade, and with the advent of next-generation sequencing (NGS)-based techniques, the genetic basis of HSP appears more complicated than previously thought. HSP forms with mixed inheritance (AD/AR) modes have been observed in six HSP forms. Moreover, a subtle mixed inheritance pattern with one dominating mode of inheritance and probable allele-dose-dependent variability in the expressed clinical phenotype was also demonstrated in some forms, adding to the genetic heterogeneity, and contributing to the complexity, of the whole picture. An example of the allele-dose-dependent variability was clear in the Kinesin encoding gene KIF1C, known to cause HSP type 58 (SPG58). KIF1C was shown to present with mild or subclinical dominant phenotype in heterozygous carriers, with the same mutations that resulted in more severe recessive phenotypes in their homozygous states (Oteyza et al., 2014). Other examples among the frequent HSP forms are related to recessive and dominantly inherited variants in SPG7 and KIF1A (Klebe et al., 2012a; 2012b) with limited phenotype-genotype correlations (Coarelli et al., 2019; Pennings et al., 2020).
Genetic Diagnosis: Approaches, Yield and Gap in Genetic Etiology
Despite the advances in genetic studies in the last two decades that resulted in a remarkable improvement of the genetic diagnostic yield, it seems that we are just seeing the tip of the iceberg. The overall diagnostic gap in HSP is currently estimated to be 40–70% on average with a smaller gap of 25–40% reported in some studies with specific ethnicities (Novarino et al., 2014; Morais et al., 2017; Bis-Brewer and Züchner, 2018; Koh et al., 2018) despite the use of combined approaches. The diagnostic yield seems to vary based on many factors that are not all well characterized. However, the approach used for genetic testing, the bioinformatic pipelines used in data analysis, the availability of DNA of other members of the family to confirm genotype-phenotype co-segregation, all seem to play roles. The types of the families included in the cohort [AD, AR or simplex/sporadic] also affect the genetic diagnostic capacity with lower yields obtained in cohorts of simplex cases. However, contradicting values have been observed (Schüle et al., 2016; Bis-Brewer and Züchner, 2018). The underlying genetic cause itself, and particularly when it is not easily analysed (novel unknown genes, variants in non-coding regions, oligogenicity), likely plays an additional role in lowering the genetic yield, together with the approach used, and its suitability to detect these types of genetic causes (Bis-Brewer and Züchner, 2018; Koh et al., 2018). Not only do these factors work individually, but also sometimes their combined interaction may result in failure to reach the genetic diagnosis in a family, and thus contributing to the genetic diagnostic gap in a cohort. The gap in the diagnostic yield of genetic studies using whole exome sequencing (WES), with the majority of non-coding region uncovered, is being gradually highlighted as exampled by the deep intronic POLR3B variant which was identified as a cause of spastic ataxia, then later being shown to account for around 3.1% of genetically unclassified AR and sporadic cases (Bis-Brewer and Züchner, 2018). This might also suggest that the heterogeneity of the phenotype can be due to, or, modified by, an unexplored non-coding region. This may question many of the results that we have about the underlying pathophysiologic mechanisms to date. NGS-based gene panels tailored to spastic disorders, movement disorders or Mendelian disorders (clinical exomes), are currently preferred for candidate gene screening approach (Méreaux et al., 2021b). However, occasionally Multiplex Ligation-dependent Probe Amplification (MLPA) and Sanger sequencing for selected genes, are performed to exclude the commonest genes causing HSP before using the NGS-based techniques. The broader the spectrum of genes covered, the higher the opportunity to identify novel HSP genes presenting with novel allelic phenotypes (Table 1). This is especially important with the remarkable overlap demonstrated in neurogenetic disorders lately (Synofzik et al., 2014; Chrestian et al., 2016; Kara et al., 2016; van de Warrenburg et al., 2016; Morais et al., 2017; Synofzik and Schüle, 2017; Elsayed et al., 2019b; Elert-Dobkowska et al., 2019). The genetic diagnostic yield using targeted gene panels and clinical exomes ranges from ≈15 to 30% when known common HSP genes have been excluded at a provisional screening. However, the yield steps up and may reach 50% or more when the panels/clinical exomes are used as the primary screening tool with common HSP genes included in them (Burguez et al., 2017; Morais et al., 2017; Elert-Dobkowska et al., 2019; Méreaux et al., 2021b). WES, and more recently genome sequencing, on the other hand, provides a better diagnostic yield of 30–60% (Novarino et al., 2014; Kara et al., 2016; Schüle et al., 2016; Bis-Brewer and Züchner, 2018). The diagnostic gap further decreases when using combined approaches tailored to the nature of the cohort (Kara et al., 2016; Koh et al., 2018). The search for rearrangements, particularly in SPAST and SPG11, using analysis of coverage in NGS-based techniques must be performed as such mutational mechanisms are relatively frequent (Méreaux et al., 2021b). Systematic reanalysis of negative panels or exomes should also be performed regularly as alignment and base-calling are constantly improving.
Very few reports have found phenotype-genotype correlations which elicit the impact of patient’s gender, the affected gene, and the nature of the mutation, in addition to other factors including ethnicity and genetic background. The impact of the patients’ gender in the phenotypic presentation has been pinpointed in a meta-analysis of 147 studies on HSP including nine of the most frequent AD HSP and AR HSP subtypes. This identified male predominance in certain subtypes [SPG7 (AR HSP) and SPG31 (AD HSP)] (Erfanian Omidvar et al., 2019). This was observed previously in SPG4 as well (Orlacchio et al., 2005b). Interestingly, a recent study identified significant lower penetrance in SPG4 female patients (Parodi et al., 2018b). Moreover, age at onset was shown to be modified by factors including ethnicity, the disease-causing genes, and the nature of the causative mutation itself (Parodi et al., 2018b; Newton et al., 2018; Erfanian Omidvar et al., 2019; Rodrigues et al., 2019). Missense mutations were found to be associated with earlier onset in SPG4 patients while the p.Ala510Val variant in SPG7 was found to be associated with a later disease onset and a more ataxic clinical presentation (Parodi et al., 2018b; Coarelli et al., 2019). Similarly, missense mutations in SPG11 are often associated with a later onset (Rubegni et al., 2015). Genetic modifiers have been reported to explain part of the variability in clinical presentation. Examples include the p.Ser44Leu and p.Pro45Gln variants in SPAST mutation carriers (Svenson et al., 2004) which were shown to have a real functional impact on spastin protein stability (Schickel et al., 2007; Pantakani et al., 2008). The p.Ser44Leu variant in SPAST was first reported as a cause of mild disease presentation then turned out to be a rare polymorphism acting as a phenotypic modifier (Lindsey et al., 2000; Svenson et al., 2004; Erichsen et al., 2007). Another example is the effect of deletions of SPAST that can sometimes extend to the nearby gene, DPY30, affecting the age at onset of SPG4 patients. This has particular importance as these deletions are relatively frequent in SPG4 (Beetz et al., 2006; Depienne et al., 2007; Newton et al., 2018). Finally, somatic mosaicism was shown to be a potential source of variant expressivity in SPAST mutation carriers (Angelini et al., 2021). However, the reduced penetrance of several variants in HSP genes still remains to be explained with the nature of the variants, their impact on protein expression/stability, the presence of compensatory protein or partners yet to be explored. This is apparently not yet the whole explanation of the great inter- and intra-familial variability of HSP patients especially with the digenic/oligogenic inheritance which is now emerging as a cause of a number of neurogenetic disorders including amyotrophic lateral sclerosis (ALS) and epilepsy. However, these models may be altered by the type of mutations as well as by the frequency of the suggested modifiers in the populations, and this might explain the effect of ethnicity and might also indicate an indirect effect of the environment over genetics (Kousi and Katsanis, 2015; Bis-Brewer and Züchner, 2018; Amin et al., 2021). Numerous studies have also suggested an impact of epigenetics in Parkinson’s, Alzheimer’s, Huntington’s diseases, cerebellar ataxias (CA), and ALS (Chestnut et al., 2011; Desplats et al., 2011; Chouliaras et al., 2013; Goodnight et al., 2019; Haertle et al., 2019; Jia et al., 2019; Lardenoije et al., 2019). A role for epigenetics in apoptosis was suggested in motor neuron pathology in ALS, with the upregulation of DNA methylase increasing DNA methylation which results in apoptosis (Chestnut et al., 2011). The role of epigenetics has also been studied in synaptic plasticity, in which decreased levels of DNA methylases, with associated synaptic degeneration, was observed in aging and degenerative disorders (Desplats et al., 2011; Chouliaras et al., 2013; Xylaki et al., 2019). However, the role of epigenetics in the HSP field, and its impact on the “expression” of the mutation, is still unclear. Identification of the factors affecting clinical presentation require international collaborative efforts in order to make this quest feasible in such rare genetic entities (Bis-Brewer and Züchner, 2018).
Ethnic differences manifest, at many levels, in the frequency of genes causing HSP, the predominant mode/s of inheritance, and the phenotypic presentation of genes. These differences reflect underlying variations which include the level of consanguinity, the ethnic genetic background, and the interaction with the environment, from which the effect of epigenetic modification of gene expression cannot be excluded. Many other factors involved are not yet well characterized. Multiple examples of these ethnic differences have been reported. For example, in many Western cohorts, KIF5A mutations (SPG10) account for about 10% of the complicated forms of AD HSP. Interestingly the first Asian (Japanese) case with a KIF5A mutation was identified only recently and broadened the clinical and electrophysiological phenotypic spectrum of KIF5A (Kaji et al., 2016). Remarkable examples also include the AR HSP forms that were first identified in highly consanguineous populations such as the KIF1A variants which were first identified in African and Palestinian families, and subsequently reported in a multitude of families from all over the world (Vecchia et al., 2021). On many occasions, genes which were the solely identified in some ethnic groups turned out later to be quite frequent causes of AR HSP; for example SPG11 and SPG15 (Boukhris et al., 2009). The impact of these phenomena needs to be carefully considered in many regions of the world, where there are not enough facilities for advanced genetic research. Interesting phenotypes and underlying genetics can be identified in these populations if studies of HSP and other neurogenetic disorders are performed using scientific methods properly adapted to the population under investigation. This has been illustrated in the infrequent neurogenetic studies that included families or cohorts from the exceedingly consanguineous African, Middle Eastern and Asian countries. Examples include Libya, Iraq, Syria, Egypt, Saudi Arabia, Sudan, Oman, Iran among others (Sridharan et al., 1985; Rainier et al., 2003; Abou Jamra et al., 2011; Alazami et al., 2011; Novarino et al., 2014; Elsayed et al., 2016a, 2016b, 2018; Ahmed et al., 2017; Yahia et al., 2018, 2021; Farazi Fard et al., 2019). The studies that included cohorts from these unexplored areas are increasing in number and despite their relative sparsity, they have contributed remarkably to the existing knowledge about HSP and other neurogenetic disorders. A good illustration is the work by Novarino et al. which reported 18 novel genes in a large set of consanguineous families from Middle East and North-Africa (Novarino et al., 2014). They illustrated the strong potential of digging into these virgin fields. Moreover, in many countries in the less developed regions of the world that are entirely unexplored from a genetic point of view, epidemiological data about most of genetic diseases are not available. This gives a hint about the fact that many of the current statistical estimates are inherently untrue. These population-based differences, highly stressed in recent research, show clearly that the overall portrait of the phenotypic presentation, the mechanisms and molecular pathways involved in the causation and pathogenicity of HSP, and the resulting cellular adaptations to the disease, will never be completely well-characterized if this gap is not overcome.
Clinical Spectrum of HSP
Assessment and Diagnostic Approach of HSP Patients
The pivotal point that strongly suggests HSP is positive family history. However, HSP remains a diagnosis of exclusion in sporadic cases, where a number of other conditions such as HTLV1 infection, food intoxication, malformations, and cancer must be excluded through appropriate investigation. In particular, the assessment of the patient requires accurate history and clinical examination using the Spastic Paraplegia Rating Scale to assess the severity of the motor symptoms (Schüle et al., 2006). However, a variety of disability scores are also used for assessment of the patient’s disability stage. Further assessment including radiological, electrophysiological and laboratory testing, will give additional evidence on the nature of the disease and help to eliminate important differential diagnoses. The principal radiological assessments utilized are magnetic resonance imaging (MRI) of the brain and spinal cord. While MRI of the spine does not show significant abnormalities most of the time, brain MRI on the contrary (or sometimes computerized tomography (CT) scan of the brain) can give critical information that can assist in the exclusion of various differential diagnoses such as leukodystrophies and leukoencephalopathies, neurodegeneration with brain iron accumulation, ALS, or infectious and dysimmune causes. In addition, it will provide direction to the category of HSP (eg. HSP with thin corpus callosum (TCC)) and probable genes beyond it with the prominent example of SPG11 being the most likely gene in TCC HSP. However, the question of the exclusion of leukodystrophies is now debated in light of the overlap of several HSP forms with this group of diseases.
Electrophysiological tests include electromyogram/nerve conduction studies (EMG/NCS), evoked potentials (somatosensory [SEP], visual, and auditory). These studies provide ample information about the underlying pathology and the clinical presentation of the patient resulting in both better diagnostic yield and delineation of the clinical spectrum of the disease phenotype.
Associated Signs and Clinical Phenotypes
There is vast heterogeneity of the phenotypes associated with HSP. In this review, we derived clinical associations that may aid in a better understanding of HSP as a model of inherited neurodegenerative disorder. The core clinical presentation of patients is well-known to be with pyramidal syndrome which consists of increased muscle tone (spasticity with scissoring and clonus in its most severe form), hyperreflexia and extensor plantar response (Babinski sign). Classically, HSP can be pure (uncomplicated) or complex (complicated) according to the absence or presence of additional neurological and extra-neurological manifestations but this distinction has most of the time no relation to the genetic etiology. Indeed, with the recent surge of genetic data, and the expansion of the associated clinical picture of several mutated genes, some forms were found to manifest with a wider clinical presentation ranging from pure to complex, with differences sometimes within the same family. Moreover, some HSP genes were linked to multiple allelic presentations that included even non-HSP phenotypes (Elsayed et al., 2019a; 2019b). Pure forms can also present with clinical features suggesting involvement of the dorsal column, with diminished or even abolished vibration sense. Sphincter involvement (principally urinary urgency) can occur in pure HSP too due to increased bladder muscle tone (Fink, 1993, 2014; Finsterer et al., 2012; Lo Giudice et al., 2014; Klebe et al., 2015; Tesson et al., 2015; de Souza et al., 2017; Boutry et al., 2019b; Elsayed et al., 2019b). Complicating neurological features include most frequently deterioration of the cognitive/mental functions (>46 SPG forms). The cognitive function shows variable levels of alteration with the AR complicated HSP forms having a higher degree of impairment. A recent large meta-analysis study found no report of cognitive disability in SPG3A patients although it was often reported in SPG4, the two AD HSP forms that were initially thought to be mostly pure. However, the same meta-analysis study found that the frequency of intellectual disability in AR HSP patients to be highest in SPG54 (89%) followed by SPG11 (86%), SPG15 (78%), SPG35 (71%), and SPG7 (8%) respectively (Erfanian Omidvar et al., 2019). The cerebral palsy-like HSP forms associated with mutations in genes encoding members of the AP4 complex (SPG47, SPG50, SPG51, SPG52) have been associated with severe cognitive impairment in agreement with their role in vesicular trafficking during the development of the central nervous system. All mutations reported in literature in the four genes - AP4B1, AP4M1, AP4E1, AP4S1 - were loss of function variants except for a missense in AP4M1 gene, that was reported in a Greek family by Bettencourt et al., in which there was compound heterozygosity with a loss of function mutation in one patient (Verkerk et al., 2009; Abou Jamra et al., 2011; Moreno-De-Luca et al., 2011; Bauer et al., 2012; Tüysüz et al., 2014; Abdollahpour et al., 2015; Hardies et al., 2015; Bettencourt et al., 2017; Jacinto-Scudeiro et al., 2019). Cognitive impairment is almost a universal feature in all complicated X-L recessive HSP forms which may suggest that it should be considered in the differential diagnosis in the cases of developmental delay and cognitive impairment in males. However, the reliability of these observations is doubtful as X-L HSP is rare and only a few families have been reported. Adducted thumbs are probably of better help in gene prognosis, at least in L1CAM mutation carriers.
Peripheral neuropathy or amyotrophy (>40 HSP forms) is the second most frequent complicating feature, followed by cerebellar signs (>37 HSP subtypes), and eye signs (cataract, retinal/macular degeneration, strabismus, and optic atrophy) taken collectively (≈22 forms). Clinical presentation can be further complicated by bulbar or pseudobulbar palsy, psychiatric symptoms, auditory neuropathy, extrapyramidal signs, stereotypic laughter, or epilepsy (Table 2). Because of the frequent association of CA with the spasticity and the frequent occurrence of pyramidal signs in patients with CA as the main complicating clinical symptom, the concept of ataxia-spasticity spectrum emerged. Patients with pyramidal or cerebellar signs can present with a phenotypic presentation varying along a continuum of clinico-genetic entities from pure CA, to spastic ataxia and to pure HSP. Spastic ataxias refer to disorders in which most patients have a clinical presentation with equal contribution of pyramidal and cerebellar involvement such as in spastic ataxia of Charlevoix-Saguenay, and in SPG7, SAX2, SPG43 and RNF170 mutation carriers ((Synofzik and Schüle, 2017). The list of clinical entities designated a spastic ataxia is progressively broadening with nine syndromes given the designation of spastic ataxia “n” [SPAX “n”] to date.

TABLE 2. Grouping of various HSP forms based on the clinical presentation.
Brain, and to a lesser extent spinal cord, imaging abnormalities are frequent. MRI of the brain may show atrophy (generalized or focal: atrophy of the cerebral cortex and/or the cerebellum, and thin corpus callosum), features of dysmyelination (hypo- and de-myelination), leukodystrophic changes (white matter hyper signal intensity lesions), and rarely brain malformations. Although normal MRI is frequently encountered, abnormal radiological features are common and can be found in combinations or one at a time with great variability (Figure 1). It is interesting how the list of radiological features associated with HSP is constantly broadening as well. Recent reports detected MRI features mimicking the “eye of a tiger” sign, most commonly associated with pantothenate kinase e-associated neurodegeneration (PKAN), in SPG7 and the closely related spastic ataxia 5 (AFG3L2-mutated) patients (Calandra et al., 2020; Rizzo et al., 2020).
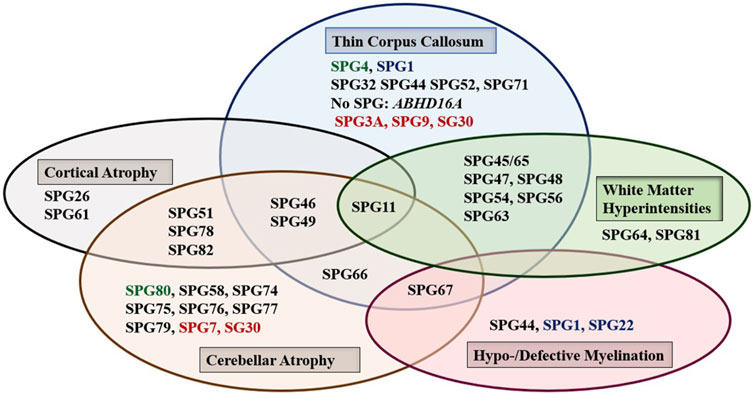
FIGURE 1. The main abnormal findings in MRI of the brain in different HSP subtypes. Font color codes correspond to various patterns of inheritance: AD HSP (green), AR HSP (violet), X-linked recessive (Blue), and Mixed AR/AD inheritance (red).
Extra-neurologic organs/systems involved include the eyes (cataract, strabismus), as well as the skeletal system, the heart, the skin, the hair and the gastrointestinal system.
The rapidly expanding genetic heterogeneity has led to an additional expansion of phenotypic heterogeneity, with the list of the reported complicating neurological and extra-neurological clinical features significantly expanding also. The spectrum of the HSP phenotype has broadened extensively to include further atypical disease presentations that were not reported previously in the earlier HSP cohorts (Harding, 1983) (Table 2). Non-exhaustive illustrative examples of atypical HSP presentations that make it worth reconsidering our working case-definition include tetraplegic cerebral palsy forms associated with AP4 complex mentioned earlier (Verkerk et al., 2009; Abou Jamra et al., 2011; Bauer et al., 2012), HSP associated with hyperbilirubinemia and persistent vomiting caused by hiatus hernia (SPG29) (Orlacchio et al., 2005a), atypical HSP presentation with areflexia, apnoea, hypoventilation, ventriculomegaly, and gastro-oesophageal reflux disease in association with SPG49/TECPR2 (Oz-Levi et al., 2012, 2013), and HSP with cutis laxa and cataract (SPG9) (Coutelier et al., 2015, 2016). On the other hand, some disorders which were considered as subtypes of HSP by the earlier HSP classification suggested by Harding in 1983, such as Sjögren-Larsson syndrome and Spastic Ataxia of Charlevoix Saguenay, are no longer classified as HSP in the recent classifications of neurogenetic disorders (Harding, 1983; Vallat et al., 2016; Bindu, 2020).
In addition to the above-mentioned examples, another dimension is highlighted by the phenotypes associated with the mitochondrial genes. MT-ATP6 and MT-TI cause an HSP-like phenotype sometimes associated with cardiomyopathy (Verny et al., 2011), whilst MT-CO3 has been linked to HSP but with Leigh syndrome-like lesions in the brain (Tiranti et al., 2000).
The greatest part of clinical heterogeneity can be attributed to the AR HSP forms which mainly present as complicated HSP in contrast to the tendency of AD HSP for pure, or relatively pure clinical presentations (Fink, 1993; Finsterer et al., 2012; Lo Giudice et al., 2014; Klebe et al., 2015; Lynch et al., 2015; Tesson et al., 2015; Elsayed et al., 2016b; de Souza et al., 2017; Boutry et al., 2019b); the more patients that are reported however, the more complex forms that are found. SPG4 and SPG11/SPG7 are the most frequent pure and complex HSP forms, respectively (Travaglini et al., 2018). These represent the commonest causes of AD HSP and AR HSP respectively in most reports (Ruano et al., 2014; Blackstone, 2018; Boutry et al., 2019a). AD HSP forms with complex presentation tend to be at low level of complexity with one or two complicating signs that are usually limited to peripheral neuropathy and amyotrophy except for SPG9, SPG29 and to a lesser extent SPG80 which show high level of complexity (Orlacchio et al., 2005a; Coutelier et al., 2015, 2016; Farazi Fard et al., 2019) (Figure 1; Table 2). On the other hand, the relatively rare X-L recessive HSP forms tend to present with complicated phenotypes except for SPG34, which was reported only in the pure form, and SPG16, which was found in pure and complex HSP forms.
Age at Onset and Disease Progression
Age at onset of HSP is widely variable ranging from birth in some HSP subtypes to more than 40 years. In some rare cases the disease can have rather a late onset up to 76 years (Parodi et al., 1993; 2018a). The variability of age at onset does not only occur between the different forms but can also be observed within single HSP forms and even within families in patients carrying the same mutations (Figure 2). This is particularly the case in dominant forms (Figure 2) where heterogeneity is complicated by the incomplete penetrance. A meta-analysis study including a large set of HSP studies illustrated a significant association of ethnicity with differences in the age at onset in some HSP forms (Erfanian Omidvar et al., 2019), suggesting the influence of the genetic backgound. This study also identified earlier mean age at onset in ATL1 mutated patients relative to patients with other AD HSP subtypes [SPG4 and SPG31] caused by mutations in SPAST and REEP1. Among the AR HSP subtypes included in the analysis, SPG35 showed a tendency for younger age at onset (Erfanian Omidvar et al., 2019).
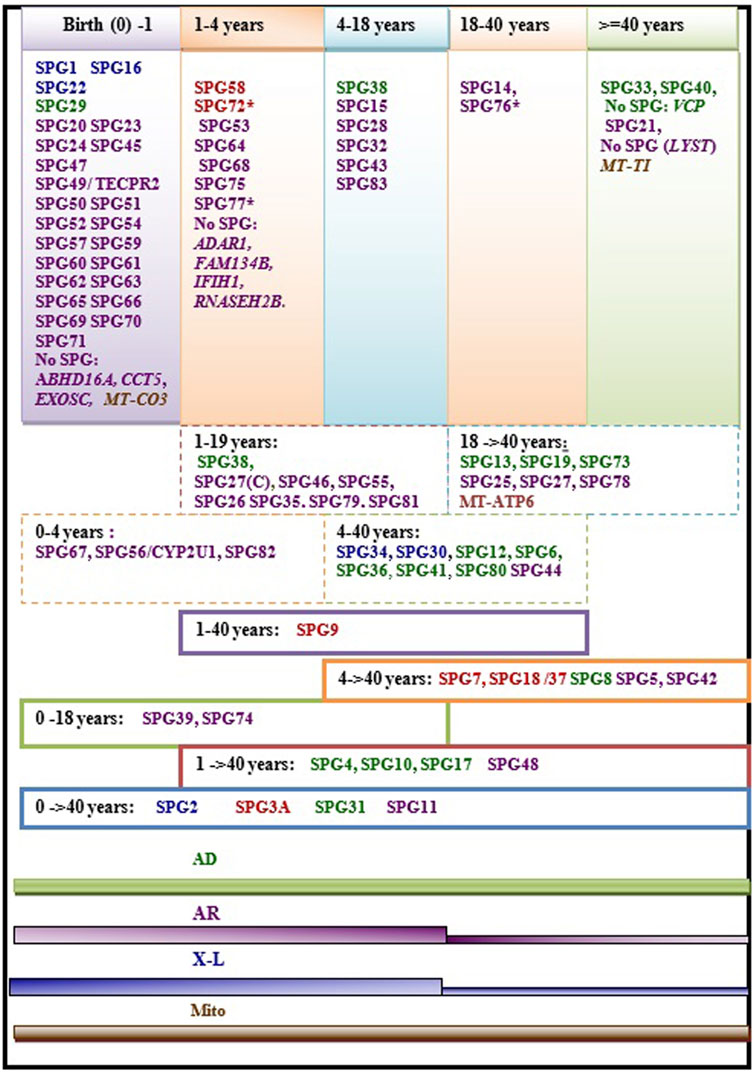
FIGURE 2. Regrouping of the age at onset of various HSP subtypes. The asterisk (*) indicates few exceptions in the age at onset range: SPG76*[one case is congenital] SPG72* and SPG77* has an age at onset range between (1–5 years)], SPG27 stands for the pure form and SPG27C for the complex form of SPG27. Font color codes represent the modes of inheritance: AD HSP (green), AR HSP (violet), X-linked recessive (Blue), Mitochondrial (brown), and Mixed AR/AD inheritance (red).
It is often difficult to assess the age at onset in a precise way in HSP. This can be sometimes attributed to the subtlety of the presenting motor symptoms, especially in complex forms. In the pure HSP forms, several patients do not complain of the disease but clinical signs may be detected at examination (Hedera, 1993; Finsterer et al., 2012; Lo Giudice et al., 2014; Klebe et al., 2015; Tesson et al., 2015; de Souza et al., 2017; Boutry et al., 2019b). This may result in both underestimation and overestimation of the incidence and the prevalence of HSP.
We found that the age at onset of AR HSP tends to cluster in childhood in 80% (≈45 forms) of the genetic forms; however, two of the most frequent forms have relatively late onset (SPG5 and SPG7). More than 34 AR HSP forms together with three X-L recessive subtypes occur in toddlers (<5 years). AD forms do not show analogous clustering but tend to have variable age at onset. Overall, several HSP subtypes show considerable variability. Of these subtypes, with widely variable age at onset, are five AD HSP subtypes [including three of the commonest AD HSP forms (SPG4, SPG3A, SPG10)], two AR HSP (SPG11 and SPG48) and an X-L recessive HSP (SPG2) (Figure 2). In addition to these variations observed in the age at onset among different forms with different modes of inheritance, two large studies found a significant difference in age at onset based on the nature of the mutation in SPG4 patients, with earlier onset in patients with missense compared to those with truncating variants (Parodi et al., 2018b; Rodrigues et al., 2019).
Most HSP subtypes are slowly progressive, particularly at the start of the disease before they reach a static plateau without improvement (Hedera, 1993; Klebe et al., 2015; de Souza et al., 2017; Boutry et al., 2019a). However, substantial variability exists regarding the disease severity and its progress. Several forms deteriorate rapidly, while many subtypes show very slowly progressive course of disease so that they can be fairly considered as non-progressive. Surprisingly, late onset forms are more often associated with a more rapidly progressive evolution. This observation was supported by analysis of large HSP patient series (Schüle et al., 2016).
Pathophysiology of HSP
Basic Pathophysiologic Mechanisms: HSPs Are Mainly Due to Neuronal Degeneration
The core phenotypic features of HSP result from axonal degeneration of neurons of the pyramidal motor system which is responsible for the voluntary movements in humans. The neurons of the pyramidal tracts extend from the layer V of the cerebral motor cortex and, after synaptic connection in the spinal cord with the secondary motor neuron, finally innervate the skeletal muscles at the neuromuscular junctions. Neurons are the principal category of cells that degenerate in HSP, even though the contribution of oligodendrocytes and other glial cells to the pathology of HSP cannot be ignored since several HSP genes are expressed in non-neuronal cells (Blackstone, 2012).
Neurons are polarized cells (Kevenaar and Hoogenraad, 2015) and the pyramidal tract neurons can be injured in a length dependent manner through a dying-back mechanism of their axons. Since the longest axons are more susceptible, this leads to the primary clinical involvement of the lower limbs (LLs) (Deluca et al., 2004; Blackstone, 2012, 2018). In the complex phenotypes, further regions of the central and peripheral nervous systems (CNS and PNS) alongside other extra-neurological organs and systems can also be involved. In these other tissues, the degeneration of cell bodies occurs in interneurons with shorter axons as well and then the dying-back hypothesis is less evident.
Pathways Involved in HSP Pathogenesis
When the function of the affected HSP protein is known or could be inferred from their loss of function in models, they can be grouped into common cellular pathways based on ontology terms. In this article, we could distinguish 11 major functional themes (Supplementary Figure, Table 3). HSP genes encode proteins of known or suspected functions mainly in intracellular trafficking (membrane traffic and energy-dependent transport), organelle shaping, myelination, development, metabolism (predominantly lipid metabolism), recycling/degradation, cytoskeleton dynamics, mitochondrial functions, and signaling pathways. However, it recently emerged that dysfunction of one cellular organelle or function can have a deep impact on other cellular functions (Figure 3). For example, proteins implicated in the cytoskeleton dynamics such as spastin, can participate in organellar shaping particularly in ER morphogenesis, or modulate the formation of lipid droplets (LD) which can be considered as the intersection of these pathways with lipid metabolism in the pathogenesis of HSP (Figure 3) (Papadopoulos et al., 2015; Tadepalle and Rugarli, 2021). Furthermore, loss of spatacsin (SPG11) that promotes tubule formation on lysosomes also affects sphingolipid metabolism and leads to impairment of cholesterol recycling from lysosomes resulting in their accumulation and decrease in the cholesterol content of the plasma membrane which allowed us to include SPG11 as the 17th HSP form involving lipid metabolism (Boutry et al., 2018; 2019a).
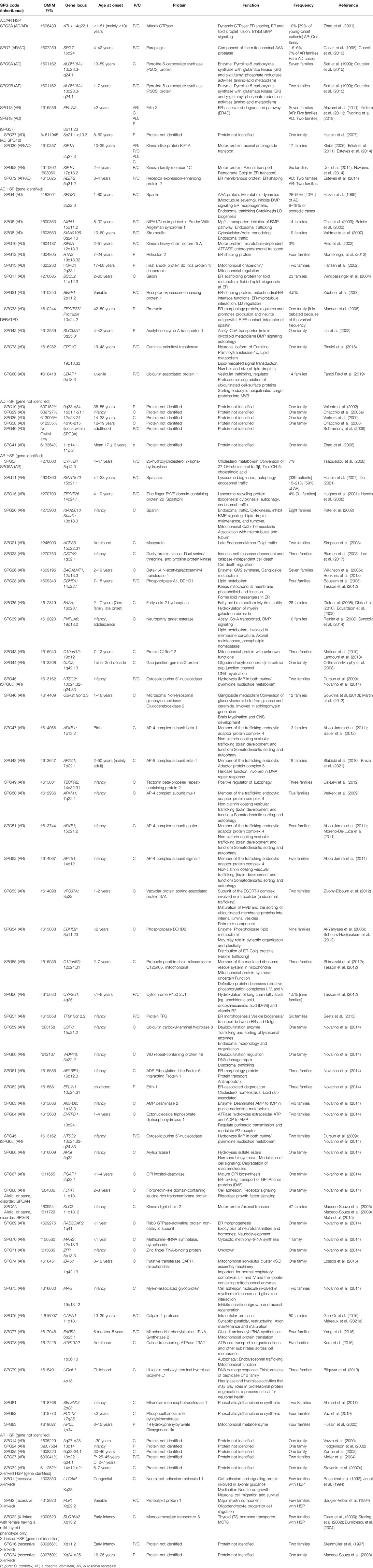
TABLE 3. Table summarizing the 88 HSP clinical-genetic entities with special focus on the functions of their proteins and our suggested primary and secondary functional categories.
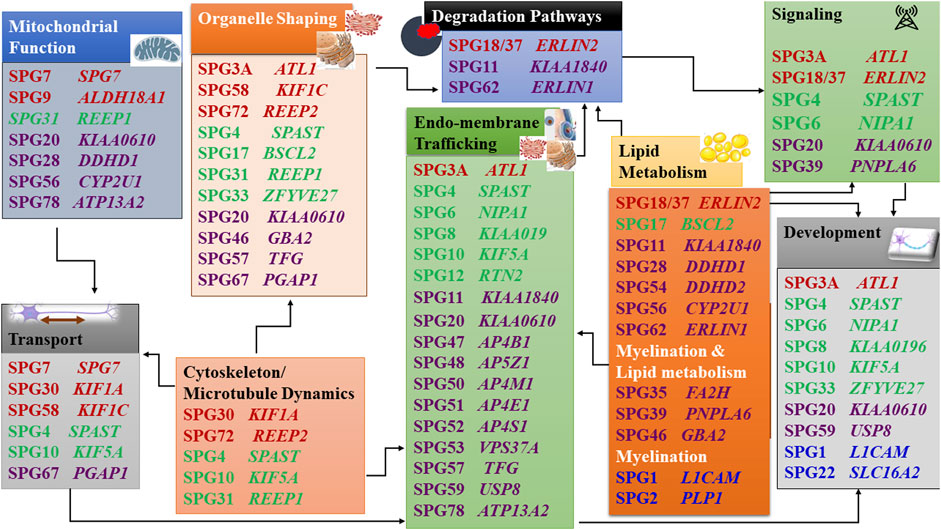
FIGURE 3. Intersections of HSP pathogenetic mechanisms highlighting the interplay between pathways. Font color codes correspond to various patterns of inheritance: AD HSP (green), AR HSP (violet), X-linked recessive (Blue), and Mixed AR/AD inheritance (red).
Another example is SPG7, in which alteration of the axonal transport due to mitochondrial impairment resulted in axonal degeneration due to traffic jam as was observed in paraplegin-deficient mice (Ferreirinha et al., 2004). In addition, numerous proteins produced from SPG genes are currently found to be connected with multiple cellular functions and are known to interact with numerous proteins which are either involved in the same pathway (or inter-digitations of pathways) or share the same gene ontology function (Figure 3) (Supplementary Figure, Table 3). It is then likely that alteration of one given HSP protein will impact multiple cellular functions. An HSP network has been produced to connect all known HSP proteins to hundreds of other potential HSP proteins (Novarino et al., 2014). This demonstrates how far we stand back from understanding the mechanisms of these disorders and make phenotype-genotype correlations difficult to establish.
One of the first identified mechanisms to be related to HSP was the disruption of the axonal transport, which was supported by the identification of several HSP genes that encode kinesin chains (KIF5A and later, KIF1A, KIF1C), essential members of the anterograde microtubule-dependent axonal transport (Crosby and Proukakis, 2002; Reid et al., 2002). Trafficking in the cell is probably the most frequent alteration found in HSP and is dependant on microtubule dynamics and mitochondrial functions, two other often affected pathways in HSP. The correct trafficking of vesicles and organelles is essential for neurons as well as is the turn-over of membranes and signaling machinery degradation. There is an obvious overlap between these functions in some forms of HSP. Trafficking involving kinesins necessitates functional mitochondria which are affected functionally and/or structurally in SPG7, SPG31, and SPG56 (Goizet et al., 2011; Tesson et al., 2012; Shanmughapriya et al., 2015; Lavie et al., 2017; Wali et al., 2020).
Another aspect of the problem in HSP is the endomembrane and autophagy/lysosomal trafficking (Toupenet Marchesi et al., 2021) which are interconnected with the signaling and receptor recycling/degradation. Cells depend on a highly regulated and dynamic membrane trafficking system for proper intracellular distribution of proteins, lipids, and complex carbohydrates. This transport relies on membrane-bound vesicles and can occur within different organelles inside the cell, or through the plasma membrane to and from the extracellular environment. Numerous HSP proteins are implicated in membrane trafficking, including regulators of the vesicle and endosomal trafficking and proteins involved in the morphogenesis of the organelles involved, mainly the endoplasmic reticulum (ER). ATL1 (SPG3A) encodes a dynamin-related GTPase, which is present in both the ER and the Golgi compartments. Disease-causing variants in this gene not only lead to altered vesicle trafficking between ER and Golgi but also to altered Golgi morphogenesis (Namekawa et al., 2007). AP4B1 (SPG47), AP4M1 (SPG50), AP4E1 (SPG51), and AP4S1 (SPG52) encode proteins that constitute the adaptor protein 4 (AP-4) complex, which mediates vesicle formation and sorting of cargo for vesicles included in the secretory and endocytic pathways (Abou Jamra et al., 2011; Hirst et al., 2013). AP5Z1 (SPG48), which encodes one of the subunits of the adaptor protein 5 (AP-5) complex, was also found to be involved in endosomal sorting, and to interact with spatacsin (SPG11) and zinc finger FYVE domain-containing protein 26 (SPG15), involved in the recycling of lysosomes (Chang et al., 2014; Renvoisé et al., 2014; Hirst et al., 2015, 2018). Magnesium transporter NIPA1 (SPG6) is a neuron-specific transmembrane protein expressed in early endosomes, cell surfaces, and the Golgi. It co-localizes with atlastin-1, and both were characterized as binding partners, with knock-down of NIPA1 expression resulting in a marked reduction of atlastin-1 in neuronal processes (Botzolakis et al., 2011). A rat model with a NIPA1 causative variant showed accumulation of tubular-vesicular organelles with endosomal features starting at axonal and dendritic terminals, followed by multifocal vacuolar degeneration in the CNS and peripheral nerves (Watanabe et al., 2013). WASHC5, formerly known as KIAA0196 (SPG8) encodes a subunit of the large protein complex WASH, which connects endosomes to the cytoskeleton, through interaction with vacuolar protein sorting-associated protein 35 (VPS35) (Harbour et al., 2012). Phospholipase DDHD1 (SPG28) and phospholipase DDHD2 (SPG54) are two enzymes making the link between membrane-sorting and lipid metabolism: DDHD2 localizes to the cis-Golgi and also to the ER-Golgi intermediate compartment suggesting that it facilitates membrane and vesicle fusion by the modification of membranes through phospholipid hydrolysis (Inoue et al., 2012; Schuurs-Hoeijmakers et al., 2012; Yadav and Rajasekharan, 2016). TECPR2 (SPG49) brought a new pathway to the HSPs, linking autophagy with the disease. Autophagy is a complex process responsible for the transport and degradation of cytoplasmic components in the lysosomes, which include compromised proteins and organelles. TECPR2 encodes a member of the tectonin β-propeller repeat-containing family, containing both TECPR and tryptophan-aspartic acid repeat (WD repeat) domains. SPG11 and ZFYVE26 (SPG15) encoded proteins are also linked to autophagy due to their role in autophagic lysosome reformation (Chang et al., 2014).
Microtubules dynamics is highly related to organellar shaping and transport. This can be well illustrated by the role that the microtubules perform in the neurons. Microtubules keep the organellar organization thus maintaining the shape of these subcellular organelles (Figure 3). They also perform a vital role in neuronal development through their function in polarized axonal transport. Spastin (SPG4) is a key element of the microtubule dynamics, important for axonal growth, cellular fluidity and organellar structure (Errico et al., 2002; Butler et al., 2010; Solowska et al., 2014; Penazzi et al., 2016; Jeong et al., 2019). Atlastin (SPG3), RTN2 and REEP1/2 (SPG31, SPG72) are good illustrations of the implication of an abnormal shaping of the ER in HSP (Park et al., 2010; Esteves et al., 2014). The ER is a multifunctional organelle involved in several essential tasks for the cell, being the most abundant membrane compartment. It is responsible for the synthesis, modification, quality control and trafficking of secreted and integral membrane proteins and has a role in the Ca2+ regulation, lipid and sterol biosynthesis and distribution, carbohydrate metabolism, peroxisome biogenesis, and the formation of enzymes involved in drug detoxification. Additionally, ER interacts with several other membranes, including mitochondria, the Golgi apparatus, the plasma membrane, and with LDs.
Causative variants in ATL1 (SPG3A) are the second most frequent cause of AD HSP. This gene encodes the protein atlastin-1, a member of the superfamily of dynamin-related GTPases that localize predominantly to the tubular ER and mediate the fusion of ER tubules (Hu et al., 2009; Orso et al., 2009). RTN2 (SPG12) encodes reticulon 2, which also localizes to the ER tubules and that acts together with DP1/Yop1 as curvature-stabilizing proteins (Voeltz et al., 2006). REEP1 (SPG31) and REEP2 (SPG72) are part of the REEP/DP1/Yop1p family of ER-shaping proteins, which like RTN2 protein has two hydrophobic domains that, when inserted into the phospholipid bilayer, are thought to form hairpin loops generating a curvature in the ER membrane. Spastin interacts with atlastin-1, REEP1 and reticulon two proteins and they all seem to contribute to the shaping of the tubular ER network (Voeltz et al., 2006; Shibata et al., 2010; Beetz et al., 2013; Esteves et al., 2014). The microtubule-based motility in the ER is important for the proper organization and distribution of the ER tubules and the impairment of this link between the tubular ER and the microtubule cytoskeleton seems to be one of the HSP pathogenic mechanisms (Blackstone, 2012). Pathogenic SPAST variants (SPG4) affecting the AAA domain lead to constitutive binding to microtubules and then a dominant negative effect, and a mutant version of the M1-spastin isoform, specific to motor neurons, seems toxic by altering dynamic trafficking (Errico et al., 2002). However, there has been a debate for years about the effects of variants in SPAST. SPAST haploinsufficiency is the most prevalent opinion because of the existence of the truncating variants and because mutant spastin has not been detected on patients’ cells when tested. However, SPAST KO in the mouse does not reproduce the disease. In addition, a mouse model overexpressing at least one missense mutation in spastin has a phenotype at the heterozygous state with accumulation in neurites and decreased microtubule stability (Solowska et al., 2010, 2014)). We can postulate that haploinsufficiency contributes to neuronal vulnerability while some missense mutations act also by additional toxicity. Other genes whose proteins are associated with ER morphogenesis include TFG (SPG57). The TFG protein is important for the organization of transitional ER and ER exit sites into larger structures and variants in this gene lead to a disruption of the peripheral ER tubules organization and collapse of the ER network (Beetz et al., 2013; Elsayed et al., 2016b; McCaughey et al., 2016). BSCL2 (SPG17), ERLIN1 (SPG62), and ERLIN2 (SPG18) encode proteins that also localize to the ER and relate to both ER and lipid metabolism pathways due to the key function of the ER in the synthesis, metabolism, and distribution of lipids and sterols (Blackstone, 2012). BSCL2 encodes an ER protein thought to act at the interface of the ER with LDs, regulating their size. ERLIN2 (SPG18) and ERLIN1 (SPG62) encode proteins that localize preferentially to cholesterol-rich domains including lipid rafts, and are involved in the regulation of cellular cholesterol homeostasis. Additionally, ERLIN proteins are associated with ER-associated degradation (ERAD) that regulates protein degradation by the ubiquitin-proteasome machinery (Browman et al., 2006; Huber et al., 2013).
Mitochondria are very important organelles, that in addition to their role in producing ATP, are involved both in the response mechanisms to oxidative stress and apoptosis and are a source of toxic-free radicals. In HSP, at least 10 genes were already associated with mitochondrial dysfunction. SPG7 encodes a protein, that together with AFG3L2, the gene which has been associated with Spastic ataxia 5 (AR) and Spinocerebellar ataxia 28 (AD), form the m-AAA metalloprotease complex of the inner mitochondrial membrane. This protease functions in protein quality control and is necessary for the regulation of ribosomal assembly (Atorino et al., 2003; Nolden et al., 2005). Fibroblasts from SPG7 patients showed reduced activity of complex I in the mitochondrial respiratory chain, and increased sensitivity to oxidative stress with increased mitochondrial DNA damage (Wedding et al., 2014). In a mouse model for SPG7, mitochondrial morphological abnormalities were observed in the synaptic terminals and distal regions of axons, correlating with the onset of motor impairment, and prior to axonal swelling and degeneration. The axonal swelling occurred due to a huge accumulation of organelles and neurofilaments, suggesting impairment of anterograde axonal transport, which may indicate that the axonal transport impairment could be secondary to mitochondrial dysfunction (Figure 3) (Ferreirinha et al., 2004; Rugarli and Langer, 2006). CYP2U1 (SPG56) encodes an enzyme involved in lipid metabolism, which seems to have a folate-dependent neurodevelopmental component followed by a deleterious impact on mitochondrial bioenergetic function with patients’ fibroblasts showing alteration of mitochondrial architecture and bioenergetics with increased oxidative stress (Tesson et al., 2012; Pujol et al., 2021).
Lipid metabolism was recently identified as a mechanism highly affected in HSP, quite predictable, as the brain is highly enriched in lipids. Lipid metabolism is affected directly in 17 different entities and their number is constantly growing. The involvement of lipids in HSP pathogenicity and neurodegeneration can be through disturbed lipid metabolism or lipid distribution affecting various subcellular organelles and their function. However, this is not the whole story; as accumulating evidence also shows that HSP genes affecting various subcellular compartments can also alter lipid metabolism. This might make the 17 SPG forms involving disturbed lipid metabolism in our classification (Supplementary Figure, Table 3) just the tip of an iceberg. Furthermore, there is a remarkable number of genes/proteins which directly involve lipid metabolism causing other neurogenetic disorders with HSP-like presentation or HSP (as an allelic presentation). This overlap zone includes a broad spectrum of spastic (non-HSP) neurogenetic entities and a remarkable variety of underlying genes: CYP27A1 [cerebrotendinous xanthomatosis (CTX)], SERAC1 (AR: 3-methylglutaconic aciduria with deafness, encephalopathy, and Leigh-like syndrome), PLA2G6 (a spectrum of AR neurodegenerative disorders including: HSP, infantile neuroaxonal dystrophy (INAD), and neurodegeneration with brain iron accumulation, parkinsonism (NBIA), GALC [AR: galactosylceramide lipidosis (Krabbe disease), ARSA [AR: metachromatic leukodystrophy (MLD)], ABCD1 [X-linked Adrenoleukodystrophy (X-ALD)], ELOVL1 [AR: ichthyotic keratoderma, spasticity, hypomyelination and dysmorphic features (IKSHD)], ELOVL4 [AR: ichthyosis, intellectual disability and spastic quadriplegia (ISQMR); AD: SCA34, Stargardt disease 3 ((STGD3)], ALDH3A2 [Sjögren-Larsson Syndrome (SLS)]. This long list of examples provides clear evidence that the existing phenotypic classes of neurogenetic disorders need to be reconsidered (Darios and Stevanin, 2020; Tadepalle and Rugarli, 2021). Various types of lipids are involved in spastic neurodegeneration including precursor lipids as cholesterol and fatty acids as well as complex lipids like phospholipids and sphingolipids. Disturbed biosynthesis and degradation of these lipids can lead to either deficiencies of molecules with functional importance, or pathogenic accumulation of precursors or undegraded damaged molecules with toxic effect, all resulting in neurodegeneration (Darios and Stevanin, 2020; Tadepalle and Rugarli, 2021).
Lipids play many different roles from storing energy, to electrical insulation in myelin, and modulation of cellular calcium signaling and other signal transduction cascades. Additionally, they are important structural components of cell membranes. The composition of the membranes in lipids and sterols controls membrane fluidity, as well as compartmentalization into specialized microdomains and functions. Altered enzymatic activities is the predominant mechanism mediating the involvement of disturbed lipid metabolism in HSP pathogenesis, facilitating the identification of biomarkers and therapeutic targets. The first gene to be associated with lipid metabolism was CYP7B1 (SPG5), which codes for a 7α-hydroxylase involved in cholesterol metabolism. More precisely, it works in two pathways: the “alternate/acidic pathway,” and the conversion of the cholesterol derivative dehydroepiandrosterone (DHEA) into 7-OH-DHEA. Additionally, there is growing evidence to suggest that this DHEA-related neurosteroids act as neuroprotective factors, and can reduce ischemia-induced neurodegeneration, which could relate CYP7B1 disease-causing variants with a loss of this neuroprotective role (Tsaousidou et al., 2008). In addition, the enzymatic blockage of the pathway by the mutated CYP7B1 enzyme led to the accumulation of accumulation of CYP7B1 substrates (three oxysterols) including 25- and 27-hydroxycholesterol [25- and 27-hydroxysterol (27-OH)] in cerebrospinal fluid and serum(Schüle et al., 2010), which were demonstrated to be toxic to neurons in cultures (Schöls et al., 2017).
The development and maintainance of axons and myelin are also affected. This is particularly well illustrated by a pathway linking multiple HSP genes: the BMP signaling cascade. BMP signaling is essential for normal axonal function and axonal guidance (pathfinding), growth, and differentiation in the nervous system in mammals and other smaller models (eg. Drosophila) implying an important impact on the pathogenesis of the axonal degeneration. Numerous HSP proteins influence the BMP signaling pathway; these include Spastin (causing SPG4, the commonest AD HSP) in addition to Magnesium transporter NIPA1 (SPG6, AD), Maspardin (SPG20, AR), Spastizin (SPG15, AR), Atlastin (SPG3A, AD/AR), and PNPLA6 (SPG39), influence the BMP signaling pathway (Charron and Tessier-Lavigne, 2007; Wang et al., 2007, 2014; Wen et al., 2007; Tsang et al., 2009; Fassier et al., 2010; Bayat et al., 2011; Hegarty et al., 2013; Nahm et al., 2013; Song et al., 2013). Some of these diseases are late-onset and this suggests that abnormal development may contribute to the sensitization of the neuronal connections which will exert their deleterious action later in life. On the other hand, neural development itself, including myelination, is affected in multiple HSP forms as shown by abnormal branching of axons in animal models (Martin et al., 2013). For some forms, the age at onset implies the direct involvement of improper development in the pathology such as in AP4 syndromes (Behne et al., 2020) or SPG11 (Stevanin et al., 2007b; Pozner et al., 2020). Another example is given by the recent study showing partial phenotypic recovery of the mouse model of SPG56 using folate supplementarion during fetal life (Pujol et al., 2021).
Are there any possible phenotype-genotype correlations that can be suggested?
Despite the marked inter-digitation of pathways and contribution of a number of HSP proteins in multiple pathways, our functional classification (Supplementary Figure) allows to observe a few connections between the pathogenic pathways and the clinical presentation, age at onset and patterns of inheritance, that can help in solving a small part of the HSP puzzle. Overall, AR forms are observed to involve principally the metabolic pathways (lipids and nucleotide metabolism), in addition to the degradation pathways, myelination and the endomembrane trafficking, in agreement with the need for a complete loss of function to alter the pathway. On the other hand, AD HSPs are largely clustering in the microtubule dynamics, organelle shaping, development, and active cellular transport (often with mixed transmission) functional categories. Age at onset is not a discriminant of the altered functions except for some tendency for childhood-onset in forms related to lipids (11/17) and nucleotides (3/3) metabolism. Early childhood-onset was also observed in 9/19 forms affecting endomembrane trafficking which is probably because of their suggested roles during development. It is worth noting that all nine forms associated with the degradation pathway, present with cerebellar ataxia. The link between cerebellar syndrome and the degradation pathways is well known in the pathogenesis of AD cerebellar ataxias. Degradation of cellular inclusions is the basis of numerous subtypes of hereditary ataxia (HA) especially the polyglutamine AD cerebellar ataxias (PolyQ: SCA). In these disorders, the proteasomal and lysosomal degradation pathways stand as the chief cellular defense mechanisms against the aggregates (the hallmark of the polyQ disorders) (Seidel et al., 2015; Mori et al., 2016). Cognitive decline and intellectual disability were reported in forms that clustered in the functional categories with an influence on endomembrane trafficking [17/19 forms], myelination [7/7 forms], lipid metabolism [11/17 forms], and degradation pathways [8/9]. It is worth noting that although SPG3A affects endomembrane trafficking with no previous association with cognitive impairment, recent studies started to detect cases with cognitive decline from different ethnic backgrounds (Fusco et al., 2012; Terada et al., 2013; Erfanian Omidvar et al., 2019). This again points to the constantly evolving capacities of the possible phenotype-genotype correlations in HSP.
Therapeutic Options and Opportunities
For all patients except those who are dopa-responsive, rehabilitation therapies, such as physical or speech therapy are the main proposed therapy. Physical therapy is based on passive tendon stretching, gait, and equilibrium rehabilitation. According to the functional repercussion of spasticity, medications used to decrease spasticity such as oral baclofen, intramuscular botulinum toxin, or intrathecal baclofen are often used. Orthopedic solutions such as special shoes for pes cavus, or Achilles tenotomy for equinovarus are also proposed to allow a longer autonomous gait. Sphincter disturbances should be confirmed by urologists and possible treatment options include oxybutynin chlorhydrate or trospium chlorure. Social solutions are proposed when patients cannot manage themselves anymore. Dancing and exergames are also proposed to preserve as much as possible motor and gait capacities (Synofzik and Ilg, 2014).
There is no curative treatment available yet for HSP patients. When dealing with rare diseases, one might want to find a common drug target for multiple HSP forms. The question which immediately arises is if there is a unifying theme. All functional subcategories are overlapping and interplay together. Lipid metabolism is important for membrane trafficking and signaling. Trafficking is dependent on ATP production provided by mitochondria. Organelle shaping is performed along cytoskeleton, and organelle functions are dependent on their shaping and trafficking. Animal models, and thus probably patients, show functional consequences of multiple pathways such as the accumulation of membranes along axons in the SPG7 KO mouse modeling the loss of function of a mitochondrial protein (Ferreirinha et al., 2004) or the abnormal mitochondrial network in SPG56 patients in which an enzyme of the lipid metabolism is affected (Tesson et al., 2012). The interplay between all these functional pathways makes it theoretically feasible to find drugs that could be active in multiple HSPs.
In practice however, multiple pathways are probably affected in all HSP, which makes it difficult to find a common cure to all aspects of the disease efficient in multiple HSP subtypes.
Another bottleneck in drug development is the lack of biomarkers available. They may play an important role in helping in the monitoring of disease progression and response to treatment in clinical trials and preclinical testing in animal models. There are very few diagnostic non-clinical and laboratory-based findings - hormonal disturbances, biochemical changes, and accumulation of various metabolites - which are associated with specific phenotypes in several instances (Table 3). We can cite the accumulation of 27- and 25-hydroxycholesterol in CSF and blood of SPG5 patients (Schöls et al., 2017; Marelli et al., 2018), testosterone levels in blood in SPG46 males (Martin et al., 2013), levels of plasmatic amino-acids in ALDH18A1 (SPG9) variant carriers (Coutelier et al., 2015), abnormal etherlipid profile in SPG82 (Vaz et al., 2019), brain imaging volumetry in SPG11 (Cardozo-Hernández et al., 2020), Coenzyme Q10/neopterin and brain calcifications in SPG56 (Pujol et al., 2021), and abnormal lipid peak at cerebral resonance spectroscopy in DDHD2 (SPG54) mutation carriers (Schuurs-Hoeijmakers et al., 2012). Most of them are found in clinico-genetic entities affecting directly the lipid metabolism (Darios and Stevanin, 2020) but the abnormal lipid profiles in SPG11 (Boutry et al., 2018; 2019b) show that the lipid homeostasis is critical for proper functioning of most cellular functions in neurons.
Numerous preclinical studies on animal models have given promising results. They include drugs which target microtubules and act to regulate their stability in SPG4 models (Trotta et al., 2004; Orso et al., 2005) and in iPSC models of SPG3 (Zhu et al., 2014). Replacement therapies gave interesting results to compensate for loss of function, including the intracerebral injection of CYP7B1 mRNA in SPG5 mimicking mouse models (Hauser et al., 2019) or viral delivery of proteins such as paraplegin in SPG7 models (Pirozzi et al., 2006) and spastin in neurons derived from iPSC of SPG4 patients (Havlicek et al., 2014). Among forms affecting directly the autophagy and lysosomal pathways, miglustat was succefully used to reduce ganglioside accumulation in SPG11 zebrafish models (Boutry et al., 2018), and tideglusib, a GSK3b inhibitor, prevented the neurite pathology in brain organoids of SPG11 patients (Pozner et al., 2018). Autophagy regulators might also be of interest in SPG48 (Breza et al., 2021) and rapamycin partially reversed the phenotype induced by Atlastin-1 loss of function in flies (Xu et al., 2017). In zebrafish mimicking SPG3, the modulation of the BMP pathway was also of interest to rescue the motor phenotype (Fassier et al., 2010). Even more promising data include the FDA approved molecules/drugs acting to decrease ER stress in SPG4 and SPG31 (Vaccaro et al., 2013; Julien et al., 2016). Folate supplementation was succefully used to improve the cognitive deficit in a SPG56 mouse model (Pujol et al., 2021). Finally, cholesterol-lowering drugs such as statins have been used in clinical trials in SPG5 patients but failed to reduce significantly the toxic 27-OH cholesterol in the CSF vs plasma (Schöls et al., 2017).
Conclusion: At the Crossroads of Molecular Pathways and Clinical Options
It is still crucial to improve the identification of the genetic causes of HSP for various reasons. First, it provides the patient with a conclusive explanation, giving closure to an often long diagnostic wandering. Furthermore, once the gene, mutation, and transmission mode have been identified, it allows for accurate genetic counselling. Another prominent reason is that discovering all genes involved is important for the identification of unifying mechanisms, which offer better hope of treatment options because they increase the number of patients available. In addition, other approaches may be useful for treatment options and may result from understanding the clinical heterogeneity. Very few initiatives have been reported to search for its aetiology. Understanding the links between the various clinical presentations and the affected molecular pathways may well be useful for better deciphering of the mechanisms implicated in the causation of HSP. This will help identify the suitable biomarkers to be used and shed light on the possible predictors of disease onset, progression and guide the choices of treatment options, for better functional outcome and quality of life.
Author Contributions
LE, IE, AA, and GS have contributed to the design of the work. LE and IE worked on the acquisition of the data. LE, IE, GS, and AA participated in drafting the review and revising it critically to bring it to the required intellectual level and all authors provided their approval for publication of the content. LE, IE, AA, and GS agreed to be responsible for the work warranting that questions associated with the integrity and accuracy of the work or any part of it are investigated and fixed appropriately.
Funding
This research was funded by the European Union through the HORIZON 2020 programme SOLVE-RD (to GS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest, except a grant received by GS from Biogen, but unrelated to this work.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.690899/full#supplementary-material
References
Abdollahpour, H., Alawi, M., Kortüm, F., Beckstette, M., Seemanova, E., Komárek, V., et al. (2015). An AP4B1 Frameshift Mutation in Siblings with Intellectual Disability and Spastic Tetraplegia Further Delineates the AP-4 Deficiency Syndrome. Eur. J. Hum. Genet. 23, 256–259. doi:10.1038/ejhg.2014.73
Abou Jamra, R., Philippe, O., Raas-Rothschild, A., Eck, S. H., Graf, E., Buchert, R., et al. (2011). Adaptor Protein Complex 4 Deficiency Causes Severe Autosomal-Recessive Intellectual Disability, Progressive Spastic Paraplegia, Shy Character, and Short Stature. Am. J. Hum. Genet. 88, 788–795. doi:10.1016/j.ajhg.2011.04.019
Ahmed, M. Y., Al-Khayat, A., Al-Murshedi, F., Al-Futaisi, A., Chioza, B. A., Pedro Fernandez-Murray, J., et al. (2017). A Mutation of EPT1 (SELENOI) Underlies a New Disorder of Kennedy Pathway Phospholipid Biosynthesis. Brain 140, 547–554. doi:10.1093/brain/aww318
Akawi, N., McRae, J., McRae, J., Ansari, M., Balasubramanian, M., Blyth, M., et al. (2015). Discovery of Four Recessive Developmental Disorders Using Probabilistic Genotype and Phenotype Matching Among 4,125 Families. Nat. Genet. 47, 1363–1369. doi:10.1038/ng.3410
Al-Yahyaee, S., Al-Gazali, L. I., De Jonghe, P., Al-Barwany, H., Al-Kindi, M., De Vriendt, E., et al. (2006). A Novel Locus for Hereditary Spastic Paraplegia with Thin Corpus Callosum and Epilepsy. Neurology 66, 1230–1234. doi:10.1212/01.wnl.0000208501.52849.dd
Alazami, A. M., Adly, N., Al Dhalaan, H., and Alkuraya, F. S. (2011). A Nullimorphic ERLIN2 Mutation Defines a Complicated Hereditary Spastic Paraplegia Locus (SPG18). Neurogenetics 12, 333–336. doi:10.1007/s10048-011-0291-8
Amin, M., Vignal, C., Hamed, A. A. A., Mohammed, I. N., Elseed, M. A., Drunat, S., et al. (2021). Novel Variants Causing Megalencephalic Leukodystrophy in Sudanese Families. J. Hum. Genet. doi:10.1038/s10038-021-00945-7
Angelini, C., Goizet, C., Said, S. A., Camu, W., Depienne, C., Heron, B., et al. (2021). Evidence of Mosaicism in SPAST Variant Carriers in Four French Families. Eur. J. Hum. Genet. 29, 1158–1163. doi:10.1038/s41431-021-00847-4
Atorino, L., Silvestri, L., Koppen, M., Cassina, L., Ballabio, A., Marconi, R., et al. (2003). Loss of M-AAA Protease in Mitochondria Causes Complex I Deficiency and Increased Sensitivity to Oxidative Stress in Hereditary Spastic Paraplegia. J. Cel Biol. 163, 777–787. doi:10.1083/jcb.200304112
Bauer, P., Leshinsky-Silver, E., Blumkin, L., Schlipf, N., Schröder, C., Schicks, J., et al. (2012). Mutation in the AP4B1 Gene Cause Hereditary Spastic Paraplegia Type 47 (SPG47). Neurogenetics 13, 73–76. doi:10.1007/s10048-012-0314-0
Bayat, V., Jaiswal, M., and Bellen, H. J. (2011). The BMP Signaling Pathway at the Drosophila Neuromuscular Junction and its Links to Neurodegenerative Diseases. Curr. Opin. Neurobiol. 21, 182–188. doi:10.1016/j.conb.2010.08.014
Beetz, C., Johnson, A., Schuh, A. L., Thakur, S., Varga, R.-E., Fothergill, T., et al. (2013). Inhibition of TFG Function Causes Hereditary Axon Degeneration by Impairing Endoplasmic Reticulum Structure. Proc. Natl. Acad. Sci. 110, 5091–5096. doi:10.1073/pnas.1217197110
Beetz, C., Nygren, A. O. H., Schickel, J., Auer-Grumbach, M., Bürk, K., Heide, G., et al. (2006). High Frequency of Partial SPAST Deletions in Autosomal Dominant Hereditary Spastic Paraplegia. Neurology 67, 1926–1930. doi:10.1212/01.wnl.0000244413.49258.f5
Behne, R., Teinert, J., Wimmer, M., D’Amore, A., Davies, A. K., Scarrott, J. M., et al. (2020). Adaptor Protein Complex 4 Deficiency: a Paradigm of Childhood-Onset Hereditary Spastic Paraplegia Caused by Defective Protein Trafficking. Hum. Mol. Genet. 29, 320–334. doi:10.1093/hmg/ddz310
Ben Hamida, M., Chaabouni, H., Madani, S., Boussen, S., Samoud, S., Letaief, F., et al. (1986). Genetic Study of Spinocerebellar Hereditary Degenerations in Tunisia. Role of Consanguinity in Their Occurrence. J. Genet. Hum. 34, 267–274.
Bettencourt, C., Salpietro, V., Efthymiou, S., Chelban, V., Hughes, D., Pittman, A. M., et al. (2017). Genotype-phenotype Correlations and Expansion of the Molecular Spectrum of AP4M1-Related Hereditary Spastic Paraplegia. Orphanet J. Rare Dis. 12, 172. doi:10.1186/s13023-017-0721-2
Bilguvar, K., Tyagi, N. K., Ozkara, C., Tuysuz, B., Bakircioglu, M., Choi, M., et al. (2013). Recessive Loss of Function of the Neuronal Ubiquitin Hydrolase UCHL1 Leads to Early-Onset Progressive Neurodegeneration. Proc. Natl. Acad. Sci. USA 110, 3489–3494. doi:10.1073/pnas.1222732110
Bindu, P. S. (2020). Sjogren-Larsson Syndrome: Mechanisms and Management. Tacg 13, 13–24. doi:10.2147/TACG.S193969
Bis-Brewer, D. M., and Züchner, S. (2018). Perspectives on the Genomics of HSP Beyond Mendelian Inheritance. Front. Neurol. 9, 958. doi:10.3389/fneur.2018.00958
Blackstone, C. (2012). Cellular Pathways of Hereditary Spastic Paraplegia. Annu. Rev. Neurosci. 35, 25–47. doi:10.1146/annurev-neuro-062111-150400
Blackstone, C. (2018). Converging Cellular Themes for the Hereditary Spastic Paraplegias. Curr. Opin. Neurobiol. 51, 139–146. doi:10.1016/j.conb.2018.04.025
Blumen, S. C., Bevan, S., Abu-Mouch, S., Negus, D., Kahana, M., Inzelberg, R., et al. (2003). A Locus for Complicated Hereditary Spastic Paraplegia Maps to Chromosome 1q24-Q32. Ann. Neurol. 54, 796–803. doi:10.1002/ana.10768
Botzolakis, E. J., Zhao, J., Gurba, K. N., Macdonald, R. L., and Hedera, P. (2011). The Effect of HSP-Causing Mutations in SPG3A and NIPA1 on the Assembly, Trafficking, and Interaction Between Atlastin-1 and NIPA1. Mol. Cell Neurosci. 46, 122–135. doi:10.1016/j.mcn.2010.08.012
Bouhouche, A., Benomar, A., Bouslam, N., Chkili, T., and Yahyaoui, M. (2006). Mutation in the Epsilon Subunit of the Cytosolic Chaperonin-Containing T-Complex Peptide-1 (Cct5) Gene Causes Autosomal Recessive Mutilating Sensory Neuropathy with Spastic Paraplegia. J. Med. Genet. 43, 441–443. doi:10.1136/jmg.2005.039230
Boukhris, A., Feki, I., Elleuch, N., Miladi, M. I., Boland-Augé, A., Truchetto, J., et al. (2010). A New Locus (SPG46) Maps to 9p21.2-q21.12 in a Tunisian Family with a Complicated Autosomal Recessive Hereditary Spastic Paraplegia with Mental Impairment and Thin Corpus Callosum. Neurogenetics 11, 441–448. doi:10.1007/s10048-010-0249-2
Boukhris, A., Schule, R., Loureiro, J. L., Lourenço, C. M., Mundwiller, E., Gonzalez, M. A., et al. (2013). Alteration of Ganglioside Biosynthesis Responsible for Complex Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 93, 118–123. doi:10.1016/j.ajhg.2013.05.006
Boukhris, A., Stevanin, G., Feki, I., Denis, E., Elleuch, N., Miladi, M. I., et al. (2008). Hereditary Spastic Paraplegia with Mental Impairment and Thin Corpus Callosum in Tunisia. Arch. Neurol. 65, 393–402. doi:10.1001/archneur.65.3.393
Boukhris, A., Stevanin, G., Feki, I., Denora, P., Elleuch, N., Miladi, M., et al. (2009). Tunisian Hereditary Spastic Paraplegias: Clinical Variability Supported by Genetic Heterogeneity. Clin. Genet. 75, 527–536. doi:10.1111/j.1399-0004.2009.01176.x
Bouslam, N., Benomar, A., Azzedine, H., Bouhouche, A., Namekawa, M., Klebe, S., et al. (2005). Mapping of a New Form of Pure Autosomal Recessive Spastic Paraplegia (SPG28). Ann. Neurol. 57, 567–571. doi:10.1002/ana.20416
Boutry, M., Branchu, J., Lustremant, C., Pujol, C., Pernelle, J., Matusiak, R., et al. (2018). Inhibition of Lysosome Membrane Recycling Causes Accumulation of Gangliosides that Contribute to Neurodegeneration. Cel Rep. 23, 3813–3826. doi:10.1016/j.celrep.2018.05.098
Boutry, M., Morais, S., and Stevanin, G. (2019a). Update on the Genetics of Spastic Paraplegias. Curr. Neurol. Neurosci. Rep. 19, 18. doi:10.1007/s11910-019-0930-2
Boutry, M., Pierga, A., Matusiak, R., Branchu, J., Houllegatte, M., Ibrahim, Y., et al. (2019b). Loss of Spatacsin Impairs Cholesterol Trafficking and Calcium Homeostasis. Commun. Biol. 2. doi:10.1038/s42003-019-0615-z
Bouwkamp, C. G., Afawi, Z., Fattal-Valevski, A., Krabbendam, I. E., Rivetti, S., Masalha, R., et al. (2018). ACO2 Homozygous Missense Mutation Associated with Complicated Hereditary Spastic Paraplegia. Neurol. Genet. 4, e223. doi:10.1212/NXG.0000000000000223
Breza, M., Hirst, J., Chelban, V., Banneau, G., Tissier, L., Kol, B., et al. (2021). Expanding the Spectrum of AP5Z1-Related Hereditary Spastic Paraplegia (HSP-SPG48): A Multicenter Study on a Rare Disease. Mov. Disord. 36, 1034–1038. doi:10.1002/mds.28487
Browman, D. T., Resek, M. E., Zajchowski, L. D., and Robbins, S. M. (2006). Erlin-1 and Erlin-2 Are Novel Members of the Prohibitin Family of Proteins that Define Lipid-raft-like Domains of the ER. J. Cel Sci. 119, 3149–3160. doi:10.1242/jcs.03060
Burguez, D., Polese-Bonatto, M., Scudeiro, L. A. J., Björkhem, I., Schöls, L., Jardim, L. B., et al. (2017). Clinical and Molecular Characterization of Hereditary Spastic Paraplegias: A Next-Generation Sequencing Panel Approach. J. Neurol. Sci. 383, 18–25. doi:10.1016/j.jns.2017.10.010
Butler, R., Wood, J. D., Landers, J. A., and Cunliffe, V. T. (2010). Genetic and Chemical Modulation of Spastin-dependent Axon Outgrowth in Zebrafish Embryos Indicates a Role for Impaired Microtubule Dynamics in Hereditary Spastic Paraplegia. Dis. Model. Mech. 3, 743–751. doi:10.1242/dmm.004002
Caballero Oteyza, A., Battalo lu, E., Ocek, L., Lindig, T., Reichbauer, J., Rebelo, A. P., et al. (2014). Motor Protein Mutations Cause a New Form of Hereditary Spastic Paraplegia. Neurology 82, 2007–2016. doi:10.1212/wnl.0000000000000479
Calandra, C. R., Buda, G., Vishnopolska, S. A., Oliveri, J., Olivieri, F. A., Pérez Millán, M. I., et al. (2020). Spastic Ataxia with Eye-of-the-tiger-like Sign in 4 Siblings Due to Novel Compound Heterozygous AFG3L2 Mutation. Parkinsonism Relat. Disord. 73, 52–54. doi:10.1016/j.parkreldis.2020.03.020
Cardozo-Hernández, A. L. d. C., Rezende, T. J. R., França, M. C., and França, M. C. (2020). Hereditary Spastic Paraplegia Type 11 (SPG11) Is Associated with Obesity and Hypothalamic Damage. J. Neurol. Sci. 416, 116982. doi:10.1016/j.jns.2020.116982
Casari, G., De Fusco, M., Ciarmatori, S., Zeviani, M., Mora, M., Fernandez, P., et al. (1998). Spastic Paraplegia and OXPHOS Impairment Caused by Mutations in Paraplegin, a Nuclear-Encoded Mitochondrial Metalloprotease. Cell 93, 973–983. doi:10.1016/s0092-8674(00)81203-9
Cassis, L., Cortès-Saladelafont, E., Molero-Luis, M., Yubero, D., González, M. J., Ormazábal, A., et al. (2015). Review and Evaluation of the Methodological Quality of the Existing Guidelines and Recommendations for Inherited Neurometabolic Disorders. Orphanet J. Rare Dis. 10, 164. doi:10.1186/s13023-015-0376-9
Chai, J.-H., Locke, D. P., Greally, J. M., Knoll, J. H. M., Ohta, T., Dunai, J., et al. (2003). Identification of Four Highly Conserved Genes Between Breakpoint Hotspots BP1 and BP2 of the Prader-Willi/Angelman Syndromes Deletion Region that Have Undergone Evolutionary Transposition Mediated by Flanking Duplicons. Am. J. Hum. Genet. 73, 898–925. doi:10.1086/378816
Chang, J., Lee, S., and Blackstone, C. (2014). Spastic Paraplegia Proteins Spastizin and Spatacsin Mediate Autophagic Lysosome Reformation. J. Clin. Invest. 124, 5249–5262. doi:10.1172/JCI77598
Charron, F., and Tessier-Lavigne, M. (2007). The Hedgehog, TGF-Beta/BMP and Wnt Families of Morphogens in Axon Guidance. Adv. Exp. Med. Biol. 621, 116–133. doi:10.1007/978-0-387-76715-4_9
Chestnut, B. A., Chang, Q., Price, A., Lesuisse, C., Wong, M., and Martin, L. J. (2011). Epigenetic Regulation of Motor Neuron Cell Death Through DNA Methylation. J. Neurosci. 31, 16619–16636. doi:10.1523/JNEUROSCI.1639-11.2011
Chouliaras, L., Mastroeni, D., Delvaux, E., Grover, A., Kenis, G., Hof, P. R., et al. (2013). Consistent Decrease in Global DNA Methylation and Hydroxymethylation in the Hippocampus of Alzheimer's Disease Patients. Neurobiol. Aging 34, 2091–2099. doi:10.1016/j.neurobiolaging.2013.02.021
Chrestian, N., Dupré, N., Gan-Or, Z., Szuto, A., Chen, S., Venkitachalam, A., et al. (2016). Clinical and Genetic Study of Hereditary Spastic Paraplegia in Canada. Neurol. Genet. 3, e122. doi:10.1212/NXG.0000000000000122
Claes, S., Devriendt, K., Van Goethem, G., Roelen, L., Meireleire, J., Raeymaekers, P., et al. (2000). Novel Syndromic Form of X-Linked Complicated Spastic Paraplegia. Am. J. Med. Genet. 94, 1–4. doi:10.1002/1096-8628(20000904)94:1<1::aid-ajmg1>3.0.co;2-v
Clarençon, F., Touzé, E., Leroy–Willig, A., Turmel, H., Naggara, O., Pavy, S., et al. (2006). Spastic Paraparesis as a Manifestation of Leber's Disease. J. Neurol. 253, 525–526. doi:10.1007/s00415-005-0994-6
Coarelli, G., Schule, R., van de Warrenburg, B. P. C., de Jonghe, P., De Jonghe, C., Martinuzzi, A., et al. (2019). Loss of Paraplegin Drives Spasticity Rather Than Ataxia in a Cohort of 241 Patients with SPG7. Neurology 92, e2679–e2690. doi:10.1212/WNL.0000000000007606
Corona, P., Lamantea, E., Greco, M., Carrara, F., Agostino, A., Guidetti, D., et al. (2002). Novel Heteroplasmic mtDNA Mutation in a Family with Heterogeneous Clinical Presentations. Ann. Neurol. 51, 118–122. doi:10.1002/ana.10059
Coutelier, M., Goizet, C., Durr, A., Habarou, F., Morais, S., Dionne-Laporte, A., et al. (2015). Alteration of Ornithine Metabolism Leads to Dominant and Recessive Hereditary Spastic Paraplegia. Brain 138, 2191–2205. doi:10.1093/brain/awv143
Coutelier, M., Mochel, F., Saudubray, J.-M., Ottolenghi, C., and Stevanin, G. (2016). Reply:ALDH18A1gene Mutations Cause Dominant Spastic Paraplegia SPG9: Loss of Function Effect and Plausibility of a Dominant Negative Mechanism. Brain 139, e4. doi:10.1093/brain/awv248
Crosby, A. H., and Proukakis, C. (2002). Is the Transportation Highway the Right Road for Hereditary Spastic Paraplegia? Am. J. Hum. Genet. 71, 1009–1016. doi:10.1086/344206
Darios, F., Mochel, F., and Stevanin, G. (2020). Lipids in the Physiopathology of Hereditary Spastic Paraplegias. Front. Neurosci. 14. doi:10.3389/fnins.2020.00074
Darios, F., and Stevanin, G. (2020). Impairment of Lysosome Function and Autophagy in Rare Neurodegenerative Diseases. J. Mol. Biol. 432, 2714–2734. doi:10.1016/j.jmb.2020.02.033
Daud, S., Kakar, N., Goebel, I., Hashmi, A. S., Yaqub, T., Nürnberg, G., et al. (2016). Identification of Two Novel ALS2 Mutations in Infantile-Onset Ascending Hereditary Spastic Paraplegia. Amyotroph. Lateral Scler. Frontotemporal Degeneration 17, 260–265. doi:10.3109/21678421.2015.1125501
de Souza, P. V. S., de Rezende Pinto, W. B. V., de Rezende Batistella, G. N., Bortholin, T., and Oliveira, A. S. B. (2017). Hereditary Spastic Paraplegia: Clinical and Genetic Hallmarks. Cerebellum 16, 525–551. doi:10.1007/s12311-016-0803-z
Deluca, G. C., Ebers, G. C., and Esiri, M. M. (2004). The Extent of Axonal Loss in the Long Tracts in Hereditary Spastic Paraplegia. Neuropathol. Appl. Neurobiol. 30, 576–584. doi:10.1111/j.1365-2990.2004.00587.x
Depienne, C., Fedirko, E., Forlani, S., Cazeneuve, C., Ribaï, P., Feki, I., et al. (2007). Exon Deletions of SPG4 Are a Frequent Cause of Hereditary Spastic Paraplegia. J. Med. Genet. 44, 281–284. doi:10.1136/jmg.2006.046425
Desplats, P., Spencer, B., Coffee, E., Patel, P., Michael, S., Patrick, C., et al. (2011). α-Synuclein Sequesters Dnmt1 from the Nucleus. J. Biol. Chem. 286, 9031–9037. doi:10.1074/jbc.C110.212589
Dick, K. J., Al-Mjeni, R., Baskir, W., Koul, R., Simpson, M. A., Patton, M. A., et al. (2008). A Novel Locus for an Autosomal Recessive Hereditary Spastic Paraplegia (SPG35) Maps to 16q21-Q23. Neurology 71, 248–252. doi:10.1212/01.wnl.0000319610.29522.8a
Dick, K. J., Eckhardt, M., Paisán-Ruiz, C., Alshehhi, A. A., Proukakis, C., Sibtain, N. A., et al. (2010). Mutation of FA2H Underlies a Complicated Form of Hereditary Spastic Paraplegia (SPG35). Hum. Mutat. 31, E1251–E1260. doi:10.1002/humu.21205
Dong, E.-L., Wang, C., Wu, S., Lu, Y.-Q., Lin, X.-H., Su, H.-Z., et al. (2018). Clinical Spectrum and Genetic Landscape for Hereditary Spastic Paraplegias in China. Mol. Neurodegeneration 13, 36. doi:10.1186/s13024-018-0269-1
Dor, T., Cinnamon, Y., Raymond, L., Shaag, A., Bouslam, N., Bouhouche, A., et al. (2014). KIF1Cmutations in Two Families with Hereditary Spastic Paraparesis and Cerebellar Dysfunction. J. Med. Genet. 51, 137–142. doi:10.1136/jmedgenet-2013-102012
Du, J. (2021). Hereditary Spastic Paraplegia Type 11: Clinicogenetic Lessons from 339 Patients. J. Clin. Neurosci. 85, 67–71. doi:10.1016/j.jocn.2020.11.036
Dumitrescu, A. M., Liao, X.-H., Best, T. B., Brockmann, K., and Refetoff, S. (2004). A Novel Syndrome Combining Thyroid and Neurological Abnormalities Is Associated with Mutations in a Monocarboxylate Transporter Gene. Am. J. Hum. Genet. 74, 168–175. doi:10.1086/380999
Dursun, U., Koroglu, C., Kocasoy Orhan, E., Ugur, S. A., and Tolun, A. (2009). Autosomal Recessive Spastic Paraplegia (SPG45) with Mental Retardation Maps to 10q24.3-q25.1. Neurogenetics 10, 325–331. doi:10.1007/s10048-009-0191-3
Edvardson, S., Hama, H., Shaag, A., Gomori, J. M., Berger, I., Soffer, D., et al. (2008). Mutations in the Fatty Acid 2-hydroxylase Gene Are Associated with Leukodystrophy with Spastic Paraparesis and Dystonia. Am. J. Hum. Genet. 83, 643–648. doi:10.1016/j.ajhg.2008.10.010
Elert-Dobkowska, E., Stepniak, I., Krysa, W., Ziora-Jakutowicz, K., Rakowicz, M., Sobanska, A., et al. (2019). Next-generation Sequencing Study Reveals the Broader Variant Spectrum of Hereditary Spastic Paraplegia and Related Phenotypes. Neurogenetics 20, 27–38. doi:10.1007/s10048-019-00565-6
Elsayed, L. E. O., Ahmed, A. E. M., and Stevanin, G. (2019a). “Neurogenetic Disorders in Africa: Hereditary Spastic Paraplegia: A Case Study,” in The Genetics of African Populations in Health and Disease Cambridge Studies in Biological and Evolutionary Anthropology. Editors C. N. Rotimi, and M. E. Ibrahim (Cambridge, UK: Cambridge University Press), 311–319. doi:10.1017/9781139680295.013
Elsayed, L. E. O., Drouet, V., Usenko, T., Mohammed, I. N., Hamed, A. A. A., Elseed, M. A., et al. (2016a). A Novel Nonsense Mutation in DNAJC 6 Expands the Phenotype of Autosomal-Recessive Juvenile-Onset Parkinson's Disease. Ann. Neurol. 79, 335–337. doi:10.1002/ana.24591
Elsayed, L. E. O., Eltazi, I. Z. M., Ahmed, A. E. M., and Stevanin, G. (2019b). Hereditary Spastic Paraplegias: Time for an Objective Case Definition and a New Nosology for Neurogenetic Disorders to Facilitate Biomarker/therapeutic Studies. Expert Rev. Neurotherapeutics 19, 409–415. doi:10.1080/14737175.2019.1608824
Elsayed, L. E. O., Mohammed, I. N., Hamed, A. A. A., Elseed, M. A., Johnson, A., Mairey, M., et al. (2016b). Hereditary Spastic Paraplegias: Identification of a Novel SPG57 Variant Affecting TFG Oligomerization and Description of HSP Subtypes in Sudan. Eur. J. Hum. Genet. 25, 100–110. doi:10.1038/ejhg.2016.108
Elsayed, L. E. O., Mohammed, I. N., Hamed, A. A. A., Elseed, M. A., Salih, M. A. M., Yahia, A., et al. (2018). Case Report of a Novel Homozygous Splice Site Mutation in PLA2G6 Gene Causing Infantile Neuroaxonal Dystrophy in a Sudanese Family. BMC Med. Genet. 19, 72. doi:10.1186/s12881-018-0592-y
Erfanian Omidvar, M., Torkamandi, S., Rezaei, S., Alipoor, B., Omrani, M. D., Darvish, H., et al. (2019). Genotype–phenotype Associations in Hereditary Spastic Paraplegia: A Systematic Review and Meta-Analysis on 13,570 Patients. J. Neurol., 2682065–2682082. doi:10.1007/s00415-019-09633-1
Erichsen, A. K., Inderhaug, E., Mattingsdal, M., Eiklid, K., and Tallaksen, C. M. E. (2007). Seven Novel Mutations and Four Exon Deletions in a Collection of Norwegian Patients with SPG4 Hereditary Spastic Paraplegia. Eur. J. Neurol. 14, 809–814. doi:10.1111/j.1468-1331.2007.01861.x
Erlich, Y., Edvardson, S., Hodges, E., Zenvirt, S., Thekkat, P., Shaag, A., et al. (2011). Exome Sequencing and Disease-Network Analysis of a Single Family Implicate a Mutation in KIF1A in Hereditary Spastic Paraparesis. Genome Res. 21, 658–664. doi:10.1101/gr.117143.110
Errico, A., Ballabio, A., and Rugarli, E. I. (2002). Spastin, the Protein Mutated in Autosomal Dominant Hereditary Spastic Paraplegia, Is Involved in Microtubule Dynamics. Hum. Mol. Genet. 11, 153–163. doi:10.1093/hmg/11.2.153
Esteves, T., Durr, A., Mundwiller, E., Loureiro, J. L., Boutry, M., Gonzalez, M. A., et al. (2014). Loss of Association of REEP2 with Membranes Leads to Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 94, 268–277. doi:10.1016/j.ajhg.2013.12.005
Eymard-Pierre, E., Lesca, G., Dollet, S., Santorelli, F. M., di Capua, M., Bertini, E., et al. (2002). Infantile-onset Ascending Hereditary Spastic Paralysis Is Associated with Mutations in the Alsin Gene. Am. J. Hum. Genet. 71, 518–527. doi:10.1086/342359
Farazi Fard, M. A., Rebelo, A. P., Buglo, E., Nemati, H., Dastsooz, H., Gehweiler, I., et al. (2019). Truncating Mutations in UBAP1 Cause Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 104, 767–773. doi:10.1016/j.ajhg.2019.03.001
Fassier, C., Hutt, J. A., Scholpp, S., Lumsden, A., Giros, B., Nothias, F., et al. (2010). Zebrafish Atlastin Controls Motility and Spinal Motor Axon Architecture via Inhibition of the BMP Pathway. Nat. Neurosci. 13, 1380–1387. doi:10.1038/nn.2662
Ferreirinha, F., Quattrini, A., Pirozzi, M., Valsecchi, V., Dina, G., Broccoli, V., et al. (2004). Axonal Degeneration in Paraplegin-Deficient Mice Is Associated with Abnormal Mitochondria and Impairment of Axonal Transport. J. Clin. Invest. 113, 231–242. doi:10.1172/JCI2013810.1172/jci200420138
Fink, J. (2014). Hereditary Spastic Paraplegia: Clinical Principles and Genetic Advances. Semin. Neurol. 34, 293–305. doi:10.1055/s-0034-1386767
Fink, J. K. (1993). “Hereditary Spastic Paraplegia Overview,” in GeneReviews(®). Editors R. A. Pagon, M. P. Adam, H. H. Ardinger, S. E. Wallace, A. Amemiya, and L. J. Bean (Seattle, WA: University of Washington). Available at: http://www.ncbi.nlm.nih.gov/books/NBK1509/ (Accessed February 22, 2016).
Finsterer, J., Löscher, W., Quasthoff, S., Wanschitz, J., Auer-Grumbach, M., and Stevanin, G. (2012). Hereditary Spastic Paraplegias with Autosomal Dominant, Recessive, X-Linked, or Maternal Trait of Inheritance. J. Neurol. Sci. 318, 1–18. doi:10.1016/j.jns.2012.03.025
Fusco, C., Frattini, D., Farnetti, E., Nicoli, D., Casali, B., and Giustina, E. D. (2012). Very Early Onset and Severe Complicated Phenotype Caused by a New Spastic Paraplegia 3A Gene Mutation. J. Child. Neurol. 27, 1348–1350. doi:10.1177/0883073811435245
Gan-Or, Z., Bouslam, N., Birouk, N., Lissouba, A., Chambers, D. B., Vérièpe, J., et al. (2016). Mutations in CAPN1 Cause Autosomal-Recessive Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 98, 1038–1046. doi:10.1016/j.ajhg.2016.04.002
Goizet, C., Depienne, C., Benard, G., Boukhris, A., Mundwiller, E., Solé, G., et al. (2011). REEP1 Mutations in SPG31: Frequency, Mutational Spectrum, and Potential Association with Mitochondrial Morpho-Functional Dysfunction. Hum. Mutat. 32, 1118–1127. doi:10.1002/humu.21542
Goodnight, A. V., Kremsky, I., Khampang, S., Jung, Y. H., Billingsley, J. M., Bosinger, S. E., et al. (2019). Chromatin Accessibility and Transcription Dynamics during In Vitro Astrocyte Differentiation of Huntington's Disease Monkey Pluripotent Stem Cells. Epigenetics & Chromatin 12, 67. doi:10.1186/s13072-019-0313-6
Haertle, L., Müller, T., Lardenoije, R., Maierhofer, A., Dittrich, M., Riemens, R. J. M., et al. (2019). Methylomic Profiling in Trisomy 21 Identifies Cognition- and Alzheimer's Disease-Related Dysregulation. Clin. Epigenet 11, 195. doi:10.1186/s13148-019-0787-x
Halevy, A., Lerer, I., Cohen, R., Kornreich, L., Shuper, A., Gamliel, M., et al. (2014). Novel EXOSC3 Mutation Causes Complicated Hereditary Spastic Paraplegia. J. Neurol. 261, 2165–2169. doi:10.1007/s00415-014-7457-x
Hanein, S., Dürr, A., Ribai, P., Forlani, S., Leutenegger, A.-L., Nelson, I., et al. (2007). A Novel Locus for Autosomal Dominant "uncomplicated" Hereditary Spastic Paraplegia Maps to Chromosome 8p21.1-q13.3. Hum. Genet. 122, 261–273. doi:10.1007/s00439-007-0396-1
Hanein, S., Martin, E., Boukhris, A., Byrne, P., Goizet, C., Hamri, A., et al. (2008). Identification of the SPG15 Gene, Encoding Spastizin, as a Frequent Cause of Complicated Autosomal-Recessive Spastic Paraplegia, Including Kjellin Syndrome. Am. J. Hum. Genet. 82, 992–1002. doi:10.1016/j.ajhg.2008.03.004
Hansen, J. J., Dürr, A., Cournu-Rebeix, I., Georgopoulos, C., Ang, D., Nielsen, M. N., et al. (2002). Hereditary Spastic Paraplegia SPG13 Is Associated with a Mutation in the Gene Encoding the Mitochondrial Chaperonin Hsp60. Am. J. Hum. Genet. 70, 1328–1332. doi:10.1086/339935
Harbour, M. E., Breusegem, S. Y., and Seaman, M. N. J. (2012). Recruitment of the Endosomal WASH Complex Is Mediated by the Extended 'tail' of Fam21 Binding to the Retromer Protein Vps35. Biochem. J. 442, 209–220. doi:10.1042/BJ20111761
Hardies, K., May, P., Djémié, T., Tarta-Arsene, O., Deconinck, T., Craiu, D., et al. (2015). Recessive Loss-Of-Function Mutations in AP4S1 Cause Mild Fever-Sensitive Seizures, Developmental Delay and Spastic Paraplegia through Loss of AP-4 Complex Assembly. Hum. Mol. Genet. 24, 2218–2227. doi:10.1093/hmg/ddu740
Harding, A. E. (1983). Classification of the Hereditary Ataxias and Paraplegias. The Lancet 321, 1151–1155. doi:10.1016/s0140-6736(83)92879-9
Hauser, S., Poenisch, M., Schelling, Y., Höflinger, P., Schuster, S., Teegler, A., et al. (2019). mRNA as a Novel Treatment Strategy for Hereditary Spastic Paraplegia Type 5. Mol. Ther. - Methods Clin. Development 15, 359–370. doi:10.1016/j.omtm.2019.10.011
Havlicek, S., Kohl, Z., Mishra, H. K., Prots, I., Eberhardt, E., Denguir, N., et al. (2014). Gene Dosage-dependent Rescue of HSP Neurite Defects in SPG4 Patients' Neurons. Hum. Mol. Genet. 23, 2527–2541. doi:10.1093/hmg/ddt644
Hazan, J., Fonknechten, N., Mavel, D., Paternotte, C., Samson, D., Artiguenave, F., et al. (1999). Spastin, a New AAA Protein, Is Altered in the Most Frequent Form of Autosomal Dominant Spastic Paraplegia. Nat. Genet. 23, 296–303. doi:10.1038/15472
Hedera, P. (1993). “Spastic Paraplegia 3A,” in GeneReviews(®). Editors R. A. Pagon, M. P. Adam, H. H. Ardinger, T. D. Bird, C. R. Dolan, and C.-T. Fong (Seattle, WA: University of Washington). Available at: http://www.ncbi.nlm.nih.gov/books/NBK45978/ (Accessed March 8, 2015).
Hegarty, S. V., O’Keeffe, G. W., and Sullivan, A. M. (2013). BMP-smad 1/5/8 Signalling in the Development of the Nervous System. Prog. Neurobiol. 109, 28–41. doi:10.1016/j.pneurobio.2013.07.002
Helal, M., Mazaheri, N., Shalbafan, B., Malamiri, R. A., Dilaver, N., Buchert, R., et al. (2018). Clinical Presentation and Natural History of Infantile-Onset Ascending Spastic Paralysis from Three Families with an ALS2 Founder Variant. Neurol. Sci. 39, 1917–1925. doi:10.1007/s10072-018-3526-8
Hirst, J., Edgar, J. R., Esteves, T., Darios, F., Madeo, M., Chang, J., et al. (2015). Loss of AP-5 Results in Accumulation of Aberrant Endolysosomes: Defining a New Type of Lysosomal Storage Disease. Hum. Mol. Genet. 24, 4984–4996. doi:10.1093/hmg/ddv220
Hirst, J., Irving, C., and Borner, G. H. H. (2013). Adaptor Protein Complexes AP-4 and AP-5: New Players in Endosomal Trafficking and Progressive Spastic Paraplegia. Traffic 14, 153–164. doi:10.1111/tra.12028
Hirst, J., Itzhak, D. N., Antrobus, R., Borner, G. H. H., and Robinson, M. S. (2018). Role of the AP-5 Adaptor Protein Complex in Late Endosome-To-Golgi Retrieval. Plos Biol. 16, e2004411. doi:10.1371/journal.pbio.2004411
Hodgkinson, C. A., Bohlega, S., Abu-Amero, S. N., Cupler, E., Kambouris, M., Meyer, B. F., et al. (2002). A Novel Form of Autosomal Recessive Pure Hereditary Spastic Paraplegia Maps to Chromosome 13q14. Neurology 59, 1905–1909. doi:10.1212/01.wnl.0000036909.49629.21
Hollstein, R., Parry, D. A., Nalbach, L., Logan, C. V., Strom, T. M., Hartill, V. L., et al. (2015). HACE1deficiency Causes an Autosomal Recessive Neurodevelopmental Syndrome. J. Med. Genet. 52, 797–803. doi:10.1136/jmedgenet-2015-103344
Hu, J., Shibata, Y., Zhu, P.-P., Voss, C., Rismanchi, N., Prinz, W. A., et al. (2009). A Class of Dynamin-like GTPases Involved in the Generation of the Tubular ER Network. Cell 138, 549–561. doi:10.1016/j.cell.2009.05.025
Huber, M. D., Vesely, P. W., Datta, K., and Gerace, L. (2013). Erlins Restrict SREBP Activation in the ER and Regulate Cellular Cholesterol Homeostasis. J. Cel Biol. 203, 427–436. doi:10.1083/jcb.201305076
Hughes, C. A., Byrne, P. C., Webb, S., McMonagle, P., Patterson, V., Hutchinson, M., et al. (2001). SPG15, A New Locus for Autosomal Recessive Complicated HSP on Chromosome 14q. Neurology 56, 1230–1233. doi:10.1212/wnl.56.9.1230
Husain, R. A., Grimmel, M., Wagner, M., Hennings, J. C., Marx, C., Feichtinger, R. G., et al. (2020). Bi-allelic HPDL Variants Cause a Neurodegenerative Disease Ranging from Neonatal Encephalopathy to Adolescent-Onset Spastic Paraplegia. Am. J. Hum. Genet. 107, 364–373. doi:10.1016/j.ajhg.2020.06.015
Ilgaz Aydinlar, E., Rolfs, A., Serteser, M., and Parman, Y. (2014). Mutation in FAM134B Causing Hereditary Sensory Neuropathy with Spasticity in a Turkish Family. Muscle Nerve 49, 774–775. doi:10.1002/mus.24145
Inoue, H., Baba, T., Sato, S., Ohtsuki, R., Takemori, A., Watanabe, T., et al. (2012). Roles of SAM and DDHD Domains in Mammalian Intracellular Phospholipase A1 KIAA0725p. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1823, 930–939. doi:10.1016/j.bbamcr.2012.02.002
Jacinto-Scudeiro, L. A., Dariva Machado, G., Ayres, A., Burguêz, D., Polese-Bonato, M., González-Salazar, C., et al. (2019). Are Cognitive Changes in Hereditary Spastic Paraplegias Restricted to Complicated Forms? Front. Neurol. 10, 508. doi:10.3389/fneur.2019.00508
Jeong, B., Kim, T. H., Kim, D.-S., Shin, W.-H., Lee, J.-R., Kim, N.-S., et al. (2019). Spastin Contributes to Neural Development through the Regulation of Microtubule Dynamics in the Primary Cilia of Neural Stem Cells. Neuroscience 411, 76–85. doi:10.1016/j.neuroscience.2019.05.024
Jia, T., Chu, C., Liu, Y., van Dongen, J., Papastergios, E., Armstrong, N. J., et al. (2019). Epigenome-wide Meta-Analysis of Blood DNA Methylation and its Association with Subcortical Volumes: Findings from the ENIGMA Epigenetics Working Group. Mol. Psychiatry. doi:10.1038/s41380-019-0605-z
Jouet, M., Rosenthal, A., Armstrong, G., MacFarlane, J., Stevenson, R., Paterson, J., et al. (1994). X-linked Spastic Paraplegia (SPG1), MASA Syndrome and X-Linked Hydrocephalus Result from Mutations in the L1 Gene. Nat. Genet. 7, 402–407. doi:10.1038/ng0794-402
Julien, C., Lissouba, A., Madabattula, S., Fardghassemi, Y., Rosenfelt, C., Androschuk, A., et al. (2016). Conserved Pharmacological Rescue of Hereditary Spastic Paraplegia-Related Phenotypes Across Model Organisms. Hum. Mol. Genet. 25, 1088–1099. doi:10.1093/hmg/ddv632
Kaji, S., Kawarai, T., Miyamoto, R., Nodera, H., Pedace, L., Orlacchio, A., et al. (2016). Late-onset Spastic Paraplegia Type 10 (SPG10) Family Presenting with Bulbar Symptoms and Fasciculations Mimicking Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 364, 45–49. doi:10.1016/j.jns.2016.03.001
Kara, E., Tucci, A., Manzoni, C., Lynch, D. S., Elpidorou, M., Bettencourt, C., et al. (2016). Genetic and Phenotypic Characterization of Complex Hereditary Spastic Paraplegia. Brain 139, 1904–1918. doi:10.1093/brain/aww111
Kevenaar, J. T., and Hoogenraad, C. C. (2015). The Axonal Cytoskeleton: From Organization to Function. Front. Mol. Neurosci. 8. doi:10.3389/fnmol.2015.00044
Klebe, S. (2006). Autosomal Recessive Spastic Paraplegia (SPG30) with Mild Ataxia and Sensory Neuropathy Maps to Chromosome 2q37.3. Brain 129, 1456–1462. doi:10.1093/brain/awl012
Klebe, S., Depienne, C., Gerber, S., Challe, G., Anheim, M., Charles, P., et al. (2012a). Spastic Paraplegia Gene 7 in Patients with Spasticity And/or Optic Neuropathy. Brain J. Neurol. 135, 2980–2993. doi:10.1093/brain/aws240
Klebe, S., Lossos, A., Azzedine, H., Mundwiller, E., Sheffer, R., Gaussen, M., et al. (2012b). KIF1A Missense Mutations in SPG30, an Autosomal Recessive Spastic Paraplegia: Distinct Phenotypes According to the Nature of the Mutations. Eur. J. Hum. Genet. 20, 645–649. doi:10.1038/ejhg.2011.261
Klebe, S., Stevanin, G., and Depienne, C. (2015). Clinical and Genetic Heterogeneity in Hereditary Spastic Paraplegias: From SPG1 to SPG72 and Still Counting. Revue Neurologique 171, 505–530. doi:10.1016/j.neurol.2015.02.017
Koh, K., Ishiura, H., Ishiura, H., Beppu, M., Shimazaki, H., Ichinose, Y., et al. (2018). Novel Mutations in the ALDH18A1 Gene in Complicated Hereditary Spastic Paraplegia with Cerebellar Ataxia and Cognitive Impairment. J. Hum. Genet. 63, 1009–1013. doi:10.1038/s10038-018-0477-0
Kousi, M., and Katsanis, N. (2015). Genetic Modifiers and Oligogenic Inheritance. Cold Spring Harbor Perspect. Med. 5, a017145. doi:10.1101/cshperspect.a017145
Landouré, G., Zhu, P.-P., Lourenço, C. M., Johnson, J. O., Toro, C., Bricceno, K. V., et al. (2013). Hereditary Spastic Paraplegia Type 43 (SPG43) Is Caused by Mutation inC19orf12. Hum. Mutat. 34, 1357–1360. doi:10.1002/humu.22378
Lardenoije, R., Roubroeks, J. A. Y., Pishva, E., Leber, M., Wagner, H., Iatrou, A., et al. (2019). Alzheimer's Disease-Associated (Hydroxy)methylomic Changes in the Brain and Blood. Clin. Epigenet 11, 164. doi:10.1186/s13148-019-0755-5
Lavie, J., Serrat, R., Bellance, N., Courtand, G., Dupuy, J.-W., Tesson, C., et al. (2017). Mitochondrial Morphology and Cellular Distribution Are Altered in SPG31 Patients and Are Linked to DRP1 Hyperphosphorylation. Hum. Mol. Genet. 26, 674–685. doi:10.1093/hmg/ddw425
Lee, J. Y. W., Hsu, C.-K., Michael, M., Nanda, A., Liu, L., McMillan, J. R., et al. (2017). Large Intragenic Deletion in DSTYK Underlies Autosomal-Recessive Complicated Spastic Paraparesis, SPG23. Am. J. Hum. Genet. 100, 364–370. doi:10.1016/j.ajhg.2017.01.014
Lin, P., Li, J., Liu, Q., Mao, F., Li, J., Qiu, R., et al. (2008). A Missense Mutation in SLC33A1, Which Encodes the Acetyl-CoA Transporter, Causes Autosomal-Dominant Spastic Paraplegia (SPG42). Am. J. Hum. Genet. 83, 752–759. doi:10.1016/j.ajhg.2008.11.003
Lindsey, J. C., Lusher, M. E., McDermott, C. J., White, K. D., Reid, E., Rubinsztein, D. C., et al. (2000). Mutation Analysis of the Spastin Gene (SPG4) in Patients with Hereditary Spastic Paraparesis. J. Med. Genet. 37, 759–765. doi:10.1136/jmg.37.10.759
Lo Giudice, T., Lombardi, F., Santorelli, F. M., Kawarai, T., and Orlacchio, A. (2014). Hereditary Spastic Paraplegia: Clinical-Genetic Characteristics and Evolving Molecular Mechanisms. Exp. Neurol. 261, 518–539. doi:10.1016/j.expneurol.2014.06.011
Lossos, A., Stümpfig, C., Stevanin, G., Gaussen, M., Zimmerman, B.-E., Mundwiller, E., et al. (2015). Fe/S Protein Assembly Gene IBA57 Mutation Causes Hereditary Spastic Paraplegia. Neurology 84, 659–667. doi:10.1212/WNL.0000000000001270
Lynch, D. S., Koutsis, G., Tucci, A., Panas, M., Baklou, M., Breza, M., et al. (2015). Hereditary Spastic Paraplegia in Greece: Characterisation of a Previously Unexplored Population Using Next-Generation Sequencing. Eur. J. Hum. Genet. 24, 857–863. doi:10.1038/ejhg.2015.200
Lynex, C. N., Carr, I. M., Leek, J. P., Achuthan, R., Mitchell, S., Maher, E. R., et al. (2004). Homozygosity for a Missense Mutation in the 67 kDa Isoform of Glutamate Decarboxylase in a Family with Autosomal Recessive Spastic Cerebral Palsy: Parallels with Stiff-Person Syndrome and Other Movement Disorders. BMC Neurol. 4, 20. doi:10.1186/1471-2377-4-20
Macedo-Souza, L. I., Kok, F., Santos, S., Amorim, S. C., Starling, A., Nishimura, A., et al. (2005). Spastic Paraplegia, Optic Atrophy, and Neuropathy Is Linked to Chromosome 11q13. Ann. Neurol. 57, 730–737. doi:10.1002/ana.20478
Macedo-Souza, L. I., Kok, F., Santos, S., Licinio, L., Lezirovitz, K., Cavaçana, N., et al. (2009). Spastic Paraplegia, Optic Atrophy, and Neuropathy: New Observations, Locus Refinement, and Exclusion of Candidate Genes. Ann. Hum. Genet. 73, 382–387. doi:10.1111/j.1469-1809.2009.00507.x
Macedo-Souza, L. I., Kok, F., Santos, S., Licinio, L., Lezirovitz, K., Nascimento, R. M. P., et al. (2008). Reevaluation of a Large Family Defines a New Locus for X-Linked Recessive Pure Spastic Paraplegia (SPG34) on Chromosome Xq25. Neurogenetics 9, 225–226. doi:10.1007/s10048-008-0130-8
Maier, A., Klopocki, E., Horn, D., Tzschach, A., Holm, T., Meyer, R., et al. (2014). De Novo Partial Deletion in GRID2 Presenting with Complicated Spastic Paraplegia. Muscle Nerve 49, 289–292. doi:10.1002/mus.24096
Mannan, A. U., Krawen, P., Sauter, S. M., Boehm, J., Chronowska, A., Paulus, W., et al. (2006). ZFYVE27 (SPG33), A Novel Spastin-Binding Protein, Is Mutated in Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 79, 351–357. doi:10.1086/504927
Marelli, C., Lamari, F., Rainteau, D., Lafourcade, A., Banneau, G., Humbert, L., et al. (2018). Plasma Oxysterols: Biomarkers for Diagnosis and Treatment in Spastic Paraplegia Type 5. Brain J. Neurol. 141, 72–84. doi:10.1093/brain/awx297
Martin, E., Schüle, R., Smets, K., Rastetter, A., Boukhris, A., Loureiro, J. L., et al. (2013). Loss of Function of Glucocerebrosidase GBA2 Is Responsible for Motor Neuron Defects in Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 92, 238–244. doi:10.1016/j.ajhg.2012.11.021
McCaughey, J., Miller, V. J., Stevenson, N. L., Brown, A. K., Budnik, A., Heesom, K. J., et al. (2016). TFG Promotes Organization of Transitional ER and Efficient Collagen Secretion. Cel Rep. 15, 1648–1659. doi:10.1016/j.celrep.2016.04.062
Meijer, I. A., Cossette, P., Roussel, J., Benard, M., Toupin, S., and Rouleau, G. A. (2004). A Novel Locus for Pure Recessive Hereditary Spastic Paraplegia Maps to 10q22.1-10q24.1. Ann. Neurol. 56, 579–582. doi:10.1002/ana.20239
Meilleur, K. G., Traoré, M., Sangaré, M., Britton, A., Landouré, G., Coulibaly, S., et al. (2010). Hereditary Spastic Paraplegia and Amyotrophy Associated with a Novel Locus on Chromosome 19. Neurogenetics 11, 313–318. doi:10.1007/s10048-009-0230-0
Melo, U. S., Macedo-Souza, L. I., Figueiredo, T., Muotri, A. R., Gleeson, J. G., Coux, G., et al. (2015). Overexpression ofKLC2due to a Homozygous Deletion in the Non-coding Region Causes SPOAN Syndrome. Hum. Mol. Genet. 24, ddv388–6885. doi:10.1093/hmg/ddv388
Méreaux, J.-L., Banneau, G., Papin, M., Coareli, G., Valter, R., Raymond, L., et al. (Forthcoming 2021b). Clinical and Genetic Spectra of 1550 index Patients with Hereditary Spastic Paraplegia. BRAIN.
Méreaux, J.-L., Firanescu, C., Coarelli, G., Kvarnung, M., Rodrigues, R., Pegoraro, E., et al. (2021a). Increasing Involvement of CAPN1 Variants in Spastic Ataxias and Phenotype-Genotype Correlations. Neurogenetics 22, 71–79. doi:10.1007/s10048-020-00633-2
Montenegro, G., Rebelo, A. P., Connell, J., Allison, R., Babalini, C., D’Aloia, M., et al. (2012). Mutations in the ER-Shaping Protein Reticulon 2 Cause the Axon-Degenerative Disorder Hereditary Spastic Paraplegia Type 12. J. Clin. Invest. 122, 538–544. doi:10.1172/JCI60560
Morais, S., Raymond, L., Mairey, M., Coutinho, P., Brandão, E., Ribeiro, P., et al. (2017). Massive Sequencing of 70 Genes Reveals a Myriad of Missing Genes or Mechanisms to Be Uncovered in Hereditary Spastic Paraplegias. Eur. J. Hum. Genet. 25, 1217–1228. doi:10.1038/ejhg.2017.124
Moreno-De-Luca, A., Helmers, S. L., Mao, H., Burns, T. G., Melton, A. M. A., Schmidt, K. R., et al. (2011). Adaptor Protein Complex-4 (AP-4) Deficiency Causes a Novel Autosomal Recessive Cerebral Palsy Syndrome with Microcephaly and Intellectual Disability. J. Med. Genet. 48, 141–144. doi:10.1136/jmg.2010.082263
Mori, F., Tanji, K., Miki, Y., Toyoshima, Y., Yoshida, M., Kakita, A., et al. (2016). G Protein-Coupled Receptor 26 Immunoreactivity in Intranuclear Inclusions Associated with Polyglutamine and Intranuclear Inclusion Body Diseases. Neuropathology 36, 50–55. doi:10.1111/neup.12237
Nahm, M., Lee, M.-J., Parkinson, W., Lee, M., Kim, H., Kim, Y.-J., et al. (2013). Spartin Regulates Synaptic Growth and Neuronal Survival by Inhibiting BMP-Mediated Microtubule Stabilization. Neuron 77, 680–695. doi:10.1016/j.neuron.2012.12.015
Namekawa, M., Muriel, M.-P., Janer, A., Latouche, M., Dauphin, A., Debeir, T., et al. (2007). Mutations in the SPG3A Gene Encoding the GTPase Atlastin Interfere with Vesicle Trafficking in the ER/Golgi Interface and Golgi Morphogenesis. Mol. Cell Neurosci. 35, 1–13. doi:10.1016/j.mcn.2007.01.012
Newton, T., Allison, R., Edgar, J. R., Lumb, J. H., Rodger, C. E., Manna, P. T., et al. (2018). Mechanistic Basis of an Epistatic Interaction Reducing Age at Onset in Hereditary Spastic Paraplegia. Brain J. Neurol. 141, 1286–1299. doi:10.1093/brain/awy034
Nolden, M., Ehses, S., Koppen, M., Bernacchia, A., Rugarli, E. I., and Langer, T. (2005). The M-AAA Protease Defective in Hereditary Spastic Paraplegia Controls Ribosome Assembly in Mitochondria. Cell 123, 277–289. doi:10.1016/j.cell.2005.08.003
Noreau, A., Dion, P. A., and Rouleau, G. A. (2014). Molecular Aspects of Hereditary Spastic Paraplegia. Exp. Cel Res. 325, 18–26. doi:10.1016/j.yexcr.2014.02.021
Novarino, G., Fenstermaker, A. G., Zaki, M. S., Hofree, M., Silhavy, J. L., Heiberg, A. D., et al. (2014). Exome Sequencing Links Corticospinal Motor Neuron Disease to Common Neurodegenerative Disorders. Science 343, 506–511. doi:10.1126/science.1247363
Oates, E. C., Rossor, A. M., Hafezparast, M., Gonzalez, M., Speziani, F., MacArthur, D. G., et al. (2013). Mutations in BICD2 Cause Dominant Congenital Spinal Muscular Atrophy and Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 92, 965–973. doi:10.1016/j.ajhg.2013.04.018
Orlacchio, A., Kawarai, T., Gaudiello, F., St George-Hyslop, P. H., Floris, R., and Bernardi, G. (2005a). New Locus for Hereditary Spastic Paraplegia Maps to Chromosome 1p31.1-1p21.1. Ann. Neurol. 58, 423–429. doi:10.1002/ana.20590
Orlacchio, A., Kawarai, T., Gaudiello, F., Totaro, A., Schillaci, O., Stefani, A., et al. (2005b). Clinical and Genetic Study of a largeSPG4 Italian Family. Mov Disord. 20, 1055–1059. doi:10.1002/mds.20494
Orlacchio, A., Patrono, C., Gaudiello, F., Rocchi, C., Moschella, V., Floris, R., et al. (2008). Silver Syndrome Variant of Hereditary Spastic Paraplegia: A Locus to 4p and Allelism with SPG4. Neurology 70, 1959–1966. doi:10.1212/01.wnl.0000294330.27058.61
Orso, G., Martinuzzi, A., Rossetto, M. G., Sartori, E., Feany, M., and Daga, A. (2005). Disease-related Phenotypes in a Drosophila Model of Hereditary Spastic Paraplegia Are Ameliorated by Treatment with Vinblastine. J. Clin. Invest. 115, 3026–3034. doi:10.1172/JCI24694
Orso, G., Pendin, D., Liu, S., Tosetto, J., Moss, T. J., Faust, J. E., et al. (2009). Homotypic Fusion of ER Membranes Requires the Dynamin-like GTPase Atlastin. Nature 460, 978–983. doi:10.1038/nature08280
Orthmann-Murphy, J. L., Salsano, E., Abrams, C. K., Bizzi, A., Uziel, G., Freidin, M. M., et al. (2008). Hereditary Spastic Paraplegia Is a Novel Phenotype for GJA12/GJC2 Mutations. Brain 132, 426–438. doi:10.1093/brain/awn328
Oz-Levi, D., Ben-Zeev, B., Ruzzo, E. K., Hitomi, Y., Gelman, A., Pelak, K., et al. (2012). Mutation in TECPR2 Reveals a Role for Autophagy in Hereditary Spastic Paraparesis. Am. J. Hum. Genet. 91, 1065–1072. doi:10.1016/j.ajhg.2012.09.015
Oz-Levi, D., Gelman, A., Elazar, Z., and Lancet, D. (2013). Tecpr2. Autophagy 9, 801–802. doi:10.4161/auto.23961
Pantakani, D., Zechner, U., Arygriou, L., Pauli, S., Sauter, S., and Mannan, A. (2008). Compound Heterozygosity in the SPG4 Gene Causes Hereditary Spastic Paraplegia. Clin. Genet. 73, 268–272. doi:10.1111/j.1399-0004.2007.00953.x
Papadopoulos, C., Orso, G., Mancuso, G., Herholz, M., Gumeni, S., Tadepalle, N., et al. (2015). Spastin Binds to Lipid Droplets and Affects Lipid Metabolism. Plos Genet. 11, e1005149. doi:10.1371/journal.pgen.1005149
Park, S. H., Zhu, P.-P., Parker, R. L., and Blackstone, C. (2010). Hereditary Spastic Paraplegia Proteins REEP1, Spastin, and Atlastin-1 Coordinate Microtubule Interactions with the Tubular ER Network. J. Clin. Invest. 120, 1097–1110. doi:10.1172/JCI40979
Parodi, L., Coarelli, G., Stevanin, G., Brice, A., and Durr, A. (2018a). Hereditary Ataxias and Paraparesias: Clinical and Genetic Update. Curr. Opin. Neurol. 31, 462–471. doi:10.1097/WCO.0000000000000585
Parodi, L., Fenu, S., Barbier, M., Banneau, G., Duyckaerts, C., Tezenas du Montcel, S., et al. (2018b). Spastic Paraplegia Due to SPAST Mutations Is Modified by the Underlying Mutation and Sex. Brain J. Neurol. 141, 3331–3342. doi:10.1093/brain/awy285
Parodi, L., Rydning, S. L., Tallaksen, C., and Durr, A. (1993). “Spastic Paraplegia 4,” in GeneReviews®. Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, and K. Stephens (Seattle, WA: University of Washington). Available at: http://www.ncbi.nlm.nih.gov/books/NBK1160/.
Patel, H., Cross, H., Proukakis, C., Hershberger, R., Bork, P., Ciccarelli, F. D., et al. (2002). SPG20 Is Mutated in Troyer Syndrome, An Hereditary Spastic Paraplegia. Nat. Genet. 31, 347–348. doi:10.1038/ng937
Penazzi, L., Bakota, L., and Brandt, R. (2016). Microtubule Dynamics in Neuronal Development, Plasticity, and Neurodegeneration. Int. Rev. Cel Mol. Biol. 321, 89–169. doi:10.1016/bs.ircmb.2015.09.004
Pennings, M., Schouten, M. I., van Gaalen, J., Meijer, R. P. P., de Bot, S. T., Kriek, M., et al. (2020). KIF1A Variants Are a Frequent Cause of Autosomal Dominant Hereditary Spastic Paraplegia. Eur. J. Hum. Genet. 28, 40–49. doi:10.1038/s41431-019-0497-z
Pirozzi, M., Quattrini, A., Andolfi, G., Dina, G., Malaguti, M. C., Auricchio, A., et al. (2006). Intramuscular Viral Delivery of Paraplegin Rescues Peripheral Axonopathy in a Model of Hereditary Spastic Paraplegia. J. Clin. Invest. 116, 202–208. doi:10.1172/JCI26210
Pozner, T., Regensburger, M., Engelhorn, T., Winkler, J., and Winner, B. (2020). Janus-faced Spatacsin (SPG11): Involvement in Neurodevelopment and Multisystem Neurodegeneration. Brain J. Neurol. 143, 2369–2379. doi:10.1093/brain/awaa099
Pozner, T., Schray, A., Regensburger, M., Lie, D. C., Schlötzer-Schrehardt, U., Winkler, J., et al. (2018). Tideglusib Rescues Neurite Pathology of SPG11 iPSC Derived Cortical Neurons. Front. Neurosci. 12, 914. doi:10.3389/fnins.2018.00914
Pujol, C., Legrand, A., Parodi, L., Thomas, P., Mochel, F., Saracino, D., et al. (2021). Implication of Folate Deficiency in CYP2U1 Loss of Function. J. Exp. Med. 218 (11), e20210846. doi:10.1084/jem.20210846
Racis, L., Tessa, A., Pugliatti, M., Storti, E., Agnetti, V., and Santorelli, F. M. (2014). Infantile-onset Ascending Hereditary Spastic Paralysis: A Case Report and Brief Literature Review. Eur. J. Paediatric Neurol. 18, 235–239. doi:10.1016/j.ejpn.2013.09.009
Rainier, S., Bui, M., Mark, E., Thomas, D., Tokarz, D., Ming, L., et al. (2008). Neuropathy Target Esterase Gene Mutations Cause Motor Neuron Disease. Am. J. Hum. Genet. 82, 780–785. doi:10.1016/j.ajhg.2007.12.018
Rainier, S., Chai, J.-H., Tokarz, D., Nicholls, R. D., and Fink, J. K. (2003). NIPA1 Gene Mutations Cause Autosomal Dominant Hereditary Spastic Paraplegia (SPG6). Am. J. Hum. Genet. 73, 967–971. doi:10.1086/378817
Reid, E., Kloos, M., Ashley-Koch, A., Hughes, L., Bevan, S., Svenson, I. K., et al. (2002). A Kinesin Heavy Chain (KIF5A) Mutation in Hereditary Spastic Paraplegia (SPG10). Am. J. Hum. Genet. 71, 1189–1194. doi:10.1086/344210
Renvoisé, B., Chang, J., Singh, R., Yonekawa, S., FitzGibbon, E. J., Mankodi, A., et al. (2014). Lysosomal Abnormalities in Hereditary Spastic Paraplegia Types SPG 15 and SPG 11. Ann. Clin. Transl. Neurol. 1, 379–389. doi:10.1002/acn3.64
Rinaldi, C., Schmidt, T., Situ, A. J., Johnson, J. O., Lee, P. R., Chen, K.-L., et al. (2015). Mutation inCPT1CAssociated with Pure Autosomal Dominant Spastic Paraplegia. JAMA Neurol. 72, 561–570. doi:10.1001/jamaneurol.2014.4769
Rizzo, G., Tonon, C., Gramegna, L. L., Bassi, M. T., Lodi, R., and Liguori, R. (2020). ''Eye of Tiger Sign" Mimic in Patients with Spastic Paraplegia Gene 7 (SPG7) Mutations. Parkinsonism Relat. Disord. 81, 158–160. doi:10.1016/j.parkreldis.2020.10.049
Rodrigues, R., Silva, R., Branco, M., Brandão, E., Alonso, I., Ruano, L., et al. (2020). Determinants of Age at Onset in a Portuguese Cohort of Autosomal Dominant Spastic Paraplegia. J. Neurol. Sci. 410, 116646. doi:10.1016/j.jns.2019.116646
Rosenthal, A., Jouet, M., and Kenwrick, S. (1992). Aberrant Splicing of Neural Cell Adhesion Molecule L1 mRNA in a Family with X-Linked Hydrocephalus. Nat. Genet. 2, 107–112. doi:10.1038/ng1092-107
Ruano, L., Melo, C., Silva, M. C., and Coutinho, P. (2014). The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence Studies. Neuroepidemiology 42, 174–183. doi:10.1159/000358801
Rubegni, A., Storti, E., Tessa, A., Federico, A., and Santorelli, F. M. (2015). Hereditary Spastic Paraplegia Type 11 with a Very Late Onset. J. Neurol. 262, 1987–1989. doi:10.1007/s00415-015-7854-9
Rugarli, E. I., and Langer, T. (2006). Translating M-AAA Protease Function in Mitochondria to Hereditary Spastic Paraplegia. Trends Mol. Med. 12, 262–269. doi:10.1016/j.molmed.2006.04.002
Rydning, S. L., Dudesek, A., Rimmele, F., Funke, C., Krüger, S., Biskup, S., et al. (2018). A Novel Heterozygous Variant inERLIN2causes Autosomal Dominant Pure Hereditary Spastic Paraplegia. Eur. J. Neurol. 25, 943–e71. doi:10.1111/ene.13625
Rydning, S. L., Koht, J., Sheng, Y., Sowa, P., Hjorthaug, H. S., Wedding, I. M., et al. (2019). Biallelic POLR3A Variants Confirmed as a Frequent Cause of Hereditary Ataxia and Spastic Paraparesis. Brain 142, e12. doi:10.1093/brain/awz041
Sambuughin, N., Goldfarb, L. G., Sivtseva, T. M., Davydova, T. K., Vladimirtsev, V. A., Osakovskiy, V. L., et al. (2015). Adult-onset Autosomal Dominant Spastic Paraplegia Linked to a GTPase-Effector Domain Mutation of Dynamin 2. BMC Neurol. 15, 223. doi:10.1186/s12883-015-0481-3
Saugier-Veber, P., Munnich, A., Bonneau, D., Rozet, J.-M., Le Merrer, M., Gil, R., et al. (1994). X-linked Spastic Paraplegia and Pelizaeus-Merzbacher Disease Are Allelic Disorders at the Proteolipid Protein Locus. Nat. Genet. 6, 257–262. doi:10.1038/ng0394-257
Schickel, J., Pamminger, T., Ehrsam, A., Münch, S., Huang, X., Klopstock, T., et al. (2007). Isoform-specific Increase of Spastin Stability by N-Terminal Missense Variants Including Intragenic Modifiers ofSPG4hereditary Spastic Paraplegia. Eur. J. Neurol. 14, 1322–1328. doi:10.1111/j.1468-1331.2007.01971.x
Schöls, L., Rattay, T. W., Martus, P., Meisner, C., Baets, J., Fischer, I., et al. (2017). Hereditary Spastic Paraplegia Type 5: Natural History, Biomarkers and a Randomized Controlled Trial. Brain J. Neurol. 140, 3112–3127. doi:10.1093/brain/awx273
Schüle, R., Holland-Letz, T., Klimpe, S., Kassubek, J., Klopstock, T., Mall, V., et al. (2006). The Spastic Paraplegia Rating Scale (SPRS): A Reliable and Valid Measure of Disease Severity. Neurology 67, 430–434. doi:10.1212/01.wnl.0000228242.53336.90
Schüle, R., Siddique, T., Deng, H.-X., Yang, Y., Donkervoort, S., Hansson, M., et al. (2010). Marked Accumulation of 27-hydroxycholesterol in SPG5 Patients with Hereditary Spastic Paresis. J. Lipid Res. 51, 819–823. doi:10.1194/jlr.M002543
Schüle, R., Wiethoff, S., Martus, P., Karle, K. N., Otto, S., Klebe, S., et al. (2016). Hereditary Spastic Paraplegia: Clinicogenetic Lessons from 608 Patients. Ann. Neurol. 79, 646–658. doi:10.1002/ana.24611
Schuurs-Hoeijmakers, J. H. M., Geraghty, M. T., Kamsteeg, E.-J., Ben-Salem, S., de Bot, S. T., Nijhof, B., et al. (2012). Mutations in DDHD2, Encoding an Intracellular Phospholipase A1, Cause a Recessive Form of Complex Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 91, 1073–1081. doi:10.1016/j.ajhg.2012.10.017
Seidel, K., Siswanto, S., Fredrich, M., Bouzrou, M., Brunt, E. R., van Leeuwen, F. W., et al. (2015). Polyglutamine Aggregation in Huntington’s Disease and Spinocerebellar Ataxia Type 3: Similar Mechanisms in Aggregate Formation. Neuropathol. Appl. Neurobiol. 42, 153–166. doi:10.1111/nan.12253
Seri, M., Cusano, R., Forabosco, P., Cinti, R., Caroli, F., Picco, P., et al. (1999). Genetic Mapping to 10q23.3-q24.2, in a Large Italian Pedigree, of a New Syndrome Showing Bilateral Cataracts, Gastroesophageal Reflux, and Spastic Paraparesis with Amyotrophy. Am. J. Hum. Genet. 64, 586–593. doi:10.1086/302241
Shanmughapriya, S., Rajan, S., Hoffman, N. E., Higgins, A. M., Tomar, D., Nemani, N., et al. (2015). SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol. Cel 60, 47–62. doi:10.1016/j.molcel.2015.08.009
Shibata, Y., Shemesh, T., Prinz, W. A., Palazzo, A. F., Kozlov, M. M., and Rapoport, T. A. (2010). Mechanisms Determining the Morphology of the Peripheral ER. Cell 143, 774–788. doi:10.1016/j.cell.2010.11.007
Shimazaki, H., Honda, J., Naoi, T., Namekawa, M., Nakano, I., Yazaki, M., et al. (2014). Autosomal-recessive Complicated Spastic Paraplegia with a Novel Lysosomal Trafficking Regulator Gene Mutation. J. Neurol. Neurosurg. Psychiatry 85, 1024–1028. doi:10.1136/jnnp-2013-306981
Shimazaki, H., Takiyama, Y., Ishiura, H., Sakai, C., Matsushima, Y., Hatakeyama, H., et al. (2012). A Homozygous Mutation ofC12orf65causes Spastic Paraplegia with Optic Atrophy and Neuropathy (SPG55). J. Med. Genet. 49, 777–784. doi:10.1136/jmedgenet-2012-101212
Simone, M., Trabacca, A., Panzeri, E., Losito, L., Citterio, A., and Bassi, M. T. (2018). KIF5A and ALS2 Variants in a Family with Hereditary Spastic Paraplegia and Amyotrophic Lateral Sclerosis. Front. Neurol. 9, 1078. doi:10.3389/fneur.2018.01078
Simpson, M. A., Cross, H., Proukakis, C., Pryde, A., Hershberger, R., Chatonnet, A., et al. (2003). Maspardin Is Mutated in Mast Syndrome, a Complicated Form of Hereditary Spastic Paraplegia Associated with Dementia. Am. J. Hum. Genet. 73, 1147–1156. doi:10.1086/379522
Słabicki, M., Theis, M., Krastev, D. B., Samsonov, S., Mundwiller, E., Junqueira, M., et al. (2010). A Genome-Scale DNA Repair RNAi Screen Identifies SPG48 as a Novel Gene Associated with Hereditary Spastic Paraplegia. Plos Biol. 8, e1000408. doi:10.1371/journal.pbio.1000408
Solowska, J. M., D'Rozario, M., Jean, D. C., Davidson, M. W., Marenda, D. R., and Baas, P. W. (2014). Pathogenic Mutation of Spastin Has Gain-Of-Function Effects on Microtubule Dynamics. J. Neurosci. 34, 1856–1867. doi:10.1523/JNEUROSCI.3309-13.2014
Solowska, J. M., Garbern, J. Y., and Baas, P. W. (2010). Evaluation of Loss of Function as an Explanation for SPG4-Based Hereditary Spastic Paraplegia. Hum. Mol. Genet. 19, 2767–2779. doi:10.1093/hmg/ddq177
Song, Y., Wang, M., Mao, F., Shao, M., Zhao, B., Song, Z., et al. (2013). Knockdown of Pnpla6 Protein Results in Motor Neuron Defects in Zebrafish. Dis. Model. Mech. 6, 404–413. doi:10.1242/dmm.009688
Sridharan, R., Radhakrishnan, K., Ashok, P. P., and Mousa, M. E. (1985). Prevalence and Pattern of Spinocerebellar Degenerations in Northeastern Libya. Brain 108 (Pt 4), 831–843. doi:10.1093/brain/108.4.831
Starling, A., Rocco, P., Cambi, F., Hobson, G. M., Passos Bueno, M. R., Zatz, M., et al. (2002). Further Evidence for a Fourth Gene Causing X-Linked Pure Spastic Paraplegia. Am. J. Med. Genet. 111, 152–156. doi:10.1002/ajmg.10551
Steinmüller, R., Lantigua-Cruz, A., Garcia-Garcia, R., Kostrzewa, M., Steinberger, D., and Müller, U. (1997). Evidence of a Third Locus in X-Linked Recessive Spastic Paraplegia. Hum. Genet. 100, 287–289. doi:10.1007/s004390050507
Stevanin, G., Paternotte, C., Coutinho, P., Klebe, S., Elleuch, N., Loureiro, J. L., et al. (2007a). A New Locus for Autosomal Recessive Spastic Paraplegia (SPG32) on Chromosome 14q12-Q21. Neurology 68, 1837–1840. doi:10.1212/01.wnl.0000262043.53386.22
Stevanin, G., Santorelli, F. M., Azzedine, H., Coutinho, P., Chomilier, J., Denora, P. S., et al. (2007b). Mutations in SPG11, Encoding Spatacsin, Are a Major Cause of Spastic Paraplegia with Thin Corpus Callosum. Nat. Genet. 39, 366–372. doi:10.1038/ng1980
Subramony, S. H., Nguyen, T. V., Langford, L., Lin, X., Parent, A. D., and Zhang, J. (2009). Identification of a New Form of Autosomal Dominant Spastic Paraplegia. Clin. Genet. 76, 113–116. doi:10.1111/j.1399-0004.2008.01122.x
Svenson, I. K., Kloos, M. T., Gaskell, P. C., Nance, M. A., Garbern, J. Y., Hisanaga, S.-i., et al. (2004). Intragenic Modifiers of Hereditary Spastic Paraplegia Due to Spastin Gene Mutations. Neurogenetics 5, 157–164. doi:10.1007/s10048-004-0186-z
Synofzik, M., Gonzalez, M. A., Lourenco, C. M., Coutelier, M., Haack, T. B., Rebelo, A., et al. (2014). PNPLA6 Mutations Cause Boucher-Neuhäuser and Gordon Holmes Syndromes as Part of a Broad Neurodegenerative Spectrum. Brain 137, 69–77. doi:10.1093/brain/awt326
Synofzik, M., and Ilg, W. (2014). Motor Training in Degenerative Spinocerebellar Disease: Ataxia-specific Improvements by Intensive Physiotherapy and Exergames. Biomed. Res. Int. 2014, 1–11. doi:10.1155/2014/583507
Synofzik, M., and Schüle, R. (2017). Overcoming the Divide Between Ataxias and Spastic Paraplegias: Shared Phenotypes, Genes, and Pathways. Mov Disord. 32, 332–345. doi:10.1002/mds.26944
Tadepalle, N., and Rugarli, E. I. (2021). Lipid Droplets in the Pathogenesis of Hereditary Spastic Paraplegia. Front. Mol. Biosci. 8, 673977. doi:10.3389/fmolb.2021.673977
Terada, T., Kono, S., Ouchi, Y., Yoshida, K., Hamaya, Y., Kanaoka, S., et al. (2013). SPG3A-linked Hereditary Spastic Paraplegia Associated with Cerebral Glucose Hypometabolism. Ann. Nucl. Med. 27, 303–308. doi:10.1007/s12149-012-0673-5
Tesson, C., Koht, J., and Stevanin, G. (2015). Delving into the Complexity of Hereditary Spastic Paraplegias: How Unexpected Phenotypes and Inheritance Modes Are Revolutionizing Their Nosology. Hum. Genet. 134, 511–538. doi:10.1007/s00439-015-1536-7
Tesson, C., Nawara, M., Salih, M. A. M., Rossignol, R., Zaki, M. S., Al Balwi, M., et al. (2012). Alteration of Fatty-Acid-Metabolizing Enzymes Affects Mitochondrial Form and Function in Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 91, 1051–1064. doi:10.1016/j.ajhg.2012.11.001
Tiranti, V., Corona, P., Greco, M., Taanman, J.-W., Carrara, F., Lamantea, E., et al. (2000). A Novel Frameshift Mutation of the mtDNA COIII Gene Leads to Impaired Assembly of Cytochrome C Oxidase in a Patient Affected by Leigh-like Syndrome. Hum. Mol. Genet. 9, 2733–2742. doi:10.1093/hmg/9.18.2733
Toupenet Marchesi, L., Leblanc, M., and Stevanin, G. (2021). Current Knowledge of Endolysosomal and Autophagy Defects in Hereditary Spastic Paraplegia. Cells 10, 1678. doi:10.3390/cells10071678
Travaglini, L., Aiello, C., Stregapede, F., D’Amico, A., Alesi, V., Ciolfi, A., et al. (2018). The Impact of Next-Generation Sequencing on the Diagnosis of Pediatric-Onset Hereditary Spastic Paraplegias: New Genotype-Phenotype Correlations for Rare HSP-Related Genes. Neurogenetics 19, 111–121. doi:10.1007/s10048-018-0545-9
Trotta, N., Orso, G., Rossetto, M. G., Daga, A., and Broadie, K. (2004). The Hereditary Spastic Paraplegia Gene, Spastin, Regulates Microtubule Stability to Modulate Synaptic Structure and Function. Curr. Biol. 14, 1135–1147. doi:10.1016/j.cub.2004.06.058
Tsang, H. T. H., Edwards, T. L., Wang, X., Connell, J. W., Davies, R. J., Durrington, H. J., et al. (2009). The Hereditary Spastic Paraplegia Proteins NIPA1, Spastin and Spartin Are Inhibitors of Mammalian BMP Signalling. Hum. Mol. Genet. 18, 3805–3821. doi:10.1093/hmg/ddp324
Tsaousidou, M. K., Ouahchi, K., Warner, T. T., Yang, Y., Simpson, M. A., Laing, N. G., et al. (2008). Sequence Alterations within CYP7B1 Implicate Defective Cholesterol Homeostasis in Motor-Neuron Degeneration. Am. J. Hum. Genet. 82, 510–515. doi:10.1016/j.ajhg.2007.10.001
Tüysüz, B., Bilguvar, K., Koçer, N., Yalçınkaya, C., Çağlayan, O., Gül, E., et al. (2014). Autosomal Recessive Spastic Tetraplegia Caused by AP4M1 and AP4B1 Gene Mutation: Expansion of the Facial and Neuroimaging Features. Am. J. Med. Genet. 164, 1677–1685. doi:10.1002/ajmg.a.36514
Vaccaro, A., Patten, S. A., Aggad, D., Julien, C., Maios, C., Kabashi, E., et al. (2013). Pharmacological Reduction of ER Stress Protects Against TDP-43 Neuronal Toxicity In Vivo. Neurobiol. Dis. 55, 64–75. doi:10.1016/j.nbd.2013.03.015
Valdmanis, P. N., Meijer, I. A., Reynolds, A., Lei, A., MacLeod, P., Schlesinger, D., et al. (2007). Mutations in the KIAA0196 Gene at the SPG8 Locus Cause Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 80, 152–161. doi:10.1086/510782
Valente, E. M., Brancati, F., Caputo, V., Bertini, E., Patrono, C., Costanti, D., et al. (2002). Novel Locus for Autosomal Dominant Pure Hereditary Spastic Paraplegia (SPG19) Maps to Chromosome 9q33-Q34. Ann. Neurol. 51, 681–685. doi:10.1002/ana.10204
Vallat, J.-M., Goizet, C., Tazir, M., Couratier, P., Magy, L., and Mathis, S. (2016). Classifications of Neurogenetic Diseases: An Increasingly Complex Problem. Revue Neurologique 172, 339–349. doi:10.1016/j.neurol.2016.04.005
van de Warrenburg, B. P., Schouten, M. I., de Bot, S. T., Vermeer, S., Meijer, R., Pennings, M., et al. (2016). Clinical Exome Sequencing for Cerebellar Ataxia and Spastic Paraplegia Uncovers Novel Gene-Disease Associations and Unanticipated Rare Disorders. Eur. J. Hum. Genet. 24, 1460–1466. doi:10.1038/ejhg.2016.42
Vavouraki, N., Tomkins, J. E., Kara, E., Houlden, H., Hardy, J., Tindall, M. J., et al. (2021). Integrating Protein Networks and Machine Learning for Disease Stratification in the Hereditary Spastic Paraplegias. iScience 24, 102484. doi:10.1016/j.isci.2021.102484
Vaz, F. M., McDermott, J. H., Alders, M., Wortmann, S. B., Kölker, S., Pras-Raves, M. L., et al. (2019). Mutations in PCYT2 Disrupt Etherlipid Biosynthesis and Cause a Complex Hereditary Spastic Paraplegia. Brain J. Neurol. 142, 3382–3397. doi:10.1093/brain/awz291
Vazza, G., Zortea, M., Boaretto, F., Micaglio, G. F., Sartori, V., and Mostacciuolo, M. L. (2000). A New Locus for Autosomal Recessive Spastic Paraplegia Associated with Mental Retardation and Distal Motor Neuropathy, SPG14, Maps to Chromosome 3q27-Q28. Am. J. Hum. Genet. 67, 504–509. doi:10.1086/303017
Vecchia, S. D., Tessa, A., Dosi, C., Baldacci, J., Pasquariello, R., Antenora, A., et al. (2021). Monoallelic KIF1A-Related Disorders: a Multicenter Cross Sectional Study and Systematic Literature Review. J. Neurol. [Epub ahead of print]. doi:10.1007/s00415-021-10792-3
Verkerk, A. J. M. H., Schot, R., Dumee, B., Schellekens, K., Swagemakers, S., Bertoli-Avella, A. M., et al. (2009). Mutation in the AP4M1 Gene Provides a Model for Neuroaxonal Injury in Cerebral Palsy. Am. J. Hum. Genet. 85, 40–52. doi:10.1016/j.ajhg.2009.06.004
Verny, C., Guegen, N., Desquiret, V., Chevrollier, A., Prundean, A., Dubas, F., et al. (2011). Hereditary Spastic Paraplegia-like Disorder Due to a Mitochondrial ATP6 Gene Point Mutation. Mitochondrion 11, 70–75. doi:10.1016/j.mito.2010.07.006
Voeltz, G. K., Prinz, W. A., Shibata, Y., Rist, J. M., and Rapoport, T. A. (2006). A Class of Membrane Proteins Shaping the Tubular Endoplasmic Reticulum. Cell 124, 573–586. doi:10.1016/j.cell.2005.11.047
Wagner, M., Osborn, D. P. S., Gehweiler, I., Nagel, M., Ulmer, U., Bakhtiari, S., et al. (2019). Bi-allelic Variants in RNF170 Are Associated with Hereditary Spastic Paraplegia. Nat. Commun. 10, 4790. doi:10.1038/s41467-019-12620-9
Wakil, S. M., Monies, D., Hagos, S., Al-Ajlan, F., Finsterer, J., Al Qahtani, A., et al. (2018). Exome Sequencing: Mutilating Sensory Neuropathy with Spastic Paraplegia Due to a Mutation in FAM134B Gene. Case Rep. Genet. 2018, 1–5. doi:10.1155/2018/9468049
Wakil, S. M., Ramzan, K., Abuthuraya, R., Hagos, S., Al-Dossari, H., Al-Omar, R., et al. (2014). Infantile-onset Ascending Hereditary Spastic Paraplegia with Bulbar Involvement Due to the Novel ALS2 Mutation c.2761C > T. Gene 536, 217–220. doi:10.1016/j.gene.2013.11.043
Wali, G., Kumar, K. R., Liyanage, E., Davis, R. L., Mackay-Sim, A., and Sue, C. M. (2020). Mitochondrial Function in Hereditary Spastic Paraplegia: Deficits in SPG7 but Not SPAST Patient-Derived Stem Cells. Front. Neurosci. 14, 820. doi:10.3389/fnins.2020.00820
Wang, R. N., Green, J., Wang, Z., Deng, Y., Qiao, M., Peabody, M., et al. (2014). Bone Morphogenetic Protein (BMP) Signaling in Development and Human Diseases. Genes Dis. 1, 87–105. doi:10.1016/j.gendis.2014.07.005
Wang, X., Shaw, W. R., Tsang, H. T. H., Reid, E., and O'Kane, C. J. (2007). Drosophila Spichthyin Inhibits BMP Signaling and Regulates Synaptic Growth and Axonal Microtubules. Nat. Neurosci. 10, 177–185. doi:10.1038/nn1841
Watanabe, F., Arnold, W. D., Hammer, R. E., Ghodsizadeh, O., Moti, H., Schumer, M., et al. (2013). Pathogenesis of Autosomal Dominant Hereditary Spastic Paraplegia (SPG6) Revealed by a Rat Model. J. Neuropathol. Exp. Neurol. 72, 1016–1028. doi:10.1097/NEN.0000000000000000
Wedding, I. M., Koht, J., Tran, G. T., Misceo, D., Selmer, K. K., Holmgren, A., et al. (2014). Spastic Paraplegia Type 7 Is Associated with Multiple Mitochondrial DNA Deletions. PLOS ONE 9, e86340. doi:10.1371/journal.pone.0086340
Wen, Z., Han, L., Bamburg, J. R., Shim, S., Ming, G.-l., and Zheng, J. Q. (2007). BMP Gradients Steer Nerve Growth Cones by a Balancing Act of LIM Kinase and Slingshot Phosphatase on ADF/cofilin. J. Cel Biol. 178, 107–119. doi:10.1083/jcb.200703055
Wilkinson, P. A., Simpson, M. A., Bastaki, L., Patel, H., Reed, J. A., Kalidas, K., et al. (2005). A New Locus for Autosomal Recessive Complicated Hereditary Spastic Paraplegia (SPG26) Maps to Chromosome 12p11.1-12q14. J. Med. Genet. 42, 80–82. doi:10.1136/jmg.2004.020172
Windpassinger, C., Auer-Grumbach, M., Irobi, J., Patel, H., Petek, E., Hörl, G., et al. (2004). Heterozygous Missense Mutations in BSCL2 Are Associated with Distal Hereditary Motor Neuropathy and Silver Syndrome. Nat. Genet. 36, 271–276. doi:10.1038/ng1313
Xu, S., Stern, M., and McNew, J. A. (2017). Beneficial Effects of Rapamycin in a Drosophila Model for Hereditary Spastic Paraplegia. J. Cel Sci. 130, 453–465. doi:10.1242/jcs.196741
Xylaki, M., Atzler, B., and Outeiro, T. F. (2019). Epigenetics of the Synapse in Neurodegeneration. Curr. Neurol. Neurosci. Rep. 19, 72. doi:10.1007/s11910-019-0995-y
Yadav, P. K., and Rajasekharan, R. (2016). Misregulation of a DDHD Domain-Containing Lipase Causes Mitochondrial Dysfunction in Yeast. J. Biol. Chem. 291, 18562–18581. doi:10.1074/jbc.M116.733378
Yahia, A., Chen, Z. S., Ahmed, A. E., Emad, S., Adil, R., Abubaker, R., et al. (2021). A Heterozygous Mutation in the CCDC88C Gene Likely Causes Early-Onset Pure Hereditary Spastic Paraplegia: A Case Report. BMC Neurol. 21, 78. doi:10.1186/s12883-021-02113-y
Yahia, A., Elsayed, L., Babai, A., Salih, M. A., El-Sadig, S. M., Amin, M., et al. (2018). Intra-familial Phenotypic Heterogeneity in a Sudanese Family with DARS2-Related Leukoencephalopathy, Brainstem and Spinal Cord Involvement and Lactate Elevation: A Case Report. BMC Neurol. 18, 175. doi:10.1186/s12883-018-1180-7
Yang, Y., Liu, W., Fang, Z., Shi, J., Che, F., He, C., et al. (2016). A Newly Identified Missense Mutation inFARS2Causes Autosomal-Recessive Spastic Paraplegia. Hum. Mutat. 37, 165–169. doi:10.1002/humu.22930
Yıldırım, Y., Kocasoy Orhan, E., Ugur Iseri, S. A., Serdaroglu-Oflazer, P., Kara, B., Solakoğlu, S., et al. (2011). A Frameshift Mutation of ERLIN2 in Recessive Intellectual Disability, Motor Dysfunction and Multiple Joint Contractures. Hum. Mol. Genet. 20, 1886–1892. doi:10.1093/hmg/ddr070
Zaki, M., Abdel-Hamid, M., Abdel-Salam, G., Boespflug-Tanguy, O., Cordeiro, N., Gleeson, J., et al. (2014). Mutations in ADAR1, IFIH1, and RNASEH2B Presenting as Spastic Paraplegia. Neuropediatrics 45, 386–391. doi:10.1055/s-0034-1389161
Zanni, G., Scotton, C., Passarelli, C., Fang, M., Barresi, S., Dallapiccola, B., et al. (2013). Exome Sequencing in a Family with Intellectual Disability, Early Onset Spasticity, and Cerebellar Atrophy Detects a Novel Mutation in EXOSC3. Neurogenetics 14, 247–250. doi:10.1007/s10048-013-0371-z
Zhao, G.-h., Hu, Z.-m., Shen, L., Jiang, H., Ren, Z.-j., Liu, X.-m., et al. (2008). A Novel Candidate Locus on Chromosome 11p14.1-p11.2 for Autosomal Dominant Hereditary Spastic Paraplegia. Chin. Med. J. 121, 430–434. doi:10.1097/00029330-200803010-00012
Zhao, X., Alvarado, D., Rainier, S., Lemons, R., Hedera, P., Weber, C. H., et al. (2001). Mutations in a Newly Identified GTPase Gene Cause Autosomal Dominant Hereditary Spastic Paraplegia. Nat. Genet. 29, 326–331. doi:10.1038/ng758
Zhu, P.-P., Denton, K. R., Pierson, T. M., Li, X.-J., and Blackstone, C. (2014). Pharmacologic Rescue of Axon Growth Defects in a Human iPSC Model of Hereditary Spastic Paraplegia SPG3A. Hum. Mol. Genet. 23, 5638–5648. doi:10.1093/hmg/ddu280
Zivony-Elboum, Y., Westbroek, W., Kfir, N., Savitzki, D., Shoval, Y., Bloom, A., et al. (2012). A Founder Mutation in Vps37A Causes Autosomal Recessive Complex Hereditary Spastic Paraparesis. J. Med. Genet. 49, 462–472. doi:10.1136/jmedgenet-2012-100742
Zortea, M., Vettori, A., Trevisan, C. P., Bellini, S., Vazza, G., Armani, M., et al. (2002). Genetic Mapping of a Susceptibility Locus for Disc Herniation and Spastic Paraplegia on 6q23.3-q24.1. J. Med. Genet. 39, 387–390. doi:10.1136/jmg.39.6.387
Keywords: spastic paraplegia, clinical spectrum, genetic heterogeneity, phenotype-genotype correlation, molecular mechanisms, diagnostic yield, diagnostic gap, allelic variants
Citation: Elsayed LEO, Eltazi IZ, Ahmed AE and Stevanin G (2021) Insights into Clinical, Genetic, and Pathological Aspects of Hereditary Spastic Paraplegias: A Comprehensive Overview. Front. Mol. Biosci. 8:690899. doi: 10.3389/fmolb.2021.690899
Received: 04 April 2021; Accepted: 19 October 2021;
Published: 26 November 2021.
Edited by:
David Pla-Martín, University Hospital of Cologne, GermanyReviewed by:
Carmen Espinós, Principe Felipe Research Center (CIPF), SpainAlan Mackay-Sim, Griffith University, Australia
Copyright © 2021 Elsayed, Eltazi, Ahmed and Stevanin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liena E. O. Elsayed, bGVlbHNheWVkQHBudS5lZHUuc2E=; Giovanni Stevanin, Z2lvdmFubmktYi5zdGV2YW5pbkBpbnNlcm0uZnI=