 Vandana Yadav
Vandana Yadav Inayat Ullah Irshad2†
Inayat Ullah Irshad2† Hemant Kumar
Hemant Kumar Ajeet K. Sharma
Ajeet K. Sharma
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Biosci., 28 June 2021
Sec. Biological Modeling and Simulation
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.688700
This article is part of the Research TopicCombining Simulations, Theory, and Experiments into Multiscale Models of Biological EventsView all 23 articles
Quantitative prediction on protein synthesis requires accurate translation initiation and codon translation rates. Ribosome profiling data, which provide steady-state distribution of relative ribosome occupancies along a transcript, can be used to extract these rate parameters. Various methods have been developed in the past few years to measure translation-initiation and codon translation rates from ribosome profiling data. In the review, we provide a detailed analysis of the key methods employed to extract the translation rate parameters from ribosome profiling data. We further discuss how these approaches were used to decipher the role of various structural and sequence-based features of mRNA molecules in the regulation of gene expression. The utilization of these accurate rate parameters in computational modeling of protein synthesis may provide new insights into the kinetic control of the process of gene expression.
Protein molecules carry out a vast array of biological functions. Indeed, almost every cellular process, from genome regulation to energy metabolism, requires a unique set of proteins with their precise concentration in a cell (Berg et al., 2002; Miyazaki and Esser, 2009). Therefore, the abundance of proteins in a cell is tightly regulated by various mechanisms acting at the level of transcription and translation (Curtis et al., 1995; Wu and Belasco, 2008). Understanding this regulation of protein synthesis remains one of the active areas of research from the last few decades (Merrick, 1992; Proud, 2006; Dever et al., 2016; Kummer and Ban, 2021). It was previously believed that cellular protein levels are primarily determined by mRNA copy number (Greenbaum et al., 2003; Lu et al., 2007). However, recent studies demonstrated that the translational regulation of protein synthesis also contributes significantly to in vivo protein abundance (Vogel et al., 2010; Li et al., 2014). Translational regulation is achieved by various structural and sequence-based features of mRNA molecules that can kinetically control the rate of protein synthesis (Kudla et al., 2009; Vogel et al., 2010; Tuller et al., 2010). Therefore, the knowledge of the rates at which different steps of translation occur and their connection with those mRNA features would provide key information concerning how the expression of an individual gene is regulated.
Single-molecule experiments provided significant insights into the details of the process of protein synthesis (Munro et al., 2008; Volkov and Johansson, 2018). However, little was known on how different mRNA features can control protein synthesis until the Next Generation Sequencing (NGS) experiments started uncovering various kinetic properties of this process (Ingolia et al., 2019). Several methods have been developed in the last decade that allows the accurate extraction of translation rate parameters using ribosome profiling data (Weinberg et al., 2016; Duc and Song, 2018; Sharma et al., 2019). The analysis of these translation rate parameters and their use in protein synthesis simulations have started unraveling the sophisticated mechanism that nature has developed to optimize the use of cellular resources (Tuller et al., 2010; Diament et al., 2018; Lyu et al., 2020).
In this mini-review, we provide the brief overview of a few recently developed methods that extract translation rate parameters from ribosome profiling data. We explain the technical details and assumptions made in these approaches, and also comment on their accuracy in several different contexts. We also highlight some of the recent results that use ribosome profiling data to provide new insights into the kinetic control of protein synthesis. This mini-review aims to promote the use of big biological data sets among biophysicists, biophysical chemists and system biologist to achieve greater accuracy and reliability in the quantitative modeling of protein synthesis.
The first mathematical model of protein synthesis was developed by MacDonald et al. (1968), and since then various similar models and their extensions were proposed to understand the different aspects of mRNA translation (Garai et al., 2009; Sharma and Chowdhury, 2012; Ciandrini et al., 2013; Margaliot and Tuller, 2013). Among these, the totally asymmetric simple exclusion process (TASEP) is a model which incorporates key steps regulating in vivo protein production and is extensively used to stimulate protein synthesis. In this model, an mRNA is considered as a one-dimensional lattice where each site in that lattice represents a single codon. A ribosome in this model is like an extended particle that covers ten consecutive codon positions of a transcript where its location is usually identified by the position of its A-site (Sharma and O’Brien, 2017; Ingolia et al., 2009). The ribosome A-site is located at the sixth codon from the 5′ end of the ribosome. Therefore, a ribosome at the
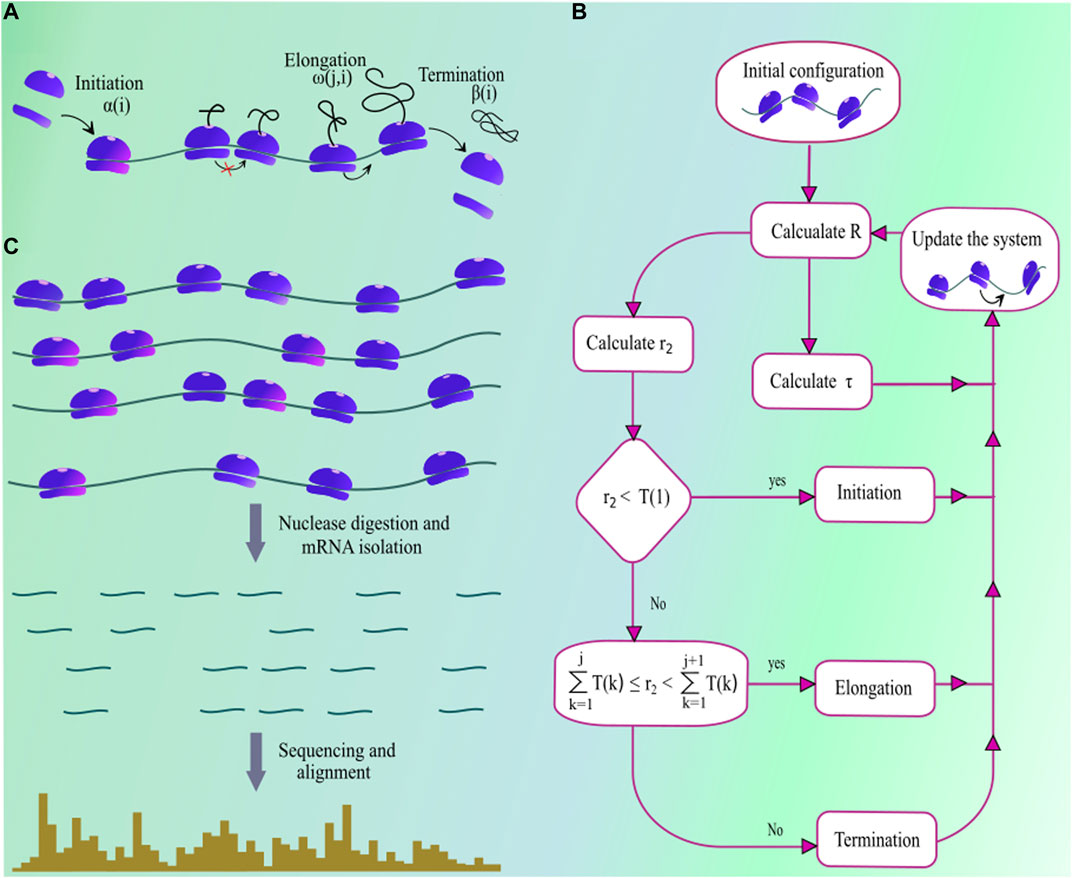
FIGURE 1. (A) A pictorial illustration of the steps involved in the process of protein synthesis. Ribosome subunits assemble at the start codon with rate
The TASEP with an uniform elongation rate, unitary particle size, and infinite lattice size is exactly solved by Derrida et al. (1992). Later, Lazarescu and Mallick (2011) extended this work by solving the TASEP with finite lattice size. Adding to this, Kolomeisky (1998) provided an analytical solution for TASEP with local inhomogeneities. For extended particles, Shaw et al. (2004) solved the TASEP model under mean-field approximation for uniform elongation rate. Recently, Szavits-Nossan et al. (2018) provided an analytical solution for the TASEP with non-uniform elongation rate and extended particle size; however, it ignores the higher-order terms of power series solution of the model. Therefore, in the absence of any exact analytical solution, computer simulation of the TASEP model is the best possible approach for making reliable quantitative predictions on protein synthesis.
Multiple approaches have been used in the past to simulate protein synthtesis on TASEP model, including kinetic Monte-Carlo (Zia et al., 2011), next reaction method (Gibson and Bruck, 2000; Duc and Song, 2018), Gillespie’s method (Gillespie, 1977), etc. The Gillespie’s method is one of the very efficient approach for studying stochastic systems and is commonly used to simulate the TASEP model (Gillespie, 1977). Solving TASEP using this method requires to calculate the parameter
which is the sum total of rates for all transitions that lead to a new state from the current state of the translation system. (Note that every unique arrangement of ribosomes on a transcript is a separate state.) In Eq. 1,
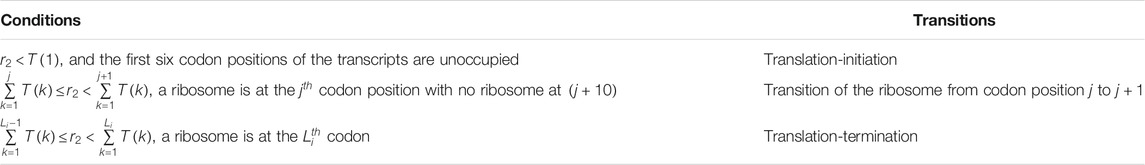
TABLE 1. A list of the types of transitions and conditions in simulation algorithm.
Accurate quantitative predictions for protein production critically depends on having precise estimates of translational rate parameters. Therefore, in this section, we discuss common approaches to extract translation rate parameters from ribosome profiles which provides the access to a plethora of information about the protein synthesis process. In ribosome profiling experiment, translation elongation is arrested by the treatment with drug cycloheximide (Figure 1C). Then, after the cell lysis, the portions of the mRNA molecule that are not protected by ribosomes are digested by nucleases. The remaining ribosome-protected mRNA fragments are subsequently sequenced and aligned to a reference genome. The typical length of a ribosome-protected mRNA fragment is 28–31 nucleotides; therefore, fragments outside this range are excluded from the analysis (Pop et al., 2014). Note that a ribosome translates the codon present at its A-site. Therefore, the position of A-site is identified on those ribosome-protected mRNA fragments (Ahmed et al., 2019), referred to as the read aligned to the A-site codon. Within a transcript, more ribosome-reads aligned to a codon means a longer translation time for that particular codon. This steady-state profile of ribosome occupancy can be used in extracting translation initiation and codon translation rates. Many computational methods have been developed in the past to extract these rate parameters from ribosome profiling data. We categorize these methods into three different groups.
These methods take advantage of some known characteristics of the translation process and extract codon translation rates by optimizing or fitting a function of ribosome profiling reads. For example, Dana and Tuller (2014) proposed a method for extracting the “typical” translation time of a codon by fitting a function that characterizes ribosome dwell time distribution. To do that, Dana and Tuller (2014) first normalize the ribosome footprint counts by the average reads aligned to the transcript. That is,
In Eq. 3,
Pop et al. (2014) proposed a similar method that calculates gene-specific and globally averaged translation time of a codon type “c” (i.e.,
The first term in the objective function represents the likelihood of observed ribosome profiles for a specific
In another optimization based approach, Szavits-Nossan and Ciandrini (2020) have used the non equilibrium analysis of ribosome profiling data to infer the ratio of elongation to initiation rate i.e.
is minimised by using the Least-Squared optimization technique.
Simulation based methods follow an iterative procedure, where in each iteration, translation rate parameters are altered until the simulation output converges to its experimentally measured counterparts. For example, Gritsenko et al. (2015) compared the experimentally measured and simulated ribosome densities on mRNA segments of different lengths. This comparison on short mRNA segments allows capturing the local variation in codon translation rates whereas in long segments it increase the reliability of measured rate parameters. These mRNA segments were constructed by dividing the whole transcript into two parts in such a way that both of them get an equal number of combined ribosome profiling and RNA-seq reads. The daughter segments were further divided recursively using the same approach until a reliable estimate of ribosome density can be made.
To implement this method, Gritsenko et al. (2015) used an experimental observation that ribosome density on a mRNA segment is distributed log-normally among all its replicates. This means the probability of finding a specific value of ribosome density in a single observation can be expressed as
Gritsenko et al. (2015) carry out the transcriptome-wide protein synthesis simulations by supplying some initial translation rate parameters to the model. Snapshots taken from protein synthesis simulations produced
In an another simulation based study, Duc and Song (2018) measured translation rate parameters by comparing the normalized in vivo ribosome profiling reads (Eq. 2) with those obtained from simulations. This method also requires initial translation rate parameters to generate in-silico ribosome profiles which will be refined in every iteration of the method. Initial codon translation rates were estimated as follows.
Authors also defined codon positions with significant error where it was larger than
Chemical kinetic-based methods do not require extensive simulations of protein synthesis. Instead, they rely on analytical expressions of translation rate parameters that use ribosome profiling and RNA-Seq data as input variables. A recent publication used the mean-field and steady-state assumptions, and derived the following analytical expressions for translation-initiation and codon translation rates (Sharma et al., 2019).
In Eq. 11, Eq. 12
Ribosome profiles provide single codon resolution to the protein synthesis process. However, these data sets are very noisy and are subjected to numerous biases associated with various steps of the ribosome profiling experiment, including the amplification of ribosome footprints by RT-PCR, nuclease digestion, cell lysis, etc. (O’Connor et al., 2016; Mohammad et al., 2019; Xiao et al., 2016; Hussmann et al., 2015). Such statistical errors and biases will also be reflected in the measured translation rate parameters. Dana and Tuller (2014) minimize their impact by ignoring any variation in the translation rate of a codon type. This approach drastically reduces the total number of parameters to be extracted from the ribosome profiling data. It minimizes the statistical uncertainty in the measurement of codon translation rates and also averages out various sequence biases. However, a major drawback of this approach is that it does not account for the context-dependent variations in codon translation rates. The other extreme approach taken by Szavits-Nossan and Ciandrini (2020) measures the translation rate for each codon in an mRNA transcript but such measurements are subjected to a higher degree of stochastic noise. A few probabilistic and machine learning models have also been applied to minimize the effect of noise and biases in the identification of A-site position on ribosome footprints (Fang et al., 2018; Tunney et al., 2018; Gobet et al., 2020). These models have successfully captured the context-dependent variation in codon translation rates and also performed well in transcripts with low abundance (Liu et al., 2020; Michel et al., 2016).
A closer look at the measured translation rate parameters unraveled the molecular determinants of translation-initiation and codon translation rates (Sharma et al., 2019; Duc et al., 2018). For example, tRNA pool hypothesis proposed codon translation rates to be proportional to the availability of cognate tRNA molecules (Ikemura, 1981; Ikemura, 1985). However, this hypothesis was never explicitly tested as there was no method that allowed translation rate measurement for all codons. Codon translation rates measured by ribosome profiling experiments supported this hypothesis in S. cerevisiae and E. coli (Dana and Tuller, 2014; Sharma et al., 2019). However, no such behaviour was observed in mouse cell lines (Gobet et al., 2020; Ingolia et al., 2011). Interestingly, a strong correlation between codon usage and tRNA abundance is observed in mammalian cells (Gobet et al., 2020; Gobet and Naef, 2017; Neelagandan et al., 2020), suggesting that tRNA levels are tuned according to their requirement in a cell.
In addition, an analysis of measured codon translation rate has shown that mRNA structures downstream to the A-site codon increase their translation time (Sharma et al., 2019). The reason for this increase is that the ribosome has to first unfold the structure to proceed to the next codon (Qu et al., 2011). The average increase of 6.7% in codon translation time was reported due to the presence of a structure in mRNA molecule. However, depending upon the stability of that structure, it may vary from one codon to another. Similarly, the presence of proline amino acid on the ribosome P-site increases the median translation time of a codon by 19% (Sharma et al., 2019) because the stereochemistry of proline amino acid delays the peptide bond formation with adjacent amino acid (Pavlov et al., 2009). Furthermore, Duc and Song (2018) have discovered that the aqueous environment inside the ribosome exit tunnel leads to a faster translation of codons when hydrophobic amino acid residues are present inside the tunnel. Electrostatic charges on the nascent-protein have been shown to modulate the translation elongation rate (Duc and Song, 2018; Riba et al., 2019). The ribosome exit tunnel is negatively charged. Therefore, the presence of positively charged amino acid into the tunnel decreases the translation rate of downstream codons (Figure 2A). The identity of the amino acid at the ribosome P-site also affects the translation rate of the A-site codon. In S. cerevisiae, eighty six different pairs of amino acids at the A- and P-sites speed up the elongation rate whereas it is slowed down in the case of eighty one other pairs of amino acids (Ahmed et al., 2020). A similar behaviour was also observed in mouse liver cells where different codon combinations at A- and P-sites, E- and P-sites, and E- and A-sites can speed up or slow down the translation elongation (Gobet et al., 2020). Post-translational modifications of tRNA molecules also enhances the rate of translation elongation in S. cerevisiae, N. crassa and C. elegans (Lyu et al., 2020; Nedialkova and Leidel, 2015). In addition to these molecular factors, patterns of slow and fast codons can cause ribosome traffic-jams on a transcript, and can significantly affect the time a ribosome spends at a given codon position (Diament et al., 2018; Duc et al., 2018).
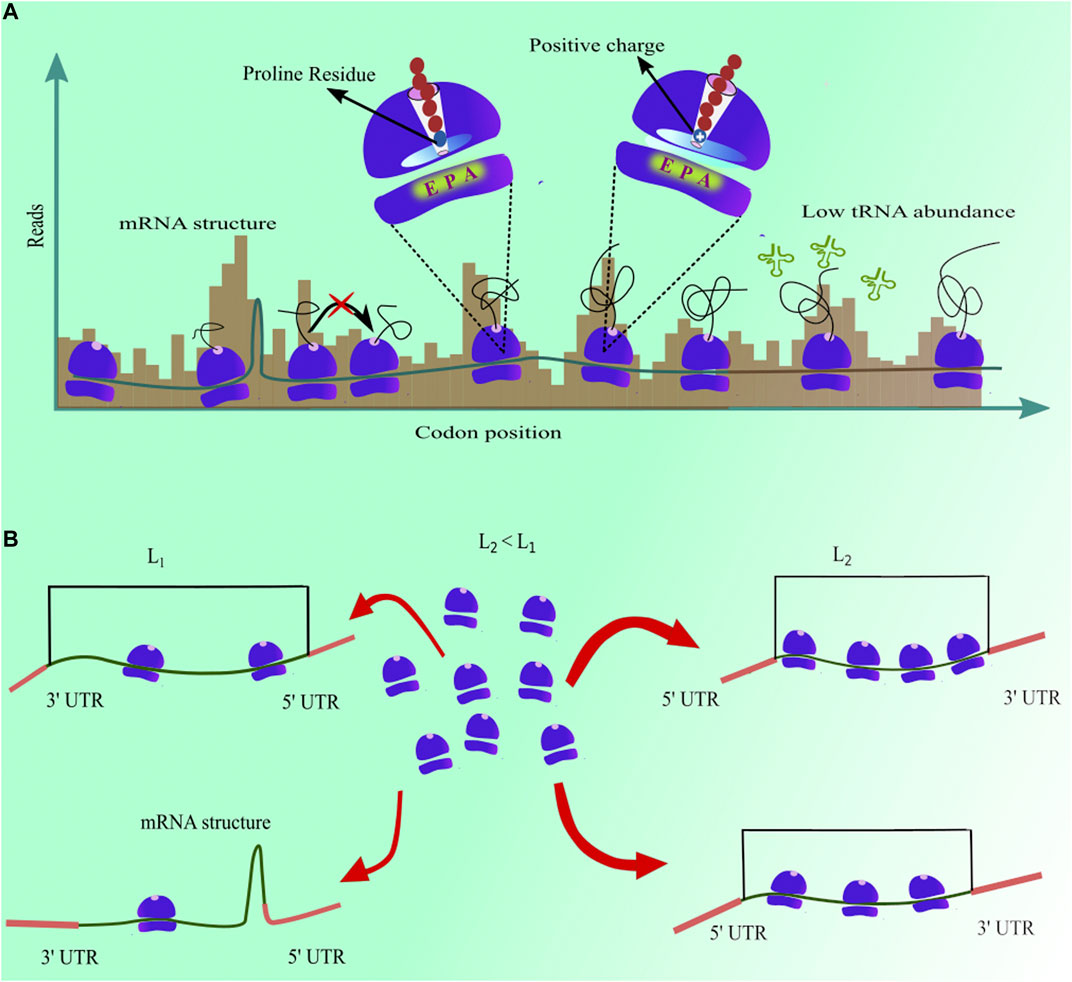
FIGURE 2. (A) mRNA structure, low tRNA abundance, and the presence of proline and positively charged amino acids in ribosome exit tunnel increase ribosome dwell time, resulting in higher ribosome profiling reads for those codon positions (B) Presence of mRNA structure at the 5′ end and length of the coding sequence in a transcript anti-correlate with initiation rate.
Translation initiation is another rate-limiting step (Shah et al., 2013) and sets an upper bound to the rate at which proteins are produced from a single transcript (Szavits-Nossan and Ciandrini, 2020). Initiation rates measured from ribosome profiling experiments also identified its molecular determinants (Figure 2B). For example, significant negative correlations of initiation rate with the free energy of mRNA folding near the start codon and transcript length were observed in multiple studies (Weinberg et al., 2016; Duc et al., 2018; Sharma et al., 2019). A stable structure near the start codon makes this region inaccessible to a ribosome for initiating the process of protein synthesis, thus decreasing the translation initiation rate. Indeed, many mRNA sequence design algorithms minimize folding energy in this region to enhance the production heterologous proteins (Salis et al., 2009; Angov, 2011). Similarly, the length of the coding sequence of a transcript is inversely proportional to the translation initiation rate (Duc and Song, 2018; Sharma et al., 2019). It is because the ribosomes completing protein synthesis at the termination end can easily diffuse to the start codon in shorter mRNA transcripts (Fernandes et al., 2017). These faster initiation rates in shorter transcripts help them in producing more proteins. Moreover, the presence of AUG codons upstream to the start codon can interfere in the recruitment of ribosomes for translation initiation, thus resulting in a decrease in the translation initiation rate (Sharma et al., 2019). Furthermore, the presence of KOZAK sequence in S. cerevisiae transcripts leads to a faster initiation as it serves as a stable binding site for the small ribosome subunit (Sharma et al., 2019). These new findings have demonstrated that a combination of several molecular factors work in tandem to finely modulate the translation-intiation and codon translation rates.
The development of ribosome profiling has allowed access to relative ribosome occupancy at single codon resolution (Ingolia et al., 2009; Ingolia et al., 2019). Many computational tools can convert this time-independent steady-state information into the kinetic rate parameters of protein synthesis (Dana and Tuller 2014; Pop et al., 2014; Szavits-Nossan and Ciandrini, 2020). Analysis of these rate parameters and their use in protein synthesis simulations give significant insight into the translational regulation of an individual gene (Shah et al., 2013; Lyu et al., 2020). These rate parameters also help identify various structural and sequence-based mRNA features that control the rate of protein synthesis (Weinberg et al., 2016; Duc et al., 2018; Sharma et al., 2019). Furthermore, the knowledge of these rates offers an unprecedented opportunity to explore and model other parallel and downstream processes influenced by translation-elongation kinetics, including co-translational protein folding, mRNA degradation, protein translocation through a membrane, chaperone binding, post-translational modifications, etc (Sharma and O’Brien, 2018; Radhakrishnan et al., 2016).
Analysis of measured translation rate parameters demonstrated that a combination of multiple molecular factors determines the translation-initiation and codon translation rates (Sharma et al., 2019). The strength with which these molecular factors act on translation rate parameters can vary from one place to another. Therefore, the same codon at two different locations can be translated at different rates. Current approaches can capture this context-dependent variation in codon translation rates (Sharma et al., 2019; Szavits-Nossan et al., 2018). However, none of them can quantify the impact of each molecular factor on translation-initiation and codon translation rates. Decoupling their effects would enable the scientists to make reliable predictions on translation rate parameters by only looking at mRNA sequence features, thus providing deeper insights into the context-dependent variation in codon translation rates.
Reliable predictions on protein synthesis, from a single transcript to the whole-cell level, are required for numerous synthetic biology applications (Purcell et al., 2013; Burke et al., 2020). For example, carefully placing the molecular determinants of translation rate parameters may help in designing heterologous genes and synthetic biological circuits. Moreover, in the absence of reliable gene expression models, synthetic biology relies heavily on the trial and error approach (Purcell et al., 2013; El Karoui et al., 2019). Therefore, the accurate predictions made by the quantitative models of protein synthesis will speed up the whole process of designing synthetic biology products with potential applications in areas such as drug delivery, cellular engineering, next-generation drugs, deployable medical devices, etc (Salis et al., 2009; Goldberg et al., 2018; Macklin et al., 2014).
AS conceptualised the project. VY, IR, HK, and AS wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ahmed, N., Sormanni, P., Ciryam, P., Vendruscolo, M., Dobson, C. M., and O'Brien, E. P. (2019). Identifying A- and P-Site Locations on Ribosome-Protected mRNA Fragments Using Integer Programming. Sci. Rep. 9, 6256. doi:10.1038/s41598-019-42348-x
Ahmed, N., Friedrich, U. A., Sormanni, P., Ciryam, P., Altman, N. S., Bukau, B., et al. (2020). Pairs of Amino Acids at the P- and A-Sites of the Ribosome Predictably and Causally Modulate Translation-Elongation Rates. J. Mol. Biol. 432, 166696. doi:10.1016/j.jmb.2020.10.030
Angov, E. (2011). Codon Usage: Nature's Roadmap to Expression and Folding of Proteins. Biotechnol. J. 6, 650–659. doi:10.1002/biot.201000332
Burke, P. E. P., Campos, C. B. d., Quiles, M. G., and Quiles, M. G. (2020). A Biochemical Network Modeling of a Whole-Cell. Sci. Rep. 10, 13303. doi:10.1038/s41598-020-70145-4
Byrd, R. H., Lu, P., Nocedal, J., and Zhu, C. (1995). A Limited Memory Algorithm for Bound Constrained Optimization. SIAM J. Sci. Comput. 16, 1190–1208. doi:10.1137/0916069
Ciandrini, L., Stansfield, I., and Romano, M. C. (2013). Ribosome Traffic on Mrnas Maps to Gene Ontology: Genome-wide Quantification of Translation Initiation Rates and Polysome Size Regulation. Plos Comput. Biol. 9, e1002866. doi:10.1371/journal.pcbi.1002866
Curtis, D., Lehmann, R., and Zamore, P. D. (1995). Translational Regulation in Development. Cell. 81, 171–178. doi:10.1016/0092-8674(95)90325-9
Dana, A., and Tuller, T. (2014). The Effect of Trna Levels on Decoding Times of Mrna Codons. Nucleic Acids Res. 42, 9171–9181. doi:10.1093/nar/gku646
Dana, A., and Tuller, T. (2015). Mean of the Typical Decoding Rates: a New Translation Efficiency index Based on the Analysis of Ribosome Profiling Data. G3: Genes, Genomes, Genet. 5, 73–80. doi:10.1534/g3.114.015099
Dao Duc, K., Saleem, Z. H., and Song, Y. S. (2018). Theoretical Analysis of the Distribution of Isolated Particles in Totally Asymmetric Exclusion Processes: Application to Mrna Translation Rate Estimation. Phys. Rev. E. 97, 012106. doi:10.1103/PhysRevE.97.012106
Dao Duc, K., and Song, Y. S. (2018). The Impact of Ribosomal Interference, Codon Usage, and Exit Tunnel Interactions on Translation Elongation Rate Variation. Plos Genet. 14, e1007166. doi:10.1371/journal.pgen.1007166
Derrida, B., Domany, E., and Mukamel, D. (1992). An Exact Solution of a One-Dimensional Asymmetric Exclusion Model with Open Boundaries. J. Stat. Phys. 69, 667–687. doi:10.1007/bf01050430
Dever, T. E., Kinzy, T. G., and Pavitt, G. D. (2016). Mechanism and Regulation of Protein Synthesis in saccharomyces Cerevisiae. Genetics. 203, 65–107. doi:10.1534/genetics.115.186221
Diament, A., Feldman, A., Schochet, E., Kupiec, M., Arava, Y., and Tuller, T. (2018). The Extent of Ribosome Queuing in Budding Yeast. Plos Comput. Biol. 14, e1005951. doi:10.1371/journal.pcbi.1005951
El Karoui, M., Hoyos-Flight, M., and Fletcher, L. (2019). Future Trends in Synthetic Biology-A Report. Front. Bioeng. Biotechnol. 7, 175. doi:10.3389/fbioe.2019.00175
Fang, H., Huang, Y.-F., Radhakrishnan, A., Siepel, A., Lyon, G. J., and Schatz, M. C. (2018). Scikit-ribo Enables Accurate Estimation and Robust Modeling of Translation Dynamics at Codon Resolution. Cel Syst. 6, 180–191. doi:10.1016/j.cels.2017.12.007
Fernandes, L. D., Moura, A. P. S., and Ciandrini, L. (2017). Gene Length as a Regulator for Ribosome Recruitment and Protein Synthesis: Theoretical Insights. Sci. Rep. 7, 17409–17411. doi:10.1038/s41598-017-17618-1
Garai, A., Chowdhury, D., Chowdhury, D., and Ramakrishnan, T. V. (2009). Stochastic Kinetics of Ribosomes: Single Motor Properties and Collective Behavior. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 80, 011908. doi:10.1103/PhysRevE.80.011908
Gibson, M. A., and Bruck, J. (2000). Efficient Exact Stochastic Simulation of Chemical Systems with many Species and many Channels. J. Phys. Chem. A. 104, 1876–1889. doi:10.1021/jp993732q
Gillespie, D. T. (1977). Exact Stochastic Simulation of Coupled Chemical Reactions. J. Phys. Chem. 81, 2340–2361. doi:10.1021/j100540a008
Gobet, C., and Naef, F. (2017). Ribosome Profiling and Dynamic Regulation of Translation in Mammals. Curr. Opin. Genet. Development. 43, 120–127. doi:10.1016/j.gde.2017.03.005
Gobet, C., Weger, B. D., Marquis, J., Martin, E., Neelagandan, N., Gachon, F., et al. (2020). Robust Landscapes of Ribosome Dwell Times and Aminoacyl-Trnas in Response to Nutrient Stress in Liver. Proc. Natl. Acad. Sci. USA. 117, 9630–9641. doi:10.1073/pnas.1918145117
Goldberg, A. P., Szigeti, B., Chew, Y. H., Sekar, J. A., Roth, Y. D., and Karr, J. R. (2018). Emerging Whole-Cell Modeling Principles and Methods. Curr. Opin. Biotechnol. 51, 97–102. doi:10.1016/j.copbio.2017.12.013
Greenbaum, D., Colangelo, C., Williams, K., and Gerstein, M. (2003). Comparing Protein Abundance and Mrna Expression Levels on a Genomic Scale. Genome Biol. 4, 117–118. doi:10.1186/gb-2003-4-9-117
Gritsenko, A. A., Hulsman, M., Reinders, M. J. T., and de Ridder, D. (2015). Unbiased Quantitative Models of Protein Translation Derived from Ribosome Profiling Data. Plos Comput. Biol. 11, e1004336. doi:10.1371/journal.pcbi.1004336
Hellen, C. U. T. (2018). Translation Termination and Ribosome Recycling in Eukaryotes. Cold Spring Harb Perspect. Biol. 10, a032656. doi:10.1101/cshperspect.a032656
Hussmann, J. A., Patchett, S., Johnson, A., Sawyer, S., and Press, W. H. (2015). Understanding Biases in Ribosome Profiling Experiments Reveals Signatures of Translation Dynamics in Yeast. Plos Genet. 11, e1005732. doi:10.1371/journal.pgen.1005732
Ikemura, T. (1981). Correlation between the Abundance of escherichia Coli Transfer Rnas and the Occurrence of the Respective Codons in its Protein Genes. J. Mol. Biol. 146, 1–21. doi:10.1016/0022-2836(81)90363-6
Ikemura, T. (1985). Codon Usage and Trna Content in Unicellular and Multicellular Organisms. Mol. Biol. Evol. 2, 13–34. doi:10.1093/oxfordjournals.molbev.a040335
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. S., and Weissman, J. S. (2009). Genome-wide Analysis In Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science 324, 218–223. doi:10.1126/science.1168978
Ingolia, N. T., Hussmann, J. A., and Weissman, J. S. (2019). Ribosome Profiling: Global Views of Translation. Cold Spring Harb. Perspect. Biol. 11, a032698. doi:10.1101/cshperspect.a032698
Ingolia, N. T., Lareau, L. F., and Weissman, J. S. (2011). Ribosome Profiling of Mouse Embryonic Stem Cells Reveals the Complexity and Dynamics of Mammalian Proteomes. Cell. 147, 789–802. doi:10.1016/j.cell.2011.10.002
Kolomeisky, A. B. (1998). Asymmetric Simple Exclusion Model with Local Inhomogeneity. J. Phys. A: Math. Gen. 31, 1153–1164. doi:10.1088/0305-4470/31/4/006
Kozak, M. (1999). Initiation of Translation in Prokaryotes and Eukaryotes. Gene. 234, 187–208. doi:10.1016/s0378-1119(99)00210-3
Kudla, G., Murray, A. W., Tollervey, D., and Plotkin, J. B. (2009). Coding-sequence Determinants of Gene Expression in escherichia Coli. Science 324, 255–258. doi:10.1126/science.1170160
Kummer, E., and Ban, N. (2021). Mechanisms and Regulation of Protein Synthesis in Mitochondria. Nat. Rev. Mol. Cel. Biol. 22, 1–19. doi:10.1038/s41580-021-00332-2
Lazarescu, A., and Mallick, K. (2011). An Exact Formula for the Statistics of the Current in the Tasep with Open Boundaries. J. Phys. A: Math. Theor. 44, 315001. doi:10.1088/1751-8113/44/31/315001
Li, G.-W., Burkhardt, D., Gross, C., and Weissman, J. S. (2014). Quantifying Absolute Protein Synthesis Rates Reveals Principles Underlying Allocation of Cellular Resources. Cell 157, 624–635. doi:10.1016/j.cell.2014.02.033
Liu, Q., Shvarts, T., Sliz, P., and Gregory, R. I. (2020). Ribotoolkit: an Integrated Platform for Analysis and Annotation of Ribosome Profiling Data to Decode Mrna Translation at Codon Resolution. Nucleic Acids Res. 48, W218–W229. doi:10.1093/nar/gkaa395
Lu, P., Vogel, C., Wang, R., Yao, X., and Marcotte, E. M. (2007). Absolute Protein Expression Profiling Estimates the Relative Contributions of Transcriptional and Translational Regulation. Nat. Biotechnol. 25, 117–124. doi:10.1038/nbt1270
Lyu, X., Yang, Q., Li, L., Dang, Y., Zhou, Z., Chen, S., et al. (2020). Adaptation of Codon Usage to Trna I34 Modification Controls Translation Kinetics and Proteome Landscape. Plos Genet. 16, e1008836. doi:10.1371/journal.pgen.1008836
MacDonald, C. T., Gibbs, J. H., and Pipkin, A. C. (1968). Kinetics of Biopolymerization on Nucleic Acid Templates. Biopolymers 6, 1–25. doi:10.1002/bip.1968.360060102
Macklin, D. N., Ruggero, N. A., and Covert, M. W. (2014). The Future of Whole-Cell Modeling. Curr. Opin. Biotechnol. 28, 111–115. doi:10.1016/j.copbio.2014.01.012
Margaliot, M., and Tuller, T. (2013). Ribosome Flow Model with Positive Feedback. J. R. Soc. Interf. 10, 20130267. doi:10.1098/rsif.2013.0267
Merrick, W. C. (1992). Mechanism and Regulation of Eukaryotic Protein Synthesis. Microbiol. Mol. Biol. Rev. 56, 291–315. doi:10.1128/mmbr.56.2.291-315.1992
Merrick, W. C., and Pavitt, G. D. (2018). Protein Synthesis Initiation in Eukaryotic Cells. Cold Spring Harb. Perspect. Biol. 10, a033092. doi:10.1101/cshperspect.a033092
Michel, A. M., Mullan, J. P. A., Velayudhan, V., O'Connor, P. B. F., Donohue, C. A., and Baranov, P. V. (2016). Ribogalaxy: a Browser Based Platform for the Alignment, Analysis and Visualization of Ribosome Profiling Data. RNA Biol. 13, 316–319. doi:10.1080/15476286.2016.1141862
Miyazaki, M., and Esser, K. A. (2009). Cellular Mechanisms Regulating Protein Synthesis and Skeletal Muscle Hypertrophy in Animals. J. Appl. Physiol. 106, 1367–1373. doi:10.1152/japplphysiol.91355.2008
Mohammad, F., Green, R., and Buskirk, A. R. (2019). A Systematically-Revised Ribosome Profiling Method for Bacteria Reveals Pauses at Single-Codon Resolution. Elife. 8, e42591. doi:10.7554/eLife.42591
Munro, J. B., Vaiana, A., Sanbonmatsu, K. Y., and Blanchard, S. C. (2008). A New View of Protein Synthesis: Mapping the Free Energy Landscape of the Ribosome Using Single-Molecule Fret. Biopolymers 89, 565–577. doi:10.1002/bip.20961
Nedialkova, D. D., and Leidel, S. A. (2015). Optimization of Codon Translation Rates via Trna Modifications Maintains Proteome Integrity. Cell 161, 1606–1618. doi:10.1016/j.cell.2015.05.022
Neelagandan, N., Lamberti, I., Carvalho, H. J. F., Gobet, C., and Naef, F. (2020). What Determines Eukaryotic Translation Elongation: Recent Molecular and Quantitative Analyses of Protein Synthesis. Open Biol. 10, 200292. doi:10.1098/rsob.200292
O’Connor, P. B., Andreev, D. E., and Baranov, P. V. (2016). Comparative Survey of the Relative Impact of Mrna Features on Local Ribosome Profiling Read Density. Nat. Commun. 7, 12915. doi:10.1038/ncomms12915
Pavlov, M. Y., Watts, R. E., Tan, Z., Cornish, V. W., Ehrenberg, M., and Forster, A. C. (2009). Slow Peptide Bond Formation by Proline and Other N-Alkylamino Acids in Translation. Proc. Natl. Acad. Sci. 106, 50–54. doi:10.1073/pnas.0809211106
Pop, C., Rouskin, S., Ingolia, N. T., Han, L., Phizicky, E. M., Weissman, J. S., et al. (2014). Causal Signals between Codon Bias, Mrna Structure, and the Efficiency of Translation and Elongation. Mol. Syst. Biol. 10, 770. doi:10.15252/msb.20145524
Proud, C. G. (2006). Regulation of Protein Synthesis by Insulin. Biochem. Soc. Trans. 34, 213–216. doi:10.1042/bst0340213
Purcell, O., Jain, B., Karr, J. R., Covert, M. W., and Lu, T. K. (2013). Towards a Whole-Cell Modeling Approach for Synthetic Biology. Chaos 23, 025112. doi:10.1063/1.4811182
Qu, X., Wen, J.-D., Lancaster, L., Noller, H. F., Bustamante, C., and Tinoco, I. (2011). The Ribosome Uses Two Active Mechanisms to Unwind Messenger Rna during Translation. Nature 475, 118–121. doi:10.1038/nature10126
Radhakrishnan, A., Chen, Y.-H., Martin, S., Alhusaini, N., Green, R., and Coller, J. (2016). The Dead-Box Protein Dhh1p Couples Mrna Decay and Translation by Monitoring Codon Optimality. Cell 167, 122–132. doi:10.1016/j.cell.2016.08.053
Riba, A., Di Nanni, N., Mittal, N., Arhné, E., Schmidt, A., and Zavolan, M. (2019). Protein Synthesis Rates and Ribosome Occupancies Reveal Determinants of Translation Elongation Rates. Proc. Natl. Acad. Sci. USA 116, 15023–15032. doi:10.1073/pnas.1817299116
Salis, H. M., Mirsky, E. A., and Voigt, C. A. (2009). Automated Design of Synthetic Ribosome Binding Sites to Control Protein Expression. Nat. Biotechnol. 27, 946–950. doi:10.1038/nbt.1568
Shah, P., Ding, Y., Niemczyk, M., Kudla, G., and Plotkin, J. B. (2013). Rate-limiting Steps in Yeast Protein Translation. Cell 153, 1589–1601. doi:10.1016/j.cell.2013.05.049
Sharma, A. K., Ahmed, N., and O'Brien, E. P. (2018). Determinants of Translation Speed Are Randomly Distributed across Transcripts Resulting in a Universal Scaling of Protein Synthesis Times. Phys. Rev. E. 97, 022409. doi:10.1103/PhysRevE.97.022409
Sharma, A. K., and Chowdhury, D. (2011). Distribution of Dwell Times of a Ribosome: Effects of Infidelity, Kinetic Proofreading and Ribosome Crowding. Phys. Biol. 8, 026005. doi:10.1088/1478-3975/8/2/026005
Sharma, A. K., and Chowdhury, D. (2012). Template-directed Biopolymerization: Tape-Copying Turing Machines. Biophys. Rev. Lett. 07, 135–175. doi:10.1142/s1793048012300083
Sharma, A. K., and O’Brien, E. P. (2017). Increasing Protein Production Rates Can Decrease the Rate at Which Functional Protein Is Produced and Their Steady-State Levels. J. Phys. Chem. B. 121, 6775–6784. doi:10.1021/acs.jpcb.7b01700
Sharma, A. K., and O’Brien, E. P. (2018). Non-equilibrium Coupling of Protein Structure and Function to Translation-Elongation Kinetics. Curr. Opin. Struct. Biol. 49, 94–103. doi:10.1016/j.sbi.2018.01.005
Sharma, A. K., Sormanni, P., Ahmed, N., Ciryam, P., Friedrich, U. A., Kramer, G., et al. (2019). A Chemical Kinetic Basis for Measuring Translation Initiation and Elongation Rates from Ribosome Profiling Data. Plos Comput. Biol. 15, e1007070. doi:10.1371/journal.pcbi.1007070
Shaw, L. B., Kolomeisky, A. B., and Lee, K. H. (2004). Local Inhomogeneity in Asymmetric Simple Exclusion Processes with Extended Objects. J. Phys. A: Math. Gen. 37, 2105–2113. doi:10.1088/0305-4470/37/6/010
Szavits-Nossan, J., and Ciandrini, L. (2020). Inferring Efficiency of Translation Initiation and Elongation from Ribosome Profiling. Nucleic Acids Res. 48, 9478–9490. doi:10.1093/nar/gkaa678
Szavits-Nossan, J., Ciandrini, L., and Romano, M. C. (2018). Deciphering Mrna Sequence Determinants of Protein Production Rate. Phys. Rev. Lett. 120, 128101. doi:10.1103/physrevlett.120.128101
Tuller, T., Carmi, A., Vestsigian, K., Navon, S., Dorfan, Y., Zaborske, J., et al. (2010). An Evolutionarily Conserved Mechanism for Controlling the Efficiency of Protein Translation. Cell. 141, 344–354. doi:10.1016/j.cell.2010.03.031
Tunney, R., McGlincy, N. J., Graham, M. E., Naddaf, N., Pachter, L., and Lareau, L. F. (2018). Accurate Design of Translational Output by a Neural Network Model of Ribosome Distribution. Nat. Struct. Mol. Biol. 25, 577–582. doi:10.1038/s41594-018-0080-2
Vogel, C., de Sousa Abreu, R., Ko, D., Le, S. Y., Shapiro, B. A., Burns, S. C., et al. (2010). Sequence Signatures and mRNA Concentration Can Explain Two‐thirds of Protein Abundance Variation in a Human Cell Line. Mol. Syst. Biol. 6, 400. doi:10.1038/msb.2010.59
Volkov, I. L., and Johansson, M. (2018). Single-molecule Tracking Approaches to Protein Synthesis Kinetics in Living Cells. Biochemistry. 58, 7–14. doi:10.1021/acs.biochem.8b00917
Weinberg, D. E., Shah, P., Eichhorn, S. W., Hussmann, J. A., Plotkin, J. B., and Bartel, D. P. (2016). Improved Ribosome-Footprint and Mrna Measurements Provide Insights into Dynamics and Regulation of Yeast Translation. Cel. Rep. 14, 1787–1799. doi:10.1016/j.celrep.2016.01.043
Wu, L., and Belasco, J. G. (2008). Let Me Count the Ways: Mechanisms of Gene Regulation by Mirnas and Sirnas. Mol. Cel. 29, 1–7. doi:10.1016/j.molcel.2007.12.010
Xiao, Z., Zou, Q., Liu, Y., and Yang, X. (2016). Genome-wide Assessment of Differential Translations with Ribosome Profiling Data. Nat. Commun. 7, 11194. doi:10.1038/ncomms11194
Keywords: ribosome profiling, ribo-seq, translation-initiation rate, codon translation rate, codon usage bias, quantitative modeling of protein synthesis
Citation: Yadav V, Ullah Irshad I, Kumar H and Sharma AK (2021) Quantitative Modeling of Protein Synthesis Using Ribosome Profiling Data. Front. Mol. Biosci. 8:688700. doi: 10.3389/fmolb.2021.688700
Received: 31 March 2021; Accepted: 25 May 2021;
Published: 28 June 2021.
Edited by:
Fabio Trovato, Freie Universität Berlin, GermanyReviewed by:
Alexander Schmidt, University of Basel, SwitzerlandCopyright © 2021 Yadav, Ullah Irshad, Kumar and Sharma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ajeet K. Sharma, YWplZXQuc2hhcm1hQGlpdGphbW11LmFjLmlu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.