 Jakub Macošek
Jakub Macošek Guillaume Mas
Guillaume Mas Sebastian Hiller
Sebastian Hiller- Biozentrum, University of Basel, Basel, Switzerland
Molecular chaperones are the key instruments of bacterial protein homeostasis. Chaperones not only facilitate folding of client proteins, but also transport them, prevent their aggregation, dissolve aggregates and resolve misfolded states. Despite this seemingly large variety, single chaperones can perform several of these functions even on multiple different clients, thus suggesting a single biophysical mechanism underlying. Numerous recently elucidated structures of bacterial chaperone–client complexes show that dynamic interactions between chaperones and their client proteins stabilize conformationally flexible non-native client states, which results in client protein denaturation. Based on these findings, we propose chaotropicity as a suitable biophysical concept to rationalize the generic activity of chaperones. We discuss the consequences of applying this concept in the context of ATP-dependent and -independent chaperones and their functional regulation.
Introduction
Most proteins need to fold into a three-dimensional structure to perform their function, as encoded in their amino acid sequence (Anfinsen et al., 1961; Haber and Anfinsen, 1962; Anfinsen, 1973). While small proteins can fold efficiently, the vast majority of nascent protein chains needs to navigate a rugged potential energy surface, driven by the hydrophobic collapse and constrained by the crowded environment of the cell (Levinthal, 1968; Bryngelson and Wolynes, 1987; Wolynes et al., 1995; Onuchic and Wolynes, 2004; Bartlett and Radford, 2009). Thus, proteins can easily become trapped in local folding minima, from where they need to overcome free energy barriers to reach the correct native conformation. Folding via such intermediate states is considered to be the rule for proteins larger than 100 amino acids (Brockwell and Radford, 2007). In addition, even proteins that are capable of spontaneously reaching their native conformation may unfold under stress conditions. Folding intermediates or unfolded proteins are dysfunctional, prone to aggregation and may lead to fatal conditions that are a threat to the health of the cell (Knowles et al., 2014; Tittelmeier et al., 2020).
To tackle this challenge, protein homeostasis networks have evolved in all kingdoms of life (Hipp et al., 2019). They comprise of different molecular chaperones, as well as the ubiquitin-proteasome system (UPS) and the autophagy system. While UPS and the autophagy system play their functional role in degradation of expired proteins, chaperones are the key instrument of protein homeostasis. Chaperones not only facilitate folding of proteins, but also transport them, prevent their aggregation, dissolve aggregates or unfold misfolded proteins (Pelham, 1986; Ellis, 1987; Hemmingsen et al., 1988; Goloubinoff et al., 1989; Walter and Buchner, 2002; Hartl and Hayer-Hartl, 2009; Balchin et al., 2016; Goloubinoff, 2016; Wentink et al., 2019; Balchin et al., 2020; Burmann et al., 2020). Interestingly, a single chaperone can often perform several of these functions. For example, heat shock protein 70 (Hsp70, or DnaK in bacteria) participates in de novo protein folding, assembly of protein complexes and translocation across membranes to protein refolding, disaggregation, and degradation (Mayer and Gierasch, 2019). The underlying mechanism allowing a single chaperone to perform functions with such drastically different outcomes remains unclear.
Here, we develop a hypothesis addressing this question. We start out by summarizing the main cellular functions of chaperones and connecting them to protein folding theory. Then, we recapitulate recent structures of chaperone–client complexes, with a focus on bacterial systems. These connect the functional understanding of chaperone activity with structural insights and identify common patterns in the client dynamics. Finally, we extrapolate from these patterns to propose chaotropicity as a concept to describe the single biophysical activity underlying the diverse cellular functions of chaperones common to many or all chaperones.
Cellular Functions of Chaperones and Their Connection to Protein Folding Theory
The traditional nomenclature to describe chaperone functions is based on their effective functionality in the cellular context. Depending on this context, a chaperone thus can act as a holdase, foldase, translocase, disaggregase, or unfoldase.
Holdase chaperones are typically ATP-independent chaperones, that merely associate with non-native client proteins for extended time periods to stabilize them and prevent their aggregation (Hall, 2020). Despite the fact that holdases do not directly fold proteins, their activity is indispensable as they protect vulnerable non-native states from aggregation. Studies revealing a broad clientome of holdases have illustrated their importance in protein folding (Haslbeck et al., 2004; Jarchow et al., 2008). Traditional representatives are the small heat shock protein (sHsp) family (Haslbeck et al., 2019), as well as a number of bacterial chaperones including cytosolic trigger factor (TF) and SecB, as well as periplasmatic Spy, Skp, and SurA (Bechtluft et al., 2010; Hoffmann et al., 2010; Goemans et al., 2014; Mas et al., 2019). Some holdases, such as TF, associate with ribosomes, thus comprising the first of the two chaperone layers participating in de novo protein folding (Frydman, 2001; Deuerling and Bukau, 2004; Kramer et al., 2004b; Kaiser et al., 2006; Merz et al., 2008). Holdases then transfer the nascent protein for active folding to the second layer of chaperones.
Active structural remodeling during de novo protein folding is the domain of ATP-dependent foldases. In bacteria, these are mainly the DnaK system and the GroEL/ES system (Hayer-Hartl et al., 2016; Rosenzweig et al., 2019). Both systems function similarly by cycling between ADP-bound and ATP-bound states that differ in affinity for non-native proteins. If the association rate of the binding to chaperone is greater than the aggregation rate and lower than the folding rate, the chaperones facilitate folding by kinetic partitioning (Diamond and Randall, 1997; Fedorov and Baldwin, 1997; De Los Rios and Barducci, 2014). Notably, DnaK and GroEL/ES systems differ in how they function mechanistically. While in the case of DnaK the folding occurs upon release, the group I chaperonin system GroEL/ES unfolds the client protein by expansion and then traps it in a cage, where the client protein collapses to fold (Hemmingsen et al., 1988; Lin and Rye, 2004; Lin et al., 2008; Sharma et al., 2008).
The third group of chaperones are translocases, which shuttle nascent proteins across membranes. Translocases are especially important in bacteria, where about a third of all proteins is exported from the cytoplasm and therefore needs to be translocated across the inner membrane. The main transport route for these proteins is the SEC pathway, using the key motor-protein SecA (Vrontou and Economou, 2004; Tsirigotaki et al., 2017). The molecular machine SecA converts chemical energy into mechanical force to translocate the unfolded nascent protein through the SecYEG membrane channel while maintaining the proteins unfolded. Nascent proteins may find SecA independently or be targeted to it by SecB and TF, but SecA associates with ribosome and interacts with nascent proteins directly as well (Huber et al., 2011; Wang et al., 2017).
The unfoldase function of chaperones is necessary to overcome free energy barriers in the case of nascent proteins trapped in local minima of their folding landscape or for the turnover of irreversibly misfolded proteins. Indeed, GroEL/ES was shown to begin its functional cycle with unfolding the client protein by expansion (Lin and Rye, 2004; Priya et al., 2013b; Mattoo and Goloubinoff, 2014), and similar unfolding by expansion was also described for DnaK (Sharma et al., 2010; Imamoglu et al., 2020). Moreover, a recent study of DnaK-assisted refolding of firefly luciferase suggests that initial unfolding is critical even for efficient folding of multi-domain proteins (Imamoglu et al., 2020). Overall, most chaperones have the capacity to destabilize protein structure (Sharma et al., 2009; Finka et al., 2016; Hiller, 2020).
If all the aforementioned activities of chaperones fall short to prevent proteins from aggregation, some chaperones still exhibit disaggregase activity, which allows them to untangle aggregates and refold the protein or target it for degradation (Sousa, 2014). Two major bacterial chaperone systems, Clp and DnaK, are capable of actively unraveling protein aggregates that would otherwise be aggregated irreversibly, and refold the proteins into their native conformation (Glover and Lindquist, 1998; Goloubinoff et al., 1999).
Two important observations support the notion that a single activity might underlie this large variety of chaperone functions in the cellular context. Firstly, for many chaperones with little client specificity – so-called general chaperones (Bose and Chakrabarti, 2017) - the major variable changing between particular cellular functions is modulation of client specificity by a co-chaperone or by subcellular localization. This implies that these general chaperones may use a single activity to perform their different cellular functions.
Secondly, functional studies indicate that different chaperones including DnaK, GroEL/ES, and Hsp90, unfold their client by expanding prior to facilitating their folding (Shtilerman et al., 1999; Ben-Zvi et al., 2004; Lin et al., 2008; Sharma et al., 2010; Walerych et al., 2010; Priya et al., 2013b; Mas et al., 2018), and some degree of unfolding is now emerging as the core aspect of the activity of many chaperones (Priya et al., 2013a; Finka et al., 2016; Jo et al., 2019). Such a chaperone activity is applicable to any of the cellular functions as unfolding the client gives it a chance to undergo renewed hydrophobic collapse.
The cellular functions of chaperones can thus be recast from the perspective of protein folding theory. Protein folding has been formulated as the problem of a nascent protein chain navigating in its conformational space on a funnel-shaped, rugged potential energy surface, with the eventual goal to attain its native conformation, a local minimum (Dill and MacCallum, 2012). Thereby, the ruggedness offers several local minima, which correspond to alternative structural states (Figure 1). From the perspective of protein folding, chaperones function in cellular folding processes by regulating transitions of the client protein between structural states on the potential energy surface. Since all cellular chaperone activities can be connected on a single folding landscape, chaperones in principle only need a single generic activity, that increases the free energy of the client. Each cellular chaperone function can then be viewed as the generic chaperone activity acting at specific positions of the potential energy surface to achieve the observed outcome.
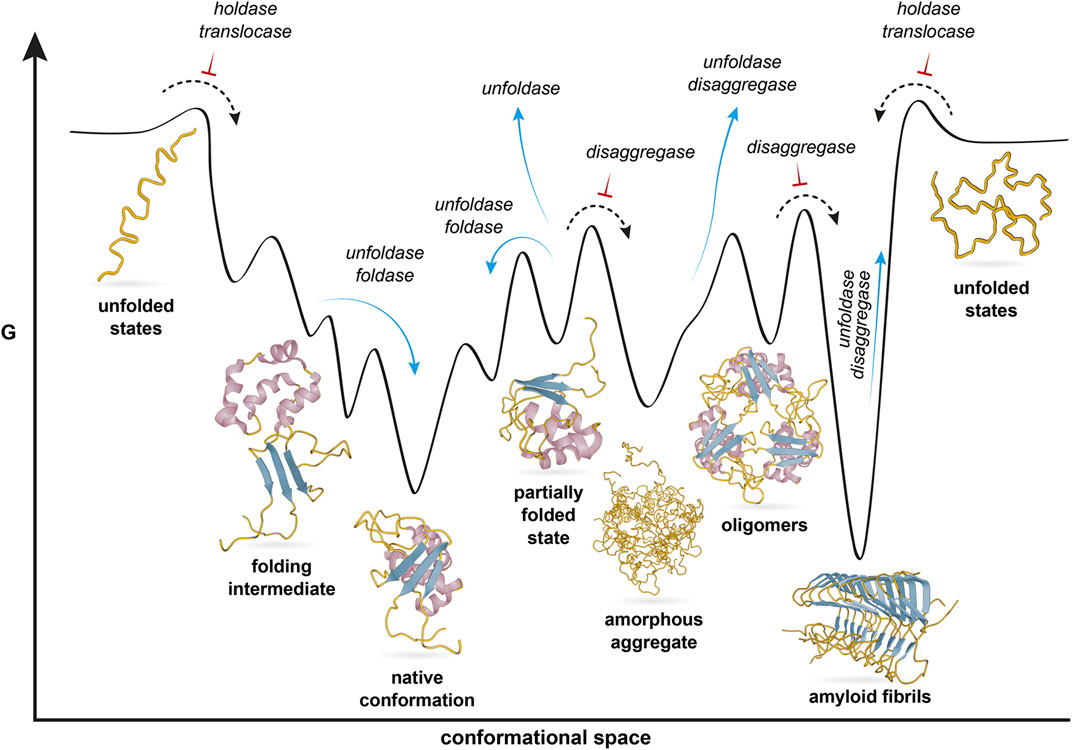
FIGURE 1. Cellular functions of chaperones in the context of a protein folding landscape. The protein polypeptide chain is shown yellow, with different secondary structure elements highlighted in purple and blue. The protein navigates a rugged free energy surface. The native conformation is one out of several local minima, representing different conformational states that are shown below the surface. Chaperones participate in a broad range of cellular processes which define a range of functions listed in italics. These navigate the protein along the energy landscape. Blue arrows indicate transitions that chaperones facilitate, whereas black arrows with red inversed T indicate transitions chaperones prevent. Figure modified from Jahn and Radford (2005).
Lessons Learnt From Structures of Bacterial Chaperone-Client Complexes
In order to understand the cellular functions of chaperones mechanistically, it is crucial to employ biophysical descriptions of chaperone activity that regulate the transitions of proteins along the potential energy surface. The ideal starting point of such investigations are detailed structural descriptions of chaperone-client complexes as they provide direct snapshots of chaperones in action. Recent technological advances, particularly in solution NMR spectroscopy, have provided atomic resolution insights such complexes. In the following, we summarize such structural descriptions, including ATP-independent and ATP-dependent chaperones in order to reveal common features (Figure 2). We thereby focus on bacterial systems, because the most detailed descriptions are available for these, and because it can be assumed that the resulting conclusion can be generalized.
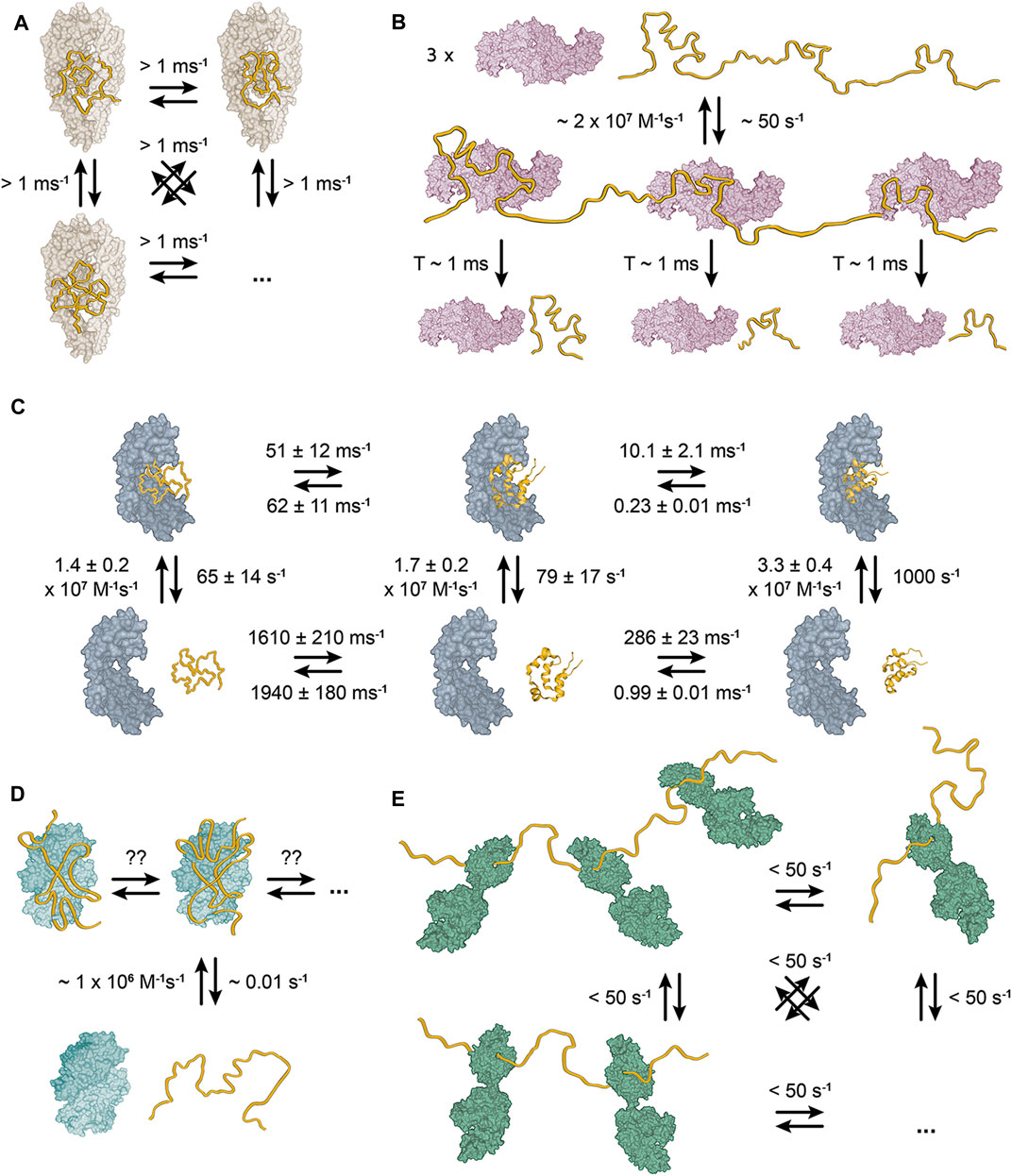
FIGURE 2. Structural models of bacterial chaperone-client complexes reveal dynamic interactions. (A) The Skp-OMP complex. The client binds as compact, flexible ensemble, which interconverts between individual conformations within 1 ms (Burmann et al., 2013). (B) The trigger factor–PhoA complex. A single molecule of PhoA interacts with three molecules of TF with short-lived interaction lifetimes of ∼1 ms (Saio et al., 2014). The kinetics reveal that the complex is also globally short-lived with a dissociation rate of ∼50 s−1. (C) The Spy-Im7 complex. The chaperone Spy binds its client Im7 as a dynamic ensemble of diverse conformations (He et al., 2016; Stull et al., 2016; Horowitz et al., 2018). The representative unfolded state (on the left), folding intermediate state (in the middle) and native state (on the right) interconvert with ms rates. However, the rates are slower for Spy-bound Im7 than for free Im7. (D) The SecB-MBP complex. One molecule of the client binds one SecB tetramer (Huang et al., 2016). No symmetry breaking of the SecB tetramer is observed upon binding of the full-length clients, which indicates that the resulting complex must be dynamic with the client rearranging on SecB surface on a very fast timescale. (E) The DnaK–hTRF1 complex. DnaK binds the client in an ensemble of globally unfolded conformations at various stoichiometric ratios (Lee et al., 2015; Sekhar et al., 2016; Rosenzweig et al., 2017).
The earliest atomic-resolution characterization of a bacterial chaperone in complex with a full-length client protein was the study of bacterial chaperone Skp and the outer membrane proteins (OMPs) tOmpA and OmpX (Burmann et al., 2013). Skp is a holdase chaperone in one of two alternative pathways to transport OMPs in the periplasm to the outer membrane (Sklar et al., 2007). It is a 3 × 17 kDa trimer, that resembles a jellyfish (Kramer et al., 2004a; Walton and Sousa, 2004). Each monomer consists of a β-strand domain, which forms the body, and an extended coiled-coil domain, which forms the tentacles. The trimerization interface is located in the body of the trimer, whereas the α-helical tentacles define a central cavity that creates a protective environment for the client proteins (Burmann et al., 2013; Callon et al., 2014). The structural characterization of the Skp-OMP complexes revealed that the client binds as compact, but structurally disordered and highly conformationally flexible ensemble, in which the individual conformations interconvert within 1 ms (Figure 2A). Individual contacts between the client and the chaperone are weak and unspecific, but their avidity results in a high-affinity complex with a lifetime of more than 2 h. Upon binding to Skp, the clients remain in a highly conformationally flexible state, which allows them to sample more than 107 conformations before their release from the chaperone (Burmann et al., 2013).
SurA is the principal chaperone in the second of the pathways to transport OMPs in the periplasm and its complexes with various OMPs have been recently structurally characterized (Marx et al., 2020). SurA is a ∼47 kDa chaperone consisting of three domains: the N and C-termini of the protein make up the core domain, while two peptidyl-prolyl isomerase domains (P1 and P2) comprise of the middle of segment of the protein chain (Bitto and McKay, 2002). The core domain and P1 form a platform connected to P2 by two flexible linkers, which allow the protein to alternate between multiple conformations (Calabrese et al., 2020). Characterization of the bound client-proteins revealed that similarly to Skp, SurA binds to the OMP in a disordered state (Calabrese et al., 2020; Marx et al., 2020).
The complex of TF and alkaline phosphatase (PhoA) is another complex that has been characterized at atomic resolution (Saio et al., 2014). PhoA is a ∼50 kDa periplasmatic enzyme, which can be unfolded and aggregation-prone in the cytosol (Valent et al., 1995). TF is a bacterial ribosome-associated holdase chaperone, that has a general function of protecting unfolded nascent proteins against aggregation (Hoffmann et al., 2010). It is a 48 kDa protein consisting of three domains adopting a dragon-like shape (Ferbitz et al., 2004). The N-terminal ribosome-binding domain (RBD) mediates ribosome interaction (Hesterkamp et al., 1997), the middle domain (PPD) has a peptidyl–prolyl isomerase activity (Hesterkamp and Bukau, 1996) and the C-terminal substrate-binding domain (SBD) carries the chaperone activity (Merz et al., 2006). TF exists in a fast monomer-dimer equilibrium, where the monomeric form is the active chaperone and the dynamic dimer is the storage form (Patzelt et al., 2002; Morgado et al., 2017). The interaction with the client PhoA causes a dissociation of the dimer, but full-length PhoA is too large for a single molecule of TF (Saio et al., 2014). On this basis, the structure of the TF–PhoA complex was determined as three individual TF bound to three fragments of PhoA (Saio et al., 2014). In the complexes, PhoA interacts predominantly with the SBD and to a lesser extent with the PPD. Each complex structure shows that a particular PhoA fragment binds in a unique conformation (Figure 2B). However, the same site of TF binds each of the PhoA fragments and NMR relaxation dispersion measurements show that the lifetime of a complex of TF and a single PhoA fragment is only ∼20 ms (Saio et al., 2014). Therefore, the TF-PhoA interaction is highly dynamic with the fragment of PhoA bound to a given molecule of TF constantly alternating.
A further milestone in our understanding of chaperone-client interactions came from the characterization of the complex of chaperone Spy and client protein colicin immunity protein 7 (Im7). Im7 is a bacterial immunity protein, which binds colicin E7 to inhibit its toxicity (James et al., 1996). Spy is a bacterial periplasmatic ATP-independent chaperone, which was identified in a screen for proteins that stabilize a mutant of Im7 (Quan et al., 2011). It is a ∼16 kDa protein that forms a cradle-shaped dimer (Kwon et al., 2010; Quan et al., 2011). Im7 binds the concave surface of Spy dimer, but determining a crystal structure of the complex did not reveal the structure of chaperone-bound Im7, because the electron density of Im7 was of insufficient quality (Horowitz et al., 2016). Subsequently, NMR spectroscopy confirmed the conformational flexibility of bound Im7 and revealed its interaction site on the concave surface of Spy (He et al., 2016). Additionally, the same binding mode was observed in the complex of Spy with the Fyn SH3 domain (He and Hiller, 2018). Spy supports the conformational flexibility of the bound client to such an extent that folding of Im7 while bound to Spy was reported (Stull et al., 2016), despite the fact that Spy is an ATP-independent chaperone that does not undergo any large conformational changes. The key aspect of Spy facilitated folding of Im7 is that the folding rate is significantly decelerated compared to the folding of free Im7 (Figure 2C).
The largest structurally well-characterized chaperone–client complex is the complex of the chaperone SecB with PhoA. SecB is a bacterial cytosolic ATP-independent holdase chaperone responsible for maintaining bacterial secretory proteins in unfolded state and delivering them to SecA in the SEC translocation pathway (Hartl et al., 1990). Additionally, SecB is also a general holdase chaperone like TF (Ullers et al., 2004). SecB exists as dimer of a dimers, where each monomer is a single-domain α/β fold protein with a molecular weight of 17.5 kDa (Hartl et al., 1990; Xu et al., 2000; Dekker et al., 2003). Dimer of dimers means that in each dimer one monomer is always equivalent to a monomer in the other dimer. In the NMR spectra of SecB, this property results in doubling of the peaks for each amino acid of the monomer (Huang et al., 2016). In contrast to TF, client binding does not induce dissociation of the SecB tetramer and so, a single molecule of PhoA binds one tetramer of SecB. In the complex, PhoA wraps around the SecB tetramer in an elongated conformation. Like in complex with TF, each PhoA binding site interacts with SecB in a unique conformation. However, different fragments of PhoA may interact with the same site of SecB and the authors do not note any further symmetry breaking in the complex of SecB with full length PhoA as well as they state that in the complex each PhoA site can bind any SecB site (Huang et al., 2016). This means that the SecB-PhoA interaction is highly dynamic with individual PhoA sites constantly alternating between the same binding sites on SecB, like PhoA sites between different molecules of TF in the TF-PhoA complex. Besides the PhoA-SecB complex the authors also characterized MBP-SecB complex, which revealed the same binding mode (Figure 2D).
DnaK (Hsp70 in eukaryotes) is one of the key general foldase chaperones in all kingdoms of life (Rosenzweig et al., 2019). To function in a large variety of cellular processes, DnaK associates with numerous nucleotide exchange factors (NEFs) and diverse co-chaperones from the Hsp40 protein family known as J domain proteins (JDPs). DnaK consists of two domains – the nucleotide binding domain (NBD), which harbors its ATPase activity (Flaherty et al., 1990), and the SBD, which consists of a β-sheet sandwich (SBDβ) and an α-helical lid (SBDα) (Zhu et al., 1996). Nucleotide binding controls the allosteric cycle of DnaK, which alternates between an open and a closed conformation of the SBD. In the ATP-bound state, SBDα is dissociated from SBDβ and both are docked on the NBD, resulting in lower affinity of SBD toward clients (Takeda and McKay, 1996; Swain et al., 2007; Zhuravleva and Gierasch, 2011; Kityk et al., 2012; Qi et al., 2013). Upon ATP hydrolysis, DnaK transitions to the ADP-bound state in which SBDα encloses the client in the cleft of SBDβ and has high affinity toward clients (McCarty et al., 1995; Zhuravleva et al., 2012). In this state SBD and NBD do not interact and tumble as independently as their connecting linker allows (Bertelsen et al., 2009). The first chaperone-client complex of DnaK, which was characterized structurally in detail was the complex of DnaK with the SH3 domain of drkN (Lee et al., 2015). Although the characterization did not result in a structural model of the chaperone–client complex, it provides the crucial observation that the SH3 domain interacts with DnaK in a dynamic ensemble of multiple globally unfolded states (Figure 2E). The interaction resembles the Skp-OMP complexes and Spy–Im7 complex, but SH3 alternates between the individual conformations on a timescale slower than for the Skp-OMP complexes (>>20 ms). Subsequently, the characterization of DnaK in complex with hTRF1 painted a similar picture (Sekhar et al., 2015; Sekhar et al., 2016; Rosenzweig et al., 2017). hTRF1 also binds DnaK in a dynamic ensemble of multiple globally unfolded states, that exchange on a slower timescale comparing to the Skp-OMP complexes. Additionally, the characterization of the DnaK-hTRF1 complex provides three more important insights. Firstly, hTRF1 binds DnaK as an ensemble regardless of the nucleotide state of DnaK, secondly, DnaK binds the client at 1:1, 1:2, and 1:3 client:DnaK stoichiometric ratios and thirdly, the DnaK residues involved in the interaction with hTRF1 are also conformationally flexible (Sekhar et al., 2015; Sekhar et al., 2016; Rosenzweig et al., 2017).
Taken together, these bacterial chaperone-client complexes characterized at atomic resolution make up a comprehensive dataset (Figure 2), which provides three key conclusions: (i) chaperone-client interactions are generally highly dynamic, with fast dissociation constants, both globally and locally, (ii) chaperone-bound clients are conformationally highly dynamic and populate interconverting conformational ensemble states on the chaperone surface. (iii) although chaperone-client interactions are widely believed to be mediated by hydrophobic contacts, this cannot be generalized from the complexes discussed above. While the complexes of SecB and TF appear dominated by hydrophobic interactions, Spy–Im7 and Skp–OMP feature both electrostatic contacts as well as hydrophobic interactions (Qu et al., 2007; Saio et al., 2014; Huang et al., 2016; Koldewey et al., 2016). The importance of both hydrophobic and electrostatic interactions in chaperone–client complexes has also been shown in a eukaryotic chaperone (Sučec et al., 2020).
The chaperone-client complexes thus demonstrate how chaperones destabilize the structure of clients by highly dynamic interactions, in line with data from functional studies (Finka et al., 2016). As seen on the examples of DnaK and Spy, the interaction is often selective for unfolded and non-native client states, which are thus stabilized relative to the native state. Notably, the binding of a chaperone to a client leads to an overall increase in stability for the resulting client – chaperone complex relative to the client alone, but this does not automatically indicate that the partially folded client is itself stabilized. Therefore, experiments that probe the result complexes tend to observe a stabilization (Mashaghi et al., 2016), while in experiments that monitor the clients selectively a destabilization is detected. In the case of Spy, which allows its client Im7 to fold while chaperone-bound, the folding rates of Im7 are significantly reduced as a result of the stabilization of the non-native states (Stull et al., 2016). Collectively, these structural and functional studies reveal as a putative general mechanism that chaperones thermodynamically destabilize protein structure by stabilizing non-native states.
Toward a Unifying Biophysical Principle Underlying Chaperone Function
During the folding process, proteins need to sample conformationally highly flexible states in order to reach a distant minimum corresponding to the native conformation on the potential energy surface. These folding transition states are thermodynamically unfavorable, because they expose hydrophobic residues to the solvent which requires ordering of the surrounding solvent molecules. Upon folding according to the hydrophobic collapse model, hydrophobic residues gather in the protein core and reduce their exposure to the solvent. The entropy of the polypeptide chain decreases upon folding, which limits its opportunity to explore its conformational space, but the overall entropy of the system increases due to the release of the solvent making hydrophobic collapse thermodynamically favorable. Chaperones provide interaction surfaces that can thermodynamically stabilize proteins in highly flexible transition states, thus delaying the hydrophobic collapse and allowing the protein to explore its conformational space better. In the characterized chaperone-client complexes (Figure 2), the interaction with chaperones selectively stabilizes conformationally flexible non-native states of the client-protein, which in turn destabilizes the highly structured states, including the native conformation or aggregated states.
Importantly, such an effect of chaperones bears a striking resemblance to the well-characterized effect of chemical chaotropes (Hiller, 2020). Urea and other small co-solutes potently disrupt native structures of biomolecules (Hamaguchi and Geiduschek, 1962). Chaotropes counteract the hydrophobic collapse by directly or indirectly increasing the solubility of the hydrophobic residues, thus destabilizing protein native structure as well as protein aggregates. Proteins dissolved in chaotropes display large conformational flexibility with high conformational entropy (Ball and Hallsworth, 2015). This entropy increase counteracts the entropy decrease from the ordering of the solvent molecules caused by solvent exposure of the hydrophobic residues. The exact mechanism of chaotropic denaturation is likely a combination of direct interactions of the chaotrope with the hydrophobic regions and tight interactions of the chaotrope with water molecules in bulk solvent, reducing the amount of available water molecules for the solvation of the protein (Bennion and Daggett, 2003; Ball and Hallsworth, 2015).
Several indications in the experimental data of chaperone-client complexes suggest that chemical chaotropes and chaperones could indeed share similar mechanisms of action. Chaperones have been shown a source of entropy to their client protein in the Im7-Spy and Skp-OMP complexes, where binding of the client increased the client’s conformational flexibility (Burmann et al., 2013; He et al., 2016). The chaperone Spy supports the conformational flexibility of the bound client, allowing it to explore the conformational space sufficiently enough to fold while bound (Stull et al., 2016) and ITC measurement of the Im7–Spy interaction clearly show that the binding is driven by entropy increase (He et al., 2016). Furthermore, the client binding site of DnaK is conformationally flexible, suggesting that DnaK may also increase conformational flexibility of the client upon binding (Rosenzweig et al., 2017; Yang et al., 2017). The chaperones can thus increase the entropy of the client, shifting the client’s equilibrium away from the native state.
Besides these similarities, we also expect functional differences between small molecule chaotropes and chaperones. Small molecule chaotropes achieve chaotropicity by modulating the entire volume of a bulk solution due to their high molar concentrations. The same effect is inconceivable for chaperones as they function at orders of magnitude lower concentrations. Chaperones form pockets and grooves in which the solution may have drastically altered physicochemical properties in comparison to the bulk solution. It is thus conceivable to imagine a pocket with chaotropic properties. The formation of chaotropicity pockets at the chaperone surface thus allows them to create highly concentrated chaotropic environment even at stoichiometric concentrations. The residues comprising the inner surface of the pockets are pivotal to formation of chaotropic pockets and the chaotropicity of these pockets could be modulated by conformational changes. Mechanisms to regulate the activation of chaperones pockets have been shown for ATP-independent chaperones, such as regulation by different transition mechanisms such as oligomer disassembly, order-to-disorder transition or coupled folding/oligomerization (Reichmann et al., 2012; Haslbeck and Vierling, 2015; Suss and Reichmann, 2015; Mas et al., 2020). Such transitions drastically modulate the surface accessibility to the client proteins, providing a potential mechanism for the regulation of chaotropicity in ATP-independent chaperones.
From these considerations, a direct step leads to chaperones that couple chaperone activity to ATP binding and hydrolysis. There are several conceivable mechanisms in which ATP hydrolysis could regulate chaotropicity of chaperones. Hsp90 is an ATP-dependent dimeric chaperone with a clamp-like structure and may provide the first example. ATP triggers large-scale conformational changes of Hsp90, which result in closing of the clamp, but interestingly that does not encapsulate the client protein, rather it creates a larger continuous bipartite binding surface (Ali et al., 2006; Street et al., 2011). Similarly, in GroEL ATP induces conformational changes that alter the client interaction surface, although not by dividing it, rather by changing the properties of the surface as a result of exchanging the residues exposed on the inner surface of the barrel due to the rotation of the chaperone monomers (Tang et al., 2006; Horwich et al., 2007; Villebeck et al., 2007; Tang et al., 2008; Horwich et al., 2009; Jewett and Shea, 2010). Thus, ATP-induced conformational changes may offer a way to directly regulate chaotropicity by perturbing the surface of the pocket where the client docks. Additionally, modular assembly of the pocket as outlined on the example of Hsp90 allows for residual chaperone activity in absence of ATP as each module would retain its chaotropicity (Figure 3). Such a residual activity in the absence of ATP was observed for many ATP-dependent chaperones (Wiech et al., 1992; Cho and Bae, 2007; Rao et al., 2010; Priya et al., 2013c; Mas et al., 2018).
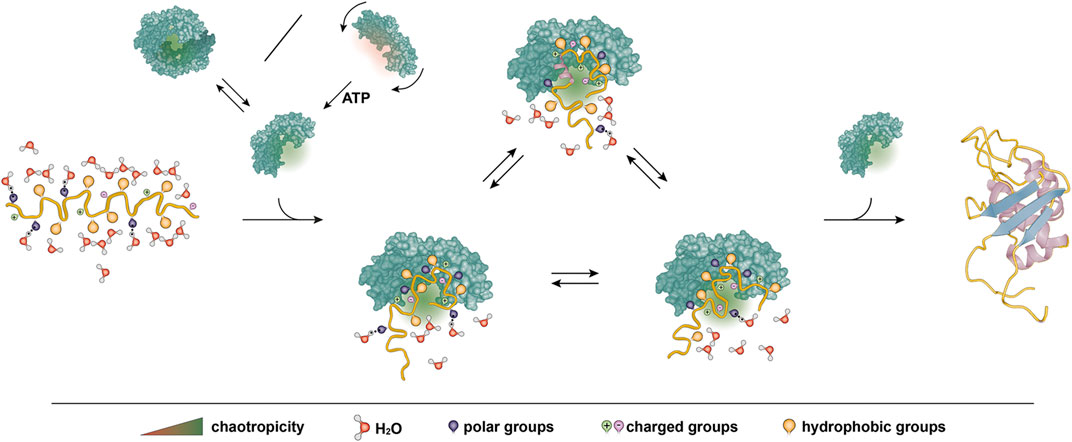
FIGURE 3. Proposed model of chaotropicity underlying generic activity of chaperones. A client protein backbone is shown in yellow with different side chain groups and solvating water molecules as indicated. A chaperone is shown in green. The chaperone features a chaotropic pocket to bind and stabilize the protein, protecting it from premature hydrophobic collapse into a misfolded state and allowing it to explore the conformational space. The protein may fold into its native structure on the chaperone surface or upon release. Chaotropicity stems from the amino acids in the pocket surface and may thus be regulated for example by altering the accessibility of the pocket through dimerization or by ATP-regulated conformational changes.
In the cases of Hsp70 and Hsp90, the ATP cycle is regulated by numerous co-chaperones, which present a potential to further regulate chaperone chaotropicity. Co-chaperones are the key element in chaperone specificity (Bose and Chakrabarti, 2017), thus providing a possibility for function-dependent regulation of chaotropicity. Chaperones such as Hsp90 are commonly found in multiple organelles as well, which may mean different physicochemical properties of the surrounding solution that would inherently alter the chaotropicity of the chaperone. Indirect regulation of chaotropicity by co-chaperones could be a potent way to regulate chaperone chaotropicity to achieve the same effect in different environments.
In summary, chaperone chaotropicity provides a theoretical framework to explain previous experimental data as well as a thermodynamic description of generic chaperone activity for ATP-dependent and -independent chaperones. In contrast to existing models explaining the mechanism of chaperone functions, chaotropicity by default describes client proteins as multistate ensembles, which reflects more accurately the dynamic nature of chaperone–client complexes. The hypothesis presumes a similarity in the molecular mechanism of chaotropicity between small molecule chaotropes and chaperones and the extent of this similarity needs to be tested experimentally. Nevertheless, due to differences in complexity and effective range of concentration between chemical chaotropes and chaperones, the chaperone chaotropicity model also postulates that chaperones exert chaotropicity in a unique form of chaotropic pockets that can be tuned upon conformational change. Considering the crucial roles of chaperones for the health of any organism, a full rationale for their biophysical principles will advance our understanding of homeostasis as well as open new avenues for translational research.
Author Contributions
JM wrote the review with contributions and input from all authors.
Funding
This work was funded by the Swiss National Science Foundation (grant 185388 to SH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Véronique Juvin (SciArtWork) for stylistic help with the Figures.
References
Ali, M. M. U., Roe, S. M., Vaughan, C. K., Meyer, P., Panaretou, B., Piper, P. W., et al. (2006). Crystal Structure of an Hsp90-Nucleotide-p23/Sba1 Closed Chaperone Complex. Nature 440 (7087), 1013–1017. doi:10.1038/nature04716
Anfinsen, C. B., Haber, E., Sela, M., and White, F. H. (1961). The Kinetics of Formation of Native Ribonuclease during Oxidation of the Reduced Polypeptide Chain. Proc. Natl. Acad. Sci. 47 (9), 1309–1314. doi:10.1073/pnas.47.9.1309
Anfinsen, C. B. (1973). Principles that Govern the Folding of Protein Chains. Science 181 (4096), 223–230. doi:10.1126/science.181.4096.223
Balchin, D., Hayer‐Hartl, M., and Hartl, F. U. (2020). Recent Advances in Understanding Catalysis of Protein Folding by Molecular Chaperones. FEBS Lett. 594 (17), 2770–2781. doi:10.1002/1873-3468.13844
Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2016). In Vivo aspects of Protein Folding and Quality Control. Science 353 (6294), aac4354. doi:10.1126/science.aac4354
Ball, P., and Hallsworth, J. E. (2015). Water Structure and Chaotropicity: Their Uses, Abuses and Biological Implications. Phys. Chem. Chem. Phys. 17 (13), 8297–8305. doi:10.1039/C4CP04564E
Bartlett, A. I., and Radford, S. E. (2009). An Expanding Arsenal of Experimental Methods Yields an Explosion of Insights into Protein Folding Mechanisms. Nat. Struct. Mol. Biol. 16 (6), 582–588. doi:10.1038/nsmb.1592
Bechtluft, P., Nouwen, N., Tans, S. J., and Driessen, A. J. M. (2010). SecB-A Chaperone Dedicated to Protein Translocation. Mol. Biosyst. 6 (4), 620–627. doi:10.1039/b915435c
Ben-Zvi, A., De Los Rios, P., Dietler, G., and Goloubinoff, P. (2004). Active Solubilization and Refolding of Stable Protein Aggregates by Cooperative Unfolding Action of Individual Hsp70 Chaperones. J. Biol. Chem. 279 (36), 37298–37303. doi:10.1074/jbc.M405627200
Bennion, B. J., and Daggett, V. (2003). The Molecular Basis for the Chemical Denaturation of Proteins by Urea. Proc. Natl. Acad. Sci. 100 (9), 5142–5147. doi:10.1073/pnas.0930122100
Bertelsen, E. B., Chang, L., Gestwicki, J. E., and Zuiderweg, E. R. P. (2009). Solution Conformation of Wild-type E. coli Hsp70 (DnaK) Chaperone Complexed with ADP and Substrate. Proc. Natl. Acad. Sci. 106 (21), 8471–8476. doi:10.1073/pnas.0903503106
Bitto, E., and McKay, D. B. (2002). Crystallographic Structure of SurA, a Molecular Chaperone that Facilitates Folding of Outer Membrane Porins. Structure 10 (11), 1489–1498. doi:10.1016/s0969-2126(02)00877-8
Bose, D., and Chakrabarti, A. (2017). Substrate Specificity in the Context of Molecular Chaperones. IUBMB Life 69 (9), 647–659. doi:10.1002/iub.1656
Brockwell, D. J., and Radford, S. E. (2007). Intermediates: Ubiquitous Species on Folding Energy Landscapes? Curr. Opin. Struct. Biol. 17 (1), 30–37. doi:10.1016/j.sbi.2007.01.003
Bryngelson, J. D., and Wolynes, P. G. (1987). Spin Glasses and the Statistical Mechanics of Protein Folding. Proc. Natl. Acad. Sci. 84 (21), 7524–7528. doi:10.1073/pnas.84.21.7524
Burmann, B. M., Gerez, J. A., Matečko-Burmann, I., Campioni, S., Kumari, P., Ghosh, D., et al. (2020). Regulation of α-synuclein by Chaperones in Mammalian Cells. Nature 577 (7788), 127–132. doi:10.1038/s41586-019-1808-9
Burmann, B. M., Wang, C., and Hiller, S. (2013). Conformation and Dynamics of the Periplasmic Membrane-Protein-Chaperone Complexes OmpX-Skp and tOmpA-Skp. Nat. Struct. Mol. Biol. 20 (11), 1265–1272. doi:10.1038/nsmb.2677
Calabrese, A. N., Schiffrin, B., Watson, M., Karamanos, T. K., Walko, M., Humes, J. R., et al. (2020). Inter-domain Dynamics in the Chaperone SurA and Multi-Site Binding to its Outer Membrane Protein Clients. Nat. Commun. 11 (1), 2155. doi:10.1038/s41467-020-15702-1
Callon, M., Burmann, B. M., and Hiller, S. (2014). Structural Mapping of a Chaperone-Substrate Interaction Surface. Angew. Chem. Int. Ed. 53 (20), 5069. doi:10.1002/anie.201310963
Cho, E.-K., and Bae, S.-J. (2007). ATP-independent Thermoprotective Activity of Nicotiana Tabacum Heat Shock Protein 70 in Escherichia coli. BMB Rep. 40 (1), 107–112. doi:10.5483/bmbrep.2007.40.1.107
De Los Rios, P., and Barducci, A. (2014). Hsp70 Chaperones Are Non-equilibrium Machines that Achieve Ultra-affinity by Energy Consumption. eLife 3, e02218. doi:10.7554/eLife.02218
Dekker, C., Kruijff, B. d., and Gros, P. (2003). Crystal Structure of SecB from Escherichia coli. J. Struct. Biol. 144 (3), 313–319. doi:10.1016/j.jsb.2003.09.012
Deuerling, E., and Bukau, B. (2004). Chaperone-assisted Folding of Newly Synthesized Proteins in the Cytosol. Crit. Rev. Biochem. Mol. Biol. 39 (5-6), 261–277. doi:10.1080/10409230490892496
Diamond, D. L., and Randall, L. L. (1997). Kinetic Partitioning. J. Biol. Chem. 272 (46), 28994–28998. doi:10.1074/jbc.272.46.28994
Dill, K. A., and MacCallum, J. L. (2012). The Protein-Folding Problem, 50 Years on. Science 338 (6110), 1042–1046. doi:10.1126/science.1219021
Ellis, J. (1987). Proteins as Molecular Chaperones. Nature 328 (6129), 378–379. doi:10.1038/328378a0
Fedorov, A. N., and Baldwin, T. O. (1997). GroE Modulates Kinetic Partitioning of Folding Intermediates between Alternative States to Maximize the Yield of Biologically Active Protein. J. Mol. Biol. 268 (4), 712–723. doi:10.1006/jmbi.1997.1007
Ferbitz, L., Maier, T., Patzelt, H., Bukau, B., Deuerling, E., and Ban, N. (2004). Trigger Factor in Complex with the Ribosome Forms a Molecular Cradle for Nascent Proteins. Nature 431 (7008), 590–596. doi:10.1038/nature02899
Finka, A., Mattoo, R. U. H., and Goloubinoff, P. (2016). Experimental Milestones in the Discovery of Molecular Chaperones as Polypeptide Unfolding Enzymes. Annu. Rev. Biochem. 85, 715–742. doi:10.1146/annurev-biochem-060815-014124
Flaherty, K. M., DeLuca-Flaherty, C., and McKay, D. B. (1990). Three-dimensional Structure of the ATPase Fragment of a 70K Heat-Shock Cognate Protein. Nature 346 (6285), 623–628. doi:10.1038/346623a0
Frydman, J. (2001). Folding of Newly Translated Proteins In Vivo: the Role of Molecular Chaperones. Annu. Rev. Biochem. 70, 603–647. doi:10.1146/annurev.biochem.70.1.603
Glover, J. R., and Lindquist, S. (1998). Hsp104, Hsp70, and Hsp40. Cell 94 (1), 73–82. doi:10.1016/s0092-8674(00)81223-4
Goemans, C., Denoncin, K., and Collet, J.-F. (2014). Folding Mechanisms of Periplasmic Proteins. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1843 (8), 1517–1528. doi:10.1016/j.bbamcr.2013.10.014
Goloubinoff, P., Christeller, J. T., Gatenby, A. A., and Lorimer, G. H. (1989). Reconstitution of Active Dimeric Ribulose Bisphosphate Carboxylase from an Unfolded State Depends on Two Chaperonin Proteins and Mg-ATP. Nature 342 (6252), 884–889. doi:10.1038/342884a0
Goloubinoff, P. (2016). Mechanisms of Protein Homeostasis in Health, Aging and Disease. Swiss Med. Wkly 146, w14306. doi:10.4414/smw.2016.14306
Goloubinoff, P., Mogk, A., Zvi, A. P. B., Tomoyasu, T., and Bukau, B. (1999). Sequential Mechanism of Solubilization and Refolding of Stable Protein Aggregates by a Bichaperone Network. Proc. Natl. Acad. Sci. 96 (24), 13732–13737. doi:10.1073/pnas.96.24.13732
Haber, E., and Anfinsen, C. B. (1962). Side-chain Interactions Governing the Pairing of Half-Cystine Residues in Ribonuclease. J. Biol. Chem. 237 (6), 1839–1844. doi:10.1016/S0021-9258(19)73945-3
Hall, D. (2020). On the Nature of the Optimal Form of the Holdase‐type Chaperone Stress Response. FEBS Lett. 594 (1), 43–66. doi:10.1002/1873-3468.13580
Hamaguchi, K., and Geiduschek, E. P. (1962). The Effect of Electrolytes on the Stability of the Deoxyribonucleate Helix. J. Am. Chem. Soc. 84 (8), 1329–1338. doi:10.1021/ja00867a001
Hartl, F.-U., Lecker, S., Schiebel, E., Hendrick, J. P., and Wickner, W. (1990). The Binding cascade of SecB to SecA to SecYE Mediates Preprotein Targeting to the E. coli Plasma Membrane. Cell 63 (2), 269–279. doi:10.1016/0092-8674(90)90160-g
Hartl, F. U., and Hayer-Hartl, M. (2009). Converging Concepts of Protein Folding In Vitro and In Vivo. Nat. Struct. Mol. Biol. 16 (6), 574–581. doi:10.1038/nsmb.1591
Haslbeck, M., Braun, N., Stromer, T., Richter, B., Model, N., Weinkauf, S., et al. (2004). Hsp42 Is the General Small Heat Shock Protein in the Cytosol of Saccharomyces cerevisiae. Embo J. 23 (3), 638–649. doi:10.1038/sj.emboj.7600080
Haslbeck, M., and Vierling, E. (2015). A First Line of Stress Defense: Small Heat Shock Proteins and Their Function in Protein Homeostasis. J. Mol. Biol. 427 (7), 1537–1548. doi:10.1016/j.jmb.2015.02.002
Haslbeck, M., Weinkauf, S., and Buchner, J. (2019). Small Heat Shock Proteins: Simplicity Meets Complexity. J. Biol. Chem. 294 (6), 2121–2132. doi:10.1074/jbc.REV118.002809
Hayer-Hartl, M., Bracher, A., and Hartl, F. U. (2016). The GroEL-GroES Chaperonin Machine: A Nano-Cage for Protein Folding. Trends Biochem. Sci. 41 (1), 62–76. doi:10.1016/j.tibs.2015.07.009
He, L., and Hiller, S. (2018). Common Patterns in Chaperone Interactions with a Native Client Protein. Angew. Chem. Int. Ed. 57 (20), 5921–5924. doi:10.1002/anie.201713064
He, L., Sharpe, T., Mazur, A., and Hiller, S. (2016). A Molecular Mechanism of Chaperone-Client Recognition. Sci. Adv. 2 (11), e1601625. doi:10.1126/sciadv.1601625
Hemmingsen, S. M., Woolford, C., van der Vies, S. M., Tilly, K., Dennis, D. T., Georgopoulos, C. P., et al. (1988). Homologous Plant and Bacterial Proteins Chaperone Oligomeric Protein Assembly. Nature 333 (6171), 330–334. doi:10.1038/333330a0
Hesterkamp, T., and Bukau, B. (1996). Identification of the Prolyl Isomerase Domain of Escherichia coli Trigger Factor. FEBS Lett. 385 (1-2), 67–71. doi:10.1016/0014-5793(96)00351-1
Hesterkamp, T., Deuerling, E., and Bukau, B. (1997). The Amino-Terminal 118 Amino Acids of Escherichia coli Trigger Factor Constitute a Domain that Is Necessary and Sufficient for Binding to Ribosomes. J. Biol. Chem. 272 (35), 21865–21871. doi:10.1074/jbc.272.35.21865
Hiller, S. (2020). Molecular Chaperones and Their Denaturing Effect on Client Proteins. J. Biomol. NMR 75, 1–8. doi:10.1007/s10858-020-00353-7
Hipp, M. S., Kasturi, P., and Hartl, F. U. (2019). The Proteostasis Network and its Decline in Ageing. Nat. Rev. Mol. Cel Biol. 20 (7), 421–435. doi:10.1038/s41580-019-0101-y
Hoffmann, A., Bukau, B., and Kramer, G. (2010). Structure and Function of the Molecular Chaperone Trigger Factor. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1803 (6), 650–661. doi:10.1016/j.bbamcr.2010.01.017
Horowitz, S., Koldewey, P., Stull, F., and Bardwell, J. C. (2018). Folding while Bound to Chaperones. Curr. Opin. Struct. Biol. 48, 1–5. doi:10.1016/j.sbi.2017.06.009
Horowitz, S., Salmon, L., Koldewey, P., Ahlstrom, L. S., Martin, R., Quan, S., et al. (2016). Visualizing Chaperone-Assisted Protein Folding. Nat. Struct. Mol. Biol. 23 (7), 691–697. doi:10.1038/nsmb.3237
Horwich, A. L., Apetri, A. C., and Fenton, W. A. (2009). The GroEL/GroES Cis Cavity as a Passive Anti-aggregation Device. FEBS Lett. 583 (16), 2654–2662. doi:10.1016/j.febslet.2009.06.049
Horwich, A. L., Fenton, W. A., Chapman, E., and Farr, G. W. (2007). Two Families of Chaperonin: Physiology and Mechanism. Annu. Rev. Cel Dev. Biol. 23, 115–145. doi:10.1146/annurev.cellbio.23.090506.123555
Huang, C., Rossi, P., Saio, T., and Kalodimos, C. G. (2016). Structural Basis for the Antifolding Activity of a Molecular Chaperone. Nature 537 (7619), 202–206. doi:10.1038/nature18965
Huber, D., Rajagopalan, N., Preissler, S., Rocco, M. A., Merz, F., Kramer, G., et al. (2011). SecA Interacts with Ribosomes in Order to Facilitate Posttranslational Translocation in Bacteria. Mol. Cel 41 (3), 343–353. doi:10.1016/j.molcel.2010.12.028
Imamoglu, R., Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2020). Bacterial Hsp70 Resolves Misfolded States and Accelerates Productive Folding of a Multi-Domain Protein. Nat. Commun. 11 (1), 365. doi:10.1038/s41467-019-14245-4
Jahn, T. R., and Radford, S. E. (2005). The Yin and Yang of Protein Folding. FEBS J. 272 (23), 5962–5970. doi:10.1111/j.1742-4658.2005.05021.x
James, R., Kleanthous, C., and Moore, G. R. (1996). The Biology of E Colicins: Paradigms and Paradoxes. Microbiology 142 (Pt 7), 1569–1580. doi:10.1099/13500872-142-7-1569
Jarchow, S., Lück, C., Görg, A., and Skerra, A. (2008). Identification of Potential Substrate Proteins for the Periplasmic Escherichia coli Chaperone Skp. Proteomics 8 (23‐24), 4987–4994. doi:10.1002/pmic.200800288
Jewett, A. I., and Shea, J.-E. (2010). Reconciling Theories of Chaperonin Accelerated Folding with Experimental Evidence. Cell. Mol. Life Sci. 67 (2), 255–276. doi:10.1007/s00018-009-0164-6
Jo, K.-S., Kim, J.-H., Ryu, K.-S., Kang, J.-S., Wang, C.-Y., Lee, Y.-S., et al. (2019). Unique Unfoldase/Aggregase Activity of a Molecular Chaperone Hsp33 in its Holding-Inactive State. J. Mol. Biol. 431 (7), 1468–1480. doi:10.1016/j.jmb.2019.02.022
Kaiser, C. M., Chang, H.-C., Agashe, V. R., Lakshmipathy, S. K., Etchells, S. A., Hayer-Hartl, M., et al. (2006). Real-time Observation of Trigger Factor Function on Translating Ribosomes. Nature 444 (7118), 455–460. doi:10.1038/nature05225
Kityk, R., Kopp, J., Sinning, I., and Mayer, M. P. (2012). Structure and Dynamics of the ATP-Bound Open Conformation of Hsp70 Chaperones. Mol. Cel 48 (6), 863–874. doi:10.1016/j.molcel.2012.09.023
Knowles, T. P. J., Vendruscolo, M., and Dobson, C. M. (2014). The Amyloid State and its Association with Protein Misfolding Diseases. Nat. Rev. Mol. Cel Biol. 15 (6), 384–396. doi:10.1038/nrm3810
Koldewey, P., Stull, F., Horowitz, S., Martin, R., and Bardwell, J. C. A. (2016). Forces Driving Chaperone Action. Cell 166 (2), 369–379. doi:10.1016/j.cell.2016.05.054
Kramer, G., Patzelt, H., Rauch, T., Kurz, T. A., Vorderwülbecke, S., Bukau, B., et al. (2004a). Trigger Factor Peptidyl-Prolyl Cis/trans Isomerase Activity Is Not Essential for the Folding of Cytosolic Proteins in Escherichia coli. J. Biol. Chem. 279 (14), 14165–14170. doi:10.1074/jbc.M313635200
Kramer, G., Rutkowska, A., Wegrzyn, R. D., Patzelt, H., Kurz, T. A., Merz, F., et al. (2004b). Functional Dissection of Escherichia coli Trigger Factor: Unraveling the Function of Individual Domains. Jb 186 (12), 3777–3784. doi:10.1128/jb.186.12.3777-3784.2004
Kwon, E., Kim, D. Y., Gross, C. A., Gross, J. D., and Kim, K. K. (2010). The crystal structureEscherichia coliSpy. Protein Sci. 19 (11), 2252–2259. doi:10.1002/pro.489
Lee, J. H., Zhang, D., Hughes, C., Okuno, Y., Sekhar, A., and Cavagnero, S. (2015). Heterogeneous Binding of the SH3 Client Protein to the DnaK Molecular Chaperone. Proc. Natl. Acad. Sci. USA. 112 (31), E4206–E4215. doi:10.1073/pnas.1505173112
Levinthal, C. (1968). Are There Pathways for Protein Folding?. J. Chim. Phys. 65, 44–45. doi:10.1051/jcp/1968650044
Lin, Z., Madan, D., and Rye, H. S. (2008). GroEL Stimulates Protein Folding through Forced Unfolding. Nat. Struct. Mol. Biol. 15 (3), 303–311. doi:10.1038/nsmb.1394
Lin, Z., and Rye, H. S. (2004). Expansion and Compression of a Protein Folding Intermediate by GroEL. Mol. Cel 16 (1), 23–34. doi:10.1016/j.molcel.2004.09.003
Marx, D. C., Plummer, A. M., Faustino, A. M., Devlin, T., Roskopf, M. A., Leblanc, M. J., et al. (2020). SurA Is a Cryptically Grooved Chaperone that Expands Unfolded Outer Membrane Proteins. Proc. Natl. Acad. Sci. USA 117 (45), 28026–28035. doi:10.1073/pnas.2008175117
Mas, G., Burmann, B. M., Sharpe, T., Claudi, B., Bumann, D., and Hiller, S. (2020). Regulation of Chaperone Function by Coupled Folding and Oligomerization. Sci. Adv. 6 (43), eabc5822. doi:10.1126/sciadv.abc5822
Mas, G., Guan, J.-Y., Crublet, E., Debled, E. C., Moriscot, C., Gans, P., et al. (2018). Structural Investigation of a Chaperonin in Action Reveals How Nucleotide Binding Regulates the Functional Cycle. Sci. Adv. 4 (9), eaau4196, doi:10.1126/sciadv.aau4196
Mas, G., Thoma, J., and Hiller, S. (2019). The Periplasmic Chaperones Skp and SurA. Subcellular Biochem. 92, 169–186. doi:10.1007/978-3-030-18768-2_6
Mashaghi, A., Bezrukavnikov, S., Minde, D. P., Wentink, A. S., Kityk, R., Zachmann-Brand, B., et al. (2016). Alternative Modes of Client Binding Enable Functional Plasticity of Hsp70. Nature 539 (7629), 448–451. doi:10.1038/nature20137
Mattoo, R. U. H., and Goloubinoff, P. (2014). Molecular Chaperones Are Nanomachines that Catalytically Unfold Misfolded and Alternatively Folded Proteins. Cel. Mol. Life Sci. 71 (17), 3311–3325. doi:10.1007/s00018-014-1627-y
Mayer, M. P., and Gierasch, L. M. (2019). Recent Advances in the Structural and Mechanistic Aspects of Hsp70 Molecular Chaperones. J. Biol. Chem. 294 (6), 2085–2097. doi:10.1074/jbc.REV118.002810
McCarty, J. S., Buchberger, A., Reinstein, J., and Bukau, B. (1995). The Role of ATP in the Functional Cycle of the DnaK Chaperone System. J. Mol. Biol. 249 (1), 126–137. doi:10.1006/jmbi.1995.0284
Merz, F., Boehringer, D., Schaffitzel, C., Preissler, S., Hoffmann, A., Maier, T., et al. (2008). Molecular Mechanism and Structure of Trigger Factor Bound to the Translating Ribosome. Embo J. 27 (11), 1622–1632. doi:10.1038/emboj.2008.89
Merz, F., Hoffmann, A., Rutkowska, A., Zachmann-Brand, B., Bukau, B., and Deuerling, E. (2006). The C-Terminal Domain of Escherichia coli Trigger Factor Represents the central Module of its Chaperone Activity. J. Biol. Chem. 281 (42), 31963–31971. doi:10.1074/jbc.M60516420010.1016/s0021-9258(19)84110-8
Morgado, L., Burmann, B. M., Sharpe, T., Mazur, A., and Hiller, S. (2017). The Dynamic Dimer Structure of the Chaperone Trigger Factor. Nat. Commun. 8 (1), 1992. doi:10.1038/s41467-017-02196-7
Onuchic, J. N., and Wolynes, P. G. (2004). Theory of Protein Folding. Curr. Opin. Struct. Biol. 14 (1), 70–75. doi:10.1016/j.sbi.2004.01.009
Patzelt, H., Kramer, G., Rauch, T., Schönfeld, H.-J., Bukau, B., and Deuerling, E. (2002). Three-state Equilibrium of Escherichia coli Trigger Factor. Biol. Chem. 383 (10), 1611–1619. doi:10.1515/bc.2002.182
Pelham, H. R. B. (1986). Speculations on the Functions of the Major Heat Shock and Glucose-Regulated Proteins. Cell 46 (7), 959–961. doi:10.1016/0092-8674(86)90693-8
Priya, S., Sharma, S. K., and Goloubinoff, P. (2013a). Molecular Chaperones as Enzymes that Catalytically Unfold Misfolded Polypeptides. FEBS Lett. 587 (13), 1981–1987. doi:10.1016/j.febslet.2013.05.014
Priya, S., Sharma, S. K., Sood, V., Mattoo, R. U. H., Finka, A., Azem, A., et al. (2013b). GroEL and CCT Are Catalytic Unfoldases Mediating Out-Of-Cage Polypeptide Refolding without ATP. Proc. Natl. Acad. Sci. 110 (18), 7199–7204. doi:10.1073/pnas.1219867110
Priya, S., Sharma, S. K., Sood, V., Mattoo, R. U. H., Finka, A., Azem, A., et al. (2013c). GroEL and CCT Are Catalytic Unfoldases Mediating Out-Of-Cage Polypeptide Refolding without ATP. Proc. Natl. Acad. Sci. 110 (18), 7199–7204. doi:10.1073/pnas.1219867110
Qi, R., Sarbeng, E. B., Liu, Q., Le, K. Q., Xu, X., Xu, H., et al. (2013). Allosteric Opening of the Polypeptide-Binding Site when an Hsp70 Binds ATP. Nat. Struct. Mol. Biol. 20 (7), 900–907. doi:10.1038/nsmb.2583
Qu, J., Mayer, C., Behrens, S., Holst, O., and Kleinschmidt, J. H. (2007). The Trimeric Periplasmic Chaperone Skp of Escherichia coli Forms 1:1 Complexes with Outer Membrane Proteins via Hydrophobic and Electrostatic Interactions. J. Mol. Biol. 374 (1), 91–105. doi:10.1016/j.jmb.2007.09.020
Quan, S., Koldewey, P., Tapley, T., Kirsch, N., Ruane, K. M., Pfizenmaier, J., et al. (2011). Genetic Selection Designed to Stabilize Proteins Uncovers a Chaperone Called Spy. Nat. Struct. Mol. Biol. 18 (3), 262–269. doi:10.1038/nsmb.2016
Rao, J. L., Reddy, P. S., Mishra, R. N., Gupta, D., Sahal, D., Tuteja, N., et al. (2010). Thermo and pH Stable ATP-independent Chaperone Activity of Heat-Inducible Hsp70 from Pennisetum Glaucum. Plant Signal. Behav. 5 (2), 110–121. doi:10.4161/psb.5.2.10547
Reichmann, D., Xu, Y., Cremers, C. M., Ilbert, M., Mittelman, R., Fitzgerald, M. C., et al. (2012). Order Out of Disorder: Working Cycle of an Intrinsically Unfolded Chaperone. Cell 148 (5), 947–957. doi:10.1016/j.cell.2012.01.045
Rosenzweig, R., Nillegoda, N. B., Mayer, M. P., and Bukau, B. (2019). The Hsp70 Chaperone Network. Nat. Rev. Mol. Cel Biol. 20 (11), 665–680. doi:10.1038/s41580-019-0133-3
Rosenzweig, R., Sekhar, A., Nagesh, J., and Kay, L. E. (2017). Promiscuous Binding by Hsp70 Results in Conformational Heterogeneity and Fuzzy Chaperone-Substrate Ensembles. eLife 6, e28030. doi:10.7554/eLife.28030
Saio, T., Guan, X., Rossi, P., Economou, A., and Kalodimos, C. G. (2014). Structural Basis for Protein Antiaggregation Activity of the Trigger Factor Chaperone. Science 344 (6184), 1250494. doi:10.1126/science.1250494
Sekhar, A., Rosenzweig, R., Bouvignies, G., and Kay, L. E. (2016). Hsp70 Biases the Folding Pathways of Client Proteins. Proc. Natl. Acad. Sci. USA 113 (20), E2794–E2801. doi:10.1073/pnas.1601846113
Sekhar, A., Rosenzweig, R., Bouvignies, G., and Kay, L. E. (2015). Mapping the Conformation of a Client Protein through the Hsp70 Functional Cycle. Proc. Natl. Acad. Sci. USA 112 (33), 10395–10400. doi:10.1073/pnas.1508504112
Sharma, S., Chakraborty, K., Müller, B. K., Astola, N., Tang, Y.-C., Lamb, D. C., et al. (2008). Monitoring Protein Conformation along the Pathway of Chaperonin-Assisted Folding. Cell 133 (1), 142–153. doi:10.1016/j.cell.2008.01.048
Sharma, S., Christen, P., and Goloubinoff, P. (2009). Disaggregating Chaperones: an Unfolding story. Cpps 10 (5), 432–446. doi:10.2174/138920309789351930
Sharma, S. K., De Los Rios, P., Christen, P., Lustig, A., and Goloubinoff, P. (2010). The Kinetic Parameters and Energy Cost of the Hsp70 Chaperone as a Polypeptide Unfoldase. Nat. Chem. Biol. 6 (12), 914–920. doi:10.1038/nchembio.455
Shtilerman, M., Lorimer, G. H., and Englander, S. W. (1999). Chaperonin Function: Folding by Forced Unfolding. Science 284 (5415), 822–825. doi:10.1126/science.284.5415.822
Sklar, J. G., Wu, T., Kahne, D., and Silhavy, T. J. (2007). Defining the Roles of the Periplasmic Chaperones SurA, Skp, and DegP in Escherichia coli. Genes Develop. 21 (19), 2473–2484. doi:10.1101/gad.1581007
Sousa, R. (2014). Structural Mechanisms of Chaperone Mediated Protein Disaggregation. Front. Mol. Biosci. 1, 12. doi:10.3389/fmolb.2014.00012
Street, T. O., Lavery, L. A., and Agard, D. A. (2011). Substrate Binding Drives Large-Scale Conformational Changes in the Hsp90 Molecular Chaperone. Mol. Cel 42 (1), 96–105. doi:10.1016/j.molcel.2011.01.029
Stull, F., Koldewey, P., Humes, J. R., Radford, S. E., and Bardwell, J. C. A. (2016). Substrate Protein Folds while it Is Bound to the ATP-independent Chaperone Spy. Nat. Struct. Mol. Biol. 23 (1), 53–58. doi:10.1038/nsmb.3133
Sučec, I., Wang, Y., Dakhlaoui, O., Weinhäupl, K., Jores, T., Costa, D., et al. (2020). Structural Basis of Client Specificity in Mitochondrial Membrane-Protein Chaperones. Sci. Adv. 6 (51). doi:10.1126/sciadv.abd0263
Suss, O., and Reichmann, D. (2015). Protein Plasticity Underlines Activation and Function of ATP-independent Chaperones. Front. Mol. Biosci. 2 (43), 43. doi:10.3389/fmolb.2015.00043
Swain, J. F., Dinler, G., Sivendran, R., Montgomery, D. L., Stotz, M., and Gierasch, L. M. (2007). Hsp70 Chaperone Ligands Control Domain Association via an Allosteric Mechanism Mediated by the Interdomain Linker. Mol. Cel 26 (1), 27–39. doi:10.1016/j.molcel.2007.02.020
Takeda, S., and McKay, D. B. (1996). Kinetics of Peptide Binding to the Bovine 70 kDa Heat Shock Cognate Protein, a Molecular Chaperone†. Biochemistry 35 (14), 4636–4644. doi:10.1021/bi952903o
Tang, Y.-C., Chang, H.-C., Chakraborty, K., Hartl, F. U., and Hayer-Hartl, M. (2008). Essential Role of the Chaperonin Folding Compartment In Vivo. Embo J. 27 (10), 1458–1468. doi:10.1038/emboj.2008.77
Tang, Y.-C., Chang, H.-C., Roeben, A., Wischnewski, D., Wischnewski, N., Kerner, M. J., et al. (2006). Structural Features of the GroEL-GroES Nano-Cage Required for Rapid Folding of Encapsulated Protein. Cell 125 (5), 903–914. doi:10.1016/j.cell.2006.04.027
Tittelmeier, J., Nachman, E., and Nussbaum-Krammer, C. (2020). Molecular Chaperones: A Double-Edged Sword in Neurodegenerative Diseases. Front. Aging Neurosci. 12, 581374. doi:10.3389/fnagi.2020.581374
Tsirigotaki, A., De Geyter, J., Šoštaric´, N., Economou, A., and Karamanou, S. (2017). Protein export through the Bacterial Sec Pathway. Nat. Rev. Microbiol. 15 (1), 21–36. doi:10.1038/nrmicro.2016.161
Ullers, R. S., Luirink, J., Harms, N., Schwager, F., Georgopoulos, C., and Genevaux, P. (2004). SecB Is a Bona Fide Generalized Chaperone in Escherichia coli. Proc. Natl. Acad. Sci. 101 (20), 7583–7588. doi:10.1073/pnas.0402398101
Valent, Q. A., Kendall, D. A., High, S., Kusters, R., Oudega, B., and Luirink, J. (1995). Early Events in Preprotein Recognition in E. coli: Interaction of SRP and Trigger Factor with Nascent Polypeptides. EMBO J. 14 (22), 5494–5505. doi:10.1002/j.1460-2075.1995.tb00236.x
Villebeck, L., Persson, M., Luan, S.-L., Hammarström, P., Lindgren, M., and Jonsson, B.-H. (2007). Conformational Rearrangements of Tail-Less Complex Polypeptide 1 (TCP-1) Ring Complex (TRiC)-Bound Actin. Biochemistry 46 (17), 5083–5093. doi:10.1021/bi062093o
Vrontou, E., and Economou, A. (2004). Structure and Function of SecA, the Preprotein Translocase Nanomotor. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1694 (1), 67–80. doi:10.1016/j.bbamcr.2004.06.003
Walerych, D., Gutkowska, M., Klejman, M. P., Wawrzynow, B., Tracz, Z., Wiech, M., et al. (2010). ATP Binding to Hsp90 Is Sufficient for Effective Chaperoning of P53 Protein. J. Biol. Chem. 285 (42), 32020–32028. doi:10.1074/jbc.M110.112110
Walter, S., and Buchner, J. (2002). Molecular Chaperones-Cellular Machines for Protein Folding. Angew. Chem. Int. Ed. 41 (7), 1098–1113. doi:10.1002/1521-3773(20020402)41:7<1098::aid-anie1098>3.0.co;2-9
Walton, T. A., and Sousa, M. C. (2004). Crystal Structure of Skp, a Prefoldin-like Chaperone that Protects Soluble and Membrane Proteins from Aggregation. Mol. Cel 15 (3), 367–374. doi:10.1016/j.molcel.2004.07.023
Wang, S., Yang, C.-I., and Shan, S.-O. (2017). SecA Mediates Cotranslational Targeting and Translocation of an Inner Membrane Protein. J. Cel Biol. 216 (11), 3639–3653. doi:10.1083/jcb.201704036
Wentink, A., Nussbaum-Krammer, C., and Bukau, B. (2019). Modulation of Amyloid States by Molecular Chaperones. Cold Spring Harb Perspect. Biol. 11 (7), a033969. doi:10.1101/cshperspect.a033969
Wiech, H., Buchner, J., Zimmermann, R., and Jakob, U. (1992). Hsp90 Chaperones Protein Folding In Vitro. Nature 358 (6382), 169–170. doi:10.1038/358169a0
Wolynes, P., Onuchic, J., and Thirumalai, D. (1995). Navigating the Folding Routes. Science 267 (5204), 1619–1620. doi:10.1126/science.7886447
Xu, Z., Knafels, J. D., and Yoshino, K. (2000). Crystal Structure of the Bacterial Protein export Chaperone secB. Nat. Struct. Biol. 7 (12), 1172–1177. doi:10.1038/82040
Yang, J., Zong, Y., Su, J., Li, H., Zhu, H., Columbus, L., et al. (2017). Conformation Transitions of the Polypeptide-Binding Pocket Support an Active Substrate Release from Hsp70s. Nat. Commun. 8 (1), 1201. doi:10.1038/s41467-017-01310-z
Zhu, X., Zhao, X., Burkholder, W. F., Gragerov, A., Ogata, C. M., Gottesman, M. E., et al. (1996). Structural Analysis of Substrate Binding by the Molecular Chaperone DnaK. Science 272 (5268), 1606–1614. doi:10.1126/science.272.5268.1606
Zhuravleva, A., Clerico, E. M., and Gierasch, L. M. (2012). An Interdomain Energetic Tug-Of-War Creates the Allosterically Active State in Hsp70 Molecular Chaperones. Cell 151 (6), 1296–1307. doi:10.1016/j.cell.2012.11.002
Keywords: protein homeostasis, chaperone, biophysical mechanisms, chaotropicity, chaperone-client complexes, protein folding
Citation: Macošek J, Mas G and Hiller S (2021) Redefining Molecular Chaperones as Chaotropes. Front. Mol. Biosci. 8:683132. doi: 10.3389/fmolb.2021.683132
Received: 19 March 2021; Accepted: 20 May 2021;
Published: 14 June 2021.
Edited by:
Axel Mogk, Heidelberg University, GermanyReviewed by:
Anne Wentink, University of Heidelberg, GermanyLila Gierasch, University of Massachusetts Amherst, United States
Titus M. Franzmann, Max Planck Society (MPG), Germany
Copyright © 2021 Macošek, Mas and Hiller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastian Hiller, c2ViYXN0aWFuLmhpbGxlckB1bmliYXMuY2g=