 Nimesha Tadepalle
Nimesha Tadepalle Elena I. Rugarli
Elena I. Rugarli- 1Molecular and Cell Biology Laboratory, Salk Institute of Biological Sciences, La Jolla, CA, United States
- 2Institute for Genetics, University of Cologne, Cologne, Germany
- 3Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD), Cologne, Germany
- 4Center for Molecular Medicine (CMMC),Cologne, Germany
Hereditary spastic paraplegias (HSPs) are genetically heterogeneous conditions caused by the progressive dying back of the longest axons in the central nervous system, the corticospinal axons. A wealth of data in the last decade has unraveled disturbances of lipid droplet (LD) biogenesis, maturation, turnover and contact sites in cellular and animal models with perturbed expression and function of HSP proteins. As ubiquitous organelles that segregate neutral lipid into a phospholipid monolayer, LDs are at the cross-road of several processes including lipid metabolism and trafficking, energy homeostasis, and stress signaling cascades. However, their role in brain cells, especially in neurons remains enigmatic. Here, we review experimental findings linking LD abnormalities to defective function of proteins encoded by HSP genes, and discuss arising questions in the context of the pathogenesis of HSP.
Introduction
Hereditary Spastic Paraplegia
In humans, voluntary and fine motor movements are controlled by the corticospinal tract, which is composed of more than a million axons of the pyramidal motor neurons (Welniarz et al., 2017). The majority of these axons travel through the internal capsule and the brainstem, decussate in the caudal medulla, and descend in the lateral funiculus of the contralateral spinal cord to make mono-synaptic contacts with spinal motor neurons in the ventral horn (contralateral corticospinal tract). A small portion of corticospinal axons descent unilaterally to form the ipsilateral corticospinal tract. Progressive degeneration of the distal corticospinal axons causes weakness and spasticity (rigidity) of the lower limb. These symptoms define pure forms of hereditary spastic paraplegia, a neurological condition with a prevalence of 1–10 per 100,000 individuals (Shribman et al., 2019; Lallemant-Dudek and Durr, 2020). Complicated HSP is characterized by the association of other clinical symptoms, including ataxia, intellectual disability, cognitive impairment, epilepsy, amyotrophy, and peripheral neuropathy (Blackstone, 2018b; Shribman et al., 2019; Lallemant-Dudek and Durr, 2020). This clinical complexity is reflected in a vast genetic heterogeneity of HSP, with over 80 genes classified in the OMIM database (www.omim.org) so far. HSP can be inherited as autosomal dominant, autosomal recessive or X-linked (Schule and Schols, 2011). The age of onset spans from childhood to late life. So far, apart from treatments to reduce spasticity, there is no available strategy to block the progression of the symptoms.
Corticospinal axons projecting to the lumbar spinal motoneurons can reach the outstanding length of 1 m in adult humans, contain 99% of the neuronal cytosol, and strongly depend on efficient trafficking mechanisms to transport macromolecules and organelles at sites very distant from the oma. The extreme polarization of cortical motor neurons makes them vulnerable to even minor disturbances of a plethora of pathways. Disruption or abnormalities of endosomal trafficking and dynamics, mitochondrial quality control, endoplasmic reticulum (ER) morphogenesis, cargo-mediated transport on the microtubules, lysosomal function, and lipid metabolism play a role in the pathogenesis of HSP (Blackstone et al., 2011; Rugarli and Langer, 2012; Allison et al., 2017; Blackstone, 2018a; Darios et al., 2020). Here, we focus on the intriguing connection between several HSP proteins and the biology of lipid droplets, and discuss the possible role of LD dysfunction in the pathogenesis of the disease.
Lipid Droplets
Organelles consist of an aqueous environment harboring various macromolecules that are surrounded by a phospholipid bilayer, leading to the compartmentalization of specific biochemical reactions occurring in a cell. Phase separation of a hydrophobic lipid core from the aqueous environment of the cell by a phospholipid monolayer makes LDs unique. LDs are evolutionary conserved, ubiquitous organelles found in prokaryotes, as well as in unicellular and higher eukaryotes (Murphy, 2001; Alvarez and Steinbuchel, 2002; Fujimoto and Parton, 2011; Ohsaki et al., 2014). The neutral lipid core of LDs consists of triacylglycerols (TAGs), sterol esters (SE) and rarely retinyl esters (Brasaemle, 2007; Fujimoto and Parton, 2011; Walther and Farese, 2012; Barbosa and Siniossoglou, 2017; Walther et al., 2017). The LD surface is composed of a different combination of phospholipids. In mammalian cells, the phospholipid monolayer of LDs is highly enriched in phosphatidylcholine (PC), followed by phosphatidylethanolamine (PE) (Bartz et al., 2007). In yeast, on the other hand, phosphatidylinositol (PI) is the second most abundant phospholipid species on LDs, after PC (Grillitsch et al., 2011; Ohsaki et al., 2014; Wang, 2015). A compendium of proteins coats the LD surface, and contributes to the function of this organelle (Kory et al., 2016; Bersuker et al., 2018).
LDs play critical roles by storing fatty acids (FAs), which can then be delivered to mitochondria or peroxisomes for β-oxidation, or be utilized to synthesize new membranes and mediate lipid signaling. Therefore, LDs are key organelles in energy homeostasis, lipid metabolism, and signal transduction. Furthermore, LDs have been implicated in regulating ER stress and in clearing of protein aggregates (Walther and Farese, 2009; Fujimoto and Parton, 2011; Walther and Farese, 2012; Walther et al., 2017; Olzmann and Carvalho, 2019; Roberts and Olzmann, 2020). LD dysfunction is the hallmark of several diseases, such as obesity, diabetes, hepatic steatosis, cardiovascular disease and cancer (Krahmer et al., 2013; Welte, 2015; Gluchowski et al., 2017; Goldberg et al., 2018; Pennetta and Welte, 2018; Cruz et al., 2020; Seebacher et al., 2020). Recently, motor neuron diseases have become subjects of investigation by LD experts due to aberrant LD biology observed in neurodegenerative diseases like amyotrophic lateral sclerosis, Huntington’s disease and HSP (Farmer et al., 2020). In the following sections, we review the experimental evidence that links the function of HSP proteins to every step of the LD life cycle (Table 1), and discuss the possible impact of these observations on the pathogenesis of HSP.
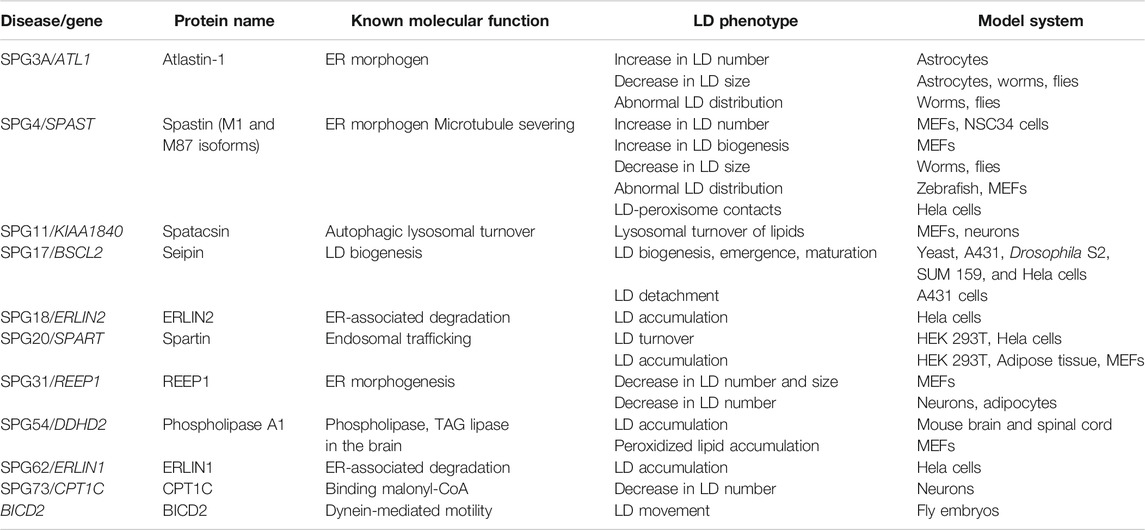
TABLE 1. Identified spastic paraplegia (SPG) genes involved in LD metabolism.
Disturbances of LD Biogenesis in HSP Pathogenesis
HSP Proteins and the Shaping of the Tubular ER
In eukaryotic cells, LDs are assembled at the ER through a series of sequential steps beginning with FA activation by esterification with coenzyme A, followed by synthesis of TAG or SE by enzymes residing in the ER. LD biogenesis begins when neutral lipids get sequestered between the two leaflets of the ER membrane, leading to the bulging of a lens-like structure, in a process that is known as the LD nucleation. With increasing neutral lipid synthesis, this lens structure expands to form a nascent LD, which further grows in a mature LD. The latter bulges toward the cytosolic side of the ER, and can ultimately detach to form a separate entity. Several excellent reviews have described in detail various steps of LD biogenesis (Walther and Farese, 2009, 2012; Pol et al., 2014; Henne et al., 2020).
The ER is composed of different compartments, the nuclear envelope, the ER sheets and the ER tubules, however LD biogenesis mainly occurs at ER tubules, at least in mammalian cells (Kassan et al., 2013; Santinho et al., 2020). One major difference between ER tubules and sheets is that ER sheets are mainly flat, while ER tubules are highly curved. Membrane curvature at the ER tubules decreases the critical concentration of TAG necessary for LD nucleation (Santinho et al., 2020). While the rough ER is only present in neuronal Soma, the tubular ER extends into axons, where it forms a long narrow continuous system (Terasaki, 2018; Öztürk et al., 2020). Maintenance of the tubular ER compartment is crucial for axonal health, as demonstrated by the involvement of several proteins that shape the tubular ER in axonopathies (Hubner and Kurth, 2014). Remarkably, genetic experiments of overexpression or depletion of these HSP proteins in various model organisms has revealed LD abnormalities, pointing to an intimate connection between ER tubule shaping and the formation of LDs (Figure 1).
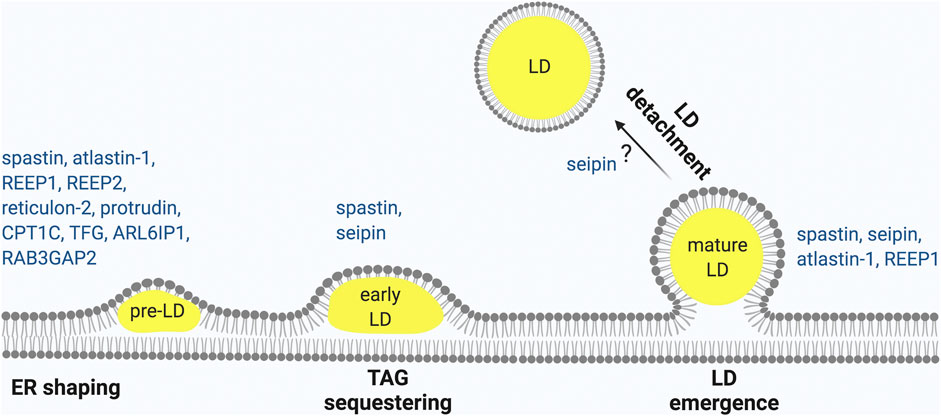
FIGURE 1. HSP proteins involved in different steps of LD biogenesis. LD biogenesis progress occurring at the ER as shown from left to right. (1) ER morphogens regulate membrane curvature required to initiate LD biogenesis. (2) Neutral lipid accumulates between the two leaflets of the ER to form nascent LDs that (3) continue to expand to early LDs (4) and finally lead to mature LDs that (5) may undergo detachment from the ER. HSP-associated proteins involved in each of these steps are indicated. Made with Biorender.com.
Atlastin-1, −2 and −3 belong to the dynamin family of GTPases and play a well-established role in homotypic ER membrane fusion (Hu et al., 2009; Orso et al., 2009; Kim et al., 2017). Atlastins localize to three-way junctions at the ER tubules and target the ER membrane using a long hydrophobic domain that insert in the membrane as a hairpin (Park et al., 2010). Mutations in ATL1 are responsible for HSP type 3 (Sauter et al., 2004), while ATL3 is mutated in hereditary sensory neuropathy (Kornak et al., 2014). Cells lacking or expressing dominant negative forms of atlastins show fragmented and unbranched ER tubules (Rismanchi et al., 2008; Hu et al., 2009; Wang et al., 2016b; Zhao et al., 2016; Behrendt et al., 2019). ER morphology defects with less three-way junctions and fragmented tubules have also been observed in flies lacking atlastin, while atlastin antibodies impaired ER network formation in vitro using a membrane fraction from Xenopus oocytes (Hu et al., 2009; Orso et al., 2009).
The C. elegans orthologue of atlastin-1 (ATLN-1) was found as a hit in a genetic screen aimed at identifying mutants with less LDs. Worms depleted or expressing mutant forms of ATLN-1 displayed smaller LDs that were abnormally distributed as compared to wildtype worms (Klemm et al., 2013). A similar phenotype was observed in Drosophila, where downregulation of the atlastin orthologue led to reduced accumulation of LDs and TAGs in the fat body, rendering fly larvae more susceptible to starvation. Furthermore, the membrane fusion activity of atlastin-1 was responsible for mediating this LD phenotype (Klemm et al., 2013). Smaller numerous LDs were recently observed in astrocytes derived from human pluripotent stem cells carrying ATL1 disease mutations (Mou et al., 2020).
Atlastins interact with members of the reticulon and the REEP/DP1/Yop1 protein families, which contain a reticulon-homology domain that target them at the ER tubules (Shibata et al., 2010). This domain consists of two hydrophobic regions, each 28–36 amino acids long, which are too long to span the membrane once and instead form a hairpin structure, embedded as a wedge and imparting curvature to the membrane. Consistently, in absence of reticulons and REEPs, the tubular ER is reduced (Shibata et al., 2010; Wang et al., 2016b; Yalcin et al., 2017). REEP1 is the causative gene of SPG31, a relatively common autosomal dominant HSP (Zuchner et al., 2006). A closely related gene, REEP2, is mutated in SPG72 (Esteves et al., 2014), while mutations in RTN2, encoding reticulon-2, lead to HSP type 12 (Montenegro et al., 2012). Reep1 null mice exhibit a progressive motor phenotype, without obvious degeneration of the corticospinal axons. Surprisingly, they also showed a mild lipoatrophy, consistent with a defect in LD formation in adipose tissue (Renvoise et al., 2016). Moreover, mouse embryonic fibroblasts (MEFs) and neurons isolated from these mice showed fewer and smaller LDs (Renvoise et al., 2016). In normal condition, REEP1 is never found on LDs, however when HSP-associated mutant variants of REEP1 were expressed in mammalian cells they were mistargeted to LDs. Notably, atlastin-1 and REEP1 cooperate in controlling LD size. In fact, concomitant overexpression of atlastin-1 and REEP1 in mammalian cells leads to the formation of bigger LDs (Falk et al., 2014).
Atlastin-1 also interacts with CPT1C, the neuronal isoform of carnitine palmitoyl-transferase (CPT1C), suggesting a role of this protein in ER morphogenesis (Rinaldi et al., 2015). Mutations in CPT1C were recently identified to cause a pure form of autosomal dominant HSP (SPG73) (Rinaldi et al., 2015). This protein does not catalyze the transfer of fatty acyl to carnitine, but rather binds malonyl-CoA and may sense lipid availability (Wolfgang et al., 2006). Interestingly, knock-out mice for CPT1C display reduced body weight and food intake, but are prone to obesity when fed a high fat diet (Wolfgang et al., 2006). Neurons isolated from CPT1C null mice possess fewer LDs (Rinaldi et al., 2015). The expression of mutant forms of CPT1C involved in HSP reduce LD number and size in a dominant negative manner (Rinaldi et al., 2015).
In conclusion, ER-resident morphogenesis proteins involved in HSP interact with one another and play essential roles in regulating LD size and formation (Figure 1). Consistently, alteration of the ratio of sheets vs. tubules by overexpression of atlastin-1, reticulon-4 or REEP-5, which induce ER tubulation, led to an increase in LD nucleation (Santinho et al., 2020), indicating that proteins involved in ensuring bending and curvature of the ER tubules assist in LD emergence (Roberts and Olzmann, 2020). This conclusion agrees with the in vivo phenotypes of atlastin-1, and REEP1 deficient models, which are all characterized by a defect in LD biogenesis. Other HSP proteins, such as TFG, ZFYVE27, RAB3GAP2, and ARL6IP1, also contribute to ER shaping and may therefore affect LD biogenesis (Hubner and Kurth, 2014).
Spastin M1, a Microtubule-Severing Protein, Is Recruited to LDs
Compelling links to LD function have been uncovered for spastin, the product of SPAST, which is the most commonly mutated autosomal dominant HSP gene (Hazan et al., 1999). Spastin is a well-characterized microtubule-severing protein (Errico et al., 2002; Evans et al., 2005; Roll-Mecak and Vale, 2008). Spastin belongs to the AAA (ATPases Associated with various cellular Activities) family of ATPases (Sauer and Baker, 2011), and uses the energy derived from ATP hydrolysis to destabilize the microtubule lattice leading ultimately to the internal breaking (severing) of the tracks (Roll-Mecak and Vale, 2008). Spastin also harbors a MIT (microtubule-interacting and trafficking) domain, which can recruit the ESCRTIII proteins IST1 and CHMP1B (Reid et al., 2005; Allison et al., 2013).
In mammalian cells, four isoforms of spastin are produced by alternative splicing of exon 4 and dual initiation of translation (Claudiani et al., 2005). When spastin translation starts at the first in frame AUG in exon 1, the resulting protein (spastin M1) comprises a stretch of 86 amino acid at the N-terminus, which contains a hydrophobic region and targets the protein at the ER (Connell et al., 2009; Park et al., 2010). When carrying mutations in the AAA domain trapping the protein on the microtubules, spastin M1 specifically bundles perinuclear microtubules associated with the ER, which are more resistant to depolymerization (Plaud et al., 2018). In contrast, spastin isoforms starting at the methionine M87 localize to the cytosol and endosomal compartments (Connell et al., 2009; Plaud et al., 2018). Spastin M87 is abundant in all cells and tissues, and has been implicated in several cellular processes, such as endosomal tubulation (Allison et al., 2013), midbody abscission and cytokinesis (Connell et al., 2009), mitotic spindle disassembly (Zhang et al., 2007; Vietri et al., 2015), and lysosomal function (Allison et al., 2019). Spastin M87 display efficient microtubule-severing activity and its overexpression disrupts the microtubule cytoskeleton (Errico et al., 2002).
Despite the fact that the expression of spastin M1 is kept low by both transcriptional and translational control mechanisms (Claudiani et al., 2005; Mancuso and Rugarli, 2008), the synthesis of M1 isoforms is evolutionary conserved not only in mammals, but also in zebrafish (Arribat et al., 2020). Furthermore, spastin M1 interacts with other HSP proteins and ER morphogens, including atlastin-1, REEP1, REEP2, reticulon-1, and protrudin (Mannan et al., 2006a; Mannan et al., 2006b; Sanderson et al., 2006; Park et al., 2010; Esteves et al., 2014), suggesting a pathogenic role of this isoform and a contribution to ER shaping. Spastin M1 behaves as a class I LD protein, i.e. proteins that are inserted in the ER membrane and can laterally diffuse from the outer leaflet of the ER to the LD phospholipid monolayer (Kory et al., 2016; Roberts and Olzmann, 2020). Consistently, the hydrophobic domain of spastin M1 adopts a hairpin conformation and mediates targeting of the protein from the ER to nascent and mature LDs when cells are loaded by oleic acids (Park et al., 2010; Papadopoulos et al., 2015; Chang et al., 2019).
What is the exact role of spastin at the LDs? Answering this question is complicated by the presence of the different spastin isoforms and the fact that manipulation of spastin levels interferes with microtubule dynamics. However, modeling spastin deficiency in vivo in several organisms has unraveled LD biogenesis defects. The Drosophila Dspastin exists as a unique form with an extended N-terminus. Loss of function of Dspastin obtained by RNA interference or by expressing a dominant-negative mutant reduced the number and size of LDs in the fat body, the skeletal muscle and the peripheral nerves (Papadopoulos et al., 2015). Decreased accumulation of LDs was also observed in the C. elegans gut, upon deletion or depletion of the spastin orthologue (Papadopoulos et al., 2015), although in the worm only a short spastin variant is described. In the zebrafish, a knock-out of all spastin isoforms by CRISPR-Cas9 gene editing led to a metabolic phenotype with reduced mitochondrial respiration and glycolysis, lipid accumulation around the swim bladder and ER abnormalities in the skeletal muscle (Arribat et al., 2020).
In apparent contrast with these findings in vivo, cultured cells lacking spastin showed an increased number of LDs accompanied by enhanced synthesis of TAGs, which were rescued by re-expressing spastin M1 and to a lesser extent spastin M87 (Tadepalle et al., 2020). This phenotype was particularly prominent upon severe starvation, a condition that stimulates LD biogenesis (Rambold et al., 2015). An increased number of smaller LDs was also observed in cells derived from spastin KO fish embryos (Arribat et al., 2020). Overexpression of spastin M1 on the other hand induced the appearance of large, but fewer LDs (Papadopoulos et al., 2015; Arribat et al., 2020). Since spastin M1 localizes to nascent LDs (Chang et al., 2019; Tadepalle et al., 2020), an explanation for these findings could be in a specific role of spastin M1-containing hexamers that are anchored to the ER in shaping the ER and the microtubule network at sites where LDs are formed, limiting LD nucleation but promoting their maturation. In line with this hypothesis, a synthetic peptide comprising the spastin M1 membrane-spanning sequence promoted phospholipid transbilayer movement (flip-flop) in artificial membranes (Nakao et al., 2016). Since reducing phospholipids at the cytoplasmic leaflet of the ER membrane could hamper LD emergence (Chorlay et al., 2019), it is possible that absence of spastin favors LD emergence. Lack of microtubule-severing could instead affect the ability of LDs to grow.
Different metabolic conditions and nutrient availability, as well as a proliferative vs. a post-mitotic status can underlie the distinct phenotypes in vivo and in vitro upon spastin depletion. It is also conceivable that the LD phenotype observed in cultured cells represents a compensatory mechanism. Spastin-deficient cells did not show abnormal ER shaping and distribution, however they depended on synthesis of TAG to preserve the general morphology of the ER upon starvation (Tadepalle et al., 2020). Similar findings have been shown in yeast cells in which LD formation is hampered (Velázquez et al., 2016). It is possible that increasing LD biogenesis simply expands the ER membrane, which has been shown to alleviate ER stress (Schuck et al., 2009). Consistent with this hypothesis is the observation that zebrafish lacking spastin are more susceptible to ER stress and displayed abnormal ER organization in sarcomeres (Arribat et al., 2020).
Together, these data obtained in various model systems, strongly support a role of spastin as a central regulator of LD behavior, but do not allow to conclusively dissect the role of spastin M1, which binds LDs, in respect to spastin M87, which can affect LDs indirectly by modulating microtubule dynamics. As we discuss in a following chapter, spastin M1 also regulates LD movement, and LD-peroxisome interaction, opening the question as to which of these multiple roles is relevant in the pathogenesis of HSP.
Seipin-Related Motoneuron Diseases
Seipin is widely recognized as a key molecular player in LD biogenesis [see (Salo et al., 2020) for a recent comprehensive review]. Seipin is an integral ER membrane protein with two transmembrane domains, exposing both N- and C-termini to the cytosol (Lundin et al., 2006). Seipin forms undecamers that localize in ER tubules and are highly mobile, but arrest at ER-LD contact sites (Yan et al., 2018; Santinho et al., 2020). Although LDs can form also in its absence (Santinho et al., 2020), seipin regulates LD assembly in multiple ways, both at early stages and during maturation. Seipin has been implicated in defining the site of LD nucleation, controlling phospholipid metabolism at the ER tubules, and mediating neutral lipids shuttling from the ER to LDs (Salo et al., 2016; Wang et al., 2016a; Yan et al., 2018; Chung et al., 2019). Consistently, lack of seipin in a variety of model systems, ranging from yeast to mammalian cells induces LD abnormalities, with several small LDs and few large LDs, while seipin overexpression efficiently inhibits LD formation (Szymanski et al., 2007; Fei et al., 2008; Fei et al., 2011). Moreover, lack of seipin leads to severe loss of ER-LD contacts (Salo et al., 2016). Thus, seipin functions as gatekeeper at ER-LD contact sites to regulate LD formation with a mechanism that is not completely understood.
Consistent with this role in LD biogenesis, human patients carrying loss-of-function mutations in BSCL2, the gene encoding for seipin, suffer from Berardinelli-Seip congenital lipodystrophy (Magre et al., 2001), a severe recessive disease characterized by abnormal fat deposition. In contrast, missense mutations in seipin at residues located in the ER lumen, N88S and S90L, are responsible of dominant forms of HSP (SPG17) and other neurological conditions, such as Silver syndrome, Charcot-Marie-Tooth neuropathy type 2, and distal hereditary motor neuropathy (dHMN) type V (Irobi et al., 2004; Windpassinger et al., 2004). These disorders, which have been dubbed seipinopathies, are all characterized by progressive axonopathy with various involvement of central and peripheral axons (Ito and Suzuki, 2009). From a pathogenic point of view, these missense mutations act via a gain-of-function mechanism, by hampering glycosylation of the protein. When expressed in yeast cells lacking Fld1, the seipin orthologue, these mutants are perfectly capably to rescue the LD phenotype (Fei et al., 2008).
What is the pathogenic mechanism of seipinopathies? Expression of seipin N88S and even more of S90L in non-neuronal cells triggers the formation of aggregates, and causes ER stress. Interestingly, the presence of these aggregates appeared to be protective and reduce apoptotic cell death (Ito et al., 2012). Later, aggregates of seipin mutants were shown to colocalize with LDs in cells incubated with oleic acid. Under this condition, cells exhibited reduced ER stress and better survival (Holtta-Vuori et al., 2013). Similarly, lipid loading rescued a swimming defect of a seipinopathy zebrafish model system (Holtta-Vuori et al., 2013). In this case, as in spastin knock-out cells, TAG loading and increased LD biogenesis may be a protective mechanism to prevent ER stress. Abnormal formations, with an ultrastructure reminiscent of LDs, were also observed in neurons of transgenic mice expressing the N88S seipin mutation (Yagi et al., 2011). It is important to stress that all these studies are based on a non-physiological overexpression of mutant seipin, however these data form a compelling case for a dysfunction of the tubular ER in BSCL2-related motoneuron diseases.
HSP Proteins and LD Turnover and Quality Control
When cells sense energy deficit, existing LDs can be utilized to provide FAs as fuel. LDs can be broken down by two major pathways: 1) lipolysis, i.e., hydrolysis of TAG or SE by respective enzymes to finally release FA, and 2) autophagy, i.e., engulfment of LDs by lysosomes leading to breakdown of LDs finally releasing FA (Hashemi and Goodman, 2015). Furthermore, LDs proteins can be targeted to degradation by ubiquitin-dependent pathways (Roberts and Olzmann, 2020). Experimental evidence has linked HSP proteins to abnormal turnover of LDs (Figure 2), raising the question of its pathogenic significance.
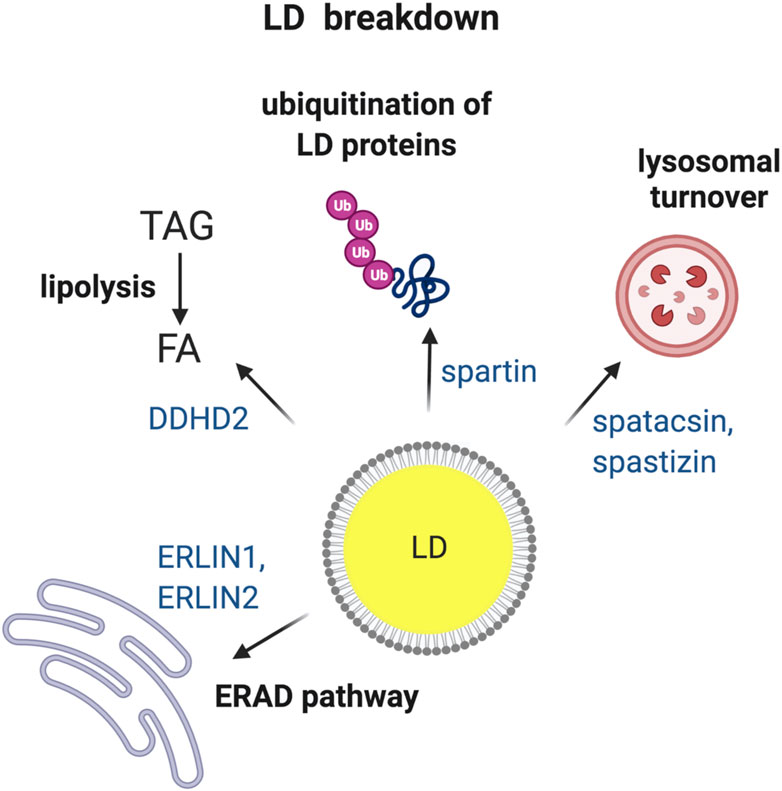
FIGURE 2. HSP proteins involved in the breakdown of LDs. Turnover of LDs via four different pathways i.e., (1) ER-associated degradation (ERAD); (2) TAG hydrolysis by lipases; (3) ubiquitin-mediated degradation of specific LD proteins and (4) breakdown of LDs by lysosomes via autophagy. HSP-associated proteins involved in each of the different pathways are indicated. Made with Biorender.com.
DDHD2: A Specific Brain TAG Lipase
The strongest claim for a defect of LD turnover in HSP is linked to mutations in DDHD2 in recessive SPG54 (Schuurs-Hoeijmakers et al., 2012). SPG54 families display early onset HSP, with thin corpus callosum and an abnormal lipid peak on cerebral proton magnetic resonance spectroscopy (Schuurs-Hoeijmakers et al., 2012). DDHD2 belong to the family of phospholipase A1, however later a role as a specific brain TAG lipase has been proposed and confirmed using the recombinant protein (Inloes et al., 2014). Disruption of Ddhd2 in mice leads to defects in motor coordination and cognition, hallmarks of HSP (Inloes et al., 2014). Remarkably, massive accumulation of TAG and LDs was observed in the brain and spinal cord of these mice, while other organs like the adipose tissue and heart remained unaffected (Inloes et al., 2014). HSP-associated disease mutants of DDHD2 failed to hydrolyze TAGs and to repress the LD accumulation in cells loaded with oleic acid (Inloes et al., 2018). Furthermore, mass spectrometric analysis of the LD proteome from brain tissue of mice lacking Ddhd2 detected enrichment of nervous system specific proteins, including several implicated in neurological conditions. Therefore, accumulation of LDs upon loss of DDHD2 could not only lead to the inability to mobilize fat stores, but also to the sequestration and disruption of the trafficking of key proteins and signaling lipids required for maintaining axonal integrity (Inloes et al., 2018).
Recent data have also implicated DDHD2 in ROS production, irrespective of its lipolysis activity (Maruyama et al., 2018). In contrast to neurons, Ddhd2 knock out MEFs do not accumulate LDs but show increased ROS, enhanced lipid peroxidation, and mitochondrial dysfunction. This role of DDHD2 would depend on a partial localization of the protein to mitochondria and be specific of peripheral tissues (Maruyama et al., 2018). Since ROS production stimulates the formation of lipids in neurons that are transported to glial cells and deposited in LDs (Liu et al., 2015; Liu et al., 2017; Ioannou et al., 2019), it is possible that several mechanisms are at play in HSP caused by deficiency of DDHD2.
HSP Proteins and Ubiquitin-dependent Degradation Pathways at LDs
The gene encoding for the protein spartin causes a complicated form of HSP called Troyer syndrome (SPG20) (Patel et al., 2002). The N-terminus of spartin contains a MIT domain that is also found in spastin and trafficking proteins involved in protein degradation through the endo-lysosomal system (Ciccarelli et al., 2003). Spartin is a cytosolic protein that can be recruited to LDs upon treatment with oleic acid, via a C-terminal region that is deleted in Troyer syndrome mutants (Eastman et al., 2009; Edwards et al., 2009; Hooper et al., 2010). Spartin colocalizes with TIP47/perilipin 3 at the LDs and competes with perilipin 2 for binding to LDs (Eastman et al., 2009). Downregulation and overexpression of spartin led to accumulation of LDs in cells (Eastman et al., 2009). Furthermore, Spg20 knock-out female mice displayed increased LD numbers and alterations in perilipin levels in fat tissues (Renvoise et al., 2012). Spartin interacts with the E3 ubiquitin ligases AIP4 and AIP5 and was found to recruit AIP4 to LDs (Eastman et al., 2009; Hooper et al., 2010). As an adaptor for AIP4, spartin regulates the ubiquitination and degradation of perilipin 2. (Hooper et al., 2010). Together, these data support the hypothesis of a key role of this protein in the turnover of crucial component of the LD proteome, and highlight the general role of LDs as sites for protein degradation (Welte, 2007; Roberts and Olzmann, 2020).
ER-associated degradation (ERAD) has been shown to regulate the turnover of proteins that shuttle from the ER tubules to the LDs (Roberts and Olzmann, 2020). The ERLIN2 gene is mutated in families with complex recessive forms of HSP (SPG18), while ERLIN1 has been implicated in autosomal recessive cases of pure HSP (SPG62) (Novarino et al., 2014) ERLIN1 and ERLIN2 are highly homologous proteins, belonging to the SPFH (stomatin/prohibitin/flotilin/HflK/c) family, which is defined by a module of ∼180–200 amino acids that has been found in some members of the family to bind cholesterol (Browman et al., 2007). The general function of SPFH proteins is to act as scaffolds for both lipids and proteins and define membrane microdomains in different cell compartments (Browman et al., 2007). Consistently, ERLIN1 and ERLIN2 have been first isolated as components of cholesterol-rich domains (lipid rafts) of the ER (Browman et al., 2006), and later shown to bind cholesterol (Huber et al., 2013). The ERLIN complex has established functions in regulating ERAD of the inositol 1,4,5 triphosphate (IP3) receptors (Pearce et al., 2007), and of 3-hydroxy-3-methylglutaryl-coenzyme reductase (HMGR), a rate-limiting enzyme for cholesterol synthesis (Jo et al., 2011), by interacting with specific E3 ubiquitin ligases. Moreover, the ERLIN complex associates with the sterol regulatory element binding proteins (SREBPs), Insig and Scap at the ER under cholesterol sufficiency. Upon downregulation of ERLINs, the degradation of Insig1 was accelerated, leading to canonical activation of SREBPs and their target genes, including HMGR. Cells depleted of ERLINs accumulated LDs, which may contain cholesterol esters, since both cholesterol and esterified cholesterol were also increased (Huber et al., 2013). Together, these data strongly suggest that ERLINs define ER subcompartments involved in regulating both Ca2+ handling and cholesterol metabolism. It is tempting to speculate that the ERLIN complex may regulate the ERAD of other proteins involved in lipid and LD metabolism.
HSP Proteins and Autophagy
Lysosomes fuse with autophagosomes to degrade cellular organelles, thus maintenance of a functional lysosomal pool in cells is required for proper functioning of autophagy. Two HSP-associated proteins, spastizin (SPG15) and spatacsin (SPG11), are involved in autophagic lysosome reformation, a pathway that generates new lysosomes (Chang et al., 2014). Mutations in spatacsin and spastizin, leads to the loss of free lysosomes and an accumulation of autolysosomes (Chang et al., 2014). Additionally, cortical neurons lacking spatacsin displayed undigested, accumulated LDs in association with lysosomes (Branchu et al., 2017). In primary mouse fibroblasts, loss of spatacsin impaired the autophagy-induced shuttling of fatty acids derived from membrane phospholipids from lysosomes to LDs. This lipid trafficking defects resulted in a decreased number of LDs not only in fibroblasts, but also in cortical neurons from Spg11 knock-out mice (Branchu et al., 2017). Spatacsin appears to be involved in clearance of gangliosides (Boutry et al., 2018) at the lysosome, suggesting that LDs in neurons may contain specific lipid class.
Recent data suggest that altered lysosomal function may be a recurrent theme in many HSP forms (Allison et al., 2017). Are defects in LD turnover in neurons, owing to impaired lysosomal function, a common pathway in HSP? Notably, atlastins emerged as novel players in mediating ER-phagy, the selective autophagy of ER tubules (Liang et al., 2018; Chen et al., 2019). One may speculate that impairment of ER-phagy in axons could not only compromise the recycling and the quality control of ER tubules, but also lead to accumulation of LDs.
HSP Proteins at the Interphase Between LDs and Other Organelles
Growing evidence shows that organelles do not reside in isolation within a cell, but they are constantly interacting with one another, either directly or indirectly. Inter-organellar communication is key to mediate processes like metabolite transfer, signal transduction, apoptosis etc. Other than the obvious and almost continuous interaction with the ER, LDs make intimate connections with almost all other organelles (Valm et al., 2017). Multiple HSP-associated proteins are involved in mediating interactions between LDs and other organelles.
HSP Proteins and LD Movement
FAs stored in LDs are mobilized by cells during energy deficiency. When cells are cultured in glucose-rich medium, most LDs are static and appear perinuclear, however glucose deprivation and AMPK activation induces dispersion of LDs that travel along detyrosinated microtubules to reach mitochondria, where FAs can be oxidized and energy converted in ATP (Herms et al., 2015). Tadepalle et al. demonstrated that this dispersion is impaired upon loss of spastin. Consistently, the ability of spastin M1 to bind to microtubules was necessary to mediate this LD dispersion (Figure 3). Furthermore, spastin depletion led to increased ER-LD contacts upon glucose starvation (Tadepalle et al., 2020), while spastin M1 overexpression reduced this contact site (Arribat et al., 2020) (Figure 3). The microtubule severing activity of spastin is governed by posttranslational modification of microtubules, especially polyglutamylation. A still speculative mechanism could involve the rewiring of the tubulin post-translational code induced by metabolic conditions, leading to spastin-mediated microtubule-severing at ER-LD contacts. This could facilitate detachment of LDs from the ER and help in movement/dispersion of LDs. However, direct demonstration of this is so far lacking. Spastin may also modulate the distribution of LDs, independent from interaction with microtubules, by simply affecting ER morphogenesis (Arribat et al., 2020).
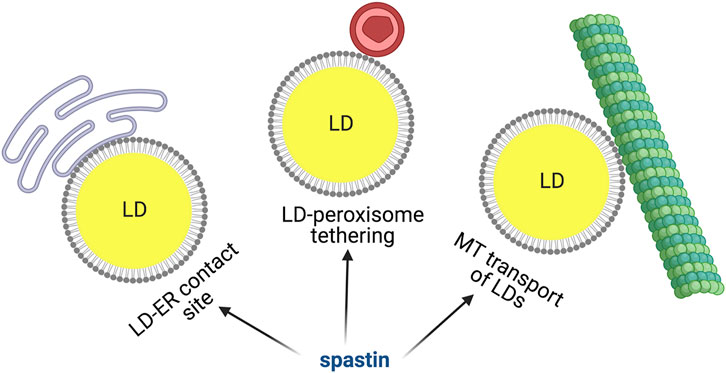
FIGURE 3. Spastin M1 and LD function. Spastin is involved in mediating ER-LD contacts, LD-peroxisome tethering and LD movement via microtubules. Made with Biorender.com.
BICD2 encodes for an important adaptor protein that interacts with dynein-dynactin motor complex and facilitates retrograde and anterograde transport of cargos along microtubules that are critical in maintaining the integrity of motor neurons (Kardon and Vale, 2009; Olenick and Holzbaur, 2019). Mutations in BICD2 have been identified in a spectrum of neurological conditions, from congenital spinal muscular atrophy to HSP (Neveling et al., 2013; Oates et al., 2013; Peeters et al., 2013). BICD2 is a homologue of the Drosophila BicD (Oh et al., 2000), which is found on LDs. Loss of BicD leads to fewer LDs with decreased movements in Drosophila embryos (Larsen et al., 2008). Whether this function is conserved in mammals is still unknown.
Spastin M1 and LD-Peroxisome Contact Sites
Once mobilized from the ER, LDs can dock on other organelles to allow trafficking of FAs. LD-mitochondria or LD-peroxisome contacts have been recently identified and the molecular machinery required for these contacts begins to be unraveled (Schuldiner and Bohnert, 2017; Henne and Hariri, 2018; Thiam and Dugail, 2019). Remarkably, a role for spastin M1 in tethering LDs to peroxisomes has been uncovered (Chang et al., 2019; Figure 3). Spastin M1 forms LD-peroxisome contacts by binding on one hand to LDs via its hairpin domain and on the other hand to peroxisomes by interacting with ABCD1. The latter is a peroxisomal ATP-binding cassette transporter mutated in X-linked adrenoleukodystrophy, a disease also characterized by spasticity of the lower limbs (Mosser et al., 1993; Chang et al., 2019). The LD-peroxisome contact site mediated by spastin M1 was important to control trafficking of FAs to peroxisome and avoid accumulation of peroxidized lipids in LDs. FA trafficking was dependent on the recruitment to LDs of IST1 and CHMP1B, proteins involved in ESCRTIII complex, by the spastin MIT domain (Chang et al., 2019). This study found that the ATPase, but not the microtubule-severing activity of spastin was implicated in the LD-peroxisome contact site. However, control experiments were conducted using nocodazole, which robustly depolymerize microtubules, and does not replicate the subtle severing effects on specific post-translationally modified species of microtubules like spastin does.
At odds with these data, overexpression of spastin M1 has been reported to reduce both the number of peroxisomes and the LD-peroxisome contacts in another study (Arribat et al., 2020). Since the ER is one of the sites of peroxisome biogenesis (Hoepfner et al., 2005; Kim et al., 2006), it is possible that over-expression of spastin M1 that is targeted to the ER and LDs alters ER-shaping thus affecting peroxisome number. Additionally, microtubule polymerization and ER tubule extension has been shown to occur simultaneously (Terasaki et al., 1986). Thus, disruption of the microtubular network due to spastin over-expression or downregulation could alter ER tubules resulting in a peroxisomal phenotype. Consistently, SPAST patient derived olfactory stem cells display aberrant peroxisome movement and distribution (Wali et al., 2016).
In conclusion, the interplay between spastin M1 at the LDs and peroxisome function is intriguing and worth elucidating. Since most of the experiments in these studies were performed in Hela cells using overexpression of spastin M1, that is detrimental for the maintenance of an intact microtubular and ER network (Errico et al., 2002; Solowska et al., 2008; Plaud et al., 2018), it will be crucial to substantiate these findings using more physiological conditions.
LDs in Neurons and Astrocytes
Apart from HSP, LD dysfunction is a common theme of other neurodegenerative disorders like Alzheimer’s, Parkinson’s and Huntington’s disease, making this organelle a new player in neurological conditions (Welte, 2015; Farmer et al., 2020). While LD abnormalities are widely reported in neurodegenerative diseases, one must be cautious in interpreting these data, since lipid regulation in the peripheral tissues is likely to be different from that in the central nervous system. So far, we know very little about control of neutral lipid metabolism and the prominence and function of LDs in different brain cells. Most of the LD abnormalities associated to HSP protein loss or mutations have been detected in cultured cells or in non-neuronal tissues, with very few exceptions, as described in the previous chapters. Thus, the relevance of these findings for cortical motor neurons remains to be elucidated.
As already discussed, axons and growth cones are enriched with ER tubules which are a local source of lipids synthesis, including PC, the major neuronal membrane phospholipid, and cholesterol that is highly enriched in synapses and growth cones (Piomelli et al., 2007; Luarte et al., 2018). However, LDs are almost never observed in axons, which does not exclude the presence of few, microscopic LDs hard to detect with common methods. LDs can instead be imaged in the cell body of aged neurons (Shimabukuro et al., 2016), often close to the endolysosomal compartment. This suggests that neurons can form LDs that probably undergo constant turnover in healthy young neurons.
Inferring the significance of LD abnormalities in central motor neurons and their axons is complicated by the apparent contradiction that some HSP mutant proteins seem to hamper LD formation, (i.e. ER shaping proteins), while others lead to their increase (the best example is DDHD2). These discrepancies are reconciled when considering the dynamic life cycle of LDs and how this affects the flux of FAs from the ER to other compartments, such as mitochondria, peroxisomes, lysosomes, and the plasma membrane. Transient LDs in axons may be used as a source of FAs to maintain the axonal membrane. Contacts between the ER and mitochondria or peroxisomes in axons may be required for trafficking of FAs from site of synthesis to their place of utilization. Moreover, lipolysis or lipophagy may control the flux of FAs. Therefore, interfering with pathways that control biogenesis, (i.e. ER shaping proteins), turnover, (i.e. DDHD2, spartin and spatacsin) or interaction of LDs with other organelles (as spastin M1) could be equally detrimental for neurons. Impairment of each of these steps could trigger an energy crisis in corticospinal neurons, culminating in the dying back of these long axons.
Notably, most studies describing LD dysfunction in neurodegeneration, report LD accumulation as a detrimental effect (Brekk et al., 2020; Marschallinger et al., 2020). Is an increased LD number just a read-out of inefficient FA catabolism or are LDs toxic for neurons? A general dogma is that neurons do not breakdown FA via β-oxidation. Thus, other cells in the brain, namely astrocytes and microglia, have been considered better candidates for LD synthesis and regulation in the brain. Accumulation of LDs in glial cells is an early sign in Drosophila models of neurodegeneration caused by mitochondrial dysfunction in neurons. This accumulation depends on neutral lipids synthesis in neurons, upon ROS production, which is followed by LD deposition in glial cells (Liu et al., 2015). It has been clearly demonstrated that this glia-neuron lipid communication ensures protection of neurons from FA toxicity. Neurons utilize lactate taken up by glial cells as a substrate for synthesis of FAs. These are then processed and transferred by FA transporter protein and apolipoproteins D and E to glial cells, where they are stored in LDs (Liu et al., 2017). Accumulation of LDs in neurons is in fact toxic, and neuronal degeneration can be alleviated by interfering with their production in neurons, for example by reducing ROS levels or the import of lactate (Liu et al., 2015; Liu et al., 2017). On the other hand, blocking the export of FAs to astrocytes precipitates neurodegeneration. This mechanism may play a role Alzheimer’s disease, since the disease linked ApoE4 allele showed a reduced capacity to transport lipids from neurons to astrocytes and induced neurodegeneration in the fly (Liu et al., 2017). Lipid metabolism coupling between neurons and astrocytes is relevant not only in pathologically conditions, but also upon neuronal stimulation, when neuronal FA metabolism is increased and lipids are then metabolized by astrocytes (Ioannou et al., 2019).
Many HSP proteins are also expressed in glial cells, however how glia cells contribute to the axonopathy is largely unknown. Recently, a LD phenotype, characterized by increased number of smaller LDs was detected in astrocytes, but not in cortical neurons, differentiated from pluripotent stem cells expressing ATL1 mutations (Mou et al., 2020). This LD defect impaired cholesterol transport from the glia to the neurons, leading to axonal degeneration defects in the cortical neurons. Both the astrocytic LD phenotype and the axonal degeneration could be rescued by treating astrocytes with GW395, a synthetic liver X receptor agonist, which increases the expression of SREBF1, a gene critical for cholesterol metabolism (Mou et al., 2020). This finding highlights the complexity of the interaction between astrocytes and neurons, when it comes to lipid metabolism, and emphasizes that non-cell autonomous LD phenotypes in astrocytes may affect neurons.
Future Directions
Several questions remain to be answered to understand the significance of LD biology in the pathogenesis of HSP. Are studies conducted in non-neuronal cells of relevance for post-mitotic neurons? What is the effect of having smaller or larger LDs but similar TAG levels? Are LD abnormalities associated with HSP protein dysfunction the signal of a common defect, or rather a read-out of different dysfunctional pathways? We do not have a convincing answer to these questions, which need to be solved to understand if targeting LD abnormalities may offer novel therapeutic opportunities.
The presence of LDs in the pyramidal motor neurons, which are distinct from other neurons in many ways, in healthy and diseased state, should be explored in depth. Furthermore, since many HSP forms are age-associated, it would be vital to study LD biology and regulation during normal healthy neuronal aging. The lack of reliable tools is a hindrance in achieving this goal. High-end and high-throughput imaging techniques would need to be modified and optimized to visualize LDs and ER microdomains in the neurons. The development of live-cell confocal and lattice light sheet spectral imaging approaches to study organelle interaction is encouraging (Valm et al., 2017), and can now be exploited more to understand LD trafficking and interaction with other organelles in neurons. Finally, lipid composition of LDs in the brain should be extensively characterized. Which are the main lipid species in brain LDs? Is there a difference between LDs in neurons and astrocytes? Imaging techniques using specific dyes to detect LDs containing FAs or cholesterol esters will be pivotal. Finally, improved mass spectrometry techniques like matrix-assisted laser desorption/ionization imaging mass spectrometry (MALDI-MS) allows for detection of lipids bound by membranes.
Apart from their role as energy storage organelles, LDs are hubs for proteins, signaling molecules, inflammatory mediators etc. Therefore, dysfunction of LDs can affect neurons in multiple pathways, which have still not been fully studied in the context of HSP. One problem in the field is that mouse models of HSP often present with a very mild in vivo phenotype. Moreover, we do not know if lipid metabolism in the mouse fully recapitulates that in humans.
Since alterations of LDs have been observed in several cellular and model systems of HSP, one might ask if restoring LD metabolism could be a unifying therapeutic strategy to pursue. In several HSP cases, we do not know if the LD abnormalities are a direct read-out of the pathogenic process or an indirect compensatory effect, and if they are recapitulated in neurons or astrocytes. As previously discussed, LDs are a dynamic storage deposit for fatty acids that traffic from the ER to other cellular membranes, and their prevalence and turnover is highly cell- and tissue-dependent. However, both the lysosome and the unfolded protein response triggered by ER stress are promising targets for pharmacological interventions in a wide range of diseases, including neurodegeneration (Bonam et al., 2019; Gonzalez-Teuber et al., 2019). In individual HSP forms, compounds that act to reduce ER stress or impinge on the autophagy-lysosome axis may be beneficial and also correct LD abnormalities. In a recent study, the flavonoid naringenin was shown to restore ER homeostasis in a fly model of REEP1-linked HSP (Napoli et al., 2019).
The fast progress in the LD field holds the promise of unraveling the basic cell biology of LDs in neurons in the near future and the mysterious connection between HSP pathogenesis and LD metabolism. Solving this puzzle will be an important step ahead to design innovative treatments for HSP patients.
Author Contributions
NT and EIR wrote the manuscripts. NT prepared the Figures. Both authors read and approved the final manuscript.
Funding
This work was supported by grants from the Tom Wahlig foundation, the Federal Ministry of Education and Research, Germany, via funding for the TreatHSP network (01GM1905), and the Center for Molecular Medicine, Cologne.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Allison, R., Edgar, J. R., Pearson, G., Rizo, T., Newton, T., Günther, S., et al. (2017). Defects in ER-Endosome Contacts Impact Lysosome Function in Hereditary Spastic Paraplegia. J. Cel Biol. 216, 1337–1355. doi:10.1083/jcb.201609033
Allison, R., Edgar, J. R., and Reid, E. (2019). Spastin MIT Domain Disease-Associated Mutations Disrupt Lysosomal Function. Front. Neurosci. 13, 1179. doi:10.3389/fnins.2019.01179
Allison, R., Lumb, J. H., Fassier, C., Connell, J. W., Ten Martin, D., Seaman, M. N. J., et al. (2013). An ESCRT-Spastin Interaction Promotes Fission of Recycling Tubules from the Endosome. J. Cel Biol. 202, 527–543. doi:10.1083/jcb.201211045
Alvarez, H. M., and Steinbüchel, A. (2002). Triacylglycerols in Prokaryotic Microorganisms. Appl. Microbiol. Biotechnol. 60, 367–376. doi:10.1007/s00253-002-1135-0
Arribat, Y., Grepper, D., Lagarrigue, S., Qi, T. M., Cohen, S., and Amati, F. (2020). Spastin Mutations Impair Coordination between Lipid Droplet Dispersion and Reticulum. Plos Genet. 16, e1008665. doi:10.1371/journal.pgen.1008665
Barbosa, A. D., and Siniossoglou, S. (2017). Function of Lipid Droplet-Organelle Interactions in Lipid Homeostasis. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1864, 1459–1468. doi:10.1016/j.bbamcr.2017.04.001
Bartz, R., Li, W.-H., Venables, B., Zehmer, J. K., Roth, M. R., Welti, R., et al. (2007). Lipidomics Reveals that Adiposomes Store Ether Lipids and Mediate Phospholipid Traffic,. J. Lipid Res. 48, 837–847. doi:10.1194/jlr.m600413-jlr200
Behrendt, L., Kurth, I., and Kaether, C. (2019). A Disease Causing ATLASTIN 3 Mutation Affects Multiple Endoplasmic Reticulum-Related Pathways. Cell. Mol. Life Sci. 76, 1433–1445. doi:10.1007/s00018-019-03010-x
Bersuker, K., Peterson, C. W. H., To, M., Sahl, S. J., Savikhin, V., Grossman, E. A., et al. (2018). A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes. Develop. Cel. 44, 97–112. doi:10.1016/j.devcel.2017.11.020
Blackstone, C. (2018a). Converging Cellular Themes for the Hereditary Spastic Paraplegias. Curr. Opin. Neurobiol. 51, 139–146. doi:10.1016/j.conb.2018.04.025
Blackstone, C. (2018b). Hereditary Spastic Paraplegia. Handb Clin. Neurol. 148, 633–652. doi:10.1016/b978-0-444-64076-5.00041-7
Blackstone, C., O'kane, C. J., and Reid, E. (2011). Hereditary Spastic Paraplegias: Membrane Traffic and the Motor Pathway. Nat. Rev. Neurosci. 12, 31–42. doi:10.1038/nrn2946
Bonam, S. R., Wang, F., and Muller, S. (2019). Lysosomes as a Therapeutic Target. Nat. Rev. Drug Discov. 18, 923–948. doi:10.1038/s41573-019-0036-1
Boutry, M., Branchu, J., Lustremant, C., Pujol, C., Pernelle, J., Matusiak, R., et al. (2018). Inhibition of Lysosome Membrane Recycling Causes Accumulation of Gangliosides that Contribute to Neurodegeneration. Cel Rep. 23, 3813–3826. doi:10.1016/j.celrep.2018.05.098
Branchu, J., Boutry, M., Sourd, L., Depp, M., Leone, C., Corriger, A., et al. (2017). Loss of Spatacsin Function Alters Lysosomal Lipid Clearance Leading to Upper and Lower Motor Neuron Degeneration. Neurobiol. Dis. 102, 21–37. doi:10.1016/j.nbd.2017.02.007
Brasaemle, D. L. (2007). Thematic Review Series: Adipocyte Biology. The Perilipin Family of Structural Lipid Droplet Proteins: Stabilization of Lipid Droplets and Control of Lipolysis. J. Lipid Res. 48, 2547–2559. doi:10.1194/jlr.r700014-jlr200
Brekk, O. R., Honey, J. R., Lee, S., Hallett, P. J., and Isacson, O. (2020). Cell Type-specific Lipid Storage Changes in Parkinson's Disease Patient Brains Are Recapitulated by Experimental Glycolipid Disturbance. Proc. Natl. Acad. Sci. USA 117, 27646–27654. doi:10.1073/pnas.2003021117
Browman, D. T., Hoegg, M. B., and Robbins, S. M. (2007). The SPFH Domain-Containing Proteins: More Than Lipid Raft Markers. Trends Cel Biol. 17, 394–402. doi:10.1016/j.tcb.2007.06.005
Browman, D. T., Resek, M. E., Zajchowski, L. D., and Robbins, S. M. (2006). Erlin-1 and Erlin-2 Are Novel Members of the Prohibitin Family of Proteins that Define Lipid-raft-like Domains of the ER. J. Cel Sci. 119, 3149–3160. doi:10.1242/jcs.03060
Chang, C.-L., Weigel, A. V., Ioannou, M. S., Pasolli, H. A., Xu, C. S., Peale, D. R., et al. (2019). Spastin Tethers Lipid Droplets to Peroxisomes and Directs Fatty Acid Trafficking through ESCRT-III. J. Cel Biol. 218, 2583–2599. doi:10.1083/jcb.201902061
Chang, J., Lee, S., and Blackstone, C. (2014). Spastic Paraplegia Proteins Spastizin and Spatacsin Mediate Autophagic Lysosome Reformation. J. Clin. Invest. 124, 5249–5262. doi:10.1172/jci77598
Chen, Q., Xiao, Y., Chai, P., Zheng, P., Teng, J., and Chen, J. (2019). ATL3 Is a Tubular ER-Phagy Receptor for GABARAP-Mediated Selective Autophagy. Curr. Biol. 29, 846–855. doi:10.1016/j.cub.2019.01.041
Chorlay, A., Monticelli, L., Veríssimo Ferreira, J., Ben M’barek, K., Ajjaji, D., Wang, S., et al. (2019). Membrane Asymmetry Imposes Directionality on Lipid Droplet Emergence from the ER. Develop. Cel. 50, 25–42. doi:10.1016/j.devcel.2019.05.003
Chung, J., Wu, X., Lambert, T. J., Lai, Z. W., Walther, T. C., and Farese, R. V. (2019). LDAF1 and Seipin Form a Lipid Droplet Assembly Complex. Develop. Cel 51, 551–563. doi:10.1016/j.devcel.2019.10.006
Ciccarelli, F. D., Proukakis, C., Patel, H., Cross, H., Azam, S., Patton, M. A., et al. (2003). The Identification of a Conserved Domain in Both Spartin and Spastin, Mutated in Hereditary Spastic Paraplegia. Genomics 81, 437–441. doi:10.1016/s0888-7543(03)00011-9
Claudiani, P., Riano, E., Errico, A., Andolfi, G., and Rugarli, E. (2005). Spastin Subcellular Localization Is Regulated through Usage of Different Translation Start Sites and Active Export from the Nucleus. Exp. Cel Res. 309, 358–369. doi:10.1016/j.yexcr.2005.06.009
Connell, J. W., Lindon, C., Luzio, J. P., and Reid, E. (2009). Spastin Couples Microtubule Severing to Membrane Traffic in Completion of Cytokinesis and Secretion. Traffic 10, 42–56. doi:10.1111/j.1600-0854.2008.00847.x
Cruz, A. L. S., Barreto, E. D. A., Fazolini, N. P. B., Viola, J. P. B., and Bozza, P. T. (2020). Lipid Droplets: Platforms with Multiple Functions in Cancer Hallmarks. Cell Death Dis. 11, 105. doi:10.1038/s41419-020-2297-3
Darios, F., Mochel, F., and Stevanin, G. (2020). Lipids in the Physiopathology of Hereditary Spastic Paraplegias. Front. Neurosci-switz 14. doi:10.3389/fnins.2020.00074
Eastman, S. W., Yassaee, M., and Bieniasz, P. D. (2009). A Role for Ubiquitin Ligases and Spartin/SPG20 in Lipid Droplet Turnover. J. Cel Biol. 184, 881–894. doi:10.1083/jcb.200808041
Edwards, T. L., Clowes, V. E., Tsang, H. T., Connell, J. W., Sanderson, C. M., Luzio, J. P., et al. (2009). Endogenous Spartin (SPG20) Is Recruited to Endosomes and Lipid Droplets and Interacts with the Ubiquitin E3 Ligases AIP4 and AIP5. Biochem. J. 423, 31–9. doi:10.1042/BJ20082398
Errico, A., Ballabio, A., and Rugarli, E. I. (2002). Spastin, the Protein Mutated in Autosomal Dominant Hereditary Spastic Paraplegia, Is Involved in Microtubule Dynamics. Hum. Mol. Genet. 11, 153–163. doi:10.1093/hmg/11.2.153
Esteves, T., Durr, A., Mundwiller, E., Loureiro, J. L., Boutry, M., Gonzalez, M. A., et al. (2014). Loss of Association of REEP2 with Membranes Leads to Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 94, 268–277. doi:10.1016/j.ajhg.2013.12.005
Evans, K. J., Gomes, E. R., Reisenweber, S. M., Gundersen, G. G., and Lauring, B. P. (2005). Linking Axonal Degeneration to Microtubule Remodeling by Spastin-Mediated Microtubule Severing. J. Cel Biol. 168, 599–606. doi:10.1083/jcb.200409058
Falk, J., Rohde, M., Bekhite, M. M., Neugebauer, S., Hemmerich, P., Kiehntopf, M., et al. (2014). Functional Mutation Analysis Provides Evidence for a Role of REEP1 in Lipid Droplet Biology. Hum. Mutat. 35, 497–504. doi:10.1002/humu.22521
Farmer, B. C., Walsh, A. E., Kluemper, J. C., and Johnson, L. A. (2020). Lipid Droplets in Neurodegenerative Disorders. Front. Neurosci. 14, 742. doi:10.3389/fnins.2020.00742
Fei, W., Shui, G., Gaeta, B., Du, X., Kuerschner, L., Li, P., et al. (2008). Fld1p, a Functional Homologue of Human Seipin, Regulates the Size of Lipid Droplets in Yeast. J. Cel Biol. 180, 473–482. doi:10.1083/jcb.200711136
Fei, W., Shui, G., Zhang, Y., Krahmer, N., Ferguson, C., Kapterian, T. S., et al. (2011). A Role for Phosphatidic Acid in the Formation of "supersized" Lipid Droplets. Plos Genet. 7, e1002201. doi:10.1371/journal.pgen.1002201
Fujimoto, T., and Parton, R. G. (2011). Not just Fat: the Structure and Function of the Lipid Droplet. Cold Spring Harb Perspect. Biol. 3, a004838. doi:10.1101/cshperspect.a004838
Gluchowski, N. L., Becuwe, M., Walther, T. C., and Farese, R. V. (2017). Lipid Droplets and Liver Disease: from Basic Biology to Clinical Implications (Vol 14, Pg 343, 2017). Nat. Rev. Gastro Hepat. 14, 343–355. doi:10.1038/nrgastro.2017.32
Goldberg, I. J., Reue, K., Abumrad, N. A., Bickel, P. E., Cohen, S., Fisher, E. A., et al. (2018). Deciphering the Role of Lipid Droplets in Cardiovascular Disease. Circulation 138, 305–315. doi:10.1161/circulationaha.118.033704
Gonzalez-Teuber, V., Albert-Gasco, H., Auyeung, V. C., Papa, F. R., Mallucci, G. R., and Hetz, C. (2019). Small Molecules to Improve ER Proteostasis in Disease. Trends Pharmacol. Sci. 40, 684–695. doi:10.1016/j.tips.2019.07.003
Grillitsch, K., Connerth, M., Köfeler, H., Arrey, T. N., Rietschel, B., Wagner, B., et al. (2011). Lipid Particles/droplets of the Yeast Saccharomyces cerevisiae Revisited: Lipidome Meets Proteome. Biochim. Biophys. Acta (Bba) - Mol. Cel Biol. Lipids 1811, 1165–1176. doi:10.1016/j.bbalip.2011.07.015
Hashemi, H. F., and Goodman, J. M. (2015). The Life Cycle of Lipid Droplets. Curr. Opin. Cel Biol. 33, 119–124. doi:10.1016/j.ceb.2015.02.002
Hazan, J., Fonknechten, N., Mavel, D., Paternotte, C., Samson, D., Artiguenave, F., et al. (1999). Spastin, a New AAA Protein, Is Altered in the Most Frequent Form of Autosomal Dominant Spastic Paraplegia. Nat. Genet. 23, 296–303. doi:10.1038/15472
Henne, M., Goodman, J. M., and Hariri, H. (2020). Spatial Compartmentalization of Lipid Droplet Biogenesis. Bba-mol Cel Biol L 1865, 158499. doi:10.1016/j.bbalip.2019.07.008
Henne, W. M., and Hariri, H. (2018). Endoplasmic Reticulum-Vacuole Contact Sites "Bloom" with Stress-Induced Lipid Droplets. Contact 1, 251525641875611. doi:10.1177/2515256418756112
Herms, A., Bosch, M., Reddy, B. J., Schieber, N. L., Fajardo, A., Ruperez, C., et al. (2015). AMPK Activation Promotes Lipid Droplet Dispersion on Detyrosinated Microtubules to Increase Mitochondrial Fatty Acid Oxidation. Nat. Commun. 6, 7176. doi:10.1038/ncomms8176
Hoepfner, D., Schildknegt, D., Braakman, I., Philippsen, P., and Tabak, H. F. (2005). Contribution of the Endoplasmic Reticulum to Peroxisome Formation. Cell 122, 85–95. doi:10.1016/j.cell.2005.04.025
Holtta-Vuori, M., Salo, V. T., Ohsaki, Y., Suster, M. L., and Ikonen, E. (2013). Alleviation of Seipinopathy-Related ER Stress by Triglyceride Storage. Hum. Mol. Genet. 22, 1157–66. doi:10.1093/hmg/dds523
Hooper, C., Puttamadappa, S. S., Loring, Z., Shekhtman, A., and Bakowska, J. C. (2010). Spartin Activates Atrophin-1-Interacting Protein 4 (AIP4) E3 Ubiquitin Ligase and Promotes Ubiquitination of Adipophilin on Lipid Droplets. BMC Biol. 8, 72. doi:10.1186/1741-7007-8-72
Hu, J., Shibata, Y., Zhu, P.-P., Voss, C., Rismanchi, N., Prinz, W. A., et al. (2009). A Class of Dynamin-like GTPases Involved in the Generation of the Tubular ER Network. Cell 138, 549–561. doi:10.1016/j.cell.2009.05.025
Huber, M. D., Vesely, P. W., Datta, K., and Gerace, L. (2013). Erlins Restrict SREBP Activation in the ER and Regulate Cellular Cholesterol Homeostasis. J. Cel Biol. 203, 427–436. doi:10.1083/jcb.201305076
Hübner, C. A., and Kurth, I. (2014). Membrane-shaping Disorders: a Common Pathway in Axon Degeneration. Brain 137, 3109–3121. doi:10.1093/brain/awu287
Inloes, J. M., Hsu, K.-L., Dix, M. M., Viader, A., Masuda, K., Takei, T., et al. (2014). The Hereditary Spastic Paraplegia-Related Enzyme DDHD2 Is a Principal Brain Triglyceride Lipase. Proc. Natl. Acad. Sci. 111, 14924–14929. doi:10.1073/pnas.1413706111
Inloes, J. M., Kiosses, W. B., Wang, H., Walther, T. C., Farese, R. V., and Cravatt, B. F. (2018). Functional Contribution of the Spastic Paraplegia-Related Triglyceride Hydrolase DDHD2 to the Formation and Content of Lipid Droplets. Biochemistry 57, 827–838. doi:10.1021/acs.biochem.7b01028
Ioannou, M. S., Jackson, J., Sheu, S.-H., Chang, C.-L., Weigel, A. V., Liu, H., et al. (2019). Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 177, 1522–1535. doi:10.1016/j.cell.2019.04.001
Irobi, J., Van Den Bergh, P., Merlini, L., Verellen, C., Van Maldergem, L., Dierick, I., et al. (2004). The Phenotype of Motor Neuropathies Associated with BSCL2 Mutations Is Broader Than Silver Syndrome and Distal HMN Type V. Brain 127, 2124–2130. doi:10.1093/brain/awh232
Ito, D., and Suzuki, N. (2009). Seipinopathy: a Novel Endoplasmic Reticulum Stress-Associated Disease. Brain 132, 8–15. doi:10.1093/brain/awn216
Ito, D., Yagi, T., Ikawa, M., and Suzuki, N. (2012). Characterization of Inclusion Bodies with Cytoprotective Properties Formed by Seipinopathy-Linked Mutant Seipin. Hum. Mol. Genet. 21, 635–646. doi:10.1093/hmg/ddr497
Jo, Y., Sguigna, P. V., and Debose-Boyd, R. A. (2011). Membrane-associated Ubiquitin Ligase Complex Containing Gp78 Mediates Sterol-Accelerated Degradation of 3-Hydroxy-3-Methylglutaryl-Coenzyme A Reductase. J. Biol. Chem. 286, 15022–15031. doi:10.1074/jbc.m110.211326
Kardon, J. R., and Vale, R. D. (2009). Regulators of the Cytoplasmic Dynein Motor. Nat. Rev. Mol. Cel Biol. 10, 854–865. doi:10.1038/nrm2804
Kassan, A., Herms, A., Fernández-Vidal, A., Bosch, M., Schieber, N. L., Reddy, B. J. N., et al. (2013). Acyl-CoA Synthetase 3 Promotes Lipid Droplet Biogenesis in ER Microdomains. J. Cel Biol. 203, 985–1001. doi:10.1083/jcb.201305142
Kim, K. T., Moon, Y., Jang, Y., Lee, K. T., Lee, C., Jun, Y., et al. (2017). Molecular Mechanisms of Atlastin-Mediated ER Membrane Fusion Revealed by a FRET-Based Single-Vesicle Fusion Assay. Sci. Rep. 7, 8700. doi:10.1038/s41598-017-09162-9
Kim, P. K., Mullen, R. T., Schumann, U., and Lippincott-Schwartz, J. (2006). The Origin and Maintenance of Mammalian Peroxisomes Involves a De Novo PEX16-dependent Pathway from the ER. J. Cel Biol. 173, 521–532. doi:10.1083/jcb.200601036
Klemm, R. W., Norton, J. P., Cole, R. A., Li, C. S., Park, S. H., Crane, M. M., et al. (2013). A Conserved Role for Atlastin GTPases in Regulating Lipid Droplet Size. Cel Rep. 3, 1465–1475. doi:10.1016/j.celrep.2013.04.015
Kornak, U., Mademan, I., Schinke, M., Voigt, M., Krawitz, P., Hecht, J., et al. (2014). Sensory Neuropathy with Bone Destruction Due to a Mutation in the Membrane-Shaping Atlastin GTPase 3. Brain 137, 683–692. doi:10.1093/brain/awt357
Kory, N., Farese, R. V., and Walther, T. C. (2016). Targeting Fat: Mechanisms of Protein Localization to Lipid Droplets. Trends Cel Biol. 26, 535–546. doi:10.1016/j.tcb.2016.02.007
Krahmer, N., Farese, R. V., and Walther, T. C. (2013). Balancing the Fat: Lipid Droplets and Human Disease. EMBO Mol. Med. 5, 973–983. doi:10.1002/emmm.201100671
Lallemant-Dudek, P., and Durr, A. (2020). Clinical and Genetic Update of Hereditary Spastic Paraparesis. Rev. Neurol. (Paris), 30628–7. doi:10.1016/j.neurol.2020.07.001
Larsen, K. S., Xu, J., Cermelli, S., Shu, Z., and Gross, S. P. (2008). BicaudalD Actively Regulates Microtubule Motor Activity in Lipid Droplet Transport. PLoS One 3, e3763. doi:10.1371/journal.pone.0003763
Liang, J. R., Lingeman, E., Ahmed, S., and Corn, J. E. (2018). Atlastins Remodel the Endoplasmic Reticulum for Selective Autophagy. J. Cel Biol. 217, 3354–3367. doi:10.1083/jcb.201804185
Liu, L., Mackenzie, K. R., Putluri, N., Maletić-Savatić, M., and Bellen, H. J. (2017). The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cel Metab. 26, 719–737. doi:10.1016/j.cmet.2017.08.024
Liu, L., Zhang, K., Sandoval, H., Yamamoto, S., Jaiswal, M., Sanz, E., et al. (2015). Glial Lipid Droplets and ROS Induced by Mitochondrial Defects Promote Neurodegeneration. Cell 160, 177–190. doi:10.1016/j.cell.2014.12.019
Luarte, A., Cornejo, V. H., Bertin, F., Gallardo, J., and Couve, A. (2018). The Axonal Endoplasmic Reticulum: One Organelle-Many Functions in Development, Maintenance, and Plasticity. Devel Neurobio. 78, 181–208. doi:10.1002/dneu.22560
Lundin, C., Nordström, R., Wagner, K., Windpassinger, C., Andersson, H., Von Heijne, G., et al. (2006). Membrane Topology of the Human Seipin Protein. FEBS Lett. 580, 2281–2284. doi:10.1016/j.febslet.2006.03.040
Magré, J., Delépine, M., Khallouf, E., Gedde-Dahl, T., Van Maldergem, L., Sobel, E., et al. (2001). Identification of the Gene Altered in Berardinelli-Seip Congenital Lipodystrophy on Chromosome 11q13. Nat. Genet. 28, 365–370. doi:10.1038/ng585
Mancuso, G., and Rugarli, E. I. (2008). A Cryptic Promoter in the First Exon of the SPG4 Gene Directs the Synthesis of the 60-kDa Spastin Isoform. BMC Biol. 6, 31. doi:10.1186/1741-7007-6-31
Mannan, A. U., Boehm, J., Sauter, S. M., Rauber, A., Byrne, P. C., Neesen, J., et al. (2006a). Spastin, the Most Commonly Mutated Protein in Hereditary Spastic Paraplegia Interacts with Reticulon 1 an Endoplasmic Reticulum Protein. Neurogenetics 7, 93–103. doi:10.1007/s10048-006-0034-4
Mannan, A. U., Krawen, P., Sauter, S. M., Boehm, J., Chronowska, A., Paulus, W., et al. (2006b). ZFYVE27 (SPG33), a Novel Spastin-Binding Protein, Is Mutated in Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 79, 351–357. doi:10.1086/504927
Marschallinger, J., Iram, T., Zardeneta, M., Lee, S. E., Lehallier, B., Haney, M. S., et al. (2020). Lipid-droplet-accumulating Microglia Represent a Dysfunctional and Proinflammatory State in the Aging Brain. Nat. Neurosci. 23, 194–208. doi:10.1038/s41593-019-0566-1
Maruyama, T., Baba, T., Maemoto, Y., Hara-Miyauchi, C., Hasegawa-Ogawa, M., Okano, H. J., et al. (2018). Loss of DDHD2, Whose Mutation Causes Spastic Paraplegia, Promotes Reactive Oxygen Species Generation and Apoptosis. Cel Death Dis. 9, 797. doi:10.1038/s41419-018-0815-3
Montenegro, G., Rebelo, A. P., Connell, J., Allison, R., Babalini, C., D’Aloia, M., et al. (2012). Mutations in the ER-Shaping Protein Reticulon 2 Cause the Axon-Degenerative Disorder Hereditary Spastic Paraplegia Type 12. J. Clin. Invest. 122, 538–544. doi:10.1172/jci60560
Mosser, J., Douar, A.-M., Sarde, C.-O., Kioschis, P., Feil, R., Moser, H., et al. (1993). Putative X-Linked Adrenoleukodystrophy Gene Shares Unexpected Homology with ABC Transporters. Nature 361, 726–730. doi:10.1038/361726a0
Mou, Y., Dong, Y., Chen, Z., Denton, K. R., Duff, M. O., Blackstone, C., et al. (2020). Impaired Lipid Metabolism in Astrocytes Underlies Degeneration of Cortical Projection Neurons in Hereditary Spastic Paraplegia. Acta Neuropathologica Commun. 8, 214. doi:10.1186/s40478-020-01088-0
Murphy, D. (2001). The Biogenesis and Functions of Lipid Bodies in Animals, Plants and Microorganisms. Prog. Lipid Res. 40, 325–438. doi:10.1016/s0163-7827(01)00013-3
Nakao, H., Ikeda, K., Ishihama, Y., and Nakano, M. (2016). Membrane-Spanning Sequences in Endoplasmic Reticulum Proteins Promote Phospholipid Flip-Flop. Biophysical J. 110, 2689–2697. doi:10.1016/j.bpj.2016.05.023
Napoli, B., Gumeni, S., Forgiarini, A., Fantin, M., De Filippis, C., Panzeri, E., et al. (2019). Naringenin Ameliorates Drosophila ReepA Hereditary Spastic Paraplegia-Linked Phenotypes. Front. Neurosci. 13, 1202. doi:10.3389/fnins.2019.01202
Neveling, K., Martinez-Carrera, L. A., Hölker, I., Heister, A., Verrips, A., Hosseini-Barkooie, S. M., et al. (2013). Mutations in BICD2, Which Encodes a Golgin and Important Motor Adaptor, Cause Congenital Autosomal-Dominant Spinal Muscular Atrophy. Am. J. Hum. Genet. 92, 946–954. doi:10.1016/j.ajhg.2013.04.011
Novarino, G., Fenstermaker, A. G., Zaki, M. S., Hofree, M., Silhavy, J. L., Heiberg, A. D., et al. (2014). Exome Sequencing Links Corticospinal Motor Neuron Disease to Common Neurodegenerative Disorders. Science 343, 506–511. doi:10.1126/science.1247363
Oates, E. C., Rossor, A. M., Hafezparast, M., Gonzalez, M., Speziani, F., Macarthur, D. G., et al. (2013). Mutations in BICD2 Cause Dominant Congenital Spinal Muscular Atrophy and Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 92, 965–973. doi:10.1016/j.ajhg.2013.04.018
Oh, J., Baksa, K., and Steward, R. (2000). Functional Domains of the Drosophila Bicaudal-D Protein. Genetics 154, 713–724.
Ohsaki, Y., Suzuki, M., and Fujimoto, T. (2014). Open Questions in Lipid Droplet Biology. Chem. Biol. 21, 86–96. doi:10.1016/j.chembiol.2013.08.009
Olenick, M. A., and Holzbaur, E. L. F. (2019). Dynein Activators and Adaptors at a Glance. J. Cel Sci. 132, jcs227132. doi:10.1242/jcs.227132
Olzmann, J. A., and Carvalho, P. (2019). Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cel Biol 20, 137–155. doi:10.1038/s41580-018-0085-z
Orso, G., Pendin, D., Liu, S., Tosetto, J., Moss, T. J., Faust, J. E., et al. (2009). Homotypic Fusion of ER Membranes Requires the Dynamin-like GTPase Atlastin. Nature 460, 978–983. doi:10.1038/nature08280
Öztürk, Z., O'kane, C. J., and Pérez-Moreno, J. J. (2020). Axonal Endoplasmic Reticulum Dynamics and its Roles in Neurodegeneration. Front. Neurosci-switz 14, 48. doi:10.3389/fnins.2020.00048
Papadopoulos, C., Orso, G., Mancuso, G., Herholz, M., Gumeni, S., Tadepalle, N., et al. (2015). Spastin Binds to Lipid Droplets and Affects Lipid Metabolism. Plos Genet. 11, e1005149. doi:10.1371/journal.pgen.1005149
Park, S. H., Zhu, P.-P., Parker, R. L., and Blackstone, C. (2010). Hereditary Spastic Paraplegia Proteins REEP1, Spastin, and Atlastin-1 Coordinate Microtubule Interactions with the Tubular ER Network. J. Clin. Invest. 120, 1097–1110. doi:10.1172/jci40979
Patel, H., Cross, H., Proukakis, C., Hershberger, R., Bork, P., Ciccarelli, F. D., et al. (2002). SPG20 Is Mutated in Troyer Syndrome, an Hereditary Spastic Paraplegia. Nat. Genet. 31, 347–348. doi:10.1038/ng937
Pearce, M. M. P., Wang, Y., Kelley, G. G., and Wojcikiewicz, R. J. H. (2007). SPFH2 Mediates the Endoplasmic Reticulum-Associated Degradation of Inositol 1,4,5-trisphosphate Receptors and Other Substrates in Mammalian Cells. J. Biol. Chem. 282, 20104–20115. doi:10.1074/jbc.m701862200
Peeters, K., Litvinenko, I., Asselbergh, B., Almeida-Souza, L., Chamova, T., Geuens, T., et al. (2013). Molecular Defects in the Motor Adaptor BICD2 Cause Proximal Spinal Muscular Atrophy with Autosomal-Dominant Inheritance. Am. J. Hum. Genet. 92, 955–964. doi:10.1016/j.ajhg.2013.04.013
Pennetta, G., and Welte, M. A. (2018). Emerging Links between Lipid Droplets and Motor Neuron Diseases. Develop. Cel 45, 427–432. doi:10.1016/j.devcel.2018.05.002
Piomelli, D., Astarita, G., and Rapaka, R. (2007). A Neuroscientist's Guide to Lipidomics. Nat. Rev. Neurosci. 8, 743–754. doi:10.1038/nrn2233
Plaud, C., Joshi, V., Kajevu, N., Pous, C., Curmi, P. A., and Burgo, A. (2018). Functional Differences of Short and Long Isoforms of Spastin Harboring Missense Mutation. Dis. Model. Mech. 11, dmm033704. doi:10.1242/dmm.033704
Pol, A., Gross, S. P., and Parton, R. G. (2014). Biogenesis of the Multifunctional Lipid Droplet: Lipids, Proteins, and Sites. J. Cel Biol. 204, 635–646. doi:10.1083/jcb.201311051
Rambold, A. S., Cohen, S., and Lippincott-Schwartz, J. (2015). Fatty Acid Trafficking in Starved Cells: Regulation by Lipid Droplet Lipolysis, Autophagy, and Mitochondrial Fusion Dynamics. Develop. Cel. 32, 678–692. doi:10.1016/j.devcel.2015.01.029
Reid, E., Connell, J., Edwards, T. L., Duley, S., Brown, S. E., and Sanderson, C. M. (2005). The Hereditary Spastic Paraplegia Protein Spastin Interacts with the ESCRT-III Complex-Associated Endosomal Protein CHMP1B. Hum. Mol. Genet. 14, 19–38. doi:10.1093/hmg/ddi003
Renvoisé, B., Malone, B., Falgairolle, M., Munasinghe, J., Stadler, J., Sibilla, C., et al. (2016). Reep1 Null Mice Reveal a Converging Role for Hereditary Spastic Paraplegia Proteins in Lipid Droplet Regulation. Hum. Mol. Genet. 25, 5111–5125. doi:10.1093/hmg/ddw315
Renvoisé, B., Stadler, J., Singh, R., Bakowska, J. C., and Blackstone, C. (2012). Spg20−/− Mice Reveal Multimodal Functions for Troyer Syndrome Protein Spartin in Lipid Droplet Maintenance, Cytokinesis and BMP Signaling. Hum. Mol. Genet. 21, 3604–3618. doi:10.1093/hmg/dds191
Rinaldi, C., Schmidt, T., Situ, A. J., Johnson, J. O., and Lee, P. R. (2015). Mutation in CPT1C Associated with Pure Autosomal Dominant Spastic Paraplegia. Jama Neurol. 72, 608. doi:10.1001/jamaneurol.2014.4769
Rismanchi, N., Soderblom, C., Stadler, J., Zhu, P. P., and Blackstone, C. (2008). Atlastin GTPases Are Required for Golgi Apparatus and ER Morphogenesis. Hum. Mol. Genet. 17, 1591–604. doi:10.1093/hmg/ddn046
Roberts, M. A., and Olzmann, J. A. (2020). Protein Quality Control and Lipid Droplet Metabolism. Annu. Rev. Cel Dev. Biol. 36, 115–139. doi:10.1146/annurev-cellbio-031320-101827
Roll-Mecak, A., and Vale, R. D. (2008). Structural Basis of Microtubule Severing by the Hereditary Spastic Paraplegia Protein Spastin. Nature 451, 363–367. doi:10.1038/nature06482
Rugarli, E. I., and Langer, T. (2012). Mitochondrial Quality Control: a Matter of Life and Death for Neurons. EMBO J. 31, 1336–1349. doi:10.1038/emboj.2012.38
Salo, V. T., Belevich, I., Li, S., Karhinen, L., Vihinen, H., Vigouroux, C., et al. (2016). Seipin Regulates ER -lipid Droplet Contacts and Cargo Delivery. EMBO J. 35, 2699–2716. doi:10.15252/embj.201695170
Salo, V. T., Hölttä-Vuori, M., and Ikonen, E. (2020). Seipin-Mediated Contacts as Gatekeepers of Lipid Flux at the Endoplasmic Reticulum–Lipid Droplet Nexus. Contact 3, 1–16. doi:10.1177/2515256420945820
Sanderson, C. M., Connell, J. W., Edwards, T. L., Bright, N. A., Duley, S., Thompson, A., et al. (2006). Spastin and Atlastin, Two Proteins Mutated in Autosomal-Dominant Hereditary Spastic Paraplegia, Are Binding Partners. Hum. Mol. Genet. 15, 307–318. doi:10.1093/hmg/ddi447
Santinho, A., Salo, V. T., Chorlay, A., Li, S., Zhou, X., Omrane, M., et al. (2020). Membrane Curvature Catalyzes Lipid Droplet Assembly. Curr. Biol. 30, 2481–2494. doi:10.1016/j.cub.2020.04.066
Sauer, R. T., and Baker, T. A. (2011). AAA+ Proteases: ATP-Fueled Machines of Protein Destruction. Annu. Rev. Biochem. 80, 587–612. doi:10.1146/annurev-biochem-060408-172623
Sauter, S. M., Engel, W., Neumann, L. M., Kunze, J., and Neesen, J. (2004). Novel Mutations in theAtlastin Gene (SPG3A) in Families with Autosomal Dominant Hereditary Spastic Paraplegia and Evidence for Late Onset Forms of HSP Linked to theSPG3A Locus. Hum. Mutat. 23, 98. doi:10.1002/humu.9205
Schuck, S., Prinz, W. A., Thorn, K. S., Voss, C., and Walter, P. (2009). Membrane Expansion Alleviates Endoplasmic Reticulum Stress Independently of the Unfolded Protein Response. J. Cel Biol. 187, 525–536. doi:10.1083/jcb.200907074
Schuldiner, M., and Bohnert, M. (2017). A Different Kind of Love - Lipid Droplet Contact Sites. Biochim. Biophys. Acta (Bba) - Mol. Cel Biol. Lipids 1862, 1188–1196. doi:10.1016/j.bbalip.2017.06.005
Schüle, R., and Schöls, L. (2011). Genetics of Hereditary Spastic Paraplegias. Semin. Neurol. 31, 484–493. doi:10.1055/s-0031-1299787
Schuurs-Hoeijmakers, J. H. M., Geraghty, M. T., Kamsteeg, E.-J., Ben-Salem, S., de Bot, S. T., Nijhof, B., et al. (2012). Mutations in DDHD2, Encoding an Intracellular Phospholipase A1, Cause a Recessive Form of Complex Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 91, 1073–1081. doi:10.1016/j.ajhg.2012.10.017
Seebacher, F., Zeigerer, A., Kory, N., and Krahmer, N. (2020). Hepatic Lipid Droplet Homeostasis and Fatty Liver Disease. Semin. Cel Develop. Biol. 108, 72–81. doi:10.1016/j.semcdb.2020.04.011
Shibata, Y., Shemesh, T., Prinz, W. A., Palazzo, A. F., Kozlov, M. M., and Rapoport, T. A. (2010). Mechanisms Determining the Morphology of the Peripheral ER. Cell 143, 774–788. doi:10.1016/j.cell.2010.11.007
Shimabukuro, M. K., Langhi, L. G. P., Cordeiro, I., Brito, J. M., Batista, C. M. D. C., Mattson, M. P., et al. (2016). Lipid-laden Cells Differentially Distributed in the Aging Brain Are Functionally Active and Correspond to Distinct Phenotypes. Sci. Rep. 6, 23795. doi:10.1038/srep23795
Shribman, S., Reid, E., Crosby, A. H., Houlden, H., and Warner, T. T. (2019). Hereditary Spastic Paraplegia: from Diagnosis to Emerging Therapeutic Approaches. Lancet Neurol. 18, 1136–1146. doi:10.1016/s1474-4422(19)30235-2
Solowska, J. M., Morfini, G., Falnikar, A., Himes, B. T., Brady, S. T., Huang, D., et al. (2008). Quantitative and Functional Analyses of Spastin in the Nervous System: Implications for Hereditary Spastic Paraplegia. J. Neurosci. 28, 2147–2157. doi:10.1523/jneurosci.3159-07.2008
Szymanski, K. M., Binns, D., Bartz, R., Grishin, N. V., Li, W.-P., Agarwal, A. K., et al. (2007). The Lipodystrophy Protein Seipin Is Found at Endoplasmic Reticulum Lipid Droplet Junctions and Is Important for Droplet Morphology. Proc. Natl. Acad. Sci. 104, 20890–20895. doi:10.1073/pnas.0704154104
Tadepalle, N., Robers, L., Veronese, M., Zentis, P., Babatz, F., Brodesser, S., et al. (2020). Microtubule-dependent and Independent Roles of Spastin in Lipid Droplet Dispersion and Biogenesis. Life Sci. Alliance 3, e202000715. doi:10.26508/lsa.202000715
Terasaki, M. (2018). Axonal Endoplasmic Reticulum Is Very Narrow. J. Cel Sci. 131, jcs210450. doi:10.1242/jcs.210450
Terasaki, M., Chen, L. B., and Fujiwara, K. (1986). Microtubules and the Endoplasmic Reticulum Are Highly Interdependent Structures. J. Cel Biol. 103, 1557–1568. doi:10.1083/jcb.103.4.1557
Thiam, A. R., and Dugail, I. (2019). Lipid Droplet-Membrane Contact Sites - from Protein Binding to Function. J. Cel Sci. 132, jcs230169. doi:10.1242/jcs.230169
Valm, A. M., Cohen, S., Legant, W. R., Melunis, J., Hershberg, U., Wait, E., et al. (2017). Applying Systems-Level Spectral Imaging and Analysis to Reveal the Organelle Interactome. Nature 546, 162–167. doi:10.1038/nature22369
Velázquez, A. P., Tatsuta, T., Ghillebert, R., Drescher, I., and Graef, M. (2016). Lipid Droplet-Mediated ER Homeostasis Regulates Autophagy and Cell Survival during Starvation. J. Cel Biol. 212, 621–631. doi:10.1083/jcb.201508102
Vietri, M., Schink, K. O., Campsteijn, C., Wegner, C. S., Schultz, S. W., Christ, L., et al. (2015). Spastin and ESCRT-III Coordinate Mitotic Spindle Disassembly and Nuclear Envelope Sealing. Nature 522, 231–235. doi:10.1038/nature14408
Wali, G., Sutharsan, R., Fan, Y., Stewart, R., Tello Velasquez, J., Sue, C. M., et al. (2016). Mechanism of Impaired Microtubule-dependent Peroxisome Trafficking and Oxidative Stress in SPAST -mutated Cells from Patients with Hereditary Spastic Paraplegia. Sci. Rep. 6, 27004. doi:10.1038/srep27004
Walther, T. C., Chung, J., and Farese, R. V. (2017). Lipid Droplet Biogenesis. Annu. Rev. Cel Dev. Biol. 33, 491–510. doi:10.1146/annurev-cellbio-100616-060608
Walther, T. C., and Farese, R. V. (2012). Lipid Droplets and Cellular Lipid Metabolism. Annu. Rev. Biochem. 81, 687–714. doi:10.1146/annurev-biochem-061009-102430
Walther, T. C., and Farese, R. V. (2009). The Life of Lipid Droplets. Biochim. Biophys. Acta (Bba) - Mol. Cel Biol. Lipids 1791, 459–466. doi:10.1016/j.bbalip.2008.10.009
Wang, C.-W. (2015). Lipid Droplet Dynamics in Budding Yeast. Cel. Mol. Life Sci. 72, 2677–2695. doi:10.1007/s00018-015-1903-5
Wang, H. J., Becuwe, M., Housden, B. E., Chitraju, C., Porras, A. J., Graham, M. M., et al. (2016). Seipin Is Required for Converting Nascent to Mature Lipid Droplets. Elife 5, e16582. doi:10.7554/elife.16582
Wang, S., Tukachinsky, H., Romano, F. B., and Rapoport, T. A. (2016). Cooperation of the ER-Shaping Proteins Atlastin, Lunapark, and Reticulons to Generate a Tubular Membrane Network. Elife 5, e18605. doi:10.7554/elife.18605
Welniarz, Q., Dusart, I., and Roze, E. (2017). The Corticospinal Tract: Evolution, Development, and Human Disorders. Devel Neurobio. 77, 810–829. doi:10.1002/dneu.22455
Welte, M. A. (2015). Expanding Roles for Lipid Droplets. Curr. Biol. 25, R470–R481. doi:10.1016/j.cub.2015.04.004
Welte, M. A. (2007). Proteins under New Management: Lipid Droplets Deliver. Trends Cel Biol. 17, 363–369. doi:10.1016/j.tcb.2007.06.004
Windpassinger, C., Auer-Grumbach, M., Irobi, J., Patel, H., Petek, E., Hörl, G., et al. (2004). Heterozygous Missense Mutations in BSCL2 Are Associated with Distal Hereditary Motor Neuropathy and Silver Syndrome. Nat. Genet. 36, 271–276. doi:10.1038/ng1313
Wolfgang, M. J., Kurama, T., Dai, Y., Suwa, A., Asaumi, M., Matsumoto, S.-i., et al. (2006). The Brain-specific Carnitine Palmitoyltransferase-1c Regulates Energy Homeostasis. Proc. Natl. Acad. Sci. 103, 7282–7287. doi:10.1073/pnas.0602205103
Yagi, T., Ito, D., Nihei, Y., Ishihara, T., and Suzuki, N. (2011). N88S Seipin Mutant Transgenic Mice Develop Features of seipinopathy/BSCL2-Related Motor Neuron Disease via Endoplasmic Reticulum Stress. Hum. Mol. Genet. 20, 3831–3840. doi:10.1093/hmg/ddr304
Yalçın, B., Zhao, L., Stofanko, M., O'sullivan, N. C., Kang, Z. H., Roost, A., et al. (2017). Modeling of Axonal Endoplasmic Reticulum Network by Spastic Paraplegia Proteins. Elife 6. e23882. doi:10.7554/eLife.23882
Yan, R., Qian, H., Lukmantara, I., Gao, M., Du, X., Yan, N., et al. (2018). Human SEIPIN Binds Anionic Phospholipids. Dev. Cel 47, 248–256. doi:10.2210/pdb6ds5/pdb
Zhang, D., Rogers, G. C., Buster, D. W., and Sharp, D. J. (2007). Three Microtubule Severing Enzymes Contribute to the "Pacman-Flux" Machinery that Moves Chromosomes. J. Cel Biol. 177, 231–242. doi:10.1083/jcb.200612011
Zhao, G., Zhu, P.-P., Renvoisé, B., Maldonado-Báez, L., Park, S. H., and Blackstone, C. (2016). Mammalian Knock Out Cells Reveal Prominent Roles for Atlastin GTPases in ER Network Morphology. Exp. Cel Res. 349, 32–44. doi:10.1016/j.yexcr.2016.09.015
Keywords: hereditary spastic paraplegia, lipid droplet, lipid metabolism, endoplasmic reticulum, spastin
Citation: Tadepalle N and Rugarli EI (2021) Lipid Droplets in the Pathogenesis of Hereditary Spastic Paraplegia. Front. Mol. Biosci. 8:673977. doi: 10.3389/fmolb.2021.673977
Received: 28 February 2021; Accepted: 26 April 2021;
Published: 10 May 2021.
Edited by:
Pilar Gonzalez-Cabo, University of Valencia, SpainReviewed by:
Giovanni Stevanin, INSERM U1127 Institut du Cerveau et de la Moelle épinière (ICM), FranceXuejun Li, University of Illinois at Chicago, United States
Rachel Allison, University of Cambridge, United Kingdom
Copyright © 2021 Tadepalle and Rugarli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena I. Rugarli, ZWxlbmEucnVnYXJsaUB1bmkta29lbG4uZGU=