 Karen M. Henkels
Karen M. Henkels Kristen M. Rehl
Kristen M. Rehl Kwang-jin Cho
Kwang-jin Cho- Department of Biochemistry and Molecular Biology, School of Boonshoft School of Medicine, Wright State University, Dayton, OH, United States
Ras proteins are membrane-bound small GTPases that promote cell proliferation, differentiation, and apoptosis. Consistent with this key regulatory role, activating mutations of Ras are present in ∼19% of new cancer cases in the United States per year. K-Ras is one of the three ubiquitously expressed isoforms in mammalian cells, and oncogenic mutations in this isoform account for ∼75% of Ras-driven cancers. Therefore, pharmacological agents that block oncogenic K-Ras activity would have great clinical utility. Most efforts to block oncogenic Ras activity have focused on Ras downstream effectors, but these inhibitors only show limited clinical benefits in Ras-driven cancers due to the highly divergent signals arising from Ras activation. Currently, four major approaches are being extensively studied to target K-Ras–driven cancers. One strategy is to block K-Ras binding to the plasma membrane (PM) since K-Ras requires the PM binding for its signal transduction. Here, we summarize recently identified molecular mechanisms that regulate K-Ras–PM interaction. Perturbing these mechanisms using pharmacological agents blocks K-Ras–PM binding and inhibits K-Ras signaling and growth of K-Ras–driven cancer cells. Together, these studies propose that blocking K-Ras–PM binding is a tractable strategy for developing anti–K-Ras therapies.
Introduction
RAS genes were initially identified as the viral oncogenes of acute transforming retroviruses, and it was designated as a mammalian proto-oncogene when mutated RAS genes were discovered in human cancer cells (Barbacid, 1987). There are three main Ras isoforms—H-, N-, and K-Ras—in mammalian cells, and each is encoded by a different gene. H-, N-, and K-RAS are situated on chromosomes 11 (11p15.1-p15.5), 1 (1p22-p32), and 12 (12p12.1-pter), respectively (Barbacid, 1987). There are four exons that code for H- and N-RAS, while in K-RAS, there are two alternative fourth exons, exons 4A and 4B, that yield two splice variants, K-Ras4A and K-Ras4B (Barbacid, 1987). While H-, N-, and K-Ras4B are ubiquitously expressed in mammalian cells, K-Ras4A is precisely and spatiotemporally expressed in the murine lung, liver, and kidney (Pells et al., 1997). Knockout studies showed that neither H- nor N-RAS individually or in concert are required for normal murine embryogenesis (Esteban et al., 2001), whereas K-RAS is unequivocally crucial to embryonic development (Johnson et al., 1997; Koera et al., 1997). Intriguingly, K-Ras knockout mice with spatiotemporally controlled expression of H-Ras by the K-Ras promoter have their embryonic lethality restored but develop dilated cardiomyopathy associated with arterial hypertension at an older age, reflecting the different molecular functions of Ras isoforms in the cell (Potenza et al., 2005).
While the three Ras isoforms are nearly identical, sharing ∼90–100% homology in their N-terminal catalytic domain sequences, there is a considerable lack of homology in the C-terminal hypervariable region (HVR) of each isoform, which accounts for <15% homology being shared between any two isoforms (Hancock, 2003). These HVRs consist of two different signal sequences that allow Ras proteins to traffic to and interact with the inner leaflet of the plasma membrane (PM) (Hancock et al., 1989). The CAAX motif, the first signal sequence, is constituted by the last four amino acid residues in the HVR and is shared in common between the different Ras isoforms. For CAAX, C is cysteine, A is an aliphatic amino acid, and X is either serine or methionine (Hancock et al., 1989). Newly synthesized Ras GTPases are cytosolic and require a series of posttranslational modifications of the CAAX motif for interacting with endomembranes. First, the CAAX motif is farnesylated by a cytosolic farnesyltransferase (FTase) that covalently attaches a farnesyl group to the cysteine residue via a thioether bond. Farnesylated Ras interacts with the cytosolic leaflet of the endoplasmic reticulum (ER), where the AAX tripeptide is removed by the Ras and a-factor–converting enzyme (Rce1). The now C-terminal cysteine is methylated by isoprenylcysteine carboxyl methyltransferase (Icmt) (Hancock et al., 1989). The CAAX motif must be processed in this series of steps in order to maintain the correct forward trafficking of Ras isoforms, since knockout of either Rce1 or Icmt results in Ras mislocalization to the cytosol (Kim et al., 1999; Lau et al., 2014).
While the correctly modified CAAX motif can direct Ras to the ER and other endomembranes, the presence of the second C-terminal signal motif is required for maximal membrane affinity and PM localization (Hancock et al., 1990). The second signal sequence situated within the HVR varies between the different Ras isoforms such that both H-Ras, N-Ras, and K-Ras4A are palmitoylated (Cys181 and Cys184 for H-RasCys181 for N-Ras and Cys180 for K-Ras4A), while K-Ras4B has a stretch of six lysine residues, forming a polybasic domain (PBD) (Lys175-180) (Hancock et al., 1990). Palmitoylation of H- and N-Ras by the Ras palmitoyltransferase takes place in the ER and Golgi complex, where H- and N-Ras are transported via the classical secretory pathway to the PM (Apolloni et al., 2000). While palmitoylation of H- or N-Ras is a short-lived modification with rapid kinetics (t1/2 of <20 min), the depalmitoylation/repalmitoylation machinery is important for delivering consistent H- and N-Ras distribution between the Golgi and the PM at a steady state (Rocks et al., 2005; Rocks et al., 2010). Palmitoylated Ras proteins diffuse from the PM to other endomembrane compartments to reach equilibrium, but depalmitoylation by poorly characterized thioesterases enhances the rate of diffusion, and thereby promotes their continuous redirection to the ER and Golgi for repalmitoylation and unidirectional trafficking back to the PM (Rocks et al., 2005; Rocks et al., 2010). The exact mechanism on how posttranslationally modified K-Ras4B (hereafter K-Ras) is transported from the ER to the PM is not fully characterized. Recent studies have demonstrated that the delta subunit of cGMP phosphodiesterase 6 (PDE6δ) functions, in part, as a K-Ras chaperone to maintain K-Ras–PM localization. PDE6δ binds the farnesyl moiety of cytosolic K-Ras, which is released in perinuclear membranes by the release factors Arl2 and 3, from where it is trapped on the recycling endosome (RE) by electrostatic interaction, and it returns to the PM via vesicular transport (Ismail et al., 2011; Chandra et al., 2012; Schmick et al., 2014). Once K-Ras is transported to the PM, it binds the PM through an electrostatic interaction of the strong positive charge of the C-terminal PBD with anionic phospholipid head groups in the inner PM leaflet (Yeung et al., 2008; Zhou et al., 2017).
K-Ras and Cancer
Oncogenic mutations in Ras are found in about 18.7% of new cancer cases in the United States per year (1.3% for H-Ras, 3.1% for N-Ras, and 14.3% for K-Ras) (Prior et al., 2020). While the oncogenic mutant K-Ras is found in approximately 88% of pancreatic, 50% of colorectal, and 32% of lung cancers (Prior et al., 2020), no anti–K-Ras drugs are currently available in clinics. Human cancer cells harboring oncogenic mutant K-Ras reprogram their signaling network so that their survival and growth depend on oncogenic K-Ras signaling, a phenomenon called K-Ras addiction (Weinstein and Joe, 2008; Singh et al., 2009; Hayes et al., 2016). RNAi-mediated knockdown of oncogenic mutant K-Ras blocks cell survival and growth in a range of pancreatic and non-small-cell lung cancers (NSCLC), which provides the rationale that blocking oncogenic K-Ras activity is a valid approach to treat K-Ras-dependent cancers. Recently, two new K-Ras direct inhibitors have shown promising outcomes in clinical trials. AMG 510 and MRTX849 are small molecules that bind to the GDP-bound inactive K-RasG12C mutant and form a covalent bond to the mutant Cys, which locks K-Ras in the inactive conformation, resulting in blocked oncogenic signaling (Ostrem et al., 2013). These compounds exhibited pronounced anticancer effects in K-RasG12C tumor mice models and clinical trials with lung and colorectal cancer patients harboring the K-RasG12C mutant (Canon et al., 2019; Hallin et al., 2020). Despite the promising clinical outcome of these inhibitors, they are specific to the K-RasG12C mutant, which is found in ∼3% of pancreatic, ∼4% of colorectal, and ∼13% of lung cancers that harbor any oncogenic mutations in K-Ras (Cox et al., 2014; Prior et al., 2020), suggesting that these inhibitors would be suitable only for a small portion of cancer patients with the oncogenic mutant K-Ras.
In addition to K-RasG12C–specific direct inhibitors, there are three other approaches that are currently being investigated for blocking all oncogenic mutant K-Ras activity. They are 1) blocking K-Ras interaction with the PM, 2) inhibiting K-Ras downstream effectors, and 3) dysregulating cell energy metabolism. This review will focus on mechanisms that regulate the PM localization of K-Ras, which could be tractable targets for developing new anti–K-Ras therapeutics.
Dissociating Ras From the Plasma Membrane Blocks its Signal Transduction
Preventing Ras Prenylation Dissociates Ras From the PM and Inhibits Ras Signaling
Point mutations in the CAAX motif, which block posttranslational modification, prevent Ras–PM localization and completely inhibit all biological activities of oncogenic mutant Ras (Willumsen et al., 1984). Thus, farnesyltransferase inhibitors (FTIs) were designed to phenotypically mimic this mode of Ras inhibition. FTIs demonstrated marked antitumor activity in H-Ras–driven in vivo and in vitro models, which allowed phase I studies on FTIs in 1999, with some progressing to phase III clinical trials in 2002 (Baines et al., 2011). However, FTIs were ineffective with regard to pancreatic cancers in phase II and III clinical trials in which oncogenic mutant K-Ras was found in 88% of all pancreatic cancers (Cohen et al., 2003; Van Cutsem et al., 2004; Macdonald et al., 2005). It is because in FTI-treated cells, an alternative prenyltransferase, geranylgeranyltransferase (GGTase), efficiently attaches the more hydrophobic C20 geranylgeranyl moiety to K- and N-Ras, allowing K- and N-Ras to interact with the PM and conduct a signal transduction that is equipotent with the farnesylated forms (Baines et al., 2011). Concomitant inhibition of FTase with GGTase to completely block prenylation of K- and N-Ras has been tested, but this approach has suffered from dose-limiting toxicities (O'Bryan, 2019). Also, there are more than 100 proteins that are prenylated, and these combined inhibitors would induce prohibitive off-target effects, preventing their clinical effectiveness. A recent study has demonstrated a promising strategy to specifically inhibit K-Ras prenylation. A modified FTI with an electrophilic moiety specifically interacts with the CAAX motif of K-Ras but not H-Ras, resulting in the blockage of K-Ras farnesylation and geranylgeranylation, trapping K-Ras in the cytosol (Novotny et al., 2017). Further improvements of this approach could lead to a more potent inhibitor of K-Ras prenylation and activity (Novotny et al., 2017; O'Bryan, 2019).
Perturbing K-Ras/PDE6δ Interaction Blocks K-Ras–PM Binding and K-Ras Signaling
Recent studies have shown that blocking PDE6δ interaction with K-Ras is a tractable strategy to inhibit K-Ras–PM localization and oncogenic K-Ras signaling. PDE6δ binds the farnesyl moiety of K-Ras via its hydrophobic pocket and acts in part as a chaperone. The release factors Arl2 and 3 unload K-Ras from PDE6δ in the perinuclear region, whence K-Ras binds to the recycling endosome (RE) for redelivery to the PM via vesicular transport (Chandra et al., 2012; Schmick et al., 2014). Deltarasin is a small molecule that binds to the hydrophobic pocket and inhibits PDE6δ/K-Ras interaction, resulting in K-Ras–PM mislocalization and abrogated signaling in K-Ras–driven cancer cells (Figure 1 and Table 1) (Zimmermann et al., 2013). Second-generation PDE6δ inhibitors, which bind PDE6δ more tightly via extra hydrogen bonds, have demonstrated greater potency for blocking the growth of K-Ras–dependent but not K-Ras–independent pancreatic cancer cells (Papke et al., 2016; Martin-Gago et al., 2017). Moreover, deltarasin does not inhibit the growth of cells transformed with the oncogenic mutant B-Raf or the overexpressed epidermal growth factor receptor (EGFR) (Klein et al., 2019), suggesting that PDE6δ inhibitors are effective against K-Ras–dependent cancer cells. In addition, deltarasin functions independent of K-Ras, where it promotes autophagy by activating the AMPK/mTOR pathway, and concomitant inhibition of autophagy and PDE6δ potentiates deltarasin-mediated cell death by elevating reactive oxygen species (ROS) (Leung et al., 2018). These observations suggest that deltarasin elevates cellular ROS, which promotes autophagy (Zhang et al., 2016), and that deltarasin in combination with an autophagy inhibitor can be a plausible strategy for treating K-Ras–driven cancers (Leung et al., 2018).
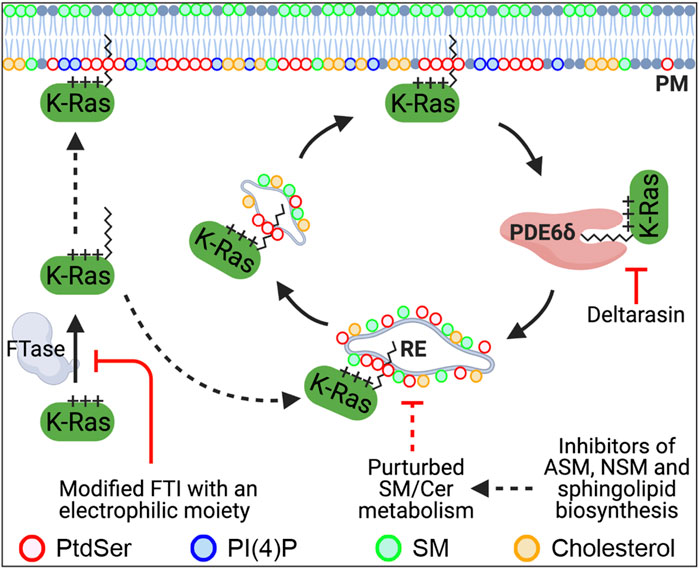
FIGURE 1. Recently identified molecular mechanisms that regulate the PM localization of K-Ras. K-Ras farnesylated by FTase localizes to the PM. Once K-Ras dissociates from the PM, PDE6δ binds K-Ras via its farnesyl moiety and releases it in the perinuclear region. K-Ras is then translocated to the recycling endosome (RE) through electrostatic interaction, where it returns to the PM via RE-mediated vesicular transport. Blocking K-Ras prenylation or the K-Ras/PDE6δ interaction mislocalizes K-Ras from the PM. Perturbed SM/ceramide metabolism is proposed to dysregulate the RE via altering its lipid composition, resulting in depletion of PtdSer and K-Ras from the PM. FTase, farnesyltransferase; FTI, FTase inhibitor; PDE6δ, phosphodiesterase 6 δ; RE, recycling endosome; PtdSer, phosphatidylserine; SM, sphingomyelin; Cer, ceramide; PI4P, phosphatidylinositol 4-phosphate; ASM, acid sphingomyelinase; NSM, neural sphingomyelinase.
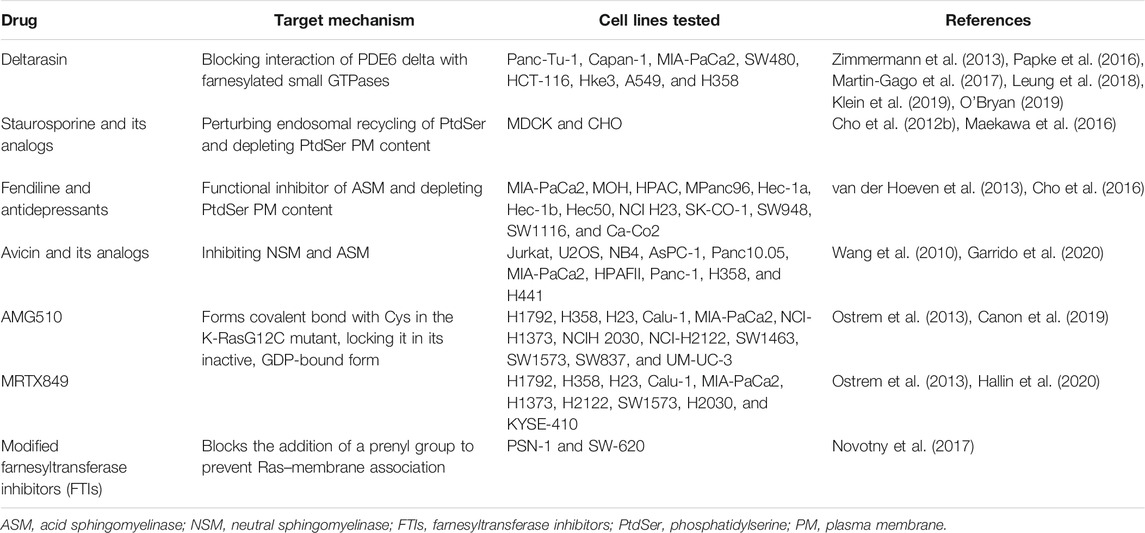
TABLE 1. Summary of the compounds that inhibit K-Ras interaction with the PM.
However, PDE6δ interacts with other prenylated small GTPases including H-Ras, N-Ras, and Rap1 (Chandra et al., 2012; Dumbacher et al., 2018), suggesting that the effect of deltarasin may not be K-Ras–specific. Moreover, K-Ras knockout mice have embryonic lethality, whereas PDE6δ knockout mice develop normally (Johnson et al., 1997; Zhang et al., 2007), indicating that K-Ras is active in the absence of PDE6δ. In sum, PDE6δ interaction with K-Ras is a tractable target to inhibit oncogenic K-Ras activity, and further validation on the K-Ras specificity of PDE6δ would promote translation into the clinic.
Reducing Phosphatidylserine Content at the Inner PM Leaflet Removes K-Ras From the PM
Phosphatidylserine (PtdSer) is an anionic phospholipid synthesized from phosphatidylcholine (PtdCho) and phosphatidylethanolamine (PtdEth) by PtdSer synthase 1 and 2, respectively, in mammalian cells. While PtdSer is found in the ER and mitochondria, it is concentrated in the inner PM via mechanisms that are not fully elucidated (Leventis and Grinstein, 2010; Kay and Fairn, 2019). PM PtdSer plays key roles in physiological processes including the clearance of apoptotic cells, coagulation cascade, and recruitment and activation of signaling proteins (Leventis and Grinstein, 2010; Kay and Fairn, 2019). The anionic head group provides a negative electrostatic potential to the inner PM leaflet, which allows interaction with a stretch of positively charged amino acid residues, called PBD, of PM-localized proteins (Yeung et al., 2008). K-Ras binds PtdSer at the inner PM leaflet through the C-terminal PBD concomitantly with the farnesyl moiety, which provides specificity for PtdSer over other anionic phospholipids (Zhou et al., 2017). Recent studies have reported a number of mechanisms that can reduce PM PtdSer content, which in turn inhibits K-Ras–PM localization and oncogenic K-Ras signaling output.
Phosphatidylinositol 4-Phosphate Regulates the PM Distribution of PtdSer and K-Ras
Phosphatidylinositol (PI) is phosphorylated to PI 4-phosphate (PI4P) by four PI 4-kinases in mammalian cells: PI4K IIα and β (PI4K2A and 2B) and PI4K IIIα and β (PI4KA and PI4KB) (Balla, 2013). PI4KA and 2B localize primarily to the PM, whereas PI4K2A and PI4KB localize to the Golgi complex (Balla, 2013). In mammalian cells, oxysterol-binding protein–related proteins (ORPs) 5 and 8 exchange newly synthesized PtdSer from the ER for PI4P from the PM at ER–PM membrane contacting sites (MCSs) (Figure 2) (Chung et al., 2015; Moser von Filseck et al., 2015). This process is maintained by PM PI4P by PI4KA and the concomitant PI4P hydrolysis by Sac1 phosphatase in the ER to keep a PI4P concentration gradient across the PM and ER (Chung et al., 2015; Moser von Filseck et al., 2015). ORP5 and 8 recruitment to ER–PM MCSs further requires additional PM PI(4,5)P2 (Ghai et al., 2017; Sohn et al., 2018). Several studies have reported that perturbing this exchange process reduces PM PtdSer content and inhibits K-Ras–PM binding and K-Ras signal output. PI(4,5)P2 reduction by the rapamycin-recruitable 5-phosphatase domain of INPP5E to the PM blocks ORP5 and 8 recruitment to ER–PM MCSs, whereas increasing the PM PI(4,5)P2 level by overexpressing PI4P 5-kinase (PIP5K) β reduces PM PI4P levels. In both cases, the exchange of ER PtdSer for PM PI4P is perturbed, resulting in PtdSer reduction in the inner PM leaflet (Ghai et al., 2017; Sohn et al., 2018). Also, the acute depletion of PM PI4P by rapamycin-recruitable Sac1 dissociates K-Ras, but not H-Ras, from the PM and inhibits K-Ras signaling (Gulyas et al., 2017). Ras proteins are spatially organized into nanoscale domains on the PM, called nanoclusters, which are critical for high-fidelity Ras signal output (Prior et al., 2003; Tian et al., 2007; Cho et al., 2012a; Cho and Hancock, 2013). PM PI4P depletion by either ORP5 or 8 knockdown or chemical inhibition redistributes PtdSer and K-Ras from the PM. It further disrupts K-Ras nanoclustering and abrogates K-Ras signal output and the growth of K-Ras–driven pancreatic cancer cells (Kattan et al., 2019). Consistently, ORP5 and 8 are highly expressed in certain types of cancer and involved in the prognosis of cancer patients. A high expression of ORP8 is observed in lung cancer tissues and hamster bile duct cancers in comparison to normal tissues (Fournier et al., 1999; Loilome et al., 2006). ORP5 overexpression enhances the invasion of pancreatic cancer cells, while ORP5 knockdown abrogates it in vitro. Moreover, the ORP5 mRNA level is significantly elevated in tumors harboring oncogenic mutant K-Ras compared with tumors with wild-type (WT) K-Ras in cohorts of pancreatic cancer, NSCLC, and 33 types of cancer in the TCGA (the Cancer Genome Atlas) database (Kattan et al., 2019). Further analysis of overall survival periods for patients in these three cohorts demonstrates that cancer patients with low ORP5 or 8 expression have better prognosis than patients with high ORP5 or 8 expression (Koga et al., 2008; Kattan et al., 2019).
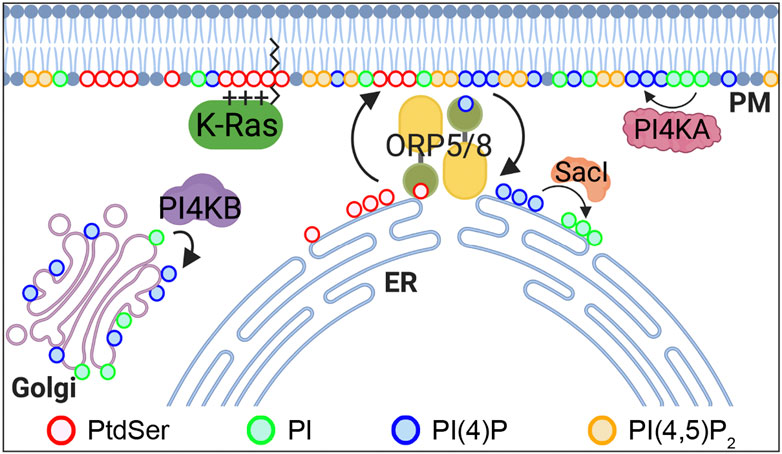
FIGURE 2. PtdSer PM enrichment is regulated by ORP5 and 8. ORP5 and 8 are lipid transporters that exchange ER PtdSer with PM PI4P. The driving force of this process is a PI4P concentration gradient, whereby PI4P levels are high in the PM by PI4KA and are kept low at the ER by Sac1 phosphatase, which converts PI4P to PI. PI4P is also generated at the Golgi complex by PI4KB. ORP, oxysterol-binding protein–related protein; PtdSer, phosphatidylserine; PI, phosphatidylinositol; PI4P, PI 4-phosphate; PI(4,5)P2, PI(4,5)-bisphosphate; PI4KA, PI 4-kinase IIIα; PI4KB, PI 4-kinase IIIβ.
In addition to PM PI4P, a recent study has demonstrated that Golgi PI4P is involved in the PM localization of PtdSer and K-Ras. Chemical inhibition of PI4KB, which depletes PI4P at the Golgi complex, but not the PM, translocates K-Ras and PtdSer from the PM to the mitochondria and endomembrane, respectively (Miller et al., 2019). Supplementation with exogenous PtdSer acutely returns K-Ras to the PM in Golgi PI4P–depleted cells, and mitochondrial PtdSer reduction by overexpressing PtdSer decarboxylase, which converts PtdSer to PtdEth at the mitochondria (Percy et al., 1983), redistributes K-Ras from the mitochondria to the endomembranes in Golgi PI4P–depleted cells (Miller et al., 2019). Furthermore, Golgi PI4P depletion inhibits Ras signaling in K-Ras–transformed but not H-Ras–transformed cells. Although the exact mechanism is yet to be elucidated, these data suggest that Golgi PI4P regulates the PM enrichment of PtdSer and thereby K-Ras–PM localization and K-Ras signaling (Miller et al., 2019). In sum, the PtdSer/PI4P exchange mechanism at the ER–PM MCSs, which regulates the PM enrichment of PtdSer and thereby K-Ras–PM localization and signaling, is a viable target for developing anti–K-Ras therapies.
Perturbing Recycling Endosomal Activity Mislocalizes PtdSer and K-Ras From the PM
In addition to the non-vesicular transport of PtdSer by ORP5 and 8, PtdSer transports via the classical vesicular trafficking. Once PM PtdSer is endocytosed, it enters the sorting endosomes, where it either returns to the PM via the RE or is transported to lysosomes for its degradation by phospholipases (Leventis and Grinstein, 2010), suggesting that recycling endosomal activity is important for maintaining PM PtdSer content. Recent studies have reported that disruption of recycling endosomal activity depletes PtdSer and K-Ras from the PM. Acylpeptide hydrolase (APEH) removes the N-terminal acylated amino acids from acetylated proteins, and regulates the ubiquitin-mediated protein degradation (Shimizu et al., 2004). APEH knockdown or inhibition blocks endocytic recycling of the transferrin receptor (TfR) and EGFR and mislocalizes K-Ras and PtdSer from the PM (Tan et al., 2019). It also reduces nanoclustering of oncogenic K-Ras that remained at the PM and prevents oncogenic K-Ras signaling and growth of pancreatic cancer cells harboring oncogenic mutant K-Ras but not WT K-Ras. This study proposes that failure to maintain PtdSer and K-Ras at the PM in APEH-depleted cells is in part induced by aberrant RE function.
A protein kinase C (PKC) inhibitor, staurosporine, and its analogs accumulate PtdSer internalized from the PM in the RE, resulting in PM PtdSer depletion in a PKC-independent manner (Cho et al., 2012b). These compounds also induce K-Ras–PM dissociation and disrupt K-Ras PM nanoclustering (Cho et al., 2012b). Consistent with this, they abrogate K-Ras signaling and cell proliferation in K-Ras–transformed cells. Taken all together, perturbing recycling endosomal activity could prevent PM PtdSer replenishment through the RE, which results in K-Ras–PM dissociation and disrupted K-Ras nanoclustering and K-Ras signaling. The perturbed recycling endosomal activity could also block the PDE6δ/RE-mediated K-Ras–PM localization, further contributing to disrupted K-Ras–PM localization and signaling.
K-Ras and PtdSer PM Localization Is Regulated by Sphingomyelin/Ceramide Biosynthesis
Recent studies have demonstrated that perturbing the enzymes involved in sphingomyelin (SM) metabolism depletes the PM localization of PtdSer and K-Ras, and blocks oncogenic K-Ras signaling. Ceramide, which is synthesized in the ER, trafficks to the Golgi complex, where it is converted to SM. SM is further transported to the PM and lysosomes, where it is reverted to ceramide by sphingomyelinases (Gault et al., 2010). Several studies have reported that the inhibition of acid or neutral sphingomyelinase (ASM and NSM, respectively) dissociates PtdSer and K-Ras from the PM and inhibits oncogenic K-Ras signal transduction (Figure 1). A wide range of ASM inhibitors including tricyclic antidepressants elevates cellular SM contents and accumulates SM in vesicular structures. They also deplete PM PtdSer content and translocate K-Ras, but not H-Ras, from the PM to endomembranes (van der Hoeven et al., 2013; Cho et al., 2016; van der Hoeven et al., 2018). Also, K-Ras is dissociated from the PM in patient-derived Niemann–Pick type A and B cell lines, in which SMPD1 gene–encoding ASM has inactivating and partial loss-of-function mutations, respectively (Cho et al., 2016; Schuchman and Desnick, 2017). These inhibitors further perturb oncogenic K-Ras PM nanoclustering and its signaling, and abrogate the growth of different types of human cancer cells expressing oncogenic mutant K-Ras but not WT K-Ras (Petersen et al., 2013; van der Hoeven et al., 2013; van der Hoeven et al., 2018). Supplementing ASM-inhibited cells with recombinant ASM returns PtdSer and K-Ras to the PM. Also, replenishing PM PtdSer content with exogenous PtdSer supplementation returns K-Ras to the PM and restores nanoclustering in ASM-inhibited cells, which indicates that K-Ras–PM dissociation occurs through PM PtdSer depletion (Cho et al., 2016). In addition, pharmacological inhibitors for enzymes in the SM/ceramide metabolic pathway redistribute PtdSer and K-Ras from the PM (van der Hoeven et al., 2018). They further perturb K-Ras nanoclustering and block the growth of pancreatic cancer cells harboring oncogenic mutant K-Ras (van der Hoeven et al., 2018). In a supplemental C. elegans study, RNAi-mediated knockdown of 14 genes encoding enzymes in the SM/ceramide biosynthesis pathway suppressed the LET-60G13D (a K-RasG13D ortholog in C. elegans)-induced multi-vulva phenotype (van der Hoeven et al., 2018).
Another approach to disrupt SM/ceramide metabolism is to alter the activity of NSM. Avicins, natural plant-derived triterpenoid saponins from Acacia victoriae, have proapoptotic, anti-inflammatory, and anticancer activities (Wang et al., 2010). A recent study demonstrated that avicin G, an isomer of avicin compounds, inhibits NSM and ASM, with a greater potency against NSM, and elevates cellular SM, ceramide, and PtdSer contents (Garrido et al., 2020). It also disrupts endosomal recycling of the EGFR and perturbs lysosomal activity by elevating the lysosomal pH (Garrido et al., 2020). Avicin G and other NSM inhibitors redistribute PtdSer from the PM, accumulate K-Ras in lysosomes, and increase the K-Ras protein level. Since K-Ras and PtdSer are proposed to be degraded in the lysosome (Lu et al., 2009; Leventis and Grinstein, 2010), the elevated K-Ras and PtdSer levels induced by avicin G, in part, account for the perturbed lysosomal activity (Garrido et al., 2020). It further perturbs K-Ras PM nanoclustering and blocks K-Ras signaling and the growth of K-Ras–addicted pancreatic and NSCLC cell lines (Garrido et al., 2020). Taken together, these studies propose that a correct SM/ceramide balance maintains the PM localization of PtdSer and K-Ras and that pharmacological agents that perturb the sphingolipid pathways could be a new strategy for developing anti–K-Ras therapies (van der Hoeven et al., 2018). One plausible mechanism of PM PtdSer depletion by altering the cellular SM contents is through perturbing recycling endosomal activity. The RE is enriched with cholesterol, SM, and PtdSer (Gagescu et al., 2000; Uchida et al., 2011), and elevating cellular sphingolipid contents blocks endosomal recycling of the glucose transporter 1 and TfR (Finicle et al., 2018). Like avicin G, staurosporine and its analogs perturb the RE activity and elevate cellular SM content in a PKC-independent manner by reducing the protein level of ORMDL, which negatively regulates serine-palmitoyltransferase, the rate-limiting enzyme for sphingolipid biosynthesis (Maekawa et al., 2016). Taken all together, it is proposed that an increased cellular SM level changes SM content at the RE, which disrupts recycling endosomal activity. This, in turn, depletes PtdSer and mislocalizes K-Ras from the PM, as discussed above.
Conclusion
Despite the essential role of oncogenic mutant K-Ras in the growth and survival of pancreatic, lung, and colorectal cancers, there are no anti–K-Ras therapies available in the clinic. Several studies have reported that knockdown of endogenous oncogenic mutant K-Ras in a range of NCSLC and pancreatic cancer cell lines blocks their growth and survival, suggesting that blocking oncogenic K-Ras activity is a valid strategy for anti–K-Ras therapies. Ras drug discovery efforts have focused largely on inhibitors of Ras downstream effectors including B-Raf, C-Raf, PI3K, and MEK (Baines et al., 2011). One example is the multikinase inhibitor, Nexavar, used against renal cell and hepatocellular carcinoma (Llovet et al., 2008; Roberts, 2008), although it is unclear to what extent the efficacy of Nexavar towards these cancers is related to the inhibition of C-Raf, B-Raf, or VEGFR (Downward, 2003; Baines et al., 2011). B-Raf–specific inhibitors produce excellent, albeit often short-lived, responses in patients with B-Raf mutant melanoma (Flaherty et al., 2010). However, further studies have shown that B-Raf–specific inhibitors paradoxically activate the MAPK cascade in melanoma cells expressing oncogenic mutant N- or K-Ras via a mechanism that involves C-Raf hyperactivation (Heidorn et al., 2010; Cho et al., 2012a). These studies illustrate that blocking MAPK signaling with Raf kinase inhibitors is a limited approach to anti-Ras therapy.
Recently, two small molecules that directly bind and inhibit the K-RasG12C mutant have shown promising outcomes in clinical trials. While the K-RasG12C mutant is found in a small fraction of K-Ras–driven human cancers, these studies demonstrate that developing anti–K-Ras therapies is feasible. One approach to inhibit all oncogenic mutant K-Ras is to block its interaction with the PM since K-Ras must localize to the PM for its signal transduction. However, the exact molecular mechanisms of K-Ras transport to and maintenance at the PM are not fully elucidated. In this review, we discussed several recently identified mechanisms that regulate K-Ras–PM interaction and thereby the K-Ras signal cascade. Compounds that perturb these mechanisms dissociate K-Ras from the PM and block K-Ras signaling and K-Ras–dependent cancer cell growth. However, this approach has pitfalls including nonspecificity and cytotoxicity since it does not specifically target K-Ras. For example, PDE6δ can bind other farnesylated small GTPases via the same hydrophobic pocket as K-Ras. Thus, blocking this binding site by PDE6δ inhibitors can dysregulate the cellular localizations and activities of K-Ras and other small GTPases. Also, PtdSer at the inner PM leaflet recruits and promotes the activity of K-Ras and other proteins containing a polybasic domain (Leventis and Grinstein, 2010; Kay and Fairn, 2019). While PM PI4P regulates the PM enrichments of PtdSer, it can be further phosphorylated to different PIPs, which activate several essential signaling proteins (Balla, 2013). Therefore, while depleting PM PtdSer or perturbing the PI4P/PtdSer exchange mechanism prevents oncogenic mutant K-Ras activity, they can also perturb other essential signaling cascades. Nevertheless, many studies have reported that disrupting these molecular mechanisms blocks the growth of human cancer cells that are K-Ras–dependent but not K-Ras–independent in vitro and in vivo, suggesting that targeting these mechanisms is a valid approach for developing anti–K-Ras therapies.
Cancer chemotherapy is most effective when a combination of drugs targeting different molecular mechanisms are applied. There are four major approaches that are currently being perused for developing anti–K-Ras therapies, and any one approach alone may not be sufficient to completely block oncogenic K-Ras signaling due to high cytotoxicity and/or nonspecificity. A recent study has demonstrated that a K-RasG12C inhibitor potentiates the anticancer effect of the MEK, mTOR, and insulin-like growth factor 1 receptor (IFG1R) inhibitors in NSCLC cells. While combined mTOR, IGF1R, and MEK inhibition shows significant tumor regression in K-RasG12C–driven lung cancer mouse models, replacing the MEK inhibitor with a K-RasG12C inhibitor in combination demonstrates greater efficacy, specificity, and tolerability (Molina-Arcas et al., 2019). Moreover, the combination of the K-RasG12C inhibitor with anti–PD-1 immune checkpoint inhibition synergistically suppresses tumor growth in K-RasG12C–driven mouse models (Canon et al., 2019). Combination therapy of K-RasG12C inhibitors with anti–PD-1 or anti–PD-L1 in patients with solid tumors harboring the K-RasG12C mutant is currently in clinical trials (ClinicalTrials.gov identifier: NCT04185883, NCT03785249). Although combination therapy with K-RasG12C inhibitors and other anticancer approaches is promising, it is limited to K-RasG12C–specific cancers, which accounts for ∼20% of K-Ras–driven cancers. Therefore, it would be worthwhile to examine the effects of combining pharmacological agents that can block all oncogenic mutant K-Ras by dissociating it from the PM with drugs developed for targeting the other approaches.
Author Contributions
KH and KR wrote the draft, and KH, KR, and K-jC edited it.
Funding
This work was supported by grants from the National Cancer Institute (Grant no. R00CA188593 to K-jC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Apolloni, A., Prior, I. A., Lindsay, M., Parton, R. G., and Hancock, J. F. (2000). H-ras but Not K-Ras Traffics to the Plasma Membrane through the Exocytic Pathway. Mol. Cel. Biol. 20 (7), 2475–2487. doi:10.1128/mcb.20.7.2475-2487.2000
Baines, A. T., Xu, D., and Der, C. J. (2011). Inhibition of Ras for Cancer Treatment: the Search Continues. Future Med. Chem. 3 (14), 1787–1808. doi:10.4155/fmc.11.121
Balla, T. (2013). Phosphoinositides: Tiny Lipids with Giant Impact on Cell Regulation. Physiol. Rev. 93 (3), 1019–1137. doi:10.1152/physrev.00028.2012
Barbacid, M. (1987). Ras Genes. Annu. Rev. Biochem. 56, 779–827. doi:10.1146/annurev.bi.56.070187.004023
Canon, J., Rex, K., Saiki, A. Y., Mohr, C., Cooke, K., Bagal, D., et al. (2019). The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-tumour Immunity. Nature. 575 (7781), 217–223. doi:10.1038/s41586-019-1694-1
Chandra, A., Grecco, H. E., Pisupati, V., Perera, D., Cassidy, L., Skoulidis, F., et al. (2012). The GDI-like Solubilizing Factor PDEδ Sustains the Spatial Organization and Signalling of Ras Family Proteins. Nat. Cel Biol. 14 (2), 148–158. doi:10.1038/ncb2394
Cho, K. J., and Hancock, J. F. (2013). Ras Nanoclusters: a New Drug Target? Small GTPases. 4 (1), 57–60. doi:10.4161/sgtp.23145
Cho, K. J., Kasai, R. S., Park, J. H., Chigurupati, S., Heidorn, S. J., van der Hoeven, D., et al. (2012). Raf Inhibitors Target Ras Spatiotemporal Dynamics. Curr. Biol. 22 (11), 945–955. doi:10.1016/j.cub.2012.03.067
Cho, K. J., Park, J. H., Piggott, A. M., Salim, A. A., Gorfe, A. A., Parton, R. G., et al. (2012). Staurosporines Disrupt Phosphatidylserine Trafficking and Mislocalize Ras Proteins. J. Biol. Chem. 287 (52), 43573–43584. doi:10.1074/jbc.M112.424457
Cho, K. J., van der Hoeven, D., Zhou, Y., Maekawa, M., Ma, X., Chen, W., et al. (2016). Inhibition of Acid Sphingomyelinase Depletes Cellular Phosphatidylserine and Mislocalizes K-Ras from the Plasma Membrane. Mol. Cel Biol. 36 (2), 363–374. doi:10.1128/MCB.00719-15
Chung, J., Torta, F., Masai, K., Lucast, L., Czapla, H., Tanner, L. B., et al. (2015). INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine Countertransport at ORP5- and ORP8-Mediated ER-Plasma Membrane Contacts. Science. 349 (6246), 428–432. doi:10.1126/science.aab1370
Cohen, S. J., Ho, L., Ranganathan, S., Abbruzzese, J. L., Alpaugh, R. K., Beard, M., et al. (2003). Phase II and Pharmacodynamic Study of the Farnesyltransferase Inhibitor R115777 as Initial Therapy in Patients with Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 21 (7), 1301–1306. doi:10.1200/JCO.2003.08.040
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J., and Der, C. J. (2014). Drugging the Undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 13 (11), 828–851. doi:10.1038/nrd4389
Downward, J. (2003). Targeting RAS Signalling Pathways in Cancer Therapy. Nat. Rev. Cancer 3 (1), 11–22. doi:10.1038/nrc969
Dumbacher, M., Van Dooren, T., Princen, K., De Witte, K., Farinelli, M., Lievens, S., et al. (2018). Modifying Rap1-Signalling by Targeting Pde6delta Is Neuroprotective in Models of Alzheimer's Disease. Mol. Neurodegener. 13 (1), 50. doi:10.1186/s13024-018-0283-3
Esteban, L. M., Vicario-Abejón, C., Fernández-Salguero, P., Fernández-Medarde, A., Swaminathan, N., Yienger, K., et al. (2001). Targeted Genomic Disruption of H-Ras and N-Ras, Individually or in Combination, Reveals the Dispensability of Both Loci for Mouse Growth and Development. Mol. Cel. Biol. 21 (5), 1444–1452. doi:10.1128/mcb.21.5.1444-1452.2001
Finicle, B. T., Ramirez, M. U., Liu, G., Selwan, E. M., McCracken, A. N., Yu, J., et al. (2018). Sphingolipids Inhibit Endosomal Recycling of Nutrient Transporters by Inactivating ARF6. J. Cel Sci. 131 (12). doi:10.1242/jcs.213314
Flaherty, K. T., Puzanov, I., Kim, K. B., Ribas, A., McArthur, G. A., Sosman, J. A., et al. (2010). Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 363 (9), 809–819. doi:10.1056/NEJMoa1002011
Fournier, M. V., Guimaraes da Costa, F., Paschoal, M. E., Ronco, L. V., Carvalho, M. G., and Pardee, A. B. (1999). Identification of a Gene Encoding a Human Oxysterol-Binding Protein-Homologue: a Potential General Molecular Marker for Blood Dissemination of Solid Tumors. Cancer Res. 59 (15), 3748–3753.
Gagescu, R., Demaurex, N., Parton, R. G., Hunziker, W., Huber, L. A., and Gruenberg, J. (2000). The Recycling Endosome of Madin-Darby Canine Kidney Cells Is a Mildly Acidic Compartment Rich in Raft Components. Mol. Biol. Cel. 11 (8), 2775–2791. doi:10.1091/mbc.11.8.2775
Garrido, C. M., Henkels, K. M., Rehl, K. M., Liang, H., Zhou, Y., Gutterman, J. U., et al. (2020). Avicin G Is a Potent Sphingomyelinase Inhibitor and Blocks Oncogenic K- and H-Ras Signaling. Sci. Rep. 10 (1), 9120. doi:10.1038/s41598-020-65882-5
Gault, C. R., Obeid, L. M., and Hannun, Y. A. (2010). An Overview of Sphingolipid Metabolism: from Synthesis to Breakdown. Adv. Exp. Med. Biol. 688, 1–23. doi:10.1007/978-1-4419-6741-1_1
Ghai, R., Du, X., Wang, H., Dong, J., Ferguson, C., Brown, A. J., et al. (2017). ORP5 and ORP8 Bind Phosphatidylinositol-4, 5-biphosphate (PtdIns(4,5)P 2) and Regulate its Level at the Plasma Membrane. Nat. Commun. 8 (1), 757. doi:10.1038/s41467-017-00861-5
Gulyas, G., Radvanszki, G., Matuska, R., Balla, A., Hunyady, L., Balla, T., et al. (2017). Plasma Membrane Phosphatidylinositol 4-phosphate and 4,5-bisphosphate Determine the Distribution and Function of K-Ras4B but Not H-Ras Proteins. J. Biol. Chem. 292 (46), 18862–18877. doi:10.1074/jbc.M117.806679
Hallin, J., Engstrom, L. D., Hargis, L., Calinisan, A., Aranda, R., Briere, D. M., et al. (2020). The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 10 (1), 54–71. doi:10.1158/2159-8290.CD-19-1167
Hancock, J. F., Magee, A. I., Childs, J. E., and Marshall, C. J. (1989). All Ras Proteins Are Polyisoprenylated but Only Some Are Palmitoylated. Cell. 57 (7), 1167–1177. doi:10.1016/0092-8674(89)90054-8
Hancock, J. F., Paterson, H., and Marshall, C. J. (1990). A Polybasic Domain or Palmitoylation Is Required in Addition to the CAAX Motif to Localize P21ras to the Plasma Membrane. Cell. 63 (1), 133–139. doi:10.1016/0092-8674(90)90294-o
Hancock, J. F. (2003). Ras Proteins: Different Signals from Different Locations. Nat. Rev. Mol. Cel Biol. 4 (5), 373–385. doi:10.1038/nrm1105
Hayes, T. K., Neel, N. F., Hu, C., Gautam, P., Chenard, M., Long, B., et al. (2016). Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell. 29 (1), 75–89. doi:10.1016/j.ccell.2015.11.011
Heidorn, S. J., Milagre, C., Whittaker, S., Nourry, A., Niculescu-Duvas, I., Dhomen, N., et al. (2010). Kinase-dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell. 140 (2), 209–221. doi:10.1016/j.cell.2009.12.040
Ismail, S. A., Chen, Y.-X., Rusinova, A., Chandra, A., Bierbaum, M., Gremer, L., et al. (2011). Arl2-GTP and Arl3-GTP Regulate a GDI-like Transport System for Farnesylated Cargo. Nat. Chem. Biol. 7 (12), 942–949. doi:10.1038/nchembio.686
Johnson, L., Greenbaum, D., Cichowski, K., Mercer, K., Murphy, E., Schmitt, E., et al. (1997). K-ras Is an Essential Gene in the Mouse with Partial Functional Overlap with N-Ras. Genes Development. 11 (19), 2468–2481. doi:10.1101/gad.11.19.2468
Kattan, W. E., Chen, W., Ma, X., Lan, T. H., van der Hoeven, D., van der Hoeven, R., et al. (2019). Targeting Plasma Membrane Phosphatidylserine Content to Inhibit Oncogenic KRAS Function. Life Sci. Alliance. 2 (5), e201900431. doi:10.26508/lsa.201900431
Kay, J. G., and Fairn, G. D. (2019). Distribution, Dynamics and Functional Roles of Phosphatidylserine within the Cell. Cell Commun Signal. 17 (1), 126. doi:10.1186/s12964-019-0438-z
Kim, E., Ambroziak, P., Otto, J. C., Taylor, B., Ashby, M., Shannon, K., et al. (1999). Disruption of the Mouse Rce1 Gene Results in Defective Ras Processing and Mislocalization of Ras within Cells. J. Biol. Chem. 274 (13), 8383–8390. doi:10.1074/jbc.274.13.8383
Klein, C. H., Truxius, D. C., Vogel, H. A., Harizanova, J., Murarka, S., Martin-Gago, P., et al. (2019). PDEdelta Inhibition Impedes the Proliferation and Survival of Human Colorectal Cancer Cell Lines Harboring Oncogenic KRas. Int. J. Cancer. 144 (4), 767–776. doi:10.1002/ijc.31859
Koera, K., Nakamura, K., Nakao, K., Miyoshi, J., Toyoshima, K., Hatta, T., et al. (1997). K-ras Is Essential for the Development of the Mouse Embryo. Oncogene. 15 (10), 1151–1159. doi:10.1038/sj.onc.1201284
Koga, Y., Ishikawa, S., Nakamura, T., Masuda, T., Nagai, Y., Takamori, H., et al. (2008). Oxysterol Binding Protein-Related Protein-5 Is Related to Invasion and Poor Prognosis in Pancreatic Cancer. Cancer Sci. 99 (12), 2387–2394. doi:10.1111/j.1349-7006.2008.00987.x
Lau, H. Y., Ramanujulu, P. M., Guo, D., Yang, T., Wirawan, M., Casey, P. J., et al. (2014). An Improved Isoprenylcysteine Carboxylmethyltransferase Inhibitor Induces Cancer Cell Death and Attenuates Tumor Growth In Vivo. Cancer Biol. Ther. 15 (9), 1280–1291. doi:10.4161/cbt.29692
Leung, E. L. H., Luo, L. X., Liu, Z. Q., Wong, V. K. W., Lu, L. L., Xie, Y., et al. (2018). Inhibition of KRAS-dependent Lung Cancer Cell Growth by Deltarasin: Blockage of Autophagy Increases its Cytotoxicity. Cell Death Dis. 9 (2), 216. doi:10.1038/s41419-017-0065-9
Leventis, P. A., and Grinstein, S. (2010). The Distribution and Function of Phosphatidylserine in Cellular Membranes. Annu. Rev. Biophys. 39, 407–427. doi:10.1146/annurev.biophys.093008.131234
Llovet, J. M., Ricci, S., Mazzaferro, V., Hilgard, P., Gane, E., Blanc, J. F., et al. (2008). Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 359 (4), 378–390. doi:10.1056/NEJMoa0708857
Loilome, W., Yongvanit, P., Wongkham, C., Tepsiri, N., Sripa, B., Sithithaworn, P., et al. (2006). Altered Gene Expression in Opisthorchis Viverrini-Associated Cholangiocarcinoma in Hamster Model. Mol. Carcinog. 45 (5), 279–287. doi:10.1002/mc.20094
Lu, A., Tebar, F., Alvarez-Moya, B., Lopez-Alcala, C., Calvo, M., Enrich, C., et al. (2009). A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J. Cel Biol. 184 (6), 863–879. doi:10.1083/jcb.200807186
Macdonald, J. S., McCoy, S., Whitehead, R. P., Iqbal, S., Wade, J. L., Giguere, J. K., et al. (2005). A Phase II Study of Farnesyl Transferase Inhibitor R115777 in Pancreatic Cancer: a Southwest Oncology Group (SWOG 9924) Study. Invest. New Drugs. 23 (5), 485–487. doi:10.1007/s10637-005-2908-y
Maekawa, M., Lee, M., Wei, K., Ridgway, N. D., and Fairn, G. D. (2016). Staurosporines Decrease ORMDL Proteins and Enhance Sphingomyelin Synthesis Resulting in Depletion of Plasmalemmal Phosphatidylserine. Sci. Rep. 6, 35762. doi:10.1038/srep35762
Martin-Gago, P., Fansa, E. K., Klein, C. H., Murarka, S., Janning, P., Schurmann, M., et al. (2017). A PDE6delta-KRas Inhibitor Chemotype with up to Seven H-Bonds and Picomolar Affinity that Prevents Efficient Inhibitor Release by Arl2. Angew. Chem. Int. Ed. Engl. 56 (9), 2423–2428. doi:10.1002/anie.201610957
Miller, T. E., Henkels, K. M., Huddleston, M., Salisbury, R., Hussain, S. M., Sasaki, A. T., et al. (2019). Depletion of Phosphatidylinositol 4-phosphate at the Golgi Translocates K-Ras to Mitochondria. J. Cel Sci. 132 (16). doi:10.1242/jcs.231886
Molina-Arcas, M., Moore, C., Rana, S., van Maldegem, F., Mugarza, E., Romero-Clavijo, P., et al. (2019). Development of Combination Therapies to Maximize the Impact of KRAS-G12c Inhibitors in Lung Cancer. Sci. Transl Med. 11 (510). doi:10.1126/scitranslmed.aaw7999
Moser von Filseck, J., Copic, A., Delfosse, V., Vanni, S., Jackson, C. L., Bourguet, W., et al. (2015). INTRACELLULAR TRANSPORT. Phosphatidylserine Transport by ORP/Osh Proteins Is Driven by Phosphatidylinositol 4-phosphate. Science. 349 (6246), 432–436. doi:10.1126/science.aab1346
Novotny, C. J., Hamilton, G. L., McCormick, F., and Shokat, K. M. (2017). Farnesyltransferase-Mediated Delivery of a Covalent Inhibitor Overcomes Alternative Prenylation to Mislocalize K-Ras. ACS Chem. Biol. 12 (7), 1956–1962. doi:10.1021/acschembio.7b00374
O'Bryan, J. P. (2019). Pharmacological Targeting of RAS: Recent success with Direct Inhibitors. Pharmacol. Res. 139, 503–511. doi:10.1016/j.phrs.2018.10.021
Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A., and Shokat, K. M. (2013). K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature. 503 (7477), 548–551. doi:10.1038/nature12796
Papke, B., Murarka, S., Vogel, H. A., Martin-Gago, P., Kovacevic, M., Truxius, D. C., et al. (2016). Identification of Pyrazolopyridazinones as PDEdelta Inhibitors. Nat. Commun. 7, 11360. doi:10.1038/ncomms11360
Pells, S., Divjak, M., Romanowski, P., Impey, H., Hawkins, N. J., Clarke, A. R., et al. (1997). Developmentally-regulated Expression of Murine K-Ras Isoforms. Oncogene. 15 (15), 1781–1786. doi:10.1038/sj.onc.1201354
Percy, A. K., Moore, J. F., Carson, M. A., and Waechter, C. J. (1983). Characterization of Brain Phosphatidylserine Decarboxylase: Localization in the Mitochondrial Inner Membrane. Arch. Biochem. Biophys. 223 (2), 484–494. doi:10.1016/0003-9861(83)90613-6
Petersen, N. H., Olsen, O. D., Groth-Pedersen, L., Ellegaard, A. M., Bilgin, M., Redmer, S., et al. (2013). Transformation-associated Changes in Sphingolipid Metabolism Sensitize Cells to Lysosomal Cell Death Induced by Inhibitors of Acid Sphingomyelinase. Cancer Cell. 24 (3), 379–393. doi:10.1016/j.ccr.2013.08.003
Potenza, N., Vecchione, C., Notte, A., De Rienzo, A., Rosica, A., Bauer, L., et al. (2005). Replacement of K‐Ras with H‐Ras Supports normal Embryonic Development Despite Inducing Cardiovascular Pathology in Adult Mice. EMBO Rep. 6 (5), 432–437. doi:10.1038/sj.embor.7400397
Prior, I. A., Hood, F. E., and Hartley, J. L. (2020). The Frequency of Ras Mutations in Cancer. Cancer Res. 80, 2969–2974. doi:10.1158/0008-5472.CAN-19-3682
Prior, I. A., Muncke, C., Parton, R. G., and Hancock, J. F. (2003). Direct Visualization of Ras Proteins in Spatially Distinct Cell Surface Microdomains. J. Cel Biol. 160 (2), 165–170. doi:10.1083/jcb.200209091
Roberts, L. R. (2008). Sorafenib in Liver Cancer-Jjust the Beginning. N. Engl. J. Med. 359 (4), 420–422. doi:10.1056/NEJMe0802241
Rocks, O., Gerauer, M., Vartak, N., Koch, S., Huang, Z.-P., Pechlivanis, M., et al. (2010). The Palmitoylation Machinery Is a Spatially Organizing System for Peripheral Membrane Proteins. Cell. 141 (3), 458–471. doi:10.1016/j.cell.2010.04.007
Rocks, O., Peyker, A., Kahms, M., Verveer, P. J., Koerner, C., Lumbierres, M., et al. (2005). An Acylation Cycle Regulates Localization and Activity of Palmitoylated Ras Isoforms. Science. 307 (5716), 1746–1752. doi:10.1126/science.1105654
Schmick, M., Vartak, N., Papke, B., Kovacevic, M., Truxius, D. C., Rossmannek, L., et al. (2014). KRas Localizes to the Plasma Membrane by Spatial Cycles of Solubilization, Trapping and Vesicular Transport. Cell. 157 (2), 459–471. doi:10.1016/j.cell.2014.02.051
Schuchman, E. H., and Desnick, R. J. (2017). Types A and B Niemann-Pick Disease. Mol. Genet. Metab. 120 (1-2), 27–33. doi:10.1016/j.ymgme.2016.12.008
Shimizu, K., Kiuchi, Y., Ando, K., Hayakawa, M., and Kikugawa, K. (2004). Coordination of Oxidized Protein Hydrolase and the Proteasome in the Clearance of Cytotoxic Denatured Proteins. Biochem. Biophys. Res. Commun. 324 (1), 140–146. doi:10.1016/j.bbrc.2004.08.231
Singh, A., Greninger, P., Rhodes, D., Koopman, L., Violette, S., Bardeesy, N., et al. (2009). A Gene Expression Signature Associated with "K-Ras Addiction" Reveals Regulators of EMT and Tumor Cell Survival. Cancer Cell. 15 (6), 489–500. doi:10.1016/j.ccr.2009.03.022
Sohn, M., Korzeniowski, M., Zewe, J. P., Wills, R. C., Hammond, G. R. V., Humpolickova, J., et al. (2018). PI(4,5)P2 Controls Plasma Membrane PI4P and PS Levels via ORP5/8 Recruitment to ER-PM Contact Sites. J. Cel Biol. 217 (5), 1797–1813. doi:10.1083/jcb.201710095
Tan, L., Cho, K. J., Kattan, W. E., Garrido, C. M., Zhou, Y., Neupane, P., et al. (2019). Acylpeptide Hydrolase Is a Novel Regulator of KRAS Plasma Membrane Localization and Function. J. Cel Sci. 132 (15). doi:10.1242/jcs.232132
Tian, T., Harding, A., Inder, K., Plowman, S., Parton, R. G., and Hancock, J. F. (2007). Plasma Membrane Nanoswitches Generate High-Fidelity Ras Signal Transduction. Nat. Cel Biol. 9 (8), 905–914. doi:10.1038/ncb1615
Uchida, Y., Hasegawa, J., Chinnapen, D., Inoue, T., Okazaki, S., Kato, R., et al. (2011). Intracellular Phosphatidylserine Is Essential for Retrograde Membrane Traffic through Endosomes. Proc. Natl. Acad. Sci. U S A. 108 (38), 15846–15851. doi:10.1073/pnas.1109101108
Van Cutsem, E., van de Velde, H., Karasek, P., Oettle, H., Vervenne, W. L., Szawlowski, A., et al. (2004). Phase III Trial of Gemcitabine Plus Tipifarnib Compared with Gemcitabine Plus Placebo in Advanced Pancreatic Cancer. J. Clin. Oncol. 22 (8), 1430–1438. doi:10.1200/JCO.2004.10.112
van der Hoeven, D., Cho, K. J., Ma, X., Chigurupati, S., Parton, R. G., and Hancock, J. F. (2013). Fendiline Inhibits K-Ras Plasma Membrane Localization and Blocks K-Ras Signal Transmission. Mol. Cel Biol. 33 (2), 237–251. doi:10.1128/MCB.00884-12
van der Hoeven, D., Cho, K. J., Zhou, Y., Ma, X., Chen, W., Naji, A., et al. (2018). Sphingomyelin Metabolism Is a Regulator of K-Ras Function. Mol. Cel Biol 38 (3), e00373. doi:10.1128/MCB.00373-17
Wang, H., Haridas, V., Gutterman, J. U., and Xu, Z. X. (2010). Natural Triterpenoid Avicins Selectively Induce Tumor Cell Death. Commun. Integr. Biol. 3 (3), 205–208. doi:10.4161/cib.3.3.11492
Weinstein, I. B., and Joe, A. (2008). Oncogene Addiction. Cancer Res. 68 (9), 3077–3080. doi:10.1158/0008-5472.CAN-07-3293
Willumsen, B. M., Christensen, A., Hubbert, N. L., Papageorge, A. G., and Lowy, D. R. (1984). The P21 Ras C-Terminus Is Required for Transformation and Membrane Association. Nature. 310 (5978), 583–586. doi:10.1038/310583a0
Yeung, T., Gilbert, G. E., Shi, J., Silvius, J., Kapus, A., and Grinstein, S. (2008). Membrane Phosphatidylserine Regulates Surface Charge and Protein Localization. Science. 319 (5860), 210–213. doi:10.1126/science.1152066
Zhang, H., Li, S., Doan, T., Rieke, F., Detwiler, P. B., Frederick, J. M., et al. (2007). Deletion of PrBP/delta Impedes Transport of GRK1 and PDE6 Catalytic Subunits to Photoreceptor Outer Segments. Proc. Natl. Acad. Sci. U S A. 104 (21), 8857–8862. doi:10.1073/pnas.0701681104
Zhang, X., Yu, L., and Xu, H. (2016). Lysosome Calcium in ROS Regulation of Autophagy. Autophagy. 12 (10), 1954–1955. doi:10.1080/15548627.2016.1212787
Zhou, Y., Prakash, P., Liang, H., Cho, K.-J., Gorfe, A. A., and Hancock, J. F. (2017). Lipid-Sorting Specificity Encoded in K-Ras Membrane Anchor Regulates Signal Output. Cell. 168 (1-2), 239–251 e16. doi:10.1016/j.cell.2016.11.059
Keywords: K-Ras, plasma membrane, mislocalization, cancer, recycing endosome, phosphatidylinositol, phosphatidylserine, sphingomyelin
Citation: Henkels KM, Rehl KM and Cho K-j (2021) Blocking K-Ras Interaction With the Plasma Membrane Is a Tractable Therapeutic Approach to Inhibit Oncogenic K-Ras Activity. Front. Mol. Biosci. 8:673096. doi: 10.3389/fmolb.2021.673096
Received: 26 February 2021; Accepted: 20 May 2021;
Published: 16 June 2021.
Edited by:
Anton A. Buzdin, I. M. Sechenov First Moscow State Medical University, RussiaReviewed by:
Sanjay Mishra, The Ohio State University, United StatesTimofey Dmitrievich Lebedev, Engelhardt Institute of Molecular Biology (RAS), Russia
Copyright © 2021 Henkels, Rehl and Cho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kwang-jin Cho, a3dhbmctamluLmNob0B3cmlnaHQuZWR1