 Ye-Chen Han1
Ye-Chen Han1 Hong-Zhi Xie
Hong-Zhi Xie Ruo-Lan Xiang
Ruo-Lan Xiang Shu-Yang Zhang
Shu-Yang Zhang- 1Department of Cardiology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 2Department of Physiology and Pathophysiology, Peking University School of Basic Medical Sciences, Beijing, China
- 3Peking University Fifth School of Clinical Medicine (Beijing Hospital), Beijing, China
N6-methyladenosine (m6A) modification plays important roles in the pathology of a variety of diseases. However, the roles of m6A modification in sepsis-induced myocardial dysfunction are not well defined. Rats were divided into control and lipopolysaccharide (LPS)-induced sepsis group. Global m6A levels of left ventricle tissue were measured by LC-MS/MS, and transcriptome-wide m6A modifications were profiled using epitranscriptomic microarrays (mRNAs and lncRNAs). Bioinformatics analysis was conducted to understand the functional implications of m6A modifications during sepsis. Methylated lncRNAs and mRNAs were measured by m6A single-base site qPCR. The global m6A levels in left ventricle tissue were significantly decreased in the LPS group. While 27 transcripts (23 mRNAs and four lncRNAs) were hypermethylated, 46 transcripts (39 mRNAs and 7 lncRNAs) were hypomethylated in the LPS group. The mRNA expression of writers and readers was significantly decreased in the LPS group. The m6A modification of Clec1b, Stk38l and Tnfrsf26 was associated with platelet activation and apoptotic pathways. Moreover, the decrease in m6A modification of lncRNA XR_346,771 may be related to cation import in cardiac tissue. Our data provide novel information regarding changes to m6A modifications in cardiac tissue during sepsis, and m6A modifications might be promising therapeutic targets.
Introduction
Sepsis is a life-threatening condition involving organ dysfunction that is caused by a dysregulated host response to infection (Cohen et al., 2015), It has been reported that approximately 50% of patients with septic shock are diagnosed with septic cardiomyopathy, which causes myocardial dysfunction, and these patients tend to have poor prognoses (Zaky et al., 2014). Myocardial dysfunction is an important factor that contributes to the high mortality rate of sepsis. Therefore, it is necessary to elucidate the pathogenesis of myocardial dysfunction in order to develop new treatment strategies to reduce the mortality rate of sepsis.
To date, a series of reversible posttranscriptional modifications located in RNAs have gained increasing attention. More than 140 chemical modifications have been discovered in RNAs. The modifications range from simple methylation or isomerisation, such as N6-methyladenosine (m6A), 5-methylcytosine (m5C), N1-methyladenosine (m1A), pseudouridine (Ψ), 5-methyluridine (m5U),1-methylguanosine, and 7-methylguanosine (m1G and m7G, respectively) and inosine (I), to complex multistep chemical modifications, such as N6-threonylcarbamoyladenosine and 5-methoxycarbonyl-methyl-2-thiouridine (mcm5s2U) (Pan, 2018). Among them, the m6A is one of the most abundant and influential modifications in eukaryotes. M6A modification is involved in regulating a variety of posttranscriptional events, including pre-miRNA processing and RNA stability, translation and alternative splicing (Chokkalla et al., 2019). M6A modifications occur via the m6A methyltransferases called “writers”; they are removed by the demethylases called “erasers” and are recognized by m6A-binding proteins called “readers”. The orchestrated interplay among writers, erasers, and readers drives the dynamics and outcomes of the m6A modification of RNAs. An increasing number of studies have reported that m6A modification plays important roles in biological and pathophysiological processes, such as tumorigenesis, embryonic stem cell differentiation, and viral infection (Kennedy et al., 2016). In mouse GC-1 SPG cells, meclofenamic acid inhibits spermatogonial proliferation by affecting CDKs expression through a m6A-dependent mRNA degradation pathway (Huang et al., 2019). In liver cancer, overexpression of METTL3 leads to the degradation of suppressor of cytokine signalling two and promotes tumor growth (Lin et al., 2019).
M6A modification is also associated with the pathophysiological processes of cardiac differentiation and a variety of cardiovascular diseases. Methyltransferase-like 3 (METTL3) is a major factor involved in abnormal m6A modification. Silencing METTL3 enhances the autophagic flux and represses apoptosis in hypoxia/reoxygenation-treated cardiomyocytes (Song et al., 2019). Overexpression METTL3 increased m6A levels in mRNAs isolated from transverse aortic constriction (TAC) mice hearts. TAC inducd pathological hypertrophic cellular growth was attenuated in hearts of METTL3-overexpressing mice, as evidenced by the crosssectional area of myocytes (Kmietczyk et al., 2019). In addition, aging caused METTL3/METTL14 down-regulation in aorta and atria in male animals while the differentiation-induced increased level of METTL16 (Arcidiacono et al., 2020). Prabhu M et al. found that downregulation of fat mass and obesity associated protein (FTO) expression in failing mammalian hearts and hypoxic cardiomyocytes increased the m6A modification of RNAs and reduced the contractile function of cardiomyocytes (Mathiyalagan et al., 2019). FTO also played a vital role in cardiac contractile function during homeostasis and remodeling. FTO overexpression attenuated the ischemia-induced elevation in m6A modification and significantly improved cardiac function of post-myocardial infraction (Mathiyalagan et al., 2019). Berulava et al. reported that differently expressed genes of m6A methylation are involved in heart failure development. Bioinformatics analysis has revealed that the differentially m6A are mainly involved in metabolism and cardiac signaling (Berulava et al., 2020). However, the regulatory role of the m6A modification in cardiac function is still unclear.
The contraction of the ventricle transports blood to the capillaries of the body or lungs, while the contraction of the atria send blood into the ventricle. In addition, the right ventricle mainly supplies blood to the lungs, and the blood in the right ventricle is venous blood. The left ventricle mainly supplies blood to the organs and tissues of the body, and the blood in the left ventricle is arterial blood. Therefore, the function of the left ventricle is very important to maintain organ perfusion. If the left ventricular function declines, it will leads to organ perfusion insufficiency and, eventually, organ failure. In this direction, we focused on the mechanism underlying left ventricle injury in sepsis to find any potential clinical significance for improving organ perfusion. The overall goal of this study was to explore a new layer of epigenetic alterations through the genome-wide screening of the altered m6A-tagged transcript profiles in lipopolysaccharide (LPS)-induced myocardial dysfunction. We successfully mapped m6A transcripts in left ventricle tissue in LPS-induced sepsis. Some potential roles of the m6A modification in the physiological and pathological mechanisms of sepsis were revealed.
Materials and Methods
Animals
Eight-week-old male Wistar rats weighing 250–350 g were used in this study. All the rats used in our study were obtained from Charles River Laboratories (Beijing, China). Water and standard laboratory food were freely available to the animals. The Experimental Animal Welfare Ethics Branch and the Biomedical Ethics Committee of Peking University approved our study protocol (LA 2020343). All the procedures for handling the animals were in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996).
After acclimating for 1 week, 10 Wistar rats were randomly divided into two groups. The rats in the LPS group (n = 5) were intraperitoneally injected with LPS (10 mg/kg, 5 mg LPS dissolved in 1 ml 0.9% saline). The rats in the control group (n = 5) were intraperitoneally injected with 0.9% saline (2 ml/kg). 24 hours after LPS injection, mean blood pressure was measured by the tail-cuff method with a noninvasive blood pressure measurement system. The left ventricle tissue samples were immediately transferred to liquid nitrogen and stored at −80°C for preservation.
RNA Extraction and Quality Control
Total RNA was isolated from the left ventricle tissues of the LPS (n = 5) and control (n = 5) groups using TRIzol Reagent (Invitrogen, United States). The quantity and purity of the total RNA samples were measured by a NanoDrop ND-1000 (ThermoFisher, United States).
Quantification of Global m6A Levels
Liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based mRNA modification detection was performed according to the Aksomics standard protocol. Briefly, 5 μg of total RNA from the heart tissues of the LPS (n = 5) and control (n = 5) groups was used to isolate mRNA using the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB, E7490, United States). The purified mRNA was quantified using the Qubit RNA HS Assay kit (ThermoFisher, United States) and digested to single dephosphorylated nucleosides by an enzyme mixture. Pretreated nucleosides solution was deproteinized using Satorius 10,000-Da MWCO spin filter. LC-MS/MS analysis was performed on an Agilent 6460 QQQ mass spectrometer with an Agilent 1260 HPLC system using Multi reaction monitoring (MRM) detection mode (Agilent, United States. The nucleosides were quantified by using retention time and the nucleoside to base ion mass transitions of 268-136 (A) and 282-150 (m6A). Quantification was performed in comparison with the standard curve obtained from pure nucleoside standards running with the same batch of samples. The m6A level was calculated as the ratio of m6A to A based on the calibrated concentrations.
Quantitative Real-Time PCR
After extraction, the total RNA was reverse-transcribed into cDNA using SuperScriptTM III Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions. Amplification and detection were performed using 2× SYBR Green PCR Master Mix (Arraysta, United States) on a ViiA 7 Real-Time PCR System (Applied Biosystems). The sequences of the gene-specific primers are listed in Supplementary Table S1. GAPDH was used as the control housekeeping gene.
Western Blot Analysis
Protein extraction of left ventricle tissue and the protein concentration was determined by Bradford method (M&C Gene Technology Ltd.). The protein samples were then subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membrane. The membranes were probed with antibodies of interest. The antibodies used were as follows: METTL3, METTL14, Wilms tumor 1-associated protein (WTAP), and YTH N6-methyladenosine RNA binding protein 1 (YTHDF1), YTHDF3 (all1:1,000; Cell Signal Technology), and β-actin (1:4,000; Abcam).
M6A mRNA&lncRNA Epitranscriptomic Microarray
Sample preparation and microarray hybridization were performed based on Arraystar’s standard protocols (Arraystar). Briefly, total RNA from the heart tissues of the LPS (n = 5) and control (n = 5) groups was immunoprecipitated with an anti-m6A antibody (Synaptic Systems, 202003). The modified RNAs immunoprecipitated by the magnetic beads were labelled as “IP”, and the unmodified RNAs in the supernatant were labelled as “Sup”. The “IP” and “Sup” RNAs were labeled with Cy5 and Cy3, respectively, as cRNAs in separate reactions using the Arraystar Super RNA Labeling Kit (ArrayStar). The cRNAs were combined and hybridized onto an Arraystar Rat mRNA&lncRNA Epitranscriptomic Microarray (4 × 44K, Arraystar) that contained 27,770 mRNA and 10,582 lncRNA degenerate probes. The hybridized arrays were scanned in two-color channels by an Agilent Scanner G2505C.
Microarray Data Analysis
Agilent Feature Extraction software (version 11.0.1.1, United States) was used to analyze the acquired array images. Each probe signal was evaluated and flagged as present, absent or marginal in at least 5 out of 10 samples. The raw intensities of the IP (immunoprecipitated, Cy5-labeled) and Sup (supernatant, Cy3-labeled) samples were normalized by the average of log2-scaled spike-in RNA intensities. The differentially m6A-methylated RNAs between the LPS and control groups were identified by filtering with thresholds of fold change >1.5 and statistical significance (p < 0.05).
Bioinformatics Analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed using some package in the R environment for statistical computing and graphics. The differentially m6A-methylated mRNAs, as well as the downstream mRNAs predicted by the competing endogenous RNA (ceRNA) network, were classified into different GO terms and enriched in certain biological pathways. Protein-protein interaction (PPI) analysis was performed using the STRING database (https://string-db.org). The SRAMP database (http://www.cuilab.cn/sramp) was used to predict the number of m6A sites on the differentially m6A-methylated transcripts (Zhou et al., 2016).
M6A Single-Base Site qPCR
Before amplification and detection, the LPS (n = 5) and control (n = 5) group samples were treated with an Escherichia coli toxin and RNA endoribonuclease, MazF. MazF is reported to be sensitive to m6A modification within the ACA motif (Imanishi et al., 2017). The MazF-digested mRNA samples and the nondigested samples were subjected to reverse transcription using SuperScriptTM III Reverse Transcriptase (Invitrogen) for qPCR as described above. The targeted lncRNAs and mRNAs were predicted by SRAMP to identify the ACA motif and m6A position. The primer sequences specific for the methylated lncRNAs and mRNAs are listed in Supplementary Table S2. The relative expression levels were calculated using the 2−△△Ct method, and the tested genes were calibrated with MazF as follows:
Competing Endogenous RNA Network Construction
The targeted lncRNAs were verified by m6A single-base site qPCR to construct the ceRNA network. Only three steps were used for ceRNA network construction. First, the potential target microRNAs of lncRNAs were predicted with Aksomics’s homemade miRNA target prediction software based on TargetScan and miRanda (Enright et al., 2003; Pasquinelli, 2012). Second, the target genes of the miRNAs involved in the lncRNA-miRNA interaction network were predicted with the database described above. Finally, the lncRNA-microRNA-mRNA interaction networks were constructed by Cytoscape v2.8.3. The targeted mRNAs were also analyzed by GO and KEGG to completely understand the ceRNA effects.
Statistical Analysis
The statistical significance of the GO and KEGG analysis of mRNAs enrichment were calculated by Fisher’s exact test p < 0.05 and -log10(p) transformed as the enrichment score as well as mRNAs predicted by the ceRNA network for GO and KEGG analysis. The significance of the differences in the expression and methylation levels between the LPS and control groups was evaluated with an unpaired two-sided t-test for LC-MS/MS, qRT-PCR, microarray analysis and m6A single-base site qPCR, and the recommended p-value threshold was less than 0.05. “*” indicates a significant difference compared with control group (p < 0.05).
Results
The Global Levels of m6A Modification Were Decreased
In about 24 h after giving intraperitoneal injection of LPS, we measured the MAP of rats. As previous paper, the rats with a MAP declined to 25–30% or below were chosen as the sepsis model (Nie et al., 2020). Furthermore, signs of shock such as lassitude, tachycardia and a sharp drop in body temperature were observed in all sepsis rats. In the control group, there was no significant change in MAP in rats injected with saline. Previous study showed that there is a positive relationship between MAP and heart function (Yang et al., 2018; Zhang et al., 2019). Therefore, we isolated heart tissue and chose the left ventricle for identification of m6A modification levels (Figure 1A). There was no measurement of cardiac function or sepsis physiology in these experiments.
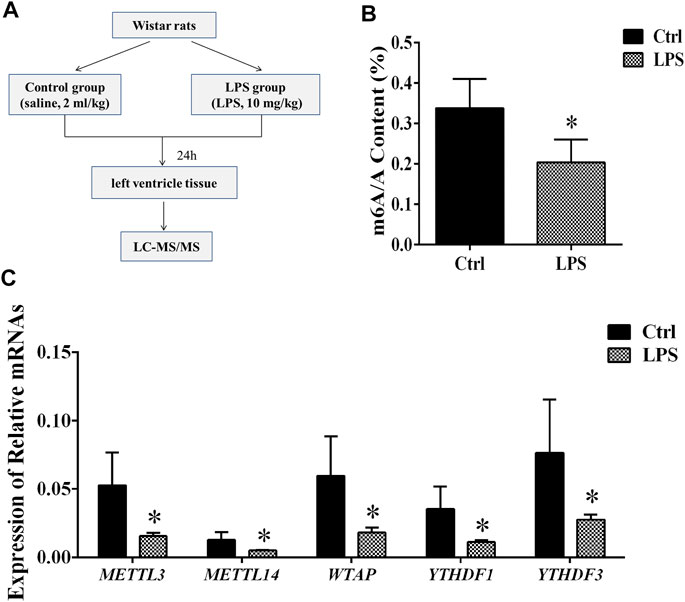
FIGURE 1. Global level of m6A modification and mRNA levels of enzymes in left ventricle tissue. (A) Flowchart of tissue collection. (B) Global level of m6A modification measured by LC-MS/MS in left ventricle tissue. (C) Relative expression mRNA levels of m6A-related enzymes in left ventricle tissue during sepsis. The mRNA levels were determined by qRT-PCR and normalized by GAPDH. *p < 0.05 versus the Ctrl group. Ctrl, control; LPS, lipopolysaccharide.
To determine the difference in the m6A modification levels between the LPS and control groups, we used LC-MS/MS to measure these levels. We found that the global m6A modification level in the rat left ventricle tissues from the LPS group was significantly decreased compared with control group (Figure 1B).
The Expression of m6A Writers and Readers Was Downregulated
The m6A methylation modification is dynamically modulated by RNA methyltransferases, RNA-binding proteins and demethylases. We analyzed the mRNAs level of RNA methyltransferases (writers: METTL3, METTL14, and WTAP), RNA-binding proteins (readers: YTHDF 1 and YTHDF 3) and a demethylase (eraser: FTO). It was found that the expression of the METTL3/METTL14/WTAP/YTHDF 1, and YTHDF 3 in the LPS group was significantly downregulated compared with control group. However, the expression of FTO has no significant difference (Figure 1C). In addition, we measured the protein expression of METTL3/14, WTAP, and YTHDF1/3. We found that protein expressions of these proteins significantly downregulated in LPS group (Supplementary Figure S1). The expressions of METTL3/14, WTAP, and YTHDF1/3 were decreased by 42.2, 64, 47.6, 49.6 and 56.6%, respectively. The results of protein expression and mRNA expression are consistent.
M6A Modification Profiles of lncRNAs and mRNAs
Probes specific for 27,770 mRNAs and 10,582 lncRNAs were used to analyze the samples from the LPS and control groups using an m6A mRNA and lncRNA Arraystar epitranscriptomic microarray. The results showed that the m6A modification levels of 62 mRNAs and 11 lncRNAs were significantly altered in the LPS group compared with control group (fold change >1.5, p-value < 0.05) (Figures 2A,B). The altered lncRNAs and mRNAs were aligned for cluster analysis (Figures 2C,D). Based on their fold changes, we selected 11 lncRNAs with significantly altered m6A modification levels and the top 10 up- and downregulated mRNAs to present the results (Tables 1, 2).
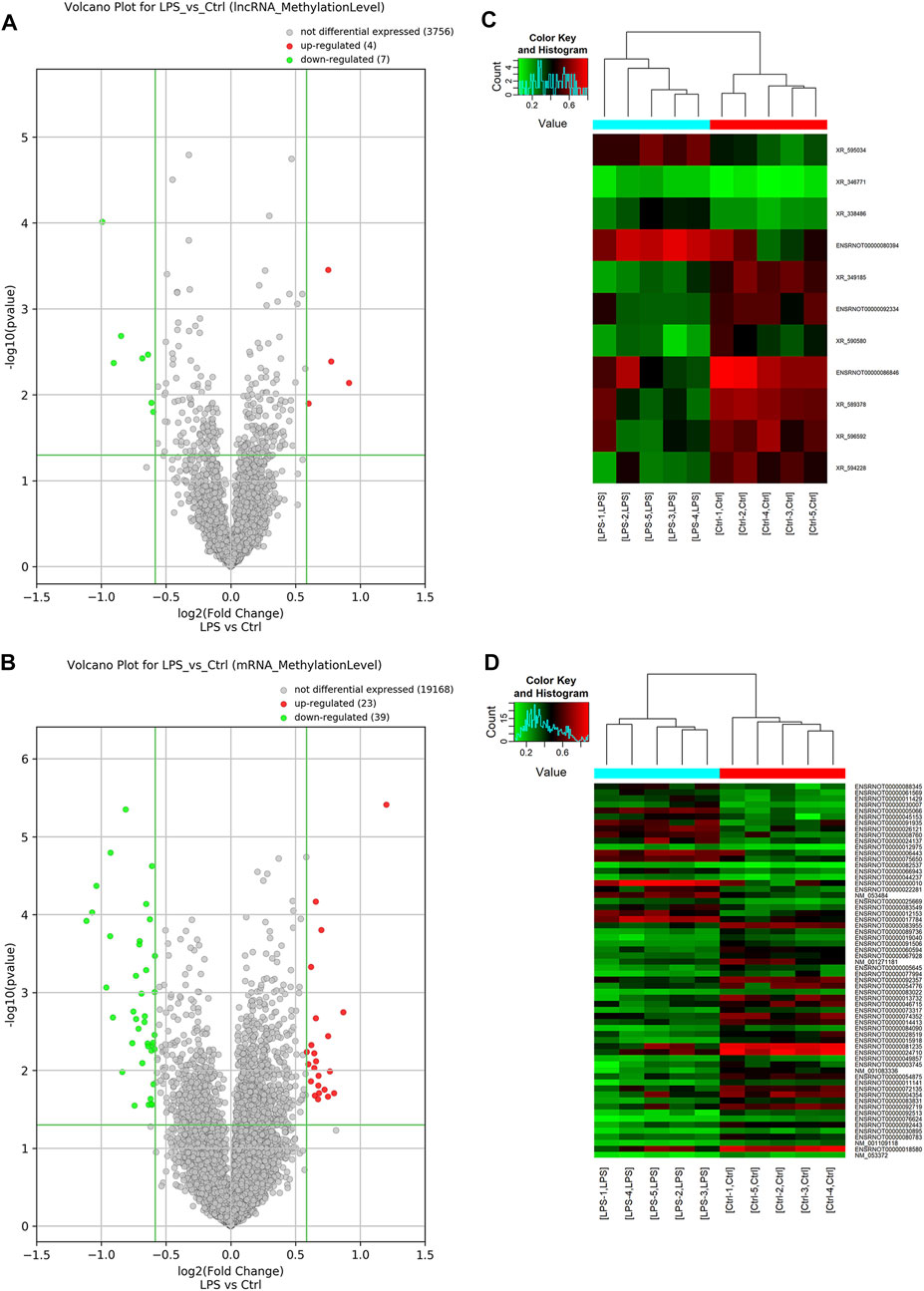
FIGURE 2. M6A modification profiles of lncRNA& mRNA in LPS and control group. (A) Volcano plots showing the lncRNAs that were differentially methylated between LPS and control group with statistical significance. (B) Volcano plots showing the mRNAs that were differentially methylated between LPS and control group with statistical significance (fold changes ≥1.5 and p < 0.05). (C) Hierarchical clustering analysis the differentially methylated lncRNAs. (D) Hierarchical clustering analysis the differentially methylated mRNAs. Ctrl, control; LPS, lipopolysaccharide.
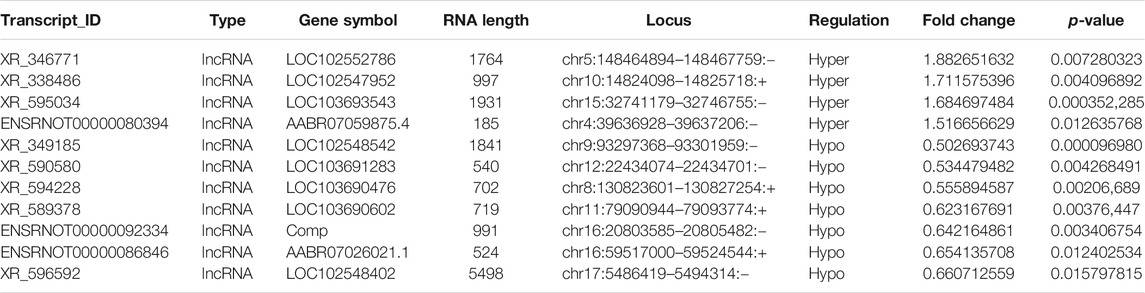
TABLE 1. The detailed information of the hyper-methylated and hypo-methylated lncRNAs.
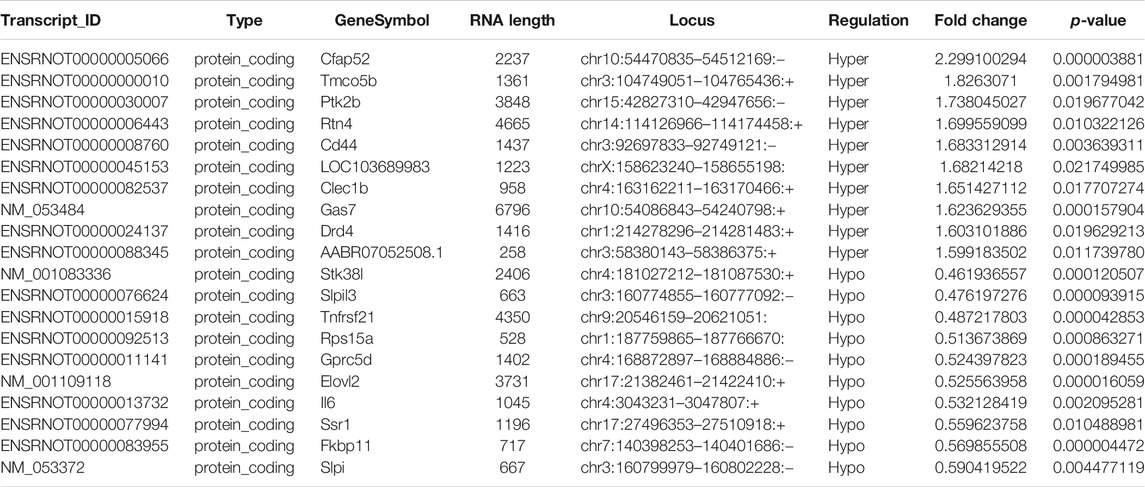
TABLE 2. The detailed information of the top ten hyper-methylated and top ten hypo-methylated mRNAs.
The data that support the findings of this study are openly available in the GenBank databases under accession number GSE159309.
GO and KEGG Analyses of Differentially Methylated mRNAs
GO analysis of the mRNAs with increased and decreased m6A modification levels was performed. Among the mRNAs with decreased m6A modification levels, the “cytokine-mediated signaling pathway” in BP, “immune response” in BP and “cytokine activity” in MF were revealed to have the maximum enrichment scores for GO terms (Figures 3A,B; Tables 3, 4).
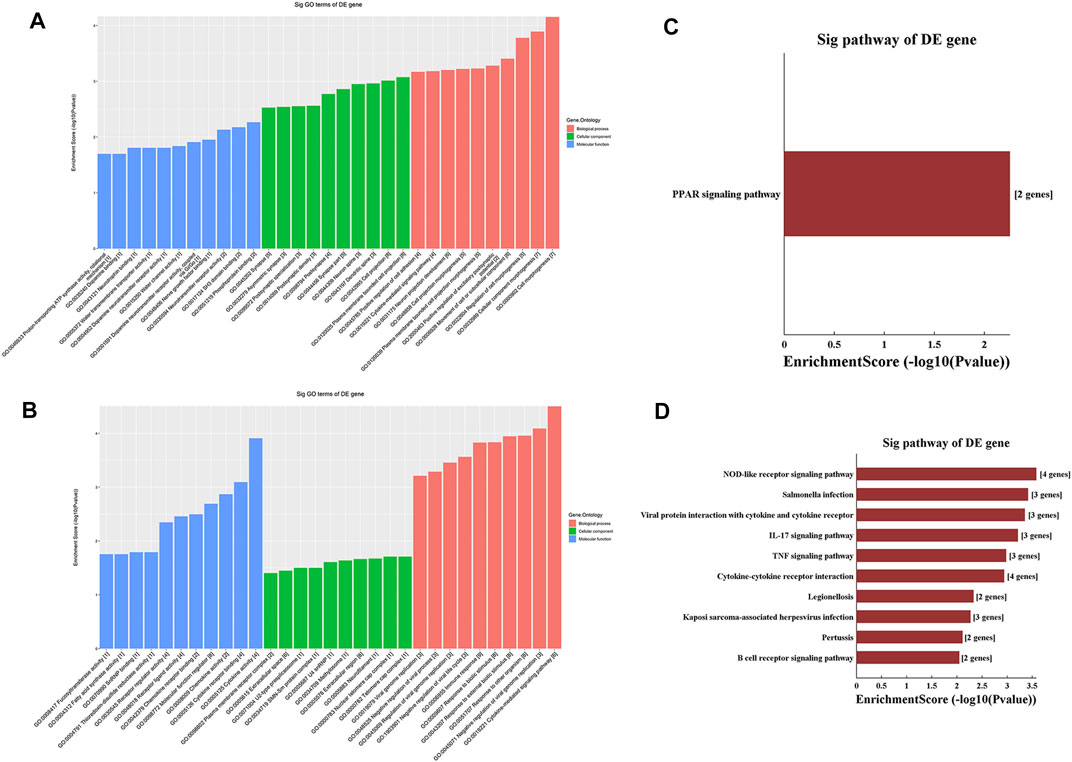
FIGURE 3. Gene ontology enrichment and Kyoto Encyclopedia of Genes and Genomes analysis of altered m6A transcripts. (A) The top ten significantly enriched GO terms of upregulation m6A transcripts. (B) The top ten significantly enriched GO terms of downregulation m6A transcripts. (C) The significantly enriched KEGG pathways of upregulation m6A transcripts. (D) The significantly enriched KEGG pathways of downregulation m6A transcripts. GO, Gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes. DE, differentially expressed.
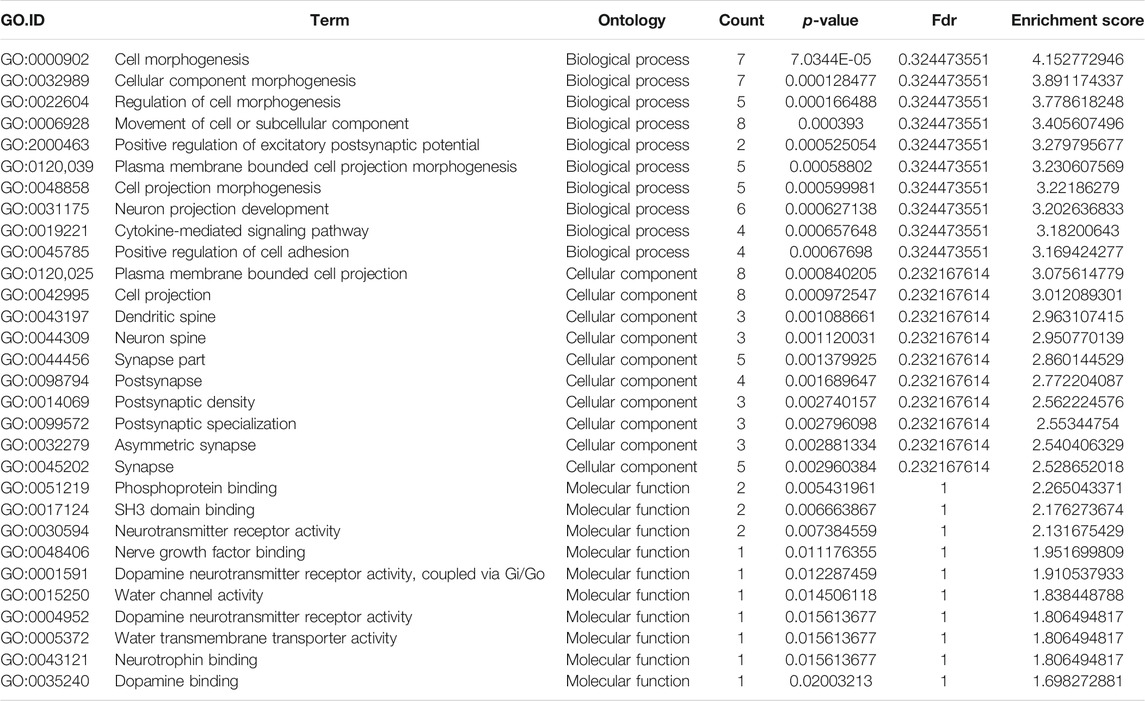
TABLE 3. Gene Ontology analysis of the top ten hyper-methylated mRNAs.
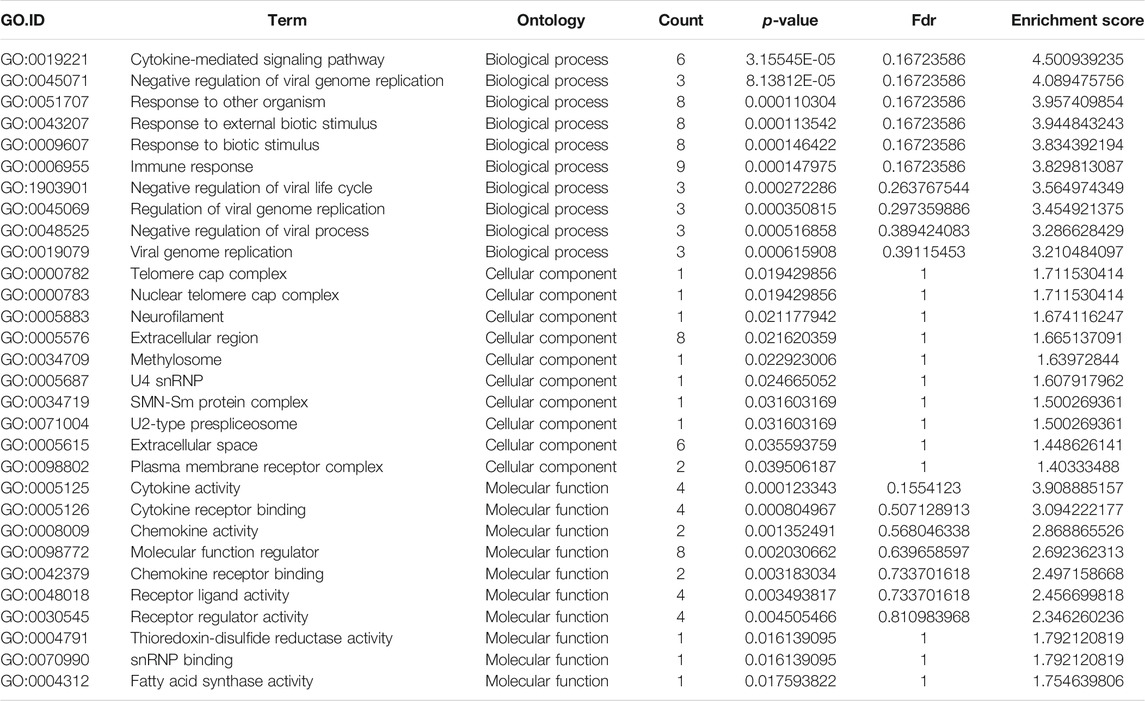
TABLE 4. Gene Ontology analysis of the top ten hypo-methylated mRNAs.
KEGG analysis of the mRNAs with increased and decreased m6A methylation modification levels were also conducted. The results showed that the mRNAs with increased m6A modification levels were enriched in only one pathway, namely, the “PPAR signaling pathway” (Figure 3C). The mRNAs with decreased m6A modification levels were enriched in 27 pathways, and the following main pathways reached higher enrichment scores: “IL-17 signaling pathway”, “TNF signaling pathway” and “Cytokine-cytokine receptor interaction” (Figure 3D).
qPCR Results of a Single-Base m6A Site
By using the SRAMP database, we predicted the mRNAs and lncRNAs with differential m6A modification in the order from high to low fold change of m6A modification. As a result, we identified four lncRNAs and six mRNAs with m6A ACA sites and with high confidence scores of qPCR validation of single-base m6A sites. The Detail information of lncRNAs and mRNAs detected by m6A single-base site qPCR was shown in Table 5. The results showed that among the lncRNAs, m6A modification levels were significantly increased in XR_346771. Among the mRNAs, m6A modification levels were significantly increased in C-type lectin domain family 1 member B (Clec1b) and tumor necrosis factor receptor superfamily member 26 (Tnfrsf26) and significantly decreased in serine/threonine kinase 38 like (STK38L) (Figure 4).

TABLE 5. Detail information of lncRNAs and mRNAs with hyper- and hypo-methylation detected by m6A single-base site qPCR.
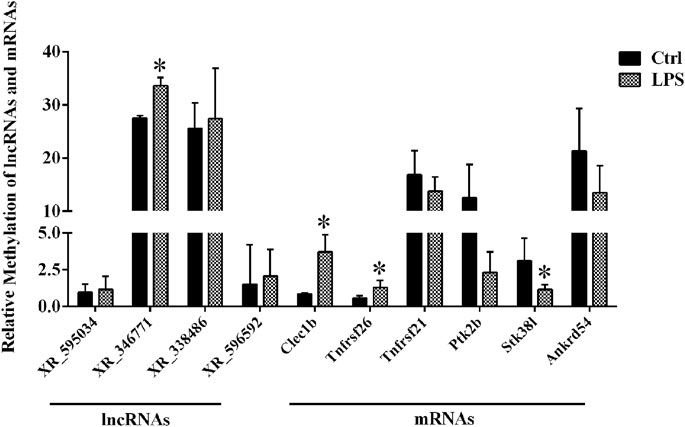
FIGURE 4. M6A single-base site qPCR was used to confirm the microarray data for the top four methylated lncRNAs and top six methylated mRNAs in left ventricle tissue between the LPS and control groups. *p < 0.05 versus the Ctrl group. Ctrl, control; LPS, lipopolysaccharide.
PPI Analysis of Targeted Genes
By employing the STRING database, we performed PPI analysis of Clec1b, Stk38l, and Tnfrsf26 (Figures 5A–C). KEGG analysis revealed that the Clec1b interaction-related proteins were mainly enriched in “platelet activation” and other pathways. The Stk38l interaction-related proteins were mainly enriched in the “hippo signaling pathway”, and the Tnfrsf26 interaction-related proteins were mainly enriched in “apoptosis” (Table 6).
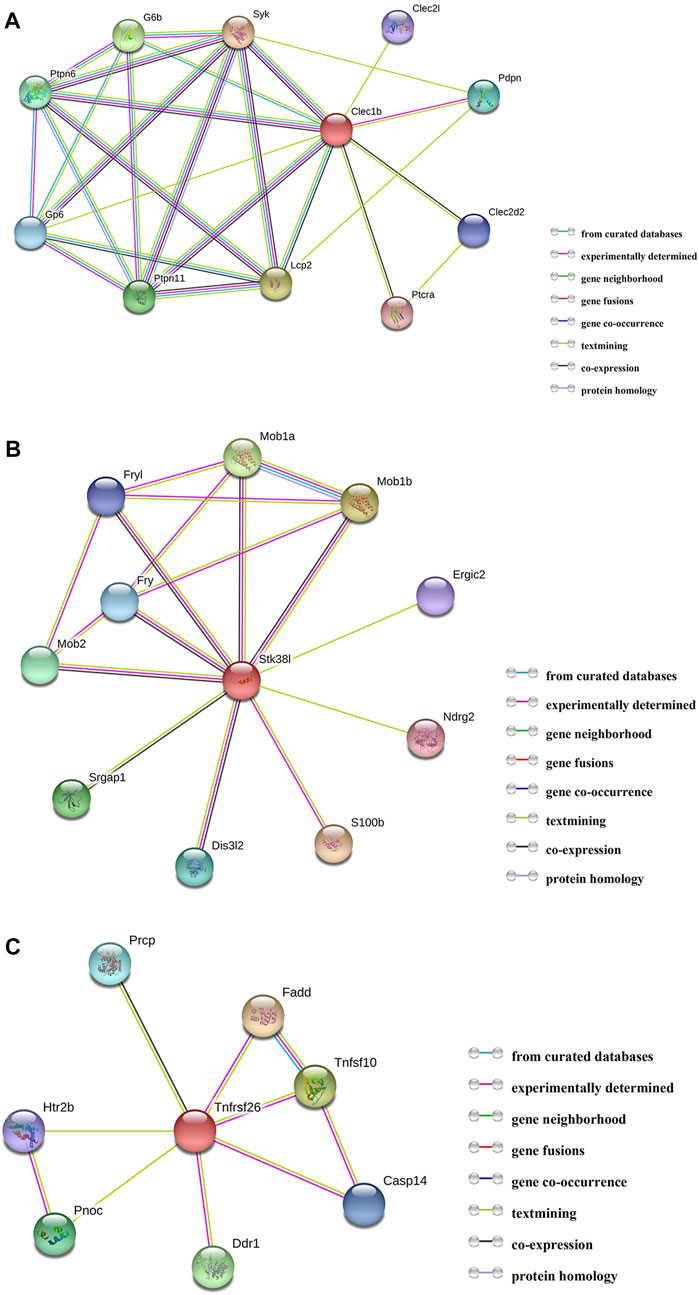
FIGURE 5. Protein–protein interaction analysis. (A) PPI network of Clec1b, (B) PPI network of Stk38l, (C) PPI network of Tnfrsf26. PPI, protein–protein interaction; Clec1b, C-type lectin domain family 1 member B; Tnfrsf26, tumor necrosis factor receptor superfamily member 26; STK38L, serine/threonine kinase 38 like.
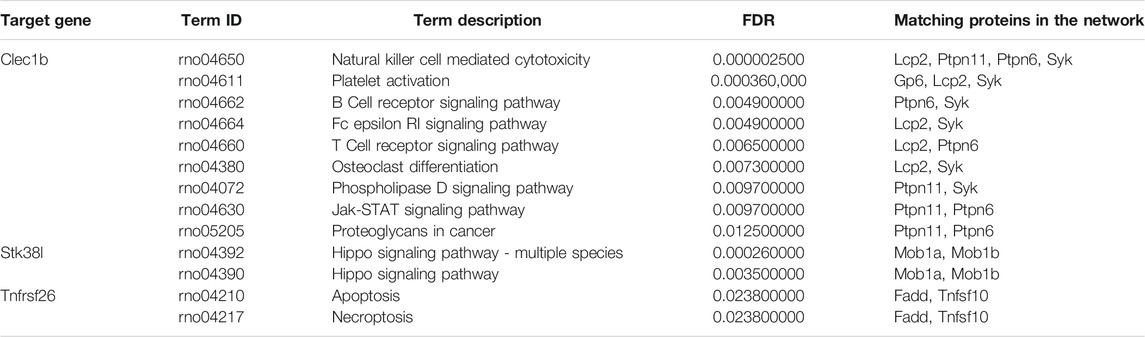
TABLE 6. Pathways of the matching proteins in the PPI network.
ceRNA Analysis of lncRNA XR_346771
A ceRNA network of the lncRNA-microRNA-mRNA interaction network was constructed for XR_346,771 (Figure 6A). GO and KEGG analyses were performed on the predicted mRNAs (Figures 6B,C). The “inorganic cation import across plasma membrane”, “transmembrane transport”, and “import across plasma membrane” in BP were significantly enriched for GO terms (Figure 6B). FAM155B and PICALM are downstream proteins related to transmembrane transport that are regulated by XR_346771, and the corresponding sponged miRNAs are shown in Table 7.
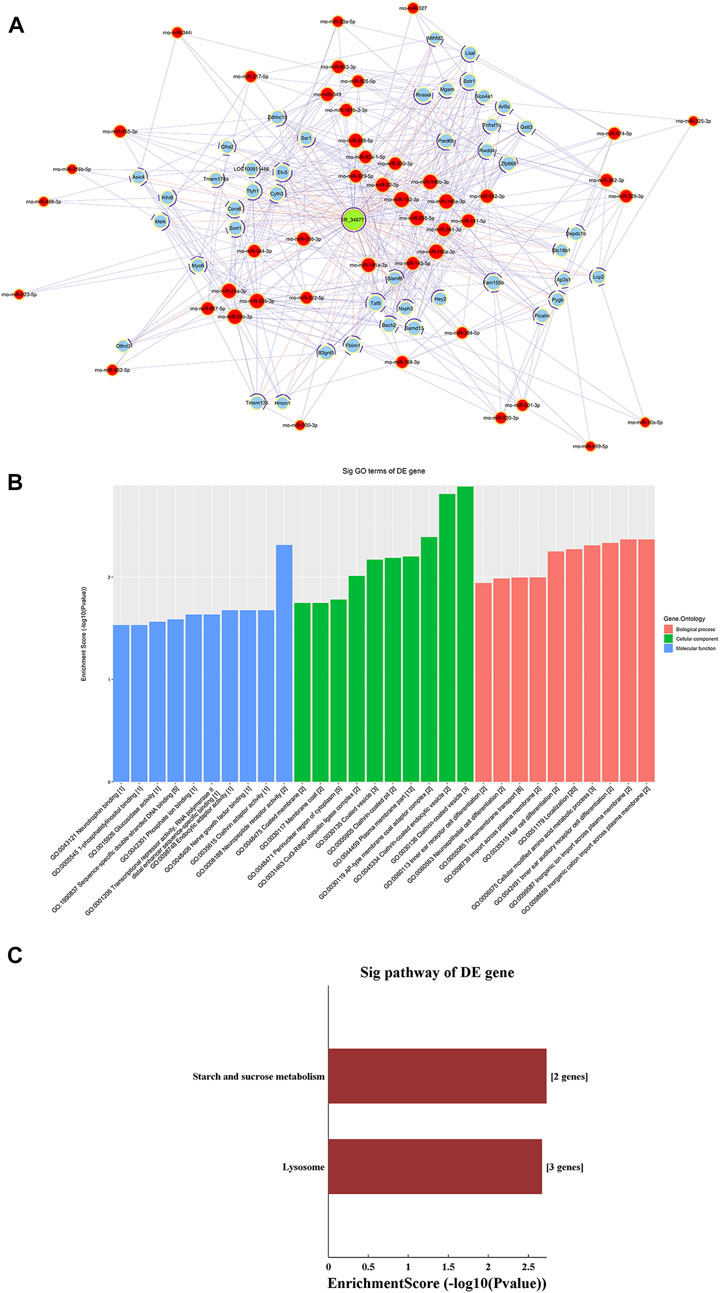
FIGURE 6. LncRNA XR_346,771-miRNA-mRNA ceRNA analysis. (A) The ceRNA network of XR_346,771–miRNA–mRNA. Red circles represent miRNAs, blue circles represent mRNAs, and green circles represent lncRNAs. (B) Ten most enriched GO terms involved in the ceRNA network. (C) KEGG pathways involved in the ceRNA network. ceRNA, competing endogenous RNA.

TABLE 7. LncRNA XR_346771targeted miRNAs and mRNAs.
Discussion
Our study suggested that the m6A modification level was decreased in septic heart tissues. Our genome-wide screening of m6A-tagged transcript profiles indicated that the m6A modification levels in 39 mRNAs and 7 lncRNAs were significantly decreased and that the m6A modification levels in 23 mRNAs and four lncRNAs were significantly increased.
Pathway analyses of mRNAs with altered m6A modification were conducted in our study. The results showed that the mRNAs with decreased m6A modification levels were enriched in many pathways related to immune and inflammatory responses. In recent years, the roles of m6A modification in autoimmune and inflammatory diseases have attracted substantial attention (Rubio et al., 2018). METTL3 is a key enzyme of m6A methylation modification and is involved in immune and inflammatory regulation. Feng et al. found that METTL3 inhibits inflammation by affecting the alternative splicing of MyD88 (Feng et al., 2018). METTL3 expression is significantly upregulated in blood samples from patients with rheumatoid arthritis. In addition, METTL3 attenuates LPS-induced macrophage-mediated inflammation through the NF-κB pathway (Wang et al., 2019a). The m6A “reader” YT521-B homology domain family 2 (YTHDF2) inhibits HCC cells and tumor vasculature by degrading the mRNAs of IL11 and serpin family E member 2 (Wang et al., 2019b). These findings indicate the important role of m6A modification in inflammatory and immune disorders. Sepsis is mainly caused by the host’s unbalanced response to infection. In sepsis, microbial infections or necrotic tissues release a large amount of harmful substances, leading to the activation of the systemic immune response and the excessive activation of immune cells. The excessive release of cytokines is destructive. There is currently no effective treatment for sepsis because in the past few decades, attempts to use anti-inflammatory treatments to limit the tissue damage caused by excessive inflammation have failed (Zeni et al., 1997). Our study suggests that alterations in m6A modification in sepsis are closely related to inflammatory and immune responses. Since we study cardiac tissue, we speculate that m6A modification plays an important role in sepsis-induced cardiac function dysfunction.
We selected four lncRNAs and six mRNAs for the qPCR validation of single-base m6A sites in our study. The results showed that among the lncRNAs, the m6A modification levels were significantly increased in XR_346771. Among the mRNAs, the m6A modification levels were significantly increased in Clec1b and Tnfrsf26 and significantly decreased in Stk38l. These results are consistent with those of high-throughput sequencing; therefore, we attempted to conduct an in-depth analysis of this lncRNA and the three mRNAs.
First, we performed PPI analysis of Clec1b, STK38L, and Tnfrsf26 to investigate the downstream possible regulatory genes and biological functions of these proteins. The Clec1b-interacting proteins are enriched in the pathway of “platelet activation”. C-type lectin-like receptor 2 (CLEC-2) is a protein encoded by the Clec1b gene. Platelets play a critical role in innate and adaptive immunity (Semple et al., 2011). Platelets can alleviate LPS-induced septic shock by regulating the ability of macrophages to engulf and kill bacteria (Xiang et al., 2013). In two mouse models of sepsis (intraperitoneal LPS injection and cecal ligation and puncture), platelet CLEC-2 reduced the severity of sepsis by controlling the migration of monocytes/macrophages to the infection site, the expression of inflammatory mediators, and damage to organs (Rayes et al., 2017). In our sepsis model, we found that the m6A modification level of Clec1b mRNA was significantly increased. Considering that the degree and pattern of m6A modification may affect the transport, splicing, storage, translation, stability, and decay of mRNAs, we speculated that the elevated level of Clec1b m6A modification affects the functions of Clec1b and associate with activation of platelets. The interacting proteins of Clec1b were glycoprotein VI (Gp6), lymphocyte cytosolic protein 2 (Lcp2), and spleen tyrosine kinase (Syk). These proteins might be used as target proteins for in-depth research.
Our study suggests that the m6A modification level of Tnfrsf26 mRNA is increased and of Stk38l mRNA is decreased. According to PPI network analysis, Stk38l-interacting proteins are enriched in the “Hippo signaling pathway” pathway. Hippo signaling pathway may promote cell differentiation and death and inhibit cell proliferation (Chang et al., 2019). Tnfrsf26-interacting proteins are mainly enriched in the pathway of apoptosis. Sepsis-induced cardiac dysfunction has been reported by many studies to be closely related to apoptosis. When LPS stimulates H9C2 cells (rat embryonic cardiomyoblasts), the overexpression of miR-146a suppresses apoptosis and helps attenuate myocardial depression (An et al., 2018). Chao et al. found that the overexpression of Tid1-S can enhance ER-α to activate p-PI3K/p-Akt, attenuating LPS-induced apoptosis in H9C2 cells (Chao et al., 2019). In recent years, as the understanding of m6A modification has gradually deepened, studies have found that m6A modification plays an important role in the process of apoptosis. In the human glioma cell line U251, lower level of m6A modification can reduce apoptosis and promote cell proliferation (Li et al., 2019). Moreover, METTL3 can regulate the expression of several important proteins that regulate the survival, apoptosis, invasion, and proliferation of lung cancer cells (Lin et al., 2016). We hypothesized that the m6A modification of Tnfrsf26 and Stk38l might be related to cardiomyocyte apoptosis.
LncRNAs are a class of noncoding RNAs that are more than 200 nucleotides in length. In recent years, m6A-modified lncRNAs have received extensive attention. M6A modification may control gene expression by regulating the translation efficiency and stability of lncRNAs (Coker et al., 2019). For example, the m6A-modified lncRNA MALAT1 is implicated in ischemia reperfusion injury-induced inflammation (Yang et al., 2020). LncRNAs contain miRNA-responsive elements, which function as ceRNAs, interact with miRNAs and indirectly regulate mRNAs. In the present study, we established and analyzed a potential ceRNA network of XR_346771 through bioinformatics analysis. We performed GO and KEGG analyses of the predicted mRNAs to further explore the function of XR_346771.
GO analysis showed that “inorganic cation import across plasma membrane”, “inorganic ion import across plasma membrane”, “transmembrane transport”, and “import across plasma membrane” were significantly enriched. Cardiac contraction requires Ca2+, so abnormal Ca2+ homeostasis may play a key role in the pathogenesis of common cardiovascular diseases. Studies have reported that m6A modification promotes neurite extension and neuronal differentiation by acting as a Ca2+ channel (Mukobata et al., 2002). In 6-OHDA-induced PC12 cells and the cerebral striatum of rats with Parkinson’s disease, decreased m6A modification induces the expression of N-methyl-d-aspartate receptor 1 and increases oxidative stress and Ca2+ influx, leading to apoptosis of dopaminergic neurons (Chen et al., 2019). These studies indicate that m6A modification is involved in Ca2+ transport. We speculated that the m6A modification of XR_346771 might associate with abnormal Ca2+ homeostasis in cardiac tissues.
It is interesting that even though the methyltransferase complex was downregulated upon sepsis induced myocardial dysfunction; there were still 27 transcripts that were significantly hypermethylated. Chokkalla et al. found that, compared with sham group, global m6A increased significantly at 12 and 24 h of reperfusion in transient middle cerebral artery occlusion in C57BL/6J mice. The FTO decreased significantly after stroke compared with sham. While 139 transcripts (122 mRNAs and 17 lncRNAs) were hypermethylated, 8 transcripts (5 mRNAs and 3 lncRNAs) were hypomethylated in the ischemic brain at 12 h reperfusion (Chokkalla et al., 2019). Wang et al. found that METTL3 was downregulated after traumatic brain injury in mice. In total, 922 m6A peaks were differentially expressed as determined by m6A-RIP-seq, with 370 upregulated and 552 downregulated methylated mRNA (Wang et al., 2019b). These studies are consistent with our research. Previous research indicated that the methylation modification of m6A has been proved to be reversible, as it modulated by RNA methyltransferases, RNA-binding proteins and demethylases. The m6A methyltransferase, also known as “Writers”, including METTL3/14, WTAP, KIAA1429 and RNA binding motifs protein 15/15B (RBM15/15B), E3 ubiquitin-protein ligase Hakai (HAKAI) and zinc finger CCCH-type containing 13 (ZC3H13). The RNA-binding proteins, also known as “readers”, including: YTHDF1/2/3, YTH domain-containing reader proteins 1/2 (YTHDC1/2), insulin-like growth factor 2 mRNA-binding proteins 1/2/3 (IGF2BP1/2/3), epithelium-specific ETS (ESE) transcription factors (ELF3), heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1). The m6A demethylase, also known as “eraser”, including: FTO, α-ketoglutarate-dependent dioxygenase alkB homologue 5 (ALKHB5). In our research we tested mRNA level of some enzymes, but not all enzymes. Therefore, there may be other enzymes involved in m6A modification upon sepsis induced myocardial dysfunction. The m6A modification status of specific RNA was catalyzed by different enzymes. METTL3 positively regulates expression of MYD88, a critical upstream regulator of NF-κB signaling, by facilitating m6A methylation modification to MYD88-RNA in mesenchymal stem cells (Yu et al., 2020). However, reduced expression of FTO increased Nanog mRNA m6A methylation under TNF-α stimulation, decreased Nanog mRNA and protein expression, and significantly inhibited mesenchymal stem cells capacity for differentiation to sweat gland cells (Wang et al., 2020). In our research, these transcripts that have undergone m6A modification might have been modified by different enzymes.
In conclusion, our experiments revealed that the level of m6A modification is significantly decreased in septic cardiac tissue. Through the genome-wide profiling of m6A-tagged mRNAs and lncRNAs and subsequent bioinformatics analysis, we revealed some potential functions of transcripts with altered m6A modification.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, GSE159309.
Ethics Statement
The animal study was reviewed and approved by The Experimental Animal Welfare Ethics Branch and the Biomedical Ethics Committee of Peking University (LA 2020343).
Author Contributions
S-YZ and Y-CH conceived and planned the experiments. Y-CH, H-ZX, and BL performed the experiment and acquired the data. R-LX, H-PZ, and J-YL analyzed and interpreted the data. Y-CH drafted the manuscript. S-YZ revised the manuscript. All authors provided critical feedback and help in shaping the research, analysis, and manuscript. All authors have read and approved the final submitted manuscript.
Funding
This work was supported by the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (No. 2016-I2M-1-011). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.670160/full#supplementary-material
Supplementary Figure 1 | Expressions of m6A-related enzymes were detected by western blot analysis. n = 3-4; *P < 0.05. Ctrl, control; LPS, lipopolysaccharide.
Supplementary Table 1 | Sequence of primers used for qRT-PCR analysis of mRNA levels.
Supplementary Table 2 | Sequence of primers used for m6A single base site qPCR analysis of relative mRNA methylation levels.
Abbreviations
BP, biological processes; CC, cellular components; ceRNA, Competing endogenous RNA; ELF3, epithelium-specific ETS (ESE) transcription factors; FTO, Fat mass and obesity-associated protein; GO, Gene Ontology; HAKAI, E3 ubiquitin-protein ligase Hakai; HCC, epatocellular carcinoma; hnRNPA2B1, heterogeneous nuclear ribonucleoprotein A2B1; IGF2BP1/2/3, insulin-like growth factor 2 mRNA-binding proteins 1/2/3; KEGG, Kyoto Encyclopedia of Genes and Genomes; LC-MS/MS, Liquid chromatography-tandem mass spectrometry; lncRNAs, long non-coding RNAs; LPS, lipopolysaccharide; m6A, N6-methyladenosine; METTL14, methyltransferase-like 14; METTL3, methyltransferase like 3; MF, molecular functions; PPI, Protein-protein interaction; RBM15/15B, RNA binding motifs protein 15/15B; WTAP, Wilms tumor 1-associated protein; YTHDC1/2, YTH domain-containing reader proteins 1/2; YTHDF 2, YTH domain family 2; ZC3H13, zinc finger CCCH-type containing 13.
References
An, R., Feng, J., Xi, C., Xu, J., and Sun, L. (2018). miR-146a Attenuates Sepsis-Induced Myocardial Dysfunction by Suppressing IRAK1 and TRAF6 via Targeting ErbB4 Expression. Oxid. Med. Cell Longev. 2018, 1–9. doi:10.1155/2018/7163057
Arcidiacono, O. A., Krejčí, J., and Bártová, E. (2020). The Distinct Function and Localization of METTL3/METTL14 and METTL16 Enzymes in Cardiomyocytes. Int. J. Mol. Sci. 21, 8139. doi:10.3390/ijms21218139
Berulava, T., Buchholz, E., Elerdashvili, V., Pena, T., Islam, M. R., Lbik, D., et al. (2020). Changes in m6A RNA Methylation Contribute to Heart Failure Progression by Modulating Translation. Eur. J. Heart Fail. 22, 54–66. doi:10.1002/ejhf.1672
Chang, Y., Fu, X. R., Cui, M., Li, W. M., Zhang, L., Li, X., et al. (2019). Activated Hippo Signal Pathway Inhibits Cell Proliferation and Promotes Apoptosis in NK/T Cell Lymphoma Cells. Cancer Med. 8, 3892–3904. doi:10.1002/cam4.2174
Chao, C. N., Lo, J. F., Khan, F. B., Day, C. H., Lai, C. H., Chen, C. H., et al. (2019). Tid1‐S Attenuates LPS‐induced Cardiac Hypertrophy and Apoptosis through ER‐a Mediated Modulation of p‐PI3K/p‐Akt Signaling cascade. J. Cel Biochem. 120, 16703–16710. doi:10.1002/jcb.28928
Chen, X., Yu, C., Guo, M., Zheng, X., Ali, S., Huang, H., et al. (2019). Down-Regulation of m6A mRNA Methylation Is Involved in Dopaminergic Neuronal Death. ACS Chem. Neurosci. 10, 2355–2363. doi:10.1021/acschemneuro.8b00657
Chokkalla, A. K., Mehta, S. L., Kim, T., Chelluboina, B., Kim, J., and Vemuganti, R. (2019). Transient Focal Ischemia Significantly Alters the M 6 A Epitranscriptomic Tagging of RNAs in the Brain. Stroke 50, 2912–2921. doi:10.1161/strokeaha.119.026433
Cohen, J., Vincent, J.-L., Adhikari, N. K. J., Machado, F. R., Angus, D. C., Calandra, T., et al. (2015). Sepsis: a Roadmap for Future Research. Lancet Infect. Dis. 15, 581–614. doi:10.1016/s1473-3099(15)70112-x
Coker, H., Wei, G., and Brockdorff, N. (2019). m6A Modification of Non-coding RNA and the Control of Mammalian Gene Expression. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 310–318. doi:10.1016/j.bbagrm.2018.12.002
Enright, A. J., John, B., Gaul, U., Tuschl, T., Sander, C., and Marks, D. S. (2003). MicroRNA Targets in Drosophila. Genome Biol. 5, R1. doi:10.1186/gb-2003-5-1-r1
Feng, Z., Li, Q., Meng, R., Yi, B., and Xu, Q. (2018). METTL 3 Regulates Alternative Splicing of MyD88 upon the Lipopolysaccharide‐induced Inflammatory Response in Human Dental Pulp Cells. J. Cel Mol. Med. 22, 2558–2568. doi:10.1111/jcmm.13491
Huang, T., Guo, J., Lv, Y., Zheng, Y., Feng, T., Gao, Q., et al. (2019). Meclofenamic Acid Represses Spermatogonial Proliferation through Modulating m6A RNA Modification. J. Anim. Sci. Biotechnol. 10, 63. doi:10.1186/s40104-019-0361-6
Imanishi, M., Tsuji, S., Suda, A., and Futaki, S. (2017). Detection ofN6-Methyladenosine Based on the Methyl-Sensitivity of MazF RNA Endonuclease. Chem. Commun. 53, 12930–12933. doi:10.1039/c7cc07699a
Kennedy, E. M., Bogerd, H. P., Kornepati, A. V. R., Kang, D., Ghoshal, D., Marshall, J. B., et al. (2016). Posttranscriptional M 6 A Editing of HIV-1 mRNAs Enhances Viral Gene Expression. Cell Host Microbe 19, 675–685. doi:10.1016/j.chom.2016.04.002
Kmietczyk, V., Riechert, E., Kalinski, L., Boileau, E., Malovrh, E., Malone, B., et al. (2019). m6A-mRNA Methylation Regulates Cardiac Gene Expression and Cellular Growth. Life Sci. Alliance 2, e201800233. doi:10.26508/lsa.201800233
Li, F., Zhang, C., and Zhang, G. (2019). m6A RNA Methylation Controls Proliferation of Human Glioma Cells by Influencing Cell Apoptosis. Cytogenet. Genome Res. 159, 119–125. doi:10.1159/000499062
Lin, S., Choe, J., Du, P., Triboulet, R., and Gregory, R. I. (2016). The M 6 A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cel 62, 335–345. doi:10.1016/j.molcel.2016.03.021
Lin, X., Chai, G., Wu, Y., Li, J., Chen, F., Liu, J., et al. (2019). RNA m6A Methylation Regulates the Epithelial Mesenchymal Transition of Cancer Cells and Translation of Snail. Nat. Commun. 10, 2065. doi:10.1038/s41467-019-09865-9
Mathiyalagan, P., Adamiak, M., Mayourian, J., Sassi, Y., Liang, Y., Agarwal, N., et al. (2019). FTO-dependent N 6 -Methyladenosine Regulates Cardiac Function during Remodeling and Repair. Circulation 139, 518–532. doi:10.1161/circulationaha.118.033794
Mukobata, S., Hibino, T., Sugiyama, A., Urano, Y., Inatomi, A., Kanai, Y., et al. (2002). M6a Acts as a Nerve Growth Factor-Gated Ca2+ Channel in Neuronal Differentiation. Biochem. Biophys. Res. Commun. 297, 722–728. doi:10.1016/s0006-291x(02)02284-2
Nie, M. W., Han, Y. C., Shen, Z. J., and Xie, H. Z. (2020). Identification of circRNA and mRNA Expression Profiles and Functional Networks of Vascular Tissue in Lipopolysaccharide‐induced Sepsis. J. Cel Mol. Med. 24, 7915–7927. doi:10.1111/jcmm.15424
Pan, T. (2018). Modifications and Functional Genomics of Human Transfer RNA. Cell Res 28, 395–404. doi:10.1038/s41422-018-0013-y
Pasquinelli, A. E. (2012). MicroRNAs and Their Targets: Recognition, Regulation and an Emerging Reciprocal Relationship. Nat. Rev. Genet. 13, 271–282. doi:10.1038/nrg3162
Rayes, J., Lax, S., Wichaiyo, S., Watson, S. K., Di, Y., Lombard, S., et al. (2017). The Podoplanin-CLEC-2 axis Inhibits Inflammation in Sepsis. Nat. Commun. 8, 2239. doi:10.1038/s41467-017-02402-6
Rubio, R. M., Depledge, D. P., Bianco, C., Thompson, L., and Mohr, I. (2018). RNA m6A Modification Enzymes Shape Innate Responses to DNA by Regulating Interferon β. Genes Dev. 32, 1472–1484. doi:10.1101/gad.319475.118
Semple, J. W., Italiano, J. E., and Freedman, J. (2011). Platelets and the Immune Continuum. Nat. Rev. Immunol. 11, 264–274. doi:10.1038/nri2956
Song, H., Feng, X., Zhang, H., Luo, Y., Huang, J., Lin, M., et al. (2019). METTL3 and ALKBH5 Oppositely Regulate m6A Modification of TFEB mRNA, Which Dictates the Fate of Hypoxia/reoxygenation-Treated Cardiomyocytes. Autophagy 15, 1419–1437. doi:10.1080/15548627.2019.1586246
Wang, J., Yan, S., Lu, H., Wang, S., and Xu, D. (2019a). METTL3 Attenuates LPS-Induced Inflammatory Response in Macrophages via NF-κB Signaling Pathway. Mediators Inflamm. 2019, 1–8. doi:10.1155/2019/3120391
Wang, Y., Mao, J., Wang, X., Lin, Y., Hou, G., Zhu, J., et al. (2019b). Genome-wide Screening of Altered m6A-Tagged Transcript Profiles in the hippocampus after Traumatic Brain Injury in Mice. Epigenomics 11, 805–819. doi:10.2217/epi-2019-0002
Wang, Y., Wang, R., Yao, B., Hu, T., Li, Z., Liu, Y., et al. (2020). TNF-α Suppresses Sweat Gland Differentiation of MSCs by Reducing FTO-Mediated m6A-Demethylation of Nanog mRNA. Sci. China Life Sci. 63, 80–91. doi:10.1007/s11427-019-9826-7
Xiang, B., Zhang, G., Guo, L., Li, X.-A., Morris, A. J., Daugherty, A., et al. (2013). Platelets Protect from Septic Shock by Inhibiting Macrophage-dependent Inflammation via the Cyclooxygenase 1 Signalling Pathway. Nat. Commun. 4, 2657. doi:10.1038/ncomms3657
Yang, C., Fan, Z., and Yang, J. (2020). m6A Modification of LncRNA MALAT1: A Novel Therapeutic Target for Myocardial Ischemia-Reperfusion Injury. Int. J. Cardiol. 306, 162. doi:10.1016/j.ijcard.2019.11.140
Yang, N., Shi, X.-L., Zhang, B.-L., Rong, J., Zhang, T.-N., Xu, W., et al. (2018). The Trend of β3-Adrenergic Receptor in the Development of Septic Myocardial Depression: A Lipopolysaccharide-Induced Rat Septic Shock Model. Cardiology 139, 234–244. doi:10.1159/000487126
Yu, J., Shen, L., Liu, Y., Ming, H., Zhu, X., Chu, M., et al. (2020). The m6A Methyltransferase METTL3 Cooperates with Demethylase ALKBH5 to Regulate Osteogenic Differentiation through NF-κB Signaling. Mol. Cel Biochem. 463, 203–210. doi:10.1007/s11010-019-03641-5
Zaky, A., Deem, S., Bendjelid, K., and Treggiari, M. M. (2014). Characterization of Cardiac Dysfunction in Sepsis. Shock 41, 12–24. doi:10.1097/shk.0000000000000065
Zeni, F., Freeman, B., and Natanson, C. (1997). Anti-inflammatory Therapies to Treat Sepsis and Septic Shock. Crit. Care Med. 25, 1095–1100. doi:10.1097/00003246-199707000-00001
Zhang, T.-N., Yang, N., Goodwin, J. E., Mahrer, K., Li, D., Xia, J., et al. (2019). Characterization of Circular RNA and microRNA Profiles in Septic Myocardial Depression: a Lipopolysaccharide-Induced Rat Septic Shock Model. Inflammation 42, 1990–2002. doi:10.1007/s10753-019-01060-8
Keywords: N6-methyladenosine, sepsis, myocardial dysfunction, epitranscriptomics, lncRNA
Citation: Han Y-C, Xie H-Z, Lu B, Xiang R-L, Zhang H-P, Li J-Y and Zhang S-Y (2021) Lipopolysaccharide Alters the m6A Epitranscriptomic Tagging of RNAs in Cardiac Tissue. Front. Mol. Biosci. 8:670160. doi: 10.3389/fmolb.2021.670160
Received: 20 February 2021; Accepted: 14 July 2021;
Published: 28 July 2021.
Edited by:
Teng Ma, Capital Medical University, ChinaReviewed by:
Sunny Sharma, The State University of New Jersey, United StatesEva Bartova, Academy of Sciences of the Czech Republic, Czechia
Copyright © 2021 Han, Xie, Lu, Xiang, Zhang, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shu-Yang Zhang, c2h1eWFuZ3poYW5nMTAzQG5yZHJzLm9yZw==