 Zahra Sadat Hashemi1
Zahra Sadat Hashemi1 Mahboubeh Zarei2
Mahboubeh Zarei2 Mohsen Karami Fath3
Mohsen Karami Fath3 Mahmoud Ganji4
Mahmoud Ganji4 Mahboube Shahrabi Farahani4
Mahboube Shahrabi Farahani4 Fatemeh Afsharnouri4
Fatemeh Afsharnouri4 Navid Pourzardosht5,6Bahman Khalesi7
Navid Pourzardosht5,6Bahman Khalesi7 Abolfazl Jahangiri8
Abolfazl Jahangiri8 Mohammad Reza Rahbar9
Mohammad Reza Rahbar9 Saeed Khalili10*
Saeed Khalili10*- 1ATMP Department, Breast Cancer Research Center, Motamed Cancer Institute, Academic Center for Education, Culture and Research, Tehran, Iran
- 2Pharmaceutical Sciences Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
- 3Department of Cellular and Molecular Biology, Faculty of Biological Sciences, Kharazmi University, Tehran, Iran
- 4Department of Medical Biotechnology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran
- 5Cellular and Molecular Research Center, Faculty of Medicine, Guilan University of Medical Sciences, Rasht, Iran
- 6Department of Biochemistry, Guilan University of Medical Sciences, Rasht, Iran
- 7Department of Research and Production of Poultry Viral Vaccine, Razi Vaccine and Serum Research Institute, Agricultural Research Education and Extension Organization, Karaj, Iran
- 8Applied Microbiology Research Center, Systems Biology and Poisonings Institute, Baqiyatallah University of Medical Sciences, Tehran, Iran
- 9Pharmaceutical Sciences Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
- 10Department of Biology Sciences, Shahid Rajaee Teacher Training University, Tehran, Iran
Large contact surfaces of protein–protein interactions (PPIs) remain to be an ongoing issue in the discovery and design of small molecule modulators. Peptides are intrinsically capable of exploring larger surfaces, stable, and bioavailable, and therefore bear a high therapeutic value in the treatment of various diseases, including cancer, infectious diseases, and neurodegenerative diseases. Given these promising properties, a long way has been covered in the field of targeting PPIs via peptide design strategies. In silico tools have recently become an inevitable approach for the design and optimization of these interfering peptides. Various algorithms have been developed to scrutinize the PPI interfaces. Moreover, different databases and software tools have been created to predict the peptide structures and their interactions with target protein complexes. High-throughput screening of large peptide libraries against PPIs; “hotspot” identification; structure-based and off-structure approaches of peptide design; 3D peptide modeling; peptide optimization strategies like cyclization; and peptide binding energy evaluation are among the capabilities of in silico tools. In the present study, the most recent advances in the field of in silico approaches for the design of interfering peptides against PPIs will be reviewed. The future perspective of the field and its advantages and limitations will also be pinpointed.
Introduction
The survival of a cell naturally depends on the connections between its proteins. In a single organism, countless cells are connected to form an interactome. The interactome is an enormous network system composed of a whole set of molecular interactions, particularly protein–protein interactions (PPIs). These interactions could be held together via electrostatic forces, hydrogen bonding, and hydrophobic effect. Approximately 130,000–600,000 PPIs are correlated with the human interactome and make up the PPI networks (Bruzzoni-Giovanelli et al., 2018). These PPIs modulate the systematic function of cells and signaling pathways of the body and are essential to understanding the medicinal chemistry and chemical biology of the cells. They catalyze the critical cellular processes such as replication, transcription, translation, and transmembrane signal transduction (Lu et al., 2020). Proteins and consequently PPIs are the functional building blocks of a living cell. The slightest error in PPIs, especially in a central node (or hub) in a network, could lead to even fatal disease (such as infectious diseases, cancer, and neurodegenerative diseases) and disturb the cell homeostasis. Therefore, focusing on aberrant PPIs holds the promise of curing various diseases and has the therapeutic potential of being attractive targets for developing new drugs and novel diagnostics.
Deliniation of the interaction between a protein domain and a peptide or another protein domain, is the central concept of PPIs (Nevola and Giralt, 2015). A linear sequence of residues, or a short protein domain, evokes the concept of peptides and peptide mimetics (Stone and Deber, 2017). Recently, various drugs have been presented that are relying on the relationship between two proteins. These drugs could mimic the 3D shape of targeted proteins and can specifically fine-tune their interactions. The peptides are capable of adapting secondary structures, which are usually α-helixes, which could also be completely disordered (Nevola and Giralt, 2015).
To provide the desired peptide or small molecule drug, clinical- and molecular-level information is needed (Bakail and Ochsenbein, 2016). Bioinformaticians widely apply high-throughput data obtained from the studies of genomics, RNAomics, proteomics, metabolomics, and glycomics. These collected data are rich sources of the molecular-level information of PPIs and could be useful for personalized treatments (Bakail and Ochsenbein, 2016). In silico methods have been widely used in various aspects of biological studies (Khalili et al., 2017; Mard-Soltani et al., 2018; Khodashenas et al., 2019; Rahbar et al., 2019). In silico methods could be used to screen for the specific topological surface of peptides capable of specific modulation of PPIs. There is a wide variety of software and algorithms for scrutinizing PPIs to design and optimize these interfering peptides.
Basic in silico research about the PPIs (from the 1990s to the 2000s) has significantly contributed to the production of a vast number of constrained peptides and peptide–drug conjugates. The first approvals of the peptide therapeutics were issued for six peptides in 2012. Pasireotide is a somatostatin analog for the treatment of Cushing’s disease. Lucinactant is a pulmonary surfactant for the treatment of infant respiratory distress syndrome. Peginesatide is an erythropoietin analog for the treatment of anemia associated with chronic kidney disease (CKD). Carfilzomib is an epoxomicin analog (proteasome inhibitor) that is used as an anticancer medication. Linaclotide is an oligo-peptide agonist of guanylate cyclase 2C used to treat irritable bowel syndrome with constipation and chronic constipation with no known causes. Teduglutide is a 33-membered polypeptide and glucagon-like peptide-2 (GLP-2) analog for the treatment of short bowel syndrome. A new field has opened up for scientists to treat different diseases with the help of various in silico algorithms, tools, and software designs by discovering therapeutic peptides and small molecules (Kaspar and Reichert, 2013). The results of in silico studies should be confirmed in vitro and in vivo to pass the phases of clinical trials. The PPI networks would be disturbed and therefore restrained the usage of these therapeutic PPI-targeting peptides (Kaspar and Reichert, 2013).
In silico methods of peptide analyses could include different approaches such as homology modeling, molecular dynamics, protein docking, and PPI targeting. Structural characterization of the peptides could be carried out by x-ray crystallography, NMR spectroscopy, and cryo-electron microscopy. The obtained structural data are stored and available in structural deposition databases like the Protein Data Bank (PDB). The advantages of computational in silico methods over empirical methods are their low cost, faster procedure speed, simple process, and reliability to target PPIs using peptides. This approach can lead to the atomic-level identification of PPIs (Murakami et al., 2017). The information about the PPIs is crucial for designing desired peptides or small molecules via virtual screening of drug candidates (Nevola and Giralt, 2015). Peptides are reported to have some advantages over small molecule drugs. Small molecules are not sufficient for the complete coverage of the large contact surfaces involved in PPIs. This surface area could practically be from 1,500 to 3,000 Å (Cunningham et al., 2017). Using small molecules, some target sites of the PPI surfaces such as pockets, grooves, or clefts could be missed. Advanced and sophisticated design and modeling of new remedial small molecules will be needed to circumvent this drawback. However, interfering peptides (IPs), natural or synthetic, are superior in this respect and would be reconciled with large or flat PPI surfaces (Bruzzoni-Giovanelli et al., 2018). The studies have shown that the ADME properties of IP therapeutic agents are better than those of small molecules. ADME is correlated with the absorption, distribution, metabolism, excretion, and toxicity of a drug molecule. IPs could be simply designed as valuable therapeutic tools to block various PPI networks. Peptides have been analyzed to block PPIs of the nervous system (Zhai et al., 2014; Shi et al., 2016), cardiovascular system (Pleasant-Jenkins et al., 2017), and even cancer (Jouaux et al., 2008; Mine et al., 2016; Ellert-Miklaszewska et al., 2017). IPs are also associated with low molecular weight, high flexibility, and minimal toxicity, which could offer a new class of biopharmaceuticals. These small peptides could also act as cargo carriers due to their cell penetration property. These cargoes could be linked via covalent or non-covalent bonds to some short peptides (5–30 amino acids). These peptides, which could have positive charges, are called cell-penetrating peptides (CPPs). CPPs facilitate cellular delivery and uptake of their cargo commonly through endocytosis (Guidotti et al., 2017). The prospect of IP drugs shows a robust pipeline and an unrivaled number of marketing opportunities for the treatment of a wide range of diseases.
Databases for Peptide Sequences and Structures
Peptides, once neglected, have now offered tremendous therapeutic applications (Antosova et al., 2009) and have obtained quite an expansion in the pharmaceutical industry. Numerous databases have already been developed to store different kinds of peptides by collecting information from public databases and published scientific articles (Table 1). These repositories can be categorized into three classes, namely antimicrobial peptide (AMP) databases, targeted delivery-related databases, and disease-specific databases.
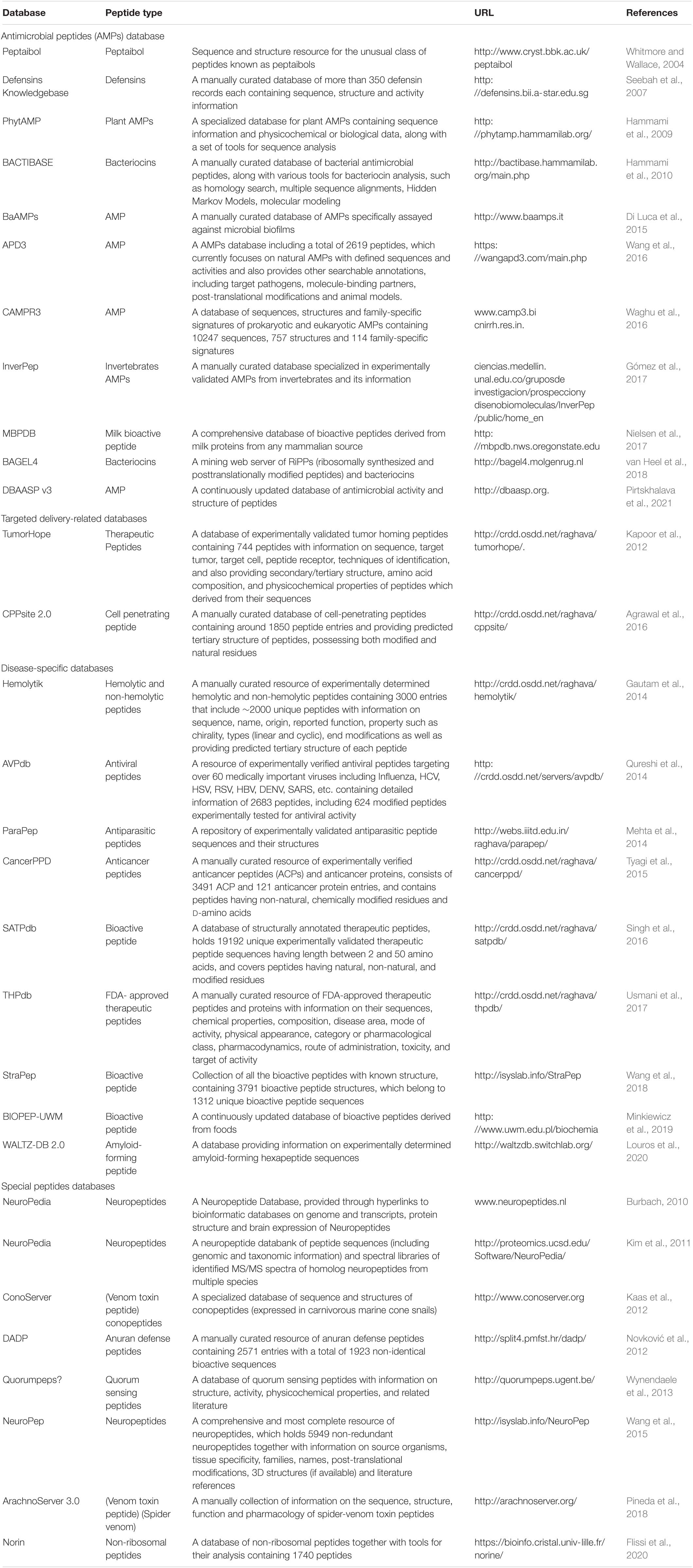
Table 1. List of peptide sequences and databases containing their structures.
Antimicrobial peptides have garnered a lot of attention for several decades due to their biological activity and their ability to make the pathogens resistant to existing drugs. The AMP databases are the most available among the peptide databases. Some AMP databases are specialized to one type of AMP family, like the Defensins Knowledgebase (defensins) (Seebah et al., 2007), BAGEL4 (bacteriocins) (van Heel et al., 2018), BACTIBASE (bacteriocins) (Hammami et al., 2010), PhytAMP (plant AMPs) (Hammami et al., 2009), the Peptaibol Database (peptaibols) (Whitmore and Wallace, 2004), and InverPep (invertebrates AMPs) (Gómez et al., 2017). Meanwhile, others are general AMP databases, such as APD (Wang et al., 2016), CAMP (Waghu et al., 2016), and DBAASP (Pirtskhalava et al., 2016). APD (antimicrobial peptide database) (Wang and Wang, 2004) initially went online in 2003 with 525 AMPs. It has been extensively accepted and referred to since then. In 2009, an updated version of APD was released (Wang et al., 2009), called APD2, which contains about 1,228 entries and has been consistently updated and further expanded into the APD3 version (Wang et al., 2016). This database is currently focused on collecting natural AMPs with defined sequences and activities. It contains 2,619 AMPs with 261 bacteriocins from bacteria, and 7, 13, 4, and 321 from protists, fungi, archaea, and plants, respectively, with an additional 1,972 animal host defense peptides. Another resource for AMPs is CAMP (collection of antimicrobial peptides) (Thomas et al., 2010). It contains information on sequences of natural as well as synthetic AMPs. The inclusion of the structure of the AMPs and family information constituted the second version of the CAMP (Waghu et al., 2014) known as the CAMPR2. Moreover, CAMPR2 contains the newly identified AMPs sequences. CAMPR3 (Waghu et al., 2016) was introduced in 2016 to include AMP family specific signatures. CAMPR3 provides comprehensive information about the sequences, structures, family signatures, activity profile, sources, target organisms, and hemolytic activity for AMPs, and also links to several external databases. DBAASP (Database of Antimicrobial Activity and Structure of Peptides) was started in 2014 with a collection of published information about the AMPs and the corresponding resources (Gogoladze et al., 2014). In 2016, it was updated to DBAASP v2, with about 8,000 entries, including structural information (Pirtskhalava et al., 2016). It also contributed to the development of several databases such as dbAMP (Jhong et al., 2019), LAMP2 (Ye et al., 2020), PlantPepDB (Das et al., 2020), ADAPTABLE (Ramos-Martín et al., 2019), and starPepDB (Aguilera-Mendoza et al., 2019). Most recently, another updated version of DBAASP (DBAASP v3) (Pirtskhalava et al., 2021) has been released to include new content and additional user services. This database has continuously developed its predictive tools to be employed in the de novo design of peptide-based drugs. Its efficacy for the design of peptide-based antimicrobial agents against both gram-positive and gram-negative bacteria has been experimentally confirmed (Vishnepolsky et al., 2019a, b). The BaAMPs database was developed for AMPs with the property of disrupting microbial biofilms (Di Luca et al., 2015). MBPDB is a database of the bioactive peptides of milk origin (Nielsen et al., 2017). CPPsite and TumorHoPe are targeted delivery-related databases. CPPsite (Gautam et al., 2012) is the first database of CPPs that contains 843 entries along with the sequence information, subcellular localization, physicochemical properties, and uptake efficiency. The updated version of CPPsite (Agrawal et al., 2016), called CPPsite 2.0, contains 1,850 entries, including the model system, cargo information, chemical modifications, predicted tertiary structure, and other information. The TumorHoPe database (Kapoor et al., 2012) contains 744 peptides that can recognize tumor tissues and tumor-associated microenvironments.
Disease-specific databases encompass peptides that can be used to design therapeutic peptides capable of targeting specific diseases. PDB (Usmani et al., 2017) is a database of FDA-approved peptides and protein therapeutics, CancerPPD (Tyagi et al., 2015) is a database of anticancer peptides (ACPs) and proteins, and AntiTbPdb (Usmani et al., 2018) is a database of experimentally verified anti-mycobacterial and anti-tubercular peptides. The HIPdb database was established to provide information about 981 HIV-inhibiting peptides (Qureshi et al., 2013), including their sequences and half-maximal inhibitory concentrations (IC50). AVPdb contains 2,683 antiviral peptides and the data about their sequences, efficacy, modifications, and predicted structures (Qureshi et al., 2014). ParaPep (Mehta et al., 2014) is a database of experimentally validated anti-parasitic peptide sequences and their structures curated and compiled from literature, patents, and various other databases. This database was created by Mehta et al. (2014) and contains 863 entries that include 519 unique peptides whose anti-parasitic activities were evaluated against multiple species of Plasmodium, Leishmania, and Trypanosoma. In ParaPep, the structures of peptides consisting of natural, and modified, amino acids have been predicted using the PEPstr software. The Hemolytik database (Gautam et al., 2014) is an information system for experimentally determined hemolytic and non-hemolytic peptides obtained by manual extraction from numerous scientific papers and various databases. This database was created by Gautam et al. (2014) and contains 3,000 entries that include 2,000 unique peptides whose hemolytic activities were assessed on erythrocytes isolated from as many as 17 different sources. WALTZ-DB (Beerten et al., 2015) is the largest available database of experimentally characterized amyloid-forming short sequences. This open-access database was created by Beerten et al. (2015). It contains 1,089 entries that provide primary information about amyloid aggregation incorporated in related databases. Louros et al. (2020) released an updated and significantly expanded version of this database, called WALTZ-DB 2.0. With WALTZ-DB 2.0, the structural model and information were added to the entries. The 3D models of the amyloid fibril cores of the entries were generated using a computational methodology developed in the Switch lab. It also provides a user-friendly option for data filtering and browsing. BIOPEP (Minkiewicz et al., 2008; Iwaniak et al., 2016), currently BIOPEP-UWM (Minkiewicz et al., 2019), is a widely used resource for the identification of bioactive peptides and bioactivity prediction as well as for in silico approaches.
The structural information of bioactive peptides is essential for the development of peptide-based drugs. Two databases, namely StraPep and SATPdb, are structural databases that are dedicated to the collection of bioactive peptides with known structures. StraPep (Wang et al., 2018) is dedicated to collecting all of the bioactive peptides with known structures. It displays the structures for 3,791 peptides and provides detailed information for each one (i.e., post-translational modification, experimental structure, secondary structure, the location of disulfide bonds, etc.). SATPdb (Singh et al., 2016) is a database of structurally annotated therapeutic peptides. It has been curated from 22 public peptide databases and has 19,192 unique, experimentally validated therapeutic peptide sequences. Several databases have been designed for unique peptides as well. Quorumpeps (Wynendaele et al., 2012) is developed for quorum sensing peptides, ConoServer (Kaas et al., 2012) and ArachnoServer (Pineda et al., 2018) are focused on venom toxin peptides; NORINE (Flissi et al., 2020) contains information on non-ribosomal peptides; DADP (Novković et al., 2012) is a database of defense peptides; and the NeuroPep, Neuropedia, and Neuropeptides1 databases contain information on neuropeptides. Besides, databases covering proteins are also considered valuable resources for peptides; for example, UniProt (Universal Protein Resource)2, which is a central resource of protein data, and protein structure databases such as PDB3 (Abes et al., 2007), which contain the 3D structure of biomolecules.
In Silico Tools and Algorithms for Peptide Design
Peptides are the most amenable modulators that can be used to tackle the high surface area of PPIs. They can easily be synthesized, closely mimic the principal features of a protein, and be modified to attain higher stability, bioavailability, and binding strength. Sequence-based design and structure-based design are two major approaches for peptide design. Sequence-based peptide designing involves various physiochemical properties of the peptides and optimizing the peptide stability, toxicity, immunogenicity, and antibody specificity. In this regard, a plethora of in silico tools have been developed for sequence-based designing of novel peptides with therapeutic properties ranging from cell-penetrating to anti-microbial, anti-parasitic, anti-cancer, and anti-hypertension (Table 2).
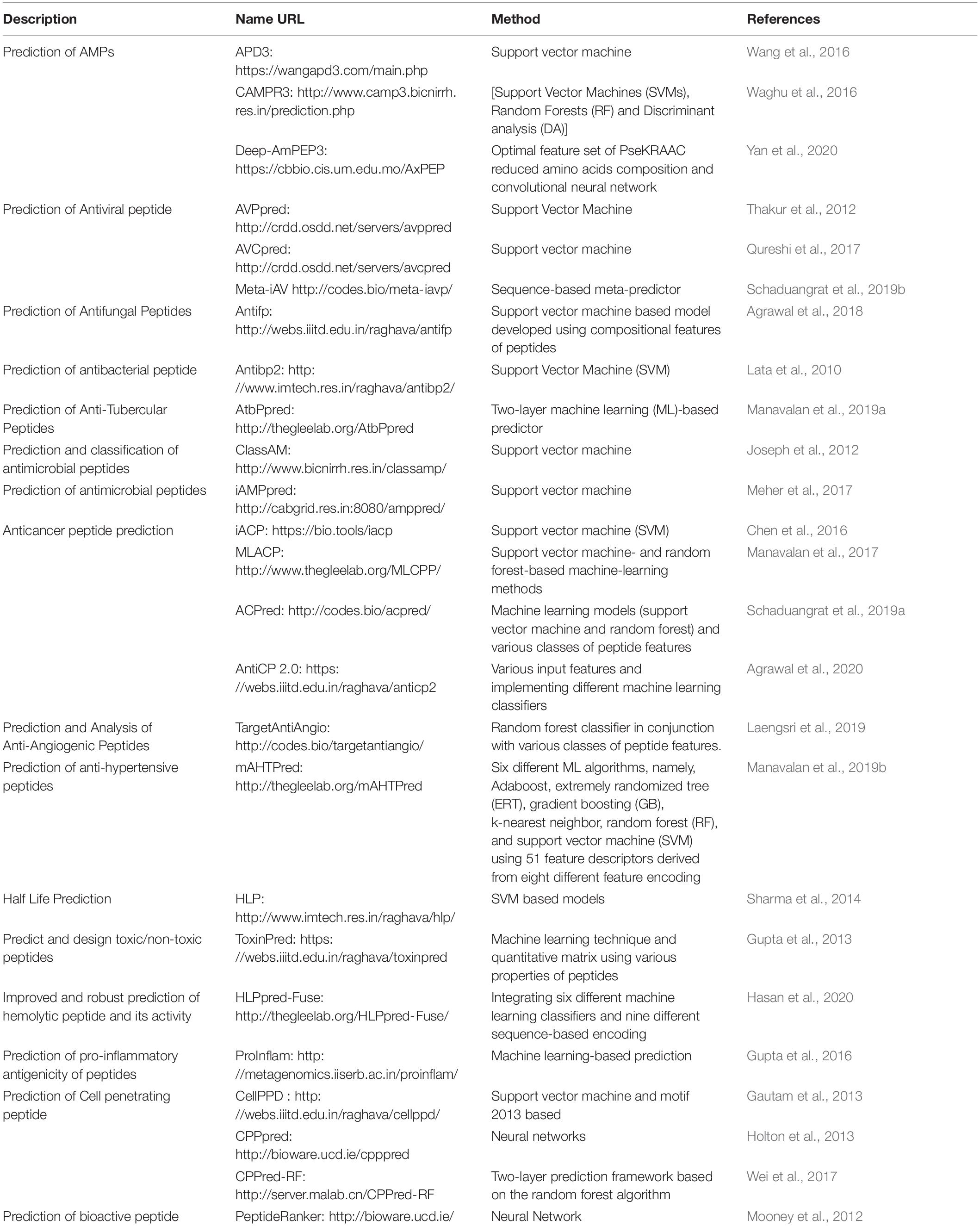
Table 2. Sequence-based peptide design tools.
Numerous methods, which are classified into general and specific methods, have been developed to predict AMPs. The APD3 (Wang et al., 2016) and CAMPR3 (Waghu et al., 2016) databases are among the general method predictors. They are designed to predict whether a given peptide is AMP or non-AMP. APD3 is developed for the classification, prediction, and design of AMPs using the parameter space defined by all available natural peptides in the database. The CAMPR3 implements four different machine-learning (ML) techniques to develop a peptide model. Deep-AmPEP30 (Yan et al., 2020) is a recently developed method that is used to predict bioactive sequences from genomes. This tool uses a short-length AMP prediction method based on the optimal feature set of PseKRAAC, reduced amino acid composition, and convolutional neural networks. The second group of methods is designed to predict AMPs, specifically on viruses, fungi, bacteria, or parasites. AVPpred (Thakur et al., 2012), AVCpred (Qureshi et al., 2017), and Meta-iAV (Schaduangrat et al., 2019b) are the more commonly used tools for the prediction of antiviral peptides. AVPpred (Thakur et al., 2012) is a web server used for the collection and detection of highly effective antiviral peptides (AVPs) using ML techniques such as support vector machine (SVM), features like the amino acid composition, and physicochemical properties. The AVCpred method is an SVM-based AVP prediction method. The experimental inhibitory percentage from ChEMBL (a large-scale bioactivity database for drug discovery) predicts the antiviral compounds against HIV, hepatitis C virus, hepatitis B virus, human herpesvirus, and 26 other viruses. Meta-iAVP is a sequence-based meta-predictor with an efficient feature representation (Schaduangrat et al., 2019b). It is designed for the accurate prediction of AVPs from peptide sequences. Antifp (Agrawal et al., 2018) is designed to predict antifungal peptides using features like amino acid composition, and similarly, Antibp2 (Lata et al., 2010) is another SVM-based method developed to predict antibacterial peptides. AtbPpred (Manavalan et al., 2019a) is a two-layer ML-based predictor for the identification of anti-Mycobacterium tuberculosis peptides. Moreover, ClassAMP (Joseph et al., 2012) and iAMPpred (Meher et al., 2017) are two methods used to predict the AMP class (e.g., antibacterial, antifungal, and antiviral). iAMPpred (Meher et al., 2017) predicts the probability of a peptide to be an antibacterial, antifungal, and antiviral agent by providing the probability score for all of the three classes.
The discovery of ACPs has provided an alternative approach to treating cancer. iACP (Chen et al., 2016), MLACP (Manavalan et al., 2017), ACPred (Schaduangrat et al., 2019a), and AntiCP 2.0 (Agrawal et al., 2020) are some famous in silico tools for the prediction and design of ACPs. The discovery of anti-angiogenic peptides is a promising therapeutic route for cancer treatment. TargetAntiAngio (Laengsri et al., 2019) was developed for the prediction and characterization of anti-angiogenic peptides using the random forest classifier in conjunction with various classes of peptide features and was demonstrated to be superior to other existing methods. mAHTPred (Manavalan et al., 2019b) was developed to predict anti-hypertension peptides using six different ML algorithms and showed superior performance compared to existing methods. HLP (half-life prediction) (Sharma et al., 2014) was developed for the prediction and design of peptides with desired half-life using SVM methods.
ToxinPred (Gupta et al., 2013) is one of the most applied SVM-based tools for predicting peptides toxicity. HLPpred-Fuse (Hasan et al., 2020) is the only tool that simultaneously identifies hemolytic peptides and their activities. It has fused six different ML classifiers in a robust hemolytic peptide prediction method. ProInflam (Gupta et al., 2016) was developed to predict the pro-inflammatory antigenicity of peptides using an ML-based prediction tool. CellPPD (Gautam et al., 2013) is an SVM-based method that has been widely used to predict CPPs. CPPpred (Holton et al., 2013) is another server used to predict CPPs based on artificial neural networks. In CPPred-RF (Wei et al., 2017), the random forest algorithm can simultaneously predict the CPPs and their uptake efficiency. PeptideRanker (Mooney et al., 2012) is also a webserver used to predict the probability of the peptides being bioactive and ranks the bioactive peptides.
In Silico Tools and Algorithms for Peptide Modeling
Bioactive peptides are critical in industrial, medical, and biological applications, and they play pivotal roles in regulating various biological processes (Gesell et al., 1997; Liu et al., 2008). There are several conventional methods to identify the tertiary structure of peptide molecules, including CD, electron paramagnetic resonance (EPR), Fourier-transform infrared (FTIR), and NMR spectroscopies and x-ray crystallography. However, these empirical methods are labor-intensive, time-consuming, and expensive to perform. Moreover, the employed solvent may have a significant influence on the peptide structure in some instances. Given these circumstances, computational methods for predicting the 3D structures of the peptide have emerged to circumvent these limitations. These methods are expected to contribute significantly to the delineation of peptide sequence to function relationships and the promotion of efficient designs for new peptide molecules. Like the methods for predicting protein 3D structure, peptides could be modeled by employing the homology, threading, and ab initio approach. The peptide modeling tools use one or a combination of these methods to predict the 3D structures of the peptides. The homology and threading methods rely on suitable template structures. Previously resolved peptide structures stored in PDB have a crucial influence on the accuracy of homology-based peptide modeling (Sali, 1995; Sanchez and Šali, 1997). The threading method is performed using the existing folds of proteins as the modeling templates (Bowie et al., 1991; Jones et al., 1992). Unlike the homology and threading methods, the ab initio approach is not template-based and exploits the physicochemical features to predict low-energy folding of the peptides (Monge et al., 1994; Bradley et al., 2005). In this regard, the bioinformatic tools involved in peptide modeling are classified into template-based and template-free methods. Various tools and servers have been developed for the prediction of peptide structures. Among the various available tools, six highly referred servers (PEPstr, Protinfo, PEP-FOLD, PEP-FOLD3, Hmmstr/Rosetta, PepLook, and PepSite & FlexPepDock server) are introduced in the following sections.
PEPstr Server
This server can predict the 3D structures of the peptide with an average length of 7–25 and sometimes 27 amino acids. It uses valuable and vital information about the beta-turns in predicting the 3D structure of the peptide. This server acts through different steps to model the peptide 3D structure. Primarily, all residues of the peptide get an extended conformation (phi = Psi = 180°). The secondary structure information from regular secondary structures (helices, beta-strands, and coil) and the beta-turns method help to detect the 2D structure of the peptide. Then, the conformational shape is created by assigning the Psi (Ψ) and phi (Φ) angles of the main chain. The standard Dunbrack backbone-dependent Rotamer library is used to determine the side chain angles. Ultimately, the obtained peptide model is refined by molecular dynamics (MD) simulation and energy minimization. The modeled 3D structure of the peptide will be stored in PDB format. Of note is that MD simulation can be accomplished in vacuum, hydrophilic, and hydrophobic states. The web server of PEPstr4 was evaluated for the modeling of short peptides (Kaur et al., 2007). This server can model natural and non-natural amino acids, D amino acids, terminal modifications, peptide cyclization, post-translational and advanced modifications of residues, and structure simulations.
Protinfo Server
This server is highly suitable for the prediction of complicated protein structures. The function of this server is based on the interolog approach for the detection of experimental samples. Using the interolog approach is helpful for the identification of similar results found in the existing databases. Following this step, this server modeled input queries based on the determined homologous samples. One of the prominent features of the Protinfo server is that it can support a wide range of templates, from small amino acid subunits to a large number of them. The Protinfo PPC web server is available at http://protinfo.compbio.washington.edu/ppc/ (Hung et al., 2005).
PEP-FOLD Server
This is a high-performance server for de novo prediction of peptide structures from amino acid sequences. It utilizes a hidden Markov model (HMM) to make the predictions. It functions through the identification of primary structural alphabet (SA) letters in each sequence. The SA letters extracted by the HMM are necessary to describe the correct conformations of four consecutive residues (Zemla, 2003). It couples the predicted series of SA letters to a greedy algorithm and a coarse-grained force field (Souza et al., 2020). PEP-FOLD can handle peptides with 9–25 amino acids (Maupetit et al., 2009, 2010). PEP-FOLD3 is an improved version of the PEP-FOLD server. It is also a de novo approach with a more advanced and faster peptide structure modeling system. It can accommodate a vast range of peptide sizes ranging from 5 to 50 amino acids (Lamiable et al., 2016). PEP-FOLD and PEP-FOLD3 are respectively accessible at https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD/ and https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/.
I-Sites/Hmmstr/Rosetta Server
This server can predict the secondary, local, super-secondary, and tertiary structures of the protein sequences. This server also uses a hidden Markov model (HMMSTR) for local and secondary structure prediction, based on the I-sites library. It has three parts: I-site, Hmmstr, and Rosetta. I-site-motifs is a library that contains an extensive collection of small motif sequences (3–19 motifs). The I-site library is connected with numerous structured databases and encompasses about one-third of database sequences (Bystroff et al., 2000). According to the literature, the combination of I-sites and Rosetta is advantageous. Rosetta ab initio 3D predictor is a Monte Carlo (MC) Fragment Insertion protein-folding program that can use the results of Critical Assessment of Fully Automated Structure Prediction (CAFASP2) during the process (Fischer et al., 2001). There are some steps in the execution process of this server. The first step is the creation of a profile sequence through multiple alignments of the input sequence using PSI-BLAST (Position-Specific Iterative Basic Local Alignment Search Tool). The second step is the prediction of I-site motifs with a determined profile sequence. The third step is the preparation of fragment movesets from predicted I-site motifs. The fourth step is the modeling of secondary and local structures via HMMstr or the analysis of fragment moveset by the Rosetta server (Bystroff and Shao, 2002). Moreover, the I-sites/Hmmstr/Rosetta web server5 is developed for 3D prediction.
PepLook Server
PepLook is another 3D peptide modeling web server that can model sequences with more than 30 residues. This server prepares many randomly produced peptide structures by altering the SA angles (Φ/Ψ). Modeling cyclic peptide conformation using distance restraint is one of the vital beneficial aspects of this server (Etchebest et al., 2005). Moreover, this server can model the 3D structure of post-translationally modified amino acids (carboxylated or hydroxylated), synthetic amino acids, and ribosomal peptides. PepLook can measure the energy factor of some features, including internal and external hydrophobicity, complete peptide structures (all atoms), and electrostatic and van der Waals interactions (Thomas et al., 2006). The PepLook server could be found at https://orbi.uliege.be/handle/2268/135016.
PepSite and FlexPepDock Server
Protein–peptide interactions are a vital part of many cellular signaling pathways. Getting a good grasp of these interactions would bring about a mechanistic understanding of how cell networks are regulated. The PepSite server can predict the binding of a given peptide onto a protein structure which unveils the details of the interaction of interest. PepSite 26 is a complete rewrite of the original PepSite, which can speed up the presentation of results to a fraction of a second. The surface position-specific scoring matrix (S-PSSMs) algorithm is used in this server to detect the binding sites for each peptide residue. Ultimately, a suitable peptide sequence can be generated against predicted biding sites considering certain distance limitations (Trabuco et al., 2012). FlexPepDock7 is a high-resolution peptide-protein docking (refinement) protocol for the modeling of peptide–protein complexes. It is implemented in the Rosetta framework, which can be combined with the result of PepSite to design atomic models for given peptides in the vicinity of binding sites (London et al., 2011; Raveh et al., 2011). The summary of the information on the mentioned servers is presented in Table 3.
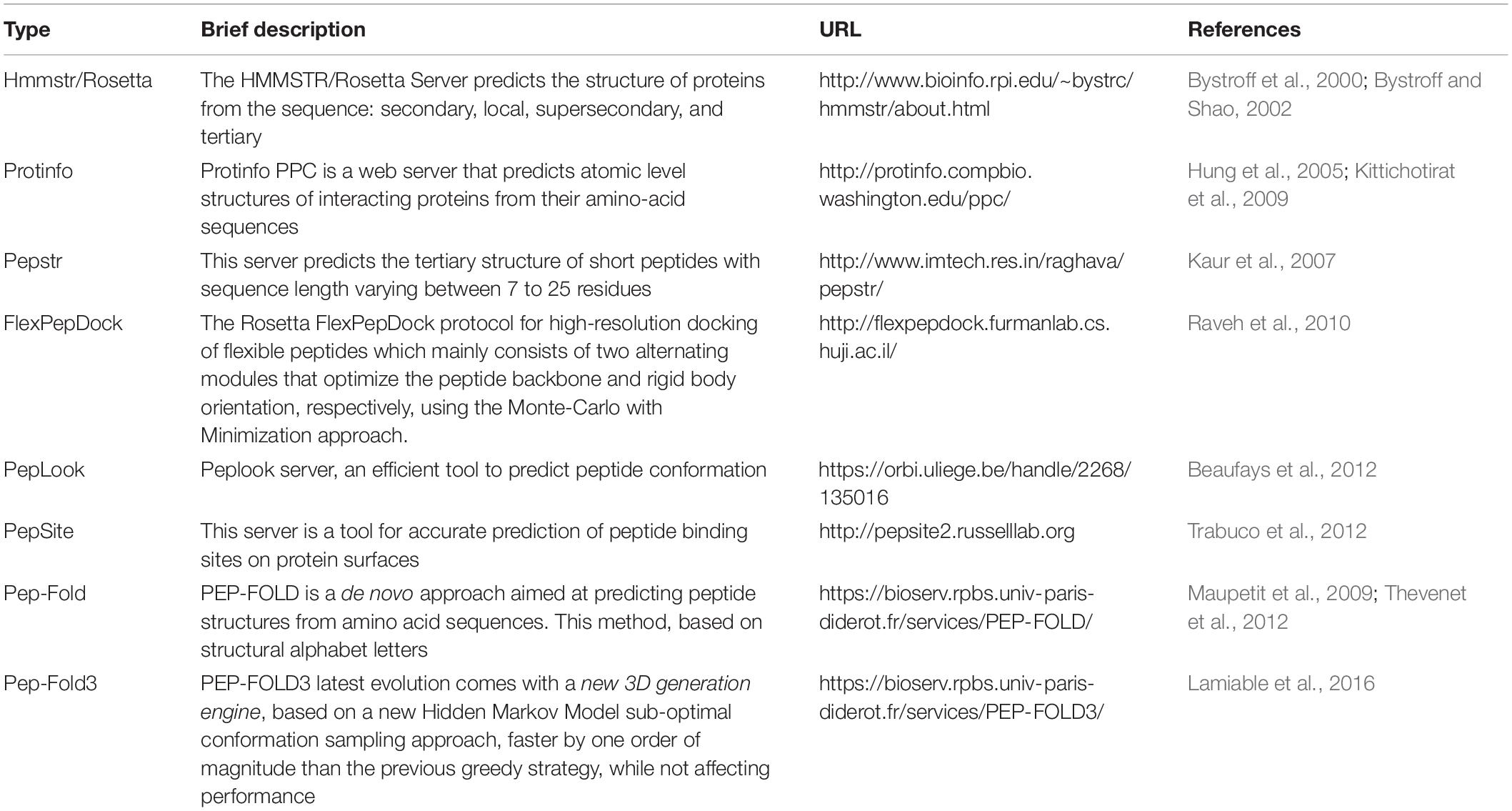
Table 3. List of important in silico peptide modeling servers.
In Silico Tools for the Prediction of Protein–Protein Interfaces
Proteins are vital agents that carry out all biological activities in the cells. PPIs play a pivotal role in the execution of these functions. Hence, investigating the changes that occur in the space between two proteins can be highly beneficial in the elucidation of disease etiology and the adaptation of appropriate treatment strategies. Various experimental methods exist for detecting biological changes in the PPI interface, including NMR, x-ray crystallography, mass spectrometry, and alanine scanning mutagenesis. NMR and x-ray crystallography can identify the interfaces at the atomic level, while the other methods like alanine scanning act at the residue level. Although the information about some of the PPI interface networks is extracted from high-throughput experiments, these experimental methods have their constraints mainly because they are expensive and time-consuming (Chen and Liu, 2005; Shoemaker and Panchenko, 2007; Casals et al., 2012; Koboldt et al., 2013). In this study, we focus on some computational methods for predicting the properties of the PPIs that can be valuable in circumventing the limitation of traditional pipelines. There are various computational approaches for the analysis of PPI interfaces. The existing strategies could be divided into information-based approaches and docking-based approaches (Figure 1). Docking is an important method that can facilitate the reconstruction of binary residue connections between two protein sections.
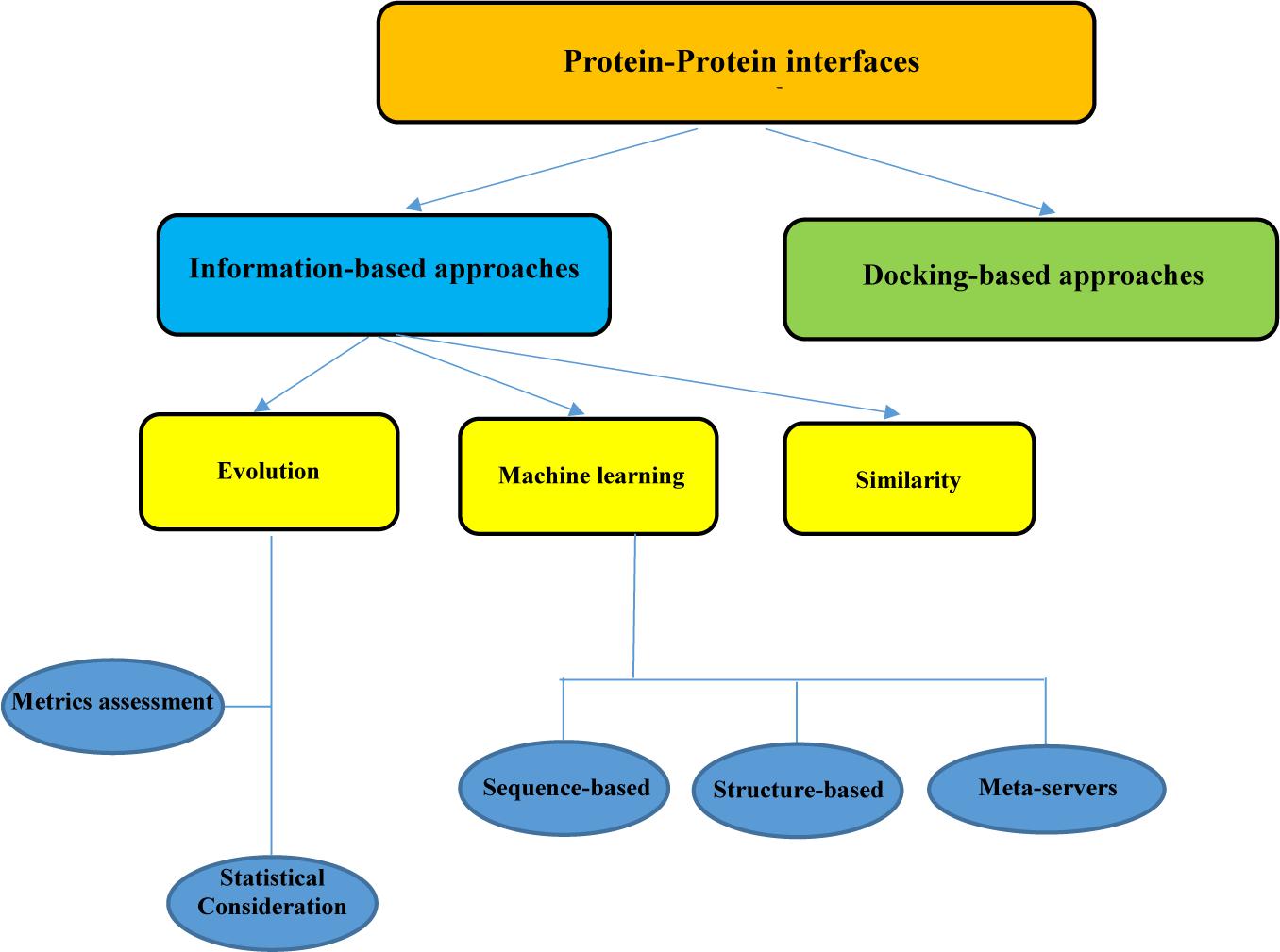
Figure 1. Summary of protein–protein interface analysis procedure.
Information-Based Approach
A wide variety of in silico methods has been developed for data-based approaches. This approach is instituted on the information that is extracted from the results of conventional experiments. Three main strategies have been established for data-based approaches to analyze the PPI interfaces, namely the similarity-based method, the ML method, and the evolutionary-based method.
Similarity-Based Method
This method, also known as the “template-based” method, is one of the most widespread searching approaches for annotating the genome function. Instead of using partial sequence and statistical features of the sequence, the method relies on the similarity of whole protein sequences. Several biological properties can be obtained by multiple sequence alignments that are practical for identifying PPIs. These properties include the conservation of amino acids, the similarity between interfacing proteins, and gene fusion. Different behaviors can be expected from the proteins using this method. For example, unstable proteins tend to utilize different interfacial binding patterns. Therefore, using homology search for the extraction of interface residues remains a controversial issue (Grishin and Phillips, 1994; Caffrey et al., 2004; Esmaielbeiki et al., 2016). A list of similarity-based prediction tools for PPI interfaces is represented in Table 4.

Table 4. Similarity-based prediction databases (information-based approach).
ML Method
Although there are beneficial aspects for the similarity-based method of predicting PPIs, it suffers from restrictions such as lack of accessible interfaces for experimental homologs and low quality (de Vries and Bonvin, 2008). ML methods operate based on a comparative pattern between interfacial protein residues and non-interfacial residues. This method could compensate for the limitations of the similarity-based methods. This approach can be categorized within the sequence and structural-based methods and consensus identifiers (Xie et al., 2020). Moreover, the use of ML methods for the prediction of PPIs has represented improved performance in comparison to other conventional approaches, such as neural networks. Thus, ML methods could play a significant role in therapeutic peptide development (Yang, 2008).
Sequence- and structural-based method
The sequence-based interface identifiers operate according to the features of the protein sequences. Most of these tools rely on evolutionary information from multiple sequence alignments (MSA) projected on the protein surfaces (Jordan et al., 2012). Therefore, these methods are inadequate in detecting interfaces of proteins with sparse homolog sequences and evolutionary variable regions. Advancements in structural proteomics have justified the establishment of structure-based automated methods for the prediction of functional surfaces of proteins (Jordan et al., 2012). The performance of this method depends on two 3D mappings and the 3D identifier. The 3D mapping is pertinent to the detection of close structures, and the 3D identifier is related to specific features such as protein surface accessibility (Hoofnagle et al., 2003; Heinig and Frishman, 2004), B-Factor (Halabi et al., 2009), the formation of the protein surface, and the 2D structure (Heinig and Frishman, 2004; Hamer et al., 2010) (Table 5).
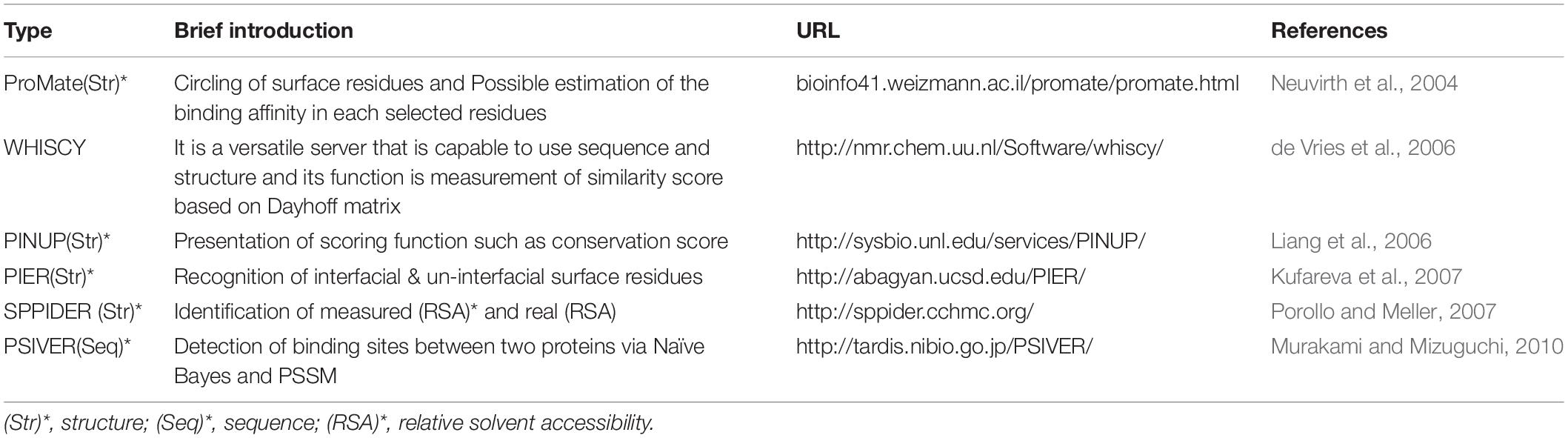
Table 5. Sequence- and structural-based databases (ML approach).
Meta-servers
Meta-servers combine several interface prediction methods into a consensus predictor to attain more reliable and stable predictions compared to the results of each predictor on its own. This means that meta-servers can act creatively to use different databases as complementary units to improve their function (Kozlowski and Bujnicki, 2012). Three common meta-servers are presented in Table 6.

Table 6. Three main meta-servers for prediction of interfaces (ML approach).
Evolutionary-Based Approach
The prediction of PPI interfaces using evolutionary data is one of the most well-known interface classifiers. This approach is deemed as a realistic approach for the identification of the biological features of PPI interfaces. The EPPIC (Evolutionary Protein–Protein Interface Classifier) server8 is a tool designed to predict the quaternary structure of proteins from their crystal structures. Primarily, it classifies the interfaces of the crystal structure to determine their biological relevance. All topologically valid assemblies are computed, and the individual interface scores are used to predict the most likely quaternary assembly (Duarte et al., 2012).
Docking-Based Approach
Docking is one of the most well-known computational methods that are highly advantageous for predicting interfaces of biological targets and molecules (Kitchen et al., 2004). This method is practical for atomic-level investigation of molecular interactions. There are several different docking methods, and the selection of each server depends on the complexity of the problem and structure sources. Some structures are extracted via NMR or x-ray crystallography, which are suitable for the available docking servers. However, the template-based approach is the best candidate for predicting complex structures (Porter et al., 2019). Testing the accuracy of computational docking algorithms in blind predictions is managed by the CAPRI (Critical Assessment of Predicted Interactions) project (Janin et al., 2003). Molecular docking can be accomplished by a vast range of tools that are already accessible. Many docking tools act on the principle of rigid-body interactions, which results in a suitable match between the surfaces of tertiary (3D) structures. However, working with them has many restrictions such as protein flexibility, which is not considered. Given the importance of molecular docking in the determination of interface residues, many efforts have been made to mend the existing limitations (Luiz Folador et al., 2015; Pinzi and Rastelli, 2019).
In Silico Hotspot Prediction Tools
It is well established that the energy distribution is not uniform in PPI interfaces, and a small number of residues have the largest share in binding free energy. Wells and Clackson, who studied the binding of the growth hormone to its receptor, discovered these residues and used the term hotspot for them (Wells, 1991; Clackson and Wells, 1995; Schreiber and Fersht, 1995; Stites, 1997; Bogan and Thorn, 1998; Clackson et al., 1998; Hu et al., 2000; Keskin et al., 2005; Kouadio et al., 2005). Subsequent studies showed that hotspots form a small number (about 9.5%) of residues of the interface area. Therefore, a more precise definition of hotspots has been proposed, which states that a hotspot is a residue whose mutation to alanine reduces at least 2 kcal/mol in binding free energy (ΔΔG = ΔG mutant type –ΔG wild type) (Bogan and Thorn, 1998; Thorn and Bogan, 2001; Moreira et al., 2007). Hotspot residues are mainly composed of tyrosine (12.3%), arginine (13.3%), and tryptophan (21%) amino acids (Lichtarge et al., 1996; Bogan and Thorn, 1998). Studies have shown that hotspots have conserved structures and predictable physicochemical properties (DeLano, 2002; Moreira et al., 2007; Kenneth Morrow and Zhang, 2012). Impaired PPIs can cause many diseases, such as neurological disorders and cancer. Moreover, the conserved structure of hotspots, as well as their great impact on the binding energy, has made them attractive medical targets for the design of inhibitor drugs. Thus, many unwanted PPIs can be avoided by the use of these inhibitors, and they could be more effective in treating various diseases (Livnah et al., 1996; Wrighton et al., 1996; Tilley et al., 1997; Li et al., 1998; DeLano et al., 2000; Sidhu et al., 2000; Arkin and Wells, 2004; Thanos et al., 2006; Moreira et al., 2007; Wells and McClendon, 2007; White et al., 2008; Blazer and Neubig, 2009). As mentioned, hotspots are predictable, and it is worth mentioning that one of the prediction methods is based on the experimental alanine scanning method. For example, when a large tryptophan residue (as one of the three main hotspot residues) mutates into alanine, this difference in size causes the formation of a cavity, resulting in instability of the complex due to binding energy reduction. Alanine scanning means that when a residue mutates into alanine, the amount of binding energy decreases; if this decreased energy is significant (10-fold or more), then that mutated residue is considered as a hotspot (Bogan and Thorn, 1998; DeLano, 2002). Mutation to alanine residue removes the side chain effect. The methyl side chain of the alanine residue is relatively neutral and also lacks the additional flexibility contribution (Cunningham and Wells, 1989; Wells, 1990; Skolnick et al., 2000). Although glycine mutagenesis removes the contribution of the side chain, it can cause flexibility in the protein backbone (Morrison and Weiss, 2001). Hence, glycine mutagenesis is not considered for hotspot detection. The Alanine Scanning Energetics Database (ASEdb) contains the results of alanine scanning experiments. The Binding Interface Database (BID) has verified experimental hotspots in the literature (Thorn and Bogan, 2001; Fischer et al., 2003). Despite their advantages, these databases also are associated with several drawbacks. The hotspots obtained from experimental studies can only be attributed to a limited number of complexes. Moreover, it is recommended to avoid these data to interpret specific residual interactions (DeLano, 2002). Experimental mutagenesis of proteins to find hotspots is not practical and useful on a large scale because individual mutated proteins must be purified and analyzed separately. It should also be noted that alanine scanning and other experimental hotspot analysis methods are highly time-consuming and costly. Given these circumstances, theoretical and computational prediction methods seem attractive for hotspot residue prediction (Kenneth Morrow and Zhang, 2012). FoldX9 is a hotspot prediction tool and server which uses the FOLDEF algorithm developed by Guerois et al. (2002). This server predicts the hotspots of PPIs by using an energy-based method. It finds the PPI energy changes with the computational alanine scanning technique (Schymkowitz et al., 2005; Kenneth Morrow and Zhang, 2012). The Robetta server10 developed by Baker and Kortemme includes various parameters such as implicit solvation and hydrogen bonding, packing interactions, solvation interactions, and Lennard-Jones interactions to calculate the interaction free energy. The method employed in this server is similar to that in FoldX (energy-based method), and the technique used is computational alanine scanning (Kim et al., 2004). Similar to FoldX, the parameters obtained from the Robetta server are based on changes in protein stability. This server mutates the side chains to alanine and then locally repacks the parts of the structure that fall in a 5-Å radius of the mutated residue. The rest of the protein structure remains unchanged. The changes in binding energies of PPIs result from these mutations and form the basis for hotspot predictions (Kortemme et al., 2004). The Robetta server can accurately predict 79% of hotspot residues with a 1.0 kcal/mol cutoff. However, it enables us to find the hotspots involved in hydrogen bonds with water molecules (Kenneth Morrow and Zhang, 2012). PP_Site is a structure-based tool with a simple algorithm technique based on three factors, namely van der Waals interactions, hydrophobic interactions, and H-Bond, developed by Gao et al. (2004) and Kenneth Morrow and Zhang (2012). FTMAP11 is an energy-based hotspot prediction tool and server. It uses a probe-based rigid-body docking with fast Fourier transform correlation. The users of FTMAP only need the PDB code or the PDB file of the protein for the prediction (Brenke et al., 2009; Kenneth Morrow and Zhang, 2012). PCRPi (Presaging Critical Residues in Protein interfaces) is a method that uses the Bayesian networks technique to unify evolutionary, structural, and energetic determinants into a common probabilistic framework. PCRPi was upgraded to PCRPi-W12 to function as a web server. The users can upload a complex or enter a PDB code and select the type of Bayesian network architecture (expert or naïve) (Assi et al., 2010; Mora et al., 2010). HotPoint13 is another hotspot prediction server that uses an empirical formula technique and simple architecture. Its prediction method is based on the contact potential of the interface residues and solvent accessibility developed by Tuncbag et al. (2009). The accuracy of this server is about 70%. The results are a table of interface residues in which the hotspots and their properties are highlighted (Tuncbag et al., 2009, 2010). Grosdidier and Fernández-Recio (2008) have developed an energy-based (Docking) tool with a normalized interface propensity technique. Higa and Tozzi (2009) have developed a tool based on structural and evolutionary methods with the SVM technique. Rajamani et al. (2004) has developed a tool based on sidechain ΔASA (accessible surface area) with the MD technique. HSPred14 is an energy-based tool with an SVM (Residue specific) technique (Lise et al., 2009, 2011). MINERVA is a tool based on molecular interaction, structure, and sequence methods with decision tree and SVM techniques (Cho et al., 2009). KFC215 is a server based on various structural features and ASA methods with the SVM technique (Zhu and Mitchell, 2011). Guharoy and Chakrabarti (2009) have developed a tool based on the interface location and H-bonding methods with the simple algorithm technique.
Peptide–Protein Docking Tools
Molecular docking is a highly applicable method in the design and discovery of small-molecule drugs. This method has also undergone drastic progress in the prediction of PPIs. The prediction of peptide–protein interactions is the subject of similar attempts to develop more amenable peptide therapeutics (Diller et al., 2015; Ciemny et al., 2018). However, docking methods generally struggle with the issue of modeling the considerably more flexible and larger peptide molecules (London et al., 2013). However, peptide therapeutics have recently garnered a lot of attention, which has led to rapid advancements in peptide-protein docking-related technologies. These efforts have resulted in advances in drug design and discovery (Fosgerau and Hoffmann, 2015; Bruzzoni-Giovanelli et al., 2018; Ciemny et al., 2018). There are three main approaches for peptide-protein docking: global docking, local docking, and template-based docking. Different methods have varying degrees of prediction accuracy, often dictated by the amount of interaction information given as input (Ciemny et al., 2018). Template-based docking methods build a model of the complex using known structures, and it can be beneficial if the template is close to the investigated complex (Kundrotas et al., 2012; Lee et al., 2015; Lensink et al., 2017; Marcu et al., 2017; Pallara et al., 2017). Local docking methods look for a peptide-binding pose that is close to a user-defined binding site. Thus, the docking accuracy relies on the input information. The more precise the defined binding site, the better the results (Ciemny et al., 2018). Global docking methods conduct a collaborative scan for the pose and peptide-binding site. The most basic procedure for this approach is considering the protein and peptide input conformations to be rigid and conducting a comprehensive rigid-body docking. Predicting the peptide conformation based on a sequence given by the user is a more complicated approach for this method. Typically, their pipeline includes three phases; primarily, the input peptide conformations should be created by various strategies [e.g., utilizing monomeric protein structure fragments (Yan et al., 2016; Porter et al., 2017), threading the sequence onto a predefined set of template conformations (de Vries et al., 2017), or peptide folding simulation in solution (Ben-Shimon and Niv, 2015)]; then, the docking of rigid bodies should be implemented; ultimately, scoring and, or refinement of the models should be performed (Ciemny et al., 2018). At least three significant challenges lie ahead in the path of efficient peptide-protein docking. The first challenge is the prediction of significant conformational changes in the protein and peptide molecules (flexibility problem) upon docking. The second challenge is selecting the structure with the highest accuracy from a large number of produced models (scoring problem). The third challenge is integrating computational predictions and experimental findings into the peptide-protein docking scheme (integrative modeling) (Ciemny et al., 2018). Table 7 includes features and descriptions of the main peptide-protein docking tools and servers that are currently available.
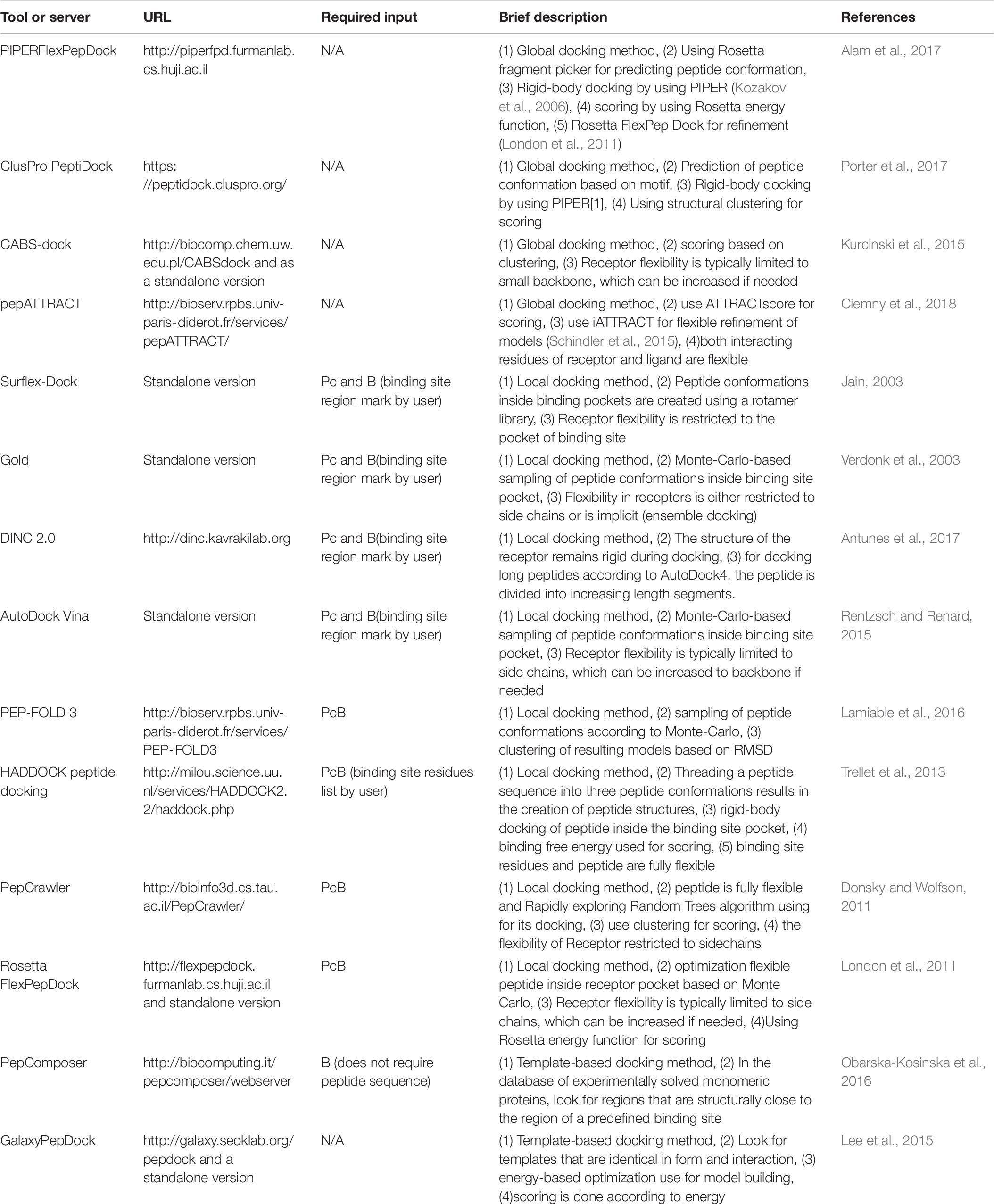
Table 7. Peptide-protein docking tools and servers.
Virtual Screening Methods for Peptide–Protein Interactions
Virtual screening (VS) is a robust computational technique capable of searching huge libraries of small molecules and identifying the most suitable ones against a protein receptor. VS has emerged as a complementary technique of high-throughput screening (HTS). Nowadays, thanks to the advancements in VS, it has become an indispensable part of the drug discovery process, leading to enormous savings in cost and time. Recent studies have established that VS can help to develop inhibitors of PPIs.
Exploration of new macromolecular structures by NMR or x-ray crystallography methods, human genome sequencing, and rapid computational methods could improve VS searches. Although increasing the numbers of the unveiled structures of protein–ligand complexes has made VS more convenient, 3D structure prediction approaches could be employed where experimentally determined structures of proteins/peptides are not available (Slater and Kontoyianni, 2019).
There are several accessible public databases for known drugs, small molecules, and chemical compounds (natural or synthetic) which could be searched when implementing VS strategies: ChemDB16, ChemBank17, NCI Open Database18, ChEMBL19, PubChem20, ZINK21, ChemSpider22, and DrugBank23 (Lavecchia and Di Giovanni, 2013). There are also some commercial databases containing data derived from patents or literature, including ACD24 and WOMBAT25 (Lavecchia and Di Giovanni, 2013). There are two main categories of VS: ligand-based VS (LBVS) and structure-based VS (SBVS).
Ligand-Based VS
The LBVS approach relies on the extracted structural and bioactivity information from an enormous small-molecule library. Three-dimensional shape matching is one of the popular LBVS methods based on seeking molecules with a similar shape to that of the known active molecules. Utilizing pharmacophore models to find the intended ligand, quantitative structure–activity relationships (QSAR), and chemical similarity analysis (to look in a database of molecules against one or more active ligand structures) are additional LBVS methods.
Similarity analysis is one of the most common techniques of VS. In the similarity searching method, the closest molecules are identified for a known active reference structure. Nevertheless, this method is simply influenced by the users because the selection of accurate input molecules is a challenging issue. However, this is a fast VS method (Lavecchia and Di Giovanni, 2013; Wu et al., 2019).
Pharmacophore modeling is another method for LBVS. A pharmacophore is a set of crucial molecular properties involved in accurate molecular recognition and interactions of a ligand with a specific biological target. Since 3D models are necessary for docking, pharmacophores could serve as vital VS processes to explore novel ligands for receptors with unknown 3D structures. There are various pharmacophore-based VS case studies for peptides. For example, a study designing the pharmacophore model for secretin resulted in gaining novel angiotensin-converting enzyme (ACE) inhibitory peptides with the desired biological activity. Angiotensin-I-converting enzyme inhibitory peptides and Intestinal Peptide Transporter hPepT1 are some of the more successful examples of pharmacophore modeling in LBVS methods (Dong et al., 2011; Wang et al., 2011; Osmulski et al., 2020). However, pharmacophore modeling has some limitations, especially when working with peptides. Some of the snags present in peptide-based pharmacophore modeling are the lack of acceptable scoring metrics, lack of clear instructor pharmacophore query, improper binding affinity evaluation, and incorrect or inadequate conformational sampling. These limitations could lead to false-positive and false-negative results (Kaserer et al., 2015; Ciemny et al., 2018; Slater and Kontoyianni, 2019).
Quantitative structure–activity relationship is one of the most popular LBVS methods. This technique could involve the interplay between biological function, the potency of active molecules, and their structural/physicochemical features. QSAR-based methods need information such as the values of IC50 and the binding affinity (Kd). QSAR modeling is classified into 2D-QSAR and 3D-QSAR; 2D-based algorithms are faster but less precise in comparison with 3D-based algorithms. The solubility, flexibility, ligand and protein conformations, and structures are not considered in both of the methods (Kanakaveti et al., 2020).
Structure-Based VS
The SBVS methods require knowledge of the 3D structures of proteins and not the biological function of known molecules. SBVS involves docking the candidate molecules with the protein target, followed by ranking the predicted binding affinity by scoring functions to detect potential lead candidates. In this approach, the 3D structure of the protein of interest should be available through x-ray crystallography, NMR, or homology modeling. Subsequently, docking could engage the small molecules as ligands of the receptor via computational algorithms; then, the top-ranked compounds could be selected for further experimental studies. Scoring of the ligands through different scoring functions (empirical, knowledge-based, and force field-based) is a critical step in SBVS. The flexibility of the target structure is another complicated aspect of SBVS, which could noticeably compromise the accuracy of the approach. In recent years, docking algorithms are seriously confronting this challenge by improving soft docking. MD simulation, which considers ligand flexibility and entropic effects, could also be combined with SBVS or LBVS to increase the information accuracy of the binding pose of candidate molecules and subsequently improve the drug development (Yan et al., 2019; Wang and Sun, 2020).
Combining Ligand-Based and Structure-Based Approaches
The drawbacks and limitations of traditional VS methods could be alleviated by combining LBVS and SBVS. This unified approach applies both structural similarity and ligand-based data. A researcher could select one of the LBVS, SBVS, or integrated approaches depending on the case study. One of the successful reports in recent years is the designing of the polo-box domain of polo-like kinase 1 (PLK1-PBD) inhibitor through a combined strategy of SBVS pharmacophore modeling and molecular-docking screening techniques (Ramezani et al., 2019; Yan et al., 2019). The study resulted in the discovery of a peptide as a potent candidate for further experimental investigation. Notwithstanding the successful results gained by the combined approach, critical improvements in the combination strategies are still needed. One of the fundamental limitations is the complicated procedure of the platform handling, so improving the performance of this process would bring new advancements soon (Wang and Sun, 2020).
Optimizing Interfering Effects of Peptides
The interfering effects of peptides could not be accessed accurately by biophysical techniques such as x-ray crystallography, NMR spectroscopy, and fluorescence spectroscopy. They have unique features such as high selectivity and immunogenicity (Bruzzoni-Giovanelli et al., 2018). Despite their advantageous features, they could exhibit limited cellular penetrability, low in vivo stability, low solubility, and low binding strength. However, various methods have been considered to improve these limitations. The peptides could be optimized by chemical, biophysical, and in silico methods (Bruzzoni-Giovanelli et al., 2018). In silico methods of peptide optimization seem appealing due to their computational nature, which have the advantages of low cost, less time consumption, and avoiding the ethical issues of empirical analyses. There are some modifications that can be made to the in silico methods which can improve the efficacy of interfering peptides.
One way to optimize the performance of interfering peptides is the utilization of peptide design tools. Peptide design tools predict the spatial and energy constraints applied to them and suggest the best folding. Two main peptide-designing approaches include the stochastical and deterministic methods. In the deterministic approach, a complete space sequence is searched to reach the sequence fold with the lowest formation energy. In contrast, the stochastical method searches for the sequence space heuristically, which includes MC and genetic algorithms. The structure of the interfering peptide is fixed by optimizing the rotation angles of the lateral chains and energy minimization is performed by tools such as Rosetta Design. This software also improves the network of hydrogen bonds and van der Waals interactions (Roy et al., 2017). It should be noted that the scaffold can be changed during the modification of amino acids. Most methods such as GRAFTER, FITSIT, Proda Match, and Scaffold selecting suitable peptide patterns require geometric constraints such as the coordinates of the pattern-representing atoms. The AUTO match tool exhibits the flexibility of backbone patterns. When a correct peptide pattern is chosen, side-chain amino acids need to be modified to improve their correctness. Side chain changes can be evaluated by various factors, such as determining the binding affinity and structural stability. Tools such as ORBIT are used to reform the side chain that can bind to the target molecule (Roy et al., 2017). PPIs have a crucial role in signal transduction processes. A rapid computational approach has been developed to predict energetically critical amino acid residues in PPIs. The input consists of a 3D structure of the protein–protein complex. The output is a list of “hot-spot” or amino acids side chains that are predicted to become unstable when mutated to alanine (Kortemme et al., 2004). In silico alanine scanning could be used to improve the efficacy of the interfering peptides.
Poor cell membrane permeability of interfering peptides is a significant hurdle. Therefore, improvement of membrane permeability or development of strategies that facilitate active intracellular uptake will be critical for successful peptide-based targeting of intracellular PPIs. Increased uptake could be achieved by the identification of unnecessary hydrophobic amino acids. These amino acids can be replaced by charged or polar residues while maintaining their native bioactivity. Recently, in silico methods have been applied to speed up this process (Lee et al., 2019). CcSOL Omics is software that is used for the prediction of proteome solubility and the identification of solvent motifs according to the given amino acid sequence. PROSO-II is another SVM-based tool that predicts the solubility of a peptide based on physicochemical properties such as hydrophobicity, hydrophilicity, and features of secondary structure (Lee et al., 2019).
One of the main impediments ahead of clinical development of interfering peptides is their high sensitivity to proteases. Their half-life is highly reduced by proteolytic cleavage. The most common structural changes that increase protein stability are chemical changes. These optimizations include the acetylation of N-terminus and C-terminus ends of the peptide scaffold, the introduction of dextrorotary (D)-amino acids, and peptide scaffold cyclization (Sorolla et al., 2020). Engineering peptides by introducing D-amino acids instead of levorotatory (L) forms is an effective strategy to avoid proteolytic degradation by proteases. These D-amino acids could cause structural changes in the target peptides, which makes them unrecognizable by proteases. Recent studies have shown that interfering peptides containing D-amino acids have a longer half-life. Many therapeutic peptides containing unnatural amino acids have been approved by the FDA, such as degarelix for prostate cancer, semaglutide for type 2 diabetes, and carbetocin (an oxytocin analog containing methyl-tyrosine) for postpartum hemorrhage (Sorolla et al., 2020). Cyclization of peptides can increase their half-lives. This cyclization can be accomplished by different methods, such as the formation of a disulfide bond between cytosine, adding an amide bond between the C- and N-terminus (head to tail), and addition of amide bond between natural amino acid side chains (side chain cyclization). Click chemistry software is a widely used tool for designing cyclic peptides. PEP-Cyclizer is another software that is used to design head-to-tail cyclization. This software has other complementary features such as the ability to search for candidate sequences compatible with the cyclization of the peptide (a facility to assist medicinal chemists), the generation of 3D models of a cyclic peptide starting from the 3D structure of the un-cyclized peptide and the sequence of the cyclized peptide, and preliminary steps for the conformational stability analysis of other peptides, or peptide-receptor docking (Pierce et al., 2014).
Peptide–Protein Binding Energy Calculation Tools
The value of determining the binding energy of protein–ligand interactions in docking studies is apparent in the field of drug design. Acquisition of sufficient knowledge at a molecular level leads to accurate simulation, a valuable tool for drug design purposes. Experimentally measuring an inhibition constant (Ki) of the enzyme or protein in the presence of both the inhibitor and the substrate can estimate the binding affinity. In theoretical calculations, evaluating the properties of the protein, ligand, and after that, the complex can estimate the free energy of the ligand. Theoretically calculated free energies of the binding are often compared with the free energy of the reaction by calculating the Kd. Molecular mechanic force fields, which play a vital role in the conformational flexibility studies, like AMBER, MMFF, CHARM, and OPLS, can calculate various interactions in non-bonded atoms (Vanommeslaeghe et al., 2014; Ciemny et al., 2018). There are several methods for calculating the binding energy, namely endpoint, pathway, and alchemical methods.
Endpoint Methods
Endpoint methods are fast; however, their accuracy is usually limited to rational ranking (with r2 starting from 0.4 to 0.9, with a median of ∼0.7). The main advantage of endpoint methods is the requirement of only the simulation of the bound and free states of the ligand compared with pathway methods, which demand the simulation of many intermediate states likewise. Molecular mechanics/generalized Born surface area (MM/GBSA) and molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) are two general similar endpoint methods employed in protein–ligand-free energy calculations (Wang et al., 2019). Changes in solvation free energy, molecular mechanical energy, and conformational entropy are utilized to calculate the free energy of binding (Hall et al., 2020).
Linear interaction energy (LIE) is another endpoint approach used to figure out binding affinities. LIE relies on the concept that the free energy of binding shows a linear dependency on the polar and non-polar variations in ligand-surrounding energies. The strategy involves calculating values for the protein–ligand binding free energy ΔG bind with conformational sampling. Efforts such as considering multiple binding poses and continuum electrostatics solvation have promoted the accuracy of the LIE method (Rifai et al., 2020).
Chemical Methods
Although physically non-realizable, there are feasible computational processes of moving a set of atoms in a system from one state to another. In such a situation, modeling by alchemical free energy calculations could be one of the helpful computational processes. In strategies based on alchemical modifications, shifting the ligand into non-interacting mock particles may offer the absolute free energies of binding. Further, calculating the difference of free energy between two ligands can test the relative free energies of binding (de Ruiter and Oostenbrink, 2011). Free energy perturbation (FEP), Bennet’s acceptance ratio (BAR), and thermodynamic integration (TI) are typical samples of alchemical methods, in which the statistical mechanics provide the demanded data. Performing more extended simulations may yield way more accurate energy calculations (Ciemny et al., 2018).
In addition to established methods, some prefer using combinations of existing approaches or try novel strategies. RETI is a successful combination of TI and replica exchange (RE). TI is known as a powerful method used to calculate free energy differences. However, when several confirmations are involved, getting accurate outcomes becomes problematic. RETI can somewhat solve the problem by enhancing sampling efficiency. Enhanced sampling-one-step perturbation (ES-OS) and linear interaction energy/one-step perturbation (LIE/OS) are among other combined approaches. The aim of using the LIE/OS method is to enhance the accuracy of energy calculation. LIE calculates the charging atoms in the interaction cavity, while OS is applied to explain the contributions from cavity formation (de Ruiter and Oostenbrink, 2011).
Path Sampling
Path sampling approaches can meet the computational aspect of a process using the separation of timescales in biomolecular systems. Thus, it is advantageous when used in process of non-clear separation timescales. Path sampling methods compute functional transition rather than stable states, by physically removing the ligand from the protein and calculating the mean force along this path. Similarly, path sampling can be used efficiently for the conformational sampling of stable states fragmented by low barriers. Transition path sampling (TPS) and MC simulations are two approaches using a complete path. TPS is helpful for chemical or physical transitions of a system from one stable state to another state (such as protein folding or chemical reactions) that infrequently occur to be observed on a computer timescale (Chong et al., 2017).
There are some vital factors to be considered in using these methods, which might remarkably affect the accuracy and computational time of calculation. Deciding on the solvent, the simulation length, and ligand orientations, following the selection of relative or absolute free binding energies, is a critical step in the calculation process (de Beer, 2012).
Common Issues
Improving the accuracy and the speed of calculations simultaneously is the main aim of the methods described above; however, it seems challenging. As an example, factoring in solvent interactions will enhance the accuracy but also will increase the required computational time. Including explicit water molecules in the binding sites will increase the accuracy for specific systems. In explicit water simulations, grand canonical MC simulations (GCMC) can determine the fluctuation of the number of water molecules in the binding site. Paying attention to polarizability, accurate force field, and charge transfer is crucial in exact free energy calculations; their effects can be considered using quantum mechanics. Quantum mechanics can efficiently calculate electrostatic interaction energies. However, it would not be affordable in the study of massive molecules. Semi-empirical quantum methods are essential in computational chemistry for treating large molecules and studying electrostatic interactions in peptide-protein interactions. Semi-empirical approaches are generally based on the Hartree–Fock formalism with some more approximations and empirical data usage (de Beer, 2012; Rifai et al., 2020).
As mentioned above, a combination of the present methods has been recently developed to overcome the sampling problems and other shortcomings of the methods on their own, while strengthening their advantages. It is assumed that these combined methods would result in more accurate energy binding calculations, consequently having more reliable outcomes for application in drug design developments.
Discussion
In silico methods are implanted into every corner of the biological analyses. The study of IPs capable of modulating the PPIs is no exception and neither are the extended in silico tools established to develop, analyze, and optimize the IPs. These methods are anticipated to grow and become more accurate by reiterating the cycles of prediction and empirical assessment of these predictions. More precise algorithms would be developed, including the parameters of new players discovered by empirical studies. The novel and more accurate in silico tools would design and optimize an almost unlimited number of IPs with a high affinity against extracellular or intracellular (CPPs) PPIs. These tools would also help develop pharmacokinetically stable, safe, and effective IPs to modulate the PPIs within the cellular membranes. Moreover, these tools would be employed to predict, exert, and analyze specific chemical modifications to improve the ADME properties of the IPs. They also would be harnessed to design multifunctional IPs and even remotely controlled PPI inhibitors. Moreover, available natural peptides, evolutionarily selected for high stability and specificity, would be employed by in silico tools for rational structure-based design of IPs harboring similar properties. The ever-growing need for more effective therapeutics would be the driving force for further growth of the in silico methods in IP development. Given the apparent advantages of the peptides over small molecule drugs, higher investment of the pharmaceutical industry into the development of IPs does not seem farfetched. In silico tools will play a pivotal role in the transition from small molecule-based modulators of PPIs to peptide-based modulators.
Modulation of PPIs using the IPs is one of the hot research topics of various scientific fields such as biochemistry, chemical biology, and pharmacology. Recent progress in developing in silico methods for the evaluation and identification of these IPs has hugely extended the field. Although some issues about the pharmacodynamics and pharmacokinetics of peptides remain to be addressed, many companies have already devoted themselves to peptide discovery, which resulted in numerous peptide drugs and homologous compounds on the market. In silico methods could offer a comprehensive and exciting portfolio of applications in IPs targeting the PPIs. The interest in these methods is reflected in the increasing number of algorithms, software, and related publications. These methods have a lot to offer toward resolving challenges like low membrane permeability, the tendency for aggregation, short half-life, fast elimination (if not stabilized), proteolytic degradation, specific targeting, toxicity, immunogenicity, and optimization of IPs. Each issue could be dealt with by using a combination of in silico tools developed based on the experimental results. Most of the peptide-related challenges require atomic-level structural information. The structural information about the peptides could be found in structural databases. The in silico methods could predict the structure of peptides lacking the previously resolved 3D structures. Given the 3D structure of an IP, its properties could be analyzed and optimized to circumvent functional limitations. These structures could also be employed to screen peptide libraries and find new targeting peptides. The efficacy of in silico tools highly depends on the accuracy of their algorithms. These algorithms are continuously revised considering the evidence obtained from experimental studies. Given their high potential to unravel the questions about IP design and optimization, in silico tools would become an inevitable part of the development of IPs that modulate the PPIs.
Author Contributions
SK, AJ, and MR contributed to the conception and design of the study. All authors contributed to the study data collection and data analysis, wrote the first draft of the manuscript, commented on previous versions of the manuscript, and read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Shahid Rajaee Teacher Training University for supporting the conduct of this research.
Footnotes
- ^ www.neuropeptides.nl
- ^ http://www.uniprot.org
- ^ https://www.rcsb.org
- ^ http://www.imtech.res.in/raghava/pepstr/
- ^ http://www.bioinfo.rpi.edu/~bystrc/hmmstr/about.html
- ^ http://pepsite2.russelllab.org
- ^ http://flexpepdock.furmanlab.cs.huji.ac.il/
- ^ http://www.eppic-web.org/ewui/
- ^ http://foldxsuite.crg.eu/
- ^ https://robetta.bakerlab.org/
- ^ http://ftmap.bu.edu/serverhelp.php
- ^ http://www.bioinsilico.org/PCRPi/
- ^ http://prism.ccbb.ku.edu.tr/hotpoint/
- ^ http://bioinf.cs.ucl.ac.uk/psipred/
- ^ https://mitchell-web.ornl.gov/KFC_Server/index.php
- ^ http://cdb.ics.uci.edu
- ^ http://chembank.broadinstitute.org
- ^ http://cactus.nci.nih.gov/ncidb2.2/
- ^ https://www.ebi.ac.uk/chembl/
- ^ http://pubchem.ncbi.nlm.nih.gov
- ^ http://zinc.docking.org
- ^ http://www.chemspider.com
- ^ http://www.drugbank.ca
- ^ http://accelrys.com/products/databases/sourcing/avaible-chemicalsdirectory.html
- ^ https://www.proofpoint.com/us/wombat-security-is-now-proofpoint
References
Abes, R., Arzumanov, A. A., Moulton, H. M., Abes, S., Ivanova, G. D., Iversen, P. L., et al. (2007). Cell-penetrating-peptide-based delivery of oligonucleotides: an overview. Biochem. Soc. Trans. 35(Pt 4), 775–779. doi: 10.1042/bst0350775
Agrawal, P., Bhagat, D., Mahalwal, M., Sharma, N., and Raghava, G. P. S. (2020). AntiCP 2.0: an updated model for predicting anticancer peptides. bioRxiv [Preprint] doi: 10.1101/2020.03.23.003780
Agrawal, P., Bhalla, S., Chaudhary, K., Kumar, R., Sharma, M., and Raghava, G. P. (2018). In silico approach for prediction of antifungal peptides. Front. Microbiol. 9:323. doi: 10.3389/fmicb.2018.00323
Agrawal, P., Bhalla, S., Usmani, S. S., Singh, S., Chaudhary, K., Raghava, G. P., et al. (2016). CPPsite 2.0: a repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 44, D1098–D1103.
Aguilera-Mendoza, L., Marrero-Ponce, Y., Beltran, J. A., Tellez Ibarra, R., Guillen-Ramirez, H. A., and Brizuela, C. A. (2019). Graph-based data integration from bioactive peptide databases of pharmaceutical interest: toward an organized collection enabling visual network analysis. Bioinformatics 35, 4739–4747. doi: 10.1093/bioinformatics/btz260
Alam, N., Goldstein, O., Xia, B., Porter, K. A., Kozakov, D., and Schueler-Furman, O. (2017). High-resolution global peptide-protein docking using fragments-based PIPER-FlexPepDock. PLoS Comput. Biol. 13:e1005905. doi: 10.1371/journal.pcbi.1005905
Antosova, Z., Mackova, M., Kral, V., and Macek, T. (2009). Therapeutic application of peptides and proteins: parenteral forever? Trends Biotechnol. 27, 628–635. doi: 10.1016/j.tibtech.2009.07.009
Antunes, D. A., Moll, M., Devaurs, D., Jackson, K. R., Lizée, G., and Kavraki, L. E. (2017). DINC 2.0: a new protein–peptide docking webserver using an incremental approach. Cancer Res. 77, e55–e57.
Arkin, M. R., and Wells, J. A. (2004). Small-molecule inhibitors of protein–protein interactions: progressing towards the dream. Nat. Rev. Drug Discov. 3, 301–317. doi: 10.1038/nrd1343
Assi, S. A., Tanaka, T., Rabbitts, T. H., and Fernandez-Fuentes, N. (2010). PCRPi: presaging critical residues in protein interfaces, a new computational tool to chart hotspots in protein interfaces. Nucleic Acids Res. 38:e86. doi: 10.1093/nar/gkp1158
Bakail, M., and Ochsenbein, F. (2016). Targeting protein–protein interactions, a wide open field for drug design. C. R. Chim. 19, 19–27. doi: 10.1016/j.crci.2015.12.004
Beaufays, J., Lins, L., Thomas, A., and Brasseur, R. (2012). In silico predictions of 3D structures of linear and cyclic peptides with natural and non-proteinogenic residues. J. Pept. Sci. 18, 17–24. doi: 10.1002/psc.1410
Beerten, J., Van Durme, J., Gallardo, R., Capriotti, E., Serpell, L., Rousseau, F., et al. (2015). WALTZ-DB: a benchmark database of amyloidogenic hexapeptides. Bioinformatics 31, 1698–1700. doi: 10.1093/bioinformatics/btv027
Ben-Shimon, A., and Niv, M. Y. (2015). AnchorDock: blind and flexible anchor-driven peptide docking. Structure 23, 929–940. doi: 10.1016/j.str.2015.03.010
Blazer, L. L., and Neubig, R. R. (2009). Small molecule protein–protein interaction inhibitors as CNS therapeutic agents: current progress and future hurdles. Neuropsychopharmacology 34, 126–141. doi: 10.1038/npp.2008.151
Bogan, A. A., and Thorn, K. S. (1998). Anatomy of hotspots in protein interfaces. J. Mol. Biol. 280, 1–9. doi: 10.1006/jmbi.1998.1843
Bowie, J. U., Luthy, R., and Eisenberg, D. (1991). A method to identify protein sequences that fold into a known three-dimensional structure. Science 253, 164–170. doi: 10.1126/science.1853201
Bradley, P., Misura, K. M., and Baker, D. (2005). Toward high-resolution de novo structure prediction for small proteins. Science 309, 1868–1871. doi: 10.1126/science.1113801
Brenke, R., Kozakov, D., Chuang, G.-Y., Beglov, D., Hall, D., Landon, M. R., et al. (2009). Fragment-based identification of druggable ‘hotspots’ of proteins using Fourier domain correlation techniques. Bioinformatics 25, 621–627. doi: 10.1093/bioinformatics/btp036
Bruzzoni-Giovanelli, H., Alezra, V., Wolff, N., Dong, C.-Z., Tuffery, P., and Rebollo, A. (2018). Interfering peptides targeting protein–protein interactions: the next generation of drugs? Drug Discov. Today 23, 272–285. doi: 10.1016/j.drudis.2017.10.016
Burbach, J. P. H. (2010). Neuropeptides from concept to online database www. neuropeptides. nl. Eur. J. Pharmacol. 626, 27–48. doi: 10.1016/j.ejphar.2009.10.015
Bystroff, C., and Shao, Y. (2002). Fully automated ab initio protein structure prediction using I-SITES, HMMSTR and ROSETTA. Bioinformatics 18(Suppl._1), S54–S61.
Bystroff, C., Thorsson, V., and Baker, D. (2000). HMMSTR: a hidden Markov model for local sequence-structure correlations in proteins. J. Mol. Biol. 301, 173–190. doi: 10.1006/jmbi.2000.3837
Caffrey, D. R., Somaroo, S., Hughes, J. D., Mintseris, J., and Huang, E. S. (2004). Are protein–protein interfaces more conserved in sequence than the rest of the protein surface? Protein Sci. 13, 190–202. doi: 10.1110/ps.03323604
Casals, F., Idaghdour, Y., Hussin, J., and Awadalla, P. (2012). Next-generation sequencing approaches for genetic mapping of complex diseases. J. Neuroimmunol. 248, 10–22. doi: 10.1016/j.jneuroim.2011.12.017
Chen, H., and Zhou, H. X. (2005). Prediction of interface residues in protein–protein complexes by a consensus neural network method: test against NMR data. Proteins 61, 21–35. doi: 10.1002/prot.20514
Chen, W., Ding, H., Feng, P., Lin, H., and Chou, K.-C. (2016). iACP: a sequence-based tool for identifying anticancer peptides. Oncotarget 7:16895. doi: 10.18632/oncotarget.7815
Chen, X.-W., and Liu, M. (2005). Prediction of protein–protein interactions using random decision forest framework. Bioinformatics 21, 4394–4400. doi: 10.1093/bioinformatics/bti721
Cho, K.-I., Kim, D., and Lee, D. (2009). A feature-based approach to modeling protein–protein interaction hotspots. Nucleic Acids Res. 37, 2672–2687. doi: 10.1093/nar/gkp132
Chong, L. T., Saglam, A. S., and Zuckerman, D. M. (2017). Path-sampling strategies for simulating rare events in biomolecular systems. Curr. Opin. Struct. Biol. 43, 88–94. doi: 10.1016/j.sbi.2016.11.019
Ciemny, M., Kurcinski, M., Kamel, K., Kolinski, A., Alam, N., Schueler-Furman, O., et al. (2018). Protein–peptide docking: opportunities and challenges. Drug Discov. Today 23, 1530–1537. doi: 10.1016/j.drudis.2018.05.006
Clackson, T., Ultsch, M. H., Wells, J. A., and de Vos, A. M. (1998). Structural and functional analysis of the 1: 1 growth hormone: receptor complex reveals the molecular basis for receptor affinity. J. Mol. Biol. 277, 1111–1128. doi: 10.1006/jmbi.1998.1669
Clackson, T., and Wells, J. A. (1995). A hotspot of binding energy in a hormone-receptor interface. Science 267, 383–386. doi: 10.1126/science.7529940
Cunningham, A. D., Qvit, N., and Mochly-Rosen, D. (2017). Peptides and peptidomimetics as regulators of protein–protein interactions. Curr. Opin. Struct. Biol. 44, 59–66. doi: 10.1016/j.sbi.2016.12.009
Cunningham, B. C., and Wells, J. A. (1989). High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244, 1081–1085. doi: 10.1126/science.2471267
Das, D., Jaiswal, M., Khan, F. N., Ahamad, S., and Kumar, S. (2020). PlantPepDB: a manually curated plant peptide database. Sci. Rep. 10:2194.
de Beer, S. B. A. (2012). Application of Free Energy Calculations for Drug Design. De Boelelaan: Vrije Universiteit Amsterdem.
de Ruiter, A., and Oostenbrink, C. (2011). Free energy calculations of protein–ligand interactions. Curr. Opin. Chem. Biol. 15, 547–552. doi: 10.1016/j.cbpa.2011.05.021
de Vries, S. J., and Bonvin, A. M. (2008). How proteins get in touch: interface prediction in the study of biomolecular complexes. Curr. Protein Pept. Sci. 9, 394–406. doi: 10.2174/138920308785132712
de Vries, S. J., and Bonvin, A. M. (2011). CPORT: a consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS One 6:e17695. doi: 10.1371/journal.pone.0017695
de Vries, S. J., Rey, J., Schindler, C. E., Zacharias, M., and Tuffery, P. (2017). The pepATTRACT web server for blind, large-scale peptide–protein docking. Nucleic Acids Res. 45, W361–W364.
de Vries, S. J., van Dijk, A. D., and Bonvin, A. M. (2006). WHISCY: what information does surface conservation yield? Application to data-driven docking. Proteins 63, 479–489. doi: 10.1002/prot.20842
DeLano, W. L. (2002). Unraveling hotspots in binding interfaces: progress and challenges. Curr. Opin. Struct. Biol. 12, 14–20. doi: 10.1016/s0959-440x(02)00283-x
DeLano, W. L., Ultsch, M. H., and Wells, J. A. (2000). Convergent solutions to binding at a protein-protein interface. Science 287, 1279–1283. doi: 10.1126/science.287.5456.1279
Di Luca, M., Maccari, G., Maisetta, G., and Batoni, G. (2015). BaAMPs: the database of biofilm-active antimicrobial peptides. Biofouling 31, 193–199. doi: 10.1080/08927014.2015.1021340
Diller, D. J., Swanson, J., Bayden, A. S., Jarosinski, M., and Audie, J. (2015). Rational, computer-enabled peptide drug design: principles, methods, applications and future directions. Future Med. Chem. 7, 2173–2193. doi: 10.4155/fmc.15.142
Dong, M., Lam, P. C.-H., Pinon, D. I., Hosohata, K., Orry, A., Sexton, P. M., et al. (2011). Molecular basis of secretin docking to its intact receptor using multiple photolabile probes distributed throughout the pharmacophore. J. Biol. Chem. 286, 23888–23899. doi: 10.1074/jbc.m111.245969
Donsky, E., and Wolfson, H. J. (2011). PepCrawler: a fast RRT-based algorithm for high-resolution refinement and binding affinity estimation of peptide inhibitors. Bioinformatics 27, 2836–2842. doi: 10.1093/bioinformatics/btr498
Duarte, J. M., Srebniak, A., Schärer, M. A., and Capitani, G. (2012). Protein interface classification by evolutionary analysis. BMC Bioinformatics 13:334. doi: 10.1186/1471-2105-13-334
Ellert-Miklaszewska, A., Poleszak, K., and Kaminska, B. (2017). Short peptides interfering with signaling pathways as new therapeutic tools for cancer treatment. Future Med Chem. 9, 199–221. doi: 10.4155/fmc-2016-0189
Esmaielbeiki, R., Krawczyk, K., Knapp, B., Nebel, J.-C., and Deane, C. M. (2016). Progress and challenges in predicting protein interfaces. Brief. Bioinform. 17, 117–131. doi: 10.1093/bib/bbv027
Esmaielbeiki, R., and Nebel, J.-C. (2014). Scoring docking conformations using predicted protein interfaces. BMC Bioinformatics 15:171.
Etchebest, C., Benros, C., Hazout, S., and de Brevern, A. G. (2005). A structural alphabet for local protein structures: improved prediction methods. Proteins 59, 810–827. doi: 10.1002/prot.20458
Fischer, D., Elofsson, A., Rychlewski, L., Pazos, F., Valencia, A., Rost, B., et al. (2001). CAFASP2: the second critical assessment of fully automated structure prediction methods. Proteins 45, 171–183. doi: 10.1002/prot.10036
Fischer, T., Arunachalam, K., Bailey, D., Mangual, V., Bakhru, S., Russo, R., et al. (2003). The binding interface database (BID): a compilation of amino acid hotspots in protein interfaces. Bioinformatics 19, 1453–1454. doi: 10.1093/bioinformatics/btg163
Flissi, A., Ricart, E., Campart, C., Chevalier, M., Dufresne, Y., Michalik, J., et al. (2020). Norine: update of the nonribosomal peptide resource. Nucleic Acids Res. 48, D465–D469.
Fosgerau, K., and Hoffmann, T. (2015). Peptide therapeutics: current status and future directions. Drug Discov. Today 20, 122–128. doi: 10.1016/j.drudis.2014.10.003
Gao, Y., Wang, R., and Lai, L. (2004). Structure-based method for analyzing protein–protein interfaces. J. Mol. Model. 10, 44–54. doi: 10.1007/s00894-003-0168-3
Gautam, A., Chaudhary, K., Kumar, R., Sharma, A., Kapoor, P., Tyagi, A., et al. (2013). In silico approaches for designing highly effective cell penetrating peptides. J. Transl. Med. 11:74. doi: 10.1186/1479-5876-11-74
Gautam, A., Chaudhary, K., Singh, S., Joshi, A., Anand, P., Tuknait, A., et al. (2014). Hemolytik: a database of experimentally determined hemolytic and non-hemolytic peptides. Nucleic Acids Res. 42, D444–D449.
Gautam, A., Singh, H., Tyagi, A., Chaudhary, K., Kumar, R., Kapoor, P., et al. (2012). CPPsite: a curated database of cell penetrating peptides. Database 2012:bas015. doi: 10.1093/database/bas015
Gesell, J., Zasloff, M., and Opella, S. J. (1997). Two-dimensional 1H NMR experiments show that the 23-residue magainin antibiotic peptide is an α-helix in dodecylphosphocholine micelles, sodium dodecylsulfate micelles, and trifluoroethanol/water solution. J. Biomol. NMR 9, 127–135.
Gogoladze, G., Grigolava, M., Vishnepolsky, B., Chubinidze, M., Duroux, P., Lefranc, M.-P., et al. (2014). DBAASP: database of antimicrobial activity and structure of peptides. FEMS Microbiol. Lett. 357, 63–68. doi: 10.1111/1574-6968.12489
Gómez, E. A., Giraldo, P., and Orduz, S. (2017). InverPep: a database of invertebrate antimicrobial peptides. J. Glob. Antimicrob. Resist. 8, 13–17. doi: 10.1016/j.jgar.2016.10.003
Grishin, N. V., and Phillips, M. A. (1994). The subunit interfaces of oligomeric enzymes are conserved to a similar extent to the overall protein sequences. Protein Sci. 3, 2455–2458. doi: 10.1002/pro.5560031231
Grosdidier, S., and Fernández-Recio, J. (2008). Identification of hot-spot residues in protein-protein interactions by computational docking. BMC Bioinformatics 9:447. doi: 10.1186/1471-2105-9-447
Guerois, R., Nielsen, J. E., and Serrano, L. (2002). Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 320, 369–387. doi: 10.1016/S0022-2836(02)00442-4
Guharoy, M., and Chakrabarti, P. (2009). Empirical estimation of the energetic contribution of individual interface residues in structures of protein–protein complexes. J. Comput. Aided Mol. Des. 23, 645–654. doi: 10.1007/s10822-009-9282-3
Guidotti, G., Brambilla, L., and Rossi, D. (2017). Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol. Sci. 38, 406–424. doi: 10.1016/j.tips.2017.01.003
Gupta, S., Kapoor, P., Chaudhary, K., Gautam, A., Kumar, R., Raghava, G. P., et al. (2013). In silico approach for predicting toxicity of peptides and proteins. PLoS One 8:e73957. doi: 10.1371/journal.pone.0073957
Gupta, S., Madhu, M. K., Sharma, A. K., and Sharma, V. K. (2016). ProInflam: a webserver for the prediction of proinflammatory antigenicity of peptides and proteins. J. Transl. Med. 14, 1–10.
Halabi, N., Rivoire, O., Leibler, S., and Ranganathan, R. (2009). Protein sectors: evolutionary units of three-dimensional structure. Cell 138, 774–786. doi: 10.1016/j.cell.2009.07.038
Hall, R., Dixon, T., and Dickson, A. (2020). On calculating free energy differences using ensembles of transition paths. Front. Mol. Biosci. 7:106. doi: 10.3389/fmolb.2020.00106
Hamer, R., Luo, Q., Armitage, J. P., Reinert, G., and Deane, C. M. (2010). i-Patch: interprotein contact prediction using local network information. Proteins 78, 2781–2797. doi: 10.1002/prot.22792
Hammami, R., Ben Hamida, J., Vergoten, G., and Fliss, I. (2009). PhytAMP: a database dedicated to antimicrobial plant peptides. Nucleic Acids Res. 37(Suppl._1), D963–D968.
Hammami, R., Zouhir, A., Le Lay, C., Hamida, J. B., and Fliss, I. (2010). BACTIBASE second release: a database and tool platform for bacteriocin characterization. BMC Microbiol. 10:22. doi: 10.1186/1471-2180-10-22
Hasan, M. M., Schaduangrat, N., Basith, S., Lee, G., Shoombuatong, W., and Manavalan, B. (2020). HLPpred-Fuse: improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics 36, 3350–3356. doi: 10.1093/bioinformatics/btaa160
Heinig, M., and Frishman, D. (2004). STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 32(Suppl._2), W500–W502.
Higa, R. H., and Tozzi, C. L. (2009). Prediction of binding hotspot residues by using structural and evolutionary parameters. Genet. Mol. Biol. 32, 626–633. doi: 10.1590/s1415-47572009000300029
Holton, T. A., Pollastri, G., Shields, D. C., and Mooney, C. (2013). CPPpred: prediction of cell penetrating peptides. Bioinformatics 29, 3094–3096. doi: 10.1093/bioinformatics/btt518
Hoofnagle, A. N., Resing, K. A., and Ahn, N. G. (2003). Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 32, 1–25.
Hu, Z., Ma, B., Wolfson, H., and Nussinov, R. (2000). Conservation of polar residues as hotspots at protein interfaces. Proteins 39, 331–342. doi: 10.1002/(sici)1097-0134(20000601)39:4<331::aid-prot60>3.0.co;2-a
Hung, L.-H., Ngan, S.-C., Liu, T., and Samudrala, R. (2005). PROTINFO: new algorithms for enhanced protein structure predictions. Nucleic Acids Res. 33(Suppl._2), W77–W80.
Iwaniak, A., Minkiewicz, P., Darewicz, M., Sieniawski, K., and Starowicz, P. (2016). BIOPEP database of sensory peptides and amino acids. Food Res. Int. 85, 155–161. doi: 10.1016/j.foodres.2016.04.031
Jain, A. N. (2003). Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 46, 499–511. doi: 10.1021/jm020406h
Janin, J., Henrick, K., Moult, J., Eyck, L. T., Sternberg, M. J., Vajda, S., et al. (2003). CAPRI: a critical assessment of predicted interactions. Proteins 52, 2–9. doi: 10.1002/prot.10381
Jhong, J.-H., Chi, Y.-H., Li, W.-C., Lin, T.-H., Huang, K.-Y., and Lee, T.-Y. (2019). dbAMP: an integrated resource for exploring antimicrobial peptides with functional activities and physicochemical properties on transcriptome and proteome data. Nucleic Acids Res. 47, D285–D297.
Jones, D. T., Taylort, W., and Thornton, J. M. (1992). A new approach to protein fold recognition. Nature 358, 86–89. doi: 10.1038/358086a0
Jordan, R. A., El-Manzalawy, Y., Dobbs, D., and Honavar, V. (2012). Predicting protein-protein interface residues using local surface structural similarity. BMC Bioinformatics 13:41. doi: 10.1186/1471-2105-13-41
Joseph, S., Karnik, S., Nilawe, P., Jayaraman, V. K., and Idicula-Thomas, S. (2012). ClassAMP: a prediction tool for classification of antimicrobial peptides. IEEE/ACM Trans. Comput. Biol. Bioinform. 9, 1535–1538. doi: 10.1109/tcbb.2012.89
Jouaux, E. M., Schmidtkunz, K., Müller, K. M., and Arndt, K. M. (2008). Targeting the c-Myc coiled coil with interfering peptides. J. Pept. Sci. 14, 1022–1031. doi: 10.1002/psc.1038
Kaas, Q., Yu, R., Jin, A.-H., Dutertre, S., and Craik, D. J. (2012). ConoServer: updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 40, D325–D330.
Kanakaveti, V., Shanmugam, A., Ramakrishnan, C., Anoosha, P., Sakthivel, R., Rayala, S. K., et al. (2020). “Chapter Two - Computational approaches for identifying potential inhibitors on targeting protein interactions in drug discovery. Adv. Protein Chem. Struct. Biol. 121, 25–47. doi: 10.1016/bs.apcsb.2019.11.013
Kapoor, P., Singh, H., Gautam, A., Chaudhary, K., Kumar, R., and Raghava, G. P. (2012). TumorHoPe: a database of tumor homing peptides. PLoS One 7:e35187. doi: 10.1371/journal.pone.0035187
Kaserer, T., Beck, K. R., Akram, M., Odermatt, A., and Schuster, D. (2015). Pharmacophore models and pharmacophore-based virtual screening: concepts and applications exemplified on hydroxysteroid dehydrogenases. Molecules 20, 22799–22832. doi: 10.3390/molecules201219880
Kaspar, A. A., and Reichert, J. M. (2013). Future directions for peptide therapeutics development. Drug Discov. Today 18, 807–817. doi: 10.1016/j.drudis.2013.05.011
Kaur, H., Garg, A., and Raghava, G. P. S. (2007). PEPstr: a de novo method for tertiary structure prediction of small bioactive peptides. Protein Pept. Lett. 14, 626–631. doi: 10.2174/092986607781483859
Kenneth Morrow, J., and Zhang, S. (2012). Computational prediction of protein hotspot residues. Curr. Pharm. Des. 18, 1255–1265. doi: 10.2174/138161212799436412
Keskin, O., Ma, B., and Nussinov, R. (2005). Hot regions in protein–protein interactions: the organization and contribution of structurally conserved hotspot residues. J. Mol. Biol. 345, 1281–1294. doi: 10.1016/j.jmb.2004.10.077
Khalili, S., Rasaee, M. J., Mousavi, S. L., Amani, J., Jahangiri, A., and Borna, H. (2017). In silico prediction and in vitro verification of a novel multi-epitope antigen for HBV detection. Mol. Genet. Microbiol. Virol. 32, 230–240. doi: 10.3103/s0891416817040097
Khodashenas, S., Khalili, S., and Moghadam, M. F. (2019). A cell ELISA based method for exosome detection in diagnostic and therapeutic applications. Biotechnol. Lett. 41, 523–531. doi: 10.1007/s10529-019-02667-5
Kim, D. E., Chivian, D., and Baker, D. (2004). Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32(Suppl._2), W526–W531.
Kim, Y., Bark, S., Hook, V., and Bandeira, N. (2011). NeuroPedia: neuropeptide database and spectral library. Bioinformatics 27, 2772–2773. doi: 10.1093/bioinformatics/btr445
Kitchen, D. B., Decornez, H., Furr, J. R., and Bajorath, J. (2004). Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 3, 935–949. doi: 10.1038/nrd1549
Kittichotirat, W., Guerquin, M., Bumgarner, R. E., and Samudrala, R. (2009). Protinfo PPC: a web server for atomic level prediction of protein complexes. Nucleic Acids Res. 37(Suppl._2), W519–W525.
Koboldt, D. C., Steinberg, K. M., Larson, D. E., Wilson, R. K., and Mardis, E. R. (2013). The next-generation sequencing revolution and its impact on genomics. Cell 155, 27–38. doi: 10.1016/j.cell.2013.09.006
Kortemme, T., Kim, D. E., and Baker, D. (2004). Computational alanine scanning of protein-protein interfaces. Sci. STKE 2004:l2.
Kouadio, J.-L. K., Horn, J. R., Pal, G., and Kossiakoff, A. A. (2005). Shotgun alanine scanning shows that growth hormone can bind productively to its receptor through a drastically minimized interface. J. Biol. Chem. 280, 25524–25532. doi: 10.1074/jbc.m502167200
Kozakov, D., Brenke, R., Comeau, S. R., and Vajda, S. (2006). PIPER: an FFT-based protein docking program with pairwise potentials. Proteins 65, 392–406. doi: 10.1002/prot.21117
Kozlowski, L. P., and Bujnicki, J. M. (2012). MetaDisorder: a meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinformatics 13:111.
Kufareva, I., Budagyan, L., Raush, E., Totrov, M., and Abagyan, R. (2007). PIER: protein interface recognition for structural proteomics. Proteins 67, 400–417. doi: 10.1002/prot.21233
Kundrotas, P. J., Zhu, Z., Janin, J., and Vakser, I. A. (2012). Templates are available to model nearly all complexes of structurally characterized proteins. Proc. Natl. Acad. Sci. U.S.A. 109, 9438–9441. doi: 10.1073/pnas.1200678109
Kurcinski, M., Jamroz, M., Blaszczyk, M., Kolinski, A., and Kmiecik, S. (2015). CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 43, W419–W424.
Laengsri, V., Nantasenamat, C., Schaduangrat, N., Nuchnoi, P., Prachayasittikul, V., and Shoombuatong, W. (2019). TargetAntiAngio: a sequence-based tool for the prediction and analysis of anti-angiogenic peptides. Int. J. Mol. Sci. 20, 2950. doi: 10.3390/ijms20122950
Lamiable, A., Thévenet, P., Rey, J., Vavrusa, M., Derreumaux, P., and Tufféry, P. (2016). PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 44, W449–W454.
Lata, S., Mishra, N. K., and Raghava, G. P. (2010). AntiBP2: improved version of antibacterial peptide prediction. BMC Bioinformatics 11:S19.
Lavecchia, A., and Di Giovanni, C. (2013). Virtual screening strategies in drug discovery: a critical review. Curr. Med. Chem. 20, 2839–2860. doi: 10.2174/09298673113209990001
Lee, A. C.-L., Harris, J. L., Khanna, K. K., and Hong, J.-H. (2019). A comprehensive review on current advances in peptide drug development and design. Int. J. Mol. Sci. 20:2383. doi: 10.3390/ijms20102383
Lee, H., Heo, L., Lee, M. S., and Seok, C. (2015). GalaxyPepDock: a protein–peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 43, W431–W435.
Lensink, M. F., Velankar, S., and Wodak, S. J. (2017). Modeling protein–protein and protein–peptide complexes: CAPRI 6th edition. Proteins 85, 359–377. doi: 10.1002/prot.25215
Li, R., Dowd, V., Stewart, D. J., Burton, S. J., and Lowe, C. R. (1998). Design, synthesis, and application of a protein A mimetic. Nat. Biotechnol. 16, 190–195. doi: 10.1038/nbt0298-190
Liang, S., Zhang, C., Liu, S., and Zhou, Y. (2006). Protein binding site prediction using an empirical scoring function. Nucleic Acids Res. 34, 3698–3707. doi: 10.1093/nar/gkl454
Lichtarge, O., Bourne, H. R., and Cohen, F. E. (1996). An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 257, 342–358. doi: 10.1006/jmbi.1996.0167
Lise, S., Archambeau, C., Pontil, M., and Jones, D. T. (2009). Prediction of hotspot residues at protein-protein interfaces by combining machine learning and energy-based methods. BMC Bioinformatics 10:365. doi: 10.1186/1471-2105-10-365
Lise, S., Buchan, D., Pontil, M., and Jones, D. T. (2011). Predictions of hotspot residues at protein-protein interfaces using support vector machines. PLoS One 6:e16774. doi: 10.1371/journal.pone.0016774
Liu, F., Baggerman, G., Schoofs, L., and Wets, G. (2008). The construction of a bioactive peptide database in Metazoa. J. Proteome Res. 7, 4119–4131. doi: 10.1021/pr800037n
Livnah, O., Stura, E. A., Johnson, D. L., Middleton, S. A., Mulcahy, L. S., Wrighton, N. C., et al. (1996). Functional mimicry of a protein hormone by a peptide agonist: the EPO receptor complex at 2.8 Å. Science 273, 464–471. doi: 10.1126/science.273.5274.464
London, N., Raveh, B., Cohen, E., Fathi, G., and Schueler-Furman, O. (2011). Rosetta FlexPepDock web server—high resolution modeling of peptide–protein interactions. Nucleic Acids Res. 39(Suppl._2), W249–W253.
London, N., Raveh, B., and Schueler-Furman, O. (2013). Druggable protein–protein interactions–from hotspots to hot segments. Curr. Opin. Chem. Biol. 17, 952–959. doi: 10.1016/j.cbpa.2013.10.011
Louros, N., Konstantoulea, K., De Vleeschouwer, M., Ramakers, M., Schymkowitz, J., and Rousseau, F. (2020). WALTZ-DB 2.0: an updated database containing structural information of experimentally determined amyloid-forming peptides. Nucleic Acids Res. 48, D389–D393.
Lu, H., Zhou, Q., He, J., Jiang, Z., Peng, C., Tong, R., et al. (2020). Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials. Signal Transduct. Target. Ther. 5, 1–23.
Luiz Folador, E., Fernandes de Oliveira Junior, A., Tiwari, S., Babar Jamal, S., Salgado Ferreira, R., Barh, D., et al. (2015). In silico protein-protein interactions: avoiding data and method biases over sensitivity and specificity. Curr. Protein Pept. Sci. 16, 689–700. doi: 10.2174/1389203716666150505235437
Manavalan, B., Basith, S., Shin, T. H., Choi, S., Kim, M. O., and Lee, G. (2017). MLACP: machine-learning-based prediction of anticancer peptides. Oncotarget 8:77121. doi: 10.18632/oncotarget.20365
Manavalan, B., Basith, S., Shin, T. H., Wei, L., and Lee, G. (2019a). AtbPpred: a robust sequence-based prediction of anti-tubercular peptides using extremely randomized trees. Comput. Struct. Biotechnol. J. 17, 972–981. doi: 10.1016/j.csbj.2019.06.024
Manavalan, B., Basith, S., Shin, T. H., Wei, L., and Lee, G. (2019b). mAHTPred: a sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics 35, 2757–2765. doi: 10.1093/bioinformatics/bty1047
Marcu, O., Dodson, E. J., Alam, N., Sperber, M., Kozakov, D., Lensink, M. F., et al. (2017). FlexPepDock lessons from CAPRI peptide–protein rounds and suggested new criteria for assessment of model quality and utility. Proteins 85, 445–462. doi: 10.1002/prot.25230
Mard-Soltani, M., Rasaee, M. J., Khalili, S., Sheikhi, A.-K., Hedayati, M., Ghaderi-Zefrehi, H., et al. (2018). The effect of differentially designed fusion proteins to elicit efficient anti-human thyroid stimulating hormone immune responses. Iran. J. Allergy Asthma Immunol. 17, 158–170.
Maupetit, J., Derreumaux, P., and Tuffery, P. (2009). PEP-FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res. 37(Suppl._2), W498–W503.
Maupetit, J., Derreumaux, P., and Tufféry, P. (2010). A fast method for large-scale De Novo peptide and miniprotein structure prediction. J. Comput. Chem. 31, 726–738.
Meher, P. K., Sahu, T. K., Saini, V., and Rao, A. R. (2017). Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 7: 42362.
Mehta, D., Anand, P., Kumar, V., Joshi, A., Mathur, D., Singh, S., et al. (2014). ParaPep: a web resource for experimentally validated antiparasitic peptide sequences and their structures. Database 2014:bau051. doi: 10.1093/database/bau051
Mine, Y., Munir, H., Nakanishi, Y., and Sugiyama, D. (2016). Biomimetic peptides for the treatment of cancer. Anticancer. Res. 36, 3565–3570.
Minkiewicz, P., Dziuba, J., Iwaniak, A., Dziuba, M., and Darewicz, M. (2008). BIOPEP database and other programs for processing bioactive peptide sequences. J. AOAC Int. 91, 965–980. doi: 10.1093/jaoac/91.4.965
Minkiewicz, P., Iwaniak, A., and Darewicz, M. (2019). BIOPEP-UWM database of bioactive peptides: current opportunities. Int. J. Mol. Sci. 20:5978. doi: 10.3390/ijms20235978
Monge, A., Friesner, R. A., and Honig, B. (1994). An algorithm to generate low-resolution protein tertiary structures from knowledge of secondary structure. Proc. Natl. Acad. Sci. U.S.A. 91, 5027–5029. doi: 10.1073/pnas.91.11.5027
Mooney, C., Haslam, N. J., Pollastri, G., and Shields, D. C. (2012). Towards the improved discovery and design of functional peptides: common features of diverse classes permit generalized prediction of bioactivity. PLoS One 7:e45012. doi: 10.1371/journal.pone.0045012
Mora, J. S., Assi, S. A., and Fernandez-Fuentes, N. (2010). Presaging critical residues in protein interfaces-web server (PCRPi-W): a web server to chart hotspots in protein interfaces. PLoS One 5:e12352. doi: 10.1371/journal.pone.0012352
Moreira, I. S., Fernandes, P. A., and Ramos, M. J. (2007). hotspots—a review of the protein–protein interface determinant amino-acid residues. Proteins 68, 803–812. doi: 10.1002/prot.21396
Morrison, K. L., and Weiss, G. A. (2001). Combinatorial alanine-scanning. Curr. Opin. Chem. Biol. 5, 302–307. doi: 10.1016/s1367-5931(00)00206-4
Murakami, Y., and Mizuguchi, K. (2010). Applying the Naïve Bayes classifier with kernel density estimation to the prediction of protein–protein interaction sites. Bioinformatics 26, 1841–1848. doi: 10.1093/bioinformatics/btq302
Murakami, Y., Tripathi, L. P., Prathipati, P., and Mizuguchi, K. (2017). Network analysis and in silico prediction of protein–protein interactions with applications in drug discovery. Curr. Opin. Struct. Biol. 44, 134–142. doi: 10.1016/j.sbi.2017.02.005
Neuvirth, H., Raz, R., and Schreiber, G. (2004). ProMate: a structure based prediction program to identify the location of protein–protein binding sites. J. Mol. Biol. 338, 181–199. doi: 10.1016/j.jmb.2004.02.040
Nevola, L., and Giralt, E. (2015). Modulating protein–protein interactions: the potential of peptides. Chem. Commun. 51, 3302–3315. doi: 10.1039/c4cc08565e
Nielsen, S. D., Beverly, R. L., Qu, Y., and Dallas, D. C. (2017). Milk bioactive peptide database: a comprehensive database of milk protein-derived bioactive peptides and novel visualization. Food Chem. 232, 673–682. doi: 10.1016/j.foodchem.2017.04.056
Novković, M., Simunić, J., Bojović, V., Tossi, A., and Juretić, D. (2012). DADP: the database of anuran defense peptides. Bioinformatics 28, 1406–1407. doi: 10.1093/bioinformatics/bts141
Obarska-Kosinska, A., Iacoangeli, A., Lepore, R., and Tramontano, A. (2016). PepComposer: computational design of peptides binding to a given protein surface. Nucleic Acids Res. 44, W522–W528.
Osmulski, P. A., Karpowicz, P., Jankowska, E., Bohmann, J., Pickering, A. M., and Gaczyńska, M. (2020). New peptide-based pharmacophore activates 20S proteasome. Molecules 25:1439. doi: 10.3390/molecules25061439
Pallara, C., Jiménez-García, B., Romero, M., Moal, I. H., and Fernández-Recio, J. (2017). pyDock scoring for the new modeling challenges in docking: protein–peptide, homo-multimers, and domain–domain interactions. Proteins 85, 487–496. doi: 10.1002/prot.25184
Pierce, B. G., Wiehe, K., Hwang, H., Kim, B.-H., Vreven, T., and Weng, Z. (2014). ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773. doi: 10.1093/bioinformatics/btu097
Pineda, S. S., Chaumeil, P.-A., Kunert, A., Kaas, Q., Thang, M. W., Le, L., et al. (2018). ArachnoServer 3.0: an online resource for automated discovery, analysis and annotation of spider toxins. Bioinformatics 34, 1074–1076. doi: 10.1093/bioinformatics/btx661
Pinzi, L., and Rastelli, G. (2019). Molecular docking: shifting paradigms in drug discovery. Int. J. Mol. Sci. 20:4331. doi: 10.3390/ijms20184331
Pirtskhalava, M., Amstrong, A. A., Grigolava, M., Chubinidze, M., Alimbarashvili, E., Vishnepolsky, B., et al. (2021). DBAASP v3: database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 49, D288– D297.
Pirtskhalava, M., Gabrielian, A., Cruz, P., Griggs, H. L., Squires, R. B., Hurt, D. E., et al. (2016). DBAASP v. 2: an enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 44, D1104–D1112.
Pleasant-Jenkins, D., Reese, C., Chinnakkannu, P., Kasiganesan, H., Tourkina, E., Hoffman, S., et al. (2017). Reversal of maladaptive fibrosis and compromised ventricular function in the pressure overloaded heart by a caveolin-1 surrogate peptide. Lab. Invest. 97, 370–382. doi: 10.1038/labinvest.2016.153
Porollo, A., and Meller, J. (2007). Prediction-based fingerprints of protein–protein interactions. Proteins 66, 630–645. doi: 10.1002/prot.21248
Porter, K. A., Desta, I., Kozakov, D., and Vajda, S. (2019). What method to use for protein–protein docking? Curr. Opin. Struct. Biol. 55, 1–7. doi: 10.1016/j.sbi.2018.12.010
Porter, K. A., Xia, B., Beglov, D., Bohnuud, T., Alam, N., Schueler-Furman, O., et al. (2017). ClusPro PeptiDock: efficient global docking of peptide recognition motifs using FFT. Bioinformatics 33, 3299–3301. doi: 10.1093/bioinformatics/btx216
Qin, S., and Zhou, H.-X. (2007). meta-PPISP: a meta web server for protein-protein interaction site prediction. Bioinformatics 23, 3386–3387. doi: 10.1093/bioinformatics/btm434
Qureshi, A., Kaur, G., and Kumar, M. (2017). AVC pred: an integrated web server for prediction and design of antiviral compounds. Chem. Biol. Drug Des. 89, 74–83. doi: 10.1111/cbdd.12834
Qureshi, A., Thakur, N., and Kumar, M. (2013). HIPdb: a database of experimentally validated HIV inhibiting peptides. PLoS One 8:e54908. doi: 10.1371/journal.pone.0054908
Qureshi, A., Thakur, N., Tandon, H., and Kumar, M. (2014). AVPdb: a database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 42, D1147–D1153.
Rahbar, M. R., Zarei, M., Jahangiri, A., Khalili, S., Nezafat, N., Negahdaripour, M., et al. (2019). Trimeric autotransporter adhesins in Acinetobacter baumannii, coincidental evolution at work. Infect. Genet. Evol. 71, 116–127. doi: 10.1016/j.meegid.2019.03.023
Rajamani, D., Thiel, S., Vajda, S., and Camacho, C. J. (2004). Anchor residues in protein–protein interactions. Proc. Natl. Acad. Sci. U.S.A. 101, 11287–11292. doi: 10.1073/pnas.0401942101
Ramezani, A., Zakeri, A., Mard-Soltani, M., Mohammadian, A., Hashemi, Z. S., Mohammadpour, H., et al. (2019). Structure based screening for inhibitory therapeutics of CTLA-4 unveiled new insights about biology of ACTH. Int. J. Pept. Res. Ther. 26, 1–11.
Ramos-Martín, F., Annaval, T., Buchoux, S., Sarazin, C., and D’amelio, N. (2019). ADAPTABLE: a comprehensive web platform of antimicrobial peptides tailored to the user’s research. Life Sci. Alliance 2:e201900512. doi: 10.26508/lsa.201900512
Raveh, B., London, N., and Schueler-Furman, O. (2010). Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins 78, 2029–2040. doi: 10.1002/prot.22716
Raveh, B., London, N., Zimmerman, L., and Schueler-Furman, O. (2011). Rosetta FlexPepDock ab-initio: simultaneous folding, docking and refinement of peptides onto their receptors. PLoS One 6:e18934. doi: 10.1371/journal.pone.0018934
Rentzsch, R., and Renard, B. Y. (2015). Docking small peptides remains a great challenge: an assessment using AutoDock Vina. Brief. Bioinform. 16, 1045–1056. doi: 10.1093/bib/bbv008
Rifai, E. A., van Dijk, M., and Geerke, D. P. (2020). Recent developments in linear interaction energy based binding free energy calculations. Front. Mol. Biosci. 7:114. doi: 10.3389/fmolb.2020.00114
Roy, A., Nair, S., Sen, N., Soni, N., and Madhusudhan, M. J. M. (2017). In silico methods for design of biological therapeutics. Methods 131, 33–65. doi: 10.1016/j.ymeth.2017.09.008
Sali, A. (1995). Comparative protein modeling by satisfaction of spatial restraints. Mol. Med. Today 1, 270–277. doi: 10.1016/s1357-4310(95)91170-7
Sanchez, R., and Šali, A. (1997). Advances in comparative protein-structure modelling. Curr. Opin. Struct. Biol. 7, 206–214. doi: 10.1016/s0959-440x(97)80027-9
Schaduangrat, N., Nantasenamat, C., Prachayasittikul, V., and Shoombuatong, W. (2019a). ACPred: a computational tool for the prediction and analysis of anticancer peptides. Molecules 24:1973. doi: 10.3390/molecules24101973
Schaduangrat, N., Nantasenamat, C., Prachayasittikul, V., and Shoombuatong, W. (2019b). Meta-iAVP: a sequence-based meta-predictor for improving the prediction of antiviral peptides using effective feature representation. Int. J. Mol. Sci. 20:5743. doi: 10.3390/ijms20225743
Schindler, C. E., de Vries, S. J., and Zacharias, M. (2015). Fully blind peptide-protein docking with pepATTRACT. Structure 23, 1507–1515. doi: 10.1016/j.str.2015.05.021
Schreiber, G., and Fersht, A. R. (1995). Energetics of protein-protein interactions: analysis ofthe Barnase-Barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 248, 478–486. doi: 10.1016/s0022-2836(95)80064-6
Schymkowitz, J., Borg, J., Stricher, F., Nys, R., Rousseau, F., and Serrano, L. (2005). The FoldX web server: an online force field. Nucleic Acids Res. 33(Suppl._2), W382–W388.
Seebah, S., Suresh, A., Zhuo, S., Choong, Y. H., Chua, H., Chuon, D., et al. (2007). Defensins knowledgebase: a manually curated database and information source focused on the defensins family of antimicrobial peptides. Nucleic Acids Res. 35(Suppl._1), D265–D268.
Sharma, A., Singla, D., Rashid, M., and Raghava, G. P. S. (2014). Designing of peptides with desired half-life in intestine-like environment. BMC Bioinformatics 15:282. doi: 10.1186/1471-2105-15-282
Shi, X.-D., Sun, K., Hu, R., Liu, X.-Y., Hu, Q.-M., Sun, X.-Y., et al. (2016). Blocking the interaction between EphB2 and ADDLs by a small peptide rescues impaired synaptic plasticity and memory deficits in a mouse model of Alzheimer’s disease. J. Neurosci. 36, 11959–11973. doi: 10.1523/jneurosci.1327-16.2016
Shoemaker, B. A., and Panchenko, A. R. (2007). Deciphering protein–protein interactions. Part I. Experimental techniques and databases. PLoS Comput Biol. 3:e42. doi: 10.1371/journal.pcbi.0030042
Shoemaker, B. A., Zhang, D., Thangudu, R. R., Tyagi, M., Fong, J. H., Marchler-Bauer, A., et al. (2010). Inferred Biomolecular Interaction Server—a web server to analyze and predict protein interacting partners and binding sites. Nucleic Acids Res. 38(Suppl._1), D518–D524.
Sidhu, S. S., Lowman, H. B., Cunningham, B. C., and Wells, J. A. (2000). [21] Phage display for selection of novel binding peptides. Methods Enzymol. 328, 333–365.
Singh, S., Chaudhary, K., Dhanda, S. K., Bhalla, S., Usmani, S. S., Gautam, A., et al. (2016). SATPdb: a database of structurally annotated therapeutic peptides. Nucleic Acids Res. 44, D1119–D1126.
Skolnick, J., Fetrow, J. S., and Kolinski, A. (2000). Structural genomics and its importance for gene function analysis. Nat. Biotechnol. 18, 283–287. doi: 10.1038/73723
Slater, O., and Kontoyianni, M. (2019). The compromise of virtual screening and its impact on drug discovery. Exp. Opin. Drug Discov. 14, 619–637. doi: 10.1080/17460441.2019.1604677
Sorolla, A., Wang, E., Golden, E., Duffy, C., Henriques, S. T., Redfern, A. D., et al. (2020). Precision medicine by designer interference peptides: applications in oncology and molecular therapeutics. Oncogene 39, 1167–1184. doi: 10.1038/s41388-019-1056-3
Souza, F. R., Souza, L. M. P., and Pimentel, A. S. (2020). Recent open issues in coarse grained force fields. J. Chem. Inf. Model. 60, 5881–5884. doi: 10.1021/acs.jcim.0c01265
Stites, W. E. (1997). Protein- protein interactions: interface structure, binding thermodynamics, and mutational analysis. Chem. Rev. 97, 1233–1250. doi: 10.1021/cr960387h
Stone, T. A., and Deber, C. M. (2017). Therapeutic design of peptide modulators of protein-protein interactions in membranes. Biochim. Biophys. Acta 1859, 577–585. doi: 10.1016/j.bbamem.2016.08.013
Thakur, N., Qureshi, A., and Kumar, M. (2012). AVPpred: collection and prediction of highly effective antiviral peptides. Nucleic Acids Res. 40, W199–W204.
Thanos, C. D., DeLano, W. L., and Wells, J. A. (2006). Hot-spot mimicry of a cytokine receptor by a small molecule. Proc. Natl. Acad. Sci. U.S.A. 103, 15422–15427. doi: 10.1073/pnas.0607058103
Thevenet, P., Shen, Y., Maupetit, J., Guyon, F., Derreumaux, P., and Tuffery, P. (2012). PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 40, W288– W293.
Thomas, A., Deshayes, S., Decaffmeyer, M., Van Eyck, M. H., Charloteaux, B., and Brasseur, R. (2006). Prediction of peptide structure: how far are we? Proteins 65, 889–897. doi: 10.1002/prot.21151
Thomas, S., Karnik, S., Barai, R. S., Jayaraman, V. K., and Idicula-Thomas, S. (2010). CAMP: a useful resource for research on antimicrobial peptides. Nucleic Acids Res. 38(Suppl._1), D774–D780.
Thorn, K. S., and Bogan, A. A. (2001). ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics 17, 284–285. doi: 10.1093/bioinformatics/17.3.284
Tilley, J. W., Chen, L., Fry, D. C., Emerson, S. D., Powers, G. D., Biondi, D., et al. (1997). Identification of a small molecule inhibitor of the IL-2/IL-2Rα receptor interaction which binds to IL-2. J. Am. Chem. Soc. 119, 7589–7590. doi: 10.1021/ja970702x
Trabuco, L. G., Lise, S., Petsalaki, E., and Russell, R. B. (2012). PepSite: prediction of peptide-binding sites from protein surfaces. Nucleic Acids Res. 40, W423–W427.
Trellet, M., Melquiond, A. S., and Bonvin, A. M. (2013). A unified conformational selection and induced fit approach to protein-peptide docking. PLoS One 8:e58769. doi: 10.1371/journal.pone.0058769
Tuncbag, N., Gursoy, A., and Keskin, O. (2009). Identification of computational hotspots in protein interfaces: combining solvent accessibility and inter-residue potentials improves the accuracy. Bioinformatics 25, 1513–1520. doi: 10.1093/bioinformatics/btp240
Tuncbag, N., Keskin, O., and Gursoy, A. (2010). HotPoint: hotspot prediction server for protein interfaces. Nucleic Acids Res. 38(Suppl._2), W402– W406.
Tyagi, A., Tuknait, A., Anand, P., Gupta, S., Sharma, M., Mathur, D., et al. (2015). CancerPPD: a database of anticancer peptides and proteins. Nucleic Acids Res. 43, D837–D843.
Usmani, S. S., Bedi, G., Samuel, J. S., Singh, S., Kalra, S., Kumar, P., et al. (2017). THPdb: database of FDA-approved peptide and protein therapeutics. PLoS One 12:e0181748. doi: 10.1371/journal.pone.0181748
Usmani, S. S., Kumar, R., Kumar, V., Singh, S., and Raghava, G. P. (2018). AntiTbPdb: a knowledgebase of anti-tubercular peptides. Database 2018:bay025.
van Heel, A. J., de Jong, A., Song, C., Viel, J. H., Kok, J., and Kuipers, O. P. (2018). BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 46, W278–W281.
Vanommeslaeghe, K., Guvench, O., and MacKerell, A. D. Jr. (2014). Molecular mechanics. Curr. Pharm. Des. 20, 3281–3292. doi: 10.2174/13816128113199990600
Verdonk, M. L., Cole, J. C., Hartshorn, M. J., Murray, C. W., and Taylor, R. D. (2003). Improved protein–ligand docking using GOLD. Proteins 52, 609–623. doi: 10.1002/prot.10465
Vishnepolsky, B., Zaalishvili, G., Karapetian, M., Gabrielian, A., Rosenthal, A., Hurt, D. E., et al. (2019a). “Development of the model of in silico design of AMPs active against Staphylococcus aureus 25923,” in Proceedings of the 5th International Electronic Conference on Medicinal Chemistry session ECMC-5, (Basel: MDPI), doi: 10.3390/ECMC2019-06359
Vishnepolsky, B., Zaalishvili, G., Karapetian, M., Nasrashvili, T., Kuljanishvili, N., Gabrielian, A., et al. (2019b). De novo design and in vitro testing of antimicrobial peptides against Gram-negative bacteria. Pharmaceuticals 12:82. doi: 10.3390/ph12020082
Waghu, F. H., Barai, R. S., Gurung, P., and Idicula-Thomas, S. (2016). CAMPR3: a database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 44, D1094–D1097.
Waghu, F. H., Gopi, L., Barai, R. S., Ramteke, P., Nizami, B., and Idicula-Thomas, S. (2014). CAMP: collection of sequences and structures of antimicrobial peptides. Nucleic Acids Res. 42, D1154–D1158.
Wang, E., Sun, H., Wang, J., Wang, Z., Liu, H., Zhang, J. Z. H., et al. (2019). End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 119, 9478–9508. doi: 10.1021/acs.chemrev.9b00055
Wang, G., Li, X., and Wang, Z. (2009). APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 37(Suppl._1), D933–D937.
Wang, G., Li, X., and Wang, Z. (2016). APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44, D1087–D1093.
Wang, J., Yin, T., Xiao, X., He, D., Xue, Z., Jiang, X., et al. (2018). StraPep: a structure database of bioactive peptides. Database 2018:bay038.
Wang, Y., Wang, M., Yin, S., Jang, R., Wang, J., Xue, Z., et al. (2015). NeuroPep: a comprehensive resource of neuropeptides. Database 2015:bav038. doi: 10.1093/database/bav038
Wang, Z., and Sun, H. (2020). Combined strategies in structure-based virtual screening. Phys. Chem. Chem. Phys. 22, 3149–3159. doi: 10.1039/c9cp06303j
Wang, Z., and Wang, G. (2004). APD: the antimicrobial peptide database. Nucleic Acids Res. 32(Suppl._1), D590–D592.
Wang, Z., Zhang, S., Jin, H., Wang, W., Huo, J., Zhou, L., et al. (2011). Angiotensin-I-converting enzyme inhibitory peptides: chemical feature based pharmacophore generation. Eur. J. Med. Chem. 46, 3428–3433. doi: 10.1016/j.ejmech.2011.05.007
Wei, L., Xing, P., Su, R., Shi, G., Ma, Z. S., and Zou, Q. (2017). CPPred-RF: a sequence-based predictor for identifying cell-penetrating peptides and their uptake efficiency. J. Proteome Res. 16, 2044–2053. doi: 10.1021/acs.jproteome.7b00019
Wells, J. A. (1990). Additivity of mutational effects in proteins. Biochemistry 29, 8509–8517. doi: 10.1021/bi00489a001
Wells, J. A. (1991). ”[18] Systematic mutational analyses of protein-protein interfaces. Methods Enzymol. 202, 390–411. doi: 10.1016/0076-6879(91)02020-a
Wells, J. A., and McClendon, C. L. (2007). Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 450, 1001–1009. doi: 10.1038/nature06526
White, A. W., Westwell, A., and Brahemi, G. (2008). Protein-protein interactions as targets for small-molecule therapeutics in cancer. Expert Rev. Mol. Med. 10:e8.
Whitmore, L., and Wallace, B. (2004). The Peptaibol Database: a database for sequences and structures of naturally occurring peptaibols. Nucleic Acids Res. 32(Suppl._1), D593–D594.
Wrighton, N. C., Farrell, F. X., Chang, R., Kashyap, A. K., Barbone, F. P., Mulcahy, L. S., et al. (1996). Small peptides as potent mimetics of the protein hormone erythropoietin. Science 273, 458–463. doi: 10.1126/science.273.5274.458
Wu, K.-J., Lei, P.-M., Liu, H., Wu, C., Leung, C.-H., and Ma, D.-L. (2019). Mimicking strategy for protein-protein interaction inhibitor discovery by virtual screening. Molecules 24:4428. doi: 10.3390/molecules24244428
Wynendaele, E., Bronselaer, A., Nielandt, J., D’Hondt, M., Stalmans, S., Bracke, N., et al. (2012). Quorumpeps database: chemical space, microbial origin and functionality of quorum sensing peptides. Nucleic Acids Res. 41, D655–D659. doi: 10.1093/nar/gks1137
Wynendaele, E., Bronselaer, A., Nielandt, J., D’Hondt, M., Stalmans, S., Bracke, N., et al. (2013). Quorumpeps database: chemical space, microbial origin and functionality of quorum sensing peptides. Nucleic Acids Res. 41, D655–D659.
Xie, Z., Deng, X., and Shu, K. (2020). Prediction of protein-protein interaction sites using convolutional neural network and improved data sets. Int. J. Mol. Sci. 21:467. doi: 10.3390/ijms21020467
Yan, C., Xu, X., and Zou, X. (2016). Fully blind docking at the atomic level for protein-peptide complex structure prediction. Structure 24, 1842–1853. doi: 10.1016/j.str.2016.07.021
Yan, F., Liu, G., Chen, T., Fu, X., and Niu, M.-M. (2019). Structure-based virtual screening and biological evaluation of peptide inhibitors for polo-box domain. Molecules 25:107. doi: 10.3390/molecules25010107
Yan, J., Bhadra, P., Li, A., Sethiya, P., Qin, L., Tai, H. K., et al. (2020). Deep-AmPEP30: improve short antimicrobial peptides prediction with deep learning. Mol. Ther. Nucleic Acids. 20, 882–894. doi: 10.1016/j.omtn.2020.05.006
Yang, Z. R. (2008). Peptide bioinformatics: peptide classification using peptide machines. Methods Mol. Biol. 458, 159–183. doi: 10.1007/978-1-60327-101-1_9
Ye, G., Wu, H., Huang, J., Wang, W., Ge, K., Li, G., et al. (2020). LAMP2: a major update of the database linking antimicrobial peptides. Database 2020:baaa061.
Zemla, A. (2003). LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res. 31, 3370–3374. doi: 10.1093/nar/gkg571
Zhai, D., Chin, K., Wang, M., and Liu, F. (2014). Disruption of the nuclear p53-GAPDH complex protects against ischemia-induced neuronal damage. Mol. Brain 7:20. doi: 10.1186/1756-6606-7-20
Zhang, Q. C., Deng, L., Fisher, M., Guan, J., Honig, B., and Petrey, D. (2011). PredUs: a web server for predicting protein interfaces using structural neighbors. Nucleic Acids Res. 39(Suppl._2), W283– W287.
Keywords: peptide, protein–protein interactions, in silico, bioinformatics 3, interfering peptides
Citation: Hashemi ZS, Zarei M, Fath MK, Ganji M, Farahani MS, Afsharnouri F, Pourzardosht N, Khalesi B, Jahangiri A, Rahbar MR and Khalili S (2021) In silico Approaches for the Design and Optimization of Interfering Peptides Against Protein–Protein Interactions. Front. Mol. Biosci. 8:669431. doi: 10.3389/fmolb.2021.669431
Received: 18 February 2021; Accepted: 06 April 2021;
Published: 28 April 2021.
Edited by:
Luca Domenico D’Andrea, National Research Council, Consiglio Nazionale delle Ricerche (CNR), ItalyReviewed by:
Denis Colum Shields, University College Dublin, IrelandHelen Mott, University of Cambridge, United Kingdom
Copyright © 2021 Hashemi, Zarei, Fath, Ganji, Farahani, Afsharnouri, Pourzardosht, Khalesi, Jahangiri, Rahbar and Khalili. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saeed Khalili, c2FlZWQua2hhbGlsaUBzcnUuYWMuaXI=