 Lei Cao
Lei Cao- 1State Key Laboratory of Experimental Hematology, The Province and Ministry Co-sponsored Collaborative Innovation Center for Medical Epigenetics, Cancer Institute and Hospital, Department of Cell Biology, School of Basic Medical Sciences, Tianjin Medical University, Tianjin, China
- 2Department of Neurosurgery, Tianjin Medical University General Hospital, Tianjin, China
The interplay between cancer genome and deregulated epigenomic control is critical for cancer initiation and progression. ASXL1 (Additional Sex combs-like 1) is frequently mutated in tumors especially myeloid malignancies. However, there remains a debate whether the mutations are loss or gain-of-function. Mechanistically, ASXL1 forms a complex with BAP1 for the erasure of mono-ubiquitylation at lysine 119 on Histone H2A (H2AK119ub1), a well-known histone mark associated with transcription repression. Unexpectedly, this de-ubiquitylation complex has been genetically defined as a Polycomb Repressive complex though the regulatory mechanisms are elusive. In this review, we will discuss about the functions of ASXL1 in malignancies and reconcile seemingly paradoxical effects of ASXL1 or BAP1 loss on transcription regulation.
Introduction
Polycomb group (PcG) and trithorax group (TrxG) proteins, are conserved epigenetic regulators from Drosophila to mammals and have caught a huge amount of attention in the past decades (Schuettengruber et al., 2017; Kuroda et al., 2020). PcG proteins are critical for controlling cellular identity through maintaining appropriate gene repression profiles by forming Polycomb repressive complexes (PRC). Among the identified subgroups of PRCs, PRC2 mainly acts a histone methyltransferase for H3K27, PRC1, or more specifically non-canonical PRC1 is responsible for H2AK119 monoubiquitylation (H2AK119ub1; Di Croce and Helin, 2013). In contrast, TrxG proteins counteract PcG-mediated gene silencing, mainly by catalyzing methylation at H3K4.
Interestingly, another group of genes with dual functions in the maintenance of both transcriptional activation and repression in Drosophila has been categorized as Enhancers of Trithorax and Polycomb (ETP; Gildea et al., 2000). Asx was initially identified as one of ETP genes as Asx-null mutants exhibit phenotypes of both TrxG and PcG mutants (Sinclair et al., 1998; Milne et al., 1999). Biochemically, Asx interacts with Calypso to form a specific H2AK119 deubiquitylase complex. Loss of Calypso or Asx results in an increase of global H2AK119ub1 levels, however, unexpectedly derepression of PcG target genes. Accordingly this complex was named as Polycomb-repressive deubiquitylase (PR-DUB; Scheuermann et al., 2010). Three mammalian Additional Sex combs-like (ASXL) genes (ASXL1, ASXL2, and ASXL3) have been identified as the orthologs of Drosophila Asx. The encoding proteins share common organized regions, an ASXN domain at the N-terminal region, an ASX homology (ASXH) domain at the N-terminal adjacent region and a plant homeodomain (PHD) domain in the C-terminal region. ASXL1 directly binds to BAP1 through the ASXH domain and its adjoining region (also called deubiquitinase adaptor, DEUBAD; Sahtoe et al., 2016; Figure 1). In this mini-review, we focus on ASXL1 and discuss the current functional and mechanistic understanding of mammalian PR-DUB.
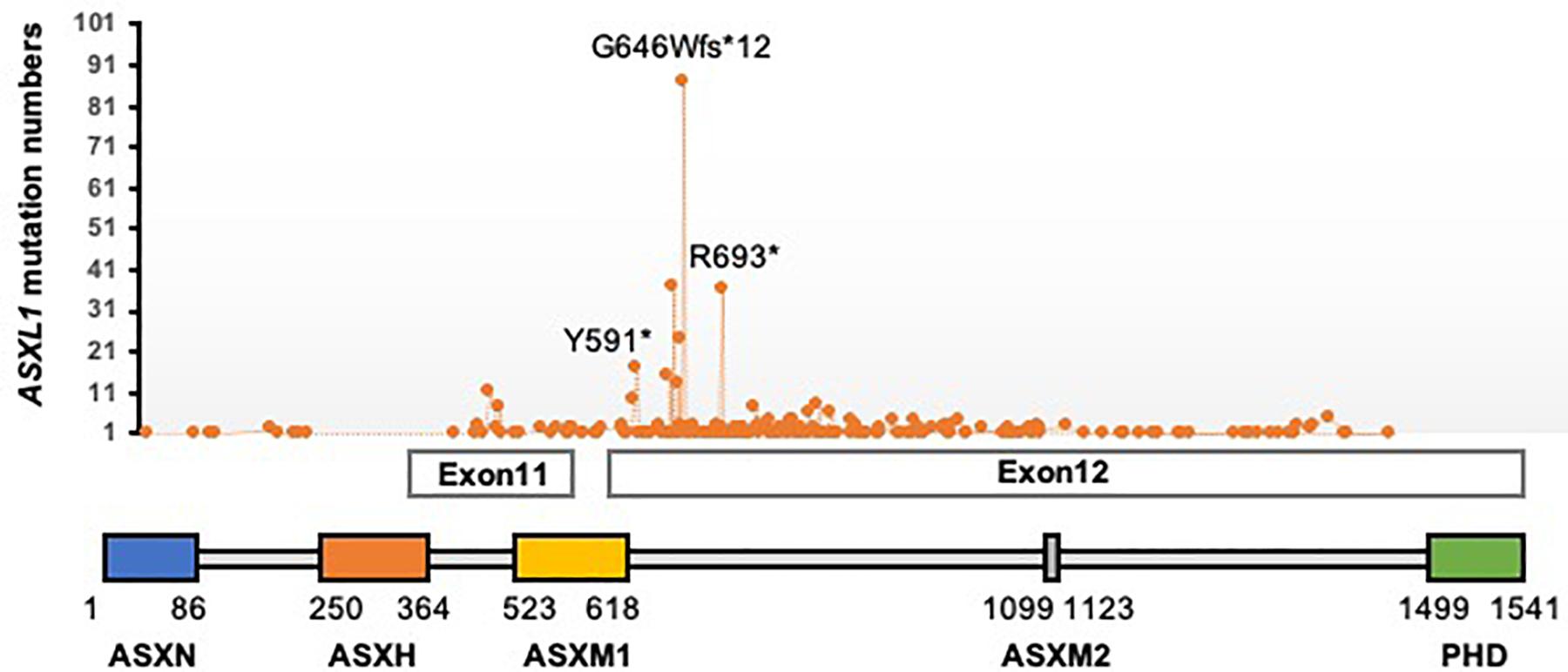
Figure 1. Schematic representation of ASXL1 protein domains and the location of amino acids affected by mutations found in cancers. ∗indicates a truncating mutation.
ASXL1 Mutations in Myeloid Malignancies: Loss or Gain-Of-Functions?
Heterozygous frame-shift or nonsense mutations of ASXL1, usually in exons 11 or 12, before the PHD domain (Figure 1), have been the most frequently found in myeloid malignancies, 10–25% in patients with myelodysplastic syndrome (MDS), 40–50% of chronic myelomonocytic leukemia (CMML) patients, 5–11% of patients with primary Acute myeloid leukemia (AML). And the presence of the mutations is associated with adverse outcomes (Gelsi-Boyer et al., 2009, 2010; Boultwood et al., 2010; Chou et al., 2010; Metzeler et al., 2011; Thol et al., 2011; Itzykson et al., 2013). In CMML patients, additional TET2 mutations were associated with shorter survival in the presence of ASXL1 mutations (Cui et al., 2015, 2016). In contrast, ASXL1 mutations are mutually exclusive with DNMT3A, NPM1, and SF3B1 mutations (Asada et al., 2019). These correlations should be taken into account for clinical prognosis as well as functional studies.
To date, a variety of mouse models have been taken to clarify roles of ASXL1 in development and diseases. It is interesting to note that mice with Asxl1 loss alone does not develop malignancies. Hematopoietic-specific deletion of Asxl1 results in progressive leukopenia and dysplasia. The numbers of hematopoietic stem/progenitor cells are increased and their differentiation is impaired (Abdel-Wahab et al., 2013). To better simulate the heterozygous patient-derived mutations, Uni et al. recently generated a mouse model with a heterozygous knock-in of Asxl1G643fs at the original Asxl1 locus. And the mutant mice developed phenotypes recapitulating human low-risk MDS and some of the mice developed MDS/myeloproliferative neoplasm-like disease after long latency (Uni et al., 2019). Consistent with the clinical prognosis (Cui et al., 2015, 2016), hematopoietic-specific deletion of both Asxl1 and Tet2 leads to a much earlier onset of MDS than the deletion of Asxl1 or Tet2 alone. In support, Asxl1 deficiency accompanied with haploinsufficiency of Nf1 or KRas oncogenic mutation accelerates the development of myeloid leukemia in mice (Abdel-Wahab et al., 2012; Zhang et al., 2018). Therefore, these models argue that loss of ASXL1 contributes to the initiation and progression of myeloid malignancies, dependent or independent of its H2AK119 deubiquitylation activity.
Notably the expression of C-terminal truncation of ASXL1 has been observed in a few leukemia cell lines (Balasubramani et al., 2015; Xia et al., 2020). Moreover, artificial or transgenic overexpression of ASXL1 C-terminal truncated mutants leads to gain of H2AK119 deubiquitylation activity and facilitates myeloid transformation (Inoue et al., 2013; Balasubramani et al., 2015; Asada et al., 2018; Nagase et al., 2018; Yang et al., 2018; Xia et al., 2020). Besides, ASXL1 mutant may synergize with hematopoietically expressed homeobox (HHEX) to promote myeloid transformation. In contrast, CRISPR/Cas9-mediated correction of ASXL1-truncated mutation decreases the proliferation of corresponding leukemia cells (Valletta et al., 2015; Takeda et al., 2020). Despite of these gain-of-function evidences for mutant ASXL1, it is yet clear how the globally decreased H2AK119ub1 contributes to malignancy.
Mechanistic Insights Into H2Ak119ub1 Deregulation in Malignancies
H2AK119ub1 has long been suggested as an important mark for PcG-mediated gene silencing. Hence it is surprising that ASXL1-BAP1 or PR-DUB is genetically defined as a repressive complex. And ASXL1 loss leads to a global reduction of H3K27me3 in certain contexts (Abdel-Wahab et al., 2012; Wang et al., 2014). However, recently, two independent studies have argued that BAP1 is required for the activation of a subset of target genes (Campagne et al., 2019; Kolovos et al., 2020), though direct evidence is lacking whether these activating functions are based on H2AK119 deubiquitylation. And we have demonstrated that ASXL1 is driven to erase H2AK119ub1 for transcription induction. Loss of ASXL1 leads to the retention of H2AK119ub1 at the regulatory loci of INK4B and PTEN and thereby the inactivation of these two well-known tumor suppressor genes in response to oncogenic signals, supporting ASXL1 as a tumor suppressor (Wu et al., 2015; Cao et al., 2020).
To resolve the discrepancy, it may require to re-examine the roles of ASXL1 and H2AK119ub1 in transcription regulation. Actually, ASXL1 but not its mutant form has been shown to be incorporated into PRC1 (Uni et al., 2019). In addition, Asxl1 mutation has been recently reported to correlate with reduced expression of PRC1 members in zebrafish (Fang et al., 2021). Hence, ASXL1 mutations may lead to loss of PRC1 functions through various mechanisms. As for H2AK119ub1, a diffuse distribution far beyond at promoters has been observed (Loubiere et al., 2020; Tamburri et al., 2020). Notably H2AK119ub1 is recognized by RYBP and JARID2 (Cooper et al., 2016; Zhao et al., 2020), and therefore it facilitates PRC2 recruitment (Kasinath et al., 2021) and the propagation of Polycomb domain (Zhao et al., 2020). Accordingly the low but detectable levels of H2AK119ub1 at non-promoter regions may function as a sponge to attract PRCs. The excessive gain of H2AK119ub1 upon loss of PR-DUB may lead to ineligible PRC relocation from repressive promoters to non-promoter regions and therefore a passive derepression of target genes (Figure 2). The observed decrease of H3K27me3 at PcG-targeted promoters in ASXL1 or BAP1-deficient cells likely follows this model (Abdel-Wahab et al., 2012; LaFave et al., 2015). Furthermore, non-canonical PRC1 and H2AK119ub1 have recently been shown to be associated with active histone marks (Yao et al., 2018; Cohen et al., 2020) and gene responsiveness in Arabidopsis (Kralemann et al., 2020; Yin et al., 2021). Considering that ubiquitin is almost half size of core histones and may affect the dynamics of chromatin architecture, we envisage that H2AK119ub1 may confer responsiveness to transcription induction for unknown reasons.
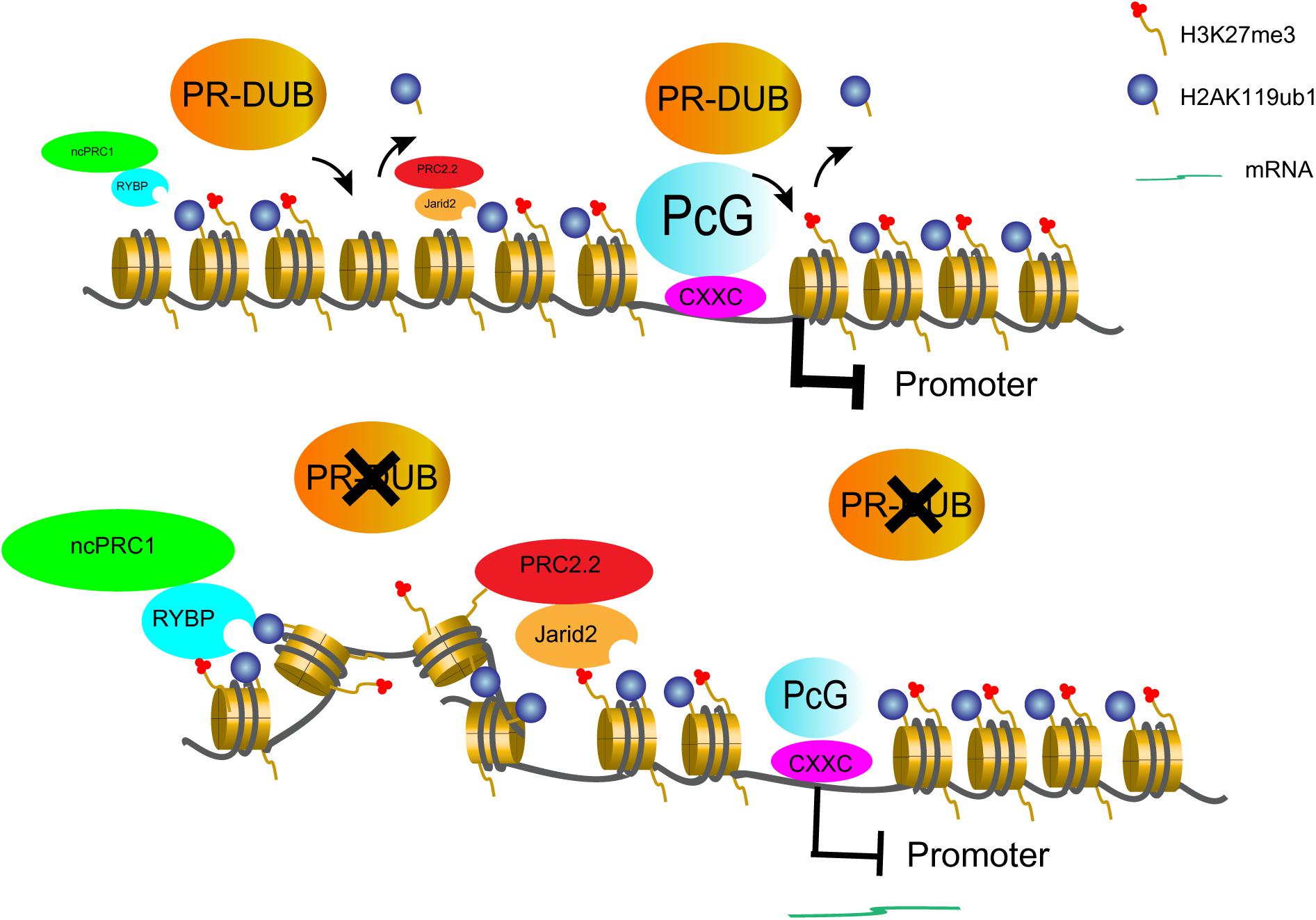
Figure 2. A prospective model to define PR-DUB as a transcription repressive complex. PR-DUB restricts H2AK119ub1 and PcG proteins mainly at promoters. However, upon PR-DUB loss, the gain of H2AK119ub1 at non-promoters attracts PcG proteins away from the target promoters and thereby leads to gene depression.
Discussion
Emerging studies have challenged the traditional viewpoint that chromatin modifications are instructive for transcription regulation (Morgan and Shilatifard, 2020). In this scenario, H2AK119ub1 does not have to determine transcriptional repression though it is associated with inactive genes. Either defining H2AK119ub1 as an active or repressive mark may be over-simplified. Hence as an eraser, PR-DUB removes H2AK119ub1 to reconfigure the chromatin landscape, rather than simply turn on or off gene transcription. It is well recognized that the epigenome function is to allow phenotypic variation in adaptation to the changing environment. ASXL1 mutations, leading to either gain or loss of H2AK119 deubiquitylation activity and therefore overly permissive or restrictive chromatin landscape, causes improper adaptation to intrinsic or extrinsic signals and malignant transformation. In a broad sense, a proper interpretation of the effects of epigenetic changes on transcription is critical for functional insight into chromatin modifiers in development and diseases settings.
Author Contributions
LC and XW wrote the manuscript. LC and RL prepare the figures. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the National key research and development program (2017YFA0504102), National Natural Science Foundation of China (31701126, 31900464), Natural Science Foundation of Tianjin Municipal Science and Technology Commission (18JCJQJC48200) and Research Project of Tianjin Education Commission (2018KJ075, 2020ZD13).
References
Abdel-Wahab, O., Adli, M., Lafave, L. M., Gao, J., Hricik, T., Shih, A. H., et al. (2012). ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 22, 180–193. doi: 10.1016/j.ccr.2012.06.032
Abdel-Wahab, O., Gao, J., Adli, M., Dey, A., Trimarchi, T., Chung, Y. R., et al. (2013). Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 210, 2641–2659. doi: 10.1084/jem.20131141
Asada, S., Fujino, T., Goyama, S., and Kitamura, T. (2019). The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell. Mol. Life Sci. 76, 2511–2523. doi: 10.1007/s00018-019-03084-7
Asada, S., Goyama, S., Inoue, D., Shikata, S., Takeda, R., Fukushima, T., et al. (2018). Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat. Commun. 9:2733.
Balasubramani, A., Larjo, A., Bassein, J. A., Chang, X., Hastie, R. B., Togher, S. M., et al. (2015). Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat. Commun. 6:7307.
Boultwood, J., Perry, J., Zaman, R., Fernandez-Santamaria, C., Littlewood, T., Kusec, R., et al. (2010). High-density single nucleotide polymorphism array analysis and ASXL1 gene mutation screening in chronic myeloid leukemia during disease progression. Leukemia 24, 1139–1145. doi: 10.1038/leu.2010.65
Campagne, A., Lee, M. K., Zielinski, D., Michaud, A., Le Corre, S., Dingli, F., et al. (2019). BAP1 complex promotes transcription by opposing PRC1-mediated H2A ubiquitylation. Nat. Commun. 10:348.
Cao, L., Xia, X., Kong, Y., Jia, F., Yuan, B., Li, R., et al. (2020). Deregulation of tumor suppressive ASXL1-PTEN/AKT axis in myeloid malignancies. J. Mol. Cell Biol. 12, 688–699. doi: 10.1093/jmcb/mjaa011
Chou, W. C., Huang, H. H., Hou, H. A., Chen, C. Y., Tang, J. L., Yao, M., et al. (2010). Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood 116, 4086–4094. doi: 10.1182/blood-2010-05-283291
Cohen, I., Bar, C., and Ezhkova, E. (2020). Activity of PRC1 and histone H2AK119 monoubiquitination: revising popular misconceptions. Bioessays 42:e1900192.
Cooper, S., Grijzenhout, A., Underwood, E., Ancelin, K., Zhang, T., Nesterova, T. B., et al. (2016). Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Commun. 7:13661.
Cui, Y., Tong, H., Du, X., Li, B., Gale, R. P., Qin, T., et al. (2015). Impact of TET2, SRSF2, ASXL1 and SETBP1 mutations on survival of patients with chronic myelomonocytic leukemia. Exp. Hematol. Oncol. 4:14.
Cui, Y., Tong, H., Du, X., Li, B., Gale, R. P., Qin, T., et al. (2016). TET2 mutations were predictive of inferior prognosis in the presence of ASXL1 mutations in patients with chronic myelomonocytic leukemia. Stem Cell Investig. 3:50. doi: 10.21037/sci.2016.09.04
Di Croce, L., and Helin, K. (2013). Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 20, 1147–1155. doi: 10.1038/nsmb.2669
Fang, X., Xu, S., Zhang, Y., Xu, J., Huang, Z., Liu, W., et al. (2021). Asxl1 C-terminal mutation perturbs neutrophil differentiation in zebrafish. Leukemia. doi: 10.1038/s41375-021-01121-8 [Epub ahead of print].
Gelsi-Boyer, V., Trouplin, V., Adelaide, J., Bonansea, J., Cervera, N., Carbuccia, N., et al. (2009). Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 145, 788–800. doi: 10.1111/j.1365-2141.2009.07697.x
Gelsi-Boyer, V., Trouplin, V., Roquain, J., Adelaide, J., Carbuccia, N., Esterni, B., et al. (2010). ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br. J. Haematol. 151, 365–375. doi: 10.1111/j.1365-2141.2010.08381.x
Gildea, J. J., Lopez, R., and Shearn, A. (2000). A screen for new trithorax group genes identified little imaginal discs, the Drosophila melanogaster homologue of human retinoblastoma binding protein 2. Genetics 156, 645–663.
Inoue, D., Kitaura, J., Togami, K., Nishimura, K., Enomoto, Y., Uchida, T., et al. (2013). Myelodysplastic syndromes are induced by histone methylation-altering ASXL1 mutations. J. Clin. Invest. 123, 4627–4640. doi: 10.1172/jci70739
Itzykson, R., Kosmider, O., Renneville, A., Gelsi-Boyer, V., Meggendorfer, M., Morabito, M., et al. (2013). Prognostic score including gene mutations in chronic myelomonocytic leukemia. J. Clin. Oncol. 31, 2428–2436. doi: 10.1200/jco.2012.47.3314
Kasinath, V., Beck, C., Sauer, P., Poepsel, S., Kosmatka, J., Faini, M., et al. (2021). JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science 371:eabc3393. doi: 10.1126/science.abc3393
Kolovos, P., Nishimura, K., Sankar, A., Sidoli, S., Cloos, P. A., Helin, K., et al. (2020). PR-DUB maintains the expression of critical genes through FOXK1/2- and ASXL1/2/3-dependent recruitment to chromatin and H2AK119ub1 deubiquitination. Genome Res. 30, 1119–1130. doi: 10.1101/gr.261016.120
Kralemann, L. E. M., Liu, S., Trejo-Arellano, M. S., Munoz-Viana, R., Kohler, C., and Hennig, L. (2020). Removal of H2Aub1 by ubiquitin-specific proteases 12 and 13 is required for stable Polycomb-mediated gene repression in Arabidopsis. Genome Biol. 21:144.
Kuroda, M. I., Kang, H., De, S., and Kassis, J. A. (2020). Dynamic competition of Polycomb and Trithorax in transcriptional programming. Annu. Rev. Biochem. 89, 235–253. doi: 10.1146/annurev-biochem-120219-103641
LaFave, L. M., Beguelin, W., Koche, R., Teater, M., Spitzer, B., Chramiec, A., et al. (2015). Loss of BAP1 function leads to EZH2-dependent transformation. Nat. Med. 21, 1344–1349. doi: 10.1038/nm.3947
Loubiere, V., Papadopoulos, G. L., Szabo, Q., Martinez, A. M., and Cavalli, G. (2020). Widespread activation of developmental gene expression characterized by PRC1-dependent chromatin looping. Sci. Adv. 6:eaax4001. doi: 10.1126/sciadv.aax4001
Metzeler, K. H., Becker, H., Maharry, K., Radmacher, M. D., Kohlschmidt, J., Mrozek, K., et al. (2011). ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN favorable genetic category. Blood 118, 6920–6929. doi: 10.1182/blood-2011-08-368225
Milne, T. A., Sinclair, D. A., and Brock, H. W. (1999). The additional sex combs gene of Drosophila is required for activation and repression of homeotic loci, and interacts specifically with Polycomb and super sex combs. Mol. Gen. Genet. 261, 753–761. doi: 10.1007/s004380050018
Morgan, M. A. J., and Shilatifard, A. (2020). Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 52, 1271–1281. doi: 10.1038/s41588-020-00736-4
Nagase, R., Inoue, D., Pastore, A., Fujino, T., Hou, H. A., Yamasaki, N., et al. (2018). Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J. Exp. Med. 215, 1729–1747. doi: 10.1084/jem.20171151
Sahtoe, D. D., van Dijk, W. J., Ekkebus, R., Ovaa, H., and Sixma, T. K. (2016). BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat. Commun. 7:10292.
Scheuermann, J. C., de Ayala Alonso, A. G., Oktaba, K., Ly-Hartig, N., McGinty, R. K., Fraterman, S., et al. (2010). Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 465, 243–247. doi: 10.1038/nature08966
Schuettengruber, B., Bourbon, H. M., Di Croce, L., and Cavalli, G. (2017). Genome regulation by Polycomb and Trithorax: 70 years and counting. Cell 171, 34–57. doi: 10.1016/j.cell.2017.08.002
Sinclair, D. A., Milne, T. A., Hodgson, J. W., Shellard, J., Salinas, C. A., Kyba, M., et al. (1998). The additional sex combs gene of Drosophila encodes a chromatin protein that binds to shared and unique Polycomb group sites on polytene chromosomes. Development 125, 1207–1216.
Takeda, R., Asada, S., Park, S. J., Yokoyama, A., Becker, H. J., Kanai, A., et al. (2020). HHEX promotes myeloid transformation in cooperation with mutant ASXL1. Blood 136, 1670–1684.
Tamburri, S., Lavarone, E., Fernandez-Perez, D., Conway, E., Zanotti, M., Manganaro, D., et al. (2020). Histone H2AK119 mono-ubiquitination is essential for Polycomb-mediated transcriptional repression. Mol. Cell 77, 840–856.e5.
Thol, F., Friesen, I., Damm, F., Yun, H., Weissinger, E. M., Krauter, J., et al. (2011). Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J. Clin. Oncol. 29, 2499–2506. doi: 10.1200/jco.2010.33.4938
Uni, M., Masamoto, Y., Sato, T., Kamikubo, Y., Arai, S., Hara, E., et al. (2019). Modeling ASXL1 mutation revealed impaired hematopoiesis caused by derepression of p16Ink4a through aberrant PRC1-mediated histone modification. Leukemia 33, 191–204. doi: 10.1038/s41375-018-0198-6
Valletta, S., Dolatshad, H., Bartenstein, M., Yip, B. H., Bello, E., Gordon, S., et al. (2015). ASXL1 mutation correction by CRISPR/Cas9 restores gene function in leukemia cells and increases survival in mouse xenografts. Oncotarget 6, 44061–44071. doi: 10.18632/oncotarget.6392
Wang, J., Li, Z., He, Y., Pan, F., Chen, S., Rhodes, S., et al. (2014). Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood 123, 541–553. doi: 10.1182/blood-2013-05-500272
Wu, X., Bekker-Jensen, I. H., Christensen, J., Rasmussen, K. D., Sidoli, S., Qi, Y., et al. (2015). Tumor suppressor ASXL1 is essential for the activation of INK4B expression in response to oncogene activity and anti-proliferative signals. Cell Res. 25, 1205–1218. doi: 10.1038/cr.2015.121
Xia, Y. K., Zeng, Y. R., Zhang, M. L., Liu, P., Liu, F., Zhang, H., et al. (2020). Tumor-derived neomorphic mutations in ASXL1 impairs the BAP1-ASXL1-FOXK1/K2 transcription network. Protein Cell. doi: 10.1007/s13238-020-00754-2 [Epub ahead of print].
Yang, H., Kurtenbach, S., Guo, Y., Lohse, I., Durante, M. A., Li, J., et al. (2018). Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 131, 328–341. doi: 10.1182/blood-2017-06-789669
Yao, M., Zhou, X., Zhou, J., Gong, S., Hu, G., Li, J., et al. (2018). PCGF5 is required for neural differentiation of embryonic stem cells. Nat. Commun. 9:1463.
Yin, X., Romero-Campero, F. J., de Los Reyes, P., Yan, P., Yang, J., Tian, G., et al. (2021). H2AK121ub in Arabidopsis associates with a less accessible chromatin state at transcriptional regulation hotspots. Nat. Commun. 12:315.
Zhang, P., He, F., Bai, J., Yamamoto, S., Chen, S., Zhang, L., et al. (2018). Chromatin regulator Asxl1 loss and Nf1 haploinsufficiency cooperate to accelerate myeloid malignancy. J. Clin. Invest. 128, 5383–5398. doi: 10.1172/jci121366
Keywords: histone modification, polycomb, H2AK119ub1, myeloid maliganancies, cancers, mutation, transcription regulation
Citation: Cao L, Li R and Wu X (2021) The Functions and Mechanisms of PR-DUB in Malignancy. Front. Mol. Biosci. 8:657150. doi: 10.3389/fmolb.2021.657150
Received: 22 January 2021; Accepted: 23 February 2021;
Published: 12 March 2021.
Edited by:
Hai-Ning Du, Wuhan University, ChinaReviewed by:
Shuhei Asada, Tokyo Women’s Medical University, JapanHongjie Yao, Chinese Academy of Sciences (CAS), China
Copyright © 2021 Cao, Li and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xudong Wu, wuxudong@tmu.edu.cn