
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci. , 22 October 2021
Sec. RNA Networks and Biology
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.652443
This article is part of the Research Topic The Role of Epithelial-Mesenchymal Transition (EMT)-Related Non-coding RNAs in Cancer View all 8 articles
 Yong Luo1*†
Yong Luo1*† Xiaobing Yang1†
Xiaobing Yang1† Spyridon P. Basourakos2
Spyridon P. Basourakos2 Xuemei Zuo2
Xuemei Zuo2 Dechao Wei1Jiahui Zhao1Mingchuan Li1Qiankun Li1Tao Feng1
Dechao Wei1Jiahui Zhao1Mingchuan Li1Qiankun Li1Tao Feng1 Pengju Guo1Yongguang Jiang1
Pengju Guo1Yongguang Jiang1Previous studies showed that CXCR7 expression was upregulated after enzalutamide (ENZ) treatment, and an increased level of CXCR7 could increase the invasion, migration, and angiogenesis of castration-resistant prostate cancer (CRPC) cells. This study demonstrated that the levels of p-JAK2, p-STAT1, C-Myc, and VEGFR2 were significantly reduced after CCX771, a specific CXCR7 inhibitor, treatment. This effect further increased after the combination treatment of ENZ and CCX771. Then, we verified that targeting the inhibition of JAK2 or STAT1 could remarkably increase apoptosis and DNA damage and decrease the migration of CRPC cells. More importantly, the combination treatment of ENZ + JAK2/STAT1 led to much greater suppression than the single-agent treatment of JAK2 or STAT1. Subcutaneous CRPC xenograft tumor growth was also reduced by single-agent ENZ treatment and single-agent FLUD, a specific STAT1 antagonist, treatment; but much superior effect was elicited by the combination treatment of ENZ + FLUD. The proliferative indices significantly decreased following combination treatment in tumor tissues compared with control-treatment tissues and single-agent-treatment tissues. Our results demonstrated that CXCR7, which signifies an androgen receptor (AR)-independent signaling pathway, caused CRPC progression via the downstream JAK2/STAT1 signal transduction cascade. Combined inhibition targeting both the AR and JAK2/STAT1 resulted in substantial tumor suppression due to the reduction in DNA damage repair ability and increment in apoptosis.
Castration-resistant prostate cancer (CRPC) is an extremely advanced state of prostate cancer progression, which indicates a complete failure of traditional androgen deprivation therapy (ADT) and has a very inadequate prognosis. ENZ, a second-generation androgen receptor (AR) antagonist, is an important treatment option for patients with CRPC, which significantly prolongs the median overall survival of CRPC to 32.4 months (Beer et al., 2014). However, about 42% of patients with CRPC are not sensitive to ENZ treatment (Azuma, 2018). Even if the treatment is effective, ENZ resistance (Beer et al., 2014) usually occurs after 11.2 months. This modest survival benefit is related to a significant variation in the rate of development of resistance pathways among individuals in clinical trials. It also indicates the need for a deeper understanding of the molecular mechanisms that lead to androgen ablation resistance and the development of more effective predictive biomarkers and therapeutic approaches for CRPC. Therefore, how to overcome ENZ resistance has become a critical research focus in this field.
Extensive research suggests that, among several risk factors, inflammation plays an important role in the development and progression of primary PCa to metastatic disease (Carver et al., 2011). Elucidating the details of inflammatory signaling pathways not only promotes the understanding of the tumor progression mechanism but also helps confirm the molecular characteristics of cases vulnerable to immunotherapy. Additionally, chemokines and chemokine receptor networks are fundamental components of the complex interactions between inflammatory cells and PCa cells. The key essence of ENZ resistance is the process in which AR activity is usually suppressed by drugs; meanwhile, the AR bypass pathways are continuously activated. Our previous study (Luo et al., 2018) confirmed that CXCR7 derepression was closely associated with ENZ resistance. When AR activity was inhibited by ENZ, CXCR7 began to derepress from AR suppression and further drove CRPC cell progression through increasing the potency of anti-apoptosis, rapid proliferation, DNA repair, and angiogenesis. After combination with the CXCR7 antagonist (CCX771), the ENZ treatment response was significantly improved in both CRPC cellular and animal models. Li also demonstrated that elevated CXCR7 expression activated MAPK/ERK signaling through ligand-independent, but β-arrestin 2–dependent, mechanisms and led to tumor progression upon enzalutamide resistance. The inhibition of MAPK/ERK activation could remarkably suppress ENZ-resistant CRPC (Li et al., 2019). Rafiei found that CXCR7 derepression induced by macrophage migration inhibitory factor could upregulate the expression of cell cycle genes through activating the Akt signaling pathway and drive cellular ENZ-resistant growth (Rafiei et al., 2019). More recent reports confirmed that CXCR7 derepression was closely involved in the androgen ablation–resistant process. However, the underlying mechanism of how CXCR7 triggers CRPC progression and survival remains unclear.
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signal transduction pathway is a master signaling pathway that always regulates cell survival, apoptosis, hypertrophy, and collagen synthesis in the heart of mammals and is activated in response to oxidative stress, inflammatory cytokines, and growth factors. The JAK/STAT signaling pathway plays an important role in various physiological processes of cell proliferation, differentiation, tumorigenesis, immune function, and hematopoiesis (Bolli et al., 2003). The present study verified that CXCR7 derepression drove CRPC cell progression and migration via the JAK2/STAT1 pathway. It also reported the synergistic effect of ENZ plus specific JAK/STAT inhibitor to inhibit the survival, invasion, and migration of CRPC cells and suppress the growth of CRPC xenograft tumors.
The human androgen-independent prostate cancer cell line C4-2B (American Type Culture Collection, VA, United States) was validated by short tandem repeat DNA fingerprinting with an AmpFLSTR Identifiler Kit (Applied Biosystems, CA, United States) at MD Anderson’s Characterized Cell Line Core Facility. The cells were cultured in DMEM containing 1 mM sodium pyruvate, 2.5 mM glutamine, 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified incubator containing 5% CO2. The serum-free medium was regularly used in the cell transfection trials. Enzalutamide (AR inhibitor), AMD3100 (CXCR4 inhibitor), and AZD1480 (ATP competitive antagonist of JAK2) were purchased from SelleckChem, and SDF-1 (CXCR4/7 activated ligand) was purchased from GenScript. CCX771 (CXCR7-specific inhibitor) and CCX704 (the control inhibitor of CXCR7, with the molecular structure similar to CCX771 but not binding to CXCR7) were provided by ChemoCentryx (CA, United States). Fludarabine (STAT1 synthesis inhibitor) and GSI-XII (Notch-1 inhibitor) were obtained from MedChemExpress. In addition, C4-2B cells were seeded at desired densities (5 × 105/well in a six-well plate for western blotting and wound-healing assay; 1 × 105/well in a 24-well plate for flow cytometry; and 1.5 × 104/well in a six-well plate for ELISA).
C4-2B cells were pretreated with 100 ng/ml SDF-1 + 1 M ENZ or 100 ng/ml SDF-1 + 800 nM CCX771 for 24 h. On the following day, the cells were again treated with 20 μg/ml AMD3100, 25 µM GSI-XII, or 1 µM CCX704 for 48 h. Additionally, the cells in the ENZ and CCX771 groups were treated with a single drug for 72 h. After treatment, the cellular protein was extracted for western blotting to assess the downstream factors of the CXCR7 signaling pathway.
C4-2B cells were transfected with siRNA to knock down JAK2 or STAT1 and further cultured for 72 h. Meanwhile, the cells were treated with 5 µM AZD1480 or 5 µM fludarabine for 72 h to inhibit the activity of JAK2 and STAT1, respectively. After treatment, cellular extraction was used for western blotting to assess the downstream factors of the JAK2/STAT1 pathway.
C4-2B cells were transfected with siRNA for 24 h. On the following day, the cells were further treated with vehicle control DMSO, 100 ng/ml SDF-1 + 800 nM CCX771, or 100 ng/ml SDF-1 + 1 µM ENZ for 48 h. After treatment, all samples were comparatively evaluated for the change profile of apoptosis, migration, and DNA damage.
The cells were seeded 1 day before siRNA transfection. JAK2 (clone ID: OHu24049, GenScript), the STAT1 (clone ID: OHu15845, GenScript) plasmid, and the control vector were transfected into cells using X-tremeGENE HP DNA transfection reagent (Roche). All samples were prepared 72 h after plasmid transfection for western blotting, wound-healing assay, flow cytometry analysis, and ELISA.
Clarified protein lysates were separated on denaturing SDS-PAGE and electro-transferred onto nitrocellulose membranes. The membranes were initially incubated with 5% non-fat dry milk in TBS for 2 h and then probed at 4°C overnight with the following antibodies: anti-CXCR7 (ab138509, 1:250), anti-DNA-PKCs (ab44815, 1:500), anti-VEGFR2 (ab39638, 1:50) (Abcam, MA, United States), anti-JAK2 (cs4040, 1:1,000), anti-P-JAK2 (cs4406, Tyr1007, 1:500), anti-STAT1 (cs9176, 1:1,000), anti-P-STAT1 (cs8826, Ser727, 1:500), anti-C-Myc (cs9402, 1:1,000), anti-cleaved-PARP-1 (cs5625, 1:500), anti-Bcl-2 (cs4223, 1:1,000), anti-γH2AX (cs7631, 1:250) (Cell Signaling, MA, United States), and anti-GAPDH (sc47724, 1:1,000) (Santa Cruz Biotechnology, CA, United States). The membranes were then hybridized with horseradish peroxidase (HRP)-conjugated secondary antibody (sc2004/2005, Santa Cruz Biotechnology) for 2 h at room temperature. An enhanced chemiluminescence system (AMRESCO, Inc., OH, United States) was used to detect the immunopositive protein bands. Additionally, the cell lysis buffer containing phosphatase inhibitors (Na3VO4/1 mM) was regularly applied during phosphorylated protein extraction and detection. Each experiment was repeated five times.
The cells were harvested and stained with propidium iodide for 30 min at room temperature. The analysis was performed at 405 nm excitation and emission with a 450/50 band-pass filter using a FACSCanto II Flow Cytometer (BD Biosciences, NJ, United States). The histograms of DNA content were analyzed using the FlowJo software (Tree Star, Inc., OR, United States) to determine cell cycle distribution. Each experiment was repeated three times.
The cells were grown to 80% confluence in six-well plates in RPMI with 10% FBS and antibiotics. A straight scratch was made using a 10 µl pipette tip at three different sites (top, middle, and bottom) in triplicate experiments. The medium was removed, and the cells were washed with the culture medium to remove the floating cell debris. After treatment, the cells were fixed and stained with crystal violet dye. Wound closure was measured using MRI Wound Healing Tool macro for ImageJ (v1.50b). Each experiment was repeated three times.
A DNA fragmentation assay was performed for apoptotic evaluation following the protocol of the Cell Death Detection ELISA Kit. Each experiment was repeated three times.
The cells (1 × 106) were resuspended in serum-free DMEM, mixed 1:1 with Matrigel (BD Biosciences, NJ, United States), and then injected subcutaneously into the left flanks of previously castrated SCID mice (Charles River Laboratories, MA, United States). When palpable tumors reached a volume of 30–50 mm3, the mice were randomly divided into different experimental groups to receive one of the following treatments for 28 days: DMSO, ENZ (10 mg/kg, oral gavage, daily), fludarabine (1 mg/kg, IP, 5 days/2 weeks), and ENZ + fludarabine. Tumor size was monitored by measuring two dimensions, and the volume was calculated as length × width2/2. When the 28-day protocol was finished, all mice were euthanized by carbon dioxide inhalation, and the tumors were harvested. The CO2 displaced the euthanized chamber at the rate of 10–30% of chamber volume per minute. The mice were also euthanized ahead of the protocol if they became severely weak or if the tumor size reached 20 mm.
Ki67 immunohistochemistry was carried out on formalin-fixed and paraffin-embedded tissue sections from the 28-day C4-2B xenografts after in vivo treatment. After tissue sections were deparaffinized and rehydrated through graded alcohol, they were heated in a microwave in 0.01 mol/L citrate buffer at pH 6.0 for 10 min to retrieve antigens. After a 30 min incubation in Dako protein blockage solution, the tissue sections were incubated in rabbit monoclonal antibody against Ki67 (1:300; Santa Cruz Biotechnology) for 90 min, followed by incubation in an HRP polymer−conjugated secondary antibody (Dako, Glostrup, Denmark) for 40 min. The immunoreaction was visualized in DAB/H2O2. The specificity of immunoreactions was verified by replacing the primary antibodies with PBS. Ten high-power fields were selected randomly in slides of each group, and the numbers of positively stained cells were counted and the percentage of positive cells was calculated.
Data were presented as mean ± standard deviation and analyzed with ANOVA with Tukey’s post hoc test. Statistical analysis was undertaken using SPSS 13.0 for Windows (SPSS, Inc., IL, United States). Two-sided p values < 0.05 indicated a statistically significant difference.
To explain how combination treatment of ENZ + CCX771 inhibited CRPC development, we detected some critical proteins of the JAK/STAT signaling pathway in C4-2B cells under different treatment conditions. Western blotting revealed a possible signaling mechanism of combined treatment. As shown in Figure 1, we demonstrated that the phosphorylation of JAK2 and STAT1 could be reduced significantly by CCX771-based treatment than ENZ-based treatment in C4-2B cells. In addition, the expression levels of C-Myc, an important proliferative regulator, and VEGFR, a critical angiogenesis producer, also markedly decreased in CCX771-based treatment cells, but the expression of cleaved-PARP-1 increased in CCX771-based treatment cells. More interestingly, the addition of ENZ to CCX771-treated cells (combination treatment group) exhibited much greater synergistic suppression of phosphorylated JAK2, phosphorylated STAT1, C-Myc, and VEGFR. ENZ treatment−induced CXCR7 upregulation was verified as the activator of the JAK2/STAT1 signaling pathway.
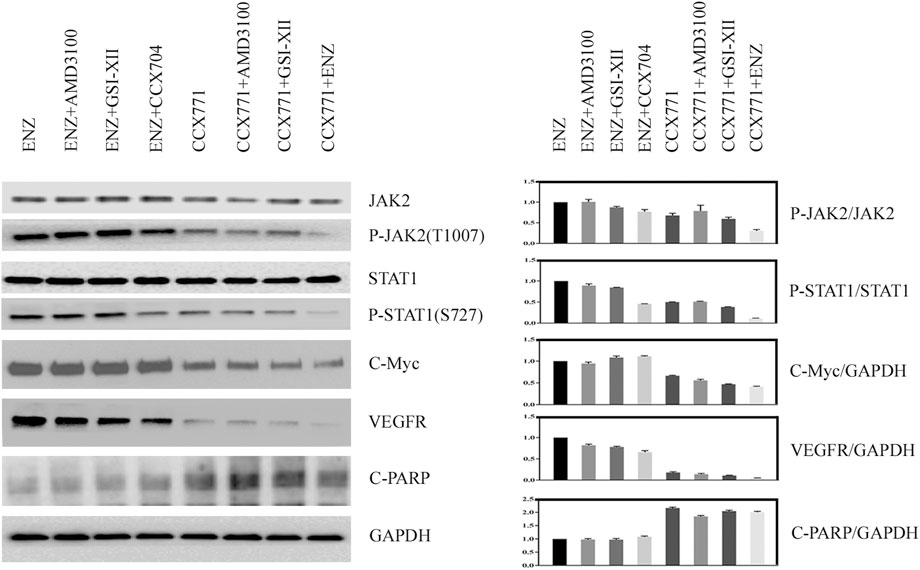
FIGURE 1. Comparison of CXCR7 downstream signaling proteins between CCX771-based treatment and ENZ-based treatment. (A) Western blotting demonstrated that the expression of phosphorylated JAK2, phosphorylated STAT1, C-Myc, and VEGFR markedly decreased, but the expression of cleaved-PARP-1 increased in CCX771-based treatment cells. The combination treatment of CCX771 + ENZ exhibited much greater synergistic suppression of phosphorylated JAK2, phosphorylated STAT1, C-Myc, and VEGFR. (B) All western blot band densities were quantitatively analyzed.
The effect of JAK2/STAT1 activity on the expression of downstream functional proteins was confirmed by western blotting. After knocking down the expression of JAK2 by siRNA transfection and repressing the activity of JAK2 with AZD1480 in C4-2B cells, the expression of CXCR7 did not display any change, but the expression of p-STAT1 decreased significantly. In addition, the expression of oncogene C-Myc and anti-apoptotic protein Bcl-2 apparently decreased, while the expression of DNA damage marker γH2AX increased. Moreover, the expression of non-homologous end-joining (NHEJ) repairing protein DNA-PKCs significantly decreased, but the expression of homologous repairing (HR) protein C-PARP-1 increased (Figure 2). Similarly, the inhibition of STAT1 activity did not affect the expression of CXCR7 and phosphorylated JAK2. The expression of C-Myc, Bcl-2, and DNA-PKCs remarkably decreased, while the expression of γH2AX and C-PARP-1 significantly increased (Figure 3).
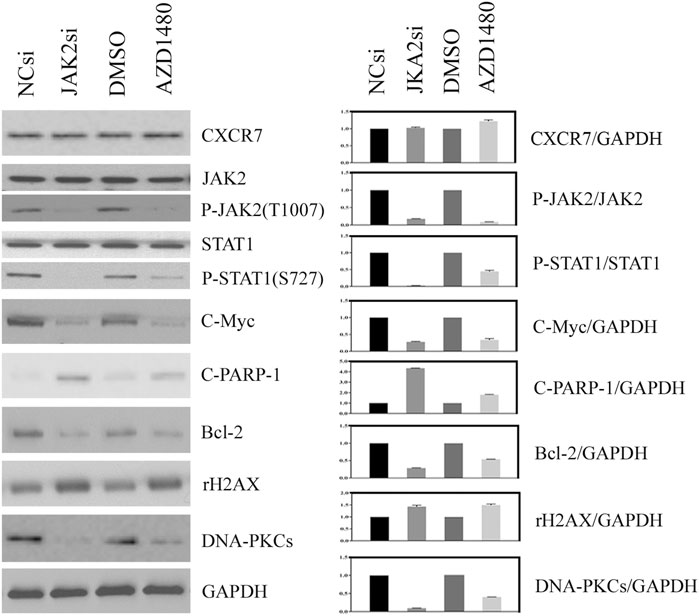
FIGURE 2. Change profile of downstream functional proteins after the inhibition of JAK2 activity. (A) Western blotting revealed that, after knocking down the expression of JAK2 by siRNA transfection and repressing the activity of JAK2 by AZD1480 in C4-2B cells, the expression of CXCR7 did not display any change, but the expression of p-STAT1, oncogene C-Myc, and anti-apoptotic protein Bcl-2 decreased significantly. In addition, the expression of DNA damage marker γH2AX increased, the expression of non-homologous end-joining (NHEJ) repairing protein DNA-PKCs significantly decreased, but the expression of homologous repairing (HR) protein C-PARP-1 increased. (B) All WB band densities were quantitatively analyzed.
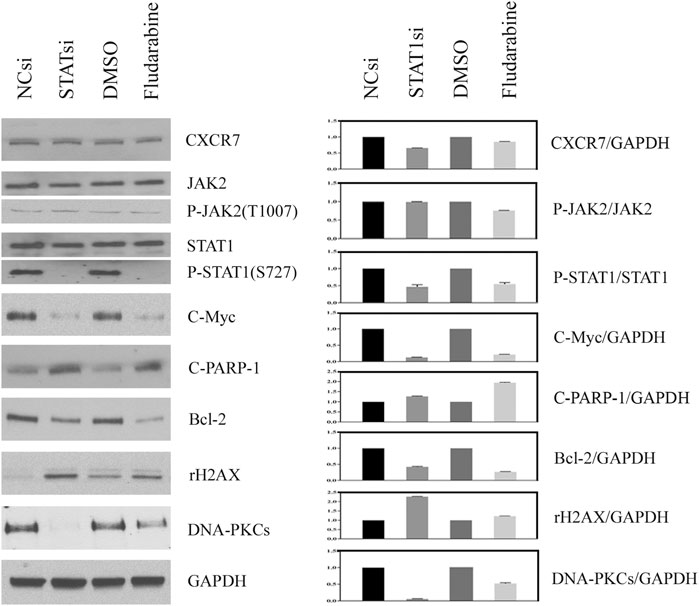
FIGURE 3. Change profile of downstream functional proteins after the inhibition of STAT1 activity. (A) Western blotting revealed that, after knocking down the expression of JAK2 by siRNA transfection and repressing the activity of JAK2 by FLUD in C4-2B cells, the expression of CXCR7 and P-JAK2 did not display any change, but the expression of C-Myc and Bcl-2 decreased remarkably. In contrast, the expression of DNA damage marker γH2AX increased apparently, the expression of homologous repairing (HR) protein C-PARP-1 increased accordingly, but the expression of non-homologous end-joining (NHEJ) repairing protein DNA-PKCs decreased measurably. (B) All WB band densities were quantitative analyzed.
We further observed the change pattern of sub-G1 cells, aggressive potency, and DNA damaging extent in CRPC C4-2B cells under different treatment conditions to analyze the specific biological effects of the combination treatment of ENZ + JAK2/STAT1si that might result from the suppression of JAK/STAT-mediated signaling. As shown in Figures 4A–D, FACS confirmed that JAK2si and STAT1si could remarkably increase the percentage of sub-G1 cells compared with the control condition in C4-2B cells (JAK2si: p = 2.85E-06; STAT1si: p = 4.29E-08). Single-agent CCX771 and ENZ treatment also increased the percentage of sub-G1 cells compared with control treatment in C4-2B cells (CCX771: p = 1.62E-05; ENZ: 8.46E-09). The combination treatment of ENZ + JAK2si showed a significantly superior effect compared with single-agent JAK2si treatment (p = 3.84E-08), single-agent CCX771 treatment (p = 4.28E-08), single-agent ENZ treatment (p = 9.91E-07), and the combination treatment of CCX771 + JAK2si (p = 1.54E-06). The combination treatment of ENZ + STAT1si also showed a much better effect compared with single-agent STAT1si treatment (p = 5.92E-09), single-agent CCX771 treatment (p = 4.21E-09), single-agent ENZ treatment (p = 2.01E-08), and the combination treatment of CCX771 + STAT1si (p = 1.20E-07).
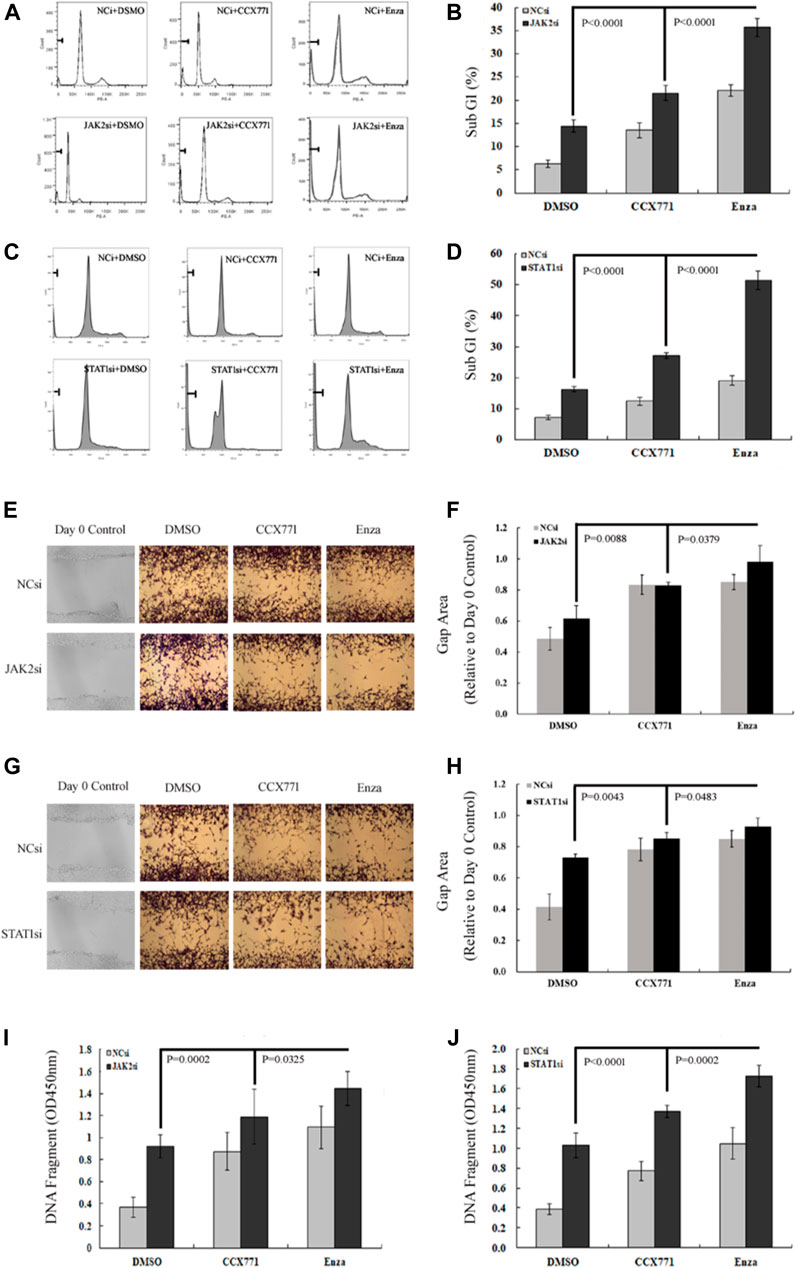
FIGURE 4. ENZ + JAK2/STAT1si synergistically suppressed survival, migration, and DNA damage repair of CRPC cells. (A,B) Flow cytometry demonstrated that single-agent JAK2si treatment remarkably increased the percentage of sub-G1 cells, and the combination treatment of ENZ + JAK2si showed a significantly superior effect compared with single-agent JAK2si treatment and the combination treatment of CCX771 + JAK2si. (C,D) Similarly, STAT1si significantly increased the percentage of sub-G1 cells, and the combination treatment of ENZ + STAT1si also showed a much greater effect compared with single-agent STAT1si treatment and the combination treatment of CCX771 + STAT1si. (E,F) Wound-healing assay results showed that the combination treatment of ENZ + JAK2si could reduce migration compared with single-agent JAK2si treatment and the combination treatment of CCX771 + JAK2si. (G,H) Wound-healing assay results showed that the combination treatment of ENZ + STAT1si also reduced migration compared with single-agent STAT1si treatment and the combination treatment of CCX771 + STAT1si. (I) ELISA results showed that the combination treatment of ENZ + JAK2si exhibited much stronger ability to induce DNA damage compared with single-agent JAK2si treatment and the combination treatment of CCX771 + JAK2si. (J) Similarly, the combination treatment of ENZ + STAT1si also led to much apparent DNA damage compared with single-agent STAT1si treatment and the combination treatment of CCX771 + STAT1si.
As displayed in Figures 4E–H, the wound-healing assay showed that C4-2B cells treated with JAK2si or STAT1si showed a significant reduction in cell migration compared with control conditions (JAK2si: p = 0.0371; STAT1si: p = 0.0031), whereas the combination treatment of ENZ + JAK2si displayed a greater effect compared with single-agent JAK2si treatment (p = 0.0088) and the combination treatment of CCX771 + JAK2si (p = 0.0379). However, a significant difference was not observed between the combination treatment of ENZ + JAK2si and single-agent ENZ/CCX771 treatment. In addition, the combination treatment of ENZ + STAT1si showed a similar suppressing effect on cell migration compared with single-agent STAT1si treatment (p = 0.0043), single-agent CCX771 treatment (p = 0.0470), single-agent ENZ treatment (p = 0.0495), and the combination treatment of CCX771 + STAT1si (p = 0.0483).
As presented in Figures 4I,J, ELISA showed that JAK2si or STAT1si treatment in C4-2B cells significantly resulted in an increase in the number of DNA fragments compared with the control condition (JAK2si: p = 1.20E-05; STAT1si: p = 5.92E-06). More importantly, the combination treatment of ENZ + JAK2si exhibited much stronger induction of DNA damage compared with single-agent JAK2si treatment (p = 0.0002), single-agent CCX771 treatment (p = 0.0006), single-agent ENZ treatment (p = 0.0117), and the combination treatment of CCX771 + JAK2si (p = 0.0325). ENZ + STAT1si also showed a better treatment effect compared with single-agent STAT1si treatment (p = 1.27E-05), single-agent CCX771 treatment (p = 4.46E-07), single-agent ENZ treatment (p = 4.62E-05), and the combination treatment of CCX771 + STAT1si (p = 0.0002). These data demonstrated that the combination treatment targeting JAK2/STAT1 and AR displayed a much stronger effect of inducing apoptosis and repressing migration in CRPC cells.
We used an AR-positive, androgen-independent, C4-2B subcutaneous CRPC model to further testify our hypothesis that the combination treatment of ENZ + JAK2/STAT1 was a promising therapy for CRPC. We treated mice bearing C4-2B subcutaneous tumors with ENZ alone, FLUD alone, and a combination of ENZ + FLUD and monitored their tumor progression following the protocol shown in Figure 5A.
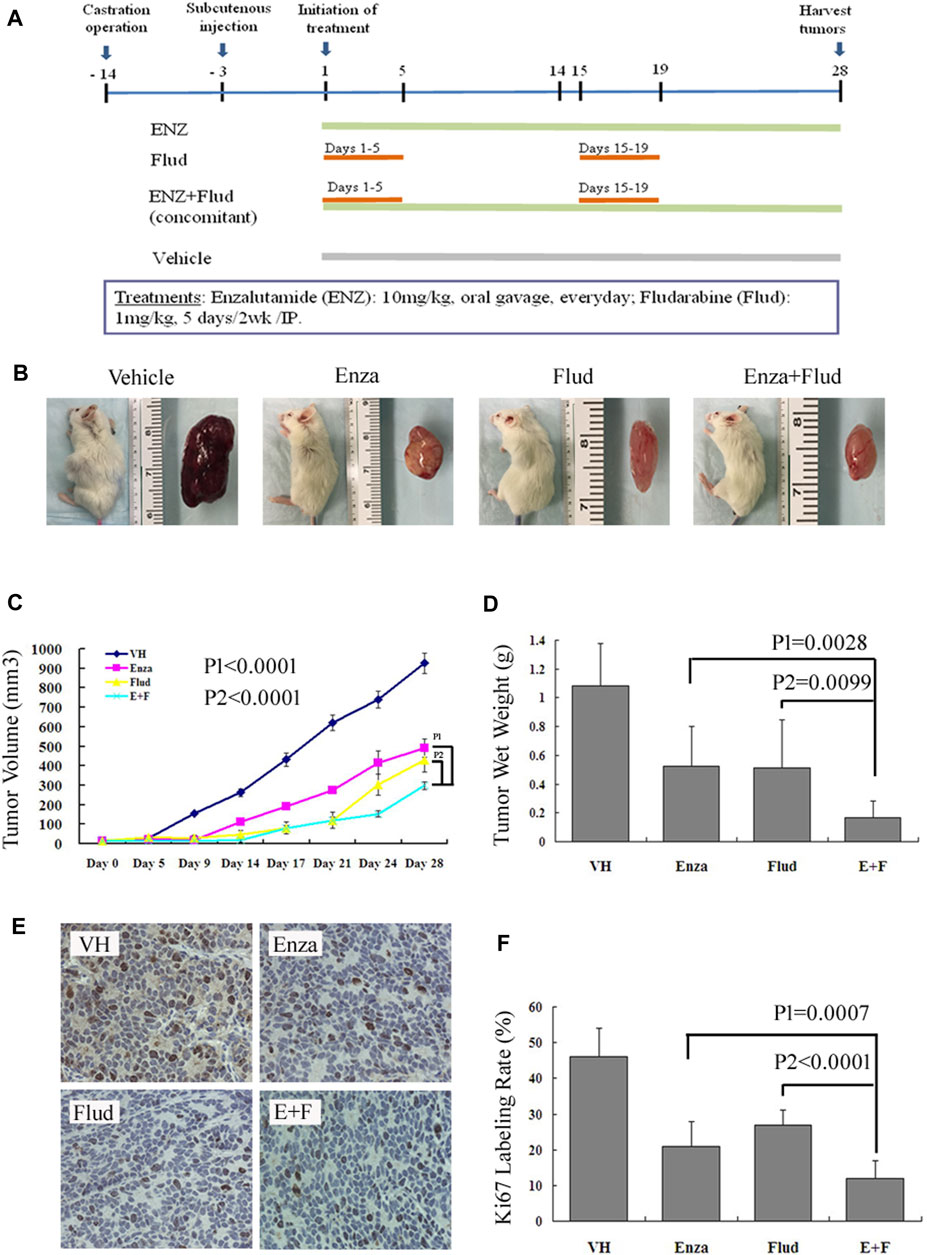
FIGURE 5. ENZ + FLUD strategy synergistically inhibited the growth of CRPC xenografts. (A) Drug protocol of each treatment group. (B) Representative C4-2B subcutaneous tumor of each treatment group. (C) Tumor volume measurements using calipers every 3–5 days. (D) Wet tumor weight at the end of 28 days of treatment. (E) Immunohistochemistry of Ki67 in each treatment group. (F) Ratio of Ki67-positive cells in each treatment group.
The AR is an extremely important regulator that drives the progression of CRPC depending on not only self-transcriptional activity but also other signaling pathways, such as Jagged1/Notch1 pathway (Wang et al., 2010), CXCR4–CXCR7 dimer (Sun et al., 2010), EGFR/Ras/ERK (Singh and Lokeshwar, 2011), EGFR/PI3K/Akt (Mittal et al., 2009), and C-Myc (Karanika et al., 2015). The crucial step of ENZ treatment is to block the binding of androgen and AR to the maximum extent in patients with CRPC. It can suppress the transcriptional activity of the AR pathway and achieve the therapeutic purpose of promoting apoptosis and destroying DNA repair. Therefore, ENZ resistance strongly suggests the presence of alternative signaling pathways beside the AR pathway to elicit and maintain the drug-resistant progression of CRPC.
The androgen receptor shear variant AR-v7 is the first drug-resistant gene confirmed in ENZ-resistant CRPC cells. After AR-v7 was knocked out, the cell proliferation and growth were remarkably inhibited (Li et al., 2013). Clinical research further demonstrated that if tumor cells of patients with CRPC positively expressed AR-v7 (Antonarakis et al., 2014), these patients would be resistant to ENZ treatment. Therefore, AR-v7 might be a breakthrough target for ENZ resistance. In addition, our previous study also demonstrated one possible ENZ-resistance–related AR bypass pathway in which the functional role of the CXCR4–CXCR7 heterodimer shifted from CXCR4-driven to CXCR7-driven after ENZ treatment (Luo et al., 2018). However, the downstream signal transduction process of the CXCR7 pathway is still unclear.
Truncated AR splice variants (AR-Vs) are mainly due to aberrant RNA splicing intragenic and DNA rearrangement (Zhu and Luo, 2020a), resulting in the loss of the ligand-binding domain (LBD) of the AR protein. The absence of LBD makes the AR protein lose the drug-binding domain, further leading to the drug resistance of AR (Kanayama et al., 2021). Several studies attempted to explore the details of how ARv7 activated the downstream survival signal. The genomics of three ENZ-resistant CRPC PDX models, which were derived from the biopsy of skin metastasis, liver metastasis, and primary cancer, were comparatively analyzed. The results showed no consistent requirement for mutations in TP53, RB1, BRCA2, PIK3CA, or MSH2, as well as the expression of SOX2 or ERG, while all drug-resistant PDX tissues lacked PTEN expression (Zhu et al., 2020b). Additionally, enzalutamide resistance requires both AR-FL expression with at least a fiftyfold increase and AR-V7 protein expression at least sevenfold to eightfold higher than AR-FL expression in the normal prostate epithelium (Zhu et al., 2020b). Another study also involved genetic analyses in three 22RV1-based models constructed by the concomitant treatment of ADT with ABI, ENZ, and their combination on 22RV1 cells. The final results in these drug-resistant cellular models confirmed that the expression of several AR target genes, such as FKBP5, PMEPA1, and TMPRSS2, significantly increased except for the general increase in the expression of AR-FL and AR-v7 (Simon et al., 2021). Our previous research screened four genes from ENZ-resistant C4-2B cells, which were CCNA2, CKAP2L, NCAPG, and NUSAP1, and confirmed that the protein levels of these four genes also remained higher in ENZ-resistant xenograft tumor tissues (Feng et al., 2021a; Feng et al., 2021b). Although several genes with significant expression variation have been screened in different ENZ-resistant models, how these genes participate in the growth of CRPC cells is still unclear. In a previous study, we found that, after the AR was inhibited by ENZ, the activity of the TopBP1–ATR–Chk1 signaling pathway increased, resulting in the enhancement of the DDR ability of cells, which weakened the therapeutic effect of ENZ. The combination treatment of AR + ENZ and Chk1 inhibition with AZD7762 demonstrated synergistic effects with regard to decreased TopBP1–ATR–Chk1 signaling and markedly increased ATM phosphorylation and apoptosis (Karanika et al., 2017). The present study did not verify the underlying correlation between the JAK2/STAT1 pathway and the TopBP1–ATR–Chk1/DDR pathway. However, we found that the inhibition of JAK2/STAT1 activity indeed promoted apoptosis and reduced DDR. Therefore, we think that a cross-regulation mechanism exists between these two pathways.
Available evidence indicated that the activation of the JAK/STAT pathway was involved in the oncogenesis and progression of PCa. Once stimuli acted on growth factors or cytokine receptors, the autophosphorylation of one of the Janus kinases, usually JAK2, occurred. Consequently, JAK2 in turn phosphorylated a STAT family member. In particular, STAT3 is generally considered an oncogene that promotes cell survival, proliferation, motility, angiogenesis, immune tolerance, and chemotherapy resistance (Kroon et al., 2013; Ham et al., 2019; Olender et al., 2019). It is not only overexpressed in pathology specimens from prostatectomy (Barton et al., 2004) but also activated in CRPC cell lines (Agarwal et al., 2007). The upregulation of the IL-6/JAK/STAT3 cascade may contribute to promoting cell growth and survival in CRPC independent of the AR (Lou et al., 2000; Lee et al., 2003; Jia et al., 2004; Tam et al., 2007). Clinical research also found that the expression levels of phosphorylated JAK and phosphorylated STAT both positively correlated with the Gleason score and clinical stage of patients with PCa but had a negative correlation with the recurrence-free survival rates (Liu et al., 2012). Once the activation of STAT3 was inhibited, significant apoptosis could be induced in CRPC cells (Agarwal et al., 2007; Kroon et al., 2013). These reports suggested that P-STAT3 was essential for cell invasion, metastasis, and progression in patients with CRPC, especially after ADT resistance.
However, our present study demonstrated that STAT1 worked as the main signal transducer in CXCR7-driven CRPC progression. STAT1 always plays opposite roles to STAT3 in tumorigenesis and mostly triggers antiproliferative and pro-apoptotic responses while enhancing anti-tumor immunity (Huang et al., 2000; Stephanou and Latchman, 2003; Olbrich and Freeman, 2018). We found that the activation of STAT1 displayed the anomalous role of tumor-promoting factors. After the targeted inhibition of STAT1 activity, the potency of cellular survival and migration remarkably decreased, and the extent of DNA damage response and apoptosis significantly increased. It may be related to the imbalance of reciprocal regulation between STAT1 and STAT3 (Avalle et al., 2012). STAT1 was derepressed after ENZ treatment (Eiro et al., 2017). In addition, the perturbation in their phosphorylation levels may re-direct cytokine/growth factor signals from proliferative to apoptotic, or from inflammatory to anti-inflammatory (Costa-Pereira et al., 2002; Maritano et al., 2004; Shim et al., 2009; Schiavone et al., 2011; Souissi et al., 2011; Avalle et al., 2012). Several research studies suggested that the functional role of STAT1 in CRPC progression also depended on other factors, such as exon deletion (Magor et al., 2016), DNA methylation status (Dellafiore et al., 2020), different JAK family proteins (Coricello et al., 2020), and CXCR4–CXCR7 activating imbalance (Tomchuck et al., 2012; Lim et al., 2018).
Although abiraterone (ABI) does not directly antagonize AR transcriptional activity, it can maximally limit androgen synthesis, reduce the stimulating effect of androgen on AR transcriptional activity, and achieve the therapeutic purpose. Therefore, the mechanism of ENZ resistance represented by the activation of the AR bypass pathway may also be the potential mechanism of ABI resistance. Systematic evaluation of a number of clinical research studies also confirmed that the proportion of ARv7-positive patients with CRPC increased significantly after receiving new hormone therapy (NHT) (Wang et al., 2020a). This suggested that ARv7 might be involved in the cross-resistance between ENZ and ABI. Although ARv7-positive patients showed much worse treatment responses to NHT (Armstrong et al., 2020; Wang et al., 2020a; Wang et al., 2020b), the clinical data analysis demonstrated that these patients could significantly benefit from taxane chemotherapy, which was possibly related to the higher activity of the E2F1/AR3 feed-forward loop (Xu et al., 2020). The CXCR7–JAK2/STAT1 signaling pathway, as an independent AR bypass pathway, may also participate in resistance to androgen target therapy. Loss-of-function mutations in BRCA1/2 lead to a deficiency in the DNA damage repairing pathway called homologous recombination, which could render cancer cells exquisitely vulnerable to the PARP inhibitor (olaparib, OLA). However, BRCA mutations are present in only ∼20% of patients with PCa, and therefore, OLA failed to achieve the satisfied effect. Our previous study (Li et al., 2017) demonstrated that CRPC cells showed increased expression of a set of HR-associated genes, including BRCA1, RAD54L, and RMI2. Although androgen-targeted therapy is not typically effective in patients with CRPC, ENZ could significantly suppress the expression of these HR genes in CRPC cells, thus creating HR deficiency and BRCA function mutation. Pharmaceutical induction of BRCA mutation could expand the use of PARP inhibitors. Therefore, the ENZ-resistant process may synchronize with the OLA-sensitive process. The “lead-in” treatment strategy of ENZ followed by OLA may be much more effective for patients with CRPC.
Exploring the details of the AR bypass pathway may help not only clarify the ENZ-resistant mechanism but also improve the treatment sensitivity of other NHT drugs, PARP inhibitors, and chemotherapy. The present study demonstrated that the JAK2/STAT1 signaling pathway was closely involved in the ENZ resistance process induced by CXCR7 derepression. Also, the inhibition of JAK2/STAT1 could notably improve the ENZ treatment effect and lead to the downregulation of C-Myc, decreasing motility and proliferation and increasing apoptosis and DNA damage. Target inhibition of the CXCR7–JAK2/STAT1–C-Myc signaling pathway may be an important and effective strategy to overcome ENZ resistance in patients with CRPC.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the Committee on Animal Research and Ethics of the Capital Medical University.
All authors contributed to the successful completion of the study. YL, XY, SB, XZ, and YGJ were involved in study design. LY and XY drafted the manuscript, and SB revised the manuscript. All authors reviewed the manuscript and approved the submission.
This study was funded by the National Natural Science Foundation of Beijing (No. 7192053).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agarwal, C., Tyagi, A., Kaur, M., and Agarwal, R. (2007). Silibinin Inhibits Constitutive Activation of Stat3, and Causes Caspase Activation and Apoptotic Death of Human Prostate Carcinoma DU145 Cells. Carcinogenesis 28, 1463–1470. doi:10.1093/carcin/bgm042
Antonarakis, E. S., Lu, C., Wang, H., Luber, B., Nakazawa, M., Roeser, J. C., et al. (2014). AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 371, 1028–1038. doi:10.1056/NEJMoa1315815
Armstrong, A. J., Luo, J., Nanus, D. M., Giannakakou, P., Szmulewitz, R. Z., Danila, D. C., et al. (2020). Prospective Multicenter Study of Circulating Tumor Cell AR-V7 and Taxane versus Hormonal Treatment Outcomes in Metastatic Castration-Resistant Prostate Cancer. JCO Precision Oncol. 4, 1285–1301. doi:10.1200/PO.20.00200
Avalle, L., Pensa, S., Regis, G., Novelli, F., and Poli, V. (2012). STAT1 and STAT3 in Tumorigenesis. JAK-STAT 1, 65–72. doi:10.4161/jkst.20045
Azuma, T. (2018). Comparing Sequencing of Abiraterone and Enzalutamide in Men with Metastatic Castration-Resistant Prostate Cancer: A Retrospective Study. Prostate 78, 278. doi:10.1002/pros.23470
Barton, B. E., Karras, J. G., Murphy, T. F., Barton, A., and Huang, H. F. (2004). Signal Transducer and Activator of Transcription 3 (STAT3) Activation in Prostate Cancer: Direct STAT3 Inhibition Induces Apoptosis in Prostate Cancer Lines. Mol. Cancer Ther. 3, 11–20.
Beer, T. M., Armstrong, A. J., Rathkopf, D. E., Loriot, Y., Sternberg, C. N., Higano, C. S., et al. (2014). Enzalutamide in Metastatic Prostate Cancer before Chemotherapy. N. Engl. J. Med. 371, 424–433. doi:10.1056/NEJMoa1405095
Bolli, R., Dawn, B., and Xuan, Y. T. (2003). Role of the JAK-STAT Pathway in protection against Myocardial Ischemia/reperfusion Injury. Trends Cardiovasc. Med. 13, 72–79. doi:10.1016/s1050-1738(02)00230-x
Carver, B. S., Chapinski, C., Wongvipat, J., Hieronymus, H., Chen, Y., Chandarlapaty, S., et al. (2011). Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 19, 575–586. doi:10.1016/j.ccr.2011.04.008
Coricello, A., Mesiti, F., Lupia, A., Maruca, A., and Alcaro, S. (2020). Inside Perspective of the Synthetic and Computational Toolbox of JAK Inhibitors: Recent Updates. Molecules 25, 3321. doi:10.3390/molecules25153321
Costa-Pereira, A. P., Tininini, S., Strobl, B., Alonzi, T., Schlaak, J. F., Is'harc, H., et al. (2002). Mutational Switch of an IL-6 Response to an Interferon- -like Response. Proc. Natl. Acad. Sci. 99, 8043–8047. doi:10.1073/pnas.122236099
Dellafiore, M., Montserrat, J. M., and Iribarren, A. M. (2020). Core Modified 8-17 DNAzymes with 2′-Deoxy-2′-C-Methylpyrimidine Nucleosides. Bioorg. Chem. 104, 104328. doi:10.1016/j.bioorg.2020.104328
Eiro, N., Fernandez-Gomez, J., Sacristán, R., Fernandez-Garcia, B., Lobo, B., Gonzalez-Suarez, J., et al. (2017). Stromal Factors Involved in Human Prostate Cancer Development, Progression and Castration Resistance. J. Cancer Res. Clin. Oncol. 143, 351–359. doi:10.1007/s00432-016-2284-3
Feng, T., Wei, D., Li, Q., Yang, X., Han, Y., Luo, Y., et al. (2021b). Four Novel Prognostic Genes Related to Prostate Cancer Identified Using Co-expression Structure Network Analysis. Front. Genet. 12, 584164. doi:10.3389/fgene.2021.584164
Feng, T., Wei, D., Zhao, J., Li, Q., Guo, P., Yang, X., et al. (2021a). Construction of Enzalutamide-Resistant Cell Model of Prostate Cancer and Preliminary Screening of Potential Drug-Resistant Genes. Exp. Biol. Med. (Maywood) 246, 1776–1787. doi:10.1177/15353702211012625
Ham, I.-H., Oh, H. J., Jin, H., Bae, C. A., Jeon, S.-M., Choi, K. S., et al. (2019). Targeting Interleukin-6 as a Strategy to Overcome Stroma-Induced Resistance to Chemotherapy in Gastric Cancer. Mol. Cancer 18, 68. doi:10.1186/s12943-019-0972-8
Huang, Y.-Q., Li, J.-J., and Karpatkin, S. (2000). Thrombin Inhibits Tumor Cell Growth in Association with Up-Regulation of P21 and Caspases via a P53-independent, STAT-1-dependent Pathway. J. Biol. Chem. 275, 6462–6468. doi:10.1074/jbc.275.9.6462
Jia, L., Choong, C. S.-Y., Ricciardelli, C., Kim, J., Tilley, W. D., and Coetzee, G. A. (2004). Androgen Receptor Signaling. Cancer Res. 64, 2619–2626. doi:10.1158/0008-5472.can-03-3486
Kanayama, M., Lu, C., Luo, J., and Antonarakis, E. S. (2021). AR Splicing Variants and Resistance to AR Targeting Agents. Cancers 13, 2563. doi:10.3390/cancers13112563
Karanika, S., Karantanos, T., Li, L., Corn, P. G., and Thompson, T. C. (2015). DNA Damage Response and Prostate Cancer: Defects, Regulation and Therapeutic Implications. Oncogene 34, 2815–2822. doi:10.1038/onc.2014.238
Karanika, S., Karantanos, T., Li, L., Wang, J., Park, S., Yang, G., et al. (2017). Targeting DNA Damage Response in Prostate Cancer by Inhibiting Androgen Receptor-CDC6-ATR-Chk1 Signaling. Cel Rep. 18, 1970–1981. doi:10.1016/j.celrep.2017.01.072
Kroon, P., Berry, P. A., Stower, M. J., Rodrigues, G., Mann, V. M., Simms, M., et al. (2013). JAK-STAT Blockade Inhibits Tumor Initiation and Clonogenic Recovery of Prostate Cancer Stem-like Cells. Cancer Res. 73, 5288–5298. doi:10.1158/0008-5472.Can-13-0874
Lee, S. O., Lou, W., Hou, M., De Miguel, F., Gerber, L., and Gao, A. C. (2003). Interleukin-6 Promotes Androgen-independent Growth in LNCaP Human Prostate Cancer Cells. Clin. Cancer Res. 9, 370–376.
Li, L., Karanika, S., Yang, G., Wang, J., Park, S., Broom, B. M., et al. (2017). Androgen Receptor Inhibitor-Induced "BRCAness" and PARP Inhibition Are Synthetically Lethal for Castration-Resistant Prostate Cancer. Sci. Signal. 10, eaam7479. doi:10.1126/scisignal.aam7479
Li, S., Fong, K. W., Gritsina, G., Zhang, A., Zhao, J. C., Kim, J., et al. (2019). Activation of MAPK Signaling by CXCR7 Leads to Enzalutamide Resistance in Prostate Cancer. Cancer Res. 79, 2580–2592. doi:10.1158/0008-5472.Can-18-2812
Li, Y., Chan, S. C., Brand, L. J., Hwang, T. H., Silverstein, K. A. T., and Dehm, S. M. (2013). Androgen Receptor Splice Variants Mediate Enzalutamide Resistance in Castration-Resistant Prostate Cancer Cell Lines. Cancer Res. 73, 483–489. doi:10.1158/0008-5472.Can-12-3630
Lim, R. Z. L., Li, L., Yong, E. L., and Chew, N. (2018). STAT-3 Regulation of CXCR4 Is Necessary for the Prenylflavonoid Icaritin to Enhance Mesenchymal Stem Cell Proliferation, Migration and Osteogenic Differentiation. Biochim. Biophys. Acta (Bba) - Gen. Subjects 1862, 1680–1692. doi:10.1016/j.bbagen.2018.04.016
Liu, X., He, Z., Li, C.-h., Huang, G., Ding, C., and Liu, H. (2012). Correlation Analysis of JAK-STAT Pathway Components on Prognosis of Patients with Prostate Cancer. Pathol. Oncol. Res. 18, 17–23. doi:10.1007/s12253-011-9410-y
Lou, W., Ni, Z., Dyer, K., Tweardy, D. J., and Gao, A. C. (2000). Interleukin-6 Induces Prostate Cancer Cell Growth Accompanied by Activation of Stat3 Signaling Pathway. Prostate 42, 239–242. doi:10.1002/(sici)1097-0045(20000215)42:3<239:aid-pros10>3.0.co;2-g
Luo, Y., Azad, A. K., Karanika, S., Basourakos, S. P., Zuo, X., Wang, J., et al. (2018). Enzalutamide and CXCR7 Inhibitor Combination Treatment Suppresses Cell Growth and Angiogenic Signaling in Castration-Resistant Prostate Cancer Models. Int. J. Cancer 142, 2163–2174. doi:10.1002/ijc.31237
Magor, G. W., Tallack, M. R., Klose, N. M., Taylor, D., Korbie, D., Mollee, P., et al. (2016). Rapid Molecular Profiling of Myeloproliferative Neoplasms Using Targeted Exon Resequencing of 86 Genes Involved in JAK-STAT Signaling and Epigenetic Regulation. J. Mol. Diagn. 18, 707–718. doi:10.1016/j.jmoldx.2016.05.006
Maritano, D., Sugrue, M. L., Tininini, S., Dewilde, S., Strobl, B., Fu, X., et al. (2004). The STAT3 Isoforms α and β Have Unique and Specific Functions. Nat. Immunol. 5, 401–409. doi:10.1038/ni1052
Mittal, S., Subramanyam, D., Dey, D., Kumar, R. V., and Rangarajan, A. (2009). Cooperation of Notch and Ras/MAPK Signaling Pathways in Human Breast Carcinogenesis. Mol. Cancer 8, 128. doi:10.1186/1476-4598-8-128
Olbrich, P., and Freeman, A. F. (2018). STAT1 and STAT3 Mutations: Important Lessons for Clinical Immunologists. Expert Rev. Clin. Immunol. 14, 1029–1041. doi:10.1080/1744666x.2018.1531704
Olender, J., Wang, B.-D., Ching, T., Garmire, L. X., Garofano, K., Ji, Y., et al. (2019). A Novel FGFR3 Splice Variant Preferentially Expressed in African American Prostate Cancer Drives Aggressive Phenotypes and Docetaxel Resistance. Mol. Cancer Res. 17, 2115–2125. doi:10.1158/1541-7786.Mcr-19-0415
Rafiei, S., Gui, B., Wu, J., Liu, X. S., Kibel, A. S., and Jia, L. (2019). Targeting the MIF/CXCR7/AKT Signaling Pathway in Castration-Resistant Prostate Cancer. Mol. Cancer Res. 17, 263–276. doi:10.1158/1541-7786.Mcr-18-0412
Schiavone, D., Avalle, L., Dewilde, S., and Poli, V. (2011). The Immediate Early Genes Fos and Egr1 Become STAT1 Transcriptional Targets in the Absence of STAT3. FEBS Lett. 585, 2455–2460. doi:10.1016/j.febslet.2011.06.020
Simon, I., Perales, S., Casado-Medina, L., Rodríguez-Martínez, A., Garrido-Navas, M. d. C., Puche-Sanz, I., et al. (2021). Cross-Resistance to Abiraterone and Enzalutamide in Castration Resistance Prostate Cancer Cellular Models Is Mediated by AR Transcriptional Reactivation. Cancers 13, 1483. doi:10.3390/cancers13061483
Singh, R. K., and Lokeshwar, B. L. (2011). The IL-8-regulated Chemokine Receptor CXCR7 Stimulates EGFR Signaling to Promote Prostate Cancer Growth. Cancer Res. 71, 3268–3277. doi:10.1158/0008-5472.Can-10-2769
Souissi, I., Najjar, I., Ah-Koon, L., Schischmanoff, P. O., Lesage, D., Le Coquil, S., et al. (2011). A STAT3-Decoy Oligonucleotide Induces Cell Death in a Human Colorectal Carcinoma Cell Line by Blocking Nuclear Transfer of STAT3 and STAT3-Bound NF-Κb. BMC Cel Biol 12, 14. doi:10.1186/1471-2121-12-14
Stephanou, A., and Latchman, D. S. (2003). STAT-1: a Novel Regulator of Apoptosis. Int. J. Exp. Pathol. 84, 239–244. doi:10.1111/j.0959-9673.2003.00363.x
Sun, X., Cheng, G., Hao, M., Zheng, J., Zhou, X., Zhang, J., et al. (2010). CXCL12/CXCR4/CXCR7 Chemokine axis and Cancer Progression. Cancer Metastasis Rev. 29, 709–722. doi:10.1007/s10555-010-9256-x
Sung, S. H., Sung, M. W., Park, S. W., and Heo, D. S. (2009). Absence of STAT1 Disturbs the Anticancer Effect Induced by STAT3 Inhibition in Head and Neck Carcinoma Cell Lines. Int. J. Mol. Med. 23, 805–810. doi:10.3892/ijmm_00000196
Tam, L., Mcglynn, L. M., Traynor, P., Mukherjee, R., Bartlett, J. M. S., and Edwards, J. (2007). Expression Levels of the JAK/STAT Pathway in the Transition from Hormone-Sensitive to Hormone-Refractory Prostate Cancer. Br. J. Cancer 97, 378–383. doi:10.1038/sj.bjc.6603871
Tomchuck, S. L., Henkle, S. L., Coffelt, S. B., and Betancourt, A. M. (2012). Toll-like Receptor 3 and Suppressor of Cytokine Signaling Proteins Regulate CXCR4 and CXCR7 Expression in Bone Marrow-Derived Human Multipotent Stromal Cells. PLoS One 7, e39592. doi:10.1371/journal.pone.0039592
Wang, J., Zhang, Y., Wei, C., Gao, X., Yuan, P., Gan, J., et al. (2020b). Prognostic Value of Androgen Receptor Splice Variant 7 in the Treatment of Metastatic Castration-Resistant Prostate Cancer: A Systematic Review and Meta-Analysis. Front. Oncol. 10, 562504. doi:10.3389/fonc.2020.562504
Wang, Z., Li, Y., Banerjee, S., Kong, D., Ahmad, A., Nogueira, V., et al. (2010). Down-regulation of Notch-1 and Jagged-1 Inhibits Prostate Cancer Cell Growth, Migration and Invasion, and Induces Apoptosis via Inactivation of Akt, mTOR, and NF-Κb Signaling Pathways. J. Cel. Biochem. 109, 726–736. doi:10.1002/jcb.22451
Wang, Z., Shen, H., Ma, N., Li, Q., Mao, Y., Wang, C., et al. (2020a). The Prognostic Value of Androgen Receptor Splice Variant 7 in Castration-Resistant Prostate Cancer Treated with Novel Hormonal Therapy or Chemotherapy: A Systematic Review and Meta-Analysis. Front. Oncol. 10, 572590. doi:10.3389/fonc.2020.572590
Xu, J., Yang, X., Deshmukh, D., Chen, H., Fang, S., and Qiu, Y. (2020). The Role of Crosstalk between AR3 and E2F1 in Drug Resistance in Prostate Cancer Cells. Cells 9, 1094. doi:10.3390/cells9051094
Zhu, Y., Dalrymple, S. L., Coleman, I., Zheng, S. L., Xu, J., Hooper, J. E., et al. (2020b). Role of Androgen Receptor Splice Variant-7 (AR-V7) in Prostate Cancer Resistance to 2nd-Generation Androgen Receptor Signaling Inhibitors. Oncogene 39, 6935–6949. doi:10.1038/s41388-020-01479-6
Keywords: castration-resistant prostate cancer, CXCR7, JAK2, STAT1, enzalutamide, resistance
Citation: Luo Y, Yang X, Basourakos SP, Zuo X, Wei D, Zhao J, Li M, Li Q, Feng T, Guo P and Jiang Y (2021) Enzalutamide-Resistant Progression of Castration-Resistant Prostate Cancer Is Driven via the JAK2/STAT1-Dependent Pathway. Front. Mol. Biosci. 8:652443. doi: 10.3389/fmolb.2021.652443
Received: 12 January 2021; Accepted: 15 September 2021;
Published: 22 October 2021.
Edited by:
Yu Xiao, Wuhan University, ChinaReviewed by:
Giuseppe Schepisi, Istituto Scientifico Romagnolo per lo Studio e il Trattamento dei Tumori (IRCCS), ItalyCopyright © 2021 Luo, Yang, Basourakos, Zuo, Wei, Zhao, Li, Li, Feng, Guo and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Luo, bHVveW9uZ2FuemhlbkAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.