 Katarzyna Banasiak
Katarzyna Banasiak Natalia A. Szulc
Natalia A. Szulc Wojciech Pokrzywa
Wojciech Pokrzywa
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Biosci. , 26 March 2021
Sec. Cellular Biochemistry
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.650730
This article is part of the Research Topic Dysregulated Protein Homeostasis in the Aging Organism View all 6 articles
The proteolytic machinery activity diminishes with age, leading to abnormal accumulation of aberrant proteins; furthermore, a decline in protein degradation capacity is associated with multiple age-related proteinopathies. Cellular proteostasis can be maintained via the removal of ubiquitin (Ub)-tagged damaged and redundant proteins by the ubiquitin-proteasome system (UPS). However, during aging, central nervous system (CNS) cells begin to express a frameshift-mutated Ub, UBB+1. Its accumulation is a neuropathological hallmark of tauopathy, including Alzheimer’s disease and polyglutamine diseases. Mechanistically, in cell-free and cell-based systems, an increase in the UBB+1 concentration disrupts proteasome processivity, leading to increased aggregation of toxic proteins. On the other hand, a low level of UBB+1 improves stress resistance and extends lifespan. Here we summarize recent findings regarding the impact of UBB+1 on Ub signaling and neurodegeneration. We also review the molecular basis of how UBB+1 affects UPS components as well as its dose-dependent switch between cytoprotective and cytotoxic roles.
Age-related impairment of protein degradation affects protein homeostasis (proteostasis) networks, causing enhanced accumulation of damaged proteins that can be cytotoxic and shorten lifespan. The primary proteolytic component of the cellular proteostasis network is the ubiquitin-proteasome system (UPS), which initiates turnover of unwanted substrates via covalent attachment of the evolutionarily conserved protein ubiquitin (Ub) (Buchberger et al., 2010; Popovic et al., 2014; Balchin et al., 2016). More than 2 decades ago, van Leeuwen and colleagues, while studying Alzheimer’s plaques in postmortem brains, identified a frameshift-mutated form of Ub currently known as UBB+1 (van Leeuwen et al., 1998). Subsequent studies confirmed the involvement of UBB+1 in several other neurodegenerative diseases (i.e., Pick disease and progressive supranuclear palsy) as well as in polyglutamine (polyQ) diseases (i.e., Huntington’s disease and spinocerebellar ataxia type 3). Furthermore, UBB+1 accumulation has been linked to disease onset and progression (Fergusson et al., 2000; Fischer et al., 2003; de Pril et al., 2004; Hol et al., 2005; Yim et al., 2014). Nonetheless, UBB+1 has also been found in healthy neurons and other cell types, including monocytes and hepatocytes (French et al., 2001; Fischer et al., 2003; Fratta et al., 2004). Recent findings highlight the positive effects associated with UBB+1 expression. For example, UBB+1 synthesis reduces amyloid-β- (Aβ-) related toxicity (Verheijen et al., 2018), and in yeast, a low level of UBB+1 expression prevents reactive oxygen species (ROS) accumulation and limits apoptosis, consequently increasing cellular life span (Muñoz-Arellano et al., 2018). Here, we discuss at least some of the many faces of UBB+1 in proteostasis maintenance.
In humans, Ub is encoded by four genes, including UB-ribosomal fusion genes, i.e., Uba52, and RPS27A, which encode a single copy of Ub fused to a ribosomal protein, and polyubiquitin genes, i.e., UBB and UBC, which consist of repeats of monoubiquitin coding units (Wiborg et al., 1985; Finley et al., 1989; Baker and Board, 1991). In addition, several Ub pseudogenes have been identified, including the recently characterized UBB pseudogene 4 (Dubois et al., 2020). Reduced Ub levels caused by UBB inactivation lead to many disorders, including adult-onset obesity and hypothalamic neurodegeneration (Ryu et al., 2008) and dysregulation of neuronal stem cell self-renewal (Ryu et al., 2014). Furthermore, abnormal transcription leads to the formation of a mutated form of ubiquitin, UBB+1. Molecular misreading leads to dinucleotide deletions (CU, GA, GU) in mRNA leading to a +1 reading frame shift, resulting in the synthesis of a “+1 protein” with abnormal extensions (Figure 1A) (van Leeuwen et al., 1998; van Leeuwen et al., 2000; van Leeuwen et al., 2002). Intensification of this molecular misreading and an accumulation of UBB+1 in multiple areas of the brain is a hallmark of neurodegeneration, including that associated with Alzheimer’s disease (van Leeuwen et al., 1998; van Leeuwen et al., 2000; van Leeuwen et al., 2002). The exact cause of these errors requires further research; however, potential mechanisms for the generation of mutant transcripts include inappropriate RNA polymerase activity at repetitive DNA sequences or ribosome-mediated frameshifting (Wills and Atkins, 2006; Verheijen et al., 2017).
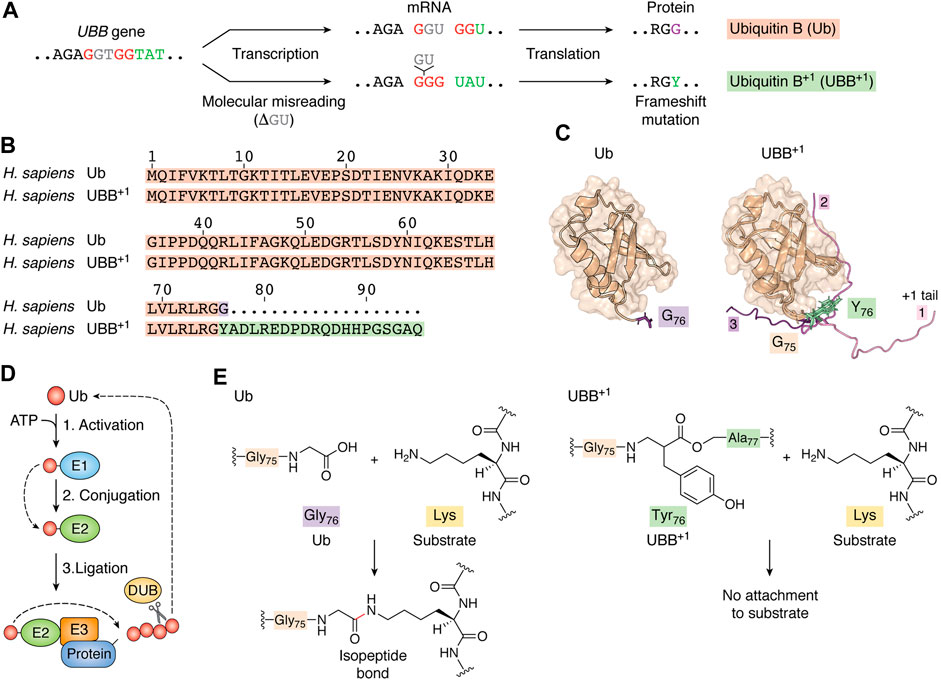
FIGURE 1. Transcription error leads to the expression of a mutated form of ubiquitin. (A) Molecular misreading of Ub mRNA sequences results in “GU” nucleotide deletion, which introduces a frameshift mutation that results in UBB+1 expression. (B) UBB+1 differs from wild-type Ub by the G76Y missense mutation and a 19-amino acid C-terminal extension. (C) The tertiary structures of both Ub (PDB ID code: 1d3z, Cornilescu et al., 1998) and UBB+1 (model built upon PDB structures with codes 1d3z and 2kx0) exhibit a compact and globular “β-grasp” fold that forms a hydrophobic core between an α-helix and five β-sheets. In our model of UBB+1, we show three distinct conformations of the extended amino acid chain. Additional 19-amino acid chains (from models 1, 2, 9 of 2kx0) were added to 1d3z structure with G76Y mutation using YASARA View (Krieger and Vriend, 2014). Each model was protonated using H++ web server version 3.2 (Gordon et al., 2005; Myers et al., 2006; Anandakrishnan et al., 2012) and optimized by running 1,500 steps of energy minimization. All calculations were performed using the AmberTools20 package (Izadi et al., 2014; Li et al., 2015; Case et al., 2020; Tian et al., 2020); optimized models were superposed and visualized in PyMOL (The PyMOL Molecular Graphics System, Version 2.2.3. Schrödinger, LLC). (D) At the start of the ubiquitylation cascade, a Ub-activating enzyme (E1) hydrolyzes ATP and forms a high-energy thioester bond between an internal cysteine residue and the C-terminus of Ub. Activated Ub is then passed on to Ub-conjugating enzymes (E2s), which form similar thioester-linked complexes with Ub. Finally, Ub is covalently attached to lysine sidechains of the substrate protein or another Ub (generating polyUb chains) with assistance from ubiquitin-protein ligases (E3s). Deubiquitylation enzymes (DUBs) modulate the length and topology of polyUb and recycle Ub. Finally, the proteasome complex recognizes ubiquitylated proteins and degrades them into short peptides via proteolysis. (E) Ub is attached to its substrate via an isopeptide bond between the C-terminal glycine residue of Ub and a lysine residue in the substrate. The G76Y mutation prevents the formation of an isopeptide bond between the UBB+1 C-terminus and lysine residues in protein substrates.
Ub attachment (ubiquitylation) is mediated by an enzymatic cascade involving Ub-activating enzymes (E1), Ub-conjugating enzymes (E2), and Ub ligases (E3). A C-terminal GG motif is necessary for Ub activation and conjugation to target proteins. PolyUb chains are assembled via an isopeptide linkage between the lysine residue of the previous Ub and the C-terminal glycine residue of the subsequent subunit. Deubiquitylation enzymes (DUBs) modulate the size and topology of polyUb (Figure 1D) (Komander and Rape, 2012). UBB+1 differs from wild-type Ub due to a G76Y mutation and a flexible 19-amino acid extension (Ko et al., 2010; Munari et al., 2018), (Figure 1B). We prepared a Ub model carrying the G76Y mutation and visualized three different conformations of the 19-amino acid extension (Figure 1C). Substitution of a glycine at residue 76 interrupts the C-terminal GG motif, which prevents its activation by E1, attachment to a substrate’s lysine, and processing by certain DUBs (Figures 1E, 2A) (Lam et al., 2000; Krutauz et al., 2014). This Ub mutant can be ubiquitylated at all lysine residues to serve as a proximal unit in polyUb chains. Studies in cells and mice have shown that UBB+1 is ubiquitously present in K29-, K48-, and K63-linked ubiquitin chains (Lam et al., 2000; Lindsten et al., 2002; van Tijn et al., 2012; Akutsu et al., 2016).
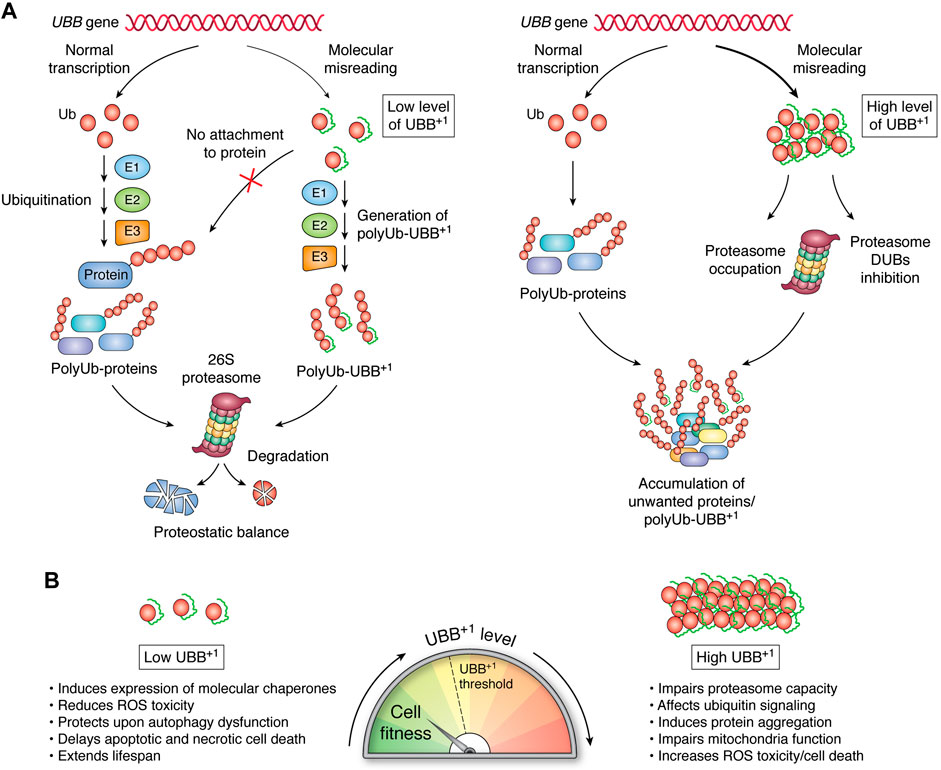
FIGURE 2. Pleiotropic effects of UBB+1 depend on its expression level. (A) E1, E2, and E3 enzymes are the sole pathway by which Ub molecules are linked to create polyUb on substrate proteins. UBB+1 is present in cells as a monomer that can be ubiquitylated at internal lysine residues via the same enzymatic cascade; however, it cannot be attached to proteins. The proteasome can efficiently recognize and proteolyze a low level of Ub-UBB+1 chains. Increased levels of Ub-UBB+1 might impair proteasomal capacity and DUB activity, leading to the accumulation of aberrant proteins designated for degradation. (B) The functionality of cellular processes correlates with the level of UBB+1 expression.
The conjugating enzyme UBE2K can directly interact with UBB+1 to initiate its ubiquitylation, which can be mediated by the E3s TRIP12 and HUWE1 (Park et al., 2009; Poulsen et al., 2009; Ko et al., 2010). In general, polyUb-UBB+1 are recognized by the proteasome, but they are efficiently degraded when UBB+1 is expressed under basal conditions (Figure 2A) (Lindsten et al., 2002; Fischer et al., 2003; van Tijn et al., 2007; van Tijn et al., 2010; Krutauz et al., 2014). Cellular UBB+1 accumulation promotes proteasome dysfunction, although not via direct inhibition of the proteosome’s hydrolytic activity. PolyUb-UBB+1 can be recognized by the substrate-shuttling factor for the proteasome, HHR23A and UBQLN1, and the proteasomal ubiquitin-receptor Rpn10 (Chojnacki et al., 2016; Munari et al., 2018). However, the 19-amino acid C-terminal extension of UBB+1 prevents the entire molecule from being effectively pulled in and reaching the 20S catalytic sites (Verhoef et al., 2008; Shabek et al., 2009). Thus, polyUb-UBB+1 can occupy proteasomes, thereby preventing the turnover of polyUb substrates (Figure 2A). UBB+1 also blocks the activity of proteasome-related DUBs, including Ubp6, which selectively cleave Lys48-linked Ub, thereby promoting substrate translocation into the 20S proteasome. Rpn11, a proteasomal subunit with DUB activity, is also inhibited by UBB+1 (Figure 2A) (Krutauz et al., 2014). Cytosolic DUBs also encounter obstacles when processing Ub-UBB+1 chains. For example, Usp5 recognizes the C-terminal glycine residue of Ub, which is absent in UBB+1; thus, Usp5 cannot remove UBB+1 subunits. YUH1/UCH-L3 DUB can hydrolyze the C-terminal extension of UBB+1, releasing, however, another conjugation-deficient ubiquitin Ub (G76Y) (Dennissen et al., 2011). Thus, the accumulation of degradation-resistant polyUb-UBB+1 and their impact on DUBs may correspond to the expansion of unwanted proteins during neurodegeneration. Moreover, Chojnacki and colleagues showed that the K63-linked Ub-UBB+1 chain is resistant to disassembly by the K63-specific DUB AMSH (Chojnacki et al., 2016). The effects that UBB+1 might have on K63-regulated processes, including DNA repair, cell signaling, and trafficking, remain to be explored.
Neuronal UBB+1 accumulation is a characteristic of neurodegenerative disorders, especially tauopathies. The link between UBB+1 and Alzheimer’s disease (AD) is the most extensively studied and is the subject of several reviews (van Leeuwen et al., 2006; Chadwick et al., 2012; Chen and Petranovic, 2016; Seynnaeve et al., 2018). Recently, UBB+1 was identified in samples from Guam island inhabitants suffering parkinsonism-dementia complex (G-PDC) (Verheijen et al., 2017). In light of current knowledge, G-PDC is the first Parkinson-related disorder with confirmed UBB+1 involvement. Inclusions of UBB+1 were accompanied by the UPS proteins and endoplasmic reticulum (ER) unfolded protein response (UPRER) regulators, i.e., pPERK and BiP/GRP78. The UPRER is an adaptive stress response pathway that counters protein misfolding and aggregation in the ER (Hetz et al., 2020). Interestingly, depletion of the UPRER sensor Ire1 in yeast ameliorates UBB+1 toxicity upon oxidative stress; however, this effect is independent of the UPRER transcriptional activator Hac1 (Braun et al., 2015). Moreover, drug-induced ER stress has been found to lead to substantial UBB+1 accumulation in HEK293 cells (Verheijen et al., 2020). It is still unclear if and how the UPRER protects against deleterious effects of UBB+1 on proteostasis and neurodegeneration.
Braun and colleagues evaluated the cytotoxic effects of UBB+1 aggregation in several yeast strains with knockouts of specific UPS genes (Braun et al., 2015). They concluded that the ratio of UBB+1 to wild-type Ub is a more reliable indicator of UBB+1 cytotoxicity than proteasomal capacity itself.
Recent data indicate that mitochondria are sensitive to the concentration-dependent cytotoxicity of UBB+1. Mitochondrial function can be evaluated by measuring cellular oxygen consumption, mitochondrial membrane potential, and ATP levels. Upon UBB+1 expression in yeast, the first two parameters increased after 2–3 days, while the cellular ATP level dropped (Braun et al., 2015). These observations indicate that even enhanced mitochondrial function is insufficient to prevent a metabolic crisis caused by UBB+1 accumulation. The cytochrome bc1 complex is a key component of the mitochondrial respiratory chain. Its alteration leads to abnormal ROS production and mitochondrial dysfunction (Crofts, 2004; Sousa et al., 2018). UBB+1 expression correlates with a significant reduction in the abundance of key components of the bc1 complex, i.e., Rip1 iron-sulfur protein and cytochrome c, which likely increases cellular ROS levels and decreases ATP production. Moreover, when accompanied by prolonged UBB+1 expression, acetate and hydrogen peroxide (H2O2) further increased the level of oxidative stress (Braun et al., 2015). Mitochondrial dynamics can also be assessed based on the expression levels of proteins related to mitochondrial fission or fusion. UBB+1 induction in primary human astrocytes and human astrocytoma cells has been shown to result in decreased expression of several fission-related proteins (Drp1, Fis1, and OPA3) while the concentrations of mitochondrial fusion proteins (Mfn1, Mfn2, and OPA1) were unchanged (Yim et al., 2014). In cells overexpressing UBB+1, mitochondria are elongated, and this phenotype has also been observed in cells treated with UPS inhibitors, which induce a significant drop in fission protein expression. Consistently, H2O2 treatment has been shown to induce astrocyte death upon UBB+1 accumulation (Yim et al., 2014). Dysfunction of the mitochondrial electron transport chain and mitochondrial network activates the mitochondrial (mt) unfolded protein response (UPRmt), a signaling cascade that aims to maintain mitochondrial protein homeostasis (Fiorese and Haynes, 2017; Shpilka and Haynes, 2018). Thus, it will be interesting to delineate the link between UBB+1 toxicity and the UPRmt regulation.
In UBB+1-overexpressing yeast cells, sixteen proteins of the mitochondrial proteome were found to have altered expression levels (e.g., Put1, Arg5, 6, Arg8, Lys1, Gpd1, and Str3). Among them, ten were previously associated with UBB+1-related pathologies (Braun et al., 2015). Deletion of ARG5,6, ARG8, and LYS1, which are involved in amino acid metabolism, rescued the clonogenic potential of cells expressing UBB+1 in response to acetate-induced oxidative stress. These findings indicate that high UBB+1 levels enhance the biosynthesis of the basic amino acids arginine, ornithine, and lysine, potentially aggravating its cytotoxicity. Therefore, Braun et al. (2015) decided to examine ROS production upon UBB+1 expression in yeast strains lacking arginine and ornithine synthesis enzymes. In the absence of upstream enzymes in the cytosolic ornithine synthesis pathway (Arg2, Arg5,6, Arg7, and Ort1), UBB+1 toxicity decreased under both normal conditions and stress conditions induced by acetate treatment. By contrast, the absence of enzymes downstream of cytosolic ornithine synthesis (Arg3, Arg1, and Arg4) did not affect UBB+1-triggered toxicity. In these phenotypes, perturbation of lysine levels appears to be irrelevant. Consequently, UBB+1 accumulation can influence mitochondria-associated overproduction of ornithine and arginine, key signaling molecules in the execution of cell death pathways (Braun et al., 2015). In aged human brains and brains from AD patients, the arginine and ornithine levels were altered (Rushaidhi et al., 2012; Inoue et al., 2013; Liu et al., 2014; Braun et al., 2015). However, the relationship between UBB+1 expression and amino acid metabolism-dependent cell death in aging requires confirmation.
In neurons, the microtubule cytoskeletal system is responsible for bidirectional mitochondria transport between the soma and synaptic terminals. While KIF1B kinesin plays a crucial role in anterograde transport, the dynein intermediate chain P74 is responsible for retrograde transport (Perlson et al., 2010; Maday et al., 2014). In intracellular organelles, impairment of this process leads to clogging by mitochondria in neuritic beads. In primary cortical neurons transfected with UBB+1, mitochondria-associated P74 levels decreased accompanied by a corresponding increase in cytosolic dynein, while KIF1B levels were unaltered (Tan et al., 2007). Consequently, UBB+1 expression can impair the interaction between mitochondria and dynein, thus leading to cargo detachment during retrograde axonal transport. Since impaired axonal mitochondrial transport promotes tau phosphorylation, which leads to its aggregation (Guo et al., 2020), it will be interesting to verify the impact of UBB+1 on this process.
Heat shock proteins (HSPs) function as chaperones that bind misfolded polypeptides and support the refolding and recovery of native protein conformations (Lindquist and Craig, 1988; Höhfeld and Hartl, 1994; Bukau et al., 2006; Morimoto, 2008). In human neuroblastoma cells, the transcript levels of several heat shock proteins (Hsp10, Hsp40, Hsp60, Hsp70, and Hsp90a) have been found to be elevated upon UBB+1 expression (Hope et al., 2003). However, this effect is only robustly reflected at the protein level for Hsp40 and Hsp70 (only a moderate increase in Hsp90a has been found). To examine how these cells manage oxidative stress upon UBB+1 expression, cell survival was measured based on mitochondrial activity after tert-butyl hydroperoxide (tBHP, a strong oxidant) treatment for 24 h. Elevated production of UBB+1, but not Ub, mitigated tBHP cytotoxicity. Similar effects were observed for 15 μM MG132 (a reversible proteasome inhibitor) and 0.1 μM lactacystin (an irreversible proteasome inhibitor) (Hope et al., 2003). These findings suggest that UBB+1 is involved in the maintenance of oxidative stability via HSPs. Moreover, the gene encoding 14-3-3 zeta (ζ), a protein with chaperone-like activity, was highly expressed in UBB+1-overexpressing mice (Irmler et al., 2012; Sluchanko et al., 2012). Due to the role of 14-3-3ζ in the regulation of the unfolded protein response in the mouse hippocampus (Brennan et al., 2013), its elevated levels associated with UBB+1 expression might ultimately protect these cells from ER stress.
Muñoz-Arellano and colleagues investigated the influence of low (low_UBB+1) or high (high_UBB+1) levels of UBB+1 expression on yeast fitness. The incorporation of azetidine-2-carboxylic acid (AZE), a proline analogue, into de novo synthesized polypeptides disrupts the flexibility of the polypeptide backbone, thereby inducing protein misfolding stress. After incubation with AZE for 3 days, high_UBB+1 cells were significantly less viable than the control cells and low_UBB+1 cells. Since lower ROS levels were detected in the low_UBB+1 cells after 9 and 14 days of culture, Annexin V staining was used to check for a corresponding decrease in apoptosis. Indeed, low_UBB+1 cells were less apoptotic than control cells and high_UBB+1 cells. Moreover, control and autophagy-deficient (∆atg1) cells were found to cope better with protein misfolding induced with 2 mM and 4 mM AZE when UBB+1 is expressed at low levels. Furthermore, when yeast cells were grown in a synthetic defined (SD) medium commonly used for chronological lifespan assays, low_UBB+1 cells showed the highest viability. After 14 days, the average viability of the low_UBB+1 cells was approximately 70%, while that of the control and high_UBB+1 cells fell below 10% (Muñoz-Arellano et al., 2018). Under H2O2-induced oxidative stress in stationary phase cells, both low_UBB+1 and high_UBB+1 cells had improved viability compared with controls, especially in the case of aging cells (Muñoz-Arellano et al., 2018). This seems to be evolutionarily conserved as a low level of UBB+1 protects astrocytic cells from oxidative stress (Yim et al., 2014). Interestingly, this effect does not require the functionality of the proteasome, as its proteolytic activity similarly decreased in the low_UBB+1 and high_UBB+1 yeast cells; however, it might be related to differences in sustained expression of certain chaperones (Muñoz-Arellano et al., 2018). Further insight is needed to understand how chaperone networks are fine-tuned to maintain the cellular proteome and support UBB+1 positive cells’ longevity.
Autosomal dominant AD is linked to mutations in the genes encoding β-amyloid precursor protein (APP), presenilin-1 (PSEN1), and presenilin-2 (PSEN2), which lead to impaired γ-secretase (PSEN1/PSEN2) function and improper processing of the amyloid precursor protein (APP), ultimately resulting in the formation of toxic forms of β-amyloid (Aβ) (Jankowsky et al., 2004; Bateman et al., 2011; O’Brien and Wong, 2011). To study the effect of UBB+1 accumulation on the development of autosomal dominant AD, mice overexpressing UBB+1 specifically in the postnatal brain neurons (tg line 3413) were crossbred with an AD mouse model, APPPS1 (line 85) (Fischer et al., 2009; Verheijen et al., 2018). Interestingly, the reduced γ-secretase activity of the APPPS1 animals was partially restored in APPPS1/UBB+1 triple transgenic animals, thus leading to fewer Aβ plaques in the brain (Chadwick et al., 2012; Verheijen et al., 2018). To determine whether UBB+1 expression might have broader beneficial effects, including modulation of contextual memory that reflects AD patients’ cognitive symptoms, behavioral tests have been performed in animal models. Both APPPS1 and APPPS1/UBB+1 mice performed worse than wild-type and UBB+1 mice in nest building (which provides information on general wellbeing). In a Y-maze spontaneous alternation test, however, all of the mice demonstrated similar eagerness to explore a new environment. By contrast, all of the transgenic mice performed more poorly in a Morris water maze (MWM) compared with the control mice. In conclusion, recovery of γ-secretase function due to UBB+1 overexpression does not appear to improve overall brain function or wellbeing. Perhaps the enhanced Aβ processing is outweighed by the negative effects of UBB+1 on the UPS.
UBB+1 can elicit pleiotropic effects depending on its expression level. A low level of UBB+1 can stimulate a chaperone-buffering capacity, which likely masks the adverse effects of UBB+1 expression while simultaneously facilitating more robust prevention of protein aggregation. By contrast, UBB+1 accumulation inhibits proteasome processivity, which might foster increased aggregation and cytotoxicity of expanded polyQ proteins (Figure 2B) (de Pril et al., 2004; de Pril et al., 2010). Drugs that effectively inhibit or induce protein aggregate removal are still under development. Perhaps a targeted-clearance strategy based on AUTAC (autophagy-targeting chimera) could be designed to target UBB+1-labeled protein aggregates to induce their removal via autophagy (Takahashi et al., 2019). On the other hand, reduced protein turnover rates correlate positively with extended lifespan in several rodent species and long-lived animals (Pérez et al., 2009; Swovick et al., 2018), while an increase in overall protein degradation can be a hallmark of accelerated aging, as manifested in Hutchinson-Gilford Progeria Syndrome (Buchwalter and Hetzer, 2017). Could reducing proteasome activity via UBB+1 expression be a strategy used by aging or damaged cells to maintain their function for as long as possible? Future studies exploring the precise spatiotemporal expression patterns of UBB+1 could help to clarify this intriguing phenomenon and its effects on young and aging tissues.
KB and WP wrote and revised the manuscript. NS prepared the three-dimensional structural models of Ub and UBB+1, wrote a figure legend, and was involved in manuscript revision. All authors contributed to the article and approved the submitted version.
This work was supported by the Foundation for the Polish Science co-financed by the European Union under the European Regional Development Fund (Grant No. POIR.04.04.00–00-5EAB/18–00).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Akutsu, M., Dikic, I., and Bremm, A. (2016). Ubiquitin chain diversity at a glance. J. Cell Sci. 129, 875–880. doi:10.1242/jcs.183954
Anandakrishnan, R., Aguilar, B., and Onufriev, A. V. (2012). H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40, W537–W541. doi:10.1093/nar/gks375
Baker, R. T., and Board, P. G. (1991). The human ubiquitin-52 amino acid fusion protein gene shares several structural features with mammalian ribosomal protein genes. Nucleic Acids Res. 19, 1035–1040. doi:10.1093/nar/19.5.1035
Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354. doi:10.1126/science.aac4354
Bateman, R. J., Aisen, P. S., De Strooper, B., Fox, N. C., Lemere, C. A., Ringman, J. M., et al. (2011). Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther. 3, 1–13. doi:10.1186/alzrt59
Braun, R. J., Sommer, C., Leibiger, C., Gentier, R. J., Dumit, V. I., Paduch, K., et al. (2015). Accumulation of basic amino acids at mitochondria dictates the cytotoxicity of aberrant ubiquitin. Cell Rep. 10, 1557–1571. doi:10.1016/j.celrep.2015.02.009
Brennan, G. P., Jimenez-Mateos, E. M., McKiernan, R. C., Engel, T., Tzivion, G., and Henshall, D. C. (2013). Transgenic overexpression of 14-3-3 zeta protects hippocampus against endoplasmic reticulum stress and status epilepticus in vivo. PLoS One 8, e54491. doi:10.1371/journal.pone.0054491
Buchberger, A., Bukau, B., and Sommer, T. (2010). Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol. Cell 40, 238–252. doi:10.1016/j.molcel.2010.10.001
Buchwalter, A., and Hetzer, M. W. (2017). Nucleolar expansion and elevated protein translation in premature aging. Nat. Commun. 8, 328–413. doi:10.1038/s41467-017-00322-z
Bukau, B., Weissman, J., and Horwich, A. (2006). Molecular chaperones and protein quality control. Cell 125, 443–451. doi:10.1016/j.cell.2006.04.014
Case, D. A., Belfon, K., Ben-Shalom, I. Y., Brozell, S. R., Cerutti, D. S., Cheatham, T. E., et al. (2020). AMBER 2020, Los Angeles, CA: University of California.
Chadwick, L., Gentle, L., Strachan, J., and Layfield, R. (2012). Review: unchained maladie—a reassessment of the role of Ubb(+1) -capped polyubiquitin chains in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 38, 118–131. doi:10.1111/j.1365-2990.2011.01236.x
Chen, X., and Petranovic, D. (2016). Role of frameshift ubiquitin b protein in Alzheimer's disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 8, 300–313. doi:10.1002/wsbm.1340
Chojnacki, M., Zhang, D., Talarowska, M., Gałecki, P., Szemraj, J., Fushman, D., et al. (2016). Characterizing polyubiquitinated forms of the neurodegenerative ubiquitin mutant UBB+1. FEBS Lett. 590, 4573–4585. doi:10.1002/1873-3468.12484
Cornilescu, G., Marquardt, J. L., Ottiger, M., and Bax, A. (1998). Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J. Am. Chem. Soc. 120, 6836–6837. doi:10.1021/ja9812610
Crofts, A. R. (2004). The cytochrome bc1 complex: function in the context of structure. Annu. Rev. Physiol. 66, 689–733. doi:10.1146/annurev.physiol.66.032102.150251
de Pril, R., Fischer, D. F., Maat-Schieman, M. L., Hobo, B., de Vos, R. A., Brunt, E. R., et al. (2004). Accumulation of aberrant ubiquitin induces aggregate formation and cell death in polyglutamine diseases. Hum. Mol. Genet. 13, 1803–1813. doi:10.1093/hmg/ddh188
de Pril, R., Hobo, B., van Tijn, P., Roos, R. A., van Leeuwen, F. W., and Fischer, D. F. (2010). Modest proteasomal inhibition by aberrant ubiquitin exacerbates aggregate formation in a Huntington disease mouse model. Mol. Cell. Neurosci. 43, 281–286. doi:10.1016/j.mcn.2009.12.001
Dennissen, F. J., Kholod, N., Hermes, D. J., Kemmerling, N., Steinbusch, H. W., Dantuma, N. P., et al. (2011). Mutant ubiquitin (UBB+1) associated with neurodegenerative disorders is hydrolyzed by ubiquitin C-terminal hydrolase L3 (UCH-L3). FEBS Lett. 585, 2568–2574. doi:10.1016/j.febslet.2011.06.037
Dubois, M. L., Meller, A., Samandi, S., Brunelle, M., Frion, J., Brunet, M. A., et al. (2020). UBB pseudogene 4 encodes functional ubiquitin variants. Nat. Commun. 11, 1306–1312. doi:10.1038/s41467-020-15090-6
Fergusson, J., Landon, M., Lowe, J., Ward, L., van Leeuwen, F. W., and Mayer, R. J. (2000). Neurofibrillary tangles in progressive supranuclear palsy brains exhibit immunoreactivity to frameshift mutant ubiquitin-b protein. Neurosci. Lett. 279, 69–72. doi:10.1016/s0304-3940(99)00917-9
Finley, D., Bartel, B., and Varshavsky, A. (1989). The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature 338, 394–401. doi:10.1038/338394a0
Fiorese, C. J., and Haynes, C. M. (2017). Integrating the UPRmt into the mitochondrial maintenance network. Crit. Rev. Biochem. Mol. Biol. 52, 304–313. doi:10.1080/10409238.2017.1291577
Fischer, D. F., De Vos, R. A., Van Dijk, R., De Vrij, F. M., Proper, E. A., Sonnemans, M. A., et al. (2003). Disease-specific accumulation of mutant ubiquitin as a marker for proteasomal dysfunction in the brain. FASEB J. 17, 2014–2024. doi:10.1096/fj.03-0205com
Fischer, D. F., van Dijk, R., van Tijn, P., Hobo, B., Verhage, M. C., van der Schors, R. C., et al. (2009). Long-term proteasome dysfunction in the mouse brain by expression of aberrant ubiquitin. Neurobiol. Aging 30, 847–863. doi:10.1016/j.neurobiolaging.2008.06.009
Fratta, P., Engel, W. K., van Leeuwen, F. W., Hol, E. M., Vattemi, G., and Askanas, V. (2004). Mutant ubiquitin UBB+1 is accumulated in sporadic inclusion-body myositis muscle fibers. Neurology 63, 1114–1117. doi:10.1212/01.WNL.0000138574.56908.5D
French, B. A., van Leeuwen, F., Riley, N. E., Yuan, Q. X., Bardag-Gorce, F., Gaal, K., et al. (2001). Aggresome formation in liver cells in response to different toxic mechanisms: role of the ubiquitin-proteasome pathway and the frameshift mutant of ubiquitin. Exp. Mol. Pathol. 71, 241–246. doi:10.1006/exmp.2001.2401
Gordon, J. C., Myers, J. B., Folta, T., Shoja, V., Heath, L. S., and Onufriev, A. (2005). H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 33, W368–W371. doi:10.1093/nar/gki464
Guo, W., Stoklund Dittlau, K., and Van Den Bosch, L. (2020). Axonal transport defects and neurodegeneration: molecular mechanisms and therapeutic implications. Semin. Cel Dev. Biol. 99, 133–150. doi:10.1016/j.semcdb.2019.07.010
Hetz, C., Zhang, K., and Kaufman, R. J. (2020). Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 21, 421–438. doi:10.1038/s41580-020-0250-z
Höhfeld, J., and Hartl, F. U. (1994). Post-translational protein import and folding. Curr. Opin. Cell Biol. 6, 499–509. doi:10.1016/0955-0674(94)90068-X
Hol, E. M., van Leeuwen, F. W., and Fischer, D. F. (2005). The proteasome in Alzheimer’s disease and Parkinson’s disease: lessons from ubiquitin B+1. Trends Mol. Med. 11, 488–495. doi:10.1016/j.molmed.2005.09.001
Hope, A. D., De Silva, R., Fischer, D. F., Hol, E. M., van Leeuwen, F. W., and Lees, A. J. (2003). Alzheimer’s associated variant ubiquitin causes inhibition of the 26S proteasome and chaperone expression. J. Neurochem. 86, 394–404. doi:10.1046/j.1471-4159.2003.01844.x
Inoue, K., Tsutsui, H., Akatsu, H., Hashizume, Y., Matsukawa, N., Yamamoto, T., et al. (2013). Metabolic profiling of Alzheimer’s disease brains. Sci. Rep. 3, 2364–2369. doi:10.1038/srep02364
Irmler, M., Gentier, R. J., Dennissen, F. J., Schulz, H., Bolle, I., Hölter, S. M., et al. (2012). Long-term proteasomal inhibition in transgenic mice by UBB(+1) expression results in dysfunction of central respiration control reminiscent of brainstem neuropathology in Alzheimer patients. Acta Neuropathol. 124, 187–197. doi:10.1007/s00401-012-1003-7
Izadi, S., Anandakrishnan, R., and Onufriev, A. V. (2014). Building water models: a different approach. J. Phys. Chem. Lett. 5, 3863–3871. doi:10.1021/jz501780a
Jankowsky, J. L., Fadale, D. J., Anderson, J., Xu, G. M., Gonzales, V., Jenkins, N. A., et al. (2004). Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 13, 159–170. doi:10.1093/hmg/ddh019
Ko, S., Kang, G. B., Song, S. M., Lee, J. G., Shin, D. Y., Yun, J. H., et al. (2010). Structural basis of E2-25K/UBB+1 interaction leading to proteasome inhibition and neurotoxicity. J. Biol. Chem. 285, 36070–36080. doi:10.1074/jbc.M110.145219
Komander, D., and Rape, M. (2012). The ubiquitin code. Annu. Rev. Biochem. 81, 203–229. doi:10.1146/annurev-biochem-060310-170328
Krieger, E., and Vriend, G. (2014). YASARA View - molecular graphics for all devices - from smartphones to workstations. Bioinformatics 30, 2981–2982. doi:10.1093/bioinformatics/btu426
Krutauz, D., Reis, N., Nakasone, M. A., Siman, P., Zhang, D., Kirkpatrick, D. S., et al. (2014). Extended ubiquitin species are protein-based DUB inhibitors. Nat. Chem. Biol. 10, 664–670. doi:10.1038/nchembio.1574
Lam, Y. A., Pickart, C. M., Alban, A., Landon, M., Jamieson, C., Ramage, R., et al. (2000). Inhibition of the ubiquitin-proteasome system in Alzheimer's disease. Proc. Natl. Acad. Sci. United States 97, 9902–9906. doi:10.1073/pnas.170173897
Li, P., Song, L. F., and Merz, K. M. (2015). Systematic parameterization of monovalent ions employing the nonbonded model. J. Chem. Theor. Comput. 11, 1645–1657. doi:10.1021/ct500918t
Lindquist, S., and Craig, E. A. (1988). The heat-shock proteins. Annu. Rev. Genet. 22, 631–677. doi:10.1146/annurev.ge.22.120188.003215
Lindsten, K., De Vrij, F. M. S., Verhoef, L. G. G. C., Fischer, D. F., Van Leeuwen, F. W., Hol, E. M., et al. (2002). Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J. Cell Biol. 157, 417–427. doi:10.1083/jcb.200111034
Liu, P., Fleete, M. S., Jing, Y., Collie, N. D., Curtis, M. A., Waldvogel, H. J., et al. (2014). Altered arginine metabolism in Alzheimer’s disease brains. Neurobiol. Aging 35, 1992–2003. doi:10.1016/j.neurobiolaging.2014.03.013
Maday, S., Twelvetrees, A. E., Moughamian, A. J., and Holzbaur, E. L. (2014). Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84, 292–309. doi:10.1016/j.neuron.2014.10.019
Morimoto, R. I. (2008). Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 22, 1427–1438. doi:10.1101/gad.1657108
Munari, F., Bortot, A., Assfalg, M., and D’Onofrio, M. (2018). Alzheimer’s disease-associated ubiquitin mutant Ubb+1: properties of the carboxy-terminal domain and its influence on biomolecular interactions. Int. J. Biol. Macromol. 108, 24–31. doi:10.1016/j.ijbiomac.2017.11.121
Muñoz-Arellano, A. J., Chen, X., Molt, A., Meza, E., and Petranovic, D. (2018). Different expression levels of human mutant ubiquitin B+1 (UBB+1) can modify chronological lifespan or stress resistance of saccharomyces cerevisiae. Front. Mol. Neurosci. 11, 1–15. doi:10.3389/fnmol.2018.00200
Myers, J., Grothaus, G., Narayanan, S., and Onufriev, A. (2006). A simple clustering algorithm can be accurate enough for use in calculations of pKs in macromolecules. Proteins 63, 928–938. doi:10.1002/prot.20922
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204. doi:10.1146/annurev-neuro-061010-113613
Park, Y., Yoon, S. K., and Yoon, J. B. (2009). The HECT domain of TRIP12 ubiquitinates substrates of the ubiquitin fusion degradation pathway. J. Biol. Chem. 284, 1540–1549. doi:10.1074/jbc.M807554200
Pérez, V. I., Buffenstein, R., Masamsetti, V., Leonard, S., Salmon, A. B., Mele, J., et al. (2009). Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent, the naked mole-rat. Proc. Natl. Acad. Sci. United States 106, 3059–3064. doi:10.1073/pnas.0809620106
Perlson, E., Maday, S., Fu, M. M., Moughamian, A. J., and Holzbaur, E. L. (2010). Retrograde axonal transport: pathways to cell death?. Trends Neurosci. 33, 335–344. doi:10.1016/j.tins.2010.03.006
Popovic, D., Vucic, D., and Dikic, I. (2014). Ubiquitination in disease pathogenesis and treatment. Nat. Med. 20, 1242–1253. doi:10.1038/nm.3739
Poulsen, E. G., Steinhauer, C., Lees, M., Lauridsen, A. M., Ellgaard, L., and Hartmann-Petersen, R. (2009). HUWE1 and TRIP12 collaborate in degradation of ubiquitin-fusion proteins and misframed ubiquitin. PLoS One 106, 11907–11912. doi:10.1371/journal.pone.0050548
Rushaidhi, M., Jing, Y., Kennard, J. T., Collie, N. D., Williams, J. M., Zhang, H., et al. (2012). Aging affects l-arginine and its metabolites in memory-associated brain structures at the tissue and synaptoneurosome levels. Neuroscience 209, 21–31. doi:10.1016/j.neuroscience.2012.02.021
Ryu, K. Y., Garza, J. C., Lu, X. Y., Barsh, G. S., and Kopito, R. R. (2008). Hypothalamic neurodegeneration and adult-onset obesity in mice lacking the Ubb polyubiquitin gene. Proc. Natl. Acad. Sci. United States 105, 4016–4021. doi:10.1073/pnas.0800096105
Ryu, H. W., Park, C. W., and Ryu, K. Y. (2014). Restoration of cellular ubiquitin reverses impairments in neuronal development caused by disruption of the polyubiquitin gene Ubb. Biochem. Biophys. Res. Commun. 453, 443–448. doi:10.1016/j.bbrc.2014.09.103
Seynnaeve, D., Del Vecchio, M., Fruhmann, G., Verelst, J., Cools, M., Beckers, J., et al. (2018). Recent insights on Alzheimer’s disease originating from yeast models. Int. J. Mol. Sci. 19, 1–26. doi:10.3390/ijms19071947
Shabek, N., Herman-Bachinsky, Y., and Ciechanover, A. (2009). Ubiquitin degradation with its substrate, or as a monomer in a ubiquitination-independent mode, provides clues to proteasome regulation. Proc. Natl. Acad. Sci. U. S. A. 106, 11907–11912. doi:10.1073/pnas.0905746106
Shpilka, T., and Haynes, C. M. (2018). The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cel Biol. 19, 109–120. doi:10.1038/nrm.2017.110
Sluchanko, N. N., Artemova, N. V., Sudnitsyna, M. V., Safenkova, I. V., Antson, A. A., Levitsky, D. I., et al. (2012). Monomeric 14-3-3ζ has a chaperone-like activity and is stabilized by phosphorylated HspB6. Biochemistry 51, 6127–6138. doi:10.1021/bi300674e
Sousa, J. S., D'Imprima, E., and Vonck, J. (2018). Mitochondrial respiratory chain complexes, Subcell Biochem. 87, 167–227. doi:10.1007/978-981-10-7757-9_7
Swovick, K., Welle, K. A., Hryhorenko, J. R., Seluanov, A., Gorbunova, V., and Ghaemmaghami, S. (2018). Cross-species comparison of proteome turnover kinetics. Mol. Cell Proteomics 17, 580–591. doi:10.1074/mcp.RA117.000574
Takahashi, D., Moriyama, J., Nakamura, T., Miki, E., Takahashi, E., Sato, A., et al. (2019). AUTACs: cargo-specific degraders using selective autophagy. Mol. Cel. 76, 797–810.e10. doi:10.1016/j.molcel.2019.09.009
Tan, Z., Sun, X., Hou, F. S., Oh, H. W., Hilgenberg, L. G., Hol, E. M., et al. (2007). Mutant ubiquitin found in Alzheimer’s disease causes neuritic beading of mitochondria in association with neuronal degeneration. Cell Death Differ. 14, 1721–1732. doi:10.1038/sj.cdd.4402180
Tian, C., Kasavajhala, K., Belfon, K. A. A., Raguette, L., Huang, H., Migues, A. N., et al. (2020). Ff19SB: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theor. Comput. 16, 528–552. doi:10.1021/acs.jctc.9b00591
van Leeuwen, F. W., Hol, E. M., and Burbach, H. P. (1998). Mutations in RNA: a first example of molecular misreading in Alzheimer’s disease. Trends Neurosci. 21, 331–335. doi:10.1016/S0166-2236(98)01280-6
van Leeuwen, F. W., Fischer, D. F., Kamel, D., Sluijs, J. A., Sonnemans, M. A., Benne, R., et al. (2000). Molecular misreading: a new type of transcript mutation expressed during aging. Neurobiol. Aging 21, 879–891. doi:10.1016/S0197-4580(00)00151-2
van Leeuwen, F. W., Gerez, L., Benne, R., and Hol, E. M. (2002). +1 Proteins and aging. Int. J. Biochem. Cell Biol. 34, 1502–1505. doi:10.1016/S1357-2725(02)00043-2
van Leeuwen, F. W., Van Tijn, P., Sonnemans, M. A., Hobo, B., Mann, D. M., Van Broeckhoven, C., et al. (2006). Frameshift proteins in autosomal dominant forms of Alzheimer disease and other tauopathies. Neurology 66, S86–S92. doi:10.1212/01.wnl.0000193882.46003.6d
van Tijn, P., de Vrij, F. M., Schuurman, K. G., Dantuma, N. P., Fischer, D. F., van Leeuwen, F. W., et al. (2007). Dose-dependent inhibition of proteasome activity by a mutant ubiquitin associated with neurodegenerative disease. J. Cel Sci. 120, 1615–1623. doi:10.1242/jcs.03438
van Tijn, P., Verhage, M. C., Hobo, B., Van Leeuwen, F. W., and Fischer, D. F. (2010). Low levels of mutant ubiquitin are degraded by the proteasome in vivo. J. Neurosci. Res. 88, 2325–2337. doi:10.1002/jnr.22396
Verheijen, B. M., Hashimoto, T., Oyanagi, K., and van Leeuwen, F. W. (2017). Deposition of mutant ubiquitin in parkinsonism-dementia complex of Guam. Acta Neuropathol. Commun. 5, 82–83. doi:10.1186/s40478-017-0490-0
Verheijen, B. M., Stevens, J. A. A., Gentier, R. J. G., Van 't Hekke, C. D., Van Den Hove, D. L. A., Hermes, D. J. H. P., et al. (2018). Paradoxical effects of mutant ubiquitin on Aβ plaque formation in an Alzheimer mouse model. Neurobiol. Aging 72, 62–71. doi:10.1016/j.neurobiolaging.2018.08.011
Verheijen, B. M., Lussier, C., Müller-Hübers, C., Garruto, R. M., Oyanagi, K., Braun, R. J., et al. (2020). Activation of the unfolded protein response and proteostasis disturbance in parkinsonism-dementia of Guam. J. Neuropathol. Exp. Neurol. 79, 34–45. doi:10.1093/jnen/nlz110
Verhoef, L. G. G. C., Heinen, C., Selivanova, A., Halff, E. F., Salomons, F. A., and Dantuma, N. P. (2009). Minimal length requirement for proteasomal degradation of ubiquitin-dependent substrates. FASEB J. 23, 123–133. doi:10.1096/fj.08-115055
Wiborg, O., Pedersen, M. S., Wind, A., Berglund, L. E., Marcker, K. A., and Vuust, J. (1985). The human ubiquitin multigene family: some genes contain multiple directly repeated ubiquitin coding sequences. EMBO J. 4, 755–759. doi:10.1002/j.1460-2075.1985.tb03693.x
Wills, N. M., and Atkins, J. F. (2006). The potential role of ribosomal frameshifting in generating aberrant proteins implicated in neurodegenerative diseases. RNA 12, 1149–1153. doi:10.1261/rna.84406
Yim, N., Ryu, S. W., Han, E. C., Yoon, J., Choi, K., and Choi, C. 2014). Mutant ubiquitin UBB+1 induces mitochondrial fusion by destabilizing mitochondrial fission-specific proteins and confers resistance to oxidative stress-induced cell death in astrocytic cells. PLoS One 9, e99937. doi:10.1371/journal.pone.0099937
Keywords: molecular misreading, UBB+1, ubiquitin proteasomal system, neurodegeneration, proteotoxic stress response, ROS generation and cytotoxicity, cellular viability
Citation: Banasiak K, Szulc NA and Pokrzywa W (2021) The Dose-Dependent Pleiotropic Effects of the UBB+1 Ubiquitin Mutant. Front. Mol. Biosci. 8:650730. doi: 10.3389/fmolb.2021.650730
Received: 07 January 2021; Accepted: 02 February 2021;
Published: 26 March 2021.
Edited by:
Heidi Olzscha, Martin Luther University of Halle-Wittenberg, GermanyReviewed by:
Nico P. Dantuma, Karolinska Institutet, SwedenCopyright © 2021 Banasiak, Szulc and Pokrzywa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wojciech Pokrzywa, d3Bva3J6eXdhQGlpbWNiLmdvdi5wbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.