 Thirumal Kumar D
Thirumal Kumar D Nishaat Shaikh
Nishaat Shaikh Udhaya Kumar S
Udhaya Kumar S George Priya Doss C
George Priya Doss C Hatem Zayed
Hatem Zayed
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci., 09 April 2021
Sec. Biological Modeling and Simulation
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.645216
This article is part of the Research TopicComputational Approaches to Understand Molecular Mechanisms of SARS-CoV-2View all 10 articles
The number of confirmed COVID-19 cases is rapidly increasing with no direct treatment for the disease. Few repurposed drugs, such as Remdesivir, Chloroquine, Hydroxychloroquine, Lopinavir, and Ritonavir, are being tested against SARS-CoV-2. Remdesivir is the drug of choice for Ebola virus disease and has been authorized for emergency use. This drug acts against SARS-CoV-2 by inhibiting the RNA-dependent-RNA-polymerase (RdRp) of SARS-CoV-2. RdRp of viruses is prone to mutations that confer drug resistance. A recent study by Pachetti et al. in 2020 identified the P323L mutation in the RdRp protein of SARS-CoV-2. In this study, we aimed to determine the potency of lead compounds similar to Remdesivir, which can be used as an alternative when variants of SARS-CoV-2 develop resistance due to RdRp mutations. The initial screening yielded 704 compounds that were 90% similar to the control drug, Remdesivir. On further evaluation through drugability and antiviral inhibition percentage analyses, we shortlisted 32 and seven compounds, respectively. These seven compounds were further analyzed for their molecular interactions, which revealed that all seven compounds interacted with RdRp with higher affinity than Remdesivir under native conditions. However, three compounds failed to interact with the mutant protein with higher affinity than Remdesivir. Dynamic cross-correlation matrix (DCCM) and vector field collective motions analyses were performed to identify the precise movements of docked complexes' residues. Furthermore, the compound SCHEMBL20144212 showed a high affinity for native and mutant proteins and might provide an alternative against SARS-CoV-2 variants that might confer resistance to Remdesivir. Further validations by in vitro and in vivo studies are needed to confirm the efficacy of our lead compounds for their inhibition against SARS-CoV-2.
COVID-19 is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Huang et al., 2020). In December 2019, China reported its first case of SARS-CoV-2 in Wuhan, Hubei province (Zhu et al., 2020). The infection was highly contagious, which led to the global spread of the virus in the following months, thus causing an outbreak (Giovanetti et al., 2020; Phan et al., 2020). It was finally declared as a Public Health Emergency of International Concern (PHEIC) by the World Health Organization (WHO) on January 30, 2020, and ultimately named Coronavirus Disease 2019 (COVID-19) on February 12, 2020. COVID-19 is responsible for high mortality worldwide (WHO, 2020a,b). Currently, there are 172, and 61 vaccines are in pre-clinical and clinical development, respectively. While it takes years to develop a new effective drug against a disease, the COVID-19 outbreak has created a global emergency. Hence, drug repositioning and repurposing are given more attention as it is a rapid solution to identify a drug to combat the disease (Li and Clercq, 2020; Morse et al., 2020). Coronaviruses are enveloped viruses about 80–120 nm in diameter with plus-strand (+) RNA as genetic material (Brian and Baric, 2005). Studies have shown that SARS-CoV-2 shares ~80% genetic similarity with SARS-CoV, while RNA-dependent RNA polymerase (RdRp) has a 96% similarity (Xu et al., 2020). RdRp and protease are encoded by the viral RNA and play an essential role in replicating and assembling the virus, and hence preexisting drugs that target these proteins are preferred. Antiviral agents that have been developed to combat coronaviruses are used here as potential drugs for the treatment of COVID-19.
A 20-kb gene encodes the replicase complex. It encompasses several viral genes encoding RdRp, RNA helicase, proteases, and some RNA processing enzymes and cellular proteins that aid in replicating and transcribing the genome of coronavirus occurring in the cytoplasm of the host cell (Koonin and Dolja, 1993; Cheng et al., 2005). RNA synthesis takes place in both a continuous and a discontinuous fashion (Gallagher, 1996). Coronaviruses are plus-strand (+) RNA viruses with a tremendously diverse genome, resulting in a variation in their RNA synthesis machinery (Goldbach, 1987). RdRps exhibit a very high transcription error rate leading to variation in the genome of the virus. During the replication process, RdRps adopts the switching mechanism resulting in the recombination of RNA. This process is also responsible for repairing deleterious mutations in the genome of the viruses, leading to gene rearrangements and new gene acquisition (Strauss and Strauss, 1988; Xu et al., 2003).
Once the virus penetrates the host cell by binding to the angiotensin-converting enzyme 2 (ACE2) receptor, the process of uncoating is initiated that releases the plus-strand (+) RNA into the cytoplasm. The ribosomes present in the host cell's cytoplasm proceed to translate the genomic RNA into a polyprotein. This polyprotein undergoes proteolytic cleavage by the protease enzyme that results in the replicase enzyme production and various viral structural proteins. Approximately 67% of the genomic RNA encodes RdRp that arbitrates the genome's synthesis (Thiel et al., 2001). The plus-strand (+) RNA can only produce viral protein products and not genetic material by replication. Thus, to achieve genome replication, RdRp first transcribes and replicates the plus-strand (+) RNA, which acts as a template to generate the minus-strand (–) RNA, and then serves as a template for the RdRPs to produce several plus-strand (+) RNAs. Some of this plus-strand (+) RNA is again translated into proteins.
In contrast, the others are enclosed into the capsid to regenerate complete virions released from the cell by exocytosis. RdRP is a popular target for a selective antiviral strategy against coronaviruses because RNA synthesis by RdRP does not occur in mammalian cells (Casais et al., 2001). Figure 1 illustrates the role of RdRp in viral replication and effect of RdRp inhibitors (Figure 1).
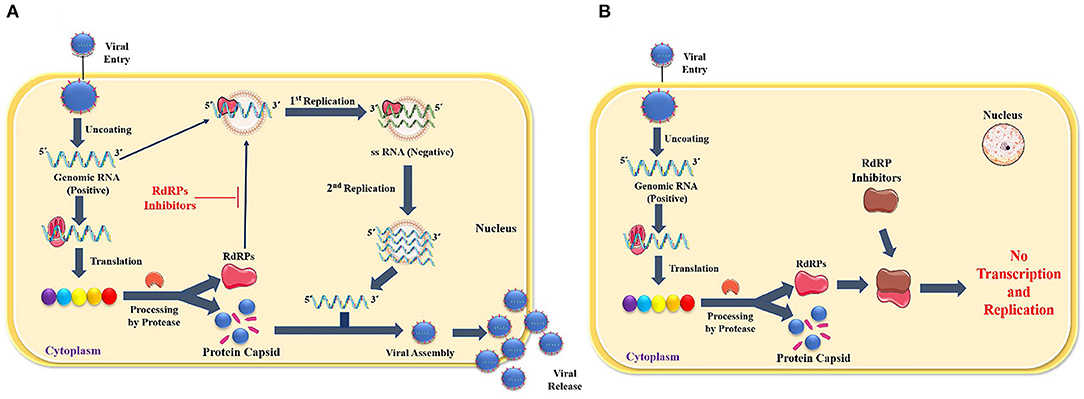
Figure 1. (A) Role of RdRp in viral replication, (B) Effect of RdRp inhibitors.
Current potential treatments against COVID-19 that have shown promising results are Lopinavir, a known protease inhibitor of coronavirus (Yao T.-T. et al., 2020), the guanosine analog, ribavirin that targets RdRp and was designed to combat the Ebola outbreak (Falzarano et al., 2013), and chloroquine, an antimalarial drug, that has shown antiviral effects by blocking viral fusion with the cell due to increased endosomal pH (Vincent et al., 2005). A derivative of chloroquine, hydroxychloroquine, is more potent than chloroquine, as reported by several studies (Yao X. et al., 2020). Several corticosteroids have also been studied for their coronavirus effects (Russell et al., 2020). Convalescent plasma transfusion is also currently being administered and has been shown to reduce mortality rate (Mair-Jenkins et al., 2015). Among the treatments, Remdesivir has shown promising results in in vitro and animal studies and is currently in phase III trials (Martinez, 2020).
Remdesivir, also known as GS-5734, is a nucleotide prodrug that is metabolized inside the cell into GS-441524 and an adenosine nucleotide analog, phosphorylated into cell-permeable di-and tri-phosphates (Varga et al., 2016; Warren et al., 2016) Viral RdRps can utilize these adenosine triphosphates during genome replication (Sheahan et al., 2017). Remdesivir causes the nascent RNA transcript to terminate prematurely and incorporates mutations into it (Agostini et al., 2018). Studies have shown that remdesivir causes RNA levels to decrease in a dose-dependent manner, and it is 4.5-fold more potent in cells lacking the ExoN proofreading activity. It has also been observed that GS-5734 is more active (~30 times) than its metabolized form, GS-441524, against the coronaviruses tested (Sheahan et al., 2017). It has been shown that in the presence of GS-441524, there was 5.6-fold resistance in coronavirus mouse hepatitis virus, which was attributed to two mutations, F476L and V553L, in the coding region of nsp12 core polymerase (Agostini et al., 2018). Similar findings were observed in SARS-CoV carrying the mutations F480L and V557L, which are not in the vicinity of the binding site of RdRp; thus, the exact mechanism behind the resistance remains undetermined (Agostini et al., 2018). This raises the concern that resistance may cause the virus to be active and functional, leading to severe disease transmission. Because of its decreased proofreading activity, RdRps have an error rate in the range of 10−4−10−6 errors/nucleotide/replication, which is very high compared to that of DNA polymerases (Eckerle et al., 2010). This high error rate is responsible for the rapid rate of virus evolution, contributing to its adaptability to the new surroundings and expediting its ability to jump between species (Pillay and Zambon, 1998). A recent study by Pachetti et al. in 2020 identified the P323L mutation in the RdRp protein of SARS-CoV-2, which may confer resistance to the drug. In this study, we utilized a virtual screening approach to identify the potential repurposed lead compound that might be efficient in treating COVID-19 due to SARS-CoV-2 variants resistant to Remdesivir.
The 2D and 3D structures of Remdesvir in canonical SMILES and SDF formats were obtained from the PubChem database with CID 121304016. These formats were also converted to PDB using the OpenBabel software (O'Boyle et al., 2011). The 3D structure of the complexes between the non-structural proteins 12, 7, and 8 and Remdesivir was obtained from the Protein Data Bank with PDB ID−7BV2, and the missing amino acids were theoretically built using the Swiss Model Server (Biasini et al., 2014; Wanchao et al., 2020). Further, the P323L mutation reported by Pachetti et al. in 2020 was introduced in the refined protein structure using the SwissPDB Viewer, and energy was minimized using the same method (Guex and Peitsch, 1997; Pachetti et al., 2020).
The inbuilt module of PubChem was used to identify compounds with 90% similarity to the control Remdesivir structure. The identified compounds were screened for absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties using the SwissADME server (Daina et al., 2017). The 90% similar compounds that satisfy the Lipinski drugability properties were further used in an in silico antiviral inhibition percentage study using the AntiViral Compound Prediction (AVCpred) server (Qureshi et al., 2017). These filtered compounds were finally subjected to molecular interaction studies against the native and mutant RdRp proteins using the AutoDock standalone package (Morris et al., 2009).
Molecular interaction studies were initiated to understand the differences between the control remdesivir and the virtually similar compounds with RdRp proteins. Hydrogen and charges were added to the RdRp protein, and the torsions were set to Remdesivir and nearly identical compounds. The grid box was placed around the active site (LYS546, SER683, ARG556, THR688, ASP624, SER760, ASN692, ASP761, and ASP762) retrieved from the published crystal structure (Wanchao et al., 2020). The Lamarckian Genetic Algorithm was used to generate the binding pockets and binding affinity of RdRp for Remdesivir or the virtually similar compounds. One hundred different pockets were developed, and those with the best binding affinity were used in the structural visualization.
The docked complexes were further subjected to the web-server DynOmics ENM to identify the residue cross-correlation matrix. The tool developed with the Elastic network models (ENM) that integrate Anisotropic Network Model (ANM) and GaussianNetwork Model (GNM) (Li et al., 2017). Time-correlated data was represented as a matrix between the protein atoms i and j (cij) in DCCM. Within the form of a map, typical fluctuations, and standardized correlations among residues are typically shown and represented with the following equation:
Cij = < ΔRi. ΔRj >
Cij (n) = < ΔRi. ΔRj >/[ < ΔRi. ΔRi > < ΔRj. ΔRj >]1/2
The range of Cij(n) varies in terms (−1, 1) and analyzes information on the cross-correlation between residue movements i and j (Rader et al., 2005). To determine how mutations affect the internal mechanics of protein conformations, the Bio3D module incorporated with the R studio was used to quantify residue-residue dynamic cross-correlation networks. The normal mode, network analysis, and correlation analysis were called with the function “nma(),” “can(),”and “dccm().” The role from these features is usually a cross-correlation matrix of residue-residue. The results were plotted by calling the functions “plot.dccm()” and “pymol.dccm()” (Grant et al., 2006; Scarabelli and Grant, 2013).
As an initiative of virtual screening, the compounds with 90% structural similarity to the control drug, Remdesivir, were screened using the inbuilt PubChem module. The screening identified 704 compounds possessing 90% similarity with Remdesivir. These 704 compounds were subjected to in silico ADME analysis using the SwissADME server (Supplementary Table 1). Based on Lipinski's drugability properties, 32 compounds were further filtered from the pool of 704 compounds. Finally, these 32 compounds were subjected to an antiviral inhibition percentage study calculated using the AVCpred server (Table 1). This analysis identified the compounds with PubChem IDs 137648734, 145074552, 58527341, 44468492, 70649275, 145074438 134502628 that showed higher antiviral inhibition percentages than Remdesivir. The inhibition percentage of Remdesivir was 51.563%. However, the inhibition percentages of compounds, 137648734, 145074552, 58527341, 44468492, 70649275, 145074438, and 134502628 were found to be 62.457, 62.213, 59.052, 57.651, 57.297, 55.189, and 54.648%, respectively. These seven compounds and Remdesivir were subjected to molecular interaction studies against the native and mutant RdRp molecules.
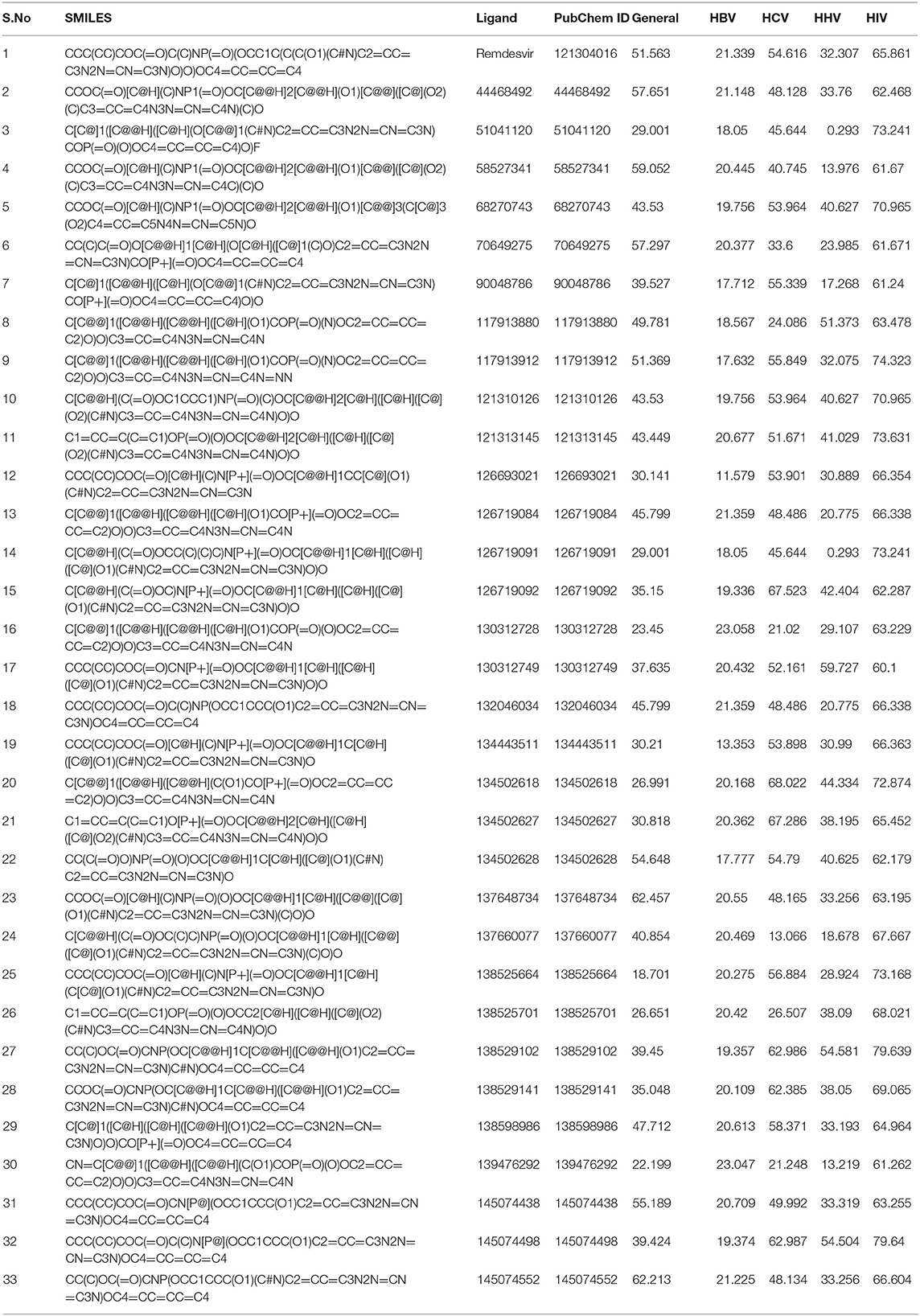
Table 1. Antiviral inhibition percentage prediction of the compounds satisfying Lipinski's drugability properties.
Molecular interaction studies were performed using AutoDock to understand the interaction pattern of Remdesivir and its similar compounds with the native and mutant RdRp proteins. It was observed that all seven compounds had a higher binding affinity than Remdesivir with native RdRp. For RdRp protein carrying the P323L mutation, we observed that the compounds 134502628, 70649275, 137648734, and 145074552 possessed higher binding affinity than Remdesivir. The binding affinity of Remdesivir for native and mutant RdRp was −4.5 and −4.2 kcal/mol, respectively. However, the compound with PubChem ID 134502628 showed the highest binding affinity of −7.5 and −7.4 kcal/mol for the native and mutant RdRp proteins, respectively (Tables 2A,B). The detailed interaction comparison between Remdesivir and top-ranked compound, 134502628 (SCHEMBL20144212), against the native and mutant RdRp is shown in Table 3, Figures 2A–D.
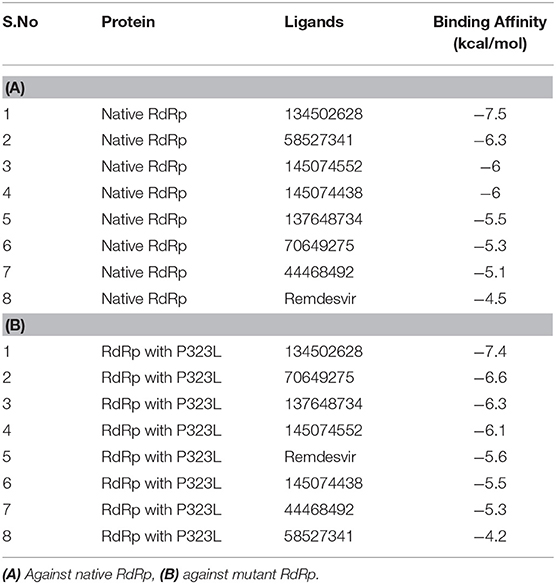
Table 2. The binding affinity of the ligands against native and mutant SARS-CoV-2 RdRp structures.
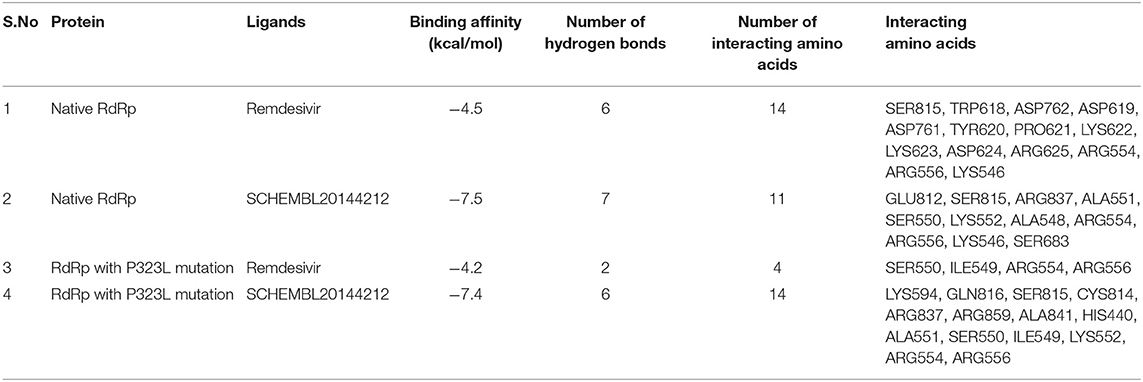
Table 3. Differences in the binding affinity (kcal/mol), number of hydrogen bonds, number of interacting amino acids, and Interacting amino acids between native and mutant RdRp against Remdesivir and the novel lead compound.
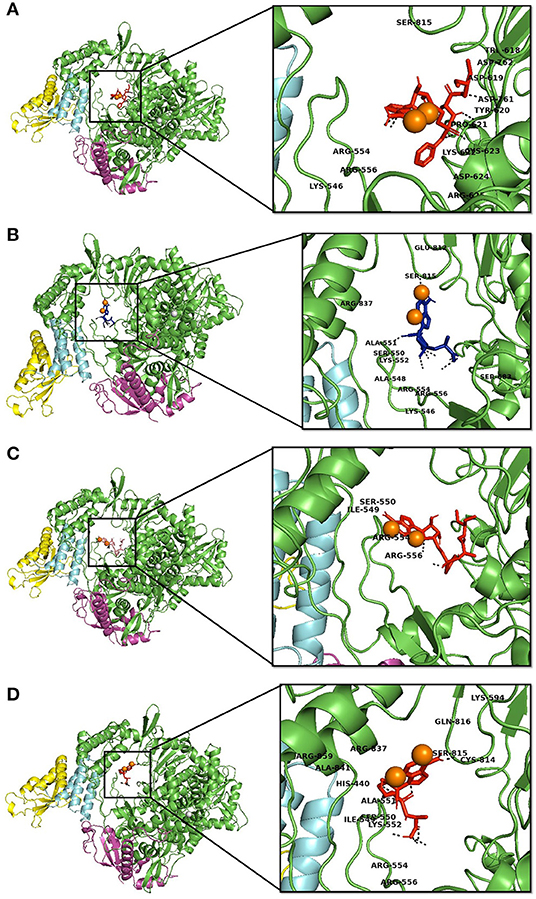
Figure 2. Interaction of Remdesvir and the novel lead compound with the native RdRp and RdRp with the mutation. (A) Native RdRp with Remdesvir, (B) Native RdRp with novel lead compound (SCHEMBL20144212), (C) Mutant RdRp with Remdesvir, (D) Mutant RdRp with novel lead compound (SCHEMBL20144212).
Firstly, we investigated the residue cross-correlation networks and vector field collective motions with the help of the Bio3D module that was integrated within R studio. All four complexes were subjected to the Bio3D module to retrieve the residue cross-correlation networks and collective motions. As a result, the residues from 5 to 120 (α1, α2, α3, β1, β2, and β-hairpin) and 830–933 (α40–45, β22, and β23) were obtained with large cross-correlation networks from RdRp-Remdesivir and SCHEMBL20144212 complexes (Figures 3A,C), whereas the P323L-Remdesivir obtained less cross-correlation networks especially at the region from 5 to 120 residues (Figure 3E). The residues from 830 to 933 exhibited similar networks for all the docked complexes. However, the differences can be seen with the P323L-Remdesivir and P323L-SCHEMBL20144212 complexes, especially at the region from 5 to 120 residues. The P323L-Remdesivir complex exhibited the least residue cross-correlation; the possible reason for this could be mutation at 323rd position in RdRp. Our results exhibited similar network cross-correlation for P323L-SCHEMBL20144212 (Figure 3G) when compared to the RdRp-SCHEMBL20144212 complex. The atomic movements of these docked complexes were investigated by representing the vector field collective motions. The collective motions were largely observed in the 5–120 and 830–933 regions, as seen in Figures 3B,D,F,H. Here, we observed the differences in the residues from 5 to 120 from the P323L-Remdesivir complex (Figure 3F) compared to the other three docked complexes. The arrows on the docked complex of proteins determine the magnitude and direction of the collective motion. Furthermore, we investigated the DCCM of RdRp (with or without mutation) with remdesivir and SCHEMBL20144212 complexes by utilizing the DynOmics tool. DCCM explores the precise movements of residues of proteins, demonstrates strongly correlated (in red) atomic movements between residues, and highlights atomic movements that are strongly anticorrelated (in blue) (Figure 4). The RdRp-SCHEMBL20144212 and P323L-SCHEMBL20144212 complexes (Figures 4B,C) illustrate the strongly correlated motions and less anticorrelation as compared to that of RdRp-Remdesivir and P323L-Remdesivir complexes (Figures 4A,C).
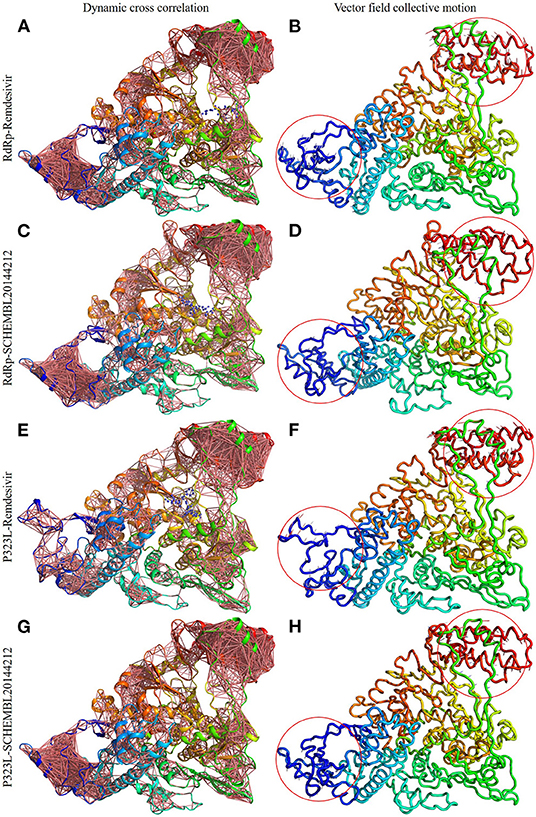
Figure 3. Dynamic cross-correlation networks and vector field collective motions of docked complexes. Depiction of cross-correlation networks: (A) RdRp-Remdesivir; (C) RdRp-SCHEMBL20144212; (E) P323L-Remdesivir; (G) P323L-SCHEMBL20144212. Depiction of vector field collective motions: (B) RdRp-Remdesivir; (D) RdRp-SCHEMBL20144212; (F) P323L-Remdesivir; (H) P323L-SCHEMBL20144212. Red circles indicate differences in the direction of collective motions by arrows.
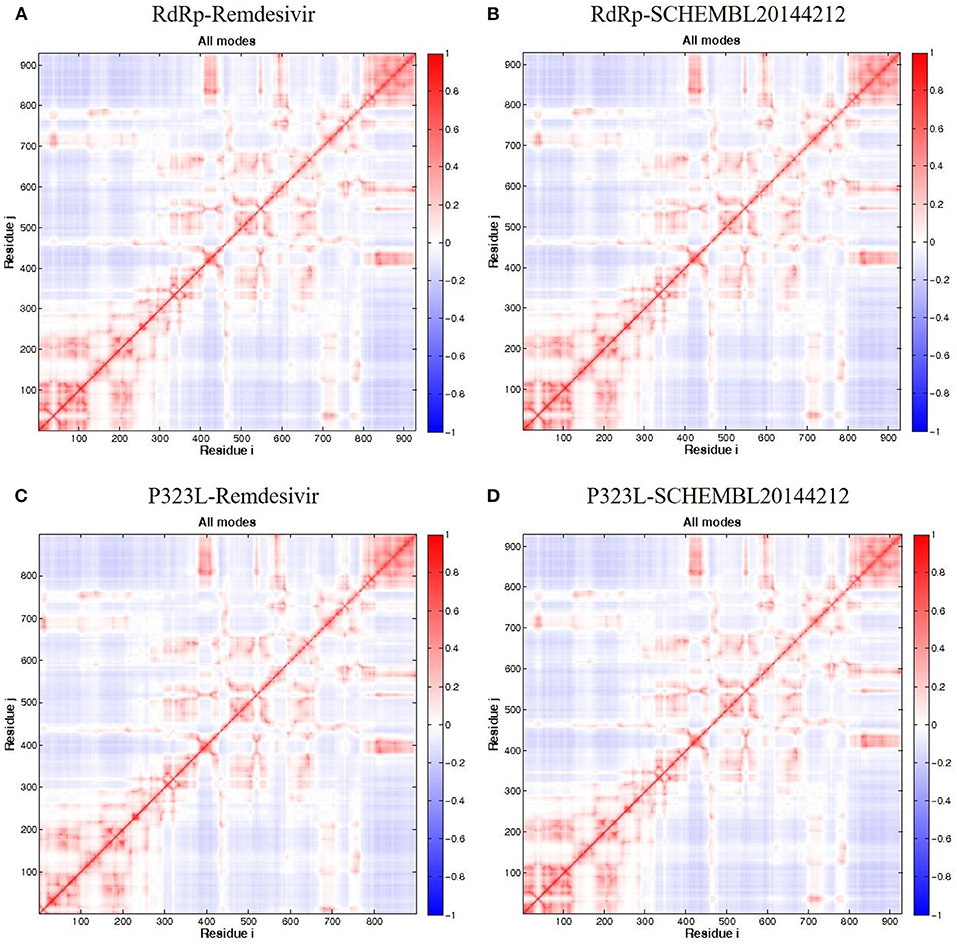
Figure 4. DCCM of the residues around their mean position for all docked RdRp complexes. (A) RdRp-Remdesivir; (B) RdRp-SCHEMBL20144212; (C) P323L-Remdesivir; (D) P323L-SCHEMBL20144212. Map size = 100, min i = 5, and min j = 5. The color ranges from −1 (blue) to +1 (red), whereas 0 remains in white color.
COVID-19, caused by SARS-CoV-2, has become a global pandemic and is responsible for about 2,71,954 deaths worldwide, with more than 3.95 million positive cases. COVID-19 has been transmitted to almost 120 countries, and discovering drugs to fight this disease is a great challenge (Worldometer, 2021). The genome of the SARS-CoV-2 virus was immediately sequenced to improve our understanding of the virus, formulate appropriate diagnostic and preventive techniques, and develop therapeutic strategies (Wang et al., 2020). The high transmission rate of the virus is responsible for the difficulty of implementing efficient preventive measures. Hence, there is an immediate requirement for a drug to inhibit the virus and eliminate the disease (Li et al., 2020). One of the emerging drugs with promising results is Remdesivir. It is a nucleotide analog, initially developed for the Ebola virus, that targets the virus's RdRp (Gordon et al., 2020). The virus's high mutation rate may introduce changes to RdRp, rendering the drug ineffective (Eckerle et al., 2010). A recent study by Pachetti et al. in 2020 identified the P323L mutation in the RdRp of SARS-CoV-2 (Pachetti et al., 2020). The possible increase in mutations highlights the need to develop a series of drugs whose efficiency is not affected by these mutations.
Thus, there is an urgent need to develop novel drugs. The conventional method for drug discovery is expensive and time-consuming, and therefore the in silico approach is a means to move forward. Our study formulated a computational pipeline to identify a series of compounds similar to Remdesivir, which could be used as an alternative if SARS-CoV-2 develops resistance to Remdesivir due to mutations. Out of 704 compounds, which were 90% similar to Remdesivir, 32 compounds were found to satisfy the rule of 5 (Supplementary Table 1). To assess drugability, these 32 compounds were subjected to in silico antiviral inhibition percentage analysis. We observed that the seven virtually similar compounds possessed a higher antiviral inhibition percentage when compared to Remdesivir. Finally, molecular interaction studies against the native and mutant RdRp were carried out with docking analysis. The interactions revealed that all the identified seven compounds could interact with higher affinity with the native protein than Remdesivir.
However, upon introducing the P323L mutation, only four compounds were found to bind with a higher binding affinity than Remdesivir. The binding efficacy of Remdesivir was also found to decrease from −4.5 kcal/mol in the native to −4.2 kcal/mol in the mutant protein. Interestingly, the compound, SCHEMBL20144212, was topmost in the binding affinity for native and mutant proteins (Table 2). Remdesivir was found to interact with the native RdRp through 14 amino acids and 6 hydrogen bonds. However, the identified novel lead compound, SCHEMBL20144212, was found to interact with native RdRp through 11 amino acids and 7 hydrogen bonds.
Similarly, in interaction with the mutant protein, Remdesivir was found to interact through four amino acids and two hydrogen bonds. However, the novel lead compound, SCHEMBL20144212, was found to interact through 14 amino acids and 6 hydrogen bonds (Table 3, Figures 2A–D). The role of hydrogen bonds and interacting amino acids is crucial for a drug to exhibit its efficacy. We observed higher binding affinity, hydrogen bond interactions, and amino acid interactions with the identified novel lead compound compared to Remdesivir. We have also depicted the detailed pharmacokinetic properties obtained from the pkCSM server in Table 4. These results suggest that SCHEMBL20144212 is more effective against the original or the mutated virus compared with Remdesivir.
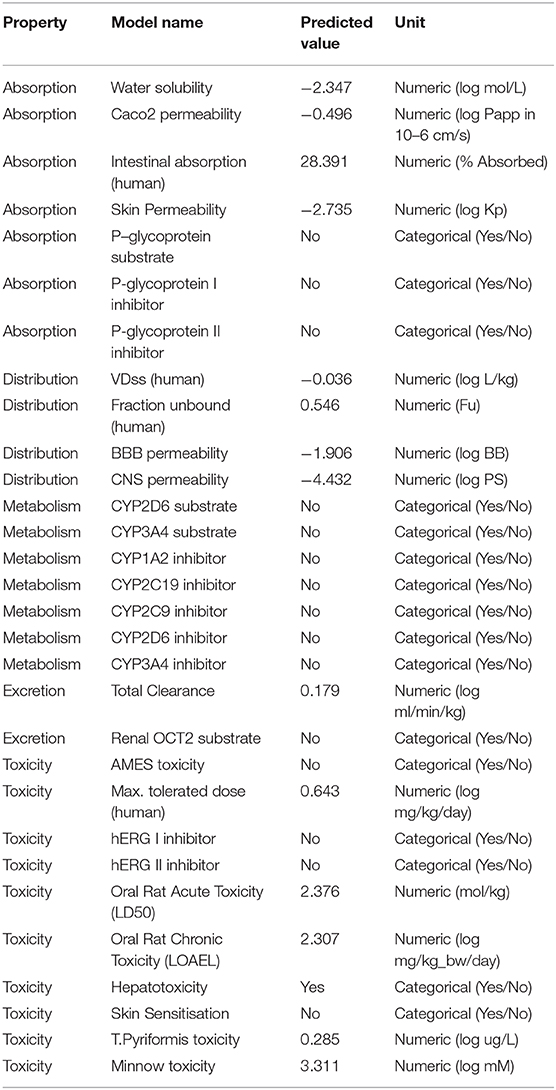
Table 4. ADMET properties of the novel lead compound obtained from the pkCSM server.
It is vital to characterize the protein folding via identifying conformational variances, change in conformational motions owing to mutations, mechanism of ion channel's opening/closing, and protein-ligand binding, as these factors directly related to the protein's stability and function (Grottesi et al., 2005; Yang et al., 2008; Kim et al., 2010; S et al., 2020; Udhaya Kumar et al., 2020). The residue cross-correlation networks were obtained for all the docked complexes, and among them, the P323L-Remdesivir complex obtained less correlative network at the region from 5 to 120 residues (Figure 3E). A possible reason for this could be a mutation that occurred at position 323; thus, complex resulted in fewer correlative networks. Also, in comparison to our identified lead compound (SCHEMBL20144212) with mutant RdRp (P323L) exhibited large correlative networks and could act as a sturdy inhibitor against mutant RdRp (Figure 3G). The vector field collective motions were identified for all the docked complexes to explore each complex's dynamic motion and magnitude (Figures 3B,D,F,H). The RdRp and mutant RdRp (P323L) with SCHEMBL20144212 lead compound exhibited a rigid complex as seen in Figures 3D,H. This clearly states that the SCHEMBL20144212 lead compound inhibits the RdRp from binding to other molecules and for the catalysis process, whereas the remdesivir complexes exhibited more flexible regions (Figures 3B,F) (Huber, 1987; Karshikoff et al., 2015). Furthermore, our results from DCCM showed the RdRp-SCHEMBL20144212 and P323L-SCHEMBL20144212 complexes exhibited strongly correlated motions (Figures 4B,D). From the above observations, we strongly believe that the molecular docking of SCHEMBL20144212 lead compound reduces the effect of P323L mutation and could act as an effective inhibitor against SARS-nCoV-2's RdRp.
This study was initiated to identify the potential repurposed lead compound to treat COVID-19 in case of possible SARS-CoV-2-virus resistance to Remdesivir due to mutations. The drug inhibits the activity of RdRp protein to a small extent. Identifying the mutation in RdRp of SARS-CoV-2 highlights the possible appearance of mutated viruses that might be resistant to current and future treatments. Therefore, it is necessary to establish a platform to identify compounds similar to but more useful than Remdesivir. Our study identified a total of 704 compounds from the PubChem database, which are 90% similar to Remdesivir. Of these 704 compounds, 32 compounds were found to possess druggability properties, out of these, seven compounds were found to possess higher antiviral inhibition percentages when compared to Remdesivir. Out of these seven compounds analyzed for molecular interaction. The SCHEMBL20144212 compound was found to possess the highest interaction affinity for both the native and mutant RdRp protein. From the DCCM and vector field collective motion analysis, we demonstrated that our lead compound's molecular docking (SCHEMBL20144212) tolerates the effect of the P323L mutation and could act as an effective inhibitor against SARS-nCoV-2's RdRp. Thus, this compound should be further validated through in vitro experiments as an alternative to Remdesivir to bypass the resistant effect of the P323L mutation. Our study offers a platform for drug repurposing during the time of viral pandemics.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
TK, UK, HZ, and GD contributed to designing the study and data acquisition, analysis, and interpretation. TK, NS, and UK involved in the acquisition, analysis, and interpreting of the results. TK, UK, and GD contributed to data interpretation, conducted, and drafting the manuscript. GD and HZ supervised the entire study and were involved in study design, acquiring, analyzing, understanding the data, and drafting the manuscript. The manuscript was reviewed and approved by all the authors.
UK, one of the authors, gratefully acknowledges the Indian Council of Medical Research (ICMR), India, for providing him a Senior Research Fellowship [ISRM/11(93)/2019].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank the Vellore Institute of Technology, India, and Qatar University, Qatar, for providing the necessary research facilities and encouragement to carry out this work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.645216/full#supplementary-material
Supplementary Table 1. The list of 90% similar compounds to the Remdesivir obtained from the PubChem database.
Agostini, M. L., Andres, E. L., Sims, A. C., Graham, R. L., Sheahan, T. P., Lu, X., et al. (2018). Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. MBio 9, 1–15. doi: 10.1128/mBio.00221-18
Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., et al. (2014). SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258. doi: 10.1093/nar/gku340
Brian, D. A., and Baric, R. S. (2005). Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 287, 1–30. doi: 10.1007/3-540-26765-4_1
Casais, R., Thiel, V., Siddell, S. G., Cavanagh, D., and Britton, P. (2001). Reverse genetics system for the avian coronavirus infectious bronchitis virus. J. Virol. 75, 12359–12369. doi: 10.1128/JVI.75.24.12359-12369.2001
Cheng, A., Zhang, W., Xie, Y., Jiang, W., Arnold, E., Sarafianos, S. G., et al. (2005). Expression, purification, and characterization of SARS coronavirus RNA polymerase. Virology 335, 165–176. doi: 10.1016/j.virol.2005.02.017
Daina, A., Michielin, O., and Zoete, V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7:42717. doi: 10.1038/srep42717
Eckerle, L. D., Becker, M. M., Halpin, R. A., Li, K., Venter, E., Lu, X., et al. (2010). Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 6, 1–15. doi: 10.1371/journal.ppat.1000896
Falzarano, D., de Wit, E., Rasmussen, A. L., Feldmann, F., Okumura, A., Scott, D. P., et al. (2013). Treatment with interferon-α2b and ribavirin improves outcome in MERS-CoV-infected rhesus macaques. Nat. Med. 19, 1313–1317. doi: 10.1038/nm.3362
Gallagher, T. M. (1996). Murine coronavirus membrane fusion is blocked by modification of thiols buried within the spike protein. J. Virol. 70, 4683–4690. doi: 10.1128/JVI.70.7.4683-4690.1996
Giovanetti, M., Benvenuto, D., Angeletti, S., and Ciccozzi, M. (2020). The first two cases of 2019-nCoV in Italy: where they come from? J. Med. Virol. 92, 518–521. doi: 10.1002/jmv.25699
Goldbach, R. (1987). Genome similarities between plant and animal RNA viruses. Microbiol. Sci. 4, 197–202.
Gordon, C. J., Tchesnokov, E. P., Feng, J. Y., Porter, D. P., and Götte, M. (2020). The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 295, 4773–4779. doi: 10.1074/jbc.AC120.013056
Grant, B. J., Rodrigues, A. P. C., ElSawy, K. M., McCammon, J. A., and Caves, L. S. D. (2006). Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 22, 2695–2696. doi: 10.1093/bioinformatics/btl461
Grottesi, A., Domene, C., Hall, B., and Sansom, M. S. P. (2005). Conformational dynamics of M2 helices in KirBac channels: helix flexibility in relation to gating via molecular dynamics simulations. Biochemistry 44, 14586–14594. doi: 10.1021/bi0510429
Guex, N., and Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723. doi: 10.1002/elps.1150181505
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. doi: 10.1016/S0140-6736(20)30183-5
Huber, R. (1987). Flexibility and rigidity, requirements for the function of proteins and protein pigment complexes. Eleventh Keilin Memorial Lecture. (Biochemical Society transactions) 15:1009–20. doi: 10.1042/bst0151009
Karshikoff, A., Nilsson, L., and Ladenstein, R. (2015). Rigidity versus flexibility: the dilemma of understanding protein thermal stability. FEBS J. 282:3899–917. doi: 10.1111/febs.13343
Kim, H. J., Choi, M. Y., Kim, H. J., and Llinás, M. (2010). Conformational dynamics and ligand binding in the multi-domain protein PDC109. PLoS ONE 5:e9180. doi: 10.1371/journal.pone.0009180
Koonin, E. V., and Dolja, V. V. (1993). Evolution and taxonomy of positive-strand RNA viruses: implications of comparative analysis of amino acid sequences. Crit. Rev. Biochem. Mol. Biol. 28, 375–430. doi: 10.3109/10409239309078440
Li, G., and Clercq, E. D. (2020). Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 19, 149–150. doi: 10.1038/d41573-020-00016-0
Li, H., Chang, Y.-Y., Lee, J. Y., Bahar, I., and Yang, L.-W. (2017). DynOmics: dynamics of structural proteome and beyond. Nucleic Acids Res. 45, W374–W380. doi: 10.1093/nar/gkx385
Li, R., Pei, S., Chen, B., Song, Y., Zhang, T., Yang, W., et al. (2020). Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV-2). Science 368, 489–493. doi: 10.1126/science.abb3221
Mair-Jenkins, J., Saavedra-Campos, M., Baillie, J. K., Cleary, P., Khaw, F.-M., Lim, W. S., et al. (2015). The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: a systematic review and exploratory meta-analysis. J. Infect. Dis. 211, 80–90. doi: 10.1093/infdis/jiu396
Martinez, M. A. (2020). Compounds with therapeutic potential against novel respiratory 2019 Coronavirus. Antimicrob. Agents Chemotherap. 64, 1–7. doi: 10.1128/AAC.00399-20
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. doi: 10.1002/jcc.21256
Morse, J. S., Lalonde, T., Xu, S., and Liu, W. R. (2020). Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. Chembiochem 21, 730–738. doi: 10.1002/cbic.202000047
O'Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., and Hutchison, G. R. (2011). Open Babel: An open chemical toolbox. J. Cheminform. 3:33. doi: 10.1186/1758-2946-3-33
Pachetti, M., Marini, B., Benedetti, F., Giudici, F., Mauro, E., Storici, P., et al. (2020). Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 18, 1–9. doi: 10.1186/s12967-020-02344-6
Phan, L. T., Nguyen, T. V., Luong, Q. C., Nguyen, T. V., Nguyen, H. T., Le, H. Q., et al. (2020). Importation and human-to-human transmission of a novel coronavirus in vietnam. N. Engl. J. Med. 382, 872–874. doi: 10.1056/NEJMc2001272
Pillay, D., and Zambon, M. (1998). Antiviral drug resistance. Br. Med. J. 317, 660–662. doi: 10.1136/bmj.317.7159.660
Qureshi, A., Kaur, G., and Kumar, M. (2017). AVCpred: an integrated web server for prediction and design of antiviral compounds. Chem. Biol. Drug Des. 89, 74–83. doi: 10.1111/cbdd.12834
Rader, A. J., Chennubhotla, C., Yang, L.-W., and Bahar, I. (2005). “The gaussian network model: theory and applications,” in Normal Mode Analysis Theory and Applications to Biological and Chemical Systems eds Cui, Q., and Bahar, I. (New York, NY: Chapman and Hall/CRC), 41–64. doi: 10.1201/9781420035070.ch3
Russell, C. D., Millar, J. E., and Baillie, J. K. (2020). Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet 395, 473–475. doi: 10.1016/S0140-6736(20)30317-2
S, U. K., R, B., D, T. K., Doss, C. G. P., and Zayed, H. (2020). Mutational landscape of K-Ras substitutions at 12th position-a systematic molecular dynamics approach. J. Biomol. Struct. Dyn. 120, 1–16. doi: 10.1080/07391102.2020.1830177
Scarabelli, G., and Grant, B. J. (2013). Mapping the structural and dynamical features of kinesin motor domains. PLoS Comput. Biol. 9:e1003329. doi: 10.1371/journal.pcbi.1003329
Sheahan, T. P., Sims, A. C., Graham, R. L., Menachery, V. D., Gralinski, L. E., Case, J. B., et al. (2017). Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 9:eaal3653. doi: 10.1126/scitranslmed.aal3653
Strauss, J. H., and Strauss, E. G. (1988). Evolution of RNA viruses. Annu. Rev. Microbiol. 42, 657–683. doi: 10.1146/annurev.mi.42.100188.003301
Thiel, V., Herold, J., Schelle, B., and Siddell, S. G. (2001). Viral replicase gene products suffice for coronavirus discontinuous transcription. J. Virol. 75, 6676–6681. doi: 10.1128/JVI.75.14.6676-6681.2001
Udhaya Kumar, S., Thirumal Kumar, D., Mandal, P. D., Sankar, S., Haldar, R., Kamaraj, B., et al. (2020). “Chapter Eight - Comprehensive in silico screening and molecular dynamics studies of missense mutations in Sjogren-Larsson syndrome associated with the ALDH3A2 gene,” in Advances in Protein Chemistry and Structural Biology Inflammatory Disorders - Part B, ed. R. Donev (Academic Press), 349–377. doi: 10.1016/bs.apcsb.2019.11.004
Varga, A., Lionne, C., and Roy, B. (2016). Intracellular metabolism of nucleoside/nucleotide analogues: a bottleneck to reach active drugs on HIV reverse transcriptase. Curr. Drug Metab. 17, 237–252. doi: 10.2174/1389200217666151210141903
Vincent, M. J., Bergeron, E., Benjannet, S., Erickson, B. R., Rollin, P. E., Ksiazek, T. G., et al. (2005). Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2:69. doi: 10.1186/1743-422X-2-69
Wanchao, Y., Chunyou, M., Xiaodong, L., Dan-Dan, S., Qingya, S., Haixia, S., et al. (2020). Structural basis for the inhibition of the RNA-Dependent RNA polymerase from SARS- CoV-2 by Remdesivir. bioRxiv 1560, 1–30. doi: 10.1101/2020.04.08.032763
Wang, P., Anderson, N., Pan, Y., Poon, L., Charlton, C., Zelyas, N., et al. (2020). The SARS-CoV-2 outbreak: diagnosis, infection prevention, and public perception. Clin. Chem. 66:hvaa080. doi: 10.1093/clinchem/hvaa080
Warren, T. K., Jordan, R., Lo, M. K., Ray, A. S., Mackman, R. L., Soloveva, V., et al. (2016). Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 531, 381–385. doi: 10.1038/nature17180
WHO (2020a). Statement on the second meeting of the International Health Regulations (2005) Emergency Committee Regarding the Outbreak of Novel Coronavirus (2019-nCoV). Available online at: https://www.who.int/news/item/30-01-2020-statement-on-the-second-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov) (accessed November 15, 2020).
WHO (2020b). WHO Director-General's Remarks at the Media Briefing on 2019-nCoV on 11 February 2020. Available online at: https://www.who.int/director-general/speeches/detail/who-director-general-s-remarks-at-the-media-briefing-on-2019-ncov-on-11-february-2020 (accessed November 15, 2020).
Worldometer (2021). Coronavirus Update (Live). Available online at: https://www.worldometers.info/coronavirus/ (accessed March 13, 2021).
Xu, J., Zhao, S., Teng, T., Abdalla, A. E., Zhu, W., Xie, L., et al. (2020). Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 12:244. doi: 10.3390/v12020244
Xu, X., Liu, Y., Weiss, S., Arnold, E., Sarafianos, S. G., and Ding, J. (2003). Molecular model of SARS coronavirus polymerase: implications for biochemical functions and drug design. Nucleic Acids Res. 31, 7117–7130. doi: 10.1093/nar/gkg916
Yang, L., Song, G., Carriquiry, A., and Jernigan, R. L. (2008). Close correspondence between the motions from principal component analysis of multiple HIV-1 protease structures and elastic network modes. Structure 16, 321–330. doi: 10.1016/j.str.2007.12.011
Yao, T.-T., Qian, J.-D., Zhu, W.-Y., Wang, Y., and Wang, G.-Q. (2020). A systematic review of lopinavir therapy for SARS coronavirus and MERS coronavirus-A possible reference for coronavirus disease-19 treatment option. J. Med. Virol. 92, 556–563. doi: 10.1002/jmv.25729
Yao, X., Ye, F., Zhang, M., Cui, C., Huang, B., Niu, P., et al. (2020). In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 71, 732–739. doi: 10.1093/cid/ciaa237
Keywords: SARS-CoV-2, COVID-19, remdesivir, virtual screening, RdRp, mutations
Citation: Kumar D T, Shaikh N, Kumar S U, Doss C GP and Zayed H (2021) Structure-Based Virtual Screening to Identify Novel Potential Compound as an Alternative to Remdesivir to Overcome the RdRp Protein Mutations in SARS-CoV-2. Front. Mol. Biosci. 8:645216. doi: 10.3389/fmolb.2021.645216
Received: 22 December 2020; Accepted: 11 March 2021;
Published: 09 April 2021.
Edited by:
Arvind Ramanathan, Argonne National Laboratory (DOE), United StatesReviewed by:
Filippo Pullara, SpIntellx Inc., United StatesCopyright © 2021 Kumar D, Shaikh, Kumar S, Doss C and Zayed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George Priya Doss C, Z2VvcmdlcHJpeWFkb3NzQHZpdC5hYy5pbg==; Hatem Zayed, aGF0ZW0uemF5ZWRAcXUuZWR1LnFh
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.