 Sutanu Bhattacharya
Sutanu Bhattacharya Rahmatullah Roche
Rahmatullah Roche Md Hossain Shuvo
Md Hossain Shuvo Debswapna Bhattacharya
Debswapna Bhattacharya
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Biosci., 11 May 2021
Sec. Structural Biology
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.643752
This article is part of the Research TopicA Journey Through 50 Years of Structural Bioinformatics in Memoriam of Cyrus ChothiaView all 11 articles
Sequence-based protein homology detection has emerged as one of the most sensitive and accurate approaches to protein structure prediction. Despite the success, homology detection remains very challenging for weakly homologous proteins with divergent evolutionary profile. Very recently, deep neural network architectures have shown promising progress in mining the coevolutionary signal encoded in multiple sequence alignments, leading to reasonably accurate estimation of inter-residue interaction maps, which serve as a rich source of additional information for improved homology detection. Here, we summarize the latest developments in protein homology detection driven by inter-residue interaction map threading. We highlight the emerging trends in distant-homology protein threading through the alignment of predicted interaction maps at various granularities ranging from binary contact maps to finer-grained distance and orientation maps as well as their combination. We also discuss some of the current limitations and possible future avenues to further enhance the sensitivity of protein homology detection.
The development of computational approaches for accurately predicting the protein three-dimensional (3D) structure directly from the sequence information is of central importance in structural biology (Jones et al., 1992; Baker and Sali, 2001; Dill and MacCallum, 2012). While ab initio modeling aims to predict the 3D structure purely from the sequence information (Marks et al., 2011; Adhikari et al., 2015; Wang et al., 2016; Adhikari and Cheng, 2018; Greener et al., 2019; Senior et al., 2019; Xu, 2019; Yang et al., 2020; Roche et al., 2021), many protein targets have evolutionary-related (homologous) structures, also known as homologous templates, already available in the Protein Data Bank (PDB) (Berman et al., 2000). Correctly identifying these templates given the sequence of a query protein and building 3D models by performing query–template alignment, a technique broadly known as homology modeling (Altschul et al., 1997; Xu et al., 2003; Wu and Zhang, 2008; Lobley et al., 2009; Wu and Zhang, 2010; Källberg et al., 2012; Ma et al., 2014) often results in highly accurate predicted structural models (Abeln et al., 2017). As such, the success of homology modeling critically depends on the ability to identify the closely homologous template on the basis of sequence similarity and generate accurate query–template alignment. Intuitively, the performance of these methods sharply deteriorates when the direct evolutionary relationship between the query and templates becomes very low, typically when the sequence similarity falls below 30%, the so-called distant-homology modeling scenarios (Bowie et al., 1991; Petrey and Honig, 2005). Protein threading, the most widely used distant-homology modeling technique, aims to address the challenge by leveraging multiple sources of information by mining the evolutionary profile of the query and templates to reveal potential distant homology and perform distant-homology modeling to predict the 3D structure of the query protein.
Existing threading methods exploit a wide range of techniques ranging from dynamic programming to profile-based comparison to machine learning (Jones, 1999; Rychlewski et al., 2000; Xu and Xu, 2000; Skolnick and Kihara, 2001; Ginalski et al., 2003; Marti et al., 2004; Jaroszewski et al., 2005; Söding, 2005; Zhou and Zhou, 2005; Cheng and Baldi, 2006; Peng and Xu, 2009; Lee and Skolnick, 2010; Peng and Xu, 2010; Yang et al., 2011; Ma et al., 2012; Ma et al., 2013; Gniewek et al., 2014). The recent advancement in predicting the inter-residue interaction maps using sequence coevolution and deep learning (Morcos et al., 2011; He et al., 2017; Wang et al., 2017; Adhikari et al., 2018; Hanson et al., 2018; Kandathil et al., 2019; Yang et al., 2020) has opened new possibilities to further improve the sensitivity of distant-homology protein threading by incorporating the predicted inter-residue interaction information. Fueled by this, several efforts have been made in the recent past to integrate interaction maps into threading. For instance, EigenTHREADER (Buchan and Jones, 2017), map_align (Ovchinnikov et al., 2017), CEthreader (Zheng et al., 2019a), CATHER (Du et al., 2020), and ThreaderAI (Zhang and Shen, 2020) have utilized predicted contact maps in protein threading. DeepThreader (Zhu et al., 2018) has exploited finer-grained distance maps for query proteins instead of using binary contacts to improve threading template selection and alignment. DisCovER (Bhattacharya et al., 2020) goes one step further by incorporating inter-residue orientation along with distance information together with topological network neighborhood (Chen et al., 2019) of query–template alignment to further improve threading performance. Here, we provide an overview of the latest advances in protein homology detection propelled by inter-residue interaction map threading.
Protein inter-residue interaction maps are predicted at various resolutions ranging from binary contact maps to finer-grained distance and orientation maps as well as their combination. A low-resolution version of inter-residue interaction is a contact map, which is a square, symmetric matrix with binary entries, where a contact indicates the spatial proximity of a residue pair at a given cutoff distance, typically set to 8Å between the Cα or Cβ carbons of the interacting residue pairs. Inter-residue distance map is finer-grained in that it captures the distribution of real-valued inter-residue spatial proximity information rather than the binary contacts at a fixed cutoff distance. Recent studies (Xu and Wang, 2019; Xu, 2019) have demonstrated the advantage of using distance maps in protein structure prediction over binary contacts as distances carry more physical constraint information of protein structures than contacts. The granularities of predicted distance maps vary from distance histograms to real-valued distances (Greener et al., 2019; Adhikari, 2020; Ding and Gong, 2020; Li and Xu, 2020; Wu et al., 2021; Yang et al., 2020). Very recently, trRosetta (Yang et al., 2020) has introduced inter-residue orientations in addition to distances to capture not only the spatial proximity information of the interacting pairs but also their relative angles and dihedrals. Collectively, inter-residue distances and orientations encapsulate the spatial positioning of the interacting pairs much better than only distances let alone binary contacts.
Figure 1 shows an overview of an interaction map threading of a query protein. Generally, threading has four components: (1) an effective scoring function to evaluate the fitness of query–template alignment; (2) efficient template searching or homology detection strategy; (3) optimal query–template alignments; and (4) building 3D models of query proteins based on alignments. One of the most important components of threading approaches is the scoring function, which is composed of standard threading features ranging from sequential features such as secondary structures, solvent accessibility, and sequence profiles to nonlinear features such as pairwise potentials (Bienkowska and Lathrop, 2005; Brylinski and Skolnick, 2010). Weights control the relative importance of different terms. An efficient scoring function should reliably differentiate a homologous template from the alternatives because the accuracy of the predicted model significantly depends on the evolutionary relatedness of the identified template. The inter-residue interaction map helps to improve the sensitivity of the threading scoring function by augmenting the standard scoring terms with additional contributions from the predicted interactions. Specifically, the score to align the
where the first term accounts for the contribution of the interaction map and the second term accounts for the standard threading features with
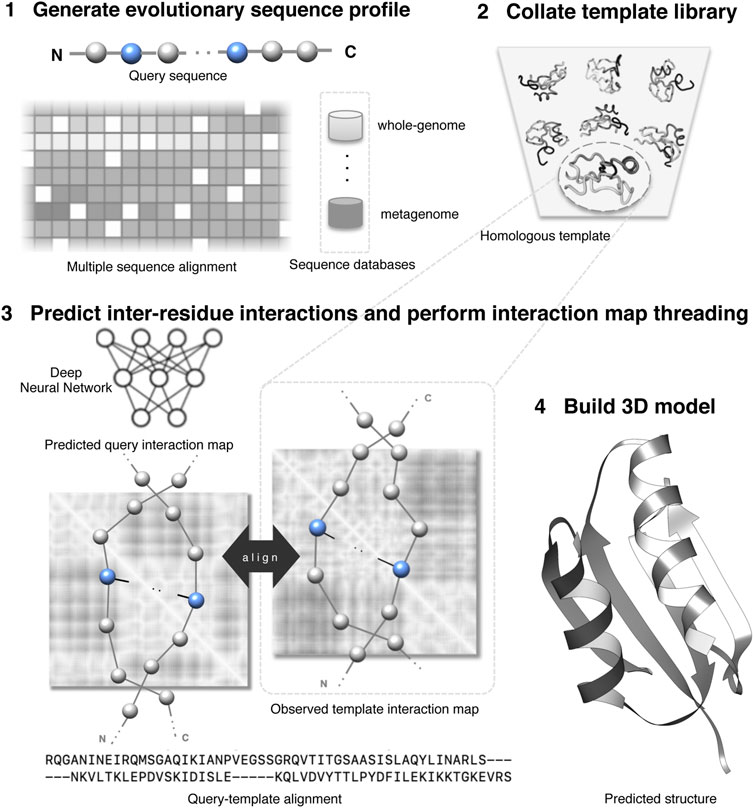
FIGURE 1. Illustration of protein interaction map threading.
With the recent advancement in contact prediction mediated by sequence coevolution and deep learning, significant research efforts have been made in the recent past to incorporate contact information as an additional scoring term into the threading scoring function for protein homology detection. For instance, Jones and coworkers developed EigenTHREADER (Buchan and Jones, 2017) that uses eigen-decomposition (Di Lena et al., 2010) of contact maps predicted using classical neural network–based predictor MetaPSICOV (Jones et al., 2015) to search a library of template contact maps for contact map threading. Baker and coworkers developed map_align (Ovchinnikov et al., 2017) that employs an iterative double dynamic programming framework (Taylor, 1999) for homology detection. map_align takes advantage of metagenomics sequence databases of microbial DNA (Söding, 2017) and uses contact maps predicted by coevolutionary contact predictor GREMLIN (Balakrishnan et al., 2011; Kamisetty et al., 2013) to perform contact map threading by maximizing the number of overlapping contacts and minimizing the number of gaps. Recently, Zhang and coworkers developed CEthreader (Zheng et al., 2019a) using contact maps predicted by deep learning–based contact map predictor ResPRE (Li et al., 2019). CEthreader also relies on eigen-decomposition and performs contact map threading through dynamic programming using a dot-product scoring function by integrating contacts as well as secondary structures and sequence profiles. Alongside, we developed a contact-assisted threading method (Bhattacharya and Bhattacharya, 2019) that incorporates contact information, predicted by deep learning–based predictor RaptorX (Wang et al., 2017), into threading using a two-stage approach. After selecting a subset of top templates from the template library using a standard profile-based threading technique in the first stage, our method subsequently uses eigen-decomposition of the contact information along with the profile-based alignment score to select the best-fit template. We further analyze the impact of contact map quality on threading performance (Bhattacharya and Bhattacharya, 2020), which reveals that incorporating high-quality contact maps having the Matthews correlation coefficient (MCC) ≥ 0.5 improves the threading performance for ∼ 30% cases in comparison to a baseline contact-free threading used as a control, while incorporating low-quality contacts with MCC <0.35 deteriorates the performance for 50% cases. Yang and coworkers developed CATHER (Du et al., 2020) by incorporating contact maps predicted by deep learning–based predictor MapPred (Wu et al., 2020) along with standard sequential information in the threading scoring function. Very recently, Shen and coworkers have developed ThreaderAI (Zhang and Shen, 2020) that implements a neural network for predicting alignments by incorporating deep learning–based contact information with conventional sequential and structural features into the scoring function.
Building on the successes of contact-assisted threading methods, Xu and coworkers developed a distance-based threading method called DeepThreader (Zhu et al., 2018). The method predicts distance maps by employing deep learning and then incorporates the predicted inter-residue distance information along with sequential features into threading through alternating direction method of multipliers (ADMM) algorithm. The inter-residue distance is binned into 12 bins: <5Å, 5–6Å, .., 14–15Å, and >15Å. Based on their reported results as well as the performance evaluation in the 13th Critical Assessment of protein Structure Prediction (CASP13), incorporating distance information boosts threading performance, particularly for distant-homology targets, outperforming contact-assisted threading methods by a large margin (Xu and Wang, 2019, 13). Zhang and coworkers have recently extended CEthreader to develop a distance-assisted threading method DEthreader introduced during the recently concluded CASP14 experiment by incorporating a distance-based scoring term into the scoring function. The method uses the Cα–Cα and Cβ–Cβ distance distribution, both are binned into 38 bins: 1 bin of <2Å, 36 bins of 2–20Å with a width of 0.5Å, and 1 bin of ≥20Å. Similarly, Yang and coworkers have extended CATHER into a distance-based threading approach by replacing contacts with distances in CASP14.
Powered by the development of the recent deep learning–based trRosetta method (Yang et al., 2020) for the prediction of inter-residue orientations and distances, our recent method DisCovER (Bhattacharya et al., 2020) goes one step further by incorporating predicted inter-residue orientations in addition to distances together with the neighborhood effect of the query–template alignment using an iterative double dynamic programming framework. The predicted distances are binned into 9 bins with a bin size of 1Å: <6Å to <14Å by summing up the likelihoods for distance bins below a distance threshold. The two orientation dihedrals (ω, θ) are binned into 24 bins with a width of 15°, and the orientation angle (ϕ) is binned into 12 bins with a width of 15°. Experimental results demonstrate the improved threading performance of DisCovER over the other state-of-the-art threading approaches on multiple benchmark datasets across various target categories, especially for distantly homologous proteins. Representative examples on CAMEO targets 6D2S_A and 6CP8_D provide some insights into the origin of the improved performance. Figure 2 shows our recent method DisCovER predicts correct folds (TM-score > 0.5) for both the targets 6D2S_A and 6CP8_D with a TM-score of 0.76 and 0.69, respectively, significantly better than the others. While the pure profile-based threading method CNFpred (Ma et al., 2012; Ma et al., 2013) and the recent contact-assisted threading method CEthreader fail to predict the correct fold for the target 6D2S_A, DisCovER and the CAMEO server RaptorX (Källberg et al., 2012; Zhu et al., 2018), employing the distance-based threading method DeepThreader (Haas et al., 2019), effectively predict the correct fold, with noticeably better performance by DisCovER (an improvement of 0.2 TM-score points) than the next best RaptorX. We also notice the superior performance of DisCovER for the target 6CP8_D where DisCovER significantly outperforms the other competing methods including the next best CEthreader by 0.18 TM-score points. It is worth mentioning both the targets are officially classified as “hard” by CAMEO (Haas et al., 2019), which warrants a distantly homologous nature in which current threading methods have limitations. Overall, the results show that the integration of the orientation information and the neighborhood effect in DisCovER results in improved threading, attaining state-of-the-art performance in (distant) homology detection.
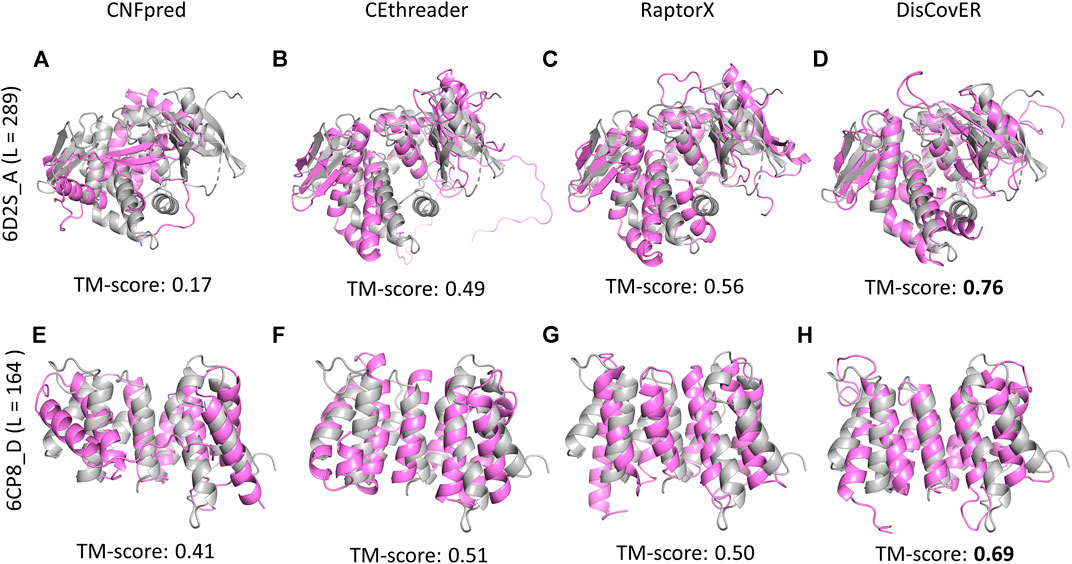
FIGURE 2. Structural superposition between predicted models using various threading methods (in violet) and the corresponding experimental structures (in gray) for representative CAMEO targets 6D2S_A of length 289 residues and 6CP8_D of length 164 residues.
The prediction of inter-residue interaction maps depends heavily on the availability of homologous sequences. As such, the role of the sequence databases is becoming increasingly important in protein homology detection via interaction map threading. In addition to the well-established whole-genome sequence databases such as the nr database from the National Center for Biotechnology Information (NCBI), UniRef (Suzek et al., 2015), UniProt (The UniProt Consortium, 2019), and Uniclust (Mirdita et al., 2017); emerging metagenome sequence databases from the European Bioinformatics Institute (EBI) Metagenomics (Markowitz et al., 2014; Mitchell et al., 2018) and Metaclust (Steinegger and Söding, 2018) are playing a prominent role. For example, Wang et al. (2019) have demonstrated the applications of marine metagenomics for improved protein structure prediction. map_align uses the Integrated Microbial Genomes (IMG) database (Markowitz et al., 2014), containing around 4 million unique protein sequences, to reliably predict high-quality models for distant-homology Pfam families of unknown structures. Another recent method for generating protein multiple sequence alignments, DeepMSA (Zhang et al., 2020), combines whole-genome and metagenome sequence databases and reports improved threading performance, particularly for distant-homology proteins. Newer sequence databases are getting larger and diverse. For example, BFD (Steinegger et al., 2019), a recent sequence database, is one of the largest sequence databases containing 2 billion protein sequences from soil samples and 292 million sequences of marine samples. Another very recent sequence database MGnify (Mitchell et al., 2020) contains around 1 billion nonredundant protein sequences. As such, the availability of evolutionary information of distant-homology proteins is getting enriched, likely leading to improved prediction accuracy of inter-residue interaction maps and hence more accurate interaction map threading for distant-homology protein modeling.
While the use of interaction maps is the main driving force behind the improved threading performance, the optimal granularity and information content of the predicted interaction maps remain elusive. Existing works consider various distance bins (Zhu et al., 2018; Bhattacharya et al., 2020) and subsets of predicted interactions either based on top predicted pairs sorted based on their likelihood values or using arbitrary likelihood cutoffs (Bhattacharya and Bhattacharya, 2019; Zheng et al., 2019a). A robust mechanism for defining and selecting interacting residue pairs can be beneficial to existing threading methods. Another challenge is how to integrate heterogeneous sources of available information from multiple interaction map predictors and/or sequence databases in a singular framework for unified interaction map threading. Finally, the use of multiple templates (Cheng, 2008; Peng and Xu, 2011; Meier and Söding, 2015) and meta-approaches (Wu and Zhang, 2007; Zheng et al., 2019b) possibly coupled with model quality assessment methods (Ray et al., 2012; Uziela et al., 2016; Uziela et al., 2017; 3; Alapati and Bhattacharya, 2018; Karasikov et al., 2019; Baldassarre et al., 2020; Eismann et al., 2020; Shuvo et al., 2020) and potentially aided by structure refinement (Bhattacharya and Cheng, 2013a; Bhattacharya and Cheng, 2013b; Bhattacharya and Cheng, 2013c; Bhattacharya et al., 2016; Bhattacharya, 2019; Wang et al., 2020; Heo and Feig, 2020) can collectively improve the accuracy of distant-homology protein modeling.
Recent CASP experiments have witnessed dramatic recent advances by DeepMind’s AlphaFold series (Senior et al., 2019; Senior et al., 2020) in ab initio protein structure prediction, significantly outperforming the other groups. The success of AlphaFold series is primarily attributed to the successful application of deep neural networks for accurately predicting inter-residue spatial proximity information coupled with end-to-end training, significantly improving the accuracy of protein structure prediction (Pearce and Zhang, 2021). The integration of deep learning into various stages of protein modeling represents an exciting future direction that shall have a transformative impact on distant-homology protein modeling via interaction map threading, complementing and supplementing ab initio protein structure prediction methods developed by DeepMind.
All authors contributed in writing and revising the manuscript under the supervision of DB.
This work was partially supported by the National Science Foundation CAREER Award DBI-1942692 to DB, the National Science Foundation grant IIS-2030722 to DB, and the National Institute of General Medical Sciences Maximizing Investigators' Research Award (MIRA) R35GM138146 to DB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was made possible in part by Auburn University Early Career Development grant to DB.
Abeln, S., Heringa, J., and Anton Feenstra, K. (2017). Introduction to protein structure prediction. arXiv [arXiv:1712.00407]. Available at: https://arxiv.org/abs/1712.00407v1.
Adhikari, B. (2020). A Fully Open-Source Framework for Deep Learning Protein Real-Valued Distances. Scientific Rep. 10 (1), 13374. doi:10.1038/s41598-020-70181-0
Adhikari, B., and Cheng, J. (2018). CONFOLD2: Improved Contact-Driven Ab Initio Protein Structure Modeling. BMC Bioinformatics 19 (1), 22. doi:10.1186/s12859-018-2032-6
Adhikari, B., Bhattacharya, D., Cao, R., and Cheng, J. (2015). CONFOLD: Residue-Residue Contact-Guidedab Initioprotein Folding. Proteins 83 (8), 1436–1449. doi:10.1002/prot.24829
Adhikari, B., Hou, J., and Cheng, J. (2018). DNCON2: Improved Protein Contact Prediction Using Two-Level Deep Convolutional Neural Networks. Bioinformatics 34 (9), 1466–1472. doi:10.1093/bioinformatics/btx781
Alapati, R., and Bhattacharya, D. (2018). “ClustQ: Efficient Protein Decoy Clustering Using Superposition-free Weighted Internal Distance Comparisons,” In Proceedings Of the 2018 ACM International Conference On Bioinformatics, Computational Biology, and Health Informatics. New York, NY, USA: Association for Computing Machinery. BCB ’18. 10.1145/3233547.3233570
Altschul, S., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 25 (17), 3389–3402. doi:10.1093/nar/25.17.3389
Baker, D., and Sali, A. (2001). Protein Structure Prediction and Structural Genomics. Science 294 (5540), 93–96. doi:10.1126/science.1065659
Balakrishnan, S., Kamisetty, H., Carbonell, J. G., Lee, S.-I., and Langmead, C. J. (2011). Carbonell, Su-In Lee, and Christopher James LangmeadLearning Generative Models for Protein Fold Families. Proteins 79 (4), 1061–1078. doi:10.1002/prot.22934
Baldassarre, F., Hurtado, D. M., Elofsson, A., and Azizpour, H. (2020). GraphQA: Protein Model Quality Assessment Using Graph Convolutional Networks. Bioinformatics, 37 (3), 360-366. doi:10.1093/bioinformatics/btaa714
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The Protein Data Bank. Nucleic Acids Res. 28 (1), 235–242. doi:10.1093/nar/28.1.235
Bhattacharya, D., and Cheng, J. (2013a). 3Drefine: Consistent Protein Structure Refinement by Optimizing Hydrogen Bonding Network and Atomic-Level Energy Minimization. Proteins 81 (1), 119–131. doi:10.1002/prot.24167
Bhattacharya, D., and Cheng, J. (2013b). I3Drefine Software for Protein 3D Structure Refinement and its Assessment in CASP10. PLOS ONE 8 (7), e69648. 10.1371/journal.pone.0069648
Bhattacharya, D., and Cheng, J. (2013c). “Protein Structure Refinement by Iterative Fragment Exchange,” in Proceedings Of the International Conference On Bioinformatics, Computational Biology And Biomedical Informatics. New York, NY, USA: BCB’13 Association for Computing Machinery.
Bhattacharya, D., Nowotny, J., Cao, R., and Cheng, J. (2016). 3Drefine: An Interactive Web Server for Efficient Protein Structure Refinement. Nucleic Acids Res. 44 (W1), W406–W409. doi:10.1093/nar/gkw336
Bhattacharya, D. (2019). RefineD: Improved Protein Structure Refinement Using Machine Learning Based Restrained Relaxation. Bioinformatics 35 (18), 3320–3328. doi:10.1093/bioinformatics/btz101
Bhattacharya, S., and Bhattacharya, D. (2019). Does Inclusion of Residue‐residue Contact Information Boost Protein Threading? Proteins 87 (7), 596–606. doi:10.1002/prot.25684
Bhattacharya, S., and Bhattacharya, D. (2020). Evaluating the Significance of Contact Maps in Low-Homology Protein Modeling Using Contact-Assisted Threading. Scientific Rep. 10 (1), 2908. 10.1038/s41598-020-59834-2
Bhattacharya, S., Roche, R., and Bhattacharya, D. (2020). DisCovER: Distance- and Orientation-Based Covariational Threading for Weakly Homologous Proteins. BioRxiv. 10.1101/2020.01.31.923409
Bienkowska, J., and Lathrop, R. (2005). “Threading Algorithms,” in Encyclopedia Of Genetics, Genomics, Proteomics and Bioinformatics. Editors L. B. Jorde, P. F. R. Little, M. J. Dunn, and S. Subramaniam (American Cancer Society). doi:10.1002/047001153X.g409202
Bowie, J., Luthy, R., and Eisenberg, D. (1991). A Method to Identify Protein Sequences that Fold into a Known Three-Dimensional Structure. Science 253 (5016), 164–170. doi:10.1126/science.1853201
Brylinski, M., and Skolnick, J. (2010). Comparison of Structure-Based and Threading-Based Approaches to Protein Functional Annotation. Proteins 78 (1), 18–134. doi:10.1002/prot.22566
Buchan, D. W. A., and Jones, D. T. (2017). EigenTHREADER: Analogous Protein Fold Recognition by Efficient Contact Map Threading. Bioinformatics 33 (17), 2684–2690. doi:10.1093/bioinformatics/btx217
Chen, C.-C., Jeong, H., Qian, X., and Yoon, B.-J. (2019). TOPAS: Network-Based Structural Alignment of RNA Sequences. Bioinformatics 35 (17), 2941–2948. doi:10.1093/bioinformatics/btz001
Cheng, J. (2008). A Multi-Template Combination Algorithm for Protein Comparative Modeling. BMC Struct. Biol. 8 (1), 18. doi:10.1186/1472-6807-8-18
Cheng, J., and Baldi, P. (2006). A Machine Learning Information Retrieval Approach to Protein Fold Recognition. Bioinformatics 22 (12), 1456–1463. doi:10.1093/bioinformatics/btl102
Di Lena, P., Fariselli, P., Margara, L., Vassura, M., and Casadio, R. (2010). Fast Overlapping of Protein Contact Maps by Alignment of Eigenvectors. Bioinformatics 26 (18), 2250–2258. doi:10.1093/bioinformatics/btq402
Dill, K. A., and MacCallum, J. L. (2012). The Protein-Folding Problem, 50 Years on. Science 338 (6110), 1042–1046. doi:10.1126/science.1219021
Ding, W., and Gong, H. (2020). Predicting the Real‐Valued Inter‐Residue Distances for Proteins. Adv. Sci. 7 (19), 2001314. doi:10.1002/advs.202001314
Du, Z., Pan, S., Wu, Q., Peng, Z., and Yang, J. (2020). CATHER: A Novel Threading Algorithm with Predicted Contacts. Bioinformatics 36 (7), 2119–2125. doi:10.1093/bioinformatics/btz876
Eismann, S., Suriana, P., Jing, B., Raphael, J., Townshend, L., and Dror, Ron. O. (2020). Protein Model Quality Assessment Using Rotation-Equivariant, Hierarchical Neural Networks [arXiv. 2011.13557]. http://arxiv.org/abs/2011.13557.
Ginalski, K., Pas, Jakub., Wyrwicz, L. S., von Grotthuss, M., Bujnicki, J. M., and Rychlewski, L. (2003). ORFeus: Detection of Distant Homology Using Sequence Profiles and Predicted Secondary Structure. Nucleic Acids Res. 31 (13), 3804–3807. doi:10.1093/nar/gkg504
Gniewek, P., Kolinski, A., Kloczkowski, A., and Gront, D. (2014). BioShell-Threading: Versatile Monte Carlo Package for Protein 3D Threading. BMC Bioinformatics 15 (1), 22. doi:10.1186/1471-2105-15-22
Greener, J. G., Kandathil, S. M., and Jones, David. T. (2019). Deep Learning Extends De Novo Protein Modelling Coverage of Genomes Using Iteratively Predicted Structural Constraints. Nat. Commun. 10 (1), 1–13. doi:10.1038/s41467-019-11994-0
Haas, J., Gumienny, R., Barbato, A., Ackermann, F., Tauriello, G., Bertoni, M., et al. (2019). Introducing “best Single Template” Models as Reference Baseline for the Continuous Automated Model Evaluation (CAMEO). Proteins 87 (12), 1378–1387. doi:10.1002/prot.25815
Hanson, J., Paliwal, K., Litfin, T., Yang, Y., and Zhou, Y. (2018). Accurate Prediction of Protein Contact Maps by Coupling Residual Two-Dimensional Bidirectional Long Short-Term Memory with Convolutional Neural Networks. Bioinformatics 34 (23), 4039–4045. doi:10.1093/bioinformatics/bty481
He, B., Mortuza, S. M., Wang, Y., Shen, H.-B., and Zhang, Y. (2017). NeBcon: Protein Contact Map Prediction Using Neural Network Training Coupled with Naïve Bayes Classifiers. Bioinformatics 33 (15), 2296–2306. doi:10.1093/bioinformatics/btx164
Heo, L., and Feig, M. (2020). High‐accuracy Protein Structures by Combining Machine‐learning with Physics‐based Refinement. Proteins 88 (5), 637–642. doi:10.1002/prot.25847
Jaroszewski, L., Rychlewski, L., Li, Z., Li, W., and Godzik, A. (2005). FFAS03: a Server for Profile-Profile Sequence Alignments. Nucleic Acids Res. 33 (Suppl. l_2), W284–W288. doi:10.1093/nar/gki418
Jones, D. T. (1999). GenTHREADER: an Efficient and Reliable Protein Fold Recognition Method for Genomic Sequences. J. Mol. Biol. 287 (4), 797–815. doi:10.1006/jmbi.1999.2583
Jones, D. T., Singh, T., Kosciolek, T., and Tetchner, S. (2015). MetaPSICOV: Combining Coevolution Methods for Accurate Prediction of Contacts and Long Range Hydrogen Bonding in Proteins. Bioinformatics 31 (7), 999–1006. doi:10.1093/bioinformatics/btu791
Jones, D. T., Taylort, W. R., and Thornton, J. M. (1992). A New Approach to Protein Fold Recognition. Nature 358 (6381), 86–89. doi:10.1038/358086a0
Källberg, M., Wang, H., Wang, S., Peng, J., Wang, Z., Lu, H., et al. (2012). Template-Based Protein Structure Modeling Using the RaptorX Web Server. Nat. Protoc. 7 (8), 1511–1522. doi:10.1038/nprot.2012.085
Kamisetty, H., Ovchinnikov, S., and Baker, D. (2013). Assessing the Utility of Coevolution-Based Residue-Residue Contact Predictions in a Sequence- and Structure-Rich Era. Proc. Natl. Acad. Sci. 110 (39), 15674–15679. doi:10.1073/pnas.1314045110
Kandathil, S. M., Greener, J. G., and Jones, D. T. (2019). Prediction of Interresidue Contacts with DeepMetaPSICOV in CASP13. Proteins 87 (12), 1092–1099. doi:10.1002/prot.25779
Karasikov, M., Pagès, G., and Grudinin, S. (2019). Smooth Orientation-dependent Scoring Function for Coarse-Grained Protein Quality Assessment. Bioinformatics 35 (16), 2801–2808. doi:10.1093/bioinformatics/bty1037
Lee, S. Y., and Skolnick, J. (2010). TASSER_WT: A Protein Structure Prediction Algorithm with Accurate Predicted Contact Restraints for Difficult Protein Targets. Biophysical J. 99 (9), 3066–3075. doi:10.1016/j.bpj.2010.09.007
Li, J., and Xu, J. (2020). “Study of Real-Valued Distance Prediction for Protein Structure Prediction with Deep Learning” BioRxiv 10.1101/2020.11.26.400523
Li, Y., Hu, J., Zhang, C., Yu, D.-J., and Zhang, Y. (2019). ResPRE: High-Accuracy Protein Contact Prediction by Coupling Precision Matrix with Deep Residual Neural Networks. Bioinformatics 35 (22), 4647–4655. doi:10.1093/bioinformatics/btz291
Lobley, A., Sadowski, M. I., and Jones, D. T. (2009). PGenTHREADER and PDomTHREADER: New Methods for Improved Protein Fold Recognition and Superfamily Discrimination. Bioinformatics 25 (14), 1761–1767. doi:10.1093/bioinformatics/btp302
Ma, Jianzhu., Wang, Sheng., Wang, Zhiyong., and Xu, Jinbo. (2014). MRFalign: Protein Homology Detection through Alignment of Markov Random Fields. PLOS Comput. Biol. 10 (3), e1003500. doi:10.1371/journal.pcbi.1003500
Ma, J., Peng, J., Wang, S., and Xu, J. (2012). A Conditional Neural Fields Model for Protein Threading. Bioinformatics 28 (12), i59–i66. doi:10.1093/bioinformatics/bts213
Ma, J., Wang, S., Zhao, F., and Xu, J. (2013). Protein Threading Using Context-specific Alignment Potential. Bioinformatics 29 (13), i257–i265. doi:10.1093/bioinformatics/btt210
Markowitz, V. M., Chen, I.-M. A., Chu, K., Szeto, E., Palaniappan, K., Pillay, M., et al. (2014). IMG/M 4 Version of the Integrated Metagenome Comparative Analysis System. Nucl. Acids Res. 42 (D1), D568–D573. doi:10.1093/nar/gkt919
Marks, D. S., Robert, S., Hopf, Ts. A., Pagnani, A., Zecchina, R., and Sander, C. (2011). Protein 3D Structure Computed from Evolutionary Sequence Variation. PLOS ONE 6 (12), e28766. doi:10.1371/journal.pone.0028766
Marti, R. M. A., Madhusudhan, M. S., and Sali, A. (2004). Alignment of Protein Sequences by Their Profiles. Protein Sci. 13 (4), 1071–1087. doi:10.1110/ps.03379804
Meier, Armin., and Söding, Johannes. (2015). Automatic Prediction of Protein 3D Structures by Probabilistic Multi-Template Homology Modeling. PLOS Comput. Biol. 11 (10), e1004343. doi:10.1371/journal.pcbi.1004343
Mirdita, M., von den Driesch, L., Galiez, C., Martin, M. J., Söding, J., and Steinegger, M. (2017). Uniclust Databases of Clustered and Deeply Annotated Protein Sequences and Alignments. Nucleic Acids Res. 45 (D1), D170–D176. doi:10.1093/nar/gkw1081
Mitchell, A. L., Almeida, A., Beracochea, M., Boland, M., Burgin, J., Cochrane, G., et al. (2020). MGnify: The Microbiome Analysis Resource in 2020. Nucleic Acids Res. 48 (D1), D570–D578. doi:10.1093/nar/gkz1035
Mitchell, A. L., Scheremetjew, M., Denise, H., Potter, S., Tarkowska, A., Qureshi, M., et al. (2018). EBI Metagenomics in 2017: Enriching the Analysis of Microbial Communities, from Sequence Reads to Assemblies. Nucleic Acids Res. 46 (D1), D726–D735. doi:10.1093/nar/gkx967
Morcos, F., Pagnani, A., Lunt, B., Bertolino, A., Marks, D. S., Sander, C., et al. (2011). Marks, Chris Sander, Riccardo Zecchina, José N. Onuchic, Terence Hwa, and Martin Weigt.Direct-Coupling Analysis of Residue Coevolution Captures Native Contacts across Many Protein Families. Proc. Natl. Acad. Sci. 108 (49), E1293–E1301. doi:10.1073/pnas.1111471108
Ovchinnikov, S., Park, H., Varghese, N., Huang, P.-S., Pavlopoulos, G. A., Kim, D. E., et al. (2017). Protein Structure Determination Using Metagenome Sequence Data. Science 355 (6322), 294–298. doi:10.1126/science.aah4043
Pearce, R., and Zhang, Y. (2021). Deep Learning Techniques Have Significantly Impacted Protein Structure Prediction and Protein Design. Curr. Opin. Struct. Biol. 68 (June), 194–207. doi:10.1016/j.sbi.2021.01.007
Peng, J., and Xu, J. (2009). “Boosting Protein Threading Accuracy,” in In Research In Computational Molecular Biology, Editor S Batzoglou, 31–45. Lecture Notes in Computer Science (Berlin, Heidelberg: Springer Berlin Heidelberg). doi:10.1007/978-3-642-02008-7_3
Peng, J., and Xu, J. (2010). Low-Homology Protein Threading. Bioinformatics 26 (12), i294–300. 10.1093/bioinformatics/btq192
Peng, J., and Xu, J. (2011). A Multiple-Template Approach to Protein Threading. Proteins: Struct. Funct. Bioinformatics 79 (6), 1930–1939. doi:10.1002/prot.23016
Petrey, D., and Honig, B. (2005). Protein Structure Prediction: Inroads to Biology. Mol. Cel 20 (6), 811–819. doi:10.1016/j.molcel.2005.12.005
Ray, A., Lindahl, E., and Wallner, B. (2012). Improved Model Quality Assessment Using ProQ2. BMC Bioinformatics 13 (1), 224. doi:10.1186/1471-2105-13-224
Roche, Rahmatullah., Bhattacharya, Sutanu., and Bhattacharya, Debswapna. (2021). Hybridized Distance- and Contact-Based Hierarchical Structure Modeling for Folding Soluble and Membrane Proteins. PLOS Comput. Biol. 17 (2), e1008753. doi:10.1371/journal.pcbi.1008753
Rychlewski, L., Jaroszewski, L., Li, W., and Godzik, A. (2000). Comparison of Sequence Profiles. Strategies for Structural Predictions Using Sequence Information. Protein Sci. 9 (2), 232–241. doi:10.1110/ps.9.2.232
Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., et al. (2019). Protein Structure Prediction Using Multiple Deep Neural Networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins 87 (12), 1141–1148. doi:10.1002/prot.25834
Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., et al. (2020). Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 577 (7792), 706–710. doi:10.1038/s41586-019-1923-7
Shuvo, M. H., Bhattacharya, S., Bhattacharya, D., and Bhattacharya, Debswapna. (2020). QDeep: Distance-Based Protein Model Quality Estimation by Residue-Level Ensemble Error Classifications Using Stacked Deep Residual Neural Networks. Bioinformatics 36 (Suppl. ment_1), i285–i291. doi:10.1093/bioinformatics/btaa455
Skolnick, J., and Kihara, D. (2001). Defrosting the Frozen Approximation: PROSPECTOR? A New Approach to Threading. Proteins 42 (3), 319–331. doi:10.1002/1097-0134(20010215)42:3<319::aid-prot30>3.0.co;2-a
Söding, J. (2017). Big-Data Approaches to Protein Structure Prediction. Science 355 (6322), 248–249. 10.1126/science.aal4512
Söding, J. (2005). Protein Homology Detection by HMM-HMM Comparison. Bioinformatics 21 (7), 951–960. doi:10.1093/bioinformatics/bti125
Steinegger, M., and Söding, J. (2018). Clustering Huge Protein Sequence Sets in Linear Time. Nat. Commun. 9 (1), 2542. doi:10.1038/s41467-018-04964-5
Steinegger, M., Mirdita, M., and Söding, J. (2019). Protein-Level Assembly Increases Protein Sequence Recovery from Metagenomic Samples Manyfold. Nat. Methods 16 (7), 603–606. doi:10.1038/s41592-019-0437-
Suzek, B. E., Wang, Y., Huang, H., McGarvey, P. B., and Wu, C. H., and the UniProt Consortium (2015). UniRef Clusters: A Comprehensive and Scalable Alternative for Improving Sequence Similarity Searches. Bioinformatics 31 (6), 926–932. doi:10.1093/bioinformatics/btu739
Taylor, William. R. (1999). Protein Structure Comparison Using Iterated Double Dynamic Programming. Protein Sci. 8 (3), 654–665. doi:10.1110/ps.8.3.654
The UniProt Consortium (2019). UniProt: A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 47 (D1), D506–D515. doi:10.1093/nar/gky1049
Uziela, K., Menéndez Hurtado, D., Shu, N., Wallner, B., and Elofsson, A. (2017). ProQ3D: Improved Model Quality Assessments Using Deep Learning. Bioinformatics 33 (10), 1578–1580. doi:10.1093/bioinformatics/btw819
Uziela, Karolis., Shu, Nanjiang., Wallner, Björn., and Elofsson, Arne. (2016). ProQ3: Improved Model Quality Assessments Using Rosetta Energy Terms. Scientific Rep. 6 (1), 33509. doi:10.1038/srep33509
Wang, D., Geng, L., Zhao, Y.-J., Yang, Y., Huang, Y., Zhang, Y., et al. (2020). Artificial Intelligence-Based Multi-Objective Optimization Protocol for Protein Structure Refinement. Bioinformatics 36 (2), 437–448. doi:10.1093/bioinformatics/btz544
Wang, Sheng., Sun, Siqi., Li, Zhen., Zhang, Renyu., and Xu, Jinbo. (2017). Accurate De Novo Prediction of Protein Contact Map by Ultra-deep Learning Model. PLOS Comput. Biol. 13 (1), e1005324. doi:10.1371/journal.pcbi.1005324
Wang, S., Li, W., Zhang, R., Liu, S., and Xu, J. (2016). CoinFold: A Web Server for Protein Contact Prediction and Contact-Assisted Protein Folding. Nucleic Acids Res. 44 (W1), W361–W366. doi:10.1093/nar/gkw307
Wang, Y., Shi, Q., Yang, P., Zhang, C., Mortuza, S. M., Xue, Z., et al. (2019). Fueling Ab Initio Folding with Marine Metagenomics Enables Structure and Function Predictions of New Protein Families. Genome Biol. 20 (1), 229. doi:10.1186/s13059-019-1823-z
Wu, Q., Peng, Z., Anishchenko, I., Cong, Q., Baker, D., and Yang, J. (2020). Protein Contact Prediction Using Metagenome Sequence Data and Residual Neural Networks. Bioinformatics 36 (1), 41–48. doi:10.1093/bioinformatics/btz477
Wu, S., and Zhang, Y. (2007). LOMETS: A Local Meta-Threading-Server for Protein Structure Prediction. Nucleic Acids Res. 35 (10), 3375–3382. doi:10.1093/nar/gkm251
Wu, S., and Zhang, Y. (2008). “MUSTER: Improving Protein Sequence Profile–Profile Alignments by Using Multiple Sources of Structure Information. Proteins: Struct. Funct. Bioinformatics 72 (2), 547–556. doi:10.1002/prot.21945
Wu, S., and Zhang, Y. (2010). Recognizing Protein Substructure Similarity Using Segmental Threading. Structure 18 (7), 858–867. doi:10.1016/j.str.2010.04.007
Wu, Tianqi., Guo, Zhiye., Hou, Jie., and Cheng, Jianlin. (2021). DeepDist: Real-Value Inter-residue Distance Prediction with Deep Residual Convolutional Network. BMC Bioinformatics 22 (1), 30. doi:10.1186/s12859-021-03960-9
Xu, J. (2019). Distance-Based Protein Folding Powered by Deep Learning. Proc. Natl. Acad. Sci. USA 116 (34), 16856–16865. doi:10.1073/pnas.1821309116
Xu, J., Li, M., Kim, D., and Xu, Y. (2003). Raptor: Optimal Protein Threading by Linear Programming. J. Bioinform. Comput. Biol. 01 (01), 95–117. doi:10.1142/s0219720003000186
Xu, J., and Wang, S. (2019). Analysis of Distance‐based Protein Structure Prediction by Deep Learning in CASP13. Proteins 87 (12), 1069–1081. doi:10.1002/prot.25810
Xu, Y., and Xu, D. (2000). Protein Threading Using PROSPECT: Design and Evaluation. Proteins 40 (3), 343–354. doi:10.1002/1097-0134(20000815)40:3<343::aid-prot10>3.0.co;2-s
Yang, J., Anishchenko, I., Park, H., Peng, Z., Ovchinnikov, S., and Baker, D. (2020). Improved Protein Structure Prediction Using Predicted Interresidue Orientations. Proc. Natl. Acad. Sci. USA 117 (3), 1496–1503. doi:10.1073/pnas.1914677117
Yang, Y., Faraggi, E., Zhao, H., and Zhou, Y. (2011). Improving Protein Fold Recognition and Template-Based Modeling by Employing Probabilistic-Based Matching between Predicted One-Dimensional Structural Properties of Query and Corresponding Native Properties of Templates. Bioinformatics 27 (15), 2076–2082. doi:10.1093/bioinformatics/btr350
Zhang, C., Zheng, W., Mortuza, S. M., Li, Y., and Zhang, Y. (2020). DeepMSA: Constructing Deep Multiple Sequence Alignment to Improve Contact Prediction and Fold-Recognition for Distant-Homology Proteins. Bioinformatics 36 (7), 2105–2112. doi:10.1093/bioinformatics/btz863
Zhang, Hg., and Shen, Y. (2020). Template-Based Prediction of Protein Structure with Deep Learning. BMC Genomics 21 (11), 878. doi:10.1186/s12864-020-07249-8
Zheng, W., Wuyun, Q., Yang, Li., Mortuza, S. M., Zhang, C., Pearce, R., et al. (2019a). Detecting Distant-Homology Protein Structures by Aligning Deep Neural-Network Based Contact Maps. PLOS Comput. Biol. 15 (10), e1007411. doi:10.1371/journal.pcbi.1007411
Zheng, W., Zhang, C., Wuyun, Q., Pearce, R., Li, Y., and Zhang, Y. (2019b). LOMETS2: Improved Meta-Threading Server for Fold-Recognition and Structure-Based Function Annotation for Distant-Homology Proteins. Nucleic Acids Res. 47 (W1), W429–W436. doi:10.1093/nar/gkz384
Zhou, H., and Zhou, Y. (2005). Fold Recognition by Combining Sequence Profiles Derived from Evolution and from Depth-dependent Structural Alignment of Fragments. Proteins 58 (2), 321–328. doi:10.1002/prot.20308
Keywords: protein homology, inter-residue interaction map, protein threading, homology modeling, protein structure prediction
Citation: Bhattacharya S, Roche R, Shuvo MH and Bhattacharya D (2021) Recent Advances in Protein Homology Detection Propelled by Inter-Residue Interaction Map Threading. Front. Mol. Biosci. 8:643752. doi: 10.3389/fmolb.2021.643752
Received: 18 December 2020; Accepted: 21 April 2021;
Published: 11 May 2021.
Edited by:
Paolo Marcatili, Technical University of Denmark, DenmarkReviewed by:
Dimitrios P. Vlachakis, Agricultural University of Athens, GreeceCopyright © 2021 Bhattacharya, Roche, Shuvo and Bhattacharya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Debswapna Bhattacharya, YmhhdHRhY2hhcnlhZEBhdWJ1cm4uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.