 A. Cassedy
A. Cassedy A. Parle-McDermott
A. Parle-McDermott R. O’Kennedy1,2,3*
R. O’Kennedy1,2,3*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci. , 20 April 2021
Sec. Molecular Diagnostics and Therapeutics
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.637559
Viruses are ubiquitous in the environment. While many impart no deleterious effects on their hosts, several are major pathogens. This risk of pathogenicity, alongside the fact that many viruses can rapidly mutate highlights the need for suitable, rapid diagnostic measures. This review provides a critical analysis of widely used methods and examines their advantages and limitations. Currently, nucleic-acid detection and immunoassay methods are among the most popular means for quickly identifying viral infection directly from source. Nucleic acid-based detection generally offers high sensitivity, but can be time-consuming, costly, and require trained staff. The use of isothermal-based amplification systems for detection could aid in the reduction of results turnaround and equipment-associated costs, making them appealing for point-of-use applications, or when high volume/fast turnaround testing is required. Alternatively, immunoassays offer robustness and reduced costs. Furthermore, some immunoassay formats, such as those using lateral-flow technology, can generate results very rapidly. However, immunoassays typically cannot achieve comparable sensitivity to nucleic acid-based detection methods. Alongside these methods, the application of next-generation sequencing can provide highly specific results. In addition, the ability to sequence large numbers of viral genomes would provide researchers with enhanced information and assist in tracing infections.
Viruses are ubiquitous entities which rely on host organisms to replicate. They exist in all habitats and are capable of infecting a wide range of life forms, from bacteria to plants and animals. Structurally, at a minimum, viruses consist of two core components. These are a nucleic acid genome, comprised of either double- or single-stranded DNA or RNA, and a capsid. The capsid is a highly symmetrical structure, formed by multiple copies of a small number of proteins and encoded by the viral genome (Domingo, 2015). The capsid packages the viral nucleic acid, protecting it. However, the capsid is also known to be multifunctional, with roles in cell-entry, genome uncoating or intracellular trafficking (Mateu, 2013; Freire et al., 2015). In addition to the capsid protein, some viruses will have an added layer of protection known as an envelope. This is generally comprised of a lipid or glycoprotein coating. Lipid envelopes are derived from the cell membrane of the infected host, whereas glycoprotein envelopes are typically coded for by the viral genome. The viral genome also encodes proteins which facilitate replication and assembly of the virus particles. An assembled, infectious, virus particle is termed a virion (Banerjee and Mukhopadhyay, 2016). While structurally, most viruses present similar characteristics, such as the symmetric capsid proteins, genetically, they are highly diverse.
This genetic diversity gives rise to a lack of understanding or an unawareness of many of the viruses present in our environment. While viruses dominate most habitats, the virome is far from complete. In conjunction with this, the interaction between host and virus remains unclear in many cases (Paez-Espino et al., 2016). While it is known that infection by some viruses imparts deleterious effects on their host, in other cases, the relationship between the two may be mutually beneficial (Pradeu, 2016; Jagdale and Joshi, 2018). It is feasible that many of these virus-host interactions, where the host remains unharmed, may persist unidentified, as infection may not present clearly recognisable symptoms. Linked to these unknown host-virus interactions is limited knowledge of the various host ranges of viruses. The host range is the number of species known to be used by a pathogen, however, defining the hosts which a virus may, or may not, infect is not easily feasible (McLeish et al., 2019). These unknown host ranges often play a role in emergence. In conjunction with this, other drivers of viral emergence are climate change, increased global transport, centralized agriculture and a high density of crops, animals and humans. These factors create an environment where the spread of viruses to hosts, which normally would not fall within the host range, is more easily facilitated. The flexibility and diversity of the viral genome itself is also a major factor driving diversification of viral host-ranges and environmental tolerances. Some viruses have the capacity to mutate at extremely rapid rates. For example, RNA viruses can have a mutation rate up to a million times higher than their hosts and can incorporate mutated nucleotides at a rate of 10–6–10–4 substitutions per nucleotide per cell infection (s/n/c) (Sanjuán and Domingo-Calap, 2016; Duffy, 2018). DNA viruses typically mutate at a slower rate than RNA viruses, with an estimated s/n/c of 10–8–10–6 (Sanjuán and Domingo-Calap, 2016). This ability of viruses to rapidly mutate and transfer between hosts makes the development of dynamic methods to detect current and emergent viral strains extremely pertinent.
Viral diagnostics are generally organised into two primary categories i.e., indirect and direct detection. This review aims to critically assess the currently employed methods for viral detection and examine their suitability, with due note to scenarios where assays may need to be rapidly developed, such as during epidemics or pandemics.
Indirect detection methods involve the propagation of virus particles via their introduction to a suitable host cell line. This is termed virus isolation. Virus isolation remains a relevant and important method, particularly as it offers a means to propagate existing and emergent viruses for further analysis and characterisation. While traditional virus cell culture is considered a relatively slow diagnostic method, sometimes taking weeks for the virus to propagate, modernised methods have allowed the turnaround to be greatly improved (Leland and Ginocchio, 2007).
Centrifuge-enhanced shell-vial techniques or well plates, termed cluster plates, facilitate rapid adsorption of the virus into the host cell line by applying low level centrifugation, greatly reducing the length of time required for inoculation of the cell line. In shell vial and cluster plate techniques, the virus infects cells growing in a monolayer, usually on a glass slide, which also facilitates easy analysis of the infected cells under a microscope (Gleaves et al., 1984; Hematian et al., 2016). Another advance in viral cell culture was the development of new cell lines. These include co-cultured cell lines, lines consisting of more than one cell type, and the use of transgenic cells. Co-cultured cells are useful for scenarios where the nature of the virus may be unknown, hence the optimal host is also unknown. Co-cultured lines can save time for diagnostics, as fewer cells lines must be maintained, infected and analysed (George et al., 2002; LaSala et al., 2007). Transgenic cell lines also aid in improved virus diagnosis. Genetically engineered cell lines are modified to contain genes which act as a reporter upon infection with a specific virus. Ideally, this reporter gene will only activate in the presence of virus-specific proteins (Storch, 2000). When using transgenic lines, the virion does not need to be fully assembled for diagnosis to be achieved, as the production of a virus-specific protein is enough to achieve expression of the reporter, thus reducing turnaround time for results (Olivo, 1996). Both co-cultured and transgenic cell lines can be used in conjunction with shell-vial techniques to further reduce time to diagnosis.
Once growing in cell-culture, there are two categories of methods by which viruses are diagnosed. They consist of those employing observation of the cytopathic effects (CPE) and those which employ molecular methods, known as pre-CPE. Infection of cells with cytopathic viruses causes damage and subsequent morphological changes to the cells which they infect, and these changes are termed CPE. Inspection of the cell cultures under light microscopy is used to determine the presence, or absence of CPE. It can take between 48 h and several weeks for the development of CPE to become apparent. Therefore, this method cannot necessarily provide a rapid turnaround time to results (Fenner et al., 1974). Furthermore, the presumptive nature of CPE-diagnosis means that it may be difficult to discern with confidence which virus is causing the damage. Another challenge is that this method is only applicable to CPE-inducing viruses, and therefore it cannot be considered a universal method for virus detection. To overcome these challenges, molecular methods can be used in conjunction with virus cell culture to provide more accurate and rapid diagnoses. This approach is termed ‘pre-CPE’ diagnosis.
Pre-CPE methods have the capacity to detect the virus post cell-entry, but before the cells present CPE, meaning they generally provide faster results. Coverslips obtained from shell vial cultures can be used directly for pre-CPE methods. Detection is usually facilitated by staining of the coverslip with an agent which detects a virus-associated protein with high sensitivity and specificity, for example an antibody (Leland and Ginocchio, 2007). An enzymatically- or fluorescently-labelled antibody, is applied to the monolayer of cells on the coverslip. The slip is inspected for a positive signal from the labelled antibody, which in turn indicates a positive confirmation of a certain virus. In general, the use of pre-CPE methods produces comparable results to traditional cell culture methods, with improved turnaround times (Peterson et al., 1988; Rabalais et al., 1992; Lipson et al., 2001). Alongside more rapid turnaround times, pre-CPE detection has the added benefit of employing virus-specific antibodies, meaning that a particular virus, or strain of virus, can be positively confirmed. A schematic of CPE- and pre-CPE-based diagnostic methods is shown in Figure 1.
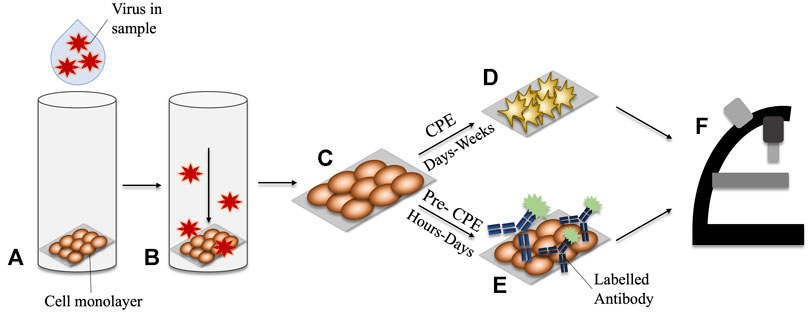
FIGURE 1. Comparison of CPE vs Pre-CPE detection in shell-vial culture. (A) Virus-containing sample is applied to the shell vial. (B) A centrifugal force is applied, encouraging inoculation of the virus into the cell monolayer. (C) The monolayer cells growing on a glass slide may be removed for interrogation. (D) CPE may take days to weeks to develop in the culturing cells. (E) Pre-CPE detection employs labelled antibodies to identify virus-specific markers; these can be detected within hours-days. (F) Microscopy can be used to inspect for morphological changes in CPE-diagnosis, or for signals arising from the presence of a labelled-antibody for pre-CPE detection.
Although pre-CPE diagnostics prove useful in reducing turnaround times when compared to traditional cell culture, the time to diagnosis can still take days in some cases. In situations where rapid diagnosis is a key factor, for example in cases of extremely damaging or virulent viruses, a turnaround of days is insufficient. While cell culture methods are important for the isolation of viruses, they do not transition well to the current demands for rapid testing. Alongside long result turnaround times, virus culture requires sampling from the viral source and removal to sterile and secure laboratories, where the virus can be grown. In the current climate, where point-of-need/point-of-care testing is highly desired, the steps required for virus culture limit it as a tool for rapid diagnostics. Hence, there has been a shift to molecular techniques which facilitate direct sampling from the source, rapid results turnaround, and amenability for quick adaption to emerging viruses. Techniques which involve the detection of the virus from the source, without any need for virus propagation, fall under the broad category of direct detection.
Direct detection methods negate the need for virus propagation. Instead, the virus is detected directly from the suspect source. Direct detection methods usually involve sophisticated techniques, including nucleic acid and immunological detection.
A key element of nucleic acid-based detection is polymerase chain reaction (PCR) which utilises multiple stepwise temperature cycles, and a polymerase, to amplify DNA strands (Mullis et al., 1986). The standard polymerase used in PCR can only synthesise from a DNA template, thus RNA amplification requires the use of an enzyme with reverse transcription activity (Bustin, 2000). PCR and reverse transcription-PCR are widely applied methods for the detection of both DNA and RNA viruses, respectively (Metcalf, 1995; Pring-Åkerblom et al., 1997; Burkhalter and Savage, 2017; Wadhwa et al., 2017; Liu et al., 2018; Lin et al., 2020). In standard PCR, the DNA products are generally visualised by gel electrophoresis, to check for the presence of expected DNA bands. Using this method, only semi-quantitative results can be achieved, facilitated by running known DNA standards alongside the unknown sample. The method can also fail when samples contain low concentrations of DNA. While high-sensitivity detection can be achieved in some cases, sensitivity can vary greatly depending on the type of DNA stain used, and the type of system used to visualise the gel (Motohashi, 2019). Gel electrophoresis can be used to analyse the results of many alternative nucleic acid amplification methods, such as those based in isothermal amplification, however, the issues remain largely the same. Given that gel electrophoresis can be considered a standard method of analysing amplified DNA products, the following sections will focus more on some of the alternative detection strategies that can be used in post-amplification nucleic acid detection.
Real time quantitative PCR (real time qPCR) measures the production of the target amplicon throughout the reaction. This is facilitated by DNA intercalating dyes, such as SYBR® green, or fluorescently labelled probes. DNA dyes will bind to all double-stranded DNA. To use these dyes, the primers must be highly optimised and produce no non-specific amplicons as these non-specific products will also produce signal, skewing quantification. Fluorescently labelled probes are a more target-specific alternative. Many types of probe are employed in qPCR, however commonplace probes include those which must bind to a specific region on the target DNA in order for fluorescence to be achieved, for example, hydrolysis or hybridization probes (Navarro et al., 2015). An illustration of the use of fluorescent dyes and probes is depicted in Figure 2. The level of fluorescence in the PCR sample is measured and is directly proportional to the initial concentration of the target in the sample, thus, allowing rapid quantification.
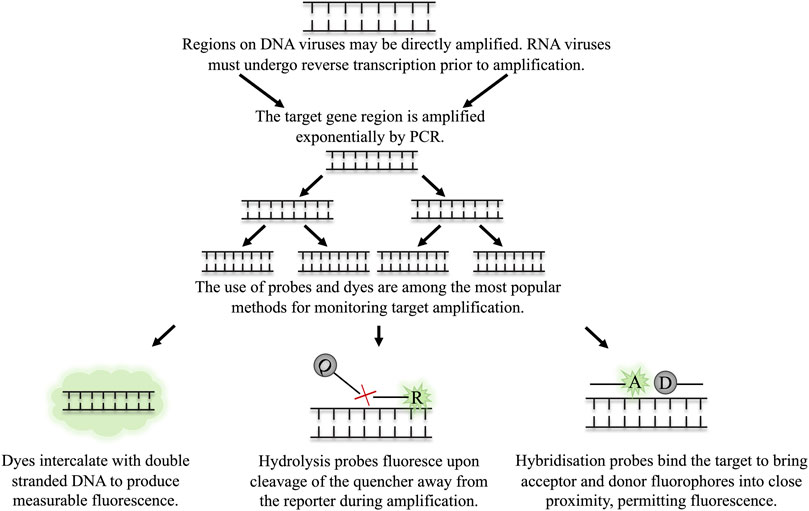
FIGURE 2. The detection of target amplicon in qPCR through dyes and probes. The target region on the virus is amplified by multiple rounds of exponential amplification in PCR. During amplification, various methods are employed to monitor the real time production of target DNA. (A) Dyes intercalate indiscriminately with double stranded DNA, causing an increase in fluorescence as the level of double stranded DNA in the sample increases. Hydrolysis and hybridisation probes require binding to a specific sequence on the target amplicon to permit fluorescence. Fluorescence is achieved either by cleavage of the probe when employing hydrolysis probes (B), or through binding of the probes to the target region in close proximity to one another, as is the case for hybridisation probes (C).
With respect to the development of rapid virus diagnostic assays, real-time qPCR has both benefits and disadvantages. One advantage is the promise of high specificity when using probes such as hydrolysis or hybridization probes, as both the primers and the probe must bind the target sequence to achieve signal. When time-pressure is an issue, an assay which produces highly specific, quantitative results over the course of a few hours is very desirable. While qPCR does offer a relatively quick results turnaround in terms of the assay itself, sample preparation, which is generally required for qPCR, can be a delaying factor. While there are several commercially available kits which facilitate the rapid isolation of DNA or RNA, they come with the disadvantage of added cost (Clark et al., 2016). This, in conjunction with the initial cost of the qPCR equipment itself, could make detection methods like qPCR inaccessible to developing regions. Another desirable trait of a rapid assay is its capacity for high-throughput testing. Given that qPCR is typically used in a 96-well assay format, the transition to high-throughput is feasible (Arya et al., 2005; Eigner et al., 2019). This is of particular interest for situations where high volume testing is vital, for example during pandemics or epidemics, or for industrial or agricultural applications where numerous samples may need to be tested simultaneously. The capacity for qPCR to be performed in multiplexed reactions complements this high-throughput feasibility. The employment of multiple pairs of specific primers and suitable probes, targeting different viruses which may be found in the same sample, facilitates testing for panels of viruses. This, in combination with the multi-well layout of qPCR assays and established high-throughput systems, provides a powerful method to test multiple samples for the presence of several viruses simultaneously (Hwang et al., 2018; Sánchez-Ponce et al., 2018; Eigner et al., 2019; Ou et al., 2020). Although, as aforementioned, the necessary sample preparation may, again, be a limiting factor to the throughput capacity of qPCR assays. This could be overcome by the continued development of novel nucleic extraction approaches such as the magnetic bead-based assays described by Xin and Chen (2012) and Clark et al. (2016). These facilitate high-throughput sample preparation in a short space of time (a few hours) at greatly reduced costs when compared to commercially available high-throughput extraction systems (Xin and Chen, 2012; Clark et al., 2016). Alternatively, sampling strategies such as composite sampling could be employed, whereby a number of samples are mixed and tested as one. The application of composite sampling can achieve higher sensitivity when compared to single sample tests (Jones et al., 2020). This would facilitate multiple sample assessment within a single assay, thereby reducing the level of sample processing required for each run. Furthermore, if one of the combined samples presents a positive result, then further analysis can be performed on single specimens to identify infected individuals. Other drawbacks to qPCR include the high cost of equipment and reagents, the need for strictly controlled temperature cycling, and the necessity for trained staff to run testing. Alongside qPCR, other nucleic acid detection methods are proposed, including those which permit isothermal amplification of DNA and RNA.
Loop-mediation isothermal amplification (LAMP), developed by Notomi et al. (2000), relies on the use of multiple primers, at minimum four, to initiate the polymerase-driven extension of the gene sequence. The amplification is facilitated by the formation of stem-loop structures created by the primers. The sequence is then amplified by a strand-displacing polymerase. This is repeated, eventually generating copious amounts of double-stranded DNA product (Notomi et al., 2000; Notomi et al., 2015). The suggested reaction temperature for amplification is 65°C, however, studies demonstrated successful amplification at temperature ranges between 57 and 67°C (Tomita et al., 2008; Francois et al., 2011). The reaction time typically ranges from 30 to 60 mins, although with optimisation of the reaction conditions, such as reaction temperature and buffer composition, it was shown to be possible to reduce the time to result to 10 mins (Tomita et al., 2008; Estrela et al., 2019). This time can be reduced by the introduction of another set of primers, termed loop primers (Nagamine et al., 2002). Additionally, swarm and stem primers can be introduced to the reaction to further reduce reaction duration (Gandelman et al., 2011; Martineau et al., 2017). Where the target is RNA-based, a reverse transcriptase can be incorporated directly into the LAMP reaction, termed RT-LAMP (Wong et al., 2018). The singular assay detection of both DNA and RNA is appealing as it reduces the “hands-on” time for target amplification, with particular note to RNA-based viruses.
The resulting amplicons from LAMP are detected through a number of means. Intercalating dyes, such as SYBR green can be added either to the end-point reaction, allowing for a rapid qualitative result, or added prior to the reaction to measure the real-time increase in fluorescence (Zhang X. et al., 2014). Some dyes may have inhibitory effects on the LAMP reaction, hence, a range of dyes should be assessed in order to identify the most suitable for each assay (Quyen et al., 2019). Fluorescently labelled probes can also be employed. However, probes developed for qPCR may be unsuitable for LAMP due to the strand displacing activity of the polymerase. To overcome this, alternative probe-based methods are proposed, including quenching of unincorporated amplification signal reporters (QUASR), detection of amplification by release of quenching (DARQ) or mediator displacement probes (Tanner et al., 2012; Ball et al., 2016; Becherer et al., 2018). Gold nanoparticles (AuNPs)-labelled with DNA, complementary to a sequence on the target, are proposed as another option for amplicon detection (Arunrut et al., 2016). Currently, there is no single optimised way to incorporate probes into the LAMP reaction and further work is required to standardise the probe-based target detection. The turbidity of the reaction is also used for both qualitative and quantitative analysis. LAMP produces large amounts of precipitated magnesium pyrophosphate as a by-product. Typically, the presence of precipitate is visible to the naked eye after a successful reaction, allowing a rapid qualitative answer (Zhang et al., 2014). Turbidity within the assay can also be measured real-time via the employment of a turbidimeter, which facilitates quantitative analysis of target DNA by comparing an unknown sample to a standard curve (Parida et al., 2008; Wang et al., 2020).
With respect to virology, LAMP offers a highly sensitive and rapid alternative to classic PCR, or qPCR. Amplification by this method does not require sophisticated apparatus as the reaction is maintained at a single temperature, typically around 65°C (Tomita et al., 2008). This temperature, however, does mean that some heating apparatus is required. This may be overcome by developments such as the employment of phosphorothioated primers, and additives such as single-stranded binding proteins and urea, which permit the performance of the LAMP assay at 40°C, albeit with a longer reaction time than was observed with traditional LAMP (Cai et al., 2018). LAMP was also shown to be robust when detecting directly from sources such as blood, urine, stool various media components and insects, meaning there was no need for additional DNA/RNA extraction equipment (Kaneko et al., 2007; Francois et al., 2011; Silva et al., 2019). As LAMP employs multiple primers the method is very specific. In order for the assay to function, the primers must bind to six regions on the sequence. However, the primers must be designed to have specific distances between their target regions, while only amplifying a product of maximum 300 bp. This can make the design of LAMP primers complicated, even with the available software to aid in their design (Parida et al., 2008; PrimerExplorer, 2020). The challenges faced with primer design may be further exacerbated by the variation in viral gene sequences across different strains, which may make the identification of conserved and suitable primer-binding sites difficult (Parida et al., 2008; Sanjuán and Domingo-Calap, 2016; Duffy, 2018). An illustration of potential issues faced with primer-binding and sequence variation is depicted in Figure 3. When validating LAMP assays, care should be taken to ensure that all strains of a virus are amplified equally with the primer set, and that no false negatives are observed.
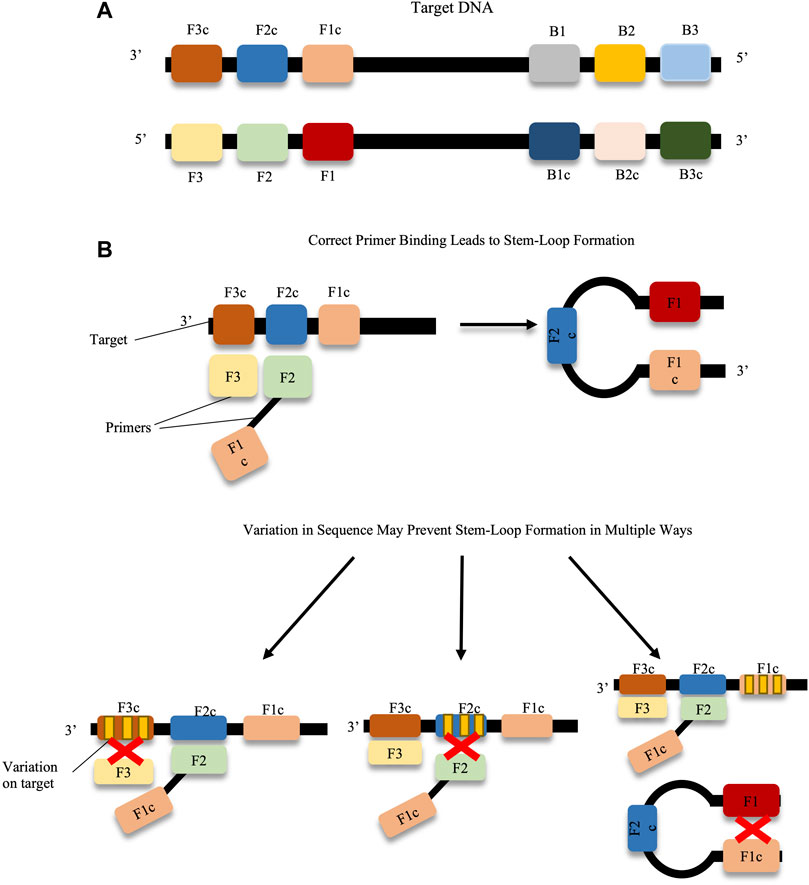
FIGURE 3. Prevention of stem-loop formation caused by target sequence variations. (A) F1-F3, F1c-F3c, B1-B3 and B1c-B3c on the target DNA represent the binding regions for the LAMP primers, e.g. primers F3 and F2-F1c as shown in the diagram. If each of the primers matches suitably with the target DNA, stem loop formation proceeds. (B) Variation across any primer-binding sites (represented by yellow bands on target regions) can prevent stem-loop formation, and hence amplification. Primer design can be challenging when trying to avoid these variable regions.
Another challenge faced by LAMP is carryover contamination. While contamination poses a threat to many amplification methods, LAMP is particular prone to the effects due to its high sensitivity. Contamination is usually caused by products from previous experiments carrying over via the environment, researchers clothing, or lab equipment. The contaminant products and are then used as templates in new reactions, causing false positives in some cases (Dhama et al., 2014; Hsieh et al., 2014). This can be mitigated by avoiding results analysis which requires the opening of reactions tubes, or by using alternative methods which incorporate dUTP, in lieu of dTTP, in the LAMP reaction. The dUTP is cleaved by uracil-DNA glycosylase prior to the next LAMP experiment, thus preventing previous amplicons from acting as templates in subsequent reactions (He and Xu, 2011; Hsieh, 2014). In a situation where the virus being tested is harm-causing, the likelihood of the outcome being a false-positive should be as low as possible to avoid the administering the wrong treatment or negative outcomes for patients (Borst et al., 2004). Further to this, in a situation of rapid viral spread such as pandemic or epidemic, a high false positive rate may put pressure on healthcare systems if tests have to be performed multiple times to ensure accuracy.
RPA was proposed by Piepenburg et al., in 2006, and since then has gained traction as a promising method for nucleic acid detection (Piepenburg et al., 2006). RPA introduces two proteins, a recombinase and a single-stranded DNA binding protein (SSBP), in the presence of other reagents such as a strand-displacing DNA polymerase and 30–35 bp long, target-specific oligonucleotides pairs (primers). RPA is initiated by the formation of a complex between the oligonucleotides and the recombinase. The complex scans double stranded DNA for sites complementary to the primers. Once target sites are identified, the complex invades the double stranded DNA, the displaced strand is stabilised by SSBPs and the recombinase dissociates. Exponential amplification is achieved through repeated cycles of this process. The RPA reaction has an extremely flexible reaction temperature, typically between 22 and 45°C. Further to this, the reaction is usually complete within 20 min (Piepenburg et al., 2006; Daher et al., 2016; Lobato and O’Sullivan, 2018). Amplification of RNA can be achieved via the addition of a reverse transcriptase directly to the RPA mixture, making the reaction time-friendly for RNA-detection (Euler et al., 2012; Daher et al., 2016).
Upon development of the RPA assay and subsequent amplification of the target, there are multiple ways in which the resulting amplicon can be detected. For quantitative, real-time results, fluorescent probes can be used, similar to qPCR. These probes aid in increasing the specificity of the technique, as only probes which bind to a complementary DNA sequence will fluoresce. Currently, RPA probes, termed exo and fpg, both require the cleavage to separate the reporter fluorophore from the quencher (Euler et al., 2013; Yang et al., 2015). Probes which facilitate detection of amplicons through lateral flow technology are also available. Detection through this means is usually in sandwich assay format and is termed nucleic acid lateral flow immunoassay (NAFLIA). In this process, throughout amplification, the amplicon is labelled with two different “antigenic” labels. These labels are then captured by label-specific antibodies on the lateral flow strip (Powell et al., 2018). The fact that RPA offers both quantitative real time detection via the use of fluorescent probes, and qualitative, or semi-quantitative, detection via agarose gel detection or NALFIA makes it appealing to the field of virology.
RPA has a quick turnaround time and provides high flexibility in terms of reaction temperature. When envisioning assays for viral detection, as with all nucleic acid amplification methods, care should be taken with primer design. However, this may be particularly relevant for RPA due to the fact that it operates at a lower temperature, which can reduce specificity (Wu et al., 1991; Luo et al., 2019). A study performed to investigate the effect of base pair mismatches between primer and target sequence found that while the RPA reaction was tolerant to some mismatches, the tolerance changed depending on the type of mismatch. For example, mismatches between bases near the 3′ end of forward and reverse primers had a larger negative impact than those elsewhere on the primers. They also showed that RPA efficiency was further reduced in situations where the mismatched sequences contained either very high or very low GC content (Daher et al., 2015). The implications of this for virus detection is that caution must be taken when choosing a target region in the viral DNA or RNA. It is established that the mutation rates and variation in viral gene sequence can be high (Sanjuán and Domingo-Calap, 2016; Duffy, 2018). While variations across target sequences can be useful in differentiating between strains, it poses a challenge if the overall goal is to detect all strains of a virus indiscriminately. For RPA it is important to ensure that there is no amplification bias between strains. Further to this, given that RPA exhibits tolerance toward mismatches, extensive analysis into any potential cross-reactive non-target sequences must be performed. Hence, when choosing a target sequence to detect, the optimal choice is one that is relatively conserved across the virus to be detected but does not show high sequence similarity to other non-target sequences. While the lower temperature reaction means that added care must be taken in primer design, it offers the benefit of a reduced equipment demands, as typically only a simple heating instrument is required. The reactions are robust to changes in temperature, allowing flexibility in situations where on-site temperature changes may cause variation. This flexibility was demonstrated by Lillis et al. (2014) where they performed successful RPA reactions at ambient air temperatures above 31°C, in the absence of any additional heating equipment. They also proposed the use of exothermic chemical heaters, such as sodium acetate trihydrate (SAT) heaters. SAT is a re-useable chemical which releases heat upon crystallisation. Chemical heaters such as these are commonplace in hand-held heaters, making this technology widely available at relatively low cost. The group demonstrated successful RPA at ambient temperatures of 10°C when the reaction was warmed with SAT-based heaters (Lillis et al., 2014). In a similar manner, successful RPA reactions were even achieved using body heat alone, with no additional heating equipment (Crannell et al., 2014). The reduced need for heating also lowers the overall cost of detection, making this technology more accessible to areas with reduced resource availability (Londoño et al., 2016). Another factor which reduces cost, and time associated with assay development, is that there is no need for RNA or DNA extraction from the viral sample. RPA functions well using crude sources, including urine and blood, albeit with a slight reduction in the sensitivity of the assay in some cases (Clancy et al., 2015; Rosser et al., 2015).
Helicase-dependant amplification (HDA) functions by employing a DNA-helicase to unwind double stranded DNA. Flanking primers anneal to the strands, where a polymerase amplifies the target region. The process is repeated to exponentially amplify the target. When initially developed, HDA was performed at 37°C and required additional proteins such as SSBPs and MutL for the amplification to commence. However, further development of the reaction has negated the need for the SSBPs and MutL. These developments also incorporate a thermo-stable helicase and polymerase, permitting the reaction to be performed at higher temperatures between 60 and 65°C (Vincent et al., 2004; An et al., 2005; Teo et al., 2015). As with RPA, the primers are longer than typically used in PCR, reaching around 30 bp for HDA. To facilitate the direct amplification of RNA viruses, a RT enzyme can be added directly to the HDA reaction in a one-step protocol (Goldmeyer et al., 2007).
The amplified products can be detected through the use of fluorescent dyes and lateral flow-based detection (Chow et al., 2008; Kim et al., 2011; Li et al., 2013; Kolm et al., 2019). As HDA employs a similar mechanism to the standard PCR reaction, with the main difference being the helicase-mediated separation of DNA rather than heat-separation, molecular probes commonly used in qPCR could also apply to HDA. Tong et al. (2008) demonstrated the application of molecular probes in conjunction with HDA to facilitate probe-based quantitative diagnostics. The fact that similar probes can be used, indicates that a transition from assays such as qPCR to HDA may be relatively easy, while at the same time providing a low-cost, isothermal alternative to traditional PCR.
As HDA only requires two primers and a small number of proteins to function, it offers a more simplistic assay design when compared to other isothermal reaction, such as LAMP. This could appeal to researchers who wish to rapidly establish diagnostic tests. Further to this, the ability to repurpose already established qPCR molecular probes would also hasten the time in which a HDA assay could be developed. However, this simplicity of design does come at the cost of reduced specificity, as issues with non-specific primer annealing can occur with HDA (Barreda-García et al., 2018). Non-specific amplification could be overcome in part by the application of molecular probes as these provide an added specificity requirement. As with other isothermal reactions, only simple heating apparatus is required, however, the higher temperature reaction of HDA negates the ability to use ambient temperatures for amplification. HDA functions in the presence of other biological agents, and can be used alongside crude samples, without the need for DNA or RNA extraction (Kim et al., 2011; Li et al., 2013; Jevšnik et al., 2020).
Rolling circle amplification (RCA) mimics a natural process by which circular DNA is replicated (Kasamatsu and Vinograd, 1974; Mohsen and Kool, 2016). This process is adapted for use in the amplification of circular DNA sequences, making it unique from the previously described methods which amplify linear DNA. At its most basic, RCA requires only one primer, from which a strand displacing polymerase initiates replication. The resulting amplicon is a linear product, consisting of repeat sequences of the circular DNA (Schweitzer and Kingsmore, 2001; Mohsen and Kool, 2016). This method of RCA does not facilitate exponential amplification, however, alterations may be made to the protocol to facilitate rapid, exponential amplification. These include the use of hyper-branched RCA, which utilises two primers. The first binds to the circular target-DNA, while the second is complementary for a separate region on the single-stranded product. As the circular DNA is replicated multiple times, this second primer continuously binds repeat regions, creating a branched amplicon (Lizardi et al., 1998; Hamidi et al., 2015b; Li et al., 2019). Multiply-primed RCA is another alternative which uses several primers. Each primer anneals to a specific region on the circular DNA template and elongation is initiated. Amplified strands are displaced by the polymerase, and secondary primer-binding can occur on the displaced strands, creating large quantities of amplified product (Dean et al., 2001; Marincevic-Zuniga et al., 2012; Fux et al., 2018). Another important variation of RCA is padlock probe-based RCA. As RCA can only act upon circularised products, these probes are used when the desired target is single-stranded, linear DNA. The padlock probe features two regions, complementary to two regions on target. Upon hybridisation of the probe and the target, the two are joined via a ligase-mediated reaction, creating a circular template on which RCA can act (Banér et al., 1998, Banér et al., 2001; Mezger et al., 2014). The RCA reaction can be performed at a range of temperatures between 30 and 65°C (Marincevic-Zuniga et al., 2012; Li et al., 2019). The various forms of RCA are compared in Figure 4.
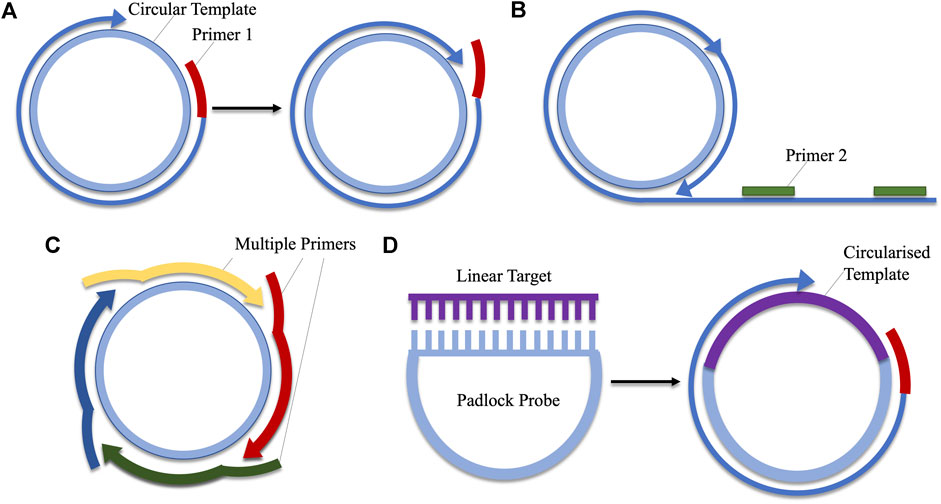
FIGURE 4. A comparison of the various types of rolling circle amplification. (A) Linear amplification using a single primer. (B) Hyper-branched RCA adds a second primer which aids in exponential amplification by binding to regions on the repeated DNA sequence. (C) Multiply-primed RCA also facilitates the production of large amounts of amplicon via the use of several primers which bind to different regions on the circular DNA. (D) Padlocks probes contain sequences complementary to linear DNA. Hybridisation of these regions facilitates ligation, producing a circularised template which can be employed in RCA.
The resulting products can be detected in real-time through the use of intercalating dyes (Kaocharoen et al., 2008). Fluorescently labelled probes can also be used to label the amplified product (Mezger et al., 2014). Real-time detection is facilitated through the use of molecular beacons (Nilsson et al., 2002). Product formation may also be measured through colorimetric means, using hydroxy naphthol blue (Hamidi and Ghourchian, 2015). Lateral flow technology can also be used to detect labelled, amplified products (Liu et al., 2019).
RCA is a rapid, sensitive and specific technique, useful for application to viral diagnostics. The assay design for RCA is relatively simple, requiring no target denaturation, and at minimum needing only one primer to amplify circular DNA or RNA (Li and Macdonald, 2015). However, this linear amplification method does not produce a high yield of product, therefore exponential RCA methods are typically used. With regards to the amplification of linear targets, for example single or double stranded linear viruses, padlock probes are useful. However, in order to perform this reaction, the target must first be denatured, and subsequently hybridised and ligated to the padlock probe. This adds to the length of time until diagnosis and the hands-on time is also increased. As RCA can take as little as 30 min, and requires minimal hands-on time, any additional reaction steps detract from the appeal of RCA (Kaocharoen et al., 2008; Mezger et al., 2014). This also contrasts to other methods such as RPA and HDA, which do not require preliminary denaturation steps. Nonetheless, there remain advantages for the use of padlock probes in viral detection. For example, the high specificity required to achieve both target hybridization and ligation means that non-specific binding is less likely to occur and produce false positives. It was also shown that multiple padlock probes, featuring varying hybridization sequences, used in the reaction, did not result in hinderance of the target-probe ligation process. This is useful for the detection of viruses which may have high levels of variation across the target region as all variants can be detected in a single reaction (Mezger et al., 2014). Alongside viral diagnostics, RCA can also be used for rapid viral genome amplification, facilitating sequencing of emergent strains, or comparisons between variants (Zanoli and Spoto, 2013). Therefore, it appeals to a number of streams within the virology field.
Cas proteins are effector proteins which play an integral role in the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas system. Cas effector proteins have the ability to cleave DNA or RNA. Their cleavage activity is controlled by guide-RNAs, which direct the Cas proteins to the correct cutting site on the target nucleic acid. The guide RNA of the CRISPR-Cas system can be modified to facilitate targeting of specific sequences, and herein lies the application to nucleic acid detection (Bhattacharyya et al., 2018). Three primary systems are thus far proposed for nucleic acid detection, named SHERLOCK (specific high-sensitivity enzymatic reporter unlocking), DETECTR (DNA Endonuclease Targeted CRISPR Trans Reporter) and HOLMES (one-HOur Low-cost Multipurpose highly Efficient System). SHERLOCK uses Cas13a to detect RNA, while HOLMES and DETECTR use Cas12a for DNA recognition (Gootenberg et al., 2017; Chen et al., 2018; Li et al., 2018). As aforementioned, the Cas nuclease cleaves the DNA or RNA at the recognition site, however, off-target cleavage can also occur with some Cas proteins. This is known as collateral cleavage. While this is a naturally occurring mechanism, collateral cleavage can be repurposed for nucleic acid detection. Normally to facilitate Cas-mediated nucleic acid detection, the target nucleic acid is first amplified via an isothermal reaction, usually RPA. Thereafter, the CRISPR-Cas system is introduced, featuring a guide RNA specific for a location on the target DNA. Binding of the guide RNA induces target cleavage, but collateral cleavage also occurs. Measurable signal in the assay can be achieved by the addition of fluorophore probes, which are cleaved by the collateral activity of the DNA-recognising Cas protein, Cas12a, resulting in measurable fluorescence (Batista and Pacheco, 2018). The reaction can be performed in a “one-pot” style, with results achieved in 1 h (Li et al., 2018; Kellner et al., 2019). The amplified and cleaved products can be detected through fluorescence measurement, or, for a simpler alternative, on a lateral flow strip (Gootenberg et al., 2018; Myhrvold et al., 2018; Broughton et al., 2019). An overview of both fluorescence- and lateral flow-based CRISPR-Cas detection is depicted in Figure 5.
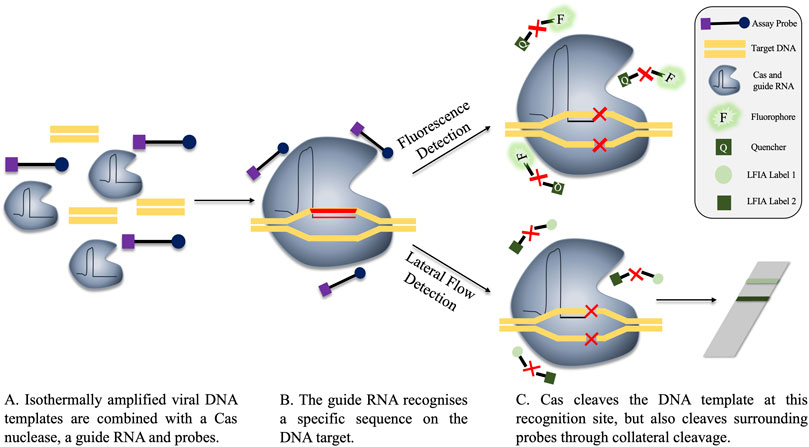
FIGURE 5. Fluorescence and lateral flow-based detection in a CRISPR-Cas detection system. (A) The components of the reaction are combined, either before or after isothermal amplification. The guide RNA is specific for a region on the target viral DNA. (B) The guide RNA recognises the target sequence, locating the Cas nuclease to this region. (C) The DNA target is cleaved by the Cas nuclease mechanism. However, Cas also performs collateral cleavage on surrounding single-stranded probes. Cleavage is represented in the diagram by a red X. These probes can be fluorescent (C, top), where the cleavage of the probe separates the quencher and the fluorophore, permitting fluorescent signal. Alternatively, probes can be used in lateral flow assays, whereby cleavage of the probe produces two distinct bands on the flow-strip, indicating a positive result. A single band on the lateral flow strip is representative of a negative result as no cleavage events occurred and the labels are not separated (C, bottom).
The dual use of isothermal amplification and CRISPR-Cas proteins provides a rapid assay with an added specificity safety net. First, primer sets must bind to the target to facilitate initial product amplification, then the guide RNA must detect a specific sequence on this product to facilitate signal generation. This dual mechanism could help alleviate issues associated with the non-specific primer binding sometimes observed in isothermal amplification. The fact that both amplification and Cas-cleavage can be performed in a single reaction is also appealing, as it reduces the risk of contamination and requires less hands-on time. Given that already existing isothermal amplification methods can be transitioned to CRISPR-Cas nucleic acid detection, these enhanced assays could be rapidly developed against a host of viruses. It is likely that with the improved specificity that this detection system offers, that its application will continue to grow in the field of virology.
The capability for high-throughput and/or multiplexed detection of viruses in a sample is an important factor to consider when choosing a suitable diagnostic method. Automated systems based on the amplification of nucleic acids from a sample are, therefore, an extremely useful tool in scenarios where multiple samples require assessment, or where a sample must be tested for the presence of a range of viruses. Currently, several systems are available for the high-throughput sensitive detection of nucleic acids, examples of which include the BioFire FilmArray, Verigene, Gen-Probe Prodesse and Cepheid systems (Babady, 2013; Van Wesenbeeck et al., 2013). Such systems are capable of running multiplexed diagnostic assays, with the number of samples used per run varying depending on the platform. Results turnaround is generally within a day, with some systems capable of rapid testing, producing results within an hour. In addition to a rapid run time, integrated sample preparation is also available on several systems, reducing hands-on time to as little as several minutes (Babady, 2013; Van Wesenbeeck et al., 2013; Huang et al., 2018). The offer of contained sample preparation within some automated systems is advantageous when the sample may contain a high-risk viral pathogen (Loftis et al., 2017). When large-scale viral detection is required, automated systems may offer an advantage over amplification methods such as isothermal methods due to their amenability to multiplexing and high-throughput analysis. However, the requirement for specialised equipment may limit the suitability of these automated assays in low resource settings. Nonetheless, the capacity to assay multiple samples simultaneously is highly useful for application in settings where large-scale testing is necessitated.
In summation, methods which rely on nucleic acid detection are useful tools for analysing viral infection. They are often very sensitive and specific, ranging in complexity of design from LAMP, which requires at least four primers to function, to RCA which at its minimum only requires one primer. The higher the number of oligonucleotides which must bind to the target for the assay to proceed generally means a lower rate of non-specific binding and non-target product formation. While qPCR is still in commonplace use in laboratory-based situations, isothermal techniques are witnessing a surge for applications such as point-of-care/use and in low-resource settings. Furthermore, many isothermal amplifications are relatively robust when it comes to the nature of the sample from which the target DNA/RNA is amplified. This is, again, appealing for on-site diagnostics but also for scenarios where time is of the essence and avoiding sample preparation would reduce the time to result. In addition to qPCR and isothermal techniques, automated DNA amplification-based platforms are an important tool in viral diagnostics as they generally offer quick turnaround times and require limited manipulation of samples. Another strength of nucleic acid-based detection systems is the speed at which they can be designed when the sequence of the virus of interest is available. Given that viruses can rapidly emerge, the ability to quickly develop assays to detect particularly damaging, pathogenic or transmissible viruses is of major interest. Therefore, nucleic acid techniques will remain essential in the future of virology. Nonetheless, there are other methods which are important for virus detection. These include methods which employ biological detection agents, such as antibodies, to identify virus-associated proteins.
Immunoassays employ antibodies as the primary means to detect viruses within a sample. Antibodies are available in polyclonal, monoclonal and recombinant formats (Ma and O’Kennedy, 2015). Polyclonal antibodies bind multiple different structures or epitopes on the target organism/molecule i.e., are polyspecific. This can be useful in situations where the desired outcome is indiscriminate detection of all viral strains, or when the goal is to capture all strains of a virus from a sample. In contrast, monoclonal antibodies, and certain recombinant forms of antibodies, present singular-epitope specificity, hence, they are monospecific. Specific antibodies are valued in diagnostics as they offer the opportunity for the targeted detection of distinct regions on the target molecule. In virus diagnosis, this is useful for differentiation between different isolates or genotypes of a single virus, or between similar viral species within a genus (Usuda et al., 2000; Yu et al., 2005; Chen et al., 2016; Lebani et al., 2017).
When the targets are highly similar, for example similar strains of a given virus, isolating non-cross-reactive antibodies can be challenging. To overcome this, antibodies can be isolated based on their ability to detect short peptide sequences. These peptides correspond to unique regions on the target protein, providing the capacity to detect strain-specific regions on an entire protein (Geysen et al., 1985). When employing this strategy, it is essential to carefully select the target peptide. Peptides can sometimes adopt conformations which do not reflect the epitope in its native conformation. Given that antibody binding is affected by both amino-acid sequence, and by structural conformation of the amino acids in the binding regions, ensuring comparable binding is essential (O’Kennedy et al., 2017). Therefore, the binding to both the peptide and the native epitope on the target protein must be validated (Conroy et al., 2009). Overall, targeting highly specific peptides, rather than an entire protein with multiple epitope regions, could result in antibodies which are less likely to cross-react with other viruses or isolates of the viral species.
The initial development of pathogen-targeting antibodies requires time to identify a valid disease-specific antigen, isolate suitable antibodies and validate the resulting antibodies and assays. However, once generated, the production of the antibody proteins is relatively fast. Furthermore, the advent of recombinant antibody technology, whereby antibodies are produced as fragments of the whole antibody protein, has made the generation of antibodies more rapid and cost-effective, as cheaper expression hosts such as yeasts and bacteria can be used in lieu of mammalian cell-culture (Jeong et al., 2011; Kunert and Reinhart, 2016).
Virus-targeting antibodies are used in virology for multiple purposes. Two major applications are for the detection of virus-associated antigens from samples, and for determining seroprevalence. Seroprevalence assays are essential techniques for identifying patients who have been exposed to a virus (Li et al., 2020). However, these assays rely on the identification of anti-virus antibodies generated by the host, whereas this review focuses on the creation, employment and design of specific antibodies to detection pathogen-associated targets. As such, the following section largely deals with the use of antibodies for identifying virus-associated antigens from samples, with only brief mention to applications for seroprevalence monitoring. The detection of virus-antigens was initially facilitated by virus propagation through cell culture methods, whereby antibodies are used to detect virus-associated proteins within a cell line, or to perform virus neutralisation assays (Leland and Ginocchio, 2007). However, as discussed, such cell culture methods are relatively slow when compared to other diagnostic means. Therefore, much focus is on the development of antibody-based assays capable of detecting the target-virus directly from source through a range of antibody-based assays, termed immunoassays.
One of the most widely applied immunoassays is the enzyme-linked immunosorbent assay (ELISA). There are a number of formats in which the ELISA assay can be performed, namely direct, indirect, competitive and sandwich (Figure 6). These formats are also used for most other immunoassays.
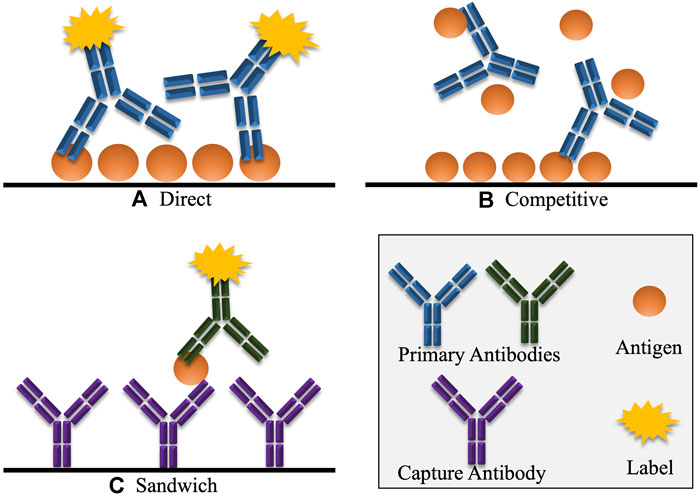
FIGURE 6. Various common formats of immunoassays, including ELISA. (A) In direct detection, the antigen is detected directly by a labelled antibody. Indirect detection is a variation of this format, where the primary antigen-specific antibody is unlabelled, and must be detected by a secondary labelled antibody that selectively binds to the primary antibody. (B) In competitive detection, free antigen in the sample competes with immobilised antigen for binding to the antibody which may be directly labelled or may be detected using a secondary labelled antibody. Hence, in competitive immunoassays, the signal is inversely proportional to the concentration of antigen. (C) In the sandwich immunoassay format, the antigen is captured by an antibody that reacts with a specific epitope on the antigen. A second labelled antibody is added which reacts with another different epitope on the captured antigen. Here the signal generated is directly proportional to the amount of antigen present.
Direct and indirect formats are similar. Initially, the antigen is coated to the surface of the ELISA well through passive adsorption, or it may be chemically linked. For direct detection, a labelled, anti-target primary antibody is applied to the well, and subsequently detected. For indirect detection, the primary antibody is unlabelled, and is detected by the addition of a labelled, secondary antibody. These formats are useful for measuring an antibody response against a given antigen. For example, with regards to virology, direct or indirect ELISA assays are useful for confirming patients who have anti-virus antibodies in their sera, having developed an immune response post-viral infection (Khudyakov and Kamili, 2011; Bréard et al., 2013; Zhang et al., 2020). However, there are some drawbacks to direct and indirect formats when transitioning to detecting viruses in crude samples such as blood, stool or other tissues. For both formats, the suspect sample may first have to be coated onto the ELISA test well. Most biological samples will contain a combination of target proteins and other proteins. These other proteins compete for adsorption to the assay well surface, meaning that the target-antigen may not be accurately represented in the sample, making quantitation challenging (He, 2013). To overcome this, formats such as competitive and sandwich are used.
Competitive assays are effective in the detection of viral-specific proteins from a given source. Competitive ELISA first involves the immobilisation of the target antigen to an ELISA well, followed by the simultaneous application of the antibody and the suspect sample to the same well. Target-antigen in the sample competes for antibody-binding, leaving less antibody available for binding to the immobilised antigen. The sample is washed away, and any remaining bound antibody is detected with a labelled, secondary antibody. Therefore, in this assay format, the signal is inversely proportional to the amount of analyte in the sample. Competitive analysis is widely used for the detection of small antigens, where the binding of multiple antibodies may not be permitted (He, 2013). This assay format is useful when considering the rapid-development of viral-detection assays as it only requires one target-specific antibody. However, the use of only one antibody also has consequences in the form of reduced assay sensitivity and specificity if any cross-reactivity with the sample matrix occurs.
The employment of a sandwich formats overcomes most issues associated with cross-reactivity as it employs two antibodies, both specific for different regions (epitopes) on the target molecule. The first of these is immobilised to the well surface. Thereafter, the sample is applied, and specific antigen is captured by the immobilised antibody. Unbound entities are washed away, and the second antibody is applied. This second antibody can be labelled directly or detected with a labelled-secondary antibody. This format offers high specificity as it requires the binding of two antibodies to produce a signal. This could be helpful for virus detection where there is a risk of cross-reactivity occurring between similar strains of virus. It is also considered the most sensitive and robust ELISA format (Ecker et al., 2013; He, 2013). Sandwich assays are less prone to cross-reactivity. Even if one of the antibodies in the pair has some cross-reactivity with the sample matrix, the likelihood is that the same cross-reactivity will not be observed with the alternate binding antibody. This makes this assay suitable for the detection of viruses directly from their sources, such as serum or tissue samples, where the matrix can be very complex (Jansen van Vuren and Paweska, 2009; Zhang et al., 2018). A drawback to the sandwich assays is the complexity of design. Both antibodies need to bind, unhindered by one another, to the target molecule. Finding a suitable binding pair can be difficult, and thorough assay validation is required to ensure no non-specific interaction occurs between the antibodies within the assay. However, the use of antibody-discovery technology, such as phage display, has led to the development of methods which facilitate the identification of binding pairs of antibodies in a rapid manner, negating the time-consuming screening associated with finding two separate binding pairs (Gorman et al., 2017). Alternatively, the previously described strategy of peptide-targeting can be employed to design antibodies which can detect distinct regions on the whole antigen molecule, facilitating sandwich detection.
ELISA continues to remain a staple platform in virus detection due to its sensitivity and robustness. However, there are drawbacks to its use. These include the risk of cross-reactivity of antibodies to other co-infecting viruses, resulting in false positives or inaccurate quantification. This highlights the need to fully validate virus-assays prior to use. In contrast to a potential for false positives, ELISA, and other immunoassays, may also be at risk of presenting false negative results. This generally occurs when testing is performed at the early window of infection. At this stage, the quantities of viral antigen may be present at low concentrations, challenging the sensitivity of the ELISA format, potentially leading to erroneous results (Tillmann, 2014; Alexander, 2016). Further to this, ELISA procedures can take several hours to perform. This limits the capacity for rapid testing and results turnaround. However, the multi-well layout of ELISA plates facilitates multiple simultaneous tests, which is ideal for scenarios where high volume testing is required, such as epidemic or pandemic scenarios. In order to improve this turnaround time other antibody-based methods are suggested for virus detection.
Blotting techniques generally involve antigen detection on the surface of a membrane. The dot blot, or slot blot, is a technique which can be used for the detection of viral antigens from a sample (Li et al., 2010; Gallagher et al., 2011). To do this, usually the suspect sample is blotted onto a membrane, allowed to dry and the membrane is then probed with an anti-virus antibody. This method is cost effective and offers advantages such as the requirement for a small sample volume, the ability to detect virus directly from the sample and the fact that blotted membranes can be stored for a number of days before testing, facilitating the testing of numerous membranes simultaneously. However, the results of dot-blots are mostly qualitative, requiring further instrumentation to perform quantitive analysis which may increase costs. Dot/slot blots can also take several hours to perform (Li et al., 2010; Thiruppathiraja et al., 2011; Fulladolsa Palma et al., 2013). Tissue blots are a variation on the dot/slot blot. These involve pressing of the infected tissue onto the membrane to facilitate virus antigen immobilisation. The dot/slot blot and tissue blot procedures are outlined in Figure 7. Blotting techniques are useful for cheap detection of multiple samples, as many samples can be blotted on the same membrane. However, the time required to produce results is a considerable drawback, as the turnaround may take several hours.
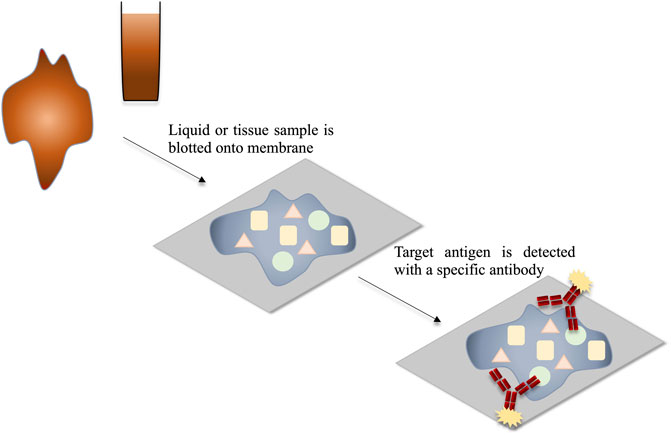
FIGURE 7. Schematic representation of the detection of viral antigens through dot blot or tissue blot. The mixture of antigens within the sample is blotted onto the membrane and thereby immobilised. The target antigens are detected using target-specific labelled antibodies.
A form of lateral flow immunoassay (LFIA), NALFIA, was discussed with respect to its usage in nucleic-acid detection, however, LFIAs are extensively used for detecting virus-associated protein directly from source. While NFLIA function based on the detection of labelled amplicons, standard LFIAs rely on labelled antibodies binding to their cognate antigens. The assays are typically formatted in either sandwich or competitive immunoassay formats (Sajid et al., 2015). The assay result is usually read by way of a colour change at a test line. In sandwich format, a colour change at this line corresponds to a positive result, whereas in competitive format no colour change is indicative of a positive result. Control lines are incorporated into both assays to ensure test validity. The colour change is facilitated by the labelling of antibodies with various molecules. Labels such as gold nanoparticles, coloured latex beads, carbon nanoparticles and magnetic particles facilitate one-step results where a colour change can be observed with the naked eye, facilitating rapid diagnosis. Other labels such as fluorophores, quantum dots and enzymatic labels can also be employed, however these may require additional equipment or steps to demonstrate results which may be qualitative or quantitative (Posthuma-Trumpie et al., 2009; Bahadır and Sezgintürk, 2016; Mak et al., 2016).
For virus detection, LFIAs offer an appealing alternative to methods such as ELISA or blotting methods. One primary advantage is that LFIAs have a turnaround time of minutes, rather than hours. Further to this, performing a LFIA requires minimal sample clean up, having been proven to efficiently detect viral antigens from crude samples, including viral transport media and blood (Ryu et al., 2016; Sastre et al., 2016; Si et al., 2019). Assays can also be performed using saliva samples providing a non-invasive and easy-to-acquire source for viral testing (Yoon et al., 2017). LFIAs are also cost effective, requiring minimal sample volumes to prepare and facilitating diagnosis with the naked eye. However, this also comes with the drawback of only offering qualitative results. To achieve quantitative results, further instrumentation is required (Mak et al., 2016). Another advantage of LFIA is their ease-of-use, which makes the technology accessible to a wide range of users. This can be helpful in scenarios where a high volume of testing is required, as non-specialists can be quickly trained to performs assays.
Overall, immunoassays in viral detection play an essential role. While they may not always offer the same sensitivity as nucleic-acid detection methods, particularly at early stages of infection, they come with the advantage of a reduced cost, reduced complexity and higher utility for use by untrained personnel. These factors are exemplified by the LFIA which can be produced cheaply, has no need for additional equipment and requires little to no training to use. Table 1 provides a comparison between the various nucleic acid detection-based and immunoassay-based methods. The specificity of antibodies in immunoassays is one of the primary challenges. This is exacerbated in virology by the presence of multiple strains of a single virus, or by similar viruses within a genus. Recombinant antibody technology facilitates rapid re-engineering of antibody characteristics such as specificity and offers a means to alter antibodies for reduced cross-reactivity (Ducancel and Muller, 2012; Ma et al., 2019). This could aid in overcoming any specificity-related obstacles. Antibody discovery using recombinant technology also has a faster turnaround when compared to methods such as hybridoma technology (Basu et al., 2019). Therefore, recombinant technology should be considered an essential tool in the development of future viral diagnostics assays for both existing and emergent viral strains.
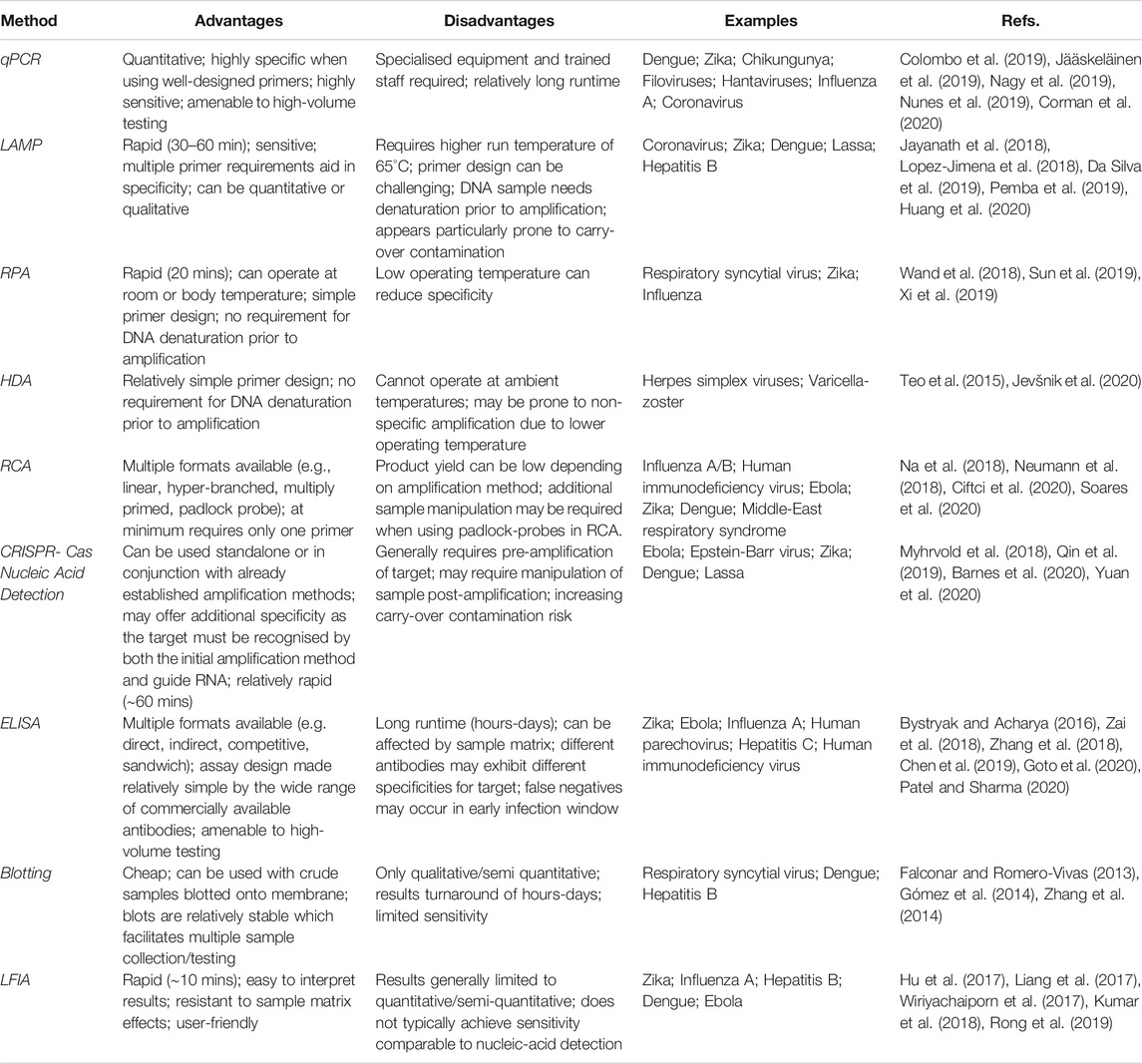
TABLE 1. Summary of the advantages, disadvantages and example applications off nucleic acid-based and immunoassay-based approaches to virus detection.
While molecular and antibody-based techniques will likely continue to dominate the field of virology, their use should be complemented by engaging with newer technologies, such as next generation sequencing (NGS) platforms. The employment of NGS benefits virology in multiple ways. Firstly, it can act as a stand-alone method for viral diagnostics, if required. NGS is used to sequence genomes, DNAs and RNAs, and can be used to differentiate host genetic information from viral sequences. This, therefore, provides a powerful tool to diagnose viruses, and would be particularly useful in scenarios where patients present with a disease and the causative-pathogen is unidentified (Barzon et al., 2013). While molecular, immuno- and cell-culture methods can be used to identifying unknown pathogens in an investigational “process of elimination” approach, NGS offers a more time-effective method to achieve the same output (Adams et al., 2009). Therefore, while currently established methods may be more rapid in diagnosing known viruses, NGS should be a promising way to reduce time-to-discovery for novel, or unexpected, viral pathogens. Instances of NGS implementation providing a diagnosis to a previously unknown virus, or identifying unexpected viruses in samples, are reported, highlighting its usefulness (Palacios et al., 2008; Prachayangprecha et al., 2014; Naccache et al., 2015; Cordey et al., 2016; Somasekar et al., 2017; Rott et al., 2018) NGS could also be applicable to diagnosing emergent viruses, where no established assays are available for the testing of the virus. Similarly, monitoring of viral levels in a population could also be facilitated by NGS, with particular note to monitoring of emergent or unexpected viral strains. Passive sampling of airborne viruses in crowded areas such as transit networks, schools, day-cares and emergency rooms could be used in conjunction with NGS to assess the viral burden and patterns in particular settings (Choi et al., 2018; Coleman et al., 2018; Prussin et al., 2019; Coleman and Sigler, 2020). In a similar manner, viral vectors or transmitters, such as mosquitoes or bats, can be sampled passively, and routinely tested, to allow surveillance of virus prevalence in the harbouring population (Eiras et al., 2018; Duarte et al., 2020). This could be helpful for predicting disease outbreaks or novel virus emergence, Another benefit of NGS in virology is the promise of expanding viral genome-sequencing data. The paucity of information with regards to viral sequences can make experimental design difficult, for example, designing broad-spectrum amplification primers or identifying conserved antigens can be challenging when not all viral strains are known (Barzon et al., 2011). The capability to sequence many viruses from a population, and hence achieve a more rounded picture of the variation in those viruses, could aid not only in the surveillance of emerging viruses, but in providing additional information to researchers looking to develop novel assays (Rosario and Breitbart, 2011; Radford et al., 2012).
While NGS is a promising technology as a tool for virus discovery, there are challenges to overcome. These include the high costs, the need for both trained staff and bioinformaticians, a potential for poor sensitivity in low viral-load infections and a lack of universal sequence data-analysis tools (Beerenwinkel et al., 2012; Barzon et al., 2013; Massart et al., 2014; Maree et al., 2018). Once addressed, NGS could work in tandem with established techniques to provide a means to identify, characterise and diagnose viral infection.
Presently, no single method meets every demand of virus detection. Nucleic acid-based detection is highly sensitive and specific. Its drawbacks include the fact that often trained staff are required, associated costs can be high, and carry-over contamination can cause issues in result analysis. In contrast, antibody-based detection is generally cheaper, more robust, and more accessible to untrained users. However, immunoassays do not typically achieve the same sensitivity as nucleic acid detection methods. As such, the decision of whether to employ nucleic acid detection or immunoassays methods will largely be dictated by the requirements of the assay. For example, considerations should include the setting in which the assay will be performed, whether it be laboratory-based or at point-of-care/use, cost of the assay, the urgency of the diagnostic, for example in situations of emergent strains which are causing an epidemic or pandemic, and the sensitivity and specificity requirements of the assay. As NGS becomes more accessible, both nucleic-acid detection and immunoassay methods will benefit from its application. As the use of NGS expands, so too will the number of assays to facilitate the detection of viruses, driven by an increased knowledge of the diversity of viral genomes and their roles.
AC authored the initial review draft. RO’K and AP-M contributed to the reading and revision of the article.
This project was funded by the Department of Agriculture, Food and the Marine's Competitive Research Funding Programme, Project Ref. 15/S/618.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adams, I. P., Glover, R. H., Monger, W. A., Mumford, R., Jackeviciene, E., Navalinskiene, M., et al. (2009). Next-generation sequencing and metagenomic analysis: a universal diagnostic tool in plant virology. Mol. Plant Pathol. 10, 537–545. doi:10.1111/j.1364-3703.2009.00545.x
Alexander, T. S. (2016). Human immunodeficiency virus diagnostic testing: 30 Years of evolution. Clin. Vaccine Immunol. 23, 249–253. doi:10.1128/CVI.00053-16
An, L., Tang, W., Ranalli, T. A., Kim, H. J., Wytiaz, J., and Kong, H. (2005). Characterization of a thermostable UvrD helicase and its participation in helicase-dependent amplification. J. Biol. Chem. 280, 28952–28958. doi:10.1074/jbc.M503096200
Arunrut, N., Kampeera, J., Sirithammajak, S., Sanguanrut, P., Proespraiwong, P., Suebsing, R., et al. (2016). Sensitive visual detection of AHPND bacteria using loop-mediated isothermal amplification combined with DNA-Functionalized gold nanoparticles as probes. PLoS One 11, e0151769. doi:10.1371/journal.pone.0151769
Arya, M., Shergill, I. S., Williamson, M., Gommersall, L., Arya, N., and Patel, H. R. (2005). Basic principles of real-time quantitative PCR. Expert Rev. Mol. Diagn. 5, 209–219. doi:10.1586/14737159.5.2.209
Babady, N. E. (2013). The FilmArray® respiratory panel: an automated, broadly multiplexed molecular test for the rapid and accurate detection of respiratory pathogens. Expert Rev. Mol. Diagn. 13, 779–788. doi:10.1586/14737159.2013.848794
Bahadır, E. B., and Sezgintürk, M. K. (2016). Lateral flow assays: principles, designs and labels. Trends Anal. Chem. 82, 286–306. doi:10.1016/j.trac.2016.06.006
Ball, C. S., Light, Y. K., Koh, C. Y., Wheeler, S. S., Coffey, L. L., and Meagher, R. J. (2016). Quenching of unincorporated amplification signal reporters in reverse-transcription loop-mediated isothermal amplification enabling bright, single-step, closed-tube, and multiplexed detection of RNA viruses. Anal. Chem. 88, 3562–3568. doi:10.1021/acs.analchem.5b04054
Banér, J., Nilsson, M., Isaksson, A., Mendel-Hartvig, M., Antson, D.-O., and Landegren, U. (2001). More keys to padlock probes : mechanisms for high-throughput nucleic acid analysis. Curr. Opin. Biotechnol. 12, 11–15. doi:10.1016/s0958-1669(00)00174-9
Banér, J., Nilsson, M., Mendel-Hartvig, M., and Landegren, U. (1998). Signal amplification of padlock probes by rolling circle replication. Nucleic Acids Res. 26, 5073–5078. doi:10.1093/nar/26.22.5073
Banerjee, N., and Mukhopadhyay, S. (2016). Viral glycoproteins: biological role and application in diagnosis. Virusdisease 27, 1–11. doi:10.1007/s13337-015-0293-5
Barnes, K. G., Lachenauer, A. E., Nitido, A., Siddiqui, S., Gross, R., Beitzel, B., et al. (2020). Deployable CRISPR-Cas13a diagnostic tools to detect and report Ebola and Lassa virus cases in real-time. Nat. Commun. 11, 4131. doi:10.1038/s41467-020-17994-9
Barreda-García, S., Miranda-Castro, R., de-los-Santos-Álvarez, N., Miranda-Ordieres, A. J., and Lobo-Castañón, M. J. (2018). Helicase-dependent isothermal amplification: a novel tool in the development of molecular-based analytical systems for rapid pathogen detection. Anal. Bioanal. Chem. 410, 679–693. doi:10.1007/s00216-017-0620-3
Barzon, L., Lavezzo, E., Costanzi, G., Franchin, E., Toppo, S., and Palù, G. (2013). Next-generation sequencing technologies in diagnostic virology. J. Clin. Virol. 58, 346–350. doi:10.1016/j.jcv.2013.03.003
Barzon, L., Lavezzo, E., Militello, V., Toppo, S., and Palù, G. (2011). Applications of next-generation sequencing technologies to diagnostic virology. Int. J. Mol. Sci. 12, 7861–7884. doi:10.3390/ijms12117861
Basu, K., Green, E. M., Cheng, Y., and Craik, C. S. (2019). Why recombinant antibodies — benefits and applications. Curr. Opin. Biotechnol. 60, 153–158. doi:10.1016/j.copbio.2019.01.012
Batista, A. C., and Pacheco, L. G. C. (2018). Detecting pathogens with Zinc-Finger, TALE and CRISPR- based programmable nucleic acid binding proteins. J. Microbiol. Methods 152, 98–104. doi:10.1016/j.mimet.2018.07.024
Becherer, L., Bakheit, M., Frischmann, S., Stinco, S., Borst, N., Zengerle, R., et al. (2018). Simplified real-time multiplex detection of loop-mediated isothermal amplification using novel mediator displacement probes with universal reporters. Anal. Chem. 90, 4741–4748. doi:10.1021/acs.analchem.7b05371
Beerenwinkel, N., Günthard, H. F., Roth, V., and Metzner, K. J. (2012). Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front. Microbiol. 3, 1–16. doi:10.3389/fmicb.2012.00329
Bhattacharyya, R. P., Thakku, S. G., and Hung, D. T. (2018). Harnessing CRISPR effectors for infectious disease diagnostics. ACS Infect. Dis. 4, 1278–1282. doi:10.1021/acsinfecdis.8b00170
Borst, A., Box, A. T. A., and Fluit, A. C. (2004). False-positive results and contamination in nucleic acid amplification assays: suggestions for a prevent and destroy strategy. Eur. J. Clin. Microbiol. Infect. Dis. 23, 289–299. doi:10.1007/s10096-004-1100-1
Bréard, E., Lara, E., Comtet, L., Viarouge, C., Doceul, V., Desprat, A., et al. (2013). Validation of a commercially available indirect ELISA using a nucleocapside recombinant protein for detection of Schmallenberg virus antibodies. PLoS One 8, e53446. doi:10.1371/journal.pone.0053446
Broughton, J. P., Deng, X., Yu, G., Fasching, C. L., Servellita, V., Singh, J., et al. (2019). CRISPR – Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38, 870–874. doi:10.1038/s41587-020-0513-4
Burkhalter, K. L., and Savage, H. M. (2017). Detection of Zika virus in desiccated mosquitoes by real-time reverse transcription PCR and plaque assay. Emerg. Infect. Dis. 23, 680–681. doi:10.3201/eid2304.161772
Bustin, S. A. (2000). Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 25, 169–193. doi:10.1677/jme.0.0250169
Bystryak, S., and Acharya, C. (2016). Detection of HIV-1 p24 antigen in patients with varying degrees of viremia using an ELISA with a photochemical signal amplification system. Clin. Chim. Acta 456, 128–136. doi:10.1016/j.cca.2016.02.022
Cai, S., Jung, C., Bhadra, S., and Ellington, A. D. (2018). Phosphorothioated primers lead to loop-mediated isothermal amplification at low temperatures. Anal. Chem. 90, 8290–8294. doi:10.1021/acs.analchem.8b02062
Chen, J. S., Ma, E., Harrington, L. B., Da Costa, M., Tian, X., Palefsky, J. M., et al. (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439. doi:10.1126/science.aar6245
Chen, L., Ruan, F., Sun, Y., Chen, H., Liu, M., Zhou, J., et al. (2019). Establishment of sandwich ELISA for detecting the H7 subtype influenza A virus. J. Med. Virol. 91, 1168–1171. doi:10.1002/jmv.25408
Chen, Y. H., Dong, J., Chien, W. C., Zheng, K., Wu, K., Yeh, S. D., et al. (2016). Monoclonal antibodies for differentiating infections of three serological-related tospoviruses prevalent in Southwestern China. Virol. J. 13, 72. doi:10.1186/s12985-016-0525-3
Choi, J. Y., Zemke, J., Philo, S. E., Bailey, E. S., Yondon, M., and Gray, G. C. (2018). Aerosol sampling in a hospital emergency room setting: a complementary surveillance method for the detection of respiratory viruses. Front. Public Heal. 6, 174. doi:10.3389/fpubh.2018.00174
Chow, W. H. A., McCloskey, C., Tong, Y., Hu, L., You, Q., Kelly, C. P., et al. (2008). Application of isothermal helicase-dependent amplification with a disposable detection device in a simple sensitive stool test for toxigenic Clostridium difficile. J. Mol. Diagn. 10, 452–458. doi:10.2353/jmoldx.2008.080008
Ciftci, S., Cánovas, R., Neumann, F., Paulraj, T., Nilsson, M., Crespo, G. A., et al. (2020). The sweet detection of rolling circle amplification: glucose-based electrochemical genosensor for the detection of viral nucleic acid. Biosens. Bioelectron. 151, 112002. doi:10.1016/j.bios.2019.112002
Clancy, E., Higgins, O., Forrest, M. S., Boo, T. W., Cormican, M., Barry, T., et al. (2015). Development of a rapid recombinase polymerase amplification assay for the detection of Streptococcus pneumoniae in whole blood. BMC Infect. Dis. 15, 481. doi:10.1186/s12879-015-1212-5
Clark, K. D., Zhang, C., and Anderson, J. L. (2016). Sample preparation for bioanalytical and pharmaceutical analysis. Anal. Chem. 88, 11262–11270. doi:10.1021/acs.analchem.6b02935
Coleman, K. K., Nguyen, T. T., Yadana, S., Hansen-Estruch, C., Lindsley, W. G., and Gray, G. C. (2018). Bioaerosol sampling for respiratory viruses in Singapore’s mass rapid transit network. Sci. Rep. 8, 17476. doi:10.1038/s41598-018-35896-1
Coleman, K. K., and Sigler, W. V. (2020). Airborne influenza A virus exposure in an elementary school. Sci. Rep. 10, 1859. doi:10.1038/s41598-020-58588-1
Colombo, T. E., Versiani, A. F., Dutra, K. R., Rubiato, J. G. D., Galvão, T. M., Negri Reis, A. F., et al. (2019). Performance of CDC Trioplex qPCR during a dengue outbreak in Brazil. J. Clin. Virol. 121, 104208. doi:10.1016/j.jcv.2019.104208
Conroy, P. J., Hearty, S., Leonard, P., and O’Kennedy, R. J. (2009). Antibody production, design and use for biosensor-based applications. Semin. Cell Dev. Biol. 20, 10–26. doi:10.1016/j.semcdb.2009.01.010
Cordey, S., Vu, D. L., Schibler, M., L’Huillier, A. G., Brito, F., Docquier, M., et al. (2016). Astrovirus MLB2, a new gastroenteric virus associated with meningitis and disseminated infection. Emerg. Infect. Dis. 22, 846–853. doi:10.3201/eid2205.151807
Corman, V. M., Landt, O., Kaiser, M., Molenkamp, R., Meijer, A., Chu, D. K., et al. (2020). Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 25, 2000045. doi:10.2807/1560-7917.ES.2020.25.3.2000045
Crannell, Z. A., Rohrman, B., and Richards-Kortum, R. (2014). Equipment-free incubation of recombinase polymerase amplification reactions using body heat. PLoS One 9, e112146. doi:10.1371/journal.pone.0112146
Da Silva, S. J. R., Pardee, K., and Pena, L. (2019). Loop-mediated isothermal amplification (LAMP) for the diagnosis of Zika virus: a review. Viruses 12, 19. doi:10.3390/v12010019
Daher, R. K., Stewart, G., Boissinot, M., and Bergeron, M. G. (2016). Recombinase polymerase amplification for diagnostic applications. Clin. Chem. 62, 947–958. doi:10.1373/clinchem.2015.245829
Daher, R. K., Stewart, G., Boissinot, M., Boudreau, D. K., and Bergeron, M. G. (2015). Influence of sequence mismatches on the specificity of recombinase polymerase amplification technology. Mol. Cell. Probes 29, 116–121. doi:10.1016/j.mcp.2014.11.005
Dean, F. B., Nelson, J. R., Giesler, T. L., and Lasken, R. S. (2001). Rapid amplification of plasmid and phage DNA using Phi29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 11, 1095–1099. doi:10.1101/gr.180501
Dhama, K., Karthik, K., Chakraborty, S., Tiwari, R., Kapoor, S., Kumar, A., et al. (2014). Loop-mediated isothermal amplification of DNA (LAMP): a new diagnostic tool lights the world of diagnosis of animal and human pathogens: a review. Pak. J. Biol. Sci. 17, 151–166. doi:10.3923/pjbs.2014.151.166
E. Domingo (Editor) (2015). “Introduction to virus origins and their role in biological evolution,” in Virus as Populations. (Amsterdam, Netherlands: Elsevier Science and Technology), 1–34.
Duarte, N. F. H., Alencar, C. H., Cavalcante, K. K. de. S., Correia, F. G. S., Romijn, P. C., Araujo, D. B., et al. (2020). Increased detection of rabies virus in bats in Ceará State (Northeast Brazil) after implementation of a passive surveillance programme. Zoonoses Public Health 67, 186–192. doi:10.1111/zph.12670
Ducancel, F., and Muller, B. H. (2012). Molecular engineering of antibodies for therapeutic and diagnostic purposes. MAbs 4, 445–457. doi:10.4161/mabs.20776
Duffy, S. (2018). Why are RNA virus mutation rates so damn high?. PLoS Biol. 16, 1–6. doi:10.1371/journal.pbio.3000003
Ecker, C., Ertl, A., Pulverer, W., Nemes, A., Szekely, P., Petrasch, A., et al. (2013). Validation and comparison of a sandwich ELISA, two competitive ELISAs and a real-time PCR method for the detection of lupine in food. Food Chem. 141, 407–418. doi:10.1016/j.foodchem.2013.02.091
Eigner, U., Reucher, S., Hefner, N., Staffa-Peichl, S., Kolb, M., Betz, U., et al. (2019). Clinical evaluation of multiplex RT-PCR assays for the detection of influenza A/B and respiratory syncytial virus using a high throughput system. J. Virol. Methods 269, 49–54. doi:10.1016/j.jviromet.2019.03.015
Eiras, A. E., Pires, S. F., Staunton, K. M., Paixão, K. S., Resende, M. C., Silva, H. A., et al. (2018). A high-risk Zika and dengue transmission hub: virus detections in mosquitoes at a Brazilian university campus. Parasit. Vectors 11, 359. doi:10.1186/s13071-018-2883-8
Estrela, P. F. N., Mendes, G. de. M., de Oliveira, K. G., Bailão, A. M., Soares, C. M. de. A., Assunção, N. A., et al. (2019). Ten-minute direct detection of Zika virus in serum samples by RT-LAMP. J. Virol. Methods 271, 113675. doi:10.1016/j.jviromet.2019.113675
Euler, M., Wang, Y., Heidenreich, D., Patel, P., Strohmeier, O., Hakenberg, S., et al. (2013). Development of a panel of recombinase polymerase amplification assays for detection of biothreat agents. J. Clin. Microbiol. 51, 1110–1117. doi:10.1128/JCM.02704-12
Euler, M., Wang, Y., Nentwich, O., Piepenburg, O., Hufert, F. T., and Weidmann, M. (2012). Recombinase polymerase amplification assay for rapid detection of Rift Valley fever virus. J. Clin. Virol. 54, 308–312. doi:10.1016/j.jcv.2012.05.006
Falconar, A. K. I., and Romero-Vivas, C. M. E. (2013). A simple, inexpensive, robust and sensitive dot-blot assay for equal detection of the nonstructural-1 glycoprotein of all dengue virus serotypes. Virol. J. 10, 126. doi:10.1186/1743-422X-10-126
Fenner, F., McAuslan, B. R., Mims, C. A., Sambrook, J., and White, D. O. (1974). “Cultivation, assay and analysis of viruses,” in The Biology of Animal Viruses. Editors F. Fenner, B. R. McAuslan, C. A. Mims, J. Sambrook, and D. O. White (New York, NY and London, United Kingdom: Academic Press), 35–57.
Francois, P., Tangomo, M., Hibbs, J., Bonetti, E. J., Boehme, C. C., Notomi, T., et al. (2011). Robustness of a loop-mediated isothermal amplification reaction for diagnostic applications. FEMS Immunol. Med. Microbiol. 62, 41–48. doi:10.1111/j.1574-695X.2011.00785.x
Freire, J. M., Santos, N. C., Veiga, A. S., Da Poian, A. T., and Castanho, M. A. R. B. (2015). Rethinking the capsid proteins of enveloped viruses: multifunctionality from genome packaging to genome transfection. FEBS J. 282, 2267–2278. doi:10.1111/febs.13274
Fu, M., Chen, G., Zhang, C., Wang, Y., Sun, R., and Zhou, J. (2019). Rapid and sensitive detection method for Karlodinium veneficum by recombinase polymerase amplification coupled with lateral flow dipstick. Harmful Algae 31, 1–9. doi:10.1016/j.hal.2019.01.011
Fulladolsa Palma, A. C., Kota, R., and Charkowski, A. O. (2013). Optimization of a chemiluminescent dot-blot immunoassay for detection of potato viruses. Am. J. Potato Res. 90, 306–312. doi:10.1007/s12230-013-9305-4
Fux, R., Söckler, C., Link, E. K., Renken, C., Krejci, R., Sutter, G., et al. (2018). Full genome characterization of porcine circovirus type 3 isolates reveals the existence of two distinct groups of virus strains. Virol. J. 15, 25. doi:10.1186/s12985-018-0929-3
Gallagher, S., Winston, S. E., Fuller, S. A., and Hurrell, J. G. (2011). Immunoblotting and immunodetection. Curr. Protoc. Cell Biol. 52, 6.2.1–6.2.28. doi:10.1002/0471143030.cb0602s52
Gandelman, O., Jackson, R., Kiddle, G., and Tisi, L. (2011). Loop-mediated amplification accelerated by stem primers. Int. J. Mol. Sci. 12, 9108–9124. doi:10.3390/ijms12129108
George, St. K., Patel, N. M., Hartwig, R. A., Scholl, D. R., Jollick, J. A., Kauffmann, L. M., et al. (2002). Rapid and sensitive detection of respiratory virus infections for directed antiviral treatment using R-Mix cultures. J. Clin. Virol. 24, 107–115. doi:10.1016/S1386-6532(01)00239-6
Geysen, H. M., Barteling, S. J., and Meloen, R. H. (1985). Small peptides induce antibodies with a sequence and structural requirement for binding antigen comparable to antibodies raised agaist the native protein. Proc. Natl. Acad. Sci. USA 82, 178–182. doi:10.1073/pnas.82.1.178
Gleaves, C. A., Smith, T. F., Shuster, E. A., and Pearson, G. R. (1984). Rapid detection of cytomegalovirus in MRC-5 cells inoculated with urine specimens by using low-speed centrifugation and monoclonal antibody to an early antigen. J. Clin. Microbiol. 19, 917–919. doi:10.1128/jcm.19.6.917-919.1984
Goldmeyer, J., Kong, H., and Tang, W. (2007). Development of a novel one-tube isothermal reverse transcription thermophilic helicase-dependent amplification platform for rapid RNA detection. J. Mol. Diagn. 9, 639–644. doi:10.2353/jmoldx.2007.070012
Gómez, R. S., Mora, J. E., Cortés, C. M., Riedel, C. A., Ferrés, M., Bueno, S. M., et al. (2014). Respiratory syncytial virus detection in cells and clinical samples by using three new monoclonal antibodies. J. Med. Virol. 86, 1256–1266. doi:10.1002/jmv.23807
Gootenberg, J. S., Abudayyeh, O. O., Kellner, M. J., Joung, J., Collins, J. J., and Zhang, F. (2018). Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a and Csm6. Science 360, 439–444. doi:10.1126/science.aaq0179
Gootenberg, J. S., Abudayyeh, O. O., Lee, J. W., Essletzbichler, P., Dy, A. J., Joung, J., et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442. doi:10.1126/science.aam9321
Gorman, K. T., Roby, L. C., Giuffre, A., Huang, R., and Kay, B. K. (2017). Tandem phage-display for the identification of non-overlapping binding pairs of recombinant Affinity reagents. Nucleic Acids Res. 45, e158. doi:10.1093/nar/gkx688
Goto, K., Yamaoka, Y., Khatun, H., Miyakawa, K., Nishi, M., Nagata, N., et al. (2020). Development of monoclonal antibodies and antigen-capture ELISA for human parechovirus type 3. Microorganisms 8, 1437. doi:10.3390/microorganisms8091437
Hamidi, S. V., and Ghourchian, H. (2015). Colorimetric monitoring of rolling circle amplification for detection of H5N1 influenza virus using metal indicator. Biosens. Bioelectron. 72, 121–126. doi:10.1016/j.bios.2015.04.078
Hamidi, S. V., Ghourchian, H., and Tavoosidana, G. (2015b). Real-time detection of H5N1 influenza virus through hyperbranched rolling circle amplification. Analyst 140, 1502–1509. doi:10.1039/c4an01954g
He, J. (2013). “Practical guide to ELISA devlopment,” in The Immunoassay Handbook: Theory and Applications of Ligand Binding, ELISA and Related Techniques. Editor D. Wild (Oxford, United Kingdom: Elsevier), 381–394.
He, L., and Xu, H. S. (2011). Development of a multiplex loop-mediated isothermal amplification (mLAMP) method for the simultaneous detection of white spot syndrome virus and infectious hypodermal and hematopoietic necrosis virus in penaeid shrimp. Aquaculture 311, 94–99. doi:10.1016/j.aquaculture.2010.11.051
Hematian, A., Sadeghifard, N., Mohebi, R., Taherikalani, M., Nasrolahi, A., Amraei, M., et al. (2016). Traditional and modern cell culture in virus diagnosis. Osong Public Heal. Res. Perspect. 7, 77–82. doi:10.1016/j.phrp.2015.11.011
Hsieh, K., Mage, P. L., Csordas, A. T., Eisenstein, M., and Tom Soh, H. (2014). Simultaneous elimination of carryover contamination and detection of DNA with uracil-DNA-glycosylase-supplemented loop-mediated isothermal amplification (UDG-LAMP). Chem. Commun. 50, 3747–3749. doi:10.1039/c4cc00540f
Hu, J., Jiang, Y. Z., Wu, L. L., Wu, Z., Bi, Y., Wong, G., et al. (2017). Dual-signal readout nanospheres for rapid point-of-care detection of Ebola virus glycoprotein. Anal. Chem. 89, 13105–13111. doi:10.1021/acs.analchem.7b02222
Huang, H. S., Tsai, C. L., Chang, J., Hsu, T. C., Lin, S., and Lee, C. C. (2018). Multiplex PCR system for the rapid diagnosis of respiratory virus infection: systematic review and meta-analysis. Clin. Microbiol. Infect. 24, 1055–1063. doi:10.1016/j.cmi.2017.11.018
Huang, W. E., Lim, B., Hsu, C. C., Xiong, D., Wu, W., Yu, Y., et al. (2020). RT-LAMP for rapid diagnosis of coronavirus SARS-CoV-2. Microb. Biotechnol. 13, 950–961. doi:10.1111/1751-7915.13586
Hwang, K. A., Ahn, J. H., and Nam, J. H. (2018). Development and validation of multiplex real-time PCR assays for rapid detection of cytomegalovirus, Epstein-Barr virus, and polyomavirus BK in whole blood from transplant candidates. J. Microbiol. 56, 593–599. doi:10.1007/s12275-018-8273-2
Jääskeläinen, A. J., Sironen, T., Diagne, C. T., Diagne, M. M., Faye, M., Faye, O., et al. (2019). Development, validation and clinical evaluation of a broad-range pan-filovirus RT-qPCR. J. Clin. Virol. 114, 26–31. doi:10.1016/j.jcv.2019.03.010
Jagdale, S. S., and Joshi, R. S. (2018). Enemies with benefits: mutualistic interactions of viruses with lower eukaryotes. Arch. Virol. 163, 821–830. doi:10.1007/s00705-017-3686-5
Jansen van Vuren, P., and Paweska, J. T. (2009). Laboratory safe detection of nucleocapsid protein of Rift Valley fever virus in human and animal specimens by a sandwich ELISA. J. Virol. Methods 157, 15–24. doi:10.1016/j.jviromet.2008.12.003
Jayanath, N. Y., Nguyen, L. T., Vu, T. T., and Tran, L. D. (2018). Development of a portable electrochemical loop mediated isothermal amplification (LAMP) device for detection of hepatitis B virus. RSC Adv. 8, 34954–34959. doi:10.1039/c8ra07235c
Jeong, K. J., Jang, S. H., and Velmurugan, N. (2011). Recombinant antibodies: engineering and production in yeast and bacterial hosts. Biotechnol. J. 6, 16–27. doi:10.1002/biot.201000381
Jevšnik, M., Lusa, L., Uršič, T., Glinšek Biškup, U., and Petrovec, M. (2020). Detection of herpes simplex and varicella-zoster virus from skin lesions: comparison of RT-PCR and isothermal amplification for rapid identification. Diagn. Microbiol. Infect. Dis. 97, 115015. doi:10.1016/j.diagmicrobio.2020.115015
Jones, C., Stewart, S., Woodworth, J., Dritz, S., and Paulk, C. (2020). Validation of sampling methods in bulk feed ingredients for detection of swine viruses. Transbound. Emerg. Dis. 67, 1–5. doi:10.1111/tbed.13326
Kaneko, H., Kawana, T., Fukushima, E., and Suzutani, T. (2007). Tolerance of loop-mediated isothermal amplification to a culture medium and biological substances. J. Biochem. Biophys. Methods 70, 499–501. doi:10.1016/j.jbbm.2006.08.008
Kaocharoen, S., Wang, B., Tsui, K. M., Trilles, L., Kong, F., and Meyer, W. (2008). Hyperbranched rolling circle amplification as a rapid and sensitive method for species identification within the Cryptococcus species complex. Electrophoresis 29, 3183–3191. doi:10.1002/elps.200700903
Kasamatsu, H., and Vinograd, J. (1974). Replication of circular DNA in eukaryotic cells. Annu. Rev. Biochem. 43, 695–719. doi:10.1146/annurev.bi.43.070174.003403
Kellner, M. J., Koob, J. G., Gootenberg, J. S., Abudayyeh, O. O., and Zhang, F. (2019). SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 14, 2986–3012. doi:10.1038/s41596-019-0210-2
Khudyakov, Y., and Kamili, S. (2011). Serological diagnostics of hepatitis E virus infection. Virus Res. 161, 84–92. doi:10.1016/j.virusres.2011.06.006
Kim, H. J., Tong, Y., Tang, W., Quimson, L., Cope, V. A., Pan, X., et al. (2011). A rapid and simple isothermal nucleic acid amplification test for detection of herpes simplex virus types 1 and 2. J. Clin. Virol. 50, 26–30. doi:10.1016/j.jcv.2010.09.006
Kolm, C., Martzy, R., Führer, M., Mach, R. L., Krska, R., Baumgartner, S., et al. (2019). Detection of a microbial source tracking marker by isothermal helicase-dependent amplification and a nucleic acid lateral-flow strip test. Sci. Rep. 9, 393. doi:10.1038/s41598-018-36749-7
Kumar, S., Bhushan, P., Krishna, V., and Bhattacharya, S. (2018). Tapered lateral flow immunoassay based point-of-care diagnostic device for ultrasensitive colorimetric detection of dengue NS1. Biomicrofluidics 12, 034104. doi:10.1063/1.5035113
Kunert, R., and Reinhart, D. (2016). Advances in recombinant antibody manufacturing. Appl. Microbiol. Biotechnol. 100, 3451–3461. doi:10.1007/s00253-016-7388-9
LaSala, P. R., Bufton, K. K., Ismail, N., and Smith, M. B. (2007). Prospective comparison of R-mixTM shell vial system with direct antigen tests and conventional cell culture for respiratory virus detection. J. Clin. Virol. 38, 210–216. doi:10.1016/j.jcv.2006.12.015
Lebani, K., Jones, M. L., Watterson, D., Ranzoni, A., Traves, R. J., Young, P. R., et al. (2017). Isolation of serotype-specific antibodies against dengue virus non-structural protein 1 using phage display and application in a multiplexed serotyping assay. PLoS One 12, e0180669. doi:10.1371/journal.pone.0180669
Leland, D. S., and Ginocchio, C. C. (2007). Role of cell culture for virus detection in the age of technology. Clin. Microbiol. Rev. 20, 49–78. doi:10.1128/CMR.00002-06
Li, C., Jaentschke, B., Song, Y., Wang, J., Cyr, T. D., Van Domselaar, G., et al. (2010). A simple slot blot for the detection of virtually all subtypes of the influenza A viral hemagglutinins using universal antibodies targeting the fusion peptide. Nat. Protoc. 5, 14–19. doi:10.1038/nprot.2009.200
Li, J., and Macdonald, J. (2015). Advances in isothermal amplification: novel strategies inspired by biological processes. Biosens. Bioelectron. 64, 196–211. doi:10.1016/j.bios.2014.08.069
Li, S. Y., Cheng, Q. X., Li, X. Y., Zhang, Z. L., Gao, S., Cao, R. B., et al. (2018). CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 4, 18–21. doi:10.1038/s41421-018-0028-z
Li, X. H., Zhang, X. L., Wu, J., Lin, N., Sun, W. M., Chen, M., et al. (2019). Hyperbranched rolling circle amplification (HRCA)-based fluorescence biosensor for ultrasensitive and specific detection of single-nucleotide polymorphism genotyping associated with the therapy of chronic hepatitis B virus infection. Talanta 191, 277–282. doi:10.1016/j.talanta.2018.08.064
Li, Y., Kumar, N., Gopalakrishnan, A., Ginocchio, C., Manji, R., Bythrow, M., et al. (2013). Detection and species identification of malaria parasites by isothermal tHDA amplification directly from human blood without sample preparation. J. Mol. Diagn. 15, 634–641. doi:10.1016/j.jmoldx.2013.05.005
Li, Z., Yi, Y., Luo, X., Xiong, N., Liu, Y., Li, S., et al. (2020). Development and clinical application of A rapid IgM‐IgG combined antibody test for SARS‐CoV‐2 infection diagnosis. J. Med. Virol. 99, 1518–1524. doi:10.1002/jmv.25727
Liang, R. L., Deng, Q. T., Chen, Z. H., Xu, X. P., Zhou, J. W., Liang, J. Y., et al. (2017). Europium (III) chelate microparticle-based lateral flow immunoassay strips for rapid and quantitative detection of antibody to hepatitis B core antigen. Sci. Rep. 7, 14093. doi:10.1038/s41598-017-14427-4
Lillis, L., Lehman, D., Singhal, M. C., Cantera, J., Singleton, J., Labarre, P., et al. (2014). Non-instrumented incubation of a recombinase polymerase amplification assay for the rapid and sensitive detection of proviral HIV-1 DNA. PLoS One 9, e108189. doi:10.1371/journal.pone.0108189
Lin, C. Y., Hwang, D., Chiu, N. C., Weng, L. C., Liu, H. F., Mu, J. J., et al. (2020). Increased detection of viruses in children with respiratory tract infection using PCR. Int. J. Environ. Res. Public Health 17, 564. doi:10.3390/ijerph17020564
Lipson, S. M., David, K., Shaikh, F., and Qian, L. (2001). Detection of precytopathic effect of enteroviruses in clinical specimens by centrifugation-enhanced antigen detection. J. Clin. Microbiol. 39, 2755–2759. doi:10.1128/JCM.39.8.2755-2759.2001
Liu, F. G., Chen, G. F., Zhang, C. Y., Wang, Y. Y., and Zhou, J. (2019). Exponential rolling circle amplification coupled with lateral flow dipstick strips as a rapid and sensitive method for the field detection of Karlodinium veneficum. J. Appl. Phycol. 31, 2423–2436. doi:10.1007/s10811-019-01762-4
Liu, S. Q., Li, X., Deng, C. L., Yuan, Z. M., and Zhang, B. (2018). Development and evaluation of one-step multiplex real-time RT-PCR assay for simultaneous detection of Zika virus and Chikungunya virus. J. Med. Virol. 90, 389–396. doi:10.1002/jmv.24970
Lizardi, P. M., Huang, X., Zhu, Z., Bray-Ward, P., Thomas, D. C., and Ward, D. C. (1998). Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat. Genet. 19, 225–232. doi:10.1038/898
Lobato, I. M., and O’Sullivan, C. K. (2018). Recombinase polymerase amplification: basics, applications and recent advances. Trends Anal. Chem. 98, 19–35. doi:10.1016/j.trac.2017.10.015
Loftis, A. J., Quellie, S., Chason, K., Sumo, E., Toukolon, M., Otieno, Y., et al. (2017). Validation of the cepheid genexpert for detecting Ebola virus in semen. J. Infect. Dis. 215, 344–350. doi:10.1093/infdis/jiw562
Londoño, M. A., Harmon, C. L., and Polston, J. E. (2016). Evaluation of recombinase polymerase amplification for detection of begomoviruses by plant diagnostic clinics. Virol. J. 13, 48. doi:10.1186/s12985-016-0504-8
Lopez-Jimena, B., Bekaert, M., Bakheit, M., Frischmann, S., Patel, P., Simon-Loriere, E., et al. (2018). Development and validation of four one-step real-time RT-LAMP assays for specific detection of each dengue virus serotype. PLoS Negl. Trop. Dis. 12, e0006381. doi:10.1371/journal.pntd.0006381
Luo, G. C., Yi, T. T., Jiang, B., Guo, X. lan., and Zhang, G. Y. (2019). Betaine-assisted recombinase polymerase assay with enhanced specificity. Anal. Biochem. 575, 36–39. doi:10.1016/j.ab.2019.03.018
Ma, H., Ó’Fágáin, C., and O’Kennedy, R. (2019). Unravelling enhancement of antibody fragment stability – role of format structure and cysteine modification. J. Immunol. Methods 464, 57–63. doi:10.1016/j.jim.2018.10.012
Ma, H., and O’Kennedy, R. (2015). The structure of natural and recombinant antibodies. Methods Mol. Biol. 1348, 7–11. doi:10.1007/978-1-4939-2999-3_2
Mak, W. C., Beni, V., and Turner, A. P. F. (2016). Lateral-flow technology: from visual to instrumental. Trends Anal. Chem. 79, 297–305. doi:10.1016/j.trac.2015.10.017
Maree, H. J., Fox, A., Al Rwahnih, M., Boonham, N., and Candresse, T. (2018). Application of HTS for routine plant virus diagnostics: state of the art and challenges. Front. Plant Sci. 9, 1–4. doi:10.3389/fpls.2018.01082
Marincevic-Zuniga, Y., Gustavsson, I., and Gyllensten, U. (2012). Multiply-primed rolling circle amplification of human papillomavirus using sequence-specific primers. Virology 432, 57–62. doi:10.1016/j.virol.2012.05.030
Martineau, R. L., Murray, S. A., Ci, S., Gao, W., Chao, S. H., and Meldrum, D. R. (2017). Improved performance of loop-mediated isothermal amplification assays via swarm priming. Anal. Chem. 89, 625–632. doi:10.1021/acs.analchem.6b02578
Massart, S., Olmos, A., Jijakli, H., and Candresse, T. (2014). Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 188, 90–96. doi:10.1016/j.virusres.2014.03.029
Mateu, M. G. (2013). Assembly, stability and dynamics of virus capsids. Arch. Biochem. Biophys. 531, 65–79. doi:10.1016/j.abb.2012.10.015
McLeish, M. J., Fraile, A., and García-Arenal, F. (2019). Evolution of plant–virus interactions: host range and virus emergence. Curr. Opin. Virol. 34, 50–55. doi:10.1016/j.coviro.2018.12.003
Metcalf, T. G. (1995). Environmental virology: from detection of virus in sewage and water by isolation to identification by molecular biology--A trip of over 50 years. Annu. Rev. Microbiol. 49, 461–487. doi:10.1146/annurev.mi.49.100195.002333
Mezger, A., Öhrmalm, C., Herthnek, D., Blomberg, J., and Nilsson, M. (2014). Detection of rotavirus using padlock probes and rolling circle amplification. PLoS One 9. doi:10.1371/journal.pone.0111874
Mohsen, M. G., and Kool, E. T. (2016). The discovery of rolling circle amplification and rolling circle transcription. Acc. Chem. Res. 49, 2540–2550. doi:10.1021/acs.accounts.6b00417
Motohashi, K. (2019). Development of highly sensitive and low-cost DNA agarose gel electrophoresis detection systems, and evaluation of non-mutagenic and loading dye-type DNA-staining reagents. PLoS One 14, 1–13. doi:10.1371/journal.pone.0222209
Mullis, K., Faloona, F., Scharf, S., Saiki, R., Horn, G., and Erlich, H. (1986). Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb. Symp. Quant. Biol. 51, 263–273. doi:10.1101/sqb.1986.051.01.032
Myhrvold, C., Freije, C. A., Gootenberg, J. S., Abudayyeh, O. O., Metsky, H. C., Durbin, A. F., et al. (2018). Field-deployable viral diagnostics using CRISPR-Cas13. Science 360, 444–448. doi:10.1126/science.aas8836
Na, W., Nam, D., Lee, H., and Shin, S. (2018). Rapid molecular diagnosis of infectious viruses in microfluidics using DNA hydrogel formation. Biosens. Bioelectron. 108, 9–13. doi:10.1016/j.bios.2018.02.040
Naccache, S. N., Peggs, K. S., Mattes, F. M., Phadke, R., Garson, J. A., Grant, P., et al. (2015). Diagnosis of neuroinvasive astrovirus infection in an immunocompromised adult with encephalitis by unbiased next-generation sequencing. Clin. Infect. Dis. 60, 919–923. doi:10.1093/cid/ciu912
Nagamine, K., Hase, T., and Notomi, T. (2002). Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell. Probes 16, 223–229. doi:10.1006/mcpr.2002.0415
Nagy, A., Jiřinec, T., Jiřincová, H., Černíková, L., and Havlíčková, M. (2019). In silico re-assessment of a diagnostic RT-qPCR assay for universal detection of influenza A viruses. Sci. Rep. 9, 1630. doi:10.1038/s41598-018-37869-w
Navarro, E., Serrano-Heras, G., Castaño, M. J., and Solera, J. (2015). Real-time PCR detection chemistry. Clin. Chim. Acta 439, 231–250. doi:10.1016/j.cca.2014.10.017
Neumann, F., Hernández-Neuta, I., Grabbe, M., Madaboosi, N., Albert, J., and Nilsson, M. (2018). Padlock probe assay for detection and subtyping of seasonal influenza. Clin. Chem. 64, 1704–1712. doi:10.1373/clinchem.2018.292979
Nilsson, M., Gullberg, M., Dahl, F., Szuhai, K., and Raap, A. K. (2002). Real-time monitoring of rolling-circle amplification using a modified molecular beacon design. Nucleic Acids Res. 30, e66. doi:10.1093/nar/gnf065
Notomi, T., Mori, Y., Tomita, N., and Kanda, H. (2015). Loop-mediated isothermal amplification (LAMP): principle, features, and future prospects. J. Microbiol. 53, 1–5. doi:10.1007/s12275-015-4656-9
Notomi, T., Okayama, H., Masubuchi, H., Yonekawa, T., Watanabe, K., Amino, N., et al. (2000). Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, e63. doi:10.1093/nar/28.12.e63
Nunes, B. T. D., De Mendonça, M. H. R., De Brito Simith, D., Moraes, A. F., Cardoso, C. C., Prazeres, I. T. E., et al. (2019). Development of RT-qPCR and semi-nested RT-PCR assays for molecular diagnosis of hantavirus pulmonary syndrome. PLoS Negl. Trop. Dis. 13, e0007884. doi:10.1371/JOURNAL.PNTD.0007884
Olivo, P. D. (1996). Transgenic cell lines for detection of animal viruses. Clin. Microbiol. Rev. 9, 321–334. doi:10.1128/cmr.9.3.321
Ou, T. P., Yun, C., Auerswald, H., In, S., Leang, R., Huy, R., et al. (2020). Improved detection of dengue and Zika viruses using multiplex RT-qPCR assays. J. Virol. Methods 282, 113862. doi:10.1016/j.jviromet.2020.113862
O’Kennedy, R., Fitzgerald, S., and Murphy, C. (2017). Don’t blame it all on antibodies – the need for exhaustive characterisation, appropriate handling, and addressing the issues that affect specificity. Trends Anal. Chem. 89, 53–59. doi:10.1016/j.trac.2017.01.009
Paez-Espino, D., Eloe-Fadrosh, E. A., Pavlopoulos, G. A., Thomas, A. D., Huntemann, M., Mikhailova, N., et al. (2016). Uncovering Earth’s virome. Nature 536, 425–430. doi:10.1038/nature19094
Palacios, G., Druce, J., Du, L., Tran, T., Birch, C., Briese, T., et al. (2008). A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med. 358, 991–998. doi:10.1056/NEJMoa073785
Parida, M., Sannarangaiah, S., Dash, P. K., Rao, P. V. L., and Morita, K. (2008). Loop mediated isothermal amplification(LAMP): a new generation of innovative geneamplification technique; perspectives in clinicaldiagnosis of infectious diseases. Rev. Med. Virol. 18, 407–421. doi:10.1002/rmv10.1002/rmv.593
Patel, J., and Sharma, P. (2020). Design of a novel rapid immunoassay for simultaneous detection of hepatitis C virus core antigen and antibodies. Arch. Virol. 165, 627–641. doi:10.1007/s00705-019-04518-0
Pemba, C. M., Kurosaki, Y., Yoshikawa, R., Oloniniyi, O. K., Urata, S., Sueyoshi, M., et al. (2019). Development of an RT-LAMP assay for the detection of Lassa viruses in southeast and south-central Nigeria. J. Virol. Methods 269, 30–37. doi:10.1016/j.jviromet.2019.04.010
Peterson, E. M., Hughes, B. L., Aarnaes, S. L., and De la Maza, L. M. (1988). Comparison of primary rabbit kidney and MRC-5 cells and two stain procedures for herpes simplex virus detection by a shell vial centrifugation method. J. Clin. Microbiol. 26, 222–224. doi:10.1128/jcm.26.2.222-224.1988
Piepenburg, O., Williams, C. H., Stemple, D. L., and Armes, N. A. (2006). DNA detection using recombination proteins. PLoS Biol. 4, 1115–1121. doi:10.1371/journal.pbio.0040204
Posthuma-Trumpie, G. A., Korf, J., and Van Amerongen, A. (2009). Lateral flow (immuno)assay: its strengths, weaknesses, opportunities and threats. A literature survey. Anal. Bioanal. Chem. 393, 569–582. doi:10.1007/s00216-008-2287-2
Powell, M. L., Bowler, F. R., Martinez, A. J., Greenwood, C. J., Armes, N., and Piepenburg, O. (2018). New Fpg probe chemistry for direct detection of recombinase polymerase amplification on lateral flow strips. Anal. Biochem. 543, 108–115. doi:10.1016/j.ab.2017.12.003
Prachayangprecha, S., Schapendonk, C. M. E., Koopmans, M. P., Osterhaus, A. D. M. E., Schürch, A. C., Pas, S. D., et al. (2014). Exploring the potential of next-generation sequencing in detection of respiratory viruses. J. Clin. Microbiol. 52, 3722–3730. doi:10.1128/JCM.01641-14
Pradeu, T. (2016). Mutualistic viruses and the heteronomy of life. Stud. Hist. Philos. Sci. Part C Stud. Hist. Philos. Biol. Biomed. Sci. 59, 80–88. doi:10.1016/j.shpsc.2016.02.007
PrimerExplorer (2020). Available at: https://primerexplorer.jp/e/ (Accessed April 15, 2020).
Pring-Åkerblom, P., Adrian, T., and Köstler, T. (1997). PCR-based detection and typing of human adenoviruses in clinical samples. Res. Virol. 148, 225–231. doi:10.1016/S0923-2516(97)83992-1
Prussin, A. J., Torres, P. J., Shimashita, J., Head, S. R., Bibby, K. J., Kelley, S. T., et al. (2019). Seasonal dynamics of DNA and RNA viral bioaerosol communities in a daycare center. Microbiome 7, 1–14. doi:10.1186/s40168-019-0672-z
Qin, P., Park, M., Alfson, K. J., Tamhankar, M., Carrion, R., Patterson, J. L., et al. (2019). Rapid and fully microfluidic Ebola virus detection with CRISPR-cas13a. ACS Sensors 4, 1048–1054. doi:10.1021/acssensors.9b00239
Quyen, T. L., Ngo, T. A., Bang, D. D., Madsen, M., and Wolff, A. (2019). Classification of multiple DNA dyes based on inhibition effects on real-time loop-mediated isothermal amplification (LAMP): prospect for point of care setting. Front. Microbiol. 10, 2234. doi:10.3389/fmicb.2019.02234
Rabalais, G. P., Stout, G. G., Ladd, K. L., and Cost, K. M. (1992). Rapid diagnosis of respiratory viral infections by using a shell vial assay and monoclonal antibody pool. J. Clin. Microbiol. 30, 1505–1508. doi:10.1128/jcm.30.6.1505-1508.1992
Radford, A. D., Chapman, D., Dixon, L., Chantrey, J., Darby, A. C., and Hall, N. (2012). Application of next-generation sequencing technologies in virology. J. Gen. Virol. 93, 1853–1868. doi:10.1099/vir.0.043182-0
Rong, Z., Wang, Q., Sun, N., Jia, X., Wang, K., Xiao, R., et al. (2019). Smartphone-based fluorescent lateral flow immunoassay platform for highly sensitive point-of-care detection of Zika virus nonstructural protein 1. Anal. Chim. Acta 1055, 140–147. doi:10.1016/j.aca.2018.12.043
Rosario, K., and Breitbart, M. (2011). Exploring the viral world through metagenomics. Curr. Opin. Virol. 1, 289–297. doi:10.1016/j.coviro.2011.06.004
Rosser, A., Rollinson, D., Forrest, M., and Webster, B. L. (2015). Isothermal Recombinase Polymerase amplification (RPA) of Schistosoma haematobium DNA and oligochromatographic lateral flow detection. Parasites and Vectors 8, 446. doi:10.1186/s13071-015-1055-3
Rott, M. E., Kesanakurti, P., Berwarth, C., Rast, H., Boyes, I., Phelan, J., et al. (2018). Discovery of negative-sense RNA viruses in trees infected with apple rubbery wood disease by next-generation sequencing. Plant Dis. 102, 1254–1263. doi:10.1094/PDIS-06-17-0851-RE
Ryu, S. W., Lee, J. H., Kim, J., Jang, M. A., Nam, J. H., Byoun, M. S., et al. (2016). Comparison of two new generation influenza rapid diagnostic tests with instrument-based digital readout systems for influenza virus detection. Br. J. Biomed. Sci. 73, 115–120. doi:10.1080/09674845.2016.1189026
Sajid, M., Kawde, A. N., and Daud, M. (2015). Designs, formats and applications of lateral flow assay: a literature review. J. Saudi Chem. Soc. 19, 689–705. doi:10.1016/j.jscs.2014.09.001
Sánchez-Ponce, Y., Varela-Fascinetto, G., Romo-Vázquez, J. C., López-Martínez, B., Sánchez-Huerta, J. L., Parra-Ortega, I., et al. (2018). Simultaneous detection of beta and gamma human herpesviruses by multiplex qPCR reveals simple infection and coinfection episodes increasing risk for graft rejection in solid organ transplantation. Viruses 10, 730. doi:10.3390/v10120730
Sanjuán, R., and Domingo-Calap, P. (2016). Mechanisms of viral mutation. Cell. Mol. Life Sci. 73, 4433–4448. doi:10.1007/s00018-016-2299-6
Sastre, P., Gallardo, C., Monedero, A., Ruiz, T., Arias, M., Sanz, A., et al. (2016). Development of a novel lateral flow assay for detection of African swine fever in blood. BMC Vet. Res. 12, 1–8. doi:10.1186/s12917-016-0831-4
Schweitzer, B., and Kingsmore, S. (2001). Combining nucleic acid amplification and detection. Curr. Opin. Biotechnol. 12, 21–27. doi:10.1016/S0958-1669(00)00172-5
Si, J., Li, J., Zhang, L., Zhang, W., Yao, J., Li, T., et al. (2019). A signal amplification system on a lateral flow immunoassay detecting for hepatitis e-antigen in human blood samples. J. Med. Virol. 91, 1301–1306. doi:10.1002/jmv.25452
Silva, S. J. R. da., Paiva, M. H. S., Guedes, D. R. D., Krokovsky, L., Melo, F. L., de, Silva, M. A. L. da., et al. (2019). Development and validation of reverse transcription loop-mediated isothermal amplification (RT-LAMP) for rapid detection of ZIKV in mosquito samples from Brazil. Sci. Rep. 9, 4494. doi:10.1038/s41598-019-40960-5
Soares, R. R. G., Varela, J. C., Neogi, U., Ciftci, S., Ashokkumar, M., Pinto, I. F., et al. (2020). Sub-attomole detection of HIV-1 using padlock probes and rolling circle amplification combined with microfluidic affinity chromatography. Biosens. Bioelectron. 166, 112442. doi:10.1016/j.bios.2020.112442
Somasekar, S., Lee, D., Rule, J., Naccache, S. N., Stone, M., Busch, M. P., et al. (2017). Viral surveillance in serum samples from patients with acute liver failure by metagenomic next-generation sequencing. Clin. Infect. Dis. 65, 1477–1485. doi:10.1093/cid/cix596
Storch, G. A. (2000). Diagnostic virology. Clin. Infect. Dis. 31, 739–751. doi:10.1086/31379010.1086/314015
Sun, N., Wang, Y., Yao, X., Chen, F., Gao, D., Wang, W., et al. (2019). Visual signal generation for the detection of influenza viruses by duplex recombinase polymerase amplification with lateral flow dipsticks. Anal. Bioanal. Chem. 411, 3591–3602. doi:10.1007/s00216-019-01840-z
Tanner, N. A., Zhang, Y., and Evans, T. C. (2012). Simultaneous multiple target detection in real-time loop-mediated isothermal amplification. Biotechniques 53, 81–89. doi:10.2144/0000113902
Teo, J. W. P., Chiang, D., Jureen, R., and Lin, R. T. P. (2015). Clinical evaluation of a helicase-dependant amplification (HDA)-based commercial assay for the simultaneous detection of HSV-1 and HSV-2. Diagn. Microbiol. Infect. Dis. 83, 261–262. doi:10.1016/j.diagmicrobio.2015.07.018
Thiruppathiraja, C., Kumar, S., Murugan, V., Adaikkappan, P., Sankaran, K., and Alagar, M. (2011). An enhanced immuno-dot blot assay for the detection of white spot syndrome virus in shrimp using antibody conjugated gold nanoparticles probe. Aquaculture 318, 262–267. doi:10.1016/j.aquaculture.2011.06.008
Tillmann, H. L. (2014). Hepatitis C virus core antigen testing: role in diagnosis, disease monitoring and treatment. World J. Gastroenterol. 20, 6701–6706. doi:10.3748/wjg.v20.i22.6701
Tomita, N., Mori, Y., Kanda, H., and Notomi, T. (2008). Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat. Protoc. 3, 877–882. doi:10.1038/nprot.2008.57
Tong, Y., Tang, W., Kim, H. J., Pan, X., Ranalli, T. A., and Kong, H. (2008). Development of isothermal TaqMan assays for detection of biothreat organisms. Biotechniques 45, 543–557. doi:10.2144/000112959
Usuda, S., Okamoto, H., Tanaka, T., Kidd-Ljunggren, K., Holland, P. V., Miyakawa, Y., et al. (2000). Differentiation of hepatitis B virus genotypes D and E by ELISA using monoclonal antibodies to epitopes on the preS2-region product. J. Virol. Methods 87, 81–89. doi:10.1016/s0166-0934(00)00153-1
Van Wesenbeeck, L., Meeuws, H., Van Immerseel, A., Ispas, G., Schmidt, K., Houspie, L., et al. (2013). Comparison of the FilmArray RP, Verigene RV+, and Prodesse ProFLU+/FAST+ multiplex platforms for detection of influenza viruses in clinical samples from the 2011-2012 influenza season in Belgium. J. Clin. Microbiol. 51, 2977–2985. doi:10.1128/JCM.00911-13
Vincent, M., Xu, Y., and Kong, H. (2004). Helicase-dependent isothermal DNA amplification. EMBO Rep. 5, 795–800. doi:10.1038/sj.embor.7400200
Wadhwa, A., Wilkins, K., Gao, J., Condori Condori, R. E., Gigante, C. M., Zhao, H., et al. (2017). A pan-lyssavirus taqman real-time RT-PCR assay for the detection of highly variable rabies virus and other lyssaviruses. PLoS Negl. Trop. Dis. 11, e0005258. doi:10.1371/journal.pntd.0005258
Wand, N. I. V., Bonney, L. C., Watson, R. J., Graham, V., and Hewson, R. (2018). Point-of-care diagnostic assay for the detection of Zika virus using the recombinase polymerase amplification method. J. Gen. Virol. 99, 1012–1026. doi:10.1099/jgv.0.001083
Wang, S., Liu, N., Zheng, L., Cai, G., and Lin, J. (2020). A lab-on-chip device for the sample-in-result-out detection of viable Salmonella using loop-mediated isothermal amplification and real-time turbidity monitoring. Lab Chip 20, 2296–2305. doi:10.1039/d0lc00290a
Wiriyachaiporn, N., Sirikett, H., Maneeprakorn, W., and Dharakul, T. (2017). Carbon nanotag based visual detection of influenza A virus by a lateral flow immunoassay. Microchim. Acta 184, 1827–1835. doi:10.1007/s00604-017-2191-6
Wong, Y. P., Othman, S., Lau, Y. L., Radu, S., and Chee, H. Y. (2018). Loop-mediated isothermal amplification (LAMP): a versatile technique for detection of micro-organisms. J. Appl. Microbiol. 124, 626–643. doi:10.1111/jam.13647
Wu, D. Y., Ugozzoli, L., Pal, B. K., Qian, J., and Wallace, R. B. (1991). The effect of temperature and oligonucleotide primer length on the specificity and efficiency of amplification by the polymerase chain reaction. DNA Cell Biol 10, 233–238. doi:10.1089/dna.1991.10.233
Xi, Y., Xu, C. Z., Xie, Z. Z., Zhu, D. L., Dong, J. M., and Xiao, G. (2019). Development of a reverse transcription recombinase polymerase amplification assay for rapid detection of human respiratory syncytial virus. Mol. Cell. Probes 45, 8–13. doi:10.1016/j.mcp.2019.03.005
Xin, Z., and Chen, J. (2012). A high throughput DNA extraction method with high yield and quality. Plant Methods 8, 26. doi:10.1186/1746-4811-8-26
Yang, Y., Qin, X., Wang, G., Zhang, Y., Shang, Y., and Zhang, Z. (2015). Development of a fluorescent probe-based recombinase polymerase amplification assay for rapid detection of Orf virus veterinary DNA viruses. Virol. J. 12, 1–7. doi:10.1186/s12985-015-0440-z
Yoon, J., Yun, S. G., Nam, J., Choi, S. H., and Lim, C. S. (2017). The use of saliva specimens for detection of influenza A and B viruses by rapid influenza diagnostic tests. J. Virol. Methods 243, 15–19. doi:10.1016/j.jviromet.2017.01.013
Yu, C., Wu, J., and Zhou, X. (2005). Detection and subgrouping of Cucumber mosaic virus isolates by TAS-ELISA and immunocapture RT-PCR. J. Virol. Methods 123, 155–161. doi:10.1016/j.jviromet.2004.09.014
Yuan, T., Mukama, O., Li, Z., Chen, W., Zhang, Y., De Dieu Habimana, J., et al. (2020). A rapid and sensitive CRISPR/Cas12a based lateral flow biosensor for the detection of Epstein-Barr virus. Analyst 145, 6388–6394. doi:10.1039/d0an00663g
Zai, J., Yi, K., Xie, L., Zhu, J., Feng, X., and Li, Y. (2018). Dual monoclonal antibody-based sandwich ELISA for detection of in vitro packaged Ebola virus. Diagn. Pathol. 13, 96. doi:10.1186/s13000-018-0773-1
Zanoli, L. M., and Spoto, G. (2013). Isothermal amplification methods for the detection of nucleic acids in microfluidic devices. Biosensors 3, 18–43. doi:10.3390/bios3010018
Zhang, L., Du, X., Chen, C., Chen, Z., Zhang, L., Han, Q., et al. (2018). Development and characterization of double-antibody sandwich ELISA for detection of Zika virus infection. Viruses 10, 634. doi:10.3390/v10110634
Zhang, P., Lu, H., Chen, J., Han, H., and Ma, W. (2014a). Simple and sensitive detection of HBsAg by using a quantum dots nanobeads based dot-blot immunoassay. Theranostics 4, 307–315. doi:10.7150/thno.8007
Zhang, W., Du, R. H., Li, B., Zheng, X. S., Yang, X. L., Hu, B., et al. (2020). Molecular and serological investigation of 2019-nCoV infected patients: implication of multiple shedding routes. Emerg. Microbes Infect. 9, 386–389. doi:10.1080/22221751.2020.1729071
Keywords: immunoassay, isothermal amplification, next generation sequencing, virus, nucleic-acid detection, sampling
Citation: Cassedy A, Parle-McDermott A and O’Kennedy R (2021) Virus Detection: A Review of the Current and Emerging Molecular and Immunological Methods. Front. Mol. Biosci. 8:637559. doi: 10.3389/fmolb.2021.637559
Received: 03 December 2020; Accepted: 01 February 2021;
Published: 20 April 2021.
Edited by:
Amit Prasad, Indian Institute of Technology Mandi, IndiaReviewed by:
Vandana Saxena, National AIDS Research Institute (ICMR), IndiaCopyright © 2021 Cassedy, Parle-McDermott and O’Kennedy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: R. O'Kennedy, cmljaGFyZG9rZW5uZWR5QGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.