 Mycal Dutta1,2
Mycal Dutta1,2 Mohammad Nezam1Subrata Chowdhury1
Mohammad Nezam1Subrata Chowdhury1 Ahmed Rakib3
Ahmed Rakib3 Arkajyoti Paul1
Arkajyoti Paul1 Saad Ahmed Sami3Md. Zia Uddin1,2Md. Sohel Rana2Shahadat Hossain4Yunus Effendi5
Saad Ahmed Sami3Md. Zia Uddin1,2Md. Sohel Rana2Shahadat Hossain4Yunus Effendi5 Rinaldi Idroes6
Rinaldi Idroes6 Trina Tallei7
Trina Tallei7 Ali M. Alqahtani8
Ali M. Alqahtani8 Talha Bin Emran1*
Talha Bin Emran1*- 1Department of Pharmacy, BGC Trust University Bangladesh, Chittagong, Bangladesh
- 2Department of Pharmacy, Jahangirnagar University, Dhaka, Bangladesh
- 3Department of Pharmacy, Faculty of Biological Sciences, University of Chittagong, Chittagong, Bangladesh
- 4Atomic Energy Centre, Chittagong, Bangladesh
- 5Department of Biology, Faculty of Science and Technology, Al-Azhar Indonesia University, Jakarta, Indonesia
- 6Department of Pharmacy, Faculty of Mathematics and Natural Sciences, Syiah Kuala University, Banda Aceh, Indonesia
- 7Department of Biology, Faculty of Mathematics and Natural Sciences, Sam Ratulangi University, Manado, Indonesia
- 8Department of Pharmacology, College of Pharmacy, King Khalid University, Abha, Saudi Arabia
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first recognized in Wuhan in late 2019 and, since then, had spread globally, eventually culminating in the ongoing pandemic. As there is a lack of targeted therapeutics, there is certain opportunity for the scientific community to develop new drugs or vaccines against COVID-19 and so many synthetic bioactive compounds are undergoing clinical trials. In most of the countries, due to the broad therapeutic spectrum and minimal side effects, medicinal plants have been used widely throughout history as traditional healing remedy. Because of the unavailability of synthetic bioactive antiviral drugs, hence all possible efforts have been focused on the search for new drugs and alternative medicines from different herbal formulations. In recent times, it has been assured that the Mpro, also called 3CLpro, is the SARS-CoV-2 main protease enzyme responsible for viral reproduction and thereby impeding the host’s immune response. As such, Mpro represents a highly specified target for drugs capable of inhibitory action against coronavirus disease 2019 (COVID-19). As there continue to be no clear options for the treatment of COVID-19, the identification of potential candidates has become a necessity. The present investigation focuses on the in silico pharmacological activity of Calotropis gigantea, a large shrub, as a potential option for COVID-19 Mpro inhibition and includes an ADME/T profile analysis of that ligand. For this study, with the help of gas chromatography–mass spectrometry analysis of C. gigantea methanolic leaf extract, a total of 30 bioactive compounds were selected. Our analyses unveiled the top four options that might turn out to be prospective anti–SARS-CoV-2 lead molecules; these warrant further exploration as well as possible application in processes of drug development to combat COVID-19.
Introduction
Coronavirus disease 2019 (COVID-19) is an infectious disease triggered by infection with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus. In late 2019, cases of a pneumonia of a then-unknown origin began to appear in the capital of China’s Hubei province, Wuhan, later progressively spreading throughout the globe, resulting in the current COVID-19 pandemic (Hui et al., 2020). COVID-19 is not the first respiratory disease that has caused an epidemic in the last two decades; previously, outbreaks of Middle East respiratory syndrome (MERS) and severe acute respiratory syndrome (SARS), both caused by other coronaviruses, have occurred (de Wit et al., 2016). Coronaviruses are enveloped positive-sense single-stranded RNA genome (26–32 kb) viruses (Su et al., 2016). To date, four genera of coronaviruses have been identified: coronavirus genera α (alpha), coronavirus genera β (beta), coronavirus genera γ (gamma), and coronavirus genera δ (delta). Human coronaviruses (HCoVs) detected thus far include the HCoV-229E and NL63 in the α-coronavirus genus and MERS-CoV, SARS-CoV, HCoV-OC43, and HCoV-HKU1 in the β-coronavirus genus (Perlman and Netland, 2009). Following virus genome sequencing in December 2019 of five hospitalized patients in Wuhan with the mysterious pneumonia, it was divulged the existence of a formerly unknown β-CoV strain existed in all of them. This isolated neoteric β-CoV manifests an 88% degree of specification to the sequence of two bat-derived SARS-like coronaviruses, bat-SL-CoVZC45 and bat-SL-CoVZXC21, and an almost 50% degree of specification to the sequence of MERS-CoV (Lu et al., 2020). Liu et al., recently determined that there is a presence of the main protease enzyme in SARS-CoV-2 (Liu et al., 2020) responsible for inhibiting SARS-CoV-2 replication. Therefore, targeting this specific enzyme may lead to potential therapeutic advantages in drug discovery and efficacy for managing this dangerous virus. Elsewhere, experiments with four drugs approved by the United States Food and Drug Administration, including praziquantel, perampanel, nelfinavir, and pitavastatin as potent inhibitors for SARS-CoV-2 Mpro enzyme, are ongoing (Yusuf et al., 1994). Recently, researchers are also reviewing medicinal plants for possible efficacy in the context of SARS-CoV-2 infection (Khaerunnisa et al., 2020).
Having relatively less non-toxic property, synthetic bioactive natural compounds have diverse bioavailability. Various phytoconstituents like phenols, steroids, flavonoid molecules are present in the natural bioactive compounds and also they have been reported and screened for their possible therapeutic effects against various viral diseases. Certain natural compounds such as digitoxigenin, crocin, withanone, β-eudesmol, caffeic acid phenethyl ester and other various bioactive molecules have also been reported to interact with SARS-CoV-2 main protease (Tripathi et al., 2020). Traditional medicine employs a holistic approach to the prevention of disease and act as a source of many components with a high therapeutic value. Globally, many studies have been conducted on these herbs and revealing unique active constituents that might activate the innate immune system through the stimulation of macrophages and lymphocytes and modulation of the cytokine profile also (Tiwari et al., 2018). The potent therapeutics that might be effective to combat SARS-CoV-2 infection include virus binding molecules, helicase inhibitors molecules or inhibitors targeting particular enzymes implicated in replication and transcription process of the virus, protease inhibitors of host cells, endocytosis inhibitors, short interfering RNA (siRNA), neutralizing antibodies and natural drugs or medicines (Dhama et al., 2020).
Various kinds of secondary metabolites such glycosides, alkaloids, tannins, lignins and terpenes are present abundantly in various folklore medicinal plants and, thus, many studies have already explored the use of these plants in the treatment of various infections and diseases with the help of these secondary metabolites. Different extractable biochemical and bioactive compounds are contained in traditional medicinal plants might cure or prevent several viral diseases and infections by targeting certain specific sites of viruses. Despite the healing histories or scientific evidences, the use of medicinal plants and natural products as prophylactics, therapeutics and their multiple beneficial applications have gained strong momentum over the last few decades (Dhama et al., 2018; Brendler et al., 2020).
Henceforth, compound isolation and subsequent further investigation for several pharmacological activities has been established as a crucial process in the areas of drug design and discovery of novel drugs as well. The present study considered some isolated phytochemicals from a medicinal plant Calotropisgigantea, a member of the Apocynaceae family, for the treatment of COVID-19 using computational biology techniques. C. gigantea, frequently called giant milkweed or crown flower, is a large shrub or small tree native to Bangladesh, India, Malaysia, Southern China, and Indonesia. The plant is also locally known as Gulancha. These large shrubs have also been extensively cultivated in tropical areas around the world and typically grow to eight to 15 feet tall.
As calotropin offers potent bioactivity, C. gigantea has been used by the folk practitioners in India for many years for a variety of purposes. In Ayurveda, the root and leaf of Calotropis procera have been being used in managing asthma and breathing problems such as shortness of breath. The bark of C. procera is also being applied in other types of diseases such as spleen and liver conditions. This plant has been reported to be effective as well in treating skin, circulatory, digestive, and neurological issues and was also used previously to treat fevers, nausea, vomiting, and diarrhea. However, these reports are usually based on folklore uses and, therefore, more scientific investigations are necessary to affirm the rational and clinical usefulness of the leaves, latex, and bark of this family. Moreover, recent research has suggested the use of calotropin as a contraceptive and as a promising medication for cancer patients (Wang et al., 2009). In one study, the extracts of C. gigantea appeared to be strongly cytotoxic against non–small-cell lung carcinoma (A549), hepatocellular carcinoma (Hep G2), and colon carcinoma (HCT 116). In other research, leaf extracts of this plant have also shown promising activities as cancer medications (Jacinto et al., 2011). One species of the Calotropis genus, C. procera, displayed potent anti–human immunodeficiency virus 1 activity when the aqueous crude extract of the leaves was analyzed (Rakshit et al., 2010). An ethanolic extract of C. gigantea exhibited activity against herpes simplex type 1 viruses and vesicular stomatitis viruses (Ali et al., 1996). A novel lignan glycoside, (+)-pinoresinol 4-O-[6″-O-vanilloyl]-β-d-glucopyranoside, isolated from the latex of C. gigantea exhibited anti-influenza activity in an in vitro study (Parhira et al., 2014). Meanwhile, several other research works have suggested the protease inhibitory effect of C. gigantea.
In alignment with these previous findings, we speculate that the phytochemicals obtained from C. gigantea might be able to impede the main protease enzyme of SARS-CoV-2 (Kaladhar et al., 2013; Gupta and Chaphalkar, 2016). Through the process of gas chromatography–mass spectrophotometry (GC-MS), several compounds have been isolated for molecular docking. Molecular docking studies are of terrific significance in establishing plans for the layout of new drug substances; specifically, their strategic aim is to explore the binding affinity and the mode of binding of a small molecule within the specific binding sites of the receptor target of interest (Chikhale et al., 2020). This methodology is based on the prediction of native ligands that pose or bind with the proper receptor binding site, i.e., to find the experimental ligand geometry associated with physicochemical molecular interactions. The aim of this study was to screen out potent bioactive ligands from the GC-MS compound list of C. gigantea that could be probable inhibitors of Mpro.
Materials and Methods
GC-MS Analysis
The methanolic leaf extract of C. gigantea was inspected in a mass spectrometer (TQ8040; Shimadzu Corporation, Kyoto, Japan) with assistance from of electron-impact ionization method and a gas chromatograph (GC-17A; Shimadzu Corporation) fused with a silica capillary column made by silica with the specifications Rxi-5 ms, 0.25 m film, 30 m long, and internal diameter 0.32 mm coated with DB-1 (J&W). The oven temperature was set at 70°C (0 min); 10°C, 150°C (5 min); 12 and 200°C (15 min); 12 and 220°C (5 min), with a hold time of 10 min for each. A temperature of 260°C was set as the inlet temperature. The flow rate was 0.6 ml/min using helium gas at a constant pressure of 90 kPa. There was an interface temperature from GC to MS, which was adjusted at a constant 280°C. The scanning range of the MS was 40–350 amu and the ionization mode (EI) was established with the mass range of 50–550 m/z. The injection volume of the sample was 1 μl. During 50 min, the total GC-MS procedure was conducted. A comparison with the National Institute of Standards and Technology (NIST) GC-MS library version 08-S was performed for the identification of compounds in the peak areas (Tareq et al., 2020).
The GC helped the blend of components to be separated into individual ones and, based on their masses, MS identified the molecules. As such, GC-MS is an analytical method combining two instruments to yield a powerful separation and identification process. From the GC-MS process, 74 compounds were isolated and 30 bioactive compounds were finally enlisted and further assessed regarding their role as ligands. Their lists as well as details of the structure and PubChem ID number are given in Table 1.
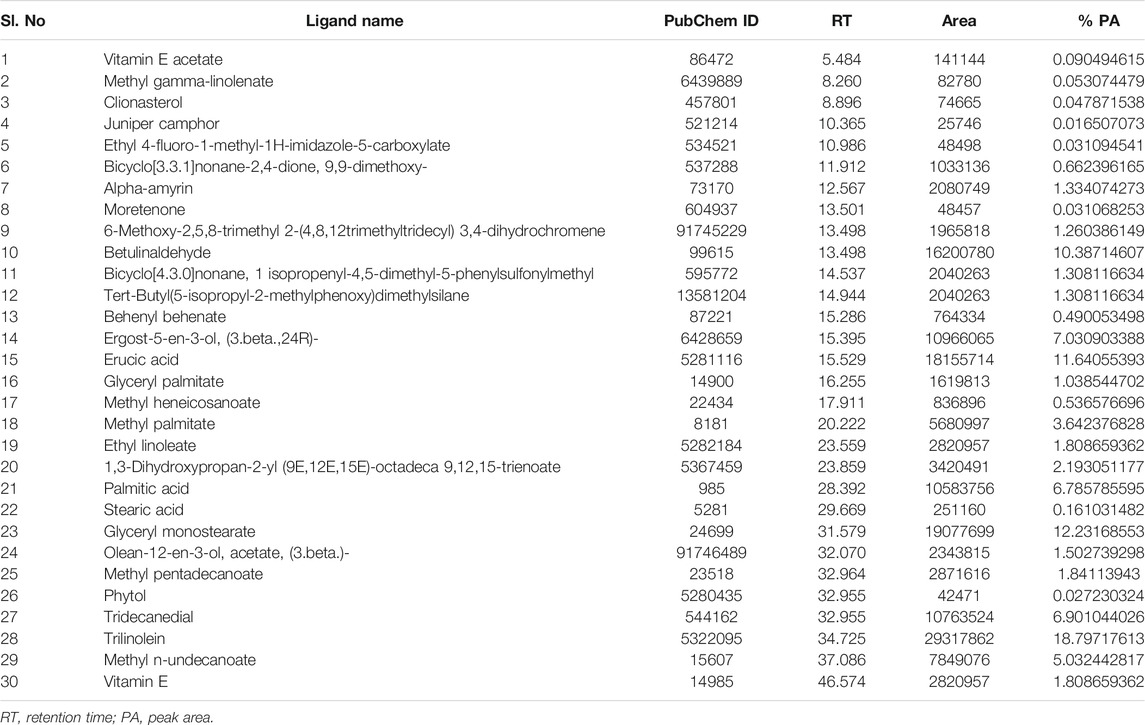
TABLE 1. List of tentative compounds identified in a methanol extract of C. gigantea leaves by GC–MS analysis.
Computational Molecular Docking Analysis
Protein and Ligand Preparation
Protein crystal structures are crucial for performing molecular docking simulation. In this study, we used the crystal structure of SARS-CoV-2 in concert with X77 (N-(4-tert- butylphenyl)-N-[(1R)-2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl]-1H-imidazole-4-carboxamide) (PDB ID: 6W63) with reference to a recent study by Dubey et al., who adopted the structure for performing docking studies by using some flavonoid compounds (Dubey and Dubey, 2020). The three-dimensional crystal structure of SARS-CoV-2 Mpro was downloaded from the Protein Data Bank in protein data bank (PDB) format having the PDB ID: 6W63 and was refined further using the protein preparation wizard of Maestro version 10.1 (Schrödinger, New York. NY, United States). Furthermore, the bond orders and charges together with the addition of hydrogen to the heavy metals; conversion of selenomethionines to the methionines; and, finally, the deletion of water were performed by using the force field OPLS_2005. Then, by setting the maximum heavy atom root mean-square-deviation to 0.30 Å, the minimization was carried out (Chowdhury et al., 2020). In addition, the structures of the chemical compounds were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov) and then ligands were prepared by neutralizing the pH to 7.0 ± 2.0 and minimized by the application of force field OPLS_2005 embedded in Maestro version 10.1 (Dash et al., 2015).
Receptor Grid Generation
For prepared proteins, the receptor grids were calculated in such a way that various ligands predicted during docking were bound within the active site. In Glide, grids were brought out by keeping the default parameters of the Van der Waals scaling factor at 1.00 and the charge cutoff at 0.25, which were exposed to OPLS_2005 force field. On the centroid, a cubic box of specific dimensions was centered at the active site residues and the bounding box was set to 17 × 17 × 17 Å for docking experiments.
Glide Standard Precision Ligand Docking
With the help of Maestro v 10.1 (Schrödinger), ligand docking was carried out, which were furthermore enforced to non-cis/trans amide bonds and the Van der Waals scaling factor; thereafter, the partial charge and cutoff values were selected (Friesner et al., 2004; Friesner et al., 2006). The ultimate scoring was executed on energy-minimized poses and represented as the glide score. For each ligand, the best-docked pose with the lowest glide score value was listed.
Visualization of Potential Interaction
The amino acids from the receptors that bind with the ligand are examined using PDBsum (http://www.ebi.ac.uk/pdbsum), a server that provides various pictorial analyses. This server integrates LIGPLOT to show the interactions of the analyzed complexes. A LIGPLOT diagram depicts the hydrogen bonds and non-bonded interactions between each ligand molecule and the protein residues in which it interacts with each ligand molecule.
Biological Activity Prediction
To assess the bioactivity of the lead compounds, the calculation of their activity scores as kinase inhibitors and enzyme inhibitors was completed. With the help of the software Molinspiration (www.molinspiration.com; Molinspiration, Slovensky Grob, Slovak Republic), all parameters were scrutinized properly (Rakib et al., 2020a; Rakib et al., 2020b; Tareq et al., 2020) and calculated drug-likeness scores of each compound were compared with the specific activity of each compound as well as those of standard drugs (i.e., nelfinavir and lopinavir).
Ligand-Based ADME/T Property Analysis
The QikProp module of the Maestro version 10.1 (Schrödinger) is an easily assessable absorption, distribution, metabolism, and excretion (ADME) prediction program that has been designed specifically to obtain certain descriptors linked to ADME. It is helpful to anticipate both the physicochemical and pharmacokinetic descriptors’ relevant properties. QikProp follows Lipinski’s rule of five for the evaluation of ADME properties, ascertaining drug-similar activities of ligand molecules. ADME/T properties of the compounds (i.e., vitamin E acetate; methyl gamma-linolenate; clionasterol; juniper camphor; ethyl 4-fluoro-1-methyl-1H-imidazole-5-carboxylate; bicyclo[3.3.1]nonane-2,4-dione; 9,9-dimethoxy-; alpha.-amyrin; moretenone; 6-methoxy-2,5,8-trimethyl 2-(4,8,12trimethyltridecyl) 3,4-dihydrochromene; betulinaldehyde; bicyclo[4.3.0]nonane; 1 isopropenyl-4,5-dimethyl-5-phenylsulfonylmethyl; tert-butyl(5-isopropyl-2-methylphenoxy)dimethylsilane; behenyl behenate; ergost-5-en-3-ol, (3.beta.,24R)-; erucic acid; glyceryl palmitate; methyl heneicosanoate; methyl palmitate; ethyl linoleate; 1,3-dihydroxypropan-2-yl (9E,12E,15E)-octadeca 9,12,15-trienoate; palmitic acid; stearic acid; glyceryl monostearate; olean-12-en-3-ol; acetate, (3.beta.)-; methyl pentadecanoate; phytol, tridecanedial; trilinolein; methyl n-undecanoate; and vitamin E) were analyzed using the QikProp 3.2 module (Rahman et al., 2020).
In Silico IC50 Calculation
Executing AutoDock
The inhibition constant in this study was determined by AutoDock 4.0 tools. AutoDock software calculates and predicts the interactions between a ligand molecule and a protein molecule, based on predefined parameters. The interactions between the molecules can be calculated at a specified user-defined region of the protein. This region must be defined by the user through the Grip map option or GridBox. Thus, the use of the GridBox at the binding site or active site, or at other essential protein regions, is essential to perform AutoDock analysis. Before executing the AutoDock, the “.pdb” files of the protein and ligand have to be moved into one folder.
Analysis in AutoDock can be divided into the following categories: a) Initialising molecules; and b) Running AutoGrid.
Initialising Molecules
Initialising the molecule involved the addition of hydrogen atoms. The ligand molecule required the addition of the Gasteiger charge, the identification of aromatic carbons, the detection of rotatable bonds, and the setting of the torsional degrees of freedom (TORSDOF) value. The protein must be initialised manually, whereas the ligand is automatically initialised when opened in the tool. The receptor and ligand files were saved in.pdbqt format. Next, the receptor was opened again using the “Grid” menu and the “Macromolecules” sub-menu, and “open” was selected. The ligand opens using the “Set Map Types” sub-menu and “Open Ligand.” Then, the GridBox is set in AutoDock to cover the identified binding sites. AutoDock only analyses the ligand molecule’s interactions and the amino acids that are present within the GridBox. The GridBox’s size can be increased or decreased using the number of points in the X/Y/Z dimension. The position of the GridBox can be adjusted to cover the binding site or binding residues, using the “Center GridBox” field that moves the GridBox in the X, Y, and Z-axis.
Running AutoGrid and AutoDock
After setting up the GridBox, the AutoGrid file was saved. When in the “Grid” menu, select the “Output” and “Save GPF” sub-menu. The file saves in.gpf format. Meanwhile, the AutoDock file saving procedure is to choose the “Docking” menu and select “Lamarckian GA” in the “Output” menu. The file saves in.dpf format. The AutoGrid and AutoDock programs was run through a command prompt directed to the folder to be docked using the command: autogrid4 -p control.gpf -l control.glg. After the AutoGrid calculation was completed successfully, AutoDock was run using the command: autodock4 -p control.dpf -l control.dlg. The results of the docking calculations are obtained in a notepad file format, reporting the values for binding energy and the estimated inhibition constant.
Results
GC-MS Analysis
From the methanolic leaf extract of C. gigantea, 52 compounds were eluted in between the retention time of 5 and 32 min (Figure 1). Thirty bioactive compounds were selected for this study (Table 1). The compound identification was carried out in comparison with the NIST GC-MS library version 08-S. The major phytochemicals included phenols, sterols, terpenoids, esters, and other organic compounds. The major compounds identified are trilinolein (18.79%), glyceryl monostearate (12.23%), erucic acid (11.64%), betulinaldehyde (10.38%), ergost-5-en-3-ol, (3.beta.,24R)- (7.03%), tridecanedial (6.90%), palmitic acid (6.78%), and methyl n-undecanoate (5.03%).
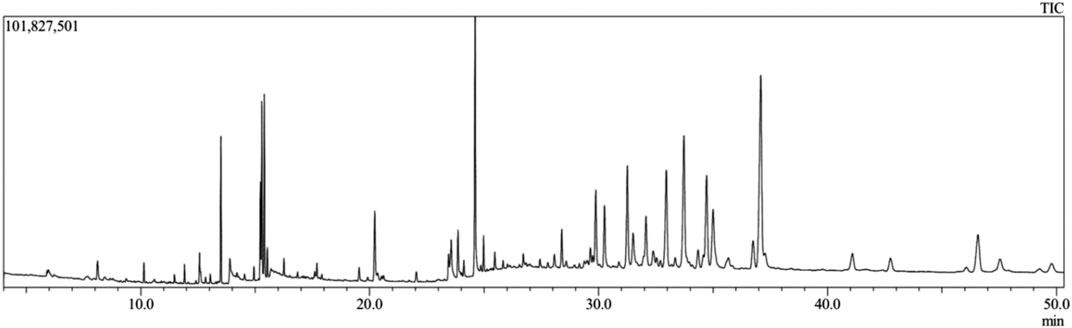
FIGURE 1. Total ionic chromatogram (TIC) of methanol extract of C. gigantea leaves by GC-MS.
Molecular Docking
A virtual screening process, including molecular docking, is a feasible approach for identifying antiviral therapeutics among diverse peptide sequences available in the online-based database. Notably, the interaction type, and the binding energy in conjunction with the bond distance of interaction between a protein substrate and a ligand, can be evaluated through the utility of molecular docking. Therefore, screening a ligand among numerous plant sources based on binding energy assessment is possible within minimum time by running molecular docking to evaluate the best ligand candidates according to the docking interaction.
Docking Interaction
In this study, we determined the interaction between several isolated compounds of C. gigantea and the Mpro enzyme. The docking results of the compounds that docked with the Mpro enzyme of SARS-CoV-2 are presented in Tables 2, 3; Figures 2 through 5. Meanwhile, the two- and three-dimensional interactions of nelfinavir and lopinavir with the active site of the SARS-CoV-2 Mpro are shown in Figures 6 and 7.

TABLE 2. Molecular docking study of major bioactive compounds from methanol extract of C. gigantea.
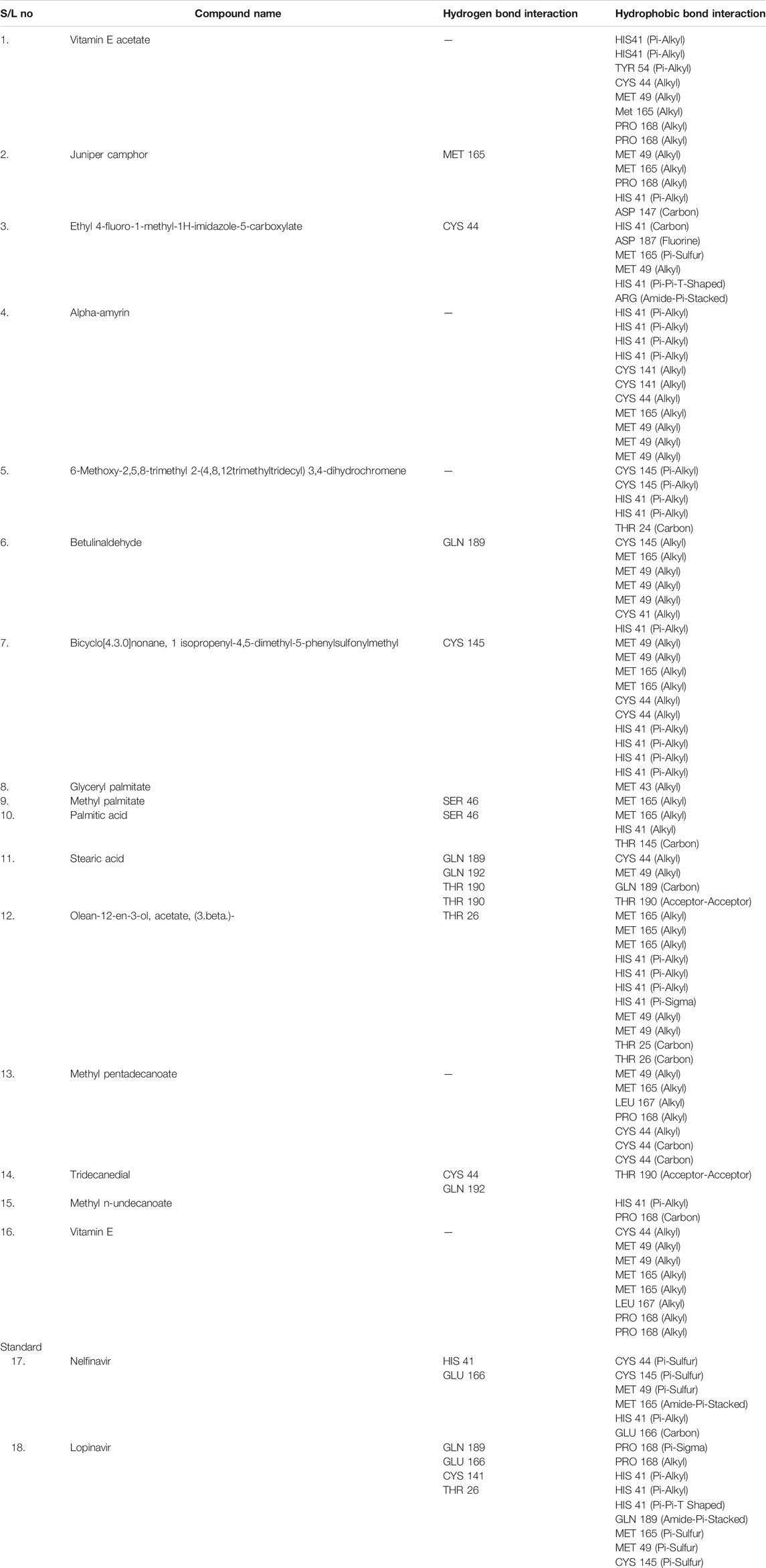
TABLE 3. Ligand interactions with amino acids of the protein active site.
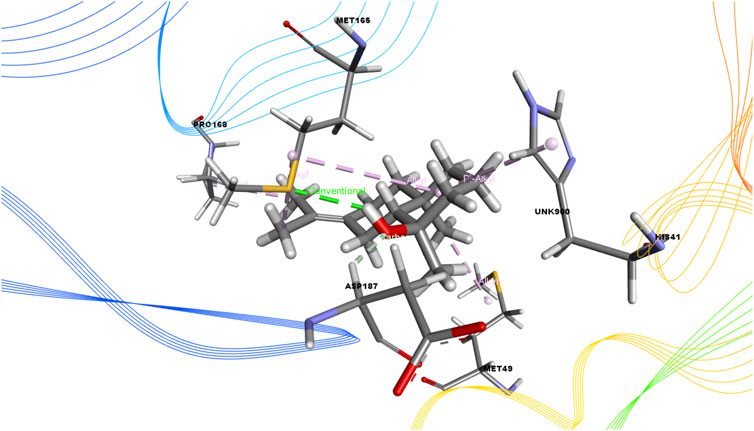
FIGURE 2. 3D interactions of Juniper camphor with the active site of SARS-CoV-2 main protease (PDB ID: 6W63).
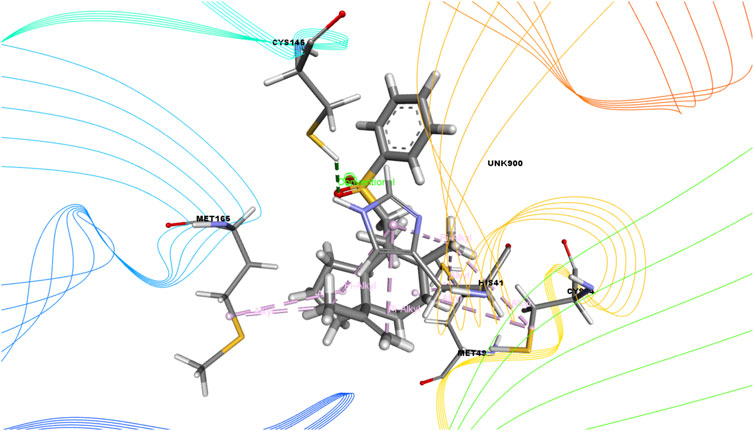
FIGURE 3. 3D interactions of Bicyclo[4.3.0]nonane, 1 isopropenyl-4,5-dimethyl-5-phenylsulfonylmethyl with the active site of SARS-CoV-2 main protease (PDB ID: 6W63).
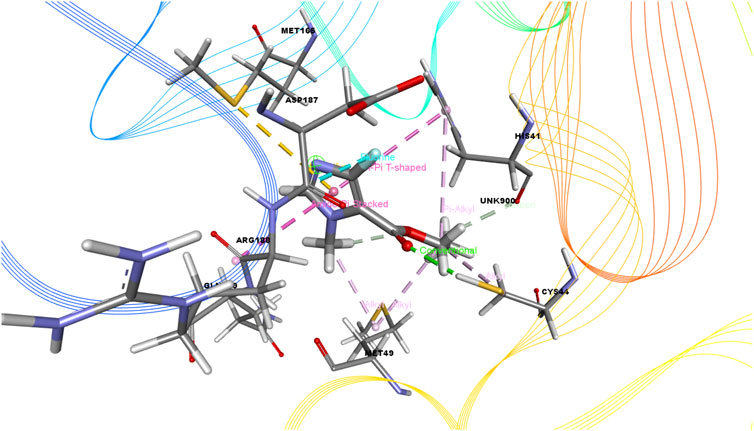
FIGURE 4. 3D interactions of Ethyl 4-fluoro-1-methyl-1H-imidazole-5-carboxylate with the active site of SARS-CoV-2 main protease (PDB ID: 6W63).
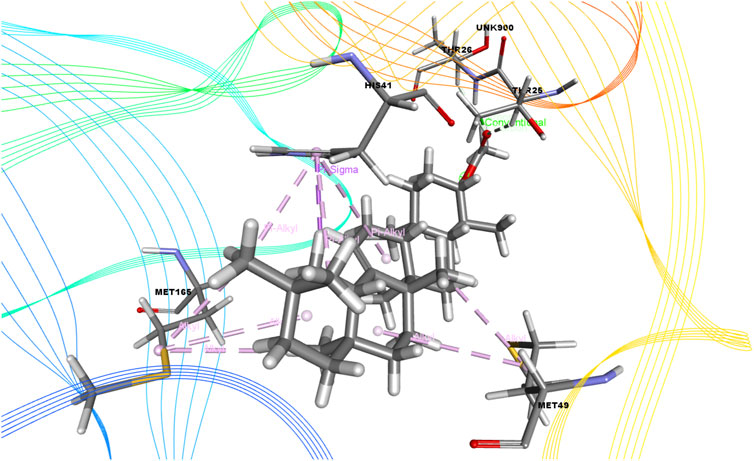
FIGURE 5. 3D interactions of Olean-12-en-3-ol, acetate, (3.beta.)- with the active site of SARS-CoV-2 main protease (PDB ID: 6W63).
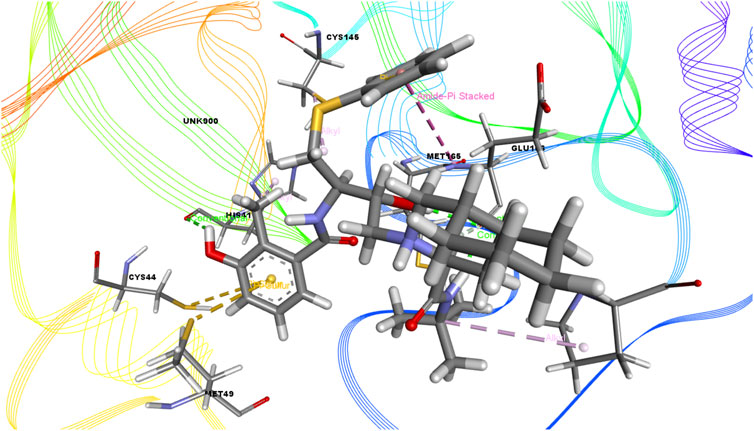
FIGURE 6. 3D interactions of Nelfinavir with the active site of SARS-CoV-2 main protease (PDB ID: 6W63).
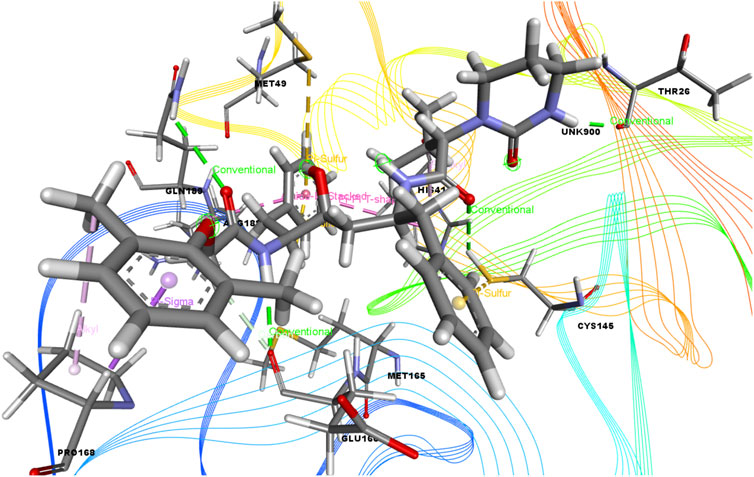
FIGURE 7. 3D interactions of Lopinavir with the active site of SARS-CoV-2 main protease (PDB ID: 6W63).
Among the molecular docking results, nelfinavir and lopinavir were confirmed to have docked with the main protease and had docking scores of −7.596 and −8.251 kcal/mol, respectively. According to the docking scores, juniper camphor was better in comparison with the standard drugs. Bicyclo[4.3.0]nonane, 1 isopropenyl-4,5-dimethyl-5-phenylsulfonylmethyl, olean-12-en-3-ol, acetate, (3.beta.)-, and ethyl 4-fluoro-1-methyl-1H-imidazole-5-carboxylate also exhibited better scores in a moderate fashion. Moreover, methyl pentadecanoate shows lowest docking score (0.806 kcal/mol). If arranging these compounds according to their scores, then the arrangement would be, from highest to lowest, juniper camphor (−6.06 kcal/mol); bicyclo[4.3.0]nonane, 1 isopropenyl-4,5-dimethyl-5-phenylsulfonylmethyl (−5.808 kcal/mol); olean-12-en-3-ol, acetate, (3.beta.)- (−5.588 kcal/mol); and ethyl 4-fluoro-1-methyl-1H-imidazole-5-carboxylate (−5.558 kcal/mol).
Again, the antiviral drug nelfinavir demonstrated interactions with HIS 41 and GLU 166 (two interactions) and three H-bonds with CYS 44, CYS 145, and MET 49 together with three pi-sulfur bonds together with one amide-pi-stacked, pi-alkyl, and carbon bond each with MET 165, HIS 41, and GLU 166, respectively. Lopinavir interacted with the protein through four H-bonds with GLN 189, GLU 166, CYS 141, and THR 26; nine hydrophobic bonds and three pi-sulfur bonds with MET 165, MET 49, and CYS 145; two pi-alkyl and pi-pi-T shaped bonds with HIS 41; one pi-sigma bond and one alkyl bond with PRO 168; and an amide-pi-stacked bond with GLN 189. Moreover, juniper camphor displayed five hydrophobic bonds, where three were alkyl bonds with MET 49, MET 165, and PRO 168; one was a pi-alkyl bond with HIS 41; and one was a carbon bond with ASP 147. These compounds also formed H-bonds with MET 165.
On the other hand, bicyclo[4.3.0]nonane, 1 isopropenyl-4,5-dimethyl-5-phenylsulfonylmethyl formed one H-bond with CYS 145; six alkyl bonds with MET 49, MET 49, MET 165, MET 165, CYS 44, and CYS 44; and four pi-alkyl bonds with HIS 41. Besides, olean-12-en-3-ol, acetate, (3.beta.)- showed a single H-bond, five alkyl bonds, three pi-alkyl bonds, two carbon bonds, and a single pi-sigma bond with THR 26, MET 165, MET 165, MET 165, MET 49, MET 49, HIS 41, HIS 41, HIS 4, HIS 41, THR 25, and THR 26, respectively.
As seen in Table 3, we also found that ethyl 4-fluoro-1-methyl-1H-imidazole-5-carboxylate interacted with the protein through a single H-bond and six hydrophobic bonds as follows: HIS 41 (carbon), ASP 187 (fluorine), MET 165 (pi-sulfur), MET 49 (alkyl), HIS 41 (pi-pi-T-shaped), and ARG (amide-pi-stacked).
Ligand-Based Biological Activity Parameter Prediction
Specific parameters, protease inhibitors, and enzyme inhibitors of all selected compounds are shown in Table 4.

TABLE 4. Biological activity parameter of the selected bioactive compounds.
Ligand-Based ADME/T Prediction
Pharmacokinetic and physicochemical properties of the 30 selected bioactive compounds were screened for drug-candidacy using the QikProp module of the Schrödinger suite-Maestro version 10.1. The ligand molecule’s similar drug attitude was determined using ADME properties by the QikProp module of Schrodinger Maestro v10.1. The ADME/T properties of the selected compounds were determined with the QikProp module of Schrodinger, shown in Table 5.
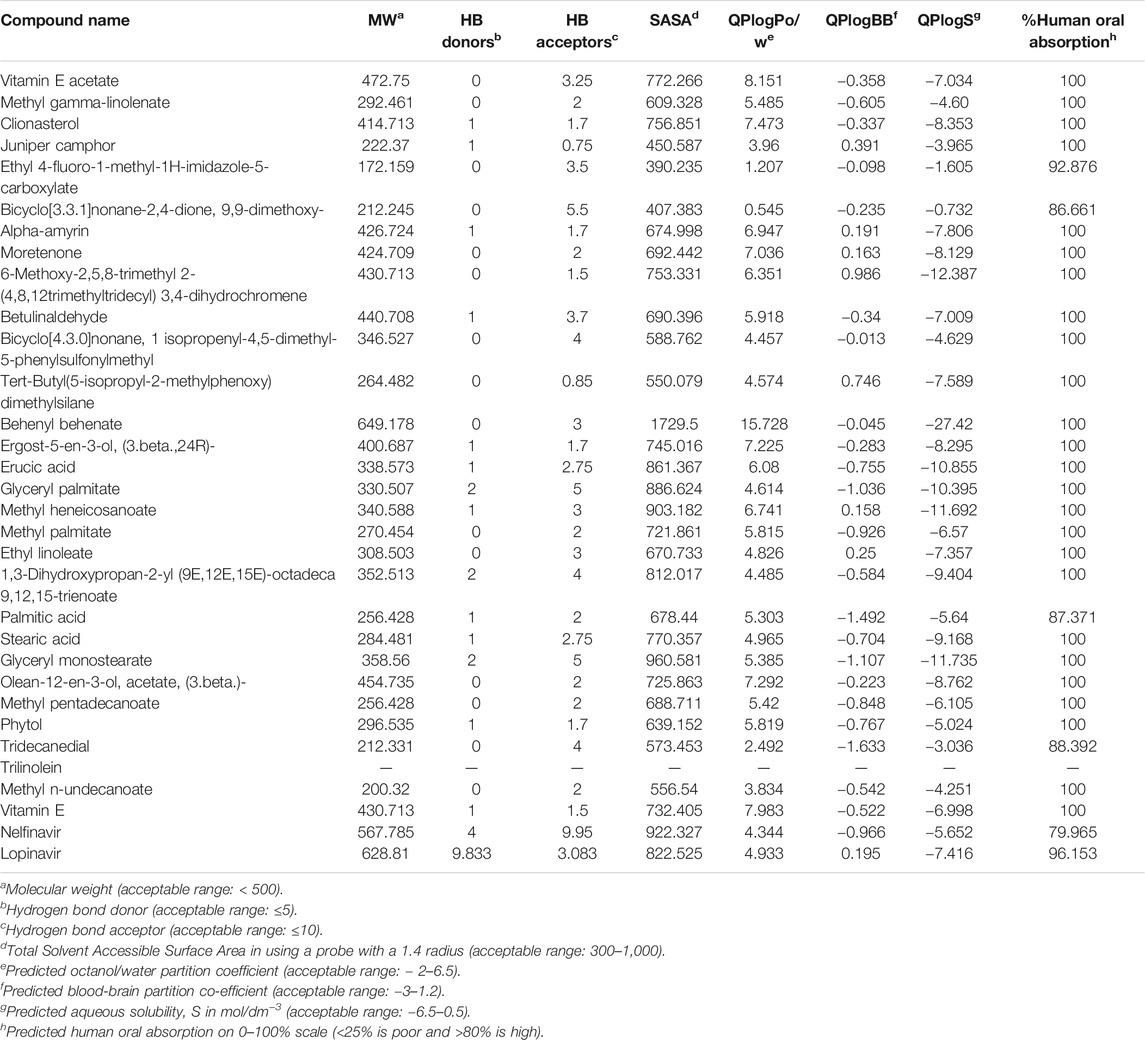
TABLE 5. ADME and drug likeness properties of selected bioactive isolated compounds from methanol extract of C. gigantea.
Visualisation of Interactions
The docking conformation of the ligands and receptors were determined by selecting the pose with the highest affinity (most negative Gibbs free energy). The results from each affinity value of the conformation were obtained in the notepad file. The visualization of three-dimensional (3D) and two-dimensional (2D) structures from the docking calculations were carried out. The visualization and potential interaction between each ligand molecule with protein residues results are presented in Supplementary Table S1; Supplementary Figure S1. The interaction of the receptor’s amino acid with the ligand in 3D and 2D were saved as image files.
In Silico Inhibition Constant Calculation
The current study determined the inhibition constant of major bioactive compounds from methanol extract of C. gigantea. toward the modeled SARS-CoV-2 Mpro. The theoretical half-maximal inhibitory concentration (IC50) was calculated using AutoDock 4.0 tools, and the results of the analysis are shown in Table 6.

TABLE 6. Inhibition constant (IC50) of major bioactive compounds from methanol extract of C. gigantea.
Discussion
Currently, SARS-CoV-2 infection has emerged as a global pandemic and requires an international public health emergence (Matveeva et al., 2020; Remali and Aizat, 2021). However, different research organizations and pharmaceutical companies are working relentlessly for the management of the infection. By far, several vaccines have been made available as immunomodulation is characterized as one of the pivotal characteristics in the maintenance of human health and productivity (Tiwari et al., 2018; Adhikari et al., 2020; Tahir Ul Qamar et al., 2020; Tallei et al., 2020 ).
Researchers are learning new things about this virus every day about the mutation of the virus (Sinha et al., 2021). Surprisingly, according to Centers for Disease Control and Prevention (CDC), multiple SARS-CoV-2 variants are available globally. In United Kingdom, the variant of SARS-CoV-2 showed a large number of mutations In South Africa, another variant emerged independently and share several characteristics with other strain. As the mutation of the virus is rising rapidly, it is crucial to identify potential therapeutic agents to overcome the pathetic situation. So far, we know that initially, some patients having attacked by COVID-19 may not figure out any symptoms, and hence they may carry the virus for two days or up to two weeks without noticing any symptoms (Tahmasbi et al., 2021). Some common symptoms have explicitly been notified to COVID-19. These include shortness of breath, a low-grade fever that gradually increases in temperature, having a cough that gets more severe over time, and fatigue. Less common symptoms include chills, repeated shaking with colds, sore throat, headache, muscle aches and pains, loss of taste, and smell loss. Some people may need to seek emergency medical services to have any of the symptoms more severe, like trouble breathing with persistent pain or pressure in the chest and excessive drowsiness (——). Although several research works are being carried out in renowned laboratories in various countries, the vaccine development for a disease generally requires a long time. Despite plenty of experimental trials related to COVID-19 treatment and medications, scientists are still seeking specific therapeutic drugs. Several previous studies have already performed for the identification of potential therapeutic agents against SARS-CoV-2 from medicinal plants (Rabaan et al., 2020; Shoaib et al., 2020; Emirik, 2020). Medicinal plants are abundant with plenty of phytocompounds and have been used to treat several kinds of diseases (Shree et al., 2020). Different secondary metabolites, including lignin’s, glycosides, alkaloids, terpenoids, and amino acids that are fundamental for the functioning and growth of plants as well as having numerous pharmacological attributes to fight against several abnormal conditions (Rahman et al., 2013; Emran et al., 2018; Diniz et al., 2021 ). We had selected the plant C. gigantea based on their traditional use and ethnomedicinal value and their contribution as an antiviral drug for certain viruses that were analyzed before. For this study, with the help of the GC-MS technique, seventy-four compounds had been isolated, among which thirty bioactive compounds were enlisted and subjected to play their role as ligands. In recent times, researchers are working to find out some potential lead compounds from medicinal plants that are active against several enzymes and other proteins responsible for viral replication and growth (Gupta et al., 2020b; Krupanidhi et al., 2020; Pendyala and Patras, 2020 ). Several studies reported the inhibitory potential of several herb derived phytochemicals against SARS-CoV-2 (Dhama et al., 2018; Gupta et al., 2020b; Jalali et al., 2020).
The advancement of numerous computational technology has affected vastly in biology and computational biology, incorporating both computational algorithms and biological data, which has emerged as a breakthrough in modern drug discovery. High-throughput screening (HTS) often requires the screening of the plenty of chemical compounds specific for a targeted receptor, utilizes molecular docking as essential tools (Brooijmans and Kuntz, 2013). We predicted the interaction between the SARS-CoV-2 Mpro and the targeted phytochemical through molecular docking simulation in the current study. The molecular docking analysis findings revealed that the selected phytocompounds were able to interact with the binding pockets of the SARS-CoV-2 Mpro. The compound Juniper camphor exhibited the most significant interaction compared with other compounds. Like two standard drugs, nelfinavir, and lopinavir, it yielded hydrogen bonding with Met165 residue. Besides, our selected compounds were able to interact with the SARS-CoV-2 Mpro, and previous other studies also suggested the residues were responsible for interacting with SARS-CoV-2 Mpro (Joshi et al., 2020; Khan et al., 2020; Wu et al., 2020).
Molinspiration is used to predict the biological activity parameters like protease inhibitory activity and enzyme inhibitory action. The increase scores specify better inhibition properties (Yamamoto et al., 2004; Chang et al., 2019; Das et al., 2020; Gentile et al., 2020; Li et al., 2020); most of the targeted compounds were shown greater inhibition properties.
QikProp module of the Schrödinger suite predicts ADMET properties related to molecular weight, partition coefficient (QPlogPo/w), stars, % oral absorption values, and Lipinski’s properties (Rule of five). Different ADME parameters were predicted, as the molecular weight ranged between 200.32 and 649.178, where only one phytochemical was outside the specified range. The estimated number of hydrogen bond donors (HB donors) by the solute to water molecules in an aqueous solution lay in the range between 1 and 2. In contrast, in the case of hydrogen bond acceptor (HB acceptors), the number was found between 0.75 and 5.5. The value of total solvent accessible surface area (SASA) ranged from 390.235 to 1729.5, where only Behenyl behenate was recorded outside the acceptable range. The value of the octanol/water partition coefficient (QPlog Po/w) lay in the range between 0.545 and 15.728. All the bioactive compounds have shown good Brain/blood partition coefficient (QPlogBB), the drug’s ability to pass through the blood-brain barrier (BBB). The QPlogBB value ranged from −1.633 to 0.986 suggested all the compounds are within the specified limits; aqueous solubility, log S (QPlogS) ranged between −27.42 and −0.732. Interestingly, all the compounds except Behenyl behenate fulfilled the Lipinski’s ROF as it was found that Lipinski violations were ≤1. The percent of human oral absorption was high for all the selected compounds. All the phytochemicals exceeded 80% and showed more oral absorption than both the standard drugs. Notably, some phytochemicals exhibited 100% oral bioavailability, which enhanced the drug candidacy of those compounds.
Enzyme activation or inhibition is the result of the binding interaction between a small molecule ligand and an enzyme protein. The protein is supposed to be a receptor, and thus binding with the ligand may result in agonism or antagonism. As a form of bioinformatics analysis, Molecular docking simulation reveals the binding affinities of ligand molecules with a specific receptor, where the lower binding energy predicts the greater binding affinity (Brooijmans and Kuntz, 2003; Lalitha and Sivakamasundari, 2010; Ahmed et al., 2019; Rakib et al., 2020c; Rakib et al., 2020d; Jahan et al., 2020; Dutta et al., 2019). Despite showing lower binding affinities than nelfinavir and lopinavir, our selected compounds interacted with the active pockets of the SARS-CoV-2 Mpro enzyme like the standard compounds. Among the selected 30 ligands, 16 ligands docked with active pockets of SARS-CoV-2 Mpro enzymes. Four of them, namely Juniper camphor, Ethyl 4-fluoro-1-methyl-1H-imidazole-5-carboxylate, Bicyclo[4.3.0]nonane, 1 isopropenyl-4,5-dimethyl-5-phenylsulfonylmethyl and Olean-12-en-3-ol, acetate, (3.beta.) possessed docking scores more likely to nelfinavir. Moreover, those compounds also followed the Lipinski’s rule of five for drug-likeness properties. Hence, these four may be potential inhibitors of COVID-19, which warrants further investigation.
While so many synthetic bioactive compounds are undergoing clinical trials, including medicinal plants derived phytocompounds, this purely bioinformatic work was limited by a lack of direct experimental evidence. We were unable to conduct follow-up experiments using other techniques, such as Western blotting or qRT-PCR, to confirm our bioactive compounds’ antiviral properties generated only from computational prediction. This study could be expanded to examine the antiviral potentiality of plant-derived bioactive compounds, specifically from several traditional medicinal plants available in the Bangladesh region. Based on results from this study, the top compounds that might turn out to be prospective anti–SARS-CoV-2 lead molecules warrant further investigation and possible application in drug development processes against COVID-19.
Conclusion
Increasing use of HTS and molecular docking simulation is emerged as a new era in the field of drug discovery. Medicinal plant derived phytochemicals are used for the treatment from since the dawn of civilization. Recently, the enriched extraction procedures and sophisticated computer-aided program have already added a new dimension in the field of drug discovery. To sum up, this study provides significant data for the use of isolated phytocompounds from C. gigantea for the treatment of COVID-19 by targeting one of the crucial enzymes that is essential replication of SARS-CoV-2. Further experimental analysis might be needed to justify the activity of the phytochemicals at the molecular level. In addition, many more such bioactive ingredients from medicinal plants existing in the rich Bangladeshi biodiversity need to be further explored. The study initiated the window toward plant-based therapy against COVID-19, though extensive research and wet lab validation need to make it usable for the patient.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
Study concept and design: MD, MN, AR, and TE. Acquisition of data: MD, SC, AR, AP, SS, and MU. Analyses and interpretation of data: MD, AR, AP, and SS. Drafting the manuscript: MD, AR, AP, SS, and TE. Critical revision of the manuscript for important intellectual content: MR, SH, YE, RI, TT, AA, and TE. Technical or material support: MR, SH, TT, AA, and TE. Study supervision: MR and TE.
Funding
This work is conducted with the individual funding of all authors.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Cambridge Proofreading® & Editing LLC. (https://proofreading.org/) for editing a draft of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.625391/full#supplementary-material
References
Adhikari, B., Marasini, B. P., Rayamajhee, B., Bhattarai, B. R., Lamichhane, G., Khadayat, K., et al. (2020). Potential Roles of Medicinal Plants for the Treatment of Viral Diseases Focusing on COVID ‐19: A Review. Phytotherapy Res. 35, 1298–1312. doi:10.1002/ptr.689310.1002/ptr.6893
Ahmed, S., Rakib, A., Islam, M. A., Khanam, B. H., Faiz, F. B., Paul, A., et al. (2019). In Vivo and In Vitro Pharmacological Activities of Tacca Integrifolia Rhizome and Investigation of Possible lead Compounds against Breast Cancer through In Silico Approaches. Clin. Phytosci. 5, 36. doi:10.1186/s40816-019-0127-x
Ali, A. M., Mackeen, M. M., El-Sharkawy, S. H., Abdul Hamid, J., Ismail, N. H., Ahmad, F., et al. (1996). Antiviral and Cytotoxic Activities of Some Plants Used in Malaysian Indigenous Medicine. Pertanika J. Trop. Agricult Sci. 19, 129–136.
Brendler, T., Al-Harrasi, A., Bauer, R., Gafner, S., Hardy, M. L., Heinrich, M., et al. (2020). Botanical Drugs and Supplements Affecting the Immune Response in the Time of COVID-19: Implications for Research and Clinical Practice. Phytother Res. 1–19. doi:10.1002/ptr.7008s
Brooijmans, N., and Kuntz, I. D. (2013). Molecular Recognition and Docking Algorithms. Annu. Rev. Biophys. Biomol. Struct. 32, 335–373. doi:10.1146/annurev.biophys.32.110601.142532
Brooijmans, N., and Kuntz, I. D. (2003). Molecular Recognition and Docking Algorithms. Annu. Rev. Biophys. Biomol. Struct. 32 (1), 335–373. doi:10.1146/annurev.biophys.32.110601.142532
Chang, K. O., Kim, Y., Lovell, S., Rathnayake, A. D., and Groutas, W. C. (2019). Antiviral Drug Discovery: Norovirus Proteases and Development of Inhibitors. Viruses 11 (2), 1–14. doi:10.3390/v11020197
Chikhale, R. V., Gurav, S. S., Patil, R. B., Sinha, S. K., Prasad, S. K., Shakya, A., et al. (2020). Sars-cov-2 Host Entry and Replication Inhibitors from Indian Ginseng: Anin-Silicoapproach. J. Biomol. Struct. Dyn., 1–12. doi:10.1080/07391102.2020.1778539
Chowdhury, K. H., Chowdhury, M. R., Mahmud, S., Tareq, A. M., Hanif, N. B., Banu, N., et al. (2020). Drug Repurposing Approach against Novel Coronavirus Disease (COVID-19) through Virtual Screening Targeting SARS-CoV-2 Main Protease. Biology 10 (1), 2. doi:10.3390/biology10010002
Das, S., Sarmah, S., Lyndem, S., and Singha Roy, A. (2020). An Investigation into the Identification of Potential Inhibitors of SARS-CoV-2 Main Protease Using Molecular Docking Study. J. Biomol. Struct. Dyn., 1–11. doi:10.1080/07391102.2020.1763201
Dash, R., Ahsan, M., Hosen, S., Rahman, M., Emran, T., and Uddin, M. (2015). Evolution of Selective COX-2 Inhibitor from Alangium Salvifolium: an In Silico Approach. J. App Pharm. Sci. 504, 089–093. doi:10.7324/JAPS.2015.50415
de Wit, E., van Doremalen, N., Falzarano, D., and Munster, V. J. (2016). SARS and MERS: Recent Insights into Emerging Coronaviruses. Nat. Rev. Microbiol. 14, 523–534. doi:10.1038/nrmicro.2016.81
Dhama, K., Karthik, K., Khandia, R., Munjal, A., Tiwari, R., Rana, R., et al. (2018). Medicinal and Therapeutic Potential of Herbs and Plant Metabolites/Extracts Countering Viral Pathogens - Current Knowledge and Future Prospects. Cdm 19 (3), 236–263. doi:10.2174/1389200219666180129145252
Dhama, K., Khan, S., Tiwari, R., Sircar, S., Bhat, S., Malik, Y. S., et al. (2020). Coronavirus Disease 2019-COVID-19. Clin. Microbiol. Rev. 33 (4), e00028–20. doi:10.1128/CMR.00028-20
Diniz, L. R. L., Perez-Castillo, Y., Elshabrawy, H. A., Filho, C. d. S. M. B., and de Sousa, D. P. (2021). Bioactive Terpenes and Their Derivatives as Potential SARS-CoV-2 Proteases Inhibitors from Molecular Modeling Studies. Biomolecules 11 (1), 74. doi:10.3390/biom11010074
Dubey, K., and Dubey, R. (2020). Computation Screening of Narcissoside a Glycosyloxyflavone for Potential Novel Coronavirus 2019 (COVID-19) Inhibitor. Biomed. J. 43 (4), 363–367. doi:10.1016/j.bj.2020.05.002
Emirik, M. (2020). Potential Therapeutic Effect of Turmeric Contents against SARS-CoV-2 Compared with Experimental COVID-19 Therapies: In Silico Study. J. Biomol. Struct. Dyn. Oct 20, 1–14. doi:10.1080/07391102.2020.1835719
Emran, T., E-Mowla, T., Ahmed, S., Zahan, S., Rakib, A., Hasan, M., et al. (2018). Sedative, Anxiolytic, Antinociceptive, Anti-inflammatory and Antipyretic Effects of a Chloroform Extract from the Leaves of Urena Sinuata in Rodents. Jalsi 16 (3), 1–19. doi:10.9734/jalsi/2018/39073
Dutta, T., Paul, A., Majumder, M., Sultan, R. A., and Emran, T. B. (2019). Pharmacological evidence for the use of Cissus assamica as a medicinal plant in the management of pain and pyrexia. Biochem. Biophys. Rep. 21, 1–8. doi:10.1016/j.bbrep.2019.100715
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., et al. (2004). Glide: a New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 47 (7), 1739–1749. doi:10.1021/jm0306430
Friesner, R. A., Murphy, R. B., Repasky, M. P., Frye, L. L., Greenwood, J. R., Halgren, T. A., et al. (2006). Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 49 (21), 6177–6196. doi:10.1021/jm051256o
Gentile, D., Patamia, V., Scala, A., Sciortino, M. T., Piperno, A., and Rescifina, A. (2020). Putative Inhibitors of SARS-CoV-2 Main Protease from a Library of marine Natural Products: A Virtual Screening and Molecular Modeling Study. Mar. Drugs 18 (4), 225. doi:10.3390/md18040225
Gupta, A., and Chaphalkar, S. R. (2016). Anti-diabetic Activity of Calotropis Gigantea in Human Whole Blood. Int. Knowld Press. 6 (3), 107–112. doi:10.22270/ikp.v7i3.1447
Gupta, S., Singh, V., Varadwaj, P. K., Chakravartty, N., Katta, A. V. S. K. M., Lekkala, S. P., et al. (2020b). Secondary Metabolites from Spice and Herbs as Potential Multitarget Inhibitors of SARS-CoV-2 Proteins. J. Biomol. Struct. Dyn. Oct 27, 1–20. doi:10.1080/07391102.2020.1837679
Hui, D. S., I Azhar, E., Madani, T. A., Ntoumi, F., Kock, R., Dar, O., et al. (2020). The Continuing 2019-nCoV Epidemic Threat of Novel Coronaviruses to Global Health - the Latest 2019 Novel Coronavirus Outbreak in Wuhan, China. Int. J. Infect. Dis. 91, 264–266. doi:10.1016/j.ijid.2020.01.009
Jacinto, S. D., Chun, E. A. C., Montuno, A. S., Shen, C. C., Espineli, D. L., and Ragasa, C. Y. (2011). Cytotoxic Cardenolide and Sterols from Calotropis Gigantea. Nat. Prod. Commun. 6 (6), 803–806. doi:10.1177/1934578x1100600614
Jahan, I., Tona, M. R., Sharmin, S., Sayeed, M. A., Tania, F. Z., Paul, A., et al. (2020). GC-MS Phytochemical Profiling, Pharmacological Properties, and In Silico Studies of Chukrasia Velutina Leaves: A Novel Source for Bioactive Agents. Molecules 25 (15), 3536. doi:10.3390/molecules25153536
Jalali, A., Dabaghian, F., Akbrialiabad, H., Foroughinia, F., and Zarshenas, M. M. (2020). A Pharmacology‐based Comprehensive Review on Medicinal Plants and Phytoactive Constituents Possibly Effective in the Management of COVID ‐19. Phytotherapy Res. 35, 1925–1938. doi:10.1002/ptr.6936
Joshi, R. S., Jagdale, S. S., Bansode, S. B., Shankar, S. S., Tellis, M. B., Pandya, V. K., et al. (2020). Discovery of Potential Multi-Target-Directed Ligands by Targeting Host-specific SARS-CoV-2 Structurally Conserved Main Protease. J. Biomol. Struct. Dyn., 1–16. doi:10.1080/07391102.2020.1760137
Kaladhar, D. S. V. G. K., Duddukuri, G. R., Ramesh, K., Varahalarao Vadlapudi, V., and Yarla, N. S. (2013). In Vitro protease Inhibition, Modulation of PLA2 Activity and Protein Interaction Studies of Calotropis Gigantea. J. Clin. Cel Immunol 4, 165. doi:10.4172/2155-9899.1000165
Khaerunnisa, S., Kurniawan, H., Awaluddin, R., Suhartati, S., and Soetjipto, S. (2020). Potential Inhibitor of COVID-19 Main Protease (Mpro) from Several Medicinal Plant Compounds by Molecular Docking Study. Prepr. 1–14. doi:10.20944/preprints202003.0226.v1
Khan, S. A., Zia, K., Ashraf, S., Uddin, R., and Ul-Haq, Z. (2020). Identification of Chymotrypsin-like Protease Inhibitors of SARS-CoV-2 via Integrated Computational Approach. J. Biomol. Struct. Dyn. 11, 1–10. doi:10.1080/07391102.2020.1751298
Krupanidhi, S., Abraham Peele, K., Venkateswarulu, T. C., Ayyagari, V. S., Nazneen BobbyD John Babu, M., John Babu, D., et al. (2020). Screening of Phytochemical Compounds ofTinospora Cordifoliafor Their Inhibitory Activity on SARS-CoV-2: Anin Silicostudy. J. Biomol. Struct. Dyn. 2020, 1–5. doi:10.1080/07391102.2020.1787226
Lalitha, P., and Sivakamasundari, S. (2010). Calculation of Molecular Lipophilicity and Drug Likeness for Few Heterocycles. Oriental J. Chem. 26 (1), 135–141.
Li, J.-Y., You, Z., Wang, Q., Zhou, Z.-J., Qiu, Y., Luo, R., et al. (2020). The Epidemic of 2019-Novel-Coronavirus (2019-nCoV) Pneumonia and Insights for Emerging Infectious Diseases in the Future. Microbes Infect. 22 (2), 80–85. doi:10.1016/j.micinf.2020.02.002
Liu, X., Zhang, B., Jin, Z., Yang, H., and Rao, Z. (2020). The Crytal Structure of 2019-NCoV Main Protease in Complex with an Inhibitor N3. RCSB Protein Data Bank. doi:10.2210/pdb7bqy/pdb
Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., et al. (2020). Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. The Lancet 395, 565–574. doi:10.1016/s0140-6736(20)30251-8
Matveeva, T., Khafizova, G., and Sokornova, S. (2020). In Search of Herbal Anti-SARS-cov2 Compounds. Front. Plant Sci. 11, 589998. doi:10.3389/fpls.2020.589998
Parhira, Supawadee., Yang, Zi-Feng., Zhu, Guo-Yuan., Chen, Qiao-Lian., Zhou, Bei-Xian., Wang, Yu-Tao., et al. (2014). Zhi-Hong Jiang. In Vitro Anti-influenza Virus Activities of a New Lignan Glycoside from the Latex of Calotropis Gigantea. Plos One 9 (8), 1–13. doi:10.1371/journal.pone.0104544
Pendyala, B., and Patras, A. (2020). In Silico Screening of Food Bioactive Compounds to Predict Potential Inhibitors of COVID-19 Main Protease (Mpro) and RNA-dependent RNA Polymerase (RdRp). Chemrxiv. Preprint. doi:10.26434/chemrxiv.12051927.v2
Perlman, S., and Netland, J. (2009). Coronaviruses post-SARS: Update on Replication and Pathogenesis. Nat. Rev. Microbiol. 7, 439–450. doi:10.1038/nrmicro2147
Rabaan, A. A., Al-Ahmed, S. H., Sah, R., Tiwari, R., Yatoo, M. I., Patel, S. K., et al. (2020). SARS-CoV-2/COVID-19 and Advances in Developing Potential Therapeutics and Vaccines to Counter This Emerging Pandemic. Ann. Clin. Microbiol. Antimicrob. 19 (1), 40. doi:10.1186/s12941-020-00384-w
Rahman, J., Tareq, A. M., Hossain, M. M., Sakib, S. A., Islam, M. N., Ali, M. H., et al. (2020). Biological Evaluation, DFT Calculations and Molecular Docking Studies on the Antidepressant and Cytotoxicity Activities of Cycas Pectinata Buch.-Ham. Compounds. Pharmaceuticals 13 (9), 232. doi:10.3390/ph13090232
Rahman, M. A., Imran, T. b., and Islam, S. (2013). Antioxidative, Antimicrobial and Cytotoxic Effects of the Phenolics of Leea Indica Leaf Extract. Saudi J. Biol. Sci. 20, 213–225. doi:10.1016/j.sjbs.2012.11.007
Rakib, A., Paul, A., Ahmed, S., Chy, M. N. U., Sami, S. A., Baral, S. K., et al. (2020a). Biochemical and Computational Approach of Phytocompounds from Tinospora Crispa in the Management of COVID-19. Molecules 25 (17), E3936. doi:10.3390/molecules25173936
Rakib, A., Ahmed, S., Islam, M. A., Haye, A., Uddin, S. M. N., Uddin, M. M. N., et al. (2020b). Antipyretic and Hepatoprotective Potential of Tinospora Crispa and Investigation of Possible lead Compounds through In Silico Approaches. Food Sci. Nutr. 8 (1), 547–556. doi:10.1002/fsn3.1339
Rakib, A., Sami, S. A., Islam, M. A., Ahmed, S., Faiz, F. B., Khanam, B. H., et al. (2020c). Epitope-Based Immunoinformatics Approach on Nucleocapsid Protein of Severe Acute Respiratory Syndrome-Coronavirus-2. Molecules 25 (21), 5088. doi:10.3390/molecules25215088
Rakib, A., Sami, S. A., Mimi, N. J., Chowdhury, M. M., Eva, T. A., Nainu, F., et al. (2020d). Immunoinformatics-guided Design of an Epitope-Based Vaccine against Severe Acute Respiratory Syndrome Coronavirus 2 Spike Glycoprotein. Comput. Biol. Med. 124, 103967. doi:10.1016/j.compbiomed.2020.103967
Rakshit, J., Nobre, M., and Mohanraj, R. (2010). Anti-HIV-1 and Antimicrobial Activity of the Leaf Extracts of Calotropis Procera. Intl J. Green. Pharm. 4, 242–246. doi:10.22377/ijgp.v4i4.154
Remali, J., and Aizat, W. M. (2021). A Review on Plant Bioactive Compounds and Their Modes of Action against Coronavirus Infection. Front. Pharmacol. 11, 589044. doi:10.3389/fphar.2020.589044
Shoaib, A., Azmi, L., Shukla, I., Alqahtani, S. S., Alsarra, I. A., and Shakeel, F. (2020). Properties of Ethno-Medicinal Plants and Their Bioactives-Possible Use for Prevention and Treatment of COVID-19: A Review. Cpd. 27, 1–9. doi:10.2174/1381612826666201106092021
Shree, P., Mishra, P., Selvaraj, C., Singh, S. K., Chaube, R., Garg, N., et al. (2020). Targeting COVID-19 (SARS-CoV-2) Main Protease through Active Phytochemicals of Ayurvedic Medicinal Plants -Withania somnifera(Ashwagandha),Tinospora cordifolia(Giloy) andOcimum sanctum(Tulsi) - a Molecular Docking Study. J. Biomol. Struct. Dyn., 1–14. doi:10.1080/07391102.2020.1810778
Sinha, S. K., Prasad, S. K., Islam, M. A., Chaudhary, S. K., Singh, S., and Shakya, A. (2021). Potential Leads from Liquorice against SARS-CoV-2 Main Protease Using Molecular Docking Simulation Studies. Cchts 24, 591–597. doi:10.2174/1386207323999200817103148
Su, S., Wong, G., Shi, W., Liu, J., Lai, A. C. K., Zhou, J., et al. (2016). Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 24, 490–502. doi:10.1016/j.tim.2016.03.003
Tahir Ul Qamar, M., Alqahtani, S. M., Alamri, M. A., and Chen, L. L. (2020). Structural Basis of SARS-CoV-2 3CLpro and Anti-COVID-19 Drug Discovery from Medicinal Plants. J. Pharm. Anal. 10 (4), 313–319. doi:10.1016/j.jpha.2020.03.009
Tahmasbi, S. F., Revell, M. A., and Tahmasebi, N. (2021). Herbal Medication to Enhance or Modulate Viral Infections. Nurs. Clin. North America 56 (1), 79–89. doi:10.1016/j.cnur.2020.10.007
Tallei, T. E., Tumilaar, S. G., Niode, N. J., Fatimawali, fnm., Kepel, B. J., Idroes, R., et al. (2020). Potential of Plant Bioactive Compounds as SARS-CoV-2 Main Protease (Mpro) and Spike (S) Glycoprotein Inhibitors: A Molecular Docking Study. Scientifica 2020, 1–18. doi:10.1155/2020/6307457
Tareq, A. M., Farhad, S., Uddin, A. B. M. N., Hoque, M., Nasrin, M. S., Uddin, M. M. R., et al. (2020). Chemical Profiles, Pharmacological Properties, and In Silico Studies Provide New Insights on Cycas Pectinata. Heliyon 6 (6), e04061. doi:10.1016/j.heliyon.2020.e04061
Tiwari, R., Latheef, S. K., Ahmed, I., Iqbal, H. M. N., Bule, M. H., Dhama, K., et al. (2018). Herbal Immunomodulators - A Remedial Panacea for Designing and Developing Effective Drugs and Medicines: Current Scenario and Future Prospects. Cdm 19 (3), 264–301. doi:10.2174/1389200219666180129125436
Tripathi, M. K., Singh, P., Sharma, S., Singh, T. P., Ethayathulla, A. S., and Kaur, P. (2020). Identification of Bioactive Molecule fromWithania somnifera(Ashwagandha) as SARS-CoV-2 Main Protease Inhibitor. J. Biomol. Struct. Dyn., 1–14. doi:10.1080/07391102.2020.1790425
Wang, S.-C., Lu, M.-C., Chen, H.-L., Tseng, H.-I., Ke, Y.-Y., Wu, Y.-C., et al. (2009). Cytotoxicity of Calotropin Is through Caspase Activation and Downregulation of Anti-apoptotic Proteins in K562 Cells. Cell Biol Int. 33 (12), 1230–1236. doi:10.1016/j.cellbi.2009.08.013
Wu, C., Liu, Y., Yang, Y., Zhang, P., Zhong, W., Wang, Y., et al. (2020). Analysis of Therapeutic Targets for SARS-CoV-2 and Discovery of Potential Drugs by Computational Methods. Acta Pharmaceutica Sinica B 10 (5), 766–788. doi:10.1016/j.apsb.2020.02.008
Keywords: COVID-19, SARS-CoV-2, Mpro, in silico, Calotropis gigantea, Mpro inhibitor
Citation: Dutta M, Nezam M, Chowdhury S, Rakib A, Paul A, Sami SA, Uddin MZ, Rana MS, Hossain S, Effendi Y, Idroes R, Tallei T, Alqahtani AM and Emran TB (2021) Appraisals of the Bangladeshi Medicinal Plant Calotropis gigantea Used by Folk Medicine Practitioners in the Management of COVID-19: A Biochemical and Computational Approach. Front. Mol. Biosci. 8:625391. doi: 10.3389/fmolb.2021.625391
Received: 02 November 2020; Accepted: 11 May 2021;
Published: 26 May 2021.
Edited by:
William C. Cho, QEH, ChinaReviewed by:
Tania Begum, Uppsala University, SwedenAnnalisa Pastore, King’s College London, United Kingdom
Copyright © 2021 Dutta, Nezam, Chowdhury, Rakib, Paul, Sami, Uddin, Rana, Hossain, Effendi, Idroes, Tallei, Alqahtani and Emran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Talha Bin Emran, dGFsaGFibWJAYmdjdHViLmFjLmJk