 Nitzan Maman
Nitzan Maman Pramod Kumar
Pramod Kumar Amarjeet Yadav
Amarjeet Yadav Mario Feingold
Mario Feingold- 1Department of Physics, Ben Gurion University of the Negev, Beer Sheva, Israel
- 2The Ilse Katz Center for Nanotechnology, Ben Gurion University of the Negev, Beer Sheva, Israel
- 3Department of Applied Physics, Babasaheb Bhimrao Ambedkar University, Lucknow, India
In the Escherichia coli, RecA plays a central role in the recombination and repair of the DNA. For homologous recombination, RecA binds to ssDNA forming a nucleoprotein filament. The RecA-ssDNA filament searches for a homologous sequence on a dsDNA and, subsequently, RecA mediates strand exchange between the ssDNA and the dsDNA. In vitro, RecA binds to both ssDNA and dsDNA. Despite a wide range of studies of the polymerization of RecA on dsDNA, both at the single molecule level and by means of biochemical methods, important aspects of this process are still awaiting a better understanding. Specifically, a detailed, quantitative description of the nucleation and growth dynamics of the RecA-dsDNA filaments is still lacking. Here, we use Optical Tweezers together with a single molecule analysis approach to measure the dynamics of the individual RecA domains on dsDNA and the corresponding growth rates for each of their fronts. We focus on the regime where the nucleation and growth rate constants, kn and kg, are comparable, leading to a coverage of the dsDNA molecule that consists of a small number of RecA domains. For the case of essentially irreversible binding (using ATPγS instead of ATP), we find that domain growth is highly asymmetric with a ratio of about 10:1 between the fast and slow fronts growth rates.
Introduction
The primary function of the RecA protein is to exchange strands as part of the homologous recombination process in Escherichia Coli (Radding, 1988; Cox, 1999; Kowalczykowski, 2000; Cox, 2007b; Prentiss et al., 2015; Bell and Kowalczykowski, 2016). In addition, RecA plays an important role in DNA repair. One of the first steps in homologous recombination is the assembly of RecA-ssDNA filaments, whereby each RecA monomer attaches to three DNA base pairs. At the next stage, a RecA-ssDNA filament searches for a homologous sequence on the target dsDNA. Once such sequence is located, RecA induced strand exchange occurs, whereby the ssDNA replaces the homologous strand of the dsDNA. In vitro, RecA can polymerize on either ssDNA or dsDNA. The RecA-DNA filament is significantly more rigid than the bare DNA. Its formation requires ATP and a divalent cation e.g. Mg2+. Polymerization of RecA on DNA takes place via the growth of domains. The first few protein monomers that bind at adjacent sites in a protein free region of the DNA form a nucleus which subsequently grows by adding monomers to each of the two sides of the new domain. The growth of domains is asymmetric, being faster in the 5′ to 3′ direction of the DNA. While for ssDNA nucleation and growth are relatively fast, when the protein binds to dsDNA the nucleation step is much slower (Pugh and Cox, 1988). In the case of dsDNA, the presence of single strand regions, e.g., a nick, accelerates the nucleation step. Nucleation on dsDNA is also accelerated by stretching the molecule or reducing the pH. In contrast, the secondary structure of dsDNA restricts the attachment of the RecA preventing full coverage of the DNA. Structurally, the effect of RecA binding is to extend the length of each group of three base pairs (bp) by a factor of about 1.5 and unwind the dsDNA from a 35° twist down to one of only 20° (Stasiak and Egelman, 1994). Proteins that are homologous to RecA are found in all species, e.g., the human Rad51 protein (Baumann and West, 1998).
The RecA protein has been widely studied using biochemical methods and many of its properties are known. More recently, single molecule methods were introduced that allow measuring directly properties of a particular molecule which are lost at the macroscopic scale (Leger et al., 1998; Hegner et al., 1999; Shivashankar et al., 1999; Sattin and Goh, 2004; van der Heijden et al., 2005; Galletto et al., 2006; Joo et al., 2006; Mine et al., 2007; van Loenhout et al., 2009; Danilowicz et al., 2012; De Vlaminck et al., 2012; Forget and Kowalczykowski, 2012; Fu et al., 2013a; Fu et al., 2013b; Lee et al., 2013; Kim et al., 2014; Kim et al., 2015; Lee et al., 2015; Qi et al., 2015; Lee et al., 2016; Danilowicz et al., 2017; Lee et al., 2017). For example, magnetic and optical traps have been used to study the effect of external force and torque on the polymerization of proteins on DNA and for testing models of homology search strategies (Leger et al., 1998; Hegner et al., 1999; Shivashankar et al., 1999; van der Heijden et al., 2005; Mine et al., 2007; van Loenhout et al., 2009; Danilowicz et al., 2012; De Vlaminck et al., 2012; Forget and Kowalczykowski, 2012; Fu et al., 2013a; Fu et al., 2013b; Lee et al., 2013). Moreover, quick dynamic changes during the RecA-DNA filament formation were measured using fluorescence resonance energy transfer (FRET) (Joo et al., 2006). FRET also allows monitoring the kinetics of the RecA mediated strand exchange process by selectively labeling each of the two strands with different fluorophores, a donor and an acceptor (Kim et al., 2014; Kim et al., 2015; Danilowicz et al., 2017).
In the assembly of the RecA-dsDNA filament, the nucleation and growth rates strongly depend on the environmental chemical and physical conditions. Measuring the rate of ATP hydrolysis, it was shown that the rate of RecA binding is larger for a dsDNA tailed with a 5′ ssDNA than one tailed with a 3′ ssDNA (Register and Griffith, 1985; Lindsley and Cox, 1990). This indicates that the growth of RecA domains on DNA is asymmetric with a strong preference to the 5′ to 3′ direction. In the light of these results, it was suggested that RecA-dsDNA filaments that form in the presence of the essentially non-hydrolysable ATPγS (Weinstock et al., 1981) should behave differently, namely, their assembly would proceed in a symmetric manner (Cox, 2007a). Apparently, this expectation relied on the assumption that the RecA-ATP complex has a different configuration when attached to DNA and this configuration change is due to the ATP hydrolysis.
Biochemical methods, as those described in the previous paragraph, cannot provide detailed information regarding the dynamics of the individual RecA domains on a particular dsDNA molecule. Such domain dynamics can be extracted using a single DNA approach that exploits the fact that RecA binding leads to the local extension of the dsDNA length. In particular, one can extract the coverage dynamics of RecA on dsDNA from the time dependent growth of the dsDNA contour length during RecA polymerization. This approach was used to determine the rate constants of nucleation,
Single molecule methods were also used to study the effect of force applied to the DNA on the rate of RecA polymerization and the efficiency of the homology search (Leger et al., 1998; van Loenhout et al., 2009; Danilowicz et al., 2012; De Vlaminck et al., 2012; Fu et al., 2013a; Fu et al., 2013b). In the experiments of Leger et al. (Leger et al., 1998), the dsDNA molecule was stretched with constant force (10–100 pN) and the elongation rate due to RecA binding was monitored. It was shown that the RecA-dsDNA complex formed more rapidly in the presence of applied force that reduced the energy barrier for protein binding. For example, stretching the dsDNA with a force of 75 pN lead to a reaction rate that was 20 times faster than that observed when a 15 pN force was applied. Fu et al. (Fu et al., 2013b) used magnetic tweezers to study the competition between RecA polymerization and de-polymerization on dsDNA for different temperatures, pH values and salt concentrations. They showed that these parameters distinguish between a regime where RecA polymerization is stable and another where it is transient reverting to naked dsDNA at the end of the process. Magnetic tweezers were also used to control the torque applied to the dsDNA and probe its effect on the assembly of RecA or Rad51 onto dsDNA (van der Heijden et al., 2005; Lee et al., 2013). van der Heijden et al. (van der Heijden et al., 2005) found that the polymerization process stalls at high torsions of the dsDNA molecule leading to dsDNA molecules that are only partially covered with RecA.
The different stages of homologous recombination were also studied at the single molecule level using fluorescence microscopy (Galletto et al., 2006; Forget and Kowalczykowski, 2012; Lee et al., 2015; Qi et al., 2015; Lee et al., 2016; Lee et al., 2017). For example, Galletto et al. (Galletto et al., 2006) analyzed the assembly of individual RecA-dsDNA filaments. They used fluorescently labeled RecA to image the segments of the dsDNA that were coated with RecA at intermediate stages of the polymerization reaction. In agreement with previous results from biochemical studies, they showed that RecA polymerization is controlled by the slow nucleation step. When ATP was used in the reaction, the corresponding critical nucleus is about four–five protein monomers. In contrast, the growth of the RecA domains is a relatively fast process for both of the cofactors that were studied, namely, ATP and ATPγS. The asymmetry of RecA domain growth was also analyzed in the presence of either ATP or ATPγS. Although for both cofactors the data indicated a certain extent of asymmetric growth, it was not sufficiently accurate to allow for the quantitative determination of the fast and slow growth rates.
In this paper, we present a new approach to measuring the growth rates of individual RecA domains on dsDNA that allows obtaining the growth rate for each of the two fronts of individual domains. This enables us to establish the extent of asymmetry between the fast and the slow fronts of each domain. To this end, we restrict our study to the case of essentially irreversible binding (ATPγS) (Lindsley and Cox, 1989). Biochemical studies indicate that ATP hydrolysis is only required in the final phase of RecA dissociating from the DNA and, accordingly, using ATPγS instead of ATP has become a standard approach in the study of the reaction between RecA and DNA (Weinstock et al., 1981). Similarly, in our study, ATPγS was used in order to separate the assembly phase of the RecA on dsDNA from that of the disassembly. Moreover, we choose the experimental parameters such that the nucleation and growth rates are comparable,
Materials and Methods
Experimental System
In our system, we use Optical Tweezers to apply and measure forces on individual dsDNA molecules. It consists of a near infrared laser beam (SDL 5422H1, 830 nm wavelength) focused through a 100X objective (Zeiss, 1.25 numerical aperture, oil immersion) together with a Quadrant Photodiode (QPD) that monitors the position of a trapped microbead (Figure 1). The power of the laser beam at the optical trap is about 30 mW. Single dsDNA molecules are attached at one end to the bottom cover glass and at the other end to a polystyrene microbead of 1.6 μm diameter (Polysciences). The experiment is performed in a flow cell that allows changing the biochemical composition of the sample. To avoid breaking the bead-dsDNA-cover glass constructs, we inject new solution at relatively low flow rates using a DC motor (Newport) to activate a syringe. Following the addition of RecA to the sample and its assembly on the dsDNA molecules, the dsDNA elongates by an extent proportional to the amount of bound protein. To monitor the change in the contour length of a particular dsDNA molecule, we extend the filament by applying an approximately constant force on the trapped bead (∼0.8 pN).
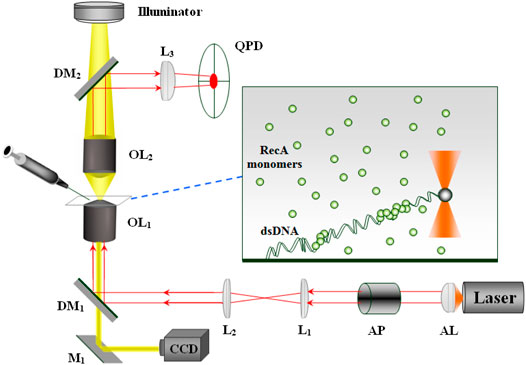
FIGURE 1. The optical system: Laser—single mode (TEM00) diode laser, AL—aspherical lens, AP—anamorphic prisms, L1, L2, L3—lenses, M1—mirror, DM1, DM2—dichoric mirrors, OL1, OL2—objectives. While OL1 focuses the laser beam forming the optical trap, OL2 collects the light scattered from the trapped bead that after reflection from DM2 reaches the QPD. The protein and the appropriate buffer are injected into the sample via a motorized syringe.
The force exerted by the dsDNA-RecA filament on the trapped bead, F, manifests as a shift in the bead position with respect to the center of the trap, Δx. Correspondingly, the value of Δx affects the distribution of the laser light scattered from the trapped bead. This variation in the distribution of the laser light transmitted through the sample is monitored by the QPD. Calibrating the QPD allows to deduce the value of Δx from the voltage difference between the appropriate quadrants. Although the system allows a sampling rate of 20 KHz, we average the data down to rates in the 1–10 Hz range to reduce the effect of Brownian motion. The force is adjusted whenever it deviates from the 0.8 pN mark by an appropriate shift of the sample. Since the optical trap is at a fixed position, we move the sample to change the end-to-end distance of the DNA molecule, leading to a corresponding change in the force. A Peltier element together with a feedback control unit (PID) is used to maintain a constant temperature in the sample at 37.0 ± 0.1°C. It is attached to a copper ring that is in thermal contact with the microscope objective.
Sample Preparation
The bead-dsDNA-glass construct is prepared using a low pH protocol for spontaneous binding (Allemand et al., 1997; Shivashankar et al., 1999). λ-DNA, 48.5 Kb, 16.5 μm (Promega) is incubated with the beads and PBS buffer at pH = 4 for 15 min to obtain the DNA-bead link. Next, the solution of DNA and beads is injected into the sample and after 2 h incubation we find that a certain fraction of the DNA molecules are tethered both to the glass bottom and to a microbead. Finally, we gently wash the sample to remove free beads and unbound DNA molecules. For a particular bead-dsDNA-glass construct, we verify that the bead is tethered by a single dsDNA molecule by measuring its force extension behavior and comparing it to the prediction of the Worm Like Chain (WLC) model (see Eq. 1) (Marko and Siggia, 1995). We then stretch the dsDNA and inject the protein together with the appropriate buffer solution into the sample cell. The final concentrations are as follows: RecA (9.33 μM), ATPγS (4.5 mM), MgCl2 (6.25 mM), DDT (6.25 mM), TrisHCl (18.75 mM), and the pH is 7.9. The pH and the protein and buffer concentrations were chosen such that the nucleation is sufficiently slow allowing to observe the dynamics due to individual nucleation events.
Although the low pH protocol for obtaining the bead-dsDNA-glass construct is not widely used in single molecule experiments, it is particularly straightforward allowing us to perform a relatively large number of experiments. On one hand, its main drawback is that it is by far less specific than, for example, the standard biotin:streptavidin tethers, leading to a fraction of bead-dsDNA-glass constructs where the dsDNA is attached at some internal site rather than at its end to either the glass or the bead. However, for our experimental approach, such constructs are equally suitable as those where the dsDNA is only tethered at its ends. To include the non-specific constructs in our experimental data, we measure the contour length of the dsDNA between tethering points for each construct using the WLC model (see Eq. 1) (Marko and Siggia, 1995). Moreover, we test that the length of the dsDNA is not affected by the increase in pH to 7.9 in preparation for the RecA reaction. On the other hand, the non-specific constructs allow obtaining data for different contour lengths of the naked dsDNA, an additional parameter that affects the polymerizarion dynamics of RecA on dsDNA.
Measuring the Length of the dsDNA-RecA Complex
To monitor the length dynamics of the dsDNA-RecA complex during the polymerization process, we need to find the way it is related to the measured time dependence of the force exerted on the trapped bead. The equilibrium behavior of polymers under tension has been extensively studied (Smith et al., 1992; Perkins et al., 1995; Cluzel et al., 1996; Simmons et al., 1996; Wang et al., 1997). Using the WLC model, it was shown that the force, F, required to stretch the polymer to a certain end-to-end distance, z, is linear in the small z regime and rapidly grows as z approaches the contour length, L (Marko and Siggia, 1995). A good approximation to the exact
where A is the persistence length, T, the temperature and
For naked dsDNA in solution of physiological ionic strength and
where

FIGURE 2. A dsDNA with a naked section (thin curve) and a single RecA domain (thick curve). Each segment of the molecule is characterized by its contour length, end-to-end distance and persistence length. In the experiment, we measure the force exerted by the dsDNA molecule on the trapped bead and the end-to-end distance of the entire molecule, z, as a function of time.
Nucleation and Growth Model for the Fast nucleation Regime
In the regime where the rate of protein nucleation,
While the rate constants of Eqs. 3a,b depend on the number of RecA binding sites on the naked DNA,
Equations. 3a,b can be easily solved leading to a sigmoidal behavior for the dynamics of the protein coverage
Moreover, the model of Eqs. 3a,b can be generalized to include the case where the average rate of growth of the domains is asymmetric, such that, the growth rate in the 3′ to 5′ direction of the dsDNA is r times slower than that in the reverse direction. For this case
where
Nucleation and Growth Model for the Slow nucleation Regime
Another regime of the nucleation and growth process that can be described via an exactly solvable model is the limit of slow nucleation,
where x · L is the position of the nucleation event and
Averaging Eq. 6 over all possible realizations, gives the ensemble averaged coverage dynamics,
Unlike the behavior in the fast nucleation regime, here
Results
Kinetics of RecA Protein Polymerization on Single dsDNA Molecules
Kinetics of the dsDNA-RecA Filament Length
In our experimental setup, we measure the force exerted on the microbead by a single stretched dsDNA-RecA filament, F. For example, in Figure 3A the time trace of F,
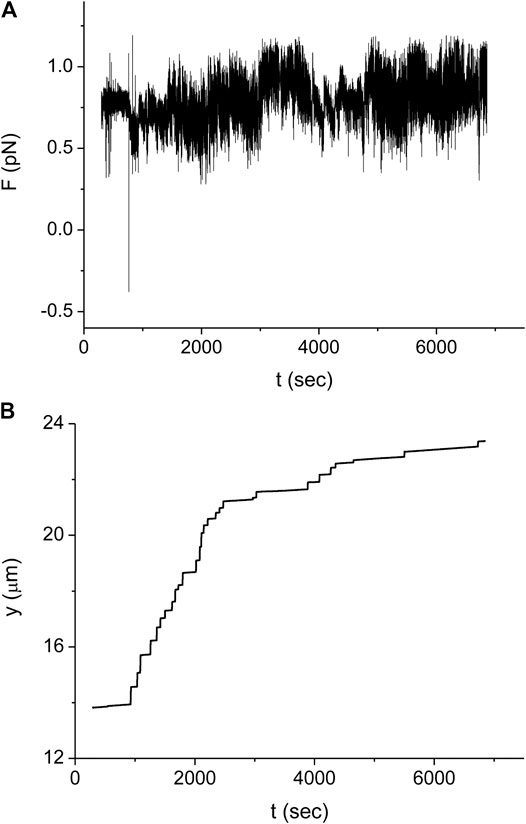
FIGURE 3. Typical experiment monitoring the RecA polymerization dynamics on a single dsDNA molecule. (A) Force as a function of time measured for a dsDNA molecule with L (0) = 16.4 μm.(B) Trap position relative to the point where the dsDNA molecule is tethered as a function of time for the same experiment as in (A). Time is measured from the moment when we start to inject the protein into the sample. Since during protein injection and for a short period afterward there is noticeable fluid flow in the sample, no measurements were made during the corresponding time interval (about 300 s).
In order to obtain the kinetics of the protein assembly on the dsDNA from the measurements of Figure 3, we use Eq. 2 where the value of the end to end distance, z, is approximated by the distance between the center of the optical trap and the dsDNA tethering point, y (see Figure 3B). The difference between these two quantities, z and y, is due to several, relatively small corrections that, moreover, almost cancel out. While the radius of the bead, 0.8 μm, and the displacement of the bead from the center of the trap due to the applied force, ∼0.2 μm, should be subtracted from the value of y, the effect due to the height of the trap above the bottom of the sample leads to an increase in the value of z relative to that of y. In our analysis, we assume that the effect of these corrections is below our experimental accuracy. In Figure 4 we show the resulting behavior of the RecA-dsDNA complex length as a function of time,
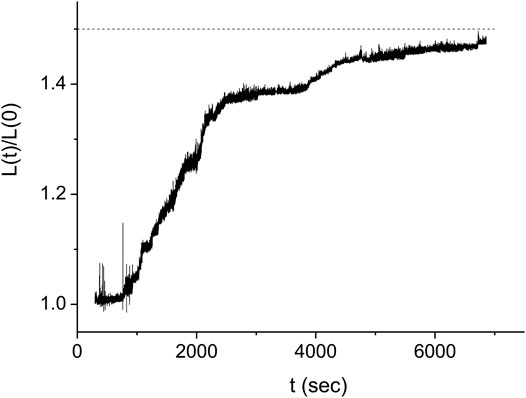
FIGURE 4. Kinetics of the DNA length as a function of time for the same experiment as in Figure 3 (full line). The dashed line indicates the expected saturation length corresponding to complete coverage of the dsDNA by RecA protein. The stepwise structure of the
Kinetics of the Individual RecA-dsDNA Domains
The multiple step behavior of
One may extend the interpretation relating the
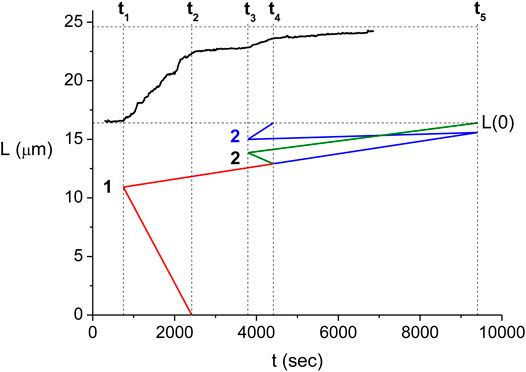
FIGURE 5. Relation between the domain kinetics and the measured
Each of the kinetic scenarios consistent with a particular
where
In the domain kinetics equations, e.g. those of Eqs. 8a–f, the values of
For the experiment of Figures 3–5, we obtain the values of
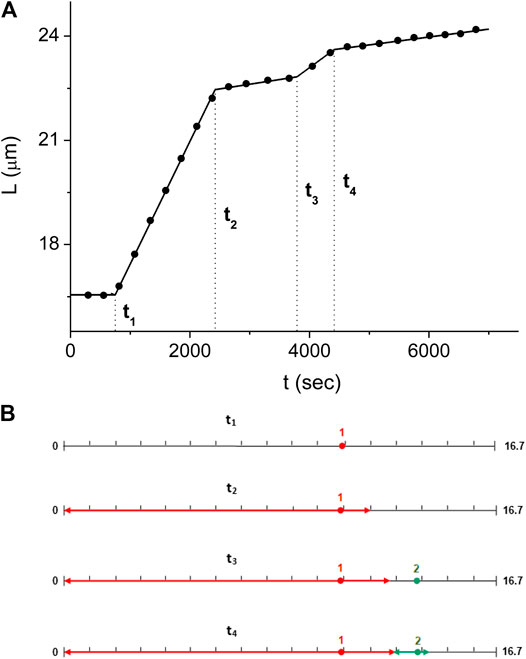
FIGURE 6. Domain kinetics corresponding to the solution of Eq. 8(A) The dsDNA length kinetics,
Statistical Analysis of the Domain Kinetics for RecA Polymerization on Single dsDNA
We have performed a series of experiments under the same conditions as those leading to the results of Figures 3–5 (see Supplementary Section S1 for details). As one would expect, the
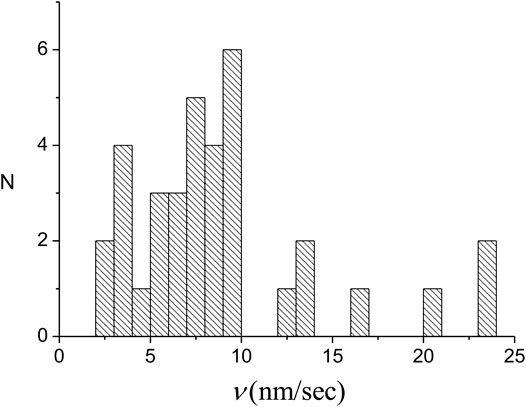
FIGURE 7. Distribution of the individual domain growth rates of 35 nuclei. For each domain the growth rate represents the sum of the rates for the slow and fast fronts.
The value of the average domain growth rate,
where
Moreover, the relation of Eq. 10 can be expanded to include the first N nucleation events, namely,
where
Equation 11 allows us to estimate the average nucleation rate density,
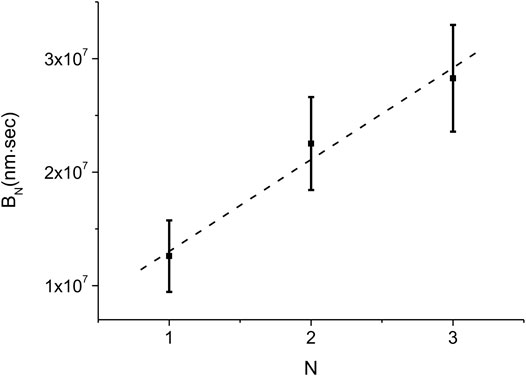
FIGURE 8. Experimental measurement of the average nucleation density rate,
For three of our experiments, the domain kinetics inferred from our model allowed to obtain the velocity of each of the individual fronts. In these experiments, the
While in the few nuclei regime where we have performed our experiments we can separately measure the average values of the nucleation and growth rates,
Nucleation and Growth Model for the Few Nuclei Regime
In the intermediate regime, where
The fluctuations of
To obtain the behavior of
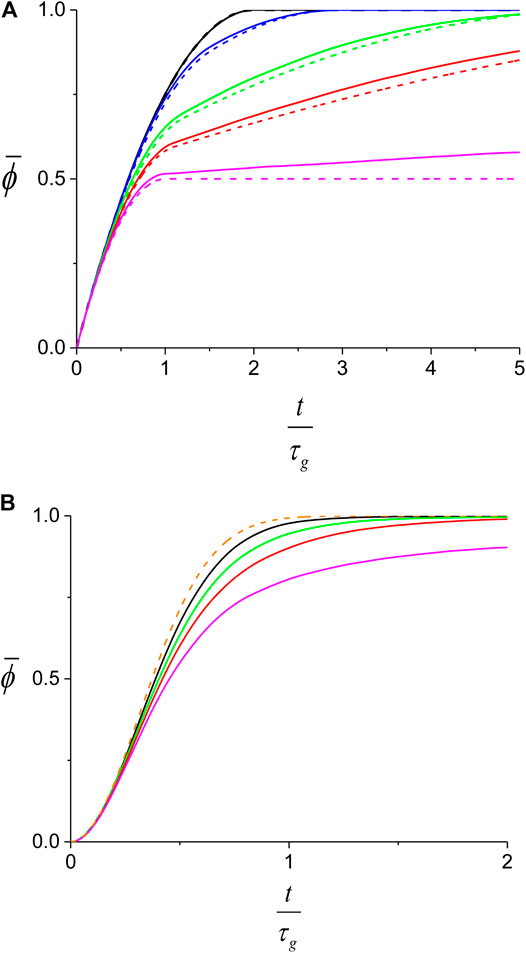
FIGURE 9. Theoretical prediction for the average protein coverage on dsDNA. While the full lines are obtained from the Monte-Carlo simulation, the dashed lines correspond to the analytical behavior described by Eqs. 5,7. (A) slow nucleation regime,
In Figure 9 we show the behavior of the average coverage,
For the slow nucleation regime, the larger the growth asymmetry, smaller r, the slower will be the process of decorating the dsDNA on the side of the slow front of the first domain. Consequently, for small r, the
The lower efficiency of protein coverage for asymmetric growth is also found in the fast nucleation regime (Figure 9B) and can be ascribed to a mechanism similar to the one presented in the previous paragraph. When the number of domains is not too large, asymmetric growth can lead to persisting, relatively large undecorated sections between two slow growing fronts. The process of protein assembly in such regions remains inefficient until a new nucleation event occurs there. In contrast, the prediction of Eq. 9 corresponds to the limit where the number of domains is infinite and therefore, the growth asymmetry has no effect on
As discussed in the previous section, our experiments were performed in the few nuclei regime where neither Eq. 5 nor Eq. 7 are valid. Instead, we can use the Monte Carlo model described above to predict the expected behavior of
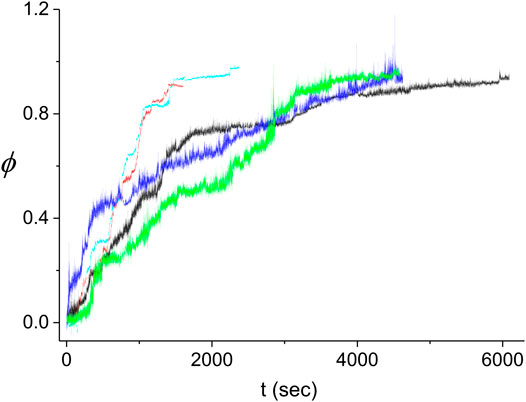
FIGURE 10. Kinetics of the decorated length fraction of the dsDNA. The curves correspond to five different experiments performed under identical conditions (see text). The black curve is the same as in Figures 4–6. The difference in the fluctuations in the light blue and red curves as compared to those in the other three curves is due to the different extent of averaging of the raw data from the QPD (sampled at 20 kHz). To obtain a similar accuracy in the decomposition of
The large variability between the experimental kinetics obtained in individual experiments indicates that these cannot be individually described by our Monte Carlo model. Instead, we expect that the model should be able to reproduce the average experimental kinetics,
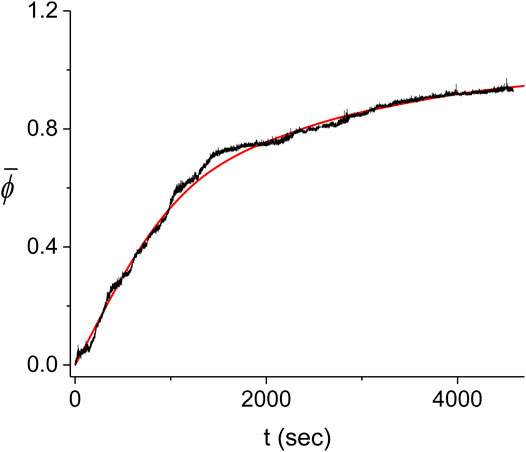
FIGURE 11. Average kinetics of the decorated length fraction of the dsDNA. The average of the five experiments shown in Figure 10 (black) is compared with the corresponding best fitting result from the Monte Carlo model (red). The best fit was obtained for
For simplicity, we also use the values of the scan steps to represent the errors of the best fitting parameters. These errors are therefore overestimated and should be regarded as upper bounds and of the same order of magnitude as the exact values. We find that the best fit to the experimental

TABLE 1. The parameters that describe the nucleation and growth process in our experiment as obtained by each our two methods: averaging over the kinetics of individual domains as obtained from the decomposition of the
Discussion
Measuring the force exerted on microbeads with accuracy better than 0.1 pN, we have monitored the polymerization of the RecA protein on individual dsDNA molecules in the presence of ATPγS. Since the RecA-dsDNA complex is about 1.5 times longer than the naked dsDNA, the polymerization process leads to a gradual increase in the contour length of the dsDNA allowing us to obtain the kinetics of the protein coverage on a particular dsDNA molecule. In the presence of ATPγS, the protein coverage is essentially irreversible (Lindsley and Cox, 1989). We show that a model which assumes a nucleation and growth mechanism exhibiting a small number of nuclei in the parameter range used in our experiments yields predictions that are consistent with our experimental data. In this regime, we can decompose the global coverage kinetics to infer the kinetics for each of the growth fronts that develop on the two sides of a nucleus. We therefore obtain the distribution of front velocities and find that each domain grows asymmetrically with a fast front that has, on average, a velocity about 10 times larger than that of the corresponding slow front.
In the few nuclei regime, the kinetics of the protein coverage on a single dsDNA,
An alternative approach that allows to obtain the rate constants relies on modeling the nucleation and growth process in the intermediate regime where
Since in our search for a low nucleation regime we used particular experimental parameters that are different from those of other studies, it is difficult to compare our results to those of previous studies. For example, our ATPγS concentration is 4.5 times larger than that used in Ref. (Shivashankar et al., 1999), the RecA concentration is almost the same, the other chemicals are at about a third of their concentration and our pH is 7.9 while theirs is 6.8. As one would expect, in the corresponding ATPγS experiment presented in Ref. (Shivashankar et al., 1999) (Figure 5B), Shivashankar et al. estimate to have 6 domains and the overall length dynamics is well approximated by the multiple domain dynamics of Eq. 3. The value of the product between the DNA length independent nucleation and growth constants,
On the Nucleation and Growth Rate Constants
Although RecA and dsDNA are complex biomolecules, we may obtain further insight on the way they assemble in the presence of ATPγS by comparing this process to a simple bimolecular reaction of the type
where the
Despite the apparent differences between the bimolecular adsobtion and reaction, in our case, the two become equivalent in the limit where the dsDNA filament is only three base pairs long, consisting of a single binding site for the
where […] denotes the concentration and we have assumed that
Since the bimolecular adsorption and simple bimolecular reaction become equivalent in the limit described above, one expects that the exponential behavior of the latter,
In the case of our adsorption experiments of
Depending on the nature of the reactants, chemical reactions can be either diffusion-limited or reaction-limited. Pugh and Cox have shown that the reaction between RecA and dsDNA is reaction-limited at saturated protein concentration (Pugh and Cox, 1987). Since our experiments are performed in this regime, the corresponding rate constants follow the Arrhenius law, that is,
and
The value of
The Asymmetry of the Domain Growth
Both the
Unlike in the biochemical studies, neither in our experiments nor in those of Galletto et al. (Galletto et al., 2006) can one establish the relation between the domain growth asymmetry and the direction along the DNA. However, we can use Eq. 17 to determine the difference between the energy barriers for
Regarding the mechanism of asymmetric domain growth of
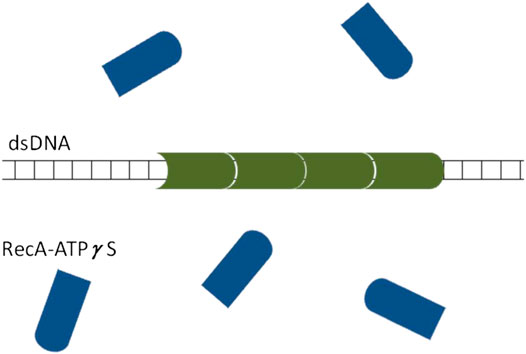
FIGURE 12. A schematic illustration of the proposed scenario leading to asymmetric
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author Contributions
NM performed the experiments. NM and MF built the experimental system, designed the experimental approach and wrote the manuscript. NM, PK and AY analyzed the data.
Funding
This research was supported in part by the Israel Academy of Science and Humanities (Grant No. 1519/18).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank I. Fishov and R. Granek for useful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.609076/full#supplementary-material.
References
Allemand, J. F., Bensimon, D., Jullien, L., Bensimon, A., and Croquette, V. (1997). pH-dependent specific binding and combing of DNA. Biophys. J. 73, 2064–2070. doi:10.1016/s0006-3495(97)78236-5
Avrami, M. (1939). Kinetics of phase change. I general theory. J. Chem. Phys. 7, 1103. doi:10.1063/1.1750380
Baumann, P., and West, S. C. (1998). Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci. 23, 247–251. doi:10.1016/s0968-0004(98)01232-8
Bell, J. C., and Kowalczykowski, S. C. (2016). RecA: regulation and mechanism of a molecular search engine. Trends Biochem. Sci. 41, 491–507. doi:10.1016/j.tibs.2016.04.002
Cluzel, P., Lebrun, A., Heller, C., Lavery, R., Viovy, J.-L., Chatenay, D., et al. (1996). DNA: an extensible molecule. Science 271, 792–794. doi:10.1126/science.271.5250.792
Cox, M. M. (1999). Recombinational DNA repair in bacteria and the RecA protein. Prog. Nucleic Acid Res. Mol. Biol. 63, 311–366. doi:10.1016/s0079-6603(08)60726-6
Cox, M. M. (2007b). Regulation of bacterial RecA protein function. Crit. Rev. Biochem. Mol. Biol. 42, 41–63. doi:10.1080/10409230701260258
Cox, M. M. (2007a). “The bacterial RecA protein: structure, function and regulation,” in Topics in current genetics. Molecular genetics of recombination. Editors A. Aguilera, and R. Rothstein (Germany: Springer-Verlag GmbH), 53–94.
Danilowicz, C., Feinstein, E., Conover, A., Coljee, V. W., Vlassakis, J., Chan, Y.-L., et al. (2012). RecA homology search is promoted by mechanical stress along the scanned duplex DNA. Nucleic Acids Res. 40, 1717–1727. doi:10.1093/nar/gkr855
Danilowicz, C., Hermans, L., Coljee, V., Prévost, C., and Prentiss, M. (2017). ATP hydrolysis provides functions that promote rejection of pairings between different copies of long repeated sequences. Nucleic Acids Res. 45, 8448–8462. doi:10.1093/nar/gkx582
De Vlaminck, I., Van Loenhout, M. T., Zweifel, L., Den Blanken, J., Hooning, K., Hage, S., et al. (2012). Mechanism of homology recognition in DNA recombination from dual-molecule experiments. Mol. Cell 46, 616–624. doi:10.1016/j.molcel.2012.03.029
Forget, A. L., and Kowalczykowski, S. C. (2012). Single-molecule imaging of DNA pairing by RecA reveals a three-dimensional homology search. Nature 482, 423–427. doi:10.1038/nature10782
Fu, H., Le, S., Chen, H., Muniyappa, K., and Yan, J. (2013a). Force and ATP hydrolysis dependent regulation of RecA nucleoprotein filament by single-stranded DNA binding protein. Nucleic Acids Res. 41, 924–932. doi:10.1093/nar/gks1162
Fu, H., Le, S., Muniyappa, K., and Yan, J. (2013b). Dynamics and regulation of RecA polymerization and de-polymerization on double-stranded DNA. PLoS One 8, e66712. doi:10.1371/journal.pone.0066712
Galletto, R., Amitani, I., Baskin, R. J., and Kowalczykowski, S. C. (2006). Direct observation of individual RecA filaments assembling on single DNA molecules. Nature 443, 875–878. doi:10.1038/nature05197
Hegner, M., Smith, S. B., and Bustamante, C. (1999). Polymerization and mechanical properties of single RecA-DNA filaments. Proc. Natl. Acad. Sci. U S A. 96, 10109–10114. doi:10.1073/pnas.96.18.10109
Joo, C., Mckinney, S. A., Nakamura, M., Rasnik, I., Myong, S., and Ha, T. (2006). Real-time observation of RecA filament dynamics with single monomer resolution. Cell 126, 515–527. doi:10.1016/j.cell.2006.06.042
Kim, S. H., Park, J., Joo, C., Kim, D., and Ha, T. (2015). Dynamic growth and shrinkage govern the pH dependence of RecA filament stability. PLoS One 10, e0115611. doi:10.1371/journal.pone.0115611
Kim, S. H., Ragunathan, K., Park, J., Joo, C., Kim, D., and Ha, T. (2014). Cooperative conformational transitions keep RecA filament active during ATPase cycle. J. Am. Chem. Soc. 136, 14796–14800. doi:10.1021/ja506363y
Kowalczykowski, S. C. (2000). Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 25, 156–165. doi:10.1016/s0968-0004(00)01569-3
Lee, J. Y., Qi, Z., and Greene, E. C. (2016). ATP hydrolysis promotes duplex DNA release by the RecA presynaptic complex. J. Biol. Chem. 291, 22218–22230. doi:10.1074/jbc.m116.740563
Lee, J. Y., Steinfeld, J. B., Qi, Z., Kwon, Y., Sung, P., and Greene, E. C. (2017). Sequence imperfections and base triplet recognition by the Rad51/RecA family of recombinases. J. Biol. Chem. 292, 11125–11135. doi:10.1074/jbc.m117.787614
Lee, J. Y., Terakawa, T., Qi, Z., Steinfeld, J. B., Redding, S., Kwon, Y., et al. (2015). DNA RECOMBINATION. Base triplet stepping by the Rad51/RecA family of recombinases. Science 349, 977–981. doi:10.1126/science.aab2666
Lee, M., Lipfert, J., Sanchez, H., Wyman, C., and Dekker, N. H. (2013). Structural and torsional properties of the RAD51-dsDNA nucleoprotein filament. Nucleic Acids Res. 41, 7023–7030. doi:10.1093/nar/gkt425
Leger, J. F., Robert, J., Bourdieu, L., Chatenay, D., and Marko, J. F. (1998). RecA binding to a single double-stranded DNA molecule: a possible role of DNA conformational fluctuations. Proc. Natl. Acad. Sci. U S A. 95, 12295–12299. doi:10.1073/pnas.95.21.12295
Lindsley, J. E., and Cox, M. M. (1990). Assembly and disassembly of RecA protein filaments occur at opposite filament ends. Relationship to DNA strand exchange. J. Biol. Chem. 265, 9043–9054. doi:10.1016/s0021-9258(19)38809-x
Lindsley, J. E., and Cox, M. M. (1989). Dissociation pathway for recA nucleoprotein filaments formed on linear duplex DNA. J. Mol. Biol. 205, 695–711. doi:10.1016/0022-2836(89)90315-x
Marko, J. F., and Siggia, E. D. (1995). Stretching DNA. Macromolecules 28, 8759–8770. doi:10.1021/ma00130a008
Mine, J., Disseau, L., Takahashi, M., Cappello, G., Dutreix, M., and Viovy, J.-L. (2007). Real-time measurements of the nucleation, growth and dissociation of single Rad51-DNA nucleoprotein filaments. Nucleic Acids Res. 35, 7171–7187. doi:10.1093/nar/gkm752
Neuhaus, J.-M., Wanger, M., Keiser, T., and Wegner, A. (1983). Treadmilling of actin. J. Muscle Res. Cell Motil. 4, 507–527. doi:10.1007/bf00712112
Perkins, T., Smith, D., Larson, R., and Chu, S. (1995). Stretching of a single tethered polymer in a uniform flow. Science 268, 83–87. doi:10.1126/science.7701345
Pierobon, P., Mine-Hattab, J., Cappello, G., Viovy, J. L., and Lagomarsino, M. C. (2010). Separation of time scales in one-dimensional directed nucleation-growth processes. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 82, 061904. doi:10.1103/physreve.82.061904
Prentiss, M., Prevost, C., and Danilowicz, C. (2015). Structure/function relationships in RecA protein-mediated homology recognition and strand exchange. Crit. Rev. Biochem. Mol. Biol. 50, 453–476. doi:10.3109/10409238.2015.1092943
Pugh, B. F., and Cox, M. M. (1988). General mechanism for RecA protein binding to duplex DNA. J. Mol. Biol. 203, 479–493. doi:10.1016/0022-2836(88)90014-9
Pugh, B. F., and Cox, M. M. (1987). Stable binding of recA protein to duplex DNA. Unraveling a paradox. J. Biol. Chem. 262, 1326–1336. doi:10.1016/s0021-9258(19)75790-1
Qi, Z., Redding, S., Lee, J. Y., Gibb, B., Kwon, Y., Niu, H., et al. (2015). DNA sequence alignment by microhomology sampling during homologous recombination. Cell 160, 856–869. doi:10.1016/j.cell.2015.01.029
Radding, C. M. (1988). “Homologous pairing and strand exchange promoted by Escherichia coli RecA,” in Genetic recombination. Editor G. R. Smith (Washington, DC: American Society for Microbiology), 193–229.
Register, J. C., and Griffith, J. (1985). The direction of RecA protein assembly onto single strand DNA is the same as the direction of strand assimilation during strand exchange. J. Biol. Chem. 260, 12308–12312. doi:10.1016/s0021-9258(17)39026-9
Rocha, M. S., Viana, N. B., and Mesquita, O. N. (2004). DNA-psoralen interaction: a single molecule experiment. J. Chem. Phys. 121, 9679–9683. doi:10.1063/1.1806817
Sattin, B. D., and Goh, M. C. (2004). Direct observation of the assembly of RecA/DNA complexes by atomic force microscopy. Biophys. J. 87, 3430–3436. doi:10.1529/biophysj.104.045567
Shaner, S. L., Flory, J., and Radding, C. M. (1987). The distribution of Escherichia coli recA protein bound to duplex DNA with single-stranded ends. J. Biol. Chem. 262, 9220–9230. doi:10.1016/s0021-9258(18)48070-2
Shaner, S. L., and Radding, C. M. (1987). Translocation of Escherichia coli recA protein from a single-stranded tail to contiguous duplex DNA. J. Biol. Chem. 262, 9211–9219. doi:10.1016/s0021-9258(18)48069-6
Shivashankar, G. V., Feingold, M., Krichevsky, O., and Libchaber, A. (1999). RecA polymerization on double-stranded DNA by using single-molecule manipulation: the role of ATP hydrolysis. Proc. Natl. Acad. Sci. U S A. 96, 7916–7921. doi:10.1073/pnas.96.14.7916
Simmons, R. M., Finer, J. T., Chu, S., and Spudich, J. A. (1996). Quantitative measurements of force and displacement using an optical trap. Biophys. J. 70, 1813–1822. doi:10.1016/s0006-3495(96)79746-1
Smith, S. B., Finzi, L., and Bustamante, C. (1992). Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science 258, 1122–1126. doi:10.1126/science.1439819
Stasiak, A., and Egelman, E. H. (1994). Structure and function of RecA-DNA complexes. Experientia 50, 192–203. doi:10.1007/bf01924002
Turner, M. S. (2000). Two time constants for the binding of proteins to DNA from micromechanical data. Biophys. J. 78, 600–607. doi:10.1016/s0006-3495(00)76620-3
van der Heijden, T., Van Noort, J., Van Leest, H., Kanaar, R., Wyman, C., Dekker, N. H., et al. (2005). Torque-limited RecA polymerization on dsDNA. Nucleic Acids Res. 33, 2099–2105. doi:10.1093/nar/gki512
van Loenhout, M. T. J., Van Der Heijden, T., Kanaar, R., Wyman, C., and Dekker, C. (2009). Dynamics of RecA filaments on single-stranded DNA. Nucleic Acids Res. 37, 4089–4099. doi:10.1093/nar/gkp326
Wang, M. D., Yin, H., Landick, R., Gelles, J., and Block, S. M. (1997). Stretching DNA with optical tweezers. Biophys. J. 72, 1335–1346. doi:10.1016/s0006-3495(97)78780-0
Keywords: single molecule, recA, optical tweezers, protein-DNA interaction, nucleation and growth
Citation: Maman N, Kumar P, Yadav A and Feingold M (2021) Single Molecule Study of the Polymerization of RecA on dsDNA: The Dynamics of Individual Domains. Front. Mol. Biosci. 8:609076. doi: 10.3389/fmolb.2021.609076
Received: 23 September 2020; Accepted: 02 February 2021;
Published: 22 March 2021.
Edited by:
Michael W. Berns, University of California, Irvine, United StatesReviewed by:
Douglas Smith, University of California, San Diego, United StatesVeikko Linko, Aalto University, Finland
Copyright © 2021 Maman, Kumar, Yadav and Feingold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario Feingold, bWFyaW9AZXhjaGFuZ2UuYmd1LmFjLmls
†These authors have contributed equally to this work