
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Biosci., 19 March 2021
Sec. Molecular Diagnostics and Therapeutics
Volume 8 - 2021 | https://doi.org/10.3389/fmolb.2021.595712
This article is part of the Research TopicPulmonary Fibrosis: from Bench to BedsideView all 9 articles
 Yilu Zhou1,2†
Yilu Zhou1,2† Charlotte Hill1†Liudi Yao1Juanjuan Li1David Hancock3Julian Downward3
Charlotte Hill1†Liudi Yao1Juanjuan Li1David Hancock3Julian Downward3 Mark G. Jones2,4,5
Mark G. Jones2,4,5 Donna E. Davies2,4,5
Donna E. Davies2,4,5 Rob M. Ewing1,2
Rob M. Ewing1,2 Paul Skipp1,2,6*
Paul Skipp1,2,6* Yihua Wang1,2,5*
Yihua Wang1,2,5*Alveolar type II (ATII) epithelial cells function as stem cells, contributing to alveolar renewal, repair and cancer. Therefore, they are a highly relevant model for studying a number of lung diseases, including acute injury, fibrosis and cancer, in which signals transduced by RAS and transforming growth factor (TGF)-β play critical roles. To identify downstream molecular events following RAS and/or TGF-β activation, we performed proteomic analysis using a quantitative label-free approach (LC-HDMSE) to provide in-depth proteome coverage and estimates of protein concentration in absolute amounts. Data are available via ProteomeXchange with identifier PXD023720. We chose ATIIER:KRASV12 as an experimental cell line in which RAS is activated by adding 4-hydroxytamoxifen (4-OHT). Proteomic analysis of ATII cells treated with 4-OHT or TGF-β demonstrated that RAS activation induces an epithelial–mesenchymal transition (EMT) signature. In contrast, under the same conditions, activation of TGF-β signaling alone only induces a partial EMT. EMT is a dynamic and reversible biological process by which epithelial cells lose their cell polarity and down-regulate cadherin-mediated cell–cell adhesion to gain migratory properties, and is involved in embryonic development, wound healing, fibrosis and cancer metastasis. Thus, these results could help to focus research on the identification of processes that are potentially driving EMT-related human disease.
Fibrosis and lung cancer present a large medical burden with lung cancer being the leading cause of death in the United States in both men and women (Torre et al., 2016). Fibrotic lung diseases, such as idiopathic pulmonary fibrosis (IPF), present a growing problem with limited treatments and poor prognosis (Richeldi et al., 2017; Hill et al., 2019a). Gas exchange in the lungs happens in alveolar sacs which are lined with two epithelial cell types (squamous alveolar type I, ATI and surfactant-secreting alveolar type II, ATII cells) (Pérez-Gil, 2008). It has been proposed that local signals are able to regulate ATII epithelial cells, which are able to function as stem cells contributing to alveolar renewal, repair and cancer (Desai et al., 2014). The integrity of these cell types is critical in the modulation of a number of diseases including cancer and fibrosis (Desai et al., 2018).
Certain signaling pathways have been demonstrated to be key in mediating a number of processes in lung-related disease. These signaling pathways have a diverse range of functions and subsequently affect a diverse range of biological processes. They are also known to interact with each other to influence their effects (Vallath et al., 2014; Aschner and Downey, 2016). Epidermal growth factor receptor (EGFR) is a transmembrane receptor tyrosine kinase activated by members of the EGF family (Linggi and Carpenter, 2006) and its pathway has been previously implicated in a number of lung pathologies, including cancer and fibrosis (Burgel and Nadel, 2008; Vallath et al., 2014; Yao et al., 2018). RAS is a downstream effector of EGFR and mutations in RAS have been widely reported in a number of cancers and other diseases (Downward, 2003; Tzouvelekis et al., 2013; Stella et al., 2014). RAS proteins switch between a GTP-bound “on-state” and a stable, GDP-bound “off-state”; this is mediated by G activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs) which convert from active to inactive and inactive to active respectively (Simanshu et al., 2017). Once active, RAS stimulates a number of downstream signaling cascades which include mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3 kinase (PI3K) (Downward, 2003). RAS signaling has been implicated in a number of cellular processes including proliferation, differentiation and apoptosis (Downward, 1998; Cox and Der, 2003; Sun et al., 2015).
Transforming growth factor (TGF)-β is a well-studied, multifunctional profibrotic cytokine known to play an important role in developmental biology (Shull et al., 1992; Kulkarni et al., 1993), cancer (Massagué, 2008), fibrotic disease (Leppäranta et al., 2012; Yao et al., 2018) and many other lung pathologies (Saito et al., 2018). Key mediators of the TGF-β family are Smads, which act together to regulate transcription (Massagué, 1998). In turn, these are involved in a number of cellular context-dependent processes including cell growth arrest (Mukherjee et al., 2010), proliferation (Carl et al., 2016; Cheng et al., 2016), apoptosis (Schuster and Krieglstein, 2002), and epithelial–mesenchymal transition (EMT) (Guo, 2017; Goldmann et al., 2018).
Our study utilized quantitative proteomic analysis to provide a comprehensive and unbiased investigation into the role of RAS and TGF-β signaling in ATII cells. In this study we identified EMT as a key pathway induced upon RAS-activation in ATII cells, but not in TGF-β-treated ATII cells in the given time. ATII cells with both RAS- and TGF-β signaling induced, leads to an EMT proteomic signature, which is mainly driven by RAS signaling. These findings are key to further understanding the mechanisms underlying lung disease.
ATIIER:KRASV12 cells (Molina-Arcas et al., 2013; Coelho et al., 2017; Yao et al., 2018) were cultured in DCCM-1 (Biological Industries Ltd.) supplemented with 10% newborn calf serum (NBCS) (Life Technologies), 1% penicillin, 1% streptomycin and 1% l-glutamine (all from Sigma Aldrich). Cells were kept at 37°C and 5% CO2. To induce RAS activation in ATIIER:KRASV12 cells, 250 nM 4-OHT (Sigma-Aldrich) was added. TGF-β1 was from PeproTech. No mycoplasma contamination was detected in the cell lines used. For wet lab experiments, treatment with both 4-OHT (250 nM) and TGF-β1 (5 ng/ml) was for 24 h, unless otherwise indicated in the figure. For proteomic cell culture, ATIIER:KRASV12 cells were cultured as indicated above. Each treatment group (control, 4-OHT, TGF-β1 and combined (TGF-β1 and 4-OHT)) was conducted in triplicate and cells were treated for 24 h with indicated treatments prior to cell lysis.
Western blot analysis was performed with lysates from cells lyzed with urea buffer (8 M Urea, 1 M Thiourea, 0.5% CHAPS, 50 mM DTT, and 24 mM Spermine). Primary antibodies were from: Santa Cruz (E-cadherin, sc-21791; ZEB1, sc-25388; Snail2, sc-10436), Abcam (β-tubulin, ab6046), Cell Signaling Technology (phospho-AKT, 9271; phospho-ERK, 9101; Snail1, 3879; Snail2, 9585; Phospho-Smad2, 3104; β-tubulin, 86298), and BD Transduction Laboratories (E-cadherin, 610405). Signals were detected using an Odyssey imaging system (LI-COR), and evaluated by ImageJ 1.42q software (National Institutes of Health).
Total RNA was isolated using RNeasy mini kit (Qiagen) according to the manufacturer’s instructions and quantified using a Nanodrop Spectrophotometer 2000c (Thermo Fisher Scientific). Real-time quantitative RT-PCR was carried out using gene-specific primers (QuantiTect Primer Assays, Qiagen) for CDH1 (E-cadherin) (QT00080143), SNAI1 (Snail1) (QT00010010), SNAI2 (Snail2) (QT00044128), ZEB1 (QT00008555), ZEB2 (QT00008554), TWIST (QT00011956), VIM (QT00095795), TGFBR1 (QT00083412), TGFBR2 (QT00014350), TGFBR3 (QT00083223) or ACTB (β-actin) (QT01680476) with QuantiNova SYBR Green RT-PCR kits (Qiagen). Relative transcript levels of target genes were normalized to ACTB (β-actin).
Samples were lyzed in 0.1 M ammonium bicarbonate containing 1% SDS followed by pulsed sonication. Lysates were then centrifuged at 13,000 x g, for 20 min at 4°C, supernatant removed and protein concentration measured using a DirectDetect spectrometer (Merck, United Kingdom).
Methanol/chloroform extraction was then performed on 25 μg of protein for each lysate. Samples were prepared in 100 μL using lysis buffer (0.1 M ammonium bicarbonate). Samples were resuspended in 100 μl of 0.1 M ammonium bicarbonate containing 0.1% SDS and reduced with 1 mM Dithiothreitol (DTT, Thermo Scientific) at 56°C for 60 min. Samples were subsequently alkylated using 5.5 mM iodoacetamide (Sigma) and incubated in the dark at room temperature for 45 min. Samples were digested by addition of 2 μg sequencing grade modified trypsin (Promega) and incubated overnight at 37°C. After digestion, samples were lyophilized in vacuo.
Enolase protein digest internal standard (Waters) was spiked into each cellular peptide sample at a concentration of 150 fmol prior to isoelectric focusing by OFFGEL fractionation using a 3–10 pH gradient (Agilent) into 12 peptide fractions. Each fraction was purified using an Empore™ C18 solid phase extraction plate (ThermoScientific Pierce) to remove residual salts, buffers and contaminants before lyophilization and resuspension in loading buffer (3% acetonitrile + 0.1% formic acid) for mass spectrometry analysis.
Separations were performed using a nanoAcquity UPLC system (Waters). Peptide digests were injected onto a Symmetry C18, 180 µm × 20 mm trapping cartridge (Waters). After 5 min washing of the trap column, peptides were separated using a 75 µm i. d. x 500 mm, 1.7 µm BEH130 C18, column (Waters) using a linear gradient of 5–40% B (buffer A = 0.1% formic acid in water, buffer B = 0.1% formic acid in acetonitrile) over 90 min with a wash to 85% B at a flow rate of 300 nL/min. All separations were automated, performed on-line and sprayed directly into the nanospray source of the mass spectrometer.
All mass spectrometry was performed using a Waters G2-Si Synapt HDMS mass spectrometer operating in MSe mode. Data was acquired from 50 to 2000 m/z with ion mobility enabled using alternate low and high collision energy (CE) scans. Low CE was 5 V and elevated, ramped from 20–40 V. The lock mass Glu-fibrinopeptide (M+2H)+2, m/z = 785.8426) was infused at 300 nl/min at a concentration of 200 fmol/μL and acquired every 13 s.
Raw mass spectra were processed using ProteinLynx Global Server Ver 3.0 (Waters, Manchester, United Kingdom) enabled using an in-house developed script and the data processed to generate reduced charge state and de-isotoped precursor and associated product ion mass lists. These peak lists were searched against the human UniProt protein sequence (downloaded March 2017). A maximum of one missed cleavage was allowed for tryptic digestion and the variable modification was set to contain oxidation of methionine and carboxyamidomethylation of cysteine was set as a fixed modification. The false discovery rate (FDR) was estimated with decoy-fusion database searches and were filtered to 1% FDR.
Raw data was imported into RStudio software (v. 1.1.456) for down-stream analysis. Expression values of proteins in femtomole (fmol) were extracted and normalized using the variance stabilization normalization (Vsn) method (Valikangas et al., 2018a). The Vsn method was implemented using the justvsn function of the Vsn package in RStudio. This method was utilized as a recent review of normalization methods for use in quantitative label-free proteomics determined that it reduced variation the most between technical replicates compared to other tested methods (Huber et al., 2002). Normalized data were transformed into log2 format. Missing values were imputed using local least squares imputation (Lls) with parameter value 150 (Valikangas et al., 2018b), using pcaMethods package (Stacklies et al., 2007).
Proteins had to be observed in at least two of three replicates to be used in the experiment. Samples were normalized and missing values imputed, before PCA (Supplementary Figure. S3) validated samples clustered appropriately, differentially expressed proteins were analyzed by hierarchal clustering and visualized on a heatmap (Supplementary Figures S1B, S2B).
The limma package (v. 3_34.2) was utilized to detect differentially expressed proteins (DEPs) (Ritchie et al., 2015). DEPs were determined by p Value <0.05 and ∣log2 [Fold Change]∣ ≥0.5. All expression data after limma is available in Supplementary Tables S1, S3, S5. These are the top table export from limma, further details about these can be found in the limma user guide (Ritchie et al., 2015). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD023720.
Prior to pathway analysis protein symbols were converted to gene symbols in RStudio (v. 3.6.1). Enrichment analyses were generated by Metascape (Zhou et al., 2019), targets were analyzed with hallmark annotation.
Proteomic experiments were performed in triplicate. Unless otherwise noted, data are presented as mean and s.d., and a two-tailed, unpaired or paired Student's t-test was used to compare two groups for independent samples, with the n number for each experiment included in the figure legends. p < 0.05 was considered statistically significant.
To characterize the proteome-wide altered expression in ATII cells, we performed quantitative mass spectrometry. We utilized a RAS-inducible model in ATIIER:KRASV12 cells (Molina-Arcas et al., 2013; Coelho et al., 2017; Yao et al., 2018). KRASV12 (containing a single amino acid mutation in KRAS, glycine to valine at position 12) fused to the estrogen receptor (ER) ligand-binding domain was introduced into ATII cells to generate ATIIER:KRASV12, where the RAS pathway is activated by the addition of 4-hydroxytamoxifen (4-OHT). Mass spectrometry identified 1844 proteins, of these 182 were determined to be differentially expressed proteins (DEPs) (p Value <0.05, ∣log2 (fold change [FC])∣ ≥ 0.5) (Supplementary Table S1).
RAS signaling is important in a number of different processes. In order to determine which pathways are altered when ATII cells undergo RAS activation, pathway analysis was conducted (Figure 1A). Hallmark epithelial-mesenchymal transition (EMT) was identified as one of the key pathways altered upon RAS activation in ATII cells (Figure 1A, q Value = 0.0007, shared genes = 8). A full list of altered pathways is included in Table 1, including shared genes identified in the analysis.
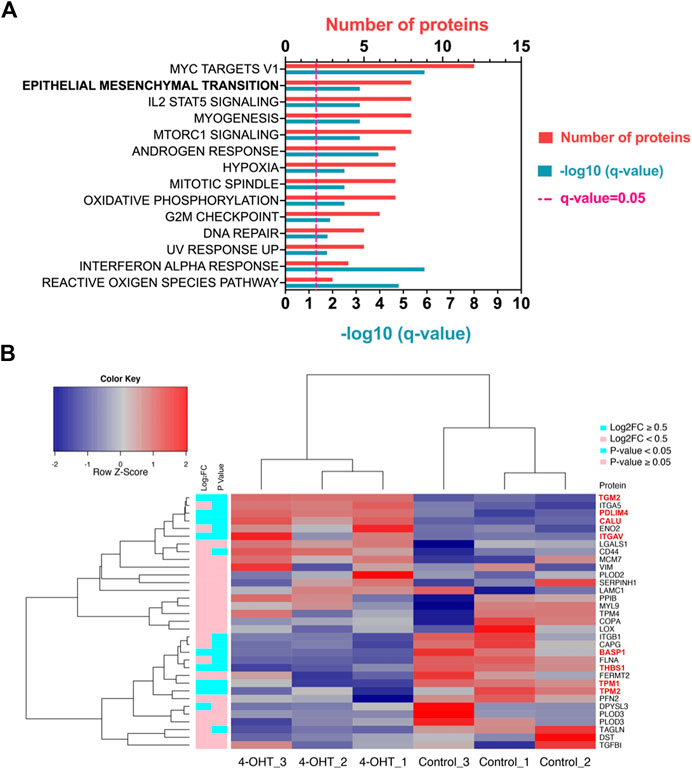
FIGURE 1. RAS-activation in ATII cells induces Hallmark EMT proteomic signature (A) Pathway analysis in Metascape (Metascape.com) analyzing differentially expressed proteins (DEPs) in ATIIER:KRASV12 cells treated with 250 nM 4-OHT or control for 24 h, all non-significant pathways have been removed from diagram. (B) Heatmap and hierarchical cluster analysis of ATIIER:KRASV12 cells treated with 250 nM 4-OHT (RAS-activated) compared with control for 24 h, a panel of hallmark EMT proteins were evaluated. p Value (blue: p Value <0.05, pink: p Value ≥ 0.05). ∣log2 fold change (blue: ∣log2FC∣ ≥ 0.5, pink: ∣log2FC∣ < 0.5). Red text indicates differentially expressed proteins (DEPs).
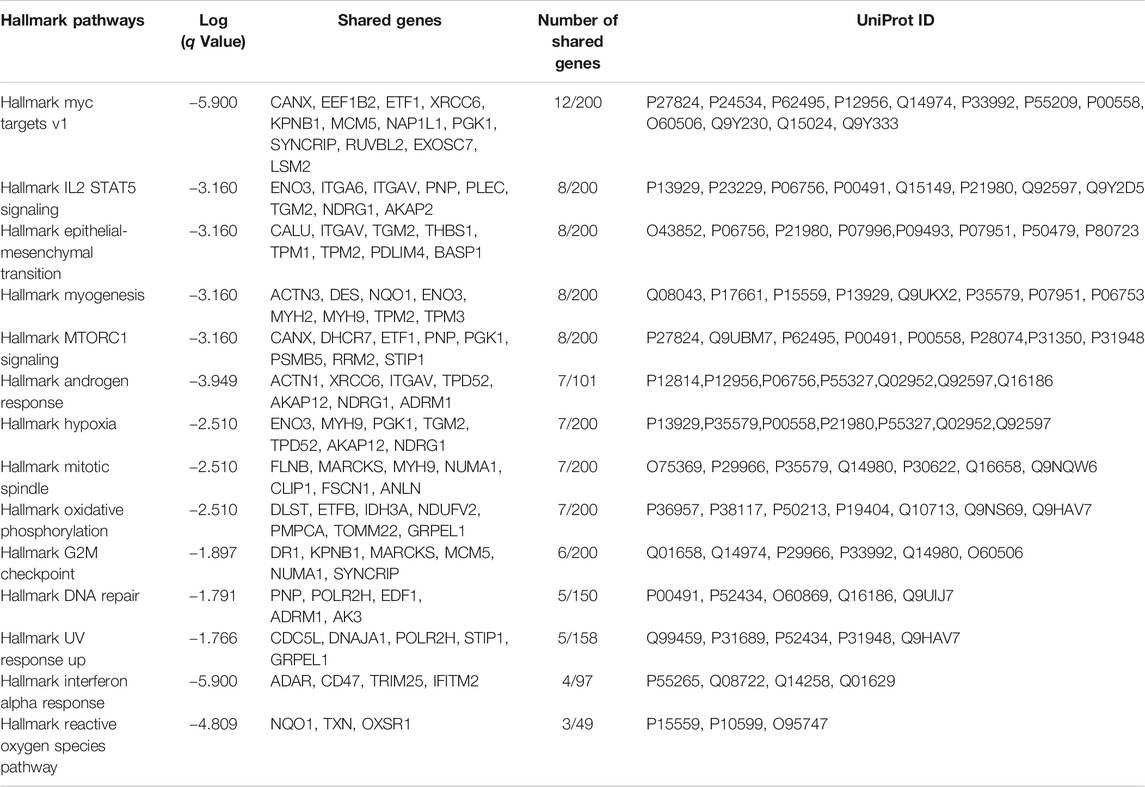
TABLE 1. Hallmark pathways identified using Metascape (Metascape.com) for differentially expressed proteins between control and RAS-activated ATIIER:KRASV12 cells.
RAS-activation has previously been demonstrated to be instrumental in the induction of EMT (Wang et al., 2010; Arner et al., 2019). To evaluate the role of RAS-activation in ATII cells on EMT processes, a selection of previously described Hallmark EMT proteins (Subramanian et al., 2005) were investigated in our proteomic dataset. Of the identified 1844 proteins, 33 identified were EMT-related proteins, as assessed by the Hallmark EMT list. Of the 33 EMT proteins found, 8 were differentially expressed between control and RAS-activated, as indicated by red text (p Value <0.05, ∣log2FC∣ ≥ 0.5) (Figure 1B). All 33 EMT-proteins identified in the proteomics data were visualized on a heatmap, a clear separation is observed between RAS-activated (4-OHT) and control (Figure 1B), suggesting a clear role for EMT in RAS-activated ATII cells. Further, all samples have been visualized on a volcano plot (Supplementary Figure S1A) and a heat map showing DEPs (Supplementary Figure S1B). Significantly up- and down-regulated EMT-proteins have been identified (Supplementary Figure S1A; Supplementary Table S2).
Given the extensive role of TGF-β in numerous lung pathologies, determining which pathways are altered in alveolar epithelial cells could be key in elucidating disease pathology. Proteomic analysis was also conducted on the ATII cells treated with and without TGF-β (5 ng/ml for 24 h). Mass spectrometry identified 1858 proteins, of which 90 were DEPs (p Value <0.05, ∣log2FC∣ ≥ 0.5) (Supplementary Table S3).
Downstream pathway analysis of TGF-β treated ATII cells did not indicate Hallmark EMT to be significantly induced (Figure 2A, q Value = 0.12). All significantly altered pathways have been indicated, in addition to Hallmark EMT. A full list of altered pathways is included in Table 2, including shared genes found in the analysis. Hallmark EMT processes did share three common proteins with the dataset, but these were not significantly altered.
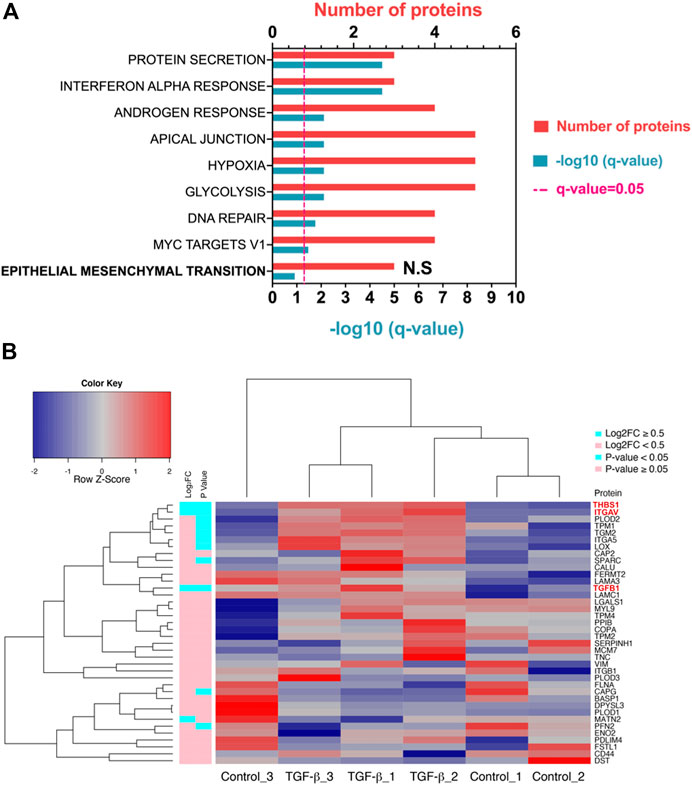
FIGURE 2. TGF-β in ATII cells does not induce a classical EMT program at a given time point. (A) Pathway analysis in Metascape (Metascape.com) analyzing differentially expressed proteins (DEPs) in ATIIER:KRASV12 cells treated with 5 ng/ml TGF-β or control for 24 h, all non-significant pathways have been removed from the diagram with the exception of epithelial-mesenchymal transition (EMT) which was not significant and has been highlighted in bold. (B) Heatmap and hierarchical cluster analysis of ATIIER:KRASV12 cells treated with 5 ng/ml TGF-β or control treated for 24 h, a panel of hallmark EMT proteins were evaluated. p Value (blue: p Value <0.05, pink: p Value ≥ 0.05). Absolute log2 fold change (blue: ∣log2FC∣ ≥ 0.5, pink: ∣log2FC∣ < 0.5). Red text indicates differentially expressed proteins (DEPs).
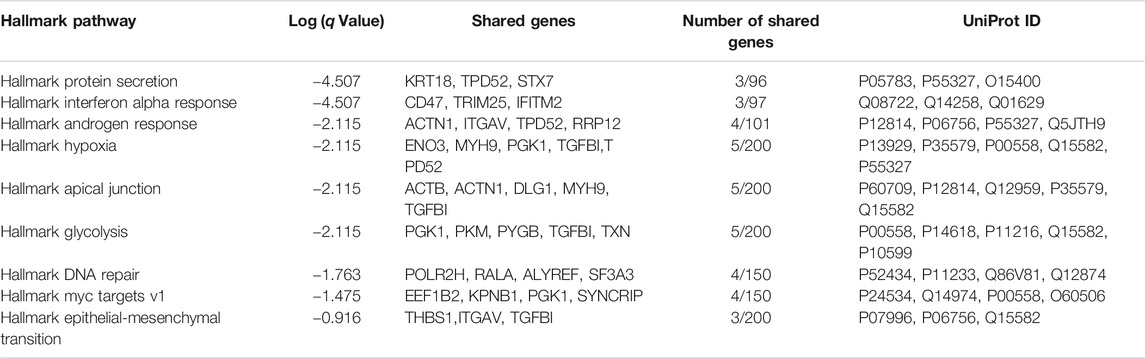
TABLE 2. Hallmark pathways identified using Metascape (Metascape.com) for differentially expressed proteins between control and TGF-β-treated ATIIER:KRASV12 cells.
The same group of Hallmark EMT proteins, as previously described, were compared to the identified proteins and 38 EMT proteins were identified, three were differentially expressed (p Value <0.05, ∣log2FC∣ ≥0.5) (all were up-regulated) (Supplementary Table S4). A heat map illustrates all Hallmark EMT proteins which were identified in the proteomic dataset based on hierarchal clustering DEPs are highlighted by red text (Figure 2B). Clustering TGF-β samples based on Hallmark EMT proteins did not cause defined separation, with Control_3 clustering with TGF-β samples. Further, there was not a defined split in up- and down-regulated EMT proteins. A volcano of all differentially expressed proteins is shown in Supplementary Figure S2A, where the three EMT proteins which were significantly altered are labeled (THBS1, ITGAV and TGFB1). Further a heat map shows all DEPs upon TGF-β treatment (Supplementary Figure S2B).
Given the different EMT response to RAS and TGF-β signaling activation we wanted to determine the effect of dual treatment and to elucidate if one pathway dominated in EMT-induction. In ATII cells with RAS and TGF-β signaling active, mass spectrometry identified 1793 proteins, of which 123 were differentially expressed between control and treatment (p Value <0.05, ∣log2FC∣ ≥ 0.5) (Supplementary Table S5).
We previously observed Hallmark EMT to be a significant pathway for RAS-activated (Figure 1A, q Value = 0.0005) but not in TGF-β treatment alone (Figure 2A, q Value = 0.12), hence we wanted to evaluate the effect of RAS-activation together with TGF-β treatment on ATIIER:KRASV12 cells. The same group of Hallmark EMT proteins, as previously described, were compared to identified proteins and 35 EMT proteins were identified, five were differentially expressed (p Value <0.05, ∣log2FC∣ ≥ 0.5) (Supplementary Table S6). Pathway analysis identified Hallmark EMT (Figure 3A, q Value = 0.01) with five shared proteins. A full list of altered pathways is included in Table 3, including shared genes found in the analysis. When all Hallmark EMT proteins are visualized using a heat map based on hierarchal clustering, a more defined separation is observed, where DEPs are signified by red text (Figure 3B), compared with TGF-β treatment alone (Figure 2B). Dendrograms show a clear divide between control and RAS-activated/TGF-β treatment, however Control_3 appears to cluster separately from other control samples. We previously confirmed a global view of sample uniformity by PCA (Supplementary Figure S3). These results suggest that hallmark EMT is also induced in RAS-activated/TGF-β treated ATIIER:KRASV12 cells.
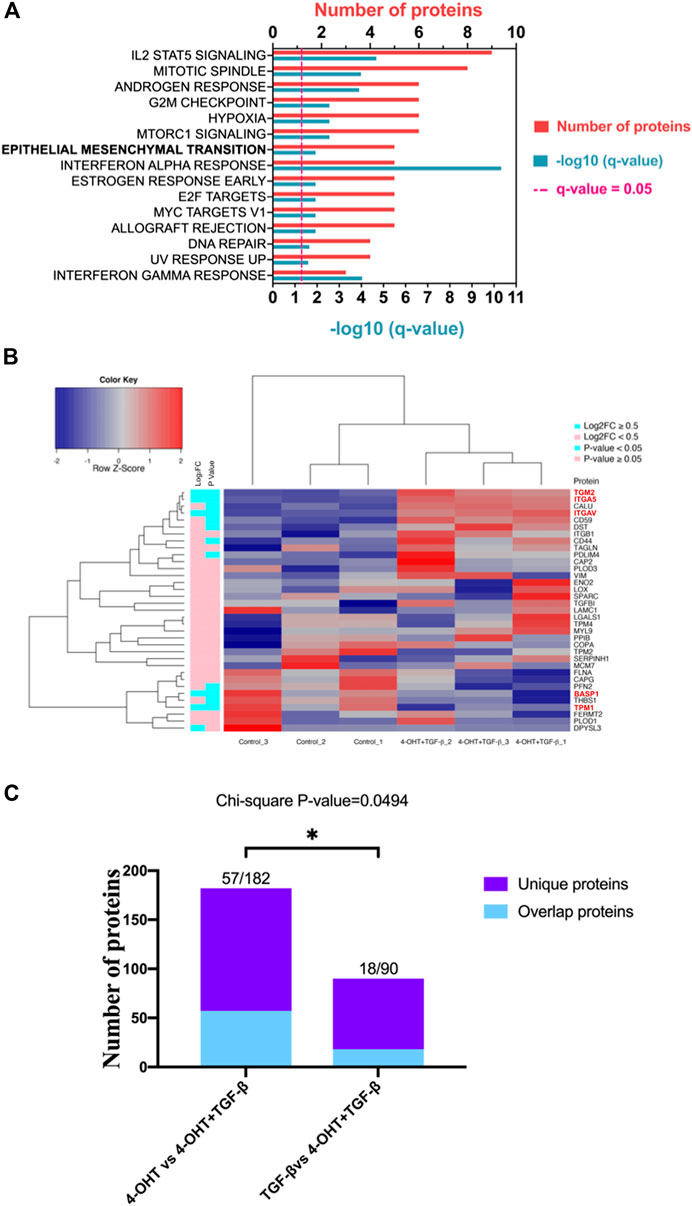
FIGURE 3. RAS-activation together with TGF-β are capable of inducing EMT and RAS signaling appears to drive EMT. (A) Pathway analysis in Metascape (Metascape.com) analyzing differentially expressed proteins (DEPs) in ATIIER:KRASV12 cells treated with a combined treatment of 250 nM 4-OHT and 5 ng/ml TGF-β or control for 24 h, all non-significant pathways have been removed from the diagram. (B) Heatmap and hierarchical cluster analysis of ATIIER:KRASV12 cells treated with a combined treatment of 250 nM 4-OHT and 5 ng/ml TGF-β or control for 24 h, a panel of hallmark EMT proteins were evaluated. p Value (blue: p Value < 0.05, pink: p Value ≥ 0.05). Absolute log2 fold change (blue: ∣log2FC∣ ≥ 0.5, pink: ∣log2FC∣ < 0.5). Red text indicates differentially expressed proteins (DEPs). (C) Diverging bar chart indicating the number of uniquely differentially expressed proteins in indicated treatments (Chi-squared test, p Value = 0.0494).
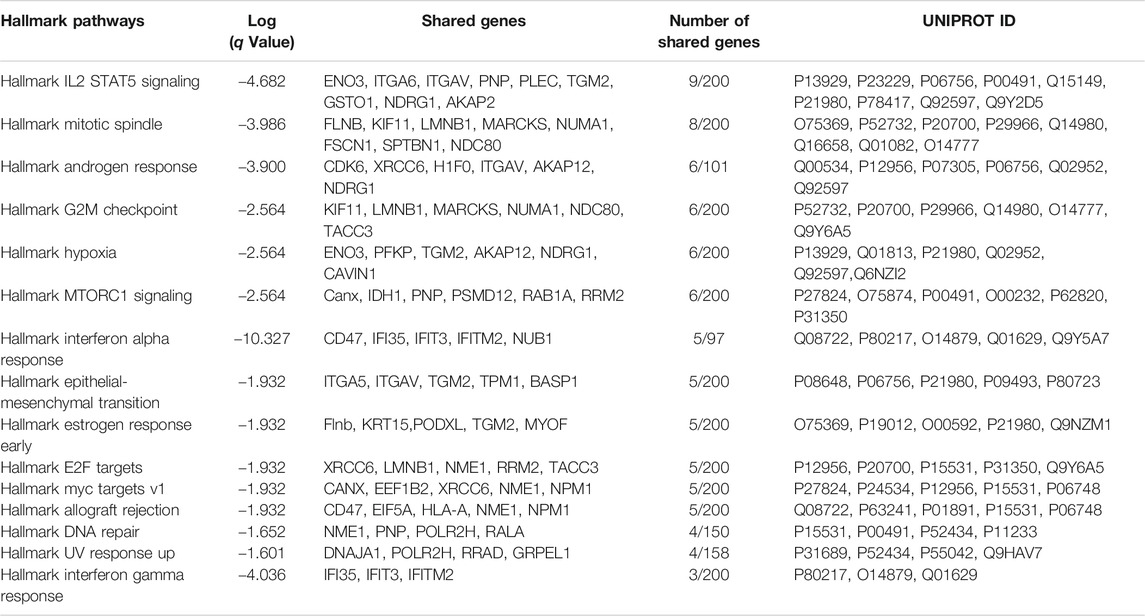
TABLE 3. Hallmark pathways identified using Metascape (Metascape.com) for differentially expressed proteins between control and TGF-β-treated RAS-activated ATIIER:KRASV12 cells.
Quantitative proteomic analysis identified different signatures of EMT in each treatment (RAS-activated, TGF-β treatment alone and combined RAS activation with TGF-β treatment) and we wanted to determine if a certain signaling pathway dominated. A chi-squared test (Figure 3C) demonstrated that when comparing A and B (A:RAS-activation vs RAS- activation with TGF-β, or B: TGF-β vs RAS-activation with TGF-β), there are a significant number of uniquely differentially expressed proteins (p Value = 0.0494). This analysis suggested that RAS-activation (4-OHT) dominates in the combined treatment of ATIIER:KRASV12 cells.
We first demonstrated the presence of TGF-β receptor 1 (TGFBR1) and 2 (TGFBR2) in ATII cells (Supplementary Figure S4). In addition, phospho-Smad2 (p-Smad2) levels were increased upon TGF-β treatment (Figure 4A). These results suggested intact TGF-β signaling in these cells. To confirm the findings from our proteomic analysis, which identified induction of Hallmark EMT in RAS-activated samples but not in TGF-β treatment alone, we sought to validate protein levels of a number of EMT markers by western blot (Figure 4A). At the time points utilized (t = 24 h) protein levels of E-cadherin, a key cell-cell adhesion molecule and an epithelial marker, was significantly reduced upon RAS-activation and RAS-activation together with TGF-β treatment. However, where ATIIER:KRASV12 cells were treated with just TGF-β, a reduction in E-cadherin was not observed. The levels of tight junction proteins ZO1 and ZO2 were also investigated in our proteomic dataset (Supplementary Figure S5). No significant differences were observed in ZO1, however ZO2 was significantly reduced upon RAS-activation and TGF-β treatment. We then examined the levels of a number of EMT-Transcription Factors (EMT-TF) altered in all treatments; as previously described (Yao et al., 2018) upon RAS-activation ZEB1 was increased, TGF-β treatment led to up-regulation of Snail1 and dual activation of by RAS and TGF-β treatment led to up-regulation of both ZEB1 and Snail1. Further, after 24 h, phase contrast images of RAS-activated ATIIER:KRASV12 cells show the change to a spindle-like morphology from the cuboidal morphology of controls, within the same time frame less pronounced changes are observed by TGF-β treatment (Figure 4B).
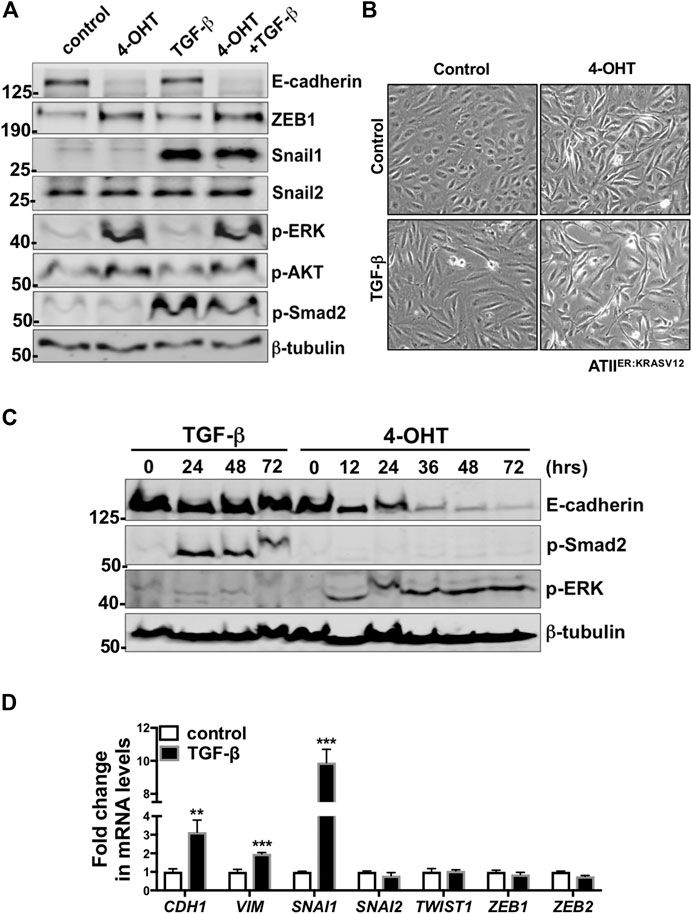
FIGURE 4. Unlike RAS-activation, TGF-β is insufficient to induce EMT-like changes at 24 h in ATIIER:KRASV12 cells. (A) Protein expression of E-cadherin, ZEB1, Snail1, Snail2, p-ERK, p-AKT, p-Smad2 in ATIIER:KRASV12 cells with indicated treatments at 24 h, β-tubulin was used as a loading control. n = 2 (B) Phase contrast images of ATIIER:KRASV12 cells with indicated treatment for 24 h. (C) Protein expression of E-cadherin, p-ERK, p-Smad2 in ATIIER:KRASV12 cells with 250 nM 4-OHT or 5 ng/ml TGF-β at indicated time points, β-tubulin was used as a loading control. n = 2 (D) Fold change in the mRNA level of CDH1 (E-cadherin), VIM (Vimentin), SNAI1, SNAI2, TWIST, ZEB1, ZEB2 in ATIIER:KRASV12 cells with indicated treatment for 24 h. ACTB (β-actin)-normalized mRNA levels in control cells were used to set the baseline value at unity. Data are mean ± s.d. n = 3 samples per group. **p < 0.01. ***p < 0.001.
Having examined the effect of each treatment at one time point (t = 24 h), we next evaluated whether alterations in E-cadherin protein levels were time-dependent. A time course of RAS-activated (4-OHT) and TGF-β treated ATIIER:KRASV12 cells determined protein levels of E-cadherin at a number of times points up to 72 h (Figure 4C). E-cadherin protein levels remain unaltered with TGF-β treatment at all time points examined, however reduction in E-cadherin is evident in RAS-activated ATIIER:KRASV12 cells from 12 h, with a large reduction observed at 36 h. To further elucidate the potential role of TGF-β in EMT in ATIIER:KRASV12 cells, mRNA levels of a number of EMT markers were evaluated (Figure 4D). Interestingly, levels of VIM (Vimentin), a structural protein mainly expressed in mesenchymal cells, were increased with TGF-β. While CDH1 (E-cadherin) levels increased upon TGF-β treatment. We evaluated a number of EMT-TFs and of these only SNAI1 (Snail1) was increased. SNAI1 is important in the direct repression of E-cadherin, however levels of CDH1 (E-cadherin) and protein levels of E-cadherin are not down-regulated, suggesting SNAI1 may mediate some form of partial EMT in these cells.
ATII epithelial cells are able to function as stem cells and subsequently contribute to a number of processes such as renewal, repair and the development of cancer (Desai et al., 2014). A number of key signaling pathways have been implicated in lung disease and to better understand their roles we performed quantitative label-free approach (LC-HDMSE), to compare the capacity of these signaling pathways. ATII cells with RAS activated and/or TGF-β treatment were utilized and hallmark pathways identified. RAS-activation alone was sufficient to induce an EMT signature. In comparison, activation of TGF-β signaling was insufficient to induce an EMT signature in the time period. When both RAS- and TGF-β- signaling were activated an EMT signature was induced and this was driven by RAS-signaling.
EMT is a well-established concept, it requires the involvement of a number of signaling pathways (Thiery and Sleeman, 2006) and the induction of EMT varies significantly in different tissues and diseases (Kalluri and Weinberg, 2009). As described, a number of signaling factors are involved in the induction of EMT, including TGF-β, fibroblast growth factor (FGF), Wnt/β-catenin and EGF. In turn, these can regulate the expression of a group of EMT specific transcription factors termed EMT-TF which are capable of orchestrating the induction of EMT, these include Snail1/2, ZEB1/2 and some basic helix-loop-helix (bHLH) factors. EMT-TFs have been demonstrated to be potent repressors of E-cadherin (Peinado et al., 2007; Nieto, 2011), which is considered to be a key step in the induction of EMT (Wheelock et al., 2008; Lamouille et al., 2014).
Previous studies have begun to compare the signaling mechanisms which drive EMT in malignancy (Taube et al., 2010), here we examine the mechanisms which drive EMT in the lung. EMT has roles in developmental biology, cancer and fibrosis and wound healing (Thiery et al., 2009; Barriere et al., 2015). In cancer, EMT has been demonstrated as a mechanism which allows cancer cells to metastasize to secondary tumor sites (Li and Balazsi, 2018). In fibrosis, the role of EMT is controversial. IPF is a chronic, interstitial lung disease which lacks both effective treatment and a clear understanding of underlying disease mechanisms (Richeldi et al., 2017; Hill et al., 2019a). A number of studies have proposed that fibroblasts are derived from epithelial cells by EMT. However, recent studies in a number of fibrotic models have suggested that although EMT may not contribute directly to the fibroblast pool, it does augment fibrosis through secreted factors (Grande et al., 2015; Lovisa et al., 2015; Yao et al., 2018; Hill et al., 2019a; Hill et al., 2019b). Both EGFR (Burgel and Nadel, 2008; Cully and Downward, 2008; Vallath et al., 2014) and TGF-β have been demonstrated as key players in lung disease (Tatler and Jenkins, 2012; Saito et al., 2018).
TGF-β is responsible for a wide range of functions (Schuster and Krieglstein, 2002; Mukherjee et al., 2010; Carl et al., 2016; Cheng et al., 2016; Guo, 2017; Goldmann et al., 2018) and elucidating pathways which are altered in the lung upon its activation could be key in determining underlying mechanisms of lung disease. A number of studies have demonstrated TGF-β-induced EMT in the lung (Kasai et al., 2005; Chen et al., 2016). These studies have modeled EMT using A549 lung cells which harbour a KRAS mutation (Yoon et al., 2010), and also utilized a longer treatment time (Kasai et al., 2005; Chen et al., 2016). Primary ATII cells treated with TGF-β for 1 week were unable to induce EMT as assessed by reduction in E-cadherin, whereas treatment with EGF led to a reduction in E-cadherin protein levels suggesting induction of EMT (Yao et al., 2018). A recent microarray study also utilizing primary alveolar epithelial cells suggested EMT induction upon TGF-β treatment, however there was no reduction in traditional EMT markers E-cadherin or tight junctions (Goldmann et al., 2018). KRAS mutations occur in around 25–35% of cases of lung cancer (Kempf et al., 2016). Taking all these findings together we suggest that in a KRAS-driven environment, as is common in many cancers, EMT is induced more potently than by TGF-β. Here, we compare RAS-activated ATIIER:KRASV12 cells together with TGF-β treatment with TGF-β treatment alone; and it appears that EMT is induced to a greater extent than with TGF-β alone. ‘Hallmark EMT’ was identified as a top hit in the RAS-active proteomic analysis, and western blot analysis suggests that EMT was induced more potently than the TGF-β treated ATIIER:KRASV12 cells. Here we demonstrated in ATII cells that in this time period (24 h) there was no significant reduction in E-cadherin protein level. Using unbiased, quantitative proteomic analysis we demonstrated that TGF-β was unable to induce complete EMT in ATII cells in the given period and pathway analysis did not identify EMT as a pathway in ATII cells with TGF-β.
In comparison, RAS pathway activation was able to induce a hallmark EMT signature in ATII cells. In lung fibrosis, the EGFR pathway, upstream of RAS, has been implicated with a number of studies demonstrating mice expressing TGF-α develop lung fibrosis (Korfhagen et al., 1994; Hardie et al., 1997) and those lacking EGFR to be resistant to bleomycin induced fibrosis (Madtes et al., 1999). Clinically, IPF patients have been demonstrated to have EGFR mutations (Stella et al., 2014) or increased expression of EGFR (Tzouvelekis et al., 2013). We have previously demonstrated in ATII cells that EMT is induced via the EGFR-RAS-ERK pathway via ZEB1, and this mediated fibrosis via paracrine signaling (Yao et al., 2018). We propose that in the same manner that RAS-induced EMT in ATII ER:KRAS V12 cells is via ZEB1, partial EMT is induced by TGF-β via Snail1 (Dang et al., 2011; Batlle et al., 2013; Li et al., 2014). As such, identifying the underlying mechanisms and more potential biomarkers/drug targets is clinically beneficial. Unbiased proteomic analysis of RAS-activated ATII cells identified a hallmark EMT signature, validated by protein and mRNA levels of a number of EMT markers. These findings are consistent with published literature on RAS-induced EMT in ATII cells (Yao et al., 2018).
Given the important roles of both RAS signaling and TGF-β in the pathogenesis of a number of lung diseases, including cancer and fibrosis, understanding the capacity of their signaling to induce different processes is particularly important (Kempf et al., 2016; Richeldi et al., 2017). Both have been proposed as potent inducers of EMT (Lamouille et al., 2014; Nieto et al., 2016), and it has been suggested in other contexts that when working together these can cooperate to have a synergistic effect through a crosstalk effect (Oft et al., 1996; Oft et al., 1998; Iglesias et al., 2000; Park et al., 2000; Gotzmann et al., 2002; Janda et al., 2002; Bates and Mercurio, 2003; Safina et al., 2009), and in the context of malignancy, RAS and TGF-β can act together to regulate epithelial cell plasticity (Janda et al., 2002). In a recent study in mammary epithelial cells, it was shown that when exogenous TGF-β was not present, inhibition of RAF/MEK/ERK was able to prevent EMT, but when exposed to TGF-β it is able to shortcut MEK to transition to mesenchymal cells (McFaline-Figueroa et al., 2019). An investigation into the transcriptomic response to TGF-β after 48 h found in human primary ATII cells both EMT and KRAS signaling to be induced (Goldmann et al., 2018). In ATII cells when RAS and TGF-β signaling are both induced, a hallmark EMT signature was identified and this was driven by RAS but a synergistic effect is not observed.
Taken together, these results indicate RAS activation to be a key inducer of EMT in the time period tested, whereas TGF-β does not seem to induce a complete EMT. When taken in the context of existing studies, it is clear that both time- and dose- of TGF-β treatment are particularly important when determining the effect on the EMT phenotype. Both TGF-β and RAS-activation are key signaling cascades in the pathogenesis of IPF, and despite studies in other contexts demonstrating a cooperation between these pathways, in ATII it appears RAS-activation drives EMT. While the exact details of these mechanism are yet to be fully elucidated in the context of IPF, understanding the interactions between these pathways will be helpful in the discovery of potential drug targets which may prevent the progression of fibrosis.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD023720.
Conceptualization: YW and PS; Investigation: YZ, CH, LY, JL, YW, and PS; Formal analysis: YZ, CH, PS, and YW; Resources: JD and DH; Writing: CH and YZ with support from MJ, DD, RE, YW, and PS; Supervision: YW and PS; Funding acquisition: YW and PS.
This project was supported by Medical Research Council (MR/S025480/1), an Academy of Medical Sciences/the Wellcome Trust Springboard Award (SBF002/1,038), Wessex Medical Trust and AAIR Charity. YZ was supported by an Institute for Life Sciences PhD Studentship. CH was supported by Gerald Kerkut Charitable Trust and University of Southampton Central VC Scholarship Scheme. LY was supported by China Scholarship Council. JD was supported by the Francis Crick Institute which receives its core funding from Cancer Research United Kingdom (FC001070), the United Kingdom Medical Research Council (FC001070) and the Wellcome Trust (FC001070). Instrumentation in the Center for Proteomic Research is supported by the BBSRC (BM/M012387/1) and the Wessex Medical Trust.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.595712/full#supplementary-material.
Arner, E. N., Du, W., and Brekken, R. A. (2019). Behind the wheel of epithelial plasticity in KRAS-driven cancers. Front Oncol. 9 (1049), 1049. doi:10.3389/fonc.2019.01049
Aschner, Y., and Downey, G. P. (2016). Transforming growth factor-β: master regulator of the respiratory system in Health and disease. Am. J. Respir. Cell Mol. Biol. 54 (5), 647–655. doi:10.1165/rcmb.2015-0391TR
Barriere, G., Fici, P., Gallerani, G., Fabbri, F., and Rigaud, M. (2015). Epithelial mesenchymal transition: a double-edged sword. Clin. Transl. Med. 4, 14. doi:10.1186/s40169-015-0055-4
Bates, R. C., and Mercurio, A. M. (2003). Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol. Biol. Cell 14, 1790–1800. doi:10.1091/mbc.E02-09-0583
Batlle, R., Alba-Castellón, L., Loubat-Casanovas, J., Armenteros, E., Francí, C., Stanisavljevic, J., et al. (2013). Snail1 controls TGF-β responsiveness and differentiation of mesenchymal stem cells. Oncogene 32 (28), 3381–3389. doi:10.1038/onc.2012.342
Burgel, P. R., and Nadel, J. A. (2008). Epidermal growth factor receptor-mediated innate immune responses and their roles in airway diseases. Eur. Respir. J 32, 1068–1081. doi:10.1183/09031936.00172007
Carl, C., Flindt, A., Hartmann, J., Dahlke, M., Rades, D., Dunst, J., et al. (2016). Ionizing radiation induces a motile phenotype in human carcinoma cells in vitro through hyperactivation of the TGF-beta signaling pathway. Cell. Mol. Life Sci. 73 (2), 427–443. doi:10.1007/s00018-015-2003-2
Chen, K. J., Li, Q., Wen, C. M., Duan, Z. X., Zhang, J. Y., Xu, C., et al. (2016). Bleomycin (BLM) induces epithelial-to-mesenchymal transition in cultured A549 cells via the TGF-β/smad signaling pathway. J. Cancer 7 (11), 1557–1564. doi:10.7150/jca.15566
Cheng, H., Wang, S., and Feng, R. (2016). STIM1 plays an important role in TGF-β-induced suppression of breast cancer cell proliferation. Oncotarget 7 (13), 16866–16878. doi:10.18632/oncotarget.7619
Coelho, M. A., de Carné Trécesson, S., Rana, S., Zecchin, D., Moore, C., Molina-Arcas, M., et al. (2017). Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity 47, 1083–1099.e6. doi:10.1016/j.immuni.2017.11.016
Cox, A. D., and Der, C. J. (2003). The dark side of Ras: regulation of apoptosis. Oncogene 22 (56), 8999–9006. doi:10.1038/sj.onc.1207111
Cully, M., and Downward, J. (2008). SnapShot: ras signaling. Cell 133, 1292.e1. doi:10.1016/j.cell.2008.06.020
Dang, H., Ding, W., Emerson, D., and Rountree, C. B. (2011). Snail1 induces epithelial-to-mesenchymal transition and tumor initiating stem cell characteristics. BMC Cancer 11 (1), 396. doi:10.1186/1471-2407-11-396
Desai, O., Winkler, J., Minasyan, M., and Herzog, E. L. (2018). The role of immune and inflammatory cells in idiopathic pulmonary fibrosis. Front. Med. 5, 43. doi:10.3389/fmed.2018.00043
Desai, T. J., Brownfield, D. G., and Krasnow, M. A. (2014). Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507, 190. doi:10.1038/nature12930
Downward, J. (1998). Ras signalling and apoptosis. Curr. Opin. Genet. Dev. 8, 49–54. doi:10.1016/S0959-437X(98)80061-0
Downward, J. (2003). Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 3, 11–22. doi:10.1038/nrc969
Goldmann, T., Zissel, G., Watz, H., Drömann, D., Reck, M., Kugler, C., et al. (2018). Human alveolar epithelial cells type II are capable of TGFβ-dependent epithelial-mesenchymal-transition and collagen-synthesis. Respir. Res. 19, 138. doi:10.1186/s12931-018-0841-9
Gotzmann, J., Huber, H., Thallinger, C., Wolschek, M., Jansen, B., Schulte-Hermann, R., et al. (2002). Hepatocytes convert to a fibroblastoid phenotype through the cooperation of TGF-beta1 and Ha-Ras: steps towards invasiveness. J. Cell Sci. 115, 1189–1202.
Grande, M. T., Sánchez-Laorden, B., López-Blau, C., De Frutos, C. A., Boutet, A., Arévalo, M., et al. (2015). Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 21, 989–997. doi:10.1038/nm.3901
Guo, Q. (2017). Changes in mitochondrial function during EMT induced by TGFβ-1 in pancreatic cancer. Oncol. Lett. 13, 1575–1580. doi:10.3892/ol.2017.5613
Hardie, W. D., Bruno, M. D., Huelsman, K. M., Iwamoto, H. S., Carrigan, P. E., Leikauf, G. D., et al. (1997). Postnatal lung function and morphology in transgenic mice expressing transforming growth factor-alpha. Am. J. Pathol 151, 1075–1083.
Hill, C., Jones, M. G., Davies, D. E., and Wang, Y. (2019a). Epithelial-mesenchymal transition contributes to pulmonary fibrosis via aberrant epithelial/fibroblastic cross-talk. J Lung Health Dis. 3, 31–35.
Hill, C., Li, J., Liu, D., Conforti, F., Brereton, C. J., Yao, L., et al. (2019b). Autophagy inhibition-mediated epithelial–mesenchymal transition augments local myofibroblasts differentiation in pulmonary fibrosis. Cell Death Dis. 10 (8), 591. doi:10.1038/s41419-019-1820-x
Huber, W., von Heydebreck, A., Sültmann, H., Poustka, A., and Vingron, M. (2002). Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18, S96–S104. doi:10.1093/bioinformatics/18.suppl_1.S96
Iglesias, M., Frontelo, P., Gamallo, C., and Quintanilla, M. (2000). Blockade of Smad4 in transformed keratinocyte containing a Ras oncogene leads to hyperactivation of the Ras-dependent Erk signalling pathway associated with progression to undifferentiated carcinomas. Oncogene 19 (36), 4134–4145. doi:10.1038/sj.onc.1203764
Janda, E., Lehmann, K., Killisch, I., Jechlinger, M., Herzig, M., Downward, J., et al. (2002). Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J. Cell Biol. 156, 299–313. doi:10.1083/jcb.200109037
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. doi:10.1172/JCI39104
Kasai, H., Allen, J. T., Mason, R. M., Kamimura, T., and Zhang, Z. (2005). TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 6, 56. doi:10.1186/1465-9921-6-56
Kempf, E., Rousseau, B., Besse, B., and Paz-Ares, L. (2016). KRAS oncogene in lung cancer: focus on molecularly driven clinical trials. Eur. Respir. Rev. 25 (139), 71. doi:10.1183/16000617.0071-2015
Korfhagen, T. R., Swantz, R. J., Wert, S. E., McCarty, J. M., Kerlakian, C. B., Glasser, S. W., et al. (1994). Respiratory epithelial cell expression of human transforming growth factor-alpha induces lung fibrosis in transgenic mice. J. Clin. Invest. 93, 1691–1699. doi:10.1172/JCI117152
Kulkarni, A. B., Huh, C. G., Becker, D., Geiser, A., Lyght, M., Flanders, K. C., et al. (1993). Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. U.S.A. 90 (2), 770–774. doi:10.1073/pnas.90.2.770
Lamouille, S., Xu, J., and Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196. doi:10.1038/nrm3758
Leppäranta, O., Sens, C., Salmenkivi, K., Kinnula, V. L., Keski-Oja, J., Myllärniemi, M., et al. (2012). Regulation of TGF-β storage and activation in the human idiopathic pulmonary fibrosis lung. Cell Tissue Res. 348, 491–503. doi:10.1007/s00441-012-1385-9
Li, C., and Balazsi, G. (2018). A landscape view on the interplay between EMT and cancer metastasis. NPJ Syst. Biol. Appl. 4 (1), 34. doi:10.1038/s41540-018-0068-x
Li, H., Li, M., Xu, D., Zhao, C., Liu, G., and Wang, F. (2014). Overexpression of Snail in retinal pigment epithelial triggered epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun 446 (1), 347–351. doi:10.1016/j.bbrc.2014.02.119
Linggi, B., and Carpenter, G. (2006). ErbB receptors: new insights on mechanisms and biology. Trends Cell Biol. 16, 649–656. doi:10.1016/j.tcb.2006.10.008
Lovisa, S., LeBleu, V. S., Tampe, B., Sugimoto, H., Vadnagara, K., Carstens, J. L., et al. (2015). Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med 21, 998–1009. doi:10.1038/nm.3902
Madtes, D. K., Elston, A. L., Hackman, R. C., Dunn, A. R., and Clark, J. G. (1999). Transforming growth factor-alpha deficiency reduces pulmonary fibrosis in transgenic mice. Am. J. Respir. Cell Mol. Biol 20, 924–934. doi:10.1165/ajrcmb.20.5.3526
Massagué, J. (1998). TGF-β signal transduction. Annu. Rev. Biochem 67, 753–791. doi:10.1146/annurev.biochem.67.1.753
McFaline-Figueroa, J. L., Hill, A. J., Qiu, X., Jackson, D., Shendure, J., and Trapnell, C. (2019). A pooled single-cell genetic screen identifies regulatory checkpoints in the continuum of the epithelial-to-mesenchymal transition. Nat. Genet. 51 (9), 1389–1398. doi:10.1038/s41588-019-0489-5
Molina-Arcas, M., Hancock, D. C., Sheridan, C., Kumar, M. S., and Downward, J. (2013). Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov. 3, 548–563. doi:10.1158/2159-8290.CD-12-0446
Mukherjee, P., Winter, S. L., and Alexandrow, M. G. (2010). Cell cycle arrest by transforming growth factor beta1 near G1/S is mediated by acute abrogation of prereplication complex activation involving an Rb-MCM interaction. Mol. Cell Biol. 30 (3), 845. doi:10.1128/MCB.01152-09
Nieto, M. A., Huang, R. Y., Jackson, R. A., and Thiery, J. P. (2016). EMT: 2016. Cell 166, 21–45. doi:10.1016/j.cell.2016.06.028
Nieto, M. A. (2011). The ins and outs of the epithelial to mesenchymal transition in Health and disease. Annu. Rev. Cell Dev. Biol. 27, 347–376. doi:10.1146/annurev-cellbio-092910-154036
Oft, M., Peli, J., Rudaz, C., Schwarz, H., Beug, H., and Reichmann, E. (1996). TGF-beta1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 10, 2462–2477. doi:10.1101/gad.10.19.2462
Oft, M., Heider, K. H., and Beug, H. (1998). TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr. Biol. 8, 1243–1252. doi:10.1016/s0960-9822(07)00533-7
Park, B. J., Park, J. I., Byun, D. S., Park, J. H., and Chi, S. G. (2000). Mitogenic conversion of transforming growth factor-beta1 effect by oncogenic Ha-Ras-induced activation of the mitogen-activated protein kinase signaling pathway in human prostate cancer. Cancer Res. 60, 3031–3038.
Peinado, H., Olmeda, D., and Cano, A. (2007). Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype?. Nat. Rev. Cancer 7, 415–428. doi:10.1038/nrc2131
Pérez-Gil, J. (2008). Structure of pulmonary surfactant membranes and films: the role of proteins and lipid-protein interactions. Biochim. Biophys. Acta. 1778, 1676–1695. doi:10.1016/J.BBAMEM.2008.05.003
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D. J., et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47 (D1), D442–D450.
Richeldi, L., Collard, H. R., and Jones, M. G. (2017). Idiopathic pulmonary fibrosis. Lancet 389, 1941–1952. doi:10.1016/S0140-6736(17)30866-8
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. doi:10.1093/nar/gkv007
Safina, A. F., Varga, A. E., Bianchi, A., Zheng, Q., Kunnev, D., Liang, P., et al. (2009). Ras alters epithelial-mesenchymal transition in response to TGFbeta by reducing actin fibers and cell-matrix adhesion. Cell Cycle 8, 284–298. doi:10.4161/cc.8.2.7590
Saito, A., Horie, M., and Nagase, T. (2018). TGF-β signaling in lung Health and disease. Int. J. Mol. Sci 19 (8), 2460. doi:10.3390/ijms19082460
Schuster, N., and Krieglstein, K. (2002). Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res 307 (1), 1–14. doi:10.1007/s00441-001-0479-6
Shull, M. M., Ormsby, I., Kier, A. B., Pawlowski, S., Diebold, R. J., Yin, M., et al. (1992). Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 359 (6397), 693–699. doi:10.1038/359693a0
Simanshu, D. K., Nissley, D. V., and McCormick, F. (2017). RAS proteins and their regulators in human disease. Cell 170 (1), 17–33. doi:10.1016/j.cell.2017.06.009
Stacklies, W., Redestig, H., Scholz, M., Walther, D., and Selbig, J. (2007). pcaMethods--a bioconductor package providing PCA methods for incomplete data. Bioinformatics 23 (9), 1164–1167. doi:10.1093/bioinformatics/btm069
Stella, G. M., Inghilleri, S., Pignochino, Y., Zorzetto, M., Oggionni, T., Morbini, P., et al. (2014). Activation of oncogenic pathways in idiopathic pulmonary fibrosis. Transl Oncol. 7, 650–655. doi:10.1016/j.tranon.2014.05.002
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102 (43), 15545–15550. doi:10.1073/pnas.0506580102
Sun, Y., Liu, W. Z., Liu, T., Feng, X., Yang, N., and Zhou, H. F. (2015). Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 35 (6), 600–604. doi:10.3109/10799893.2015.1030412
Tatler, A. L., and Jenkins, G. (2012). TGF-β activation and lung fibrosis. Proc. Am. Thorac. Soc. 9 (3), 130–136. doi:10.1513/pats.201201-003AW
Taube, J. H., Herschkowitz, J. I., Komurov, K., Zhou, A. Y., Gupta, S., Yang, J., et al. (2010). Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. U.S.A. 107 (35), 15449–15454. doi:10.1073/pnas.1004900107
Thiery, J. P., Acloque, H., Huang, R. Y., and Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890. doi:10.1016/j.cell.2009.11.007
Thiery, J. P., and Sleeman, J. P. (2006). Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142. doi:10.1038/nrm1835
Torre, L. A., Siegel, R. L., and Jemal, A. (2016). Lung cancer statistics. Adv. Exp. Med. Biol. 893, 1–19. doi:10.1007/978-3-319-24223-1_1
Tzouvelekis, A., Ntolios, P., Karameris, A., Vilaras, G., Boglou, P., Koulelidis, A., et al. (2013). Increased expression of epidermal growth factor receptor (EGF-R) in patients with different forms of lung fibrosis. BioMed Res. Int 2013, 654354. doi:10.1155/2013/654354
Valikangas, T., Suomi, T., and Elo, L. L. (2018a). A systematic evaluation of normalization methods in quantitative label-free proxemics. Briefings Bioinf 19 (1), 1–11. doi:10.1093/bib/bbw095
Valikangas, T., Suomi, T., Elo, L. L., Välikangas, T., Suomi, T., and Elo, L. L. (2018b). A comprehensive evaluation of popular proxemics software workflows for label-free proteome quantification and imputation. Briefings Bioinf. 19, 1344–1355. doi:10.1093/bib/bbx054
Vallath, S., Hynds, R. E., Succony, L., Janes, S. M., and Giangreco, A. (2014). Targeting EGFR signalling in chronic lung disease: therapeutic challenges and opportunities. Eur. Respir. J 44, 513–522. doi:10.1183/09031936.00146413
Wang, Y., Ngo, V. N., Marani, M., Yang, Y., Wright, G., Staudt, L. M., et al. (2010). Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene 29 (33), 4658–4670. doi:10.1038/onc.2010.218
Wheelock, M. J., Shintani, Y., Maeda, M., Fukumoto, Y., and Johnson, K. R. (2008). Cadherin switching. J. Cell Sci. 121, 727–735. doi:10.1242/jcs.000455
Yao, L., Conforti, F., Hill, C., Bell, J., Drawater, L., Li, J., et al. (2018). Paraffine signalling during ZEB1-mediated epithelial–mesenchymal transition augments local myofibroblasts differentiation in lung fibrosis. Cell Death Differ. 26, 1–15. doi:10.1038/s41418-018-0175-7
Yoon, Y. K., Kim, H. P., Han, S. W., Oh, D. Y., Im, S. A., Bang, Y. J., et al. (2010). KRAS mutant lung cancer cells are differentially responsive to MEK inhibitor due to AKT or STAT3 activation: implication for combinatorial approach. Mol. Carcinog 49 (4), 353–362. doi:10.1002/mc.20607
Keywords: epithelial-mesenchymal transition (EMT), RAS, TGF-β, lung disease, fibrosis, proteomics
Citation: Zhou Y, Hill C, Yao L, Li J, Hancock D, Downward J, Jones MG, Davies DE, Ewing RM, Skipp P and Wang Y (2021) Quantitative Proteomic Analysis in Alveolar Type II Cells Reveals the Different Capacities of RAS and TGF-β to Induce Epithelial–Mesenchymal Transition. Front. Mol. Biosci. 8:595712. doi: 10.3389/fmolb.2021.595712
Received: 17 August 2020; Accepted: 15 January 2021;
Published: 19 March 2021.
Edited by:
Raffaele Strippoli, Sapienza University of Rome, ItalyReviewed by:
Mohit Kumar Jolly, Indian Institute of Science (IISc), IndiaCopyright © 2021 Zhou, Hill, Yao, Li, Hancock, Downward, Jones, Davies, Ewing, Skipp and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yihua Wang, eWlodWEud2FuZ0Bzb3Rvbi5hYy51aw==; Paul Skipp, cGpzc0Bzb3Rvbi5hYy51aw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.