
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Mol. Biosci. , 29 September 2020
Sec. Biological Modeling and Simulation
Volume 7 - 2020 | https://doi.org/10.3389/fmolb.2020.556481
This article is part of the Research Topic Coronavirus Disease (COVID-19): Molecular Mechanisms, Translational Approaches and Therapeutics View all 118 articles
 Zhongren Xu1†
Zhongren Xu1† Lixiang Yang2†
Lixiang Yang2† Xinghao Zhang1
Xinghao Zhang1 Qiling Zhang1
Qiling Zhang1 Zhibin Yang1
Zhibin Yang1 Yuanhao Liu1
Yuanhao Liu1 Shuang Wei3
Shuang Wei3 Wukun Liu1*
Wukun Liu1*The outbreak of 2019 novel coronavirus (COVID-19) has caused serious threat to public health. Discovery of new anti-COVID-19 drugs is urgently needed. Fortunately, the crystal structure of COVID-19 3CL proteinase was recently resolved. The proteinase has been identified as a promising target for drug discovery in this crisis. Here, a dataset including 2030 natural compounds was screened and refined based on the machine learning and molecular docking. The performance of six machine learning (ML) methods of predicting active coronavirus inhibitors had achieved satisfactory accuracy, especially, the AUC (Area Under ROC Curve) scores with fivefold cross-validation of Logistic Regression (LR) reached up to 0.976. Comprehensive ML prediction and molecular docking results accounted for the compound Rutin, which was approved by NMPA (National Medical Products Administration), exhibited the best AUC and the most promising binding affinity compared to other compounds. Therefore, Rutin might be a promising agent in anti-COVID-19 drugs development.
At the end of 2019, the pneumonia of unknown cause was detected. A few weeks later a coronavirus was newly isolated, and it was first identified and regarded as the seventh member of beta coronavirus (Zhu et al., 2020). COVID-19 is an infectious disease caused by the most recently discovered coronavirus by WHO (World Health Organization). Globally, as of 9 Aug, more than 19,000,000 confirmed and 720,000 deaths were reported to WHO. Anxiously, the number of infections worldwide is still rising (Wang and Zhang, 2020). Therefore, this is a rapidly evolving emergency. It is imperative to discover and develop potential agents to treat the outbreak.
The sequence alignment demonstrated that the COVID-19 has 82% nucleotide sequence identity with human SARS-CoV (severe acute respiratory syndrome) (Chan et al., 2020; Lee and Hsueh, 2020; Xu et al., 2020). Recently, Prof. Zihe Rao’s research team successfully expressed the 3C-Like Proteinase of COVID-19. The crystal structure of COVID-19 3CLpro (PDB: 6LU7) (Liu et al., 2020) was identified, resolved in a very short time, and available in PDB (protein data bank). It is well-known that chymotrypsin-like protease is crucial in the life cycle of virus, and the protease is stable inside the coronaviruses. Thus, the COVID-19 3CLpro is a potential target for developing the new anti-COVID-19 drugs.
Since the COVID-19 3CLpro has successfully resolved, guaranteed accurate virtual screen strategies are generally considered as a rapid progress of discovering potential drugs, especially in the public health crisis. Experimental screening programs are generally considered to be time-consuming and laborious. Thus, to expedite screening possible drug molecules and prevent the outbreak, the procedure of machine learning and molecular docking were performed to narrow down the potential candidates before experimental assays (Chen et al., 2005; Berry et al., 2015). In this work, a natural compounds library including 2030 compounds (Chinese medicine compounds mostly) was chosen as the database. Some Chinese medicine compounds in the dataset are wildly used in clinic. Their mechanisms, side effects, and safety were investigated. Encouragingly, the NHC (National Health Commission of People’s Republic of China) recently issued a statement that several Chinese medicine formulas were suggested curing the patients in the early stage of infections. Of note, more and more patients were discharged from hospital after being cured by integrated treatment by combining Chinese with Western medicine in China (Cao et al., 2020; Qiu et al., 2020). In addition, it also should be mentioned that many Chinese herbal formulas such as Le-Cao-Shi, JieZe-1, and San Wu Huang Qin decoctions were used to prevent virus infection and cure the viral diseases in China (Ma et al., 2018; Zhao et al., 2019; Duan et al., 2020). The natural compounds are appropriate to be selected as a source of prototype inhibitors against COVID-19 3CLpro.
Chemical fingerprint recognition is a method to convert the drawn molecules into 0 and 1-bit streams. The old fingerprint type was MACCS key, which was developed by the former MDL as a fast method for substructure screening in molecular databases (Polton, 1982). Another available fingerprint is the Morgan fingerprint, a circular fingerprint (Morgan, 1965). The environment and connectivity of each atom are analyzed to a given radius, and each possibility is encoded. Therefore, Morgan fingerprint was applied to this virtual screen project as a molecular representation for machine learning. Morgan fingerprint set a molecular fingerprint by setting a radius from a specific atom to count the number of molecular structures within this radius. We can set the number of radius and bits to get different molecular fingerprints and lengths (Xue et al., 2004). Ultimately, the fingerprint length (128-bit, 256-bit, 526-bit, 1024-bit, 2048-bit) was selected in our study.
Since the binding cavity of COVID-19 3CLpro and SARS 3CLpro is extremely similar (Supplementary Figure S1), it is rational to make the learning model based on the data of inhibitors of above two proteins. The 66 active compounds and 66 inactive compounds were collected as the training and testing set data (patent: US7495011 B2) (Yamamoto et al., 2004; Chen et al., 2006; Wang et al., 2007; Jacobs et al., 2013; Adedeji and Sarafianos, 2014; García-Fernández et al., 2016; Konno et al., 2017; Hu et al., 2020; Jo et al., 2020; Yoshizawa et al., 2020). The chemical structures of compounds were drawn by ChemDraw software and translated into canonical SMILES by the RDKit python package (Shi and Borchardt, 2017). These active compounds and inactive compounds were prepared as positive samples and negative samples respectively during model training.
Six machine learning classifiers were evaluated in this study for comparison.
Random forest (RF) or random decision forests 9 are ensemble learning methods for classification, regression, and other tasks. The methods are operated by constructing a multitude of decision trees at training time, and they output the classes, which consist of the mode of the classes (classification) or mean prediction (regression) of the individual trees 10.
The support-vector machines (SVMs, also support-vector networks) 11 are supervised learning models with associated learning algorithms, analyzing data for the classification and regression. Given a set of training examples, each marked as belonging to one or the other of two categories, the SVM training algorithm builds a model that assigns new examples to one category or the other, making a non-probabilistic binary linear classifier.
The K-nearest neighbors algorithm (K-NN) 12 is a non-parametric method for the classification and regression. In both cases, the input consists of the K closest training examples in the feature space. The output depends on whether K-NN is used for the classification or regression: (1) In the K-NN classification, the output is a class membership. (2) In the K-NN regression, the output is a property value for the object. This value is the average of the values of K nearest neighbors. The K-NN is a type of instance-based learning, or lazy learning, where the function is only approximated locally and all computation is deferred until the classification.
Naïve Bayes 13 is a simple technique for constructing classifiers: models that assign class labels to the problem instances, representing as vectors of feature values, where the class labels are drawn from some finite sets. There is not a single algorithm for training such classifiers, but a family of algorithms based on a common principle: all naïve Bayes classifiers assume that the value of a particular feature is independent of the value of any other feature, given the class variable.
A decision tree 14 is a flowchart-like structure in which each internal node represents a “test” on an attribute (e.g., whether a coin flip comes up heads or tails), each branch represents the outcome of the test, and each leaf node represents a class label (decision taken after computing all attributes). The paths from the root to the leaf represent classification rules.
Logistic regression 15 is a statistical model for using a logistic function to form a model of binary dependent variable. In the regression analysis and logistic regression (or logit regression), it is always used to estimate the parameters of a logistic model (a form of binary regression).
Above all, Random Forest, Support Vector Machine, K-nearest neighbors, Naïve Bayes, Decision Tree, and Logistic Regression were realized by a machine learning package in scikit-learn (Pedregosa et al., 2011).
As for the evaluation of machine learning classifier, the AUC describes the performance of the classifier and presents the comparison between the true positive rate or sensitivity of a given model and the false positive rates (Hand, 2009). The increase in sensitivity is at the expense of the false positive rate. The AUC is a measure of the accuracy of the model. An AUC >0.5 means that the classifier could differ between the positive and negative samples effectively. A perfect classifier should be with AUC = 1.0.
The crystal structure of COVID-19 3CLpro complex was downloaded from protein data bank (PDB ID: 6LU7) (Liu et al., 2020). A commercial database including 2030 approved natural compounds was used as the screening library (Selleck Chemicals, Houston, TX, United States).
The virtual screen procedure and refinement were conducted by Glide module in Schrödinger Maestro software (Berry et al., 2015). The 6LU7 (COVID-19 3CLpro) was performed as the acceptor and prepared in the Protein Preparation Wizard. The receptor was preprocessed, optimized, and minimized (the restrained minimization using the OPLS2005 force field). All compounds were prepared by the default settings of LigPre module. For the screening in the Glide module, the prepared receptor was imported to specify the suitable position in the Receptor Grid Generation. The grid box was generated 10 Å in X, Y, and Z direction, and the residue of His41 and Cys145 was selected as the centroid of the grid box. The Glide Ligand docking was subsequently carried out with the default settings applied (standard precision) and XP (extra precision) templates. The dataset was screened through SP docking first; the SP docking template is appropriate for screening compounds in large numbers. Subsequently, the top ligands that had been determined to be high-scoring using SP were screened by XP docking template, and the XP method is to provide a better correlation between good poses and good scores. All other settings remained default to the Ligand docking wizard.
The phase module of pharmacophore was used to generate pharmacophore hypothesis and define the pharmacophoric features using the receptor-original ligand complex 6LU7. Conformation of each hit was generated using Confgen by applying the OPLS-2005 force field (Watts et al., 2010; Rohini and Shanthi, 2018). Features were set based on the original inhibitor binding model, which contained essential binding interactions with key residues including His41, Phe140, Gln142, Cys145, His164, Glu166, Gln189, and Thr190. The phase ligand screening was subsequently carried out using the above pharmacophore hypothesis based on Glide XP scoring terms. The visualization results and the Phase Screen Score were used to examine the alignment between initial ligands and pharmacophore features.
The binding free energy of each complex was calculated using the panel of MM-GBSA technology available with Prime. This panel can also be used to calculate ligand strain energies for a set of ligands and a single receptor. The ligands and the receptor must be properly prepared beforehand, and it is common to prepare by using LigPrep and the Protein Preparation Wizard. The calculated difference between the minimized receptor-ligand complex and the minimized unbound ligand-receptor was scored by the MM-GBSA with the VGSB solution model.
Based on ML prediction and molecular docking procedures, six flavonoids were presumably considered as potential inhibitors of COVID-19 3CL proteinase. Among these inhibitors, the most likely one is the compound Rutin, depending on the comprehensive assessment. The summary of the results was shown in Figure 1, and the details were described and discussed as below.
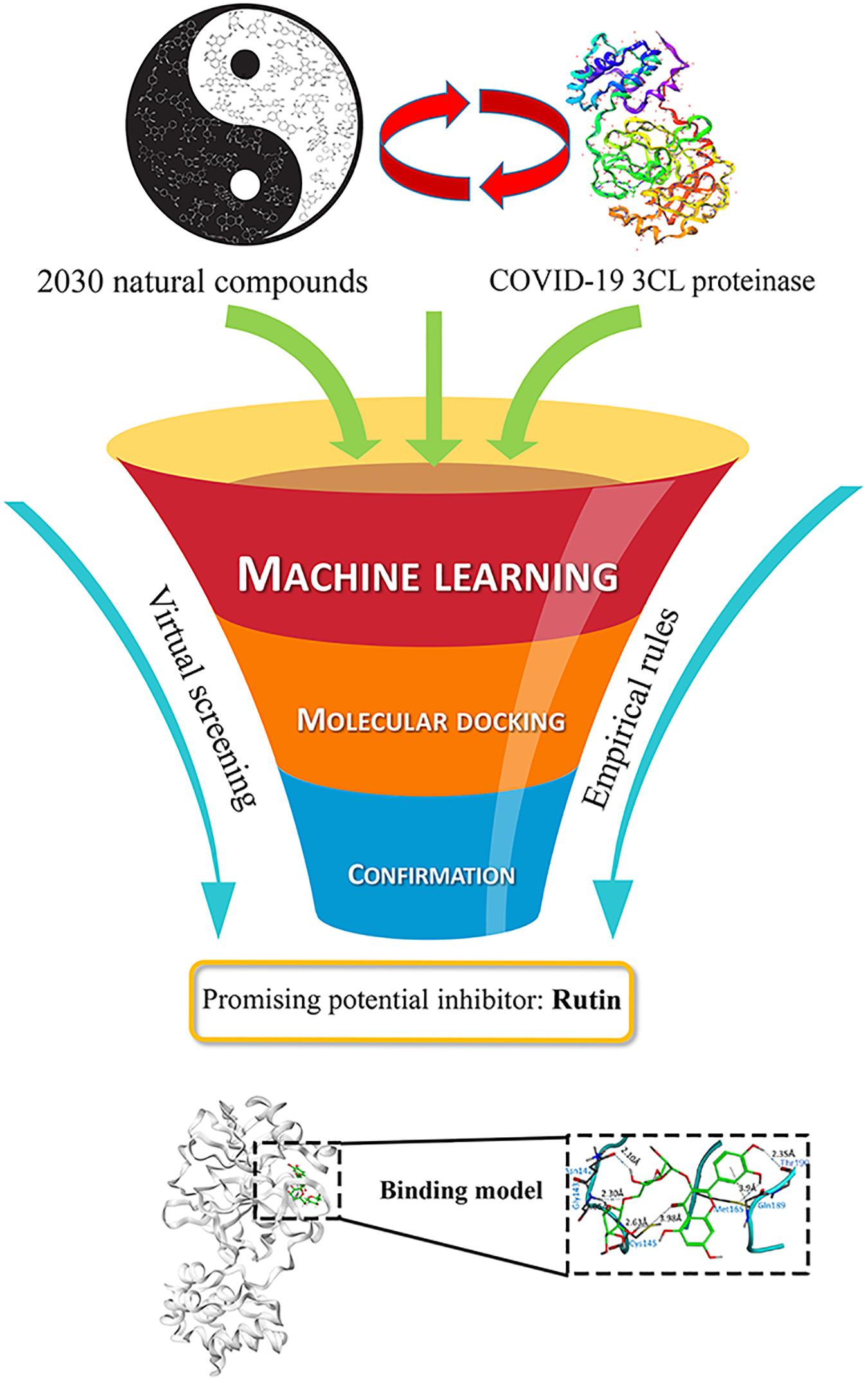
Figure 1. Schematic illustration of the anti-COVID-19 discover procedure of virtual screening.
To evaluate the performance of the training model, two factors were considered in a combinational way. These factors were (1) machine learning methods (LR, NB, DT, KNN, SVM, RF) and (2) fingerprint bits (128-bit, 256-bit, 526-bit, 1024-bit, 2048-bit). The AUC under different conditions were summarized (Figure 2 and Supplementary Table S1). On average, the performance of LR was best compared with the other five ML classifiers in the different fingerprint length. The AUC (0.976) of LR in the case of 2048-bit fingerprint was best compared with the case of other bits fingerprint. Finally, the Logistic Regression with 2048-bit fingerprint was selected to calculate the probability values (AUC) for the screening library.
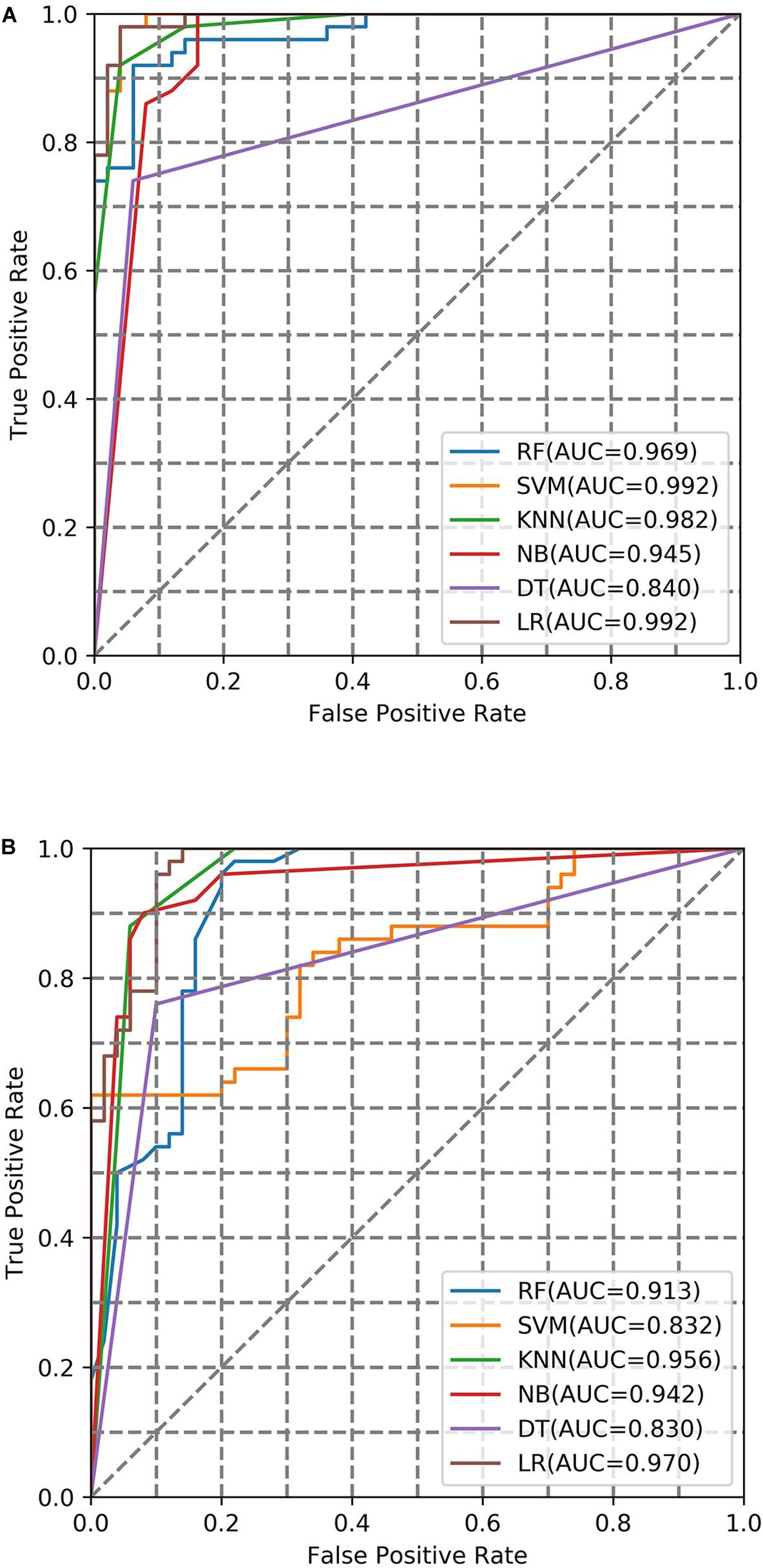
Figure 2. AUC curve of fivefold cross-validation with negative samples from inactive compounds in the case of different ML models. (A,B) are the AUC curve in the 1024-bit and 2048-bit length fingerprints, respectively.
In general, the molecular docking settings are often calibrated based on the experimental ligand-acceptor. Especially, the accuracy of docking protocol needs to be cross-checked by the training set used. However, it wasn’t long before the crystal structure of COVID-19 3CLpro complex analyzed and resolved. We found the exciting truth that the catalytic pocket of SARS 3CLpro (PDB: 3IWM) and COVID-19 3CLpro are exactly similar (Yang et al., 2006; Zhang et al., 2010; Supplementary Figure S1). This phenomenon highlights the key to confirm the binding mode of COVID-19 3CLpro complex, imitate the interaction between SARS 3CLpro and its approved inhibitors, and replicate the above binding mode to discover the potential COVID-19 3CLpro inhibitors.
The 6LU7 three-dimensional structure as well as the peptide inhibitor was analyzed. The inhibitor forms hydrogen bonds with different residues including His143, His164, Glu166, Gln189, and Thr190, and a weakly hydrogen binding with the residue Phe140. In addition, the possible formations of π-π stacking interaction with His41 and covalent bond with Cys145 were also observed. These observations further demonstrated that the original inhibitor would interact with key residues of COVID-19 3CLpro, in a similar way to that of the screening potential inhibitors against COVID-19. To confirm the proper docking protocol used, the original inhibitor was redocked five times, and the binding pose was in line with the previous crystal conformation.
The Glide ligand docking module was used to conduct the initial screening based on the templates of SP and XP. As the screening dataset, 2030 natural compounds (Chinese Medicine compounds mostly) were added. We assumed that the predicted binding affinity reflected the real binding model. The screening results were ranked based on the docking score as an initial selection, and score section showed the clear properties between binding complexes and no-binding complexes (Wilsey et al., 2013). In other words, the lowest binding score means the best binding affinity. The results yielded 1340 compounds in the entry of Workspace. Further refinement of the above initial selected compounds were performed by analyzing and scoring with the MM-GBSA and the Pharmacophore Modeling.
The pharmacophore model was subsequently generated and optimized slightly. We clearly observed the main binding model between COVID-19 3CLpro and its original inhibitor. Especially, the generated model consisted of several features including one aromatic feature projecting to the residue Thr26, H-bond acceptors targeting Gly143, Cys145, and Glu166, and two H-bond donors targeting Glu166 and Thr190. Then, the optimized pharmacophore model was used to rescore the initial screened 1340 compounds. In this case, 98 hits were already reported to give reliability to our pharmacophore model. Since the catalytic pocket of COVID-19 3CLpro is extremely similar to SARS, the binding model is highly similar. The relevant factors of docking model should possibly follow the model of inhibitors binding with SARS-CoV 3CL protease (Chen et al., 2005; Jo et al., 2020). According to the comprehensive evaluation, the docking score and AUC of molecules were better than −7.0 kcal/mol and 0.70, respectively, which were considered more reliable. As mentioned above, the 32 hits were tentatively considered as the potential inhibitors.
After visualizing the docked complexes carefully, we found that six agents with better docking scores compared to the others in the refinement list (Table 1 and Supplementary Table S2). Therefore, the six compounds were subsequently applied for further analysis. According to the binding sites of 6LU7 complex, the original inhibitor could form hydrogen bonds with surrounding residues in the active pocket. In addition, we noted that these compounds all presented electrostatic interaction with the residue Cys145, which is the key residue in the catalytic center of COVID-19 3CLpro complex. Besides, the residue Cys145 has also been approved to be the crucial residue forming within a radius of 6 Å around the catalytic center of SARS-CoV 3CLpro (Jacobs et al., 2013; Park et al., 2016). The key residues surrounding the active center of SARS-CoV 3CLpro include Hid41, Leu141, Gly143, Cys145, Glu166, and Asp187 (Li et al., 2016). Recently, the flavonoids have been approved to be the inhibitor targeting with the SARS-CoV 3CL protease (Jo et al., 2020), and they occupy the main pockets of the catalytic center including Asn 142, Glu166, and Gln189 residues. Interestingly, six screened compounds of our findings are also flavonoids. As mentioned above, both coronaviruses have the similar binding affinity, which means that the screened compounds could potentially occupy the catalytic active center of the protease very well, inhibiting the activity of protease to reduce the ability of virus copy.

Table 1. Six selected natural compounds according to the docked results and AUC.
According to the above analysis results, the compound Rutin (docking score: −9.16 kcal/mol and AUC: 0.990) was considered to be the most potential inhibitor compared with others. This compound was predicted to form the hydrogen bonds involving Cys145 (2.63Å), Asn142 (2.1 Å), Gly143 (2.3 Å), and Thr190 (2.35 Å), with additionally the possible formation of σ-π stacking interaction with Gln189 (Figures 3A,B). Notably, the major binding affinity was based on the presence of hydroxyl group, which presented the key in anchoring and blocking the substrate into the active pocket of catalytic center. Overall, the Rutin matched very well with 6LU7 binding pocket, indicating that it may be a potential inhibitor.
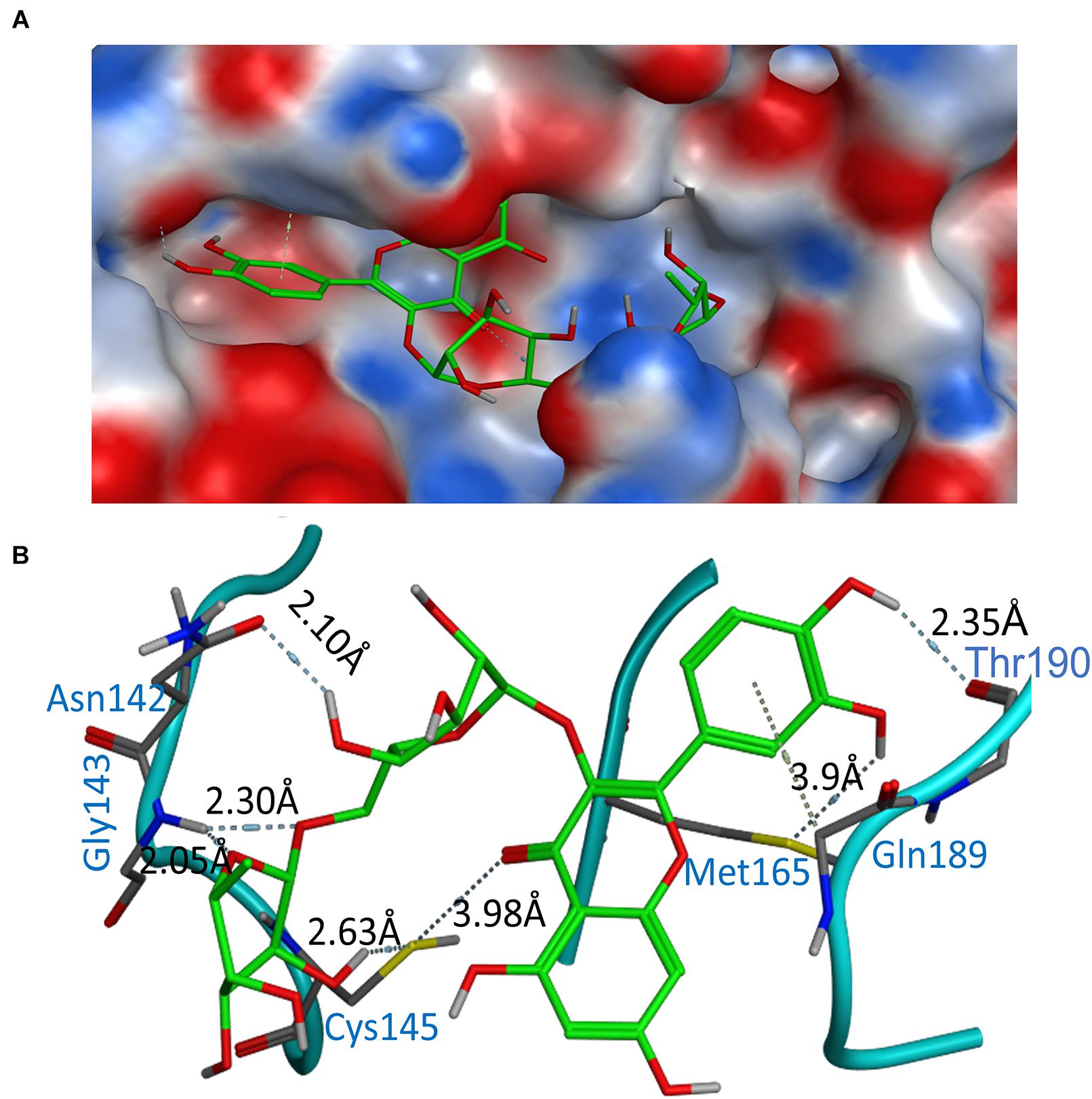
Figure 3. Binding modes of Rutin with 6LU7. (A) Electrostatic interaction between Rutin and crystal structure (6LU7) of COVID-19 3CLpro. (B) Interactions between Rutin and associated residues in the crystal structure (6LU7) of COVID-19 3CLpro. Blue and gray labels shown in the figure are residue names and interaction distance respectively.
In summary, the rapid and efficient drug discovery procedure of virtual screening combined ML methods with molecule docking was performed. Based on the further evaluation and refinement, the most potential compound Rutin was highly screened, suggesting the compound might be active against the COVID-19 3CLpro. Moreover, two flavonoids, baicalin and baicalein, have recently been identified as the novel, natural product inhibitors of 3CL protease in vitro (Su et al., 2020), and the flavonoids could be potential anti-COVID-19 inhibitors (Liu et al., 2020). In addition, the Rutin has been proved to be against the flu viruses, and Rutin tablets have been used in clinic for many years in China. Therefore, Rutin may be a potential inhibitor against COVID-19 3CLpro. There are also some limits of our approach. The number of compounds used in the machine learning procedure is not enough and we still contribute to this database. In addition, we are still keeping an eye on the latest development of COVID-19 researches. We will collect more related compounds and update our machine learning training data. Simultaneously, there is still room for progress in machine learning procedure, and the deep machine learning used in the drug screening would be perfect for filling the gap. This article is just the beginning of the research on the combination of machine learning and molecular docking. We believe that the combination of molecular docking and machine learning will bring more surprises for the discovery of drugs. In vitro and in vivo studies will be designed and carried out to validate the therapeutic effects of Rutin for COVID-19 in the future (Chen et al., 2020).
All datasets generated for this study are included in the article/Supplementary Material.
ZX and LY were responsible for the molecular docking, machine learning, and data analysis. XZ, QZ, ZY, YL, and SW devoted themselves to the compounds data collection. WL committed himself to the manuscript to revise and polish. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (No. 81703337, WL; Nos. 81772477 and 81201848, SW), the Jiangsu Province Graduate Practice Innovation Program (KYCX20_1489). The Priority Academic Program Development of Jiangsu Higher Education Institutions (Integration of Chinese and Western Medicine) and the Jiangsu Specially-Appointed Professors program and the Six Talent Peaks Project in Jiangsu Province of China (No. SWYY-069).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2020.556481/full#supplementary-material
FIGURE S1 | The superpose structure of COVID-19 3CLpro (gray) and SARS-3CLpro (green). The catalytic dyad (His41 and Cys145) are shown as sticks.
TABLE S1 | The AUC score of fivefold cross-validation.
TABLE S2 | Analyzed results of six selected compounds.
Adedeji, A. O., and Sarafianos, S. G. (2014). Antiviral drugs specific for coronaviruses in preclinical development. Curr. Opin. Virol. 8, 45–53. doi: 10.1016/j.coviro.2014.06.002
Altman, N. S. (1992). An introduction to kernel and nearest-neighbor nonparametric regression. Am. Stat. 46, 175–185. doi: 10.2307/2685209
Berry, M., Fielding, B. C., and Gamieldien, J. (2015). Potential broad spectrum inhibitors of the coronavirus 3CLpro: a virtual screening and structure-based drug design study. Viruses 7, 6642–6660. doi: 10.3390/v7122963
Biau, G., Devroye, L., and Lugosi, G. (2008). Consistency of random forests and other averaging classifiers. J. Mach. Learn. Res. 9, 2015–2033. doi: 10.1145/1390681.1442799
Cao, P., Wu, S., Wu, T., Deng, Y., Zhang, Q., Wang, K., et al. (2020). The important role of polysaccharides from a traditional Chinese medicine-lung cleansing and detoxifying decoction against the COVID-19 pandemic. Carbohydr. Polym. 240:116346. doi: 10.1016/j.carbpol.2020.116346
Chan, J. F., Kok, K. H., Zhu, Z., Chu, H., To, K. K., Yuan, S., et al. (2020). Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes. Infect. 9, 221–236. doi: 10.1080/22221751.2020.1719902
Chen, L., Gui, C., Luo, X., Yang, Q., Günther, S., Scandella, E., et al. (2005). Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J. Viorl. 79, 7095–7103. doi: 10.1128/JVI.79.11.7095-7103.2005
Chen, L., Li, J., Luo, C., Liu, H., Xu, W., Chen, G., et al. (2006). Binding interaction of quercetin-3-β-galactoside and its synthetic derivatives with SARS-CoV 3CLpro: structure–activity relationship studies reveal salient pharmacophore features. Bioorgan. Med. Chem. 14, 8295–8306. doi: 10.1016/j.bmc.2006.09.014
Chen, L., Liu, H., Liu, W., Liu, J., Liu, K., Shang, J., et al. (2020). Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Chinese. J. Tuberc. Respir. Dis. 43:E005. doi: 10.3760/cma.j.issn.1001-0939.2020.0005
Cortes, C., and Vapnik, V. (1995). Support-vector networks. Mach. Learn. 20, 273–297. doi: 10.1023/A:1022627411411
Duan, Q., Liu, T., Yuan, P., Huang, C., Shao, Q., Xu, L., et al. (2020). Antiviral effect of Chinese herbal prescription JieZe-1 on adhesion and penetration of VK2/E6E7 with herpes simplex viruses type 2. J. Ethnopharmacol. 249:112405. doi: 10.1016/j.jep.2019.112405
García-Fernández, R., Ziegelmüller, P., González, L., Mansur, M., Machado, Y., Redecke, L., et al. (2016). Two variants of the major serine protease inhibitor from the sea anemone Stichodactyla helianthus, expressed in Pichia pastoris. Protein Express. Purif. 123, 42–50. doi: 10.1016/j.pep.2016.03.003
Hand, D. J. (2009). Measuring classifier performance: a coherent alternative to the area under the ROC curve. Mach. Learn. 77, 103–123. doi: 10.1007/s10994-009-5119-5
Hu, X., Cai, X., Song, X., Li, C., Zhao, J., Luo, W., et al. (2020). Possible SARS-coronavirus 2 inhibitor revealed by simulated molecular docking to viral main protease and host toll-like receptor. Future Virol. 15:99. doi: 10.2217/fvl-2020-0099
Jacobs, J., Grum-Tokars, V., Zhou, Y., Turlington, M., Saldanha, S. A., Chase, P., et al. (2013). Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 56, 534–546. doi: 10.1021/jm301580n
Jo, S., Kim, S., Shin, D. H., and Kim, M.-S. (2020). Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzyme Inhib. Med. Chem. 35, 145–151. doi: 10.1080/14756366.2019.1690480
Konno, H., Onuma, T., Nitanai, I., Wakabayashi, M., Yano, S., Teruya, K., et al. (2017). Synthesis and evaluation of phenylisoserine derivatives for the SARS-CoV 3CL protease inhibitor. Bioorgan. Med. Chem. Lett. 27, 2746–2751. doi: 10.1016/j.bmcl.2017.04.056
Lee, P., and Hsueh, P. (2020). Emerging threats from zoonotic coronaviruses-from SARS and MERS to 2019-nCoV. J. Microbiol. Immunol. Infect. 53, 365–367. doi: 10.1016/j.jmii.2020.02.001
Li, C., Teng, X., Qi, Y., Tang, B., Shi, H., Ma, X., et al. (2016). Conformational flexibility of a short loop near the active site of the SARS-3CLpro is essential to maintain catalytic activity. Sci. Rep. 6:20918. doi: 10.1038/srep20918
Liu, X., Zhang, B., Jin, Z., Yang, H., and Rao, Z. (2020). The crystal structure of COVID-19 main protease in complex with an inhibitor N3. Protein DataBank
Ma, Q., Yu, Q., Xing, X., Liu, S., Shi, C., and Luo, J. (2018). San wu huangqin decoction, a chinese herbal formula, inhibits influenza a/PR/8/34 (H1N1) virus infection in vitro and in vivo. Viruses 10:117. doi: 10.3390/v10030117
Morgan, H. (1965). The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service. Chem. Doc. J. 5, 107–113. doi: 10.1021/c160017a018
Park, J., Ko, J., Kim, D. W., Kim, Y. M., Kwon, H., Jeong, H. J., et al. (2016). Chalcones isolated from Angelica keiskei inhibit cysteine proteases of SARS-CoV. J. Enzyme. Inhib. Med. Chem. 31, 23–30. doi: 10.3109/14756366.2014.1003215
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., et al. (2011). Scikit-learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830. doi: 10.1524/auto.2011.0951
Polton, D. (1982). Installation and operational experiences with MACCS (molecular access system). Online Rev. 6, 235–242. doi: 10.1108/eb024099
Qiu, R., Zhao, C., Liang, T., Hao, X., Huang, Y., Zhang, X., et al. (2020). Core outcome set for clinical trials of COVID-19 based on traditional chinese and western medicine. Front. Pharmacol. 11:781. doi: 10.3389/fphar.2020.00781
Rohini, K., and Shanthi, V. (2018). Discovery of potent neuraminidase inhibitors using a combination of pharmacophore-based virtual screening and molecular simulation approach. Appl. Biochem. Biotech. 184, 1421–1440. doi: 10.1007/s12010-017-2625-y
Safavian, S. R., and Landgrebe, D. (1991). A survey of decision tree classifier methodology. IEEE T. Syst. Man. 21, 660–674. doi: 10.1109/21.97458
Shi, C., and Borchardt, T. B. (2017). JRgui: a Python program of Joback and Reid method. ACS Omega 2, 8682–8688. doi: 10.1021/acsomega.7b01464
Su, H., Yao, S., Zhao, W., Li, M., Liu, J., Shang, W., et al. (2020). Discovery of baicalin and baicalein as novel, natural product inhibitors of SARS-CoV-2 3CL protease in vitro. BioRxiv [Preprint]. doi: 10.1101/2020.04.13.038687
Tolles, J., and Meurer, W. J. (2016). Logistic regression: relating patient characteristics to outcomes. JAMA 316, 533–534. doi: 10.1001/jama.2016.7653
Wang, F., and Zhang, C. (2020). What to do next to control the 2019-nCoV epidemic? Lancet 395, 391–393. doi: 10.1016/S0140-6736(20)30300-7
Wang, S., Du, Q., Zhao, K., Li, A., Wei, D., and Chou, K. (2007). Virtual screening for finding natural inhibitor against cathepsin-L for SARS therapy. Amino Acids 33, 129–135. doi: 10.1007/s00726-006-0403-1
Watson, P. (2008). Naive Bayes classification using 2D pharmacophore feature triplet vectors. J. Chem. Inf. Model. 48, 166–178. doi: 10.1021/ci7003253
Watts, K. S., Dalal, P., Murphy, R. B., Sherman, W., Friesner, R. A., and Shelley, J. C. (2010). ConfGen: a conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 50, 534–546. doi: 10.1021/ci100015j
Wilsey, C., Gurka, J., Toth, D., and Franco, J. (2013). A large scale virtual screen of DprE1. Comput. Biol. Chem. 47, 121–125. doi: 10.1016/j.compbiolchem.2013.08.006
Xu, X., Chen, P., Wang, J., Feng, J., Zhou, H., Li, X., et al. (2020). Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 63, 457–460. doi: 10.1007/s11427-020-1637-5
Xue, L., Stahura, F. L., and Bajorath, J. (2004). Similarity search profiling reveals effects of fingerprint scaling in virtual screening. J. Chem. Inf. Comput. Sci. 44, 2032–2039. doi: 10.1021/ci0400819
Yamamoto, N., Yang, R., Yoshinaka, Y., Amari, S., Nakano, T., Cinatl, J., et al. (2004). HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem. Biophys. Res. Commun. 318, 719–725. doi: 10.1016/j.bbrc.2004.04.083
Yang, H., Bartlam, M., and Rao, Z. (2006). Drug design targeting the main protease, the Achilles’ heel of coronaviruses. Curr. Pharm. Des. 12, 4573–4590. doi: 10.2174/138161206779010369
Yoshizawa, S.-I., Hattori, Y., Kobayashi, K., and Akaji, K. (2020). Evaluation of an octahydroisochromene scaffold used as a novel SARS 3CL protease inhibitor. Bioorgan. Med. Chem. 28:115273. doi: 10.1016/j.bmc.2019.115273
Zhang, S., Zhong, N., Xue, F., Kang, X., Ren, X., Chen, J., et al. (2010). Three-dimensional domain swapping as a mechanism to lock the active conformation in a super-active octamer of SARS-CoV main protease. Protein Cell. 1, 371–383. doi: 10.1007/s13238-010-0044-8
Zhao, Q., Ren, X., Chen, M., Yue, S. J., Zhang, M. Q., Chen, K. X., et al. (2019). Effects of traditional Chinese medicine formula Le-Cao-Shi on hepatitis B: in vivo and in vitro studies. J. Ethnopharmacol. 244:112132. doi: 10.1016/j.jep.2019.112132
Keywords: COVID-19 3CL proteinase, flavonoids, rutin, virtual screening, machine learning, molecular docking
Citation: Xu Z, Yang L, Zhang X, Zhang Q, Yang Z, Liu Y, Wei S and Liu W (2020) Discovery of Potential Flavonoid Inhibitors Against COVID-19 3CL Proteinase Based on Virtual Screening Strategy. Front. Mol. Biosci. 7:556481. doi: 10.3389/fmolb.2020.556481
Received: 30 April 2020; Accepted: 20 August 2020;
Published: 29 September 2020.
Edited by:
Francesco Luigi Gervasio, University College London, United KingdomReviewed by:
Yassmine Chebaro, Centre National de la Recherche Scientifique (CNRS), FranceCopyright © 2020 Xu, Yang, Zhang, Zhang, Yang, Liu, Wei and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wukun Liu, bGl1d3VrdW4wMDAwQGhvdG1haWwuY29t; bGl1d3VrdW4wMDAwQG5qdWNtLmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.