 Anwar Ullah
Anwar Ullah Rehana Masood
Rehana Masood- 1Department of Biosciences, COMSATS University Islamabad, Islamabad, Pakistan
- 2Department of Biochemistry, Shaheed Benazir Bhutto Women University Peshawar, Peshawar, Pakistan
Snake venom phospholipases B (SVPLBs) are the least studied enzymes. They constitute about 1% of Bothrops crude venoms, however, in other snake venoms, it is present in less than 1%. These enzymes are considered the most potent hemolytic agent in the venom. Currently, no structural information is available about these enzymes from snake venom. To better understand its three-dimensional structure and mechanisms of envenomation, the current work describes the first model-based structure report of this enzyme from Bothrops moojeni venom named as B. moojeni phospholipase B (PLB_Bm). The structure model of PLB_Bm was generated using model building software like I-TESSER, MODELLER 9v19, and Swiss-Model. The build PLB_Bm model was validated using validation tools (PROCHECK, ERRAT, and Verif3D). The analysis of the PLB_Bm modeled structure indicates that it contains 491 amino acid residues that form a well-defined four-layer αββα sandwich core and has a typical fold of the N-terminal nucleophile aminohydrolase (Ntn-hydrolase). The overall structure of PLB_Bm contains 18 β-strands and 17 α-helices with many connecting loops. The structure divides into two chains (A and B) after maturation. The A chain is smaller and contains 207 amino acid residues, whereas the B chain is larger and contains 266 amino acid residues. The sequence and structural comparison among homologous snake venom, bacterial, and mammals PLBs indicate that differences in the length and sequence composition may confer variable substrate specificity to these enzymes. Moreover, the surface charge distribution, average volume, and depth of the active site cavity also vary in these enzymes. The present work will provide more information about the structure–function relationship and mechanism of action of these enzymes in snakebite envenomation.
Introduction
Phospholipases B (PLBs) or lysophospholipases (EC3.1.1.5) are high-molecular-mass enzymes that break ester linkages of glycerophospholipids of membranes at both positions sn-1 and sn-2 (Shiloah et al., 1973; Rokyta et al., 2011; Chapeaurouge et al., 2015). These enzymes have been named as PLBs (Doery and Pearson, 1964), phospholipase B-like (Doery and Pearson, 1964; Aird et al., 2013), lysophospholipases (Takasaki and Tamiya, 1982), and Ntn-hydrolases (Oinonen and Rouvinen, 2000). These are reported to exist in the venom proteomes of various snakes, bee, scorpions, and insects), fungi, bacteria, animal tissues, and rice bran (Table 1).

Table 1. Occurrence of PLBs in various organisms.
Currently, little is known about the pathological and physiological effects of these enzymes in snake venom (Rokyta et al., 2011; Chapeaurouge et al., 2015; Oliveira et al., 2019; Tang et al., 2019). Upon snakebite envenomation, snake venom PLBs (SVPLBs) display strong hemolytic and cytotoxic activities and cause myoglobinuria and cytotoxicity (Takasaki and Tamiya, 1982; Bernheimer et al., 1986, 1987). The hemolytic activity of these enzymes is related to the hydrolysis of phosphatidylcholine (Bernheimer et al., 1986).
The relative abundance of PLBs varies in snake venoms, and generally, it constitutes a small percentage of the crude venoms; for example, PLBs constitute about 0.34% of the crude venom of Elapidae (Margres et al., 2013) and in the Viperidae, this percentage varies from 0.23 to 2.5 (Sousa et al., 2013). In Botrops species, the highest percentage has been reported in Rhinocerophis cotiara (2.5% of the crude venom) (Sousa et al., 2013).
SVPLBs are high-molecular-mass proteins (∼55 kDa) (Rokyta et al., 2011; Chapeaurouge et al., 2015; Wiezel et al., 2015) with a pI of 6.2 (Bernheimer et al., 1987). These enzymes display maximum catalytic activity in the pH range from 8.5 to 10 (Doery and Pearson, 1964).
Research about the SVPLBs is in nascent stage, and the first sequence report (based on transcriptomic analysis) about this protein came out in 2011 (Chatrath et al., 2011; Rokyta et al., 2011). The primary structure of SVPLB contains 553 amino acids in which the first 36 amino acids form the signal peptides and the remaining 526 make the PLB domain (Rokyta et al., 2011). Both monomeric and dimeric forms of PLBs have been reported to exist in snake venoms (Bernheimer et al., 1987; Chatrath et al., 2011). SVPLBs are stable proteins and show full enzymatic activity in the temperature range of 0–60°C; however, some of these retain 47% of the biological activity even at a temperature of 100°C (Bernheimer et al., 1987).
Although the primary amino acid sequence of PLBs from a number of snake venoms is present in the protein sequence database (UniProt databank), there is no report about their three-dimensional (3D) structure. Owing to this, it is difficult to co-relate their structural properties with the function. Keeping this in view, the current work reports model-based structural characterization of PLBs from Bothrops moojeni venom.
Results and Discussion
Sequence Alignment Analysis
The primary amino acid sequence of PLB_Bm contains 553 amino acid residues in the precursor form and 491 amino acid residues in the mature form (Amorim et al., 2017). The sequence alignment analysis indicates a high sequence identity (70–97%) among SVPLBs, moderate sequence identity (63–67%) with cow PLB, and very low sequence identity (34%) with mouse PLB (Figure 1 and Table 2). The primary amino acid sequence of PLB_Bm contains seven cysteine residues in the precursor form and five cysteine residues in the mature form (Figure 1). Of the five cysteine residues, four make two disulfide bonds (Cys88–Cys500 and Cys499–Cys523), whereas one cysteine (Cys237) remains in the free form. This free cysteine functions as one of the main amino acids in the active site of these enzymes, and they are also called cysteine proteinases (Verma et al., 2016). This cysteine residue is fully conserved in all SVPLBs and also PLBs of cow and mouse (Figure 1). The four other cysteine residues are fully conserved among SVPLB and mouse PLB; however, in the cow PLB, the cysteine at positions 501, 502, and 523 are not conserved (Figure 1). The amino acid residues belonging to the active sites (Asp303, Lys527, Cys237, His254, and Arg265) are also fully conserved among SVPLBs and mouse and cow PLBs. SVPLBs contain one glycosylation site (Asn69), which is fully conserved with the mouse PLB. The concurrence (consensus) lipase sequence GXSXG is fully conserved among all the aligned PLBs (Figure 1). The analysis of the phylogenetic tree generated from the aligned sequence shows a close relationship among SVPLBs and PLBs from mouse and bovine kidneys (Supplementary Figure S1).

Figure 1. Sequence alignment among snake venom phospholipases B (SVPLBs), PLB from bovine kidneys, and 66.3 kDa protein from Mus musculus. PLB_Bm, phospholipase B from Bothrops moojeni; PLB_Pf, phospholipase B from Protobothrops flavoviridis; PLB_Ca, phospholipase B from Crotalus atrox; PLB_Oo, phospholipase B from Ovophis okinavensis; PLB_Cj, phospholipase B from Coturnix japonica; PLB_Ss, phospholipase B from Spilotes sulphureus; PLB_Ns, phospholipase B from Notechis scutatus; 4BWC, phospholipase B like protein 1 from bovine kidneys; 3FGW, 66.3 kDa protein from M. musculus. The amino acid residues involved in catalysis are underlined with blue, and the cysteine residues are underlined with yellow. The consensus lipase sequence is underlined with gold color. The cysteine residues that make disulfide bridges are linked (yellow lines). The putative N-glycosylation amino acid residues are underlined in green. The secondary structure elements (α-helices and β-strands) are shown above the sequence.
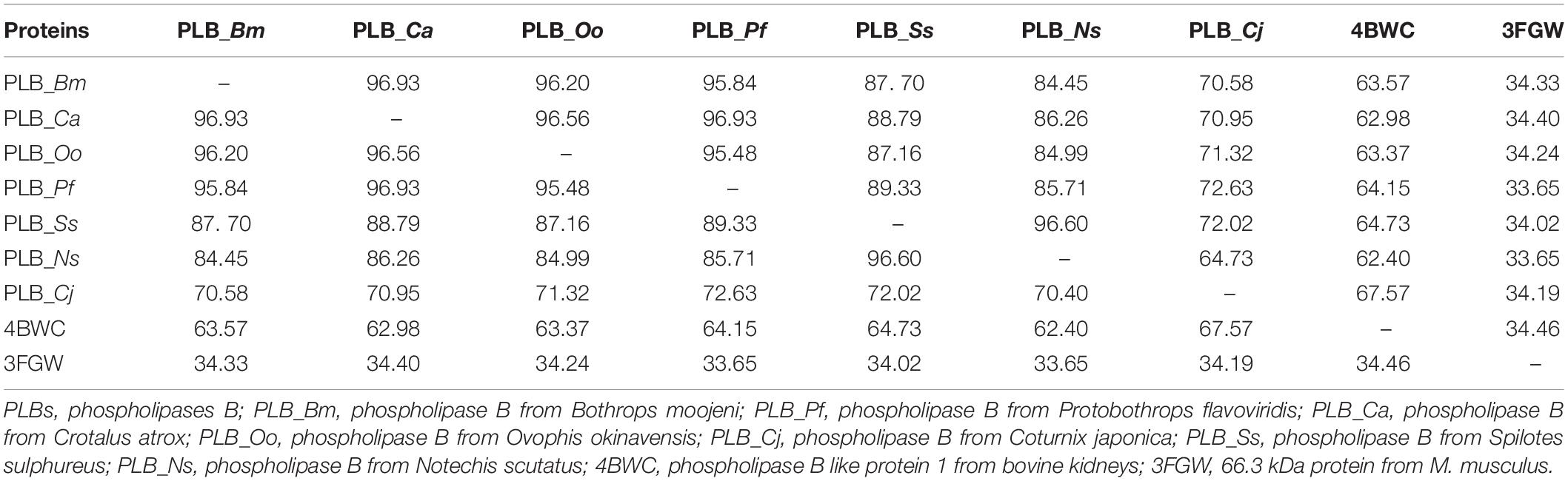
Table 2. Percent sequence identity among snake venom PLBs, PLB-like protein 1 from bovine kidneys, and 66.3 kDa protein from Mus musculus.
Domain Analysis
The ThreaDom (Threading-based Protein Domain Prediction) (Xue et al., 2013) analysis indicates that PLB_Bm is a single-domain protein. The molecular weights (calculated through ProtParam (Gasteiger et al., 2005) of the precursor and mature protein were 63.88 and 57.09 kDa with the corresponding pI of 8.80 and 7.71, respectively. These results indicate that the pI of PLB_Bm changes from highly basic to slightly basic upon maturation. The theoretically calculated molecular weights and pIs agree with the experimentally observed molecular weights and pIs of these enzymes (Doery and Pearson, 1964; Takasaki and Tamiya, 1982; Bernheimer et al., 1986, 1987; Chatrath et al., 2011).
Homology Modeling
For the 3D structure characterization of PLB_Bm, the homology model was generated using the online modeling servers like the SWISS Model (Waterhouse et al., 2018), I-TESSER (Laskowski et al., 2001), and MODELLER 9v19 program (Webb and Sali, 2016). The atomic coordinates of phospholipase B-like protein 1 from Bos taurus (PDB ID: 4BWC; 70% amino acid sequence identity with PLB_Bm) (Repo et al., 2014), were applied as a template.
Model Validation
The generated model of PLB_Bm was validated using programs like PROCHECK, ERRAT, and Verif3D software (Bowie et al., 1991; Lüthy et al., 1992; Colovos and Yeates, 1993; Laskowski et al., 1993). The best model was selected based on the analysis coming from these programs. The PROCHECK analysis of the best 3D structure model of PLB_Bm shows that 95.7% (468 amino acid residues) were in the favored region and 4.3% (21 amino acid residues) were in the allowed region with no amino acid residue in the outlier region of the Ramachandran plot (Lovell et al., 2003; Supplementary Figure S2). The overall quality factor of the ERRAT analysis was 96 (Supplementary Figure S3), which lies for the best structure quality of the proteins 3D structure according to the writers of the program (Colovos and Yeates, 1993).
Molecular Dynamics Simulation
The programs used for the molecular dynamics (MD) simulation includes GROMACS (Berendsen et al., 1995; Maier et al., 2015), AMBER16 (Case et al., 2005; Salomon-Ferrer et al., 2013), MDWeb, and MDMobby (Hospital et al., 2012). The analysis of the MD simulation coming from all these programs indicates the same results for the modeled structure of PLB_Bm (Supplementary Figures S4A–D). The important 3D structure parameters like chirality, disulfide bonds, and unusual cis/trans configuration were correct, and there were no steric clashes in the modeled PLB_Bm structure (Supplementary Figure S4A). The analysis of the root-mean-square deviation (RMSD) and radius of gyration (RG), the two essential parameters for 3D structure validation, have shown that the PLB_Bm has not undergone substantial changes during the modeling process. The RMSD value did not diverge more than 1 Å (Supplementary Figure S4B), and the radius of gyration was constant (kept around 21.5 Å) throughout the MD simulation process (Supplementary Figure S4C). The B-factor per residue was ∼17 Å (Supplementary Figure S4D), which lies in the average B-factor range for the proteins with X-ray resolution (1.8–2.1 Å) (Carugo, 2018).
The Overall Structure of Snake Venom Phospholipase B
The mature protein of PLB_Bm contains 491 amino acid residues that fold into a well-defined 3D structure, which contains four-layer αββα sandwich core and has a typical fold of the N-terminal nucleophile aminohydrolase (Ntn-hydrolase) (Figures 2A,B; Oinonen and Rouvinen, 2000; Lakomek et al., 2009; Repo et al., 2014). The overall structure of PLB_Bm contains 18 β-strands and 17 α-helices with many connecting loops (Figures 2A,B and Supplementary Figure S5). The structure divides into two chains (A and B) after maturation (Oinonen and Rouvinen, 2000; Repo et al., 2014). The A chain is small and contains 207 amino acid residues, whereas the B chain is large and contains 266 amino acid residues (Supplementary Figure S5).
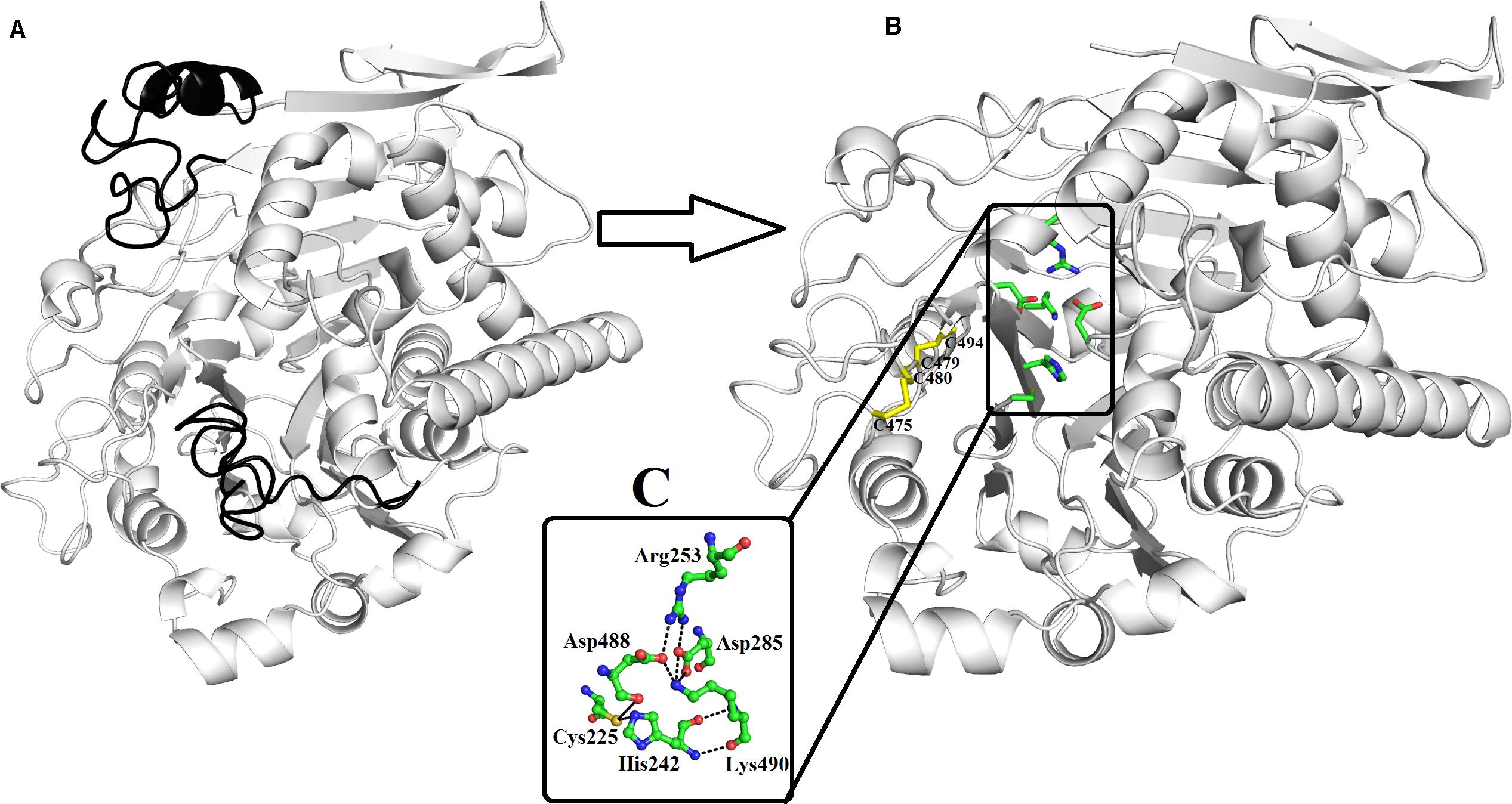
Figure 2. Overall structure of phospholipase B from Bothrops moojeni (PLB_Bm) cartoon representation of (A) zymogen and (B) mature protein. The signal and the inner peptides are shown in black color. The active site residues and the disulfide bridges are shown as green and yellow sticks. (C) The residues involved in catalysis are highlighted.
The A chain has four β-strands numbered 1 through 4 and five α-helices named A to E (Supplementary Figure S5). The β-strands are anti-parallel to each other. The N-terminal of this chain starts with long β-strands, and the C-terminal ends at α-helix (Figure 2B and Supplementary Figure S5). This chain is stabilized by four intrachain salt bridges (His110–Asp69, Arg144–Asp167, Arg144–Asp140, and Arg159–Asp55) and two interchain salt bridges (Lys82–Asp259 and Arg157–Asp264) (Table 3).
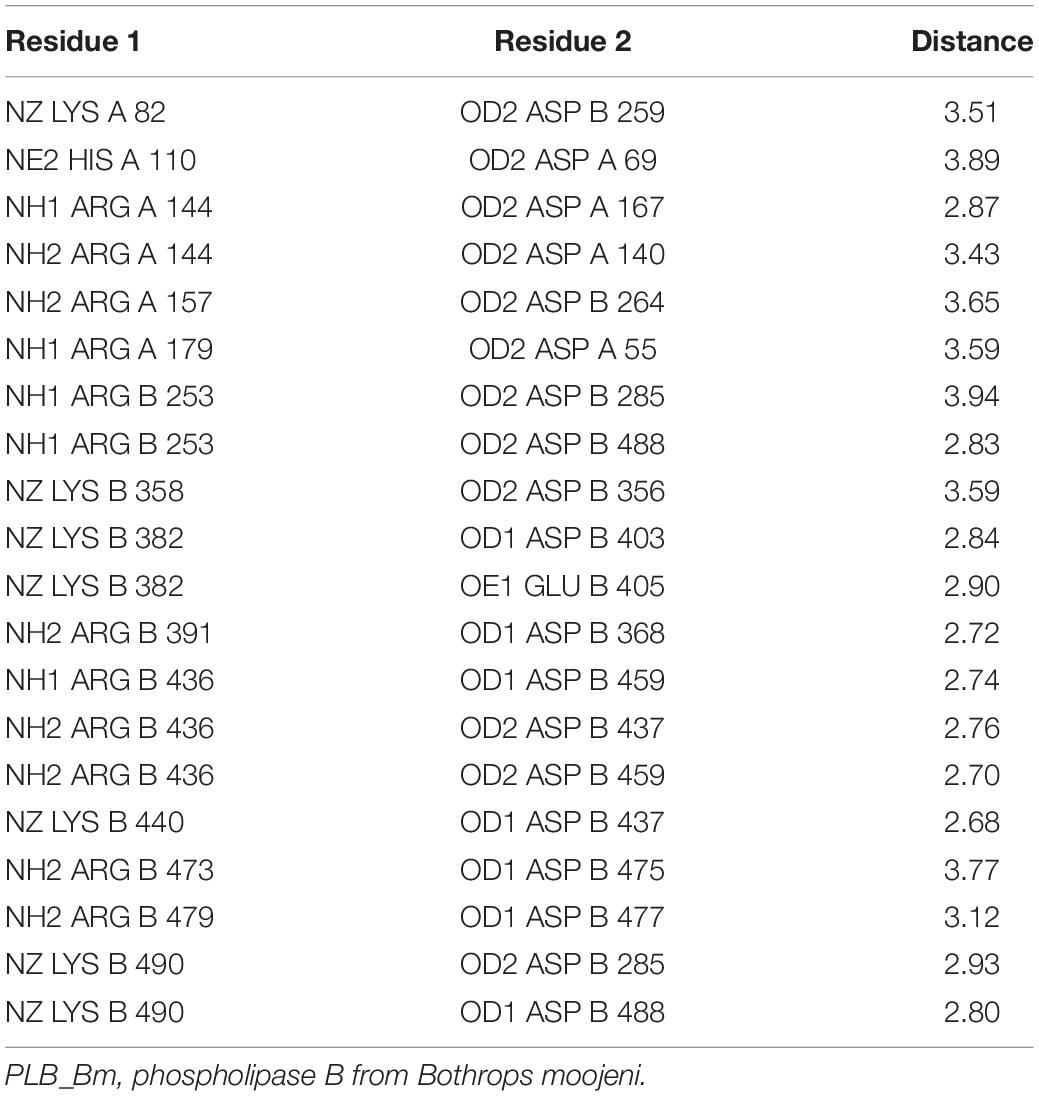
Table 3. Salt bridges in the PLB_Bm three-dimensional structure.
The B chain is more compact, and it contains 14 β-strands and 12 α-helices. Of the 14 β-strands, seven are parallel, whereas the other seven are antiparallel. The N-terminal of this chain starts with a long β-strand, and the C-terminal ends with a short β-strand. The active site is located in this chain (Figure 2B). This chain is stabilized by two interchain salt bridges (Lys82–Asp259 and Arg157–Asp264) and 14 intrachain salt bridges (Arg253–Asp285, Arg253–Asp488, Lys358–Asp356, Lys382–Asp403, Lys382–Glu405, Arg391–Asp368, Arg436–Asp459, Lys440–Asp437, Arg473–Asp475, Arg479–Asp477, Lys490–Asp285, and Lys490–Asp488). This chain is further stabilized by two intrachain disulfide bridges (Cys88–Cys500 and Cys499–Cys523) (Figures 1, 2B).
Active Site
A 16-amino-acid-residue segment (208–224) is removed autocatalytically between chains A and B, which opens the active site and produces a cavity that facilitates the entry of a substrate to the active site (Figure 2B). The free cysteine residue (Cys225), which is situated between the key β-sheets in chain B, forms the active site of this enzyme. This cysteine residue functions as both a nucleophile and a general base during catalysis. It is further supported by His242 and Lys490, which is in turn assisted by Asp285, Asp488, and Arg253 (Figure 2C). These active site residues are conserved in the PLBs from other organisms as well (Figure 1 and Supplementary Figure S6). The sequence logo produced from the aligned sequence of SVPLBs and PLBs of mouse and bovine kidneys display high sequence identity around the active site (Supplementary Figure S7).
Glycosylation
PLB_Bm contains carbohydrate moiety like PLBs from other organisms (Lakomek et al., 2009; Repo et al., 2014). The NetNGlyc server1 (Gupta et al., 2004) found a single glycosylation site for this enzyme at Asn69. In the primary amino acid structure of Drysdalia coronoides PLB, two putative glycosylation sites have been found (Chatrath et al., 2011). In bovine lysosomal phospholipase B-like protein (PDB ID: 4WBC) (Repo et al., 2014), six glycosylation sites were identified, which include Asn68, Asn211, Asn305, Asn363, Asn408, and Asn523. Of these, only Asn69 that is conserved between PLB_Bm and bovine lysosomal phospholipase B-like protein contains N-acetylglucosamine (NAG). Although Asn211, Asn305, Asn408, and Asn523 are conserved with PLB_Bm, these were found without carbohydrate moiety. In the structure of lysosomal 66.3 kDa protein from mouse (PDB ID: 3FGR) (Lakomek et al., 2009), seven NAG molecules were found, which were attached to Asn93, Asn236, and Asn520 (one NAG each) and Asn115 and Asn441 (two NAGs each). Only Asn93, which corresponds to Asn69 of PLB_Bm, is conserved between the two proteins and contains NAG. In SVPLBs, the specific function of the glycan moiety is not fully known; however, it may help the enzyme to specifically bind to the cell surface, thereby facilitating the hydrolysis processes.
Substrate Specificity
SVPLBs have been shown to hydrolyze phosphatidylcholine, phosphatidylethanolamine, and lysophosphatidylcholine, however, they are not active against phosphatidylinositol, phosphatidylserine, sphingomyelin, and cardiolipin (Supplementary Figure S8; Bernheimer et al., 1986, 1987).
On the other hand, PLBs of fungi, bacteria, and mammals have been shown to hydrolyze a broad range of substrates like phosphatidylcholine, phosphatidylinositol, phosphatidylserine, phosphatidylethanolamine, phosphatidic acid, lysophosphatidyl- choline, and lysophosphatidylethanolamine (Supplementary Figure S8; Morgan et al., 2004; Lakomek et al., 2009; Repo et al., 2014). An explanation for the observed specificity of SVPLBs and PLBs from other organisms can be made on the basis of surface charge distribution around the active site cavity (Ullah et al., 2018,2019; Ullah, 2020). In SVPLBs, the active site cavity is negatively charged, whereas the entry to the active site is positively charged (Figure 3A). In the case of bovine lysosomal phospholipase B-like protein, the active site cavity and its entrance are both neutral and positively charged, respectively (Figure 3B), whereas lysosomal 66.3 kDa protein from mouse that is also a PLB has an active site cavity that is highly negatively charged, and its entrance is also negatively charged (Figure 3C). A second factor that may involve this substrate specificity is the volume of the active site cavity. The SVPLBs have large active site cavity volumes with long average depth (Table 4). Owing to this, the phospholipids with the large polar head group easily reach their active sites, whereas the PLBs from other organisms have relatively small cavity volume with small depth and can accommodate phospholipids with a small polar head group (Table 4). The size of the active site cavity gradually decreases in PLB_Bm while going from the surface to the interior of the protein (Figure 3A). From the above discussion, it is clear that the enzymes showing specificity for phosphatidylcholine, phosphatidylethanolamine, and lysophosphatidylcholine (SVPLBs) have negatively charged active site with a large volume, which can accommodate the phospholipids with large and positively charged head groups (Supplementary Figure S8). The other PLBs (fungi, bacteria, and mammals) having broad specificity display surface charge distribution (partially positive and neutral and highly negative), and the active site with a relatively small volume can accommodate phospholipids with head group that is positively and negatively charged or neutral (Supplementary Figure S8).
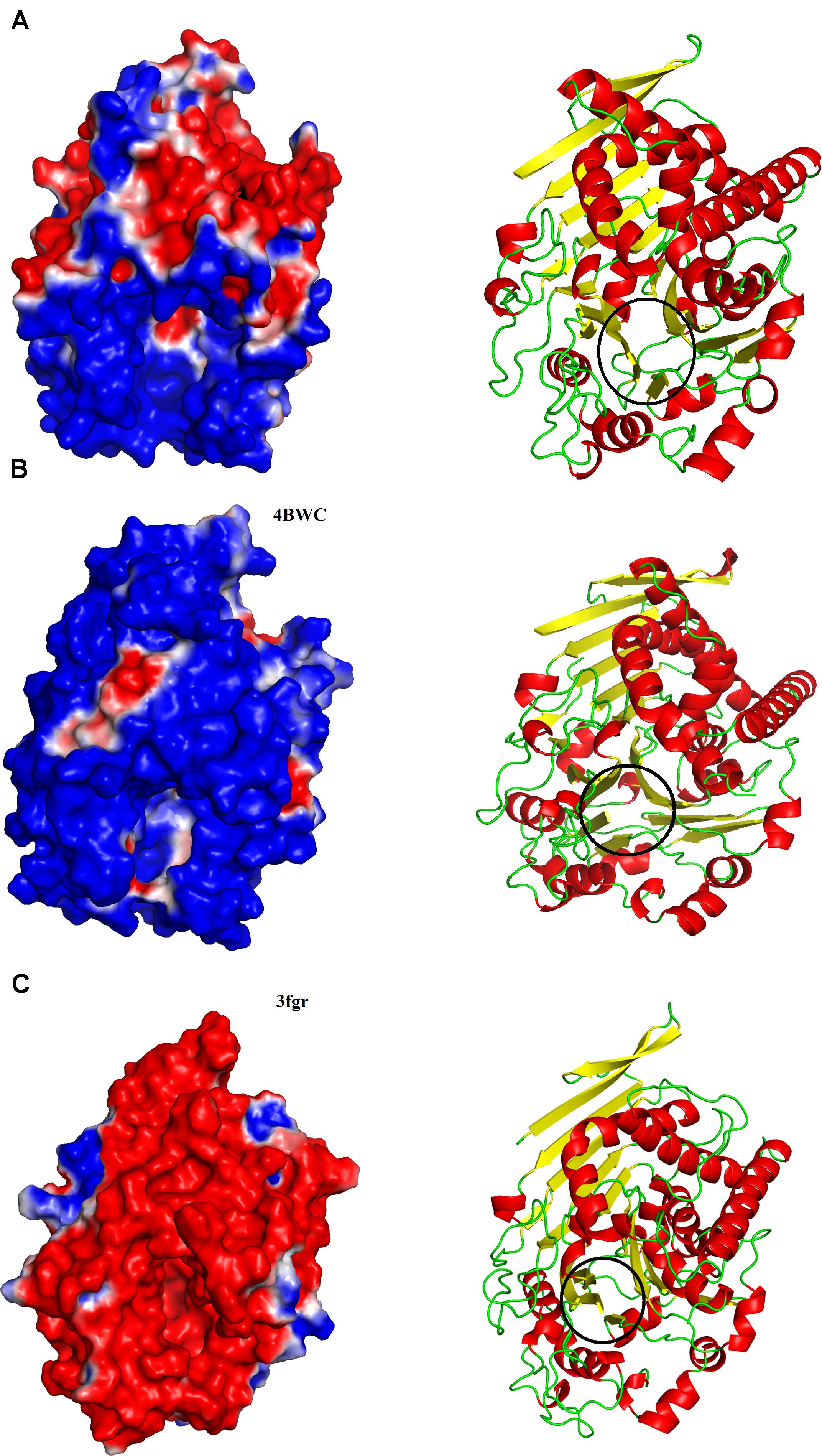
Figure 3. Surface charge distributions of (A) phospholipase B from Bothrops moojeni (PLB_Bm), (B) phospholipase B like protein 1 from bovine kidneys, and (C) 66.3 kDa protein from Mus musculus highlighting the contrasts at the catalytic interface. Black circles indicate the location of the active-site pocket. Blue, red, and white represent positive, negative, and neutral charges, respectively.

Table 4. Average active site cavity volumes and average active site cavity depths of PLB_Bm and their mammalian counterparts.
Maturation of Snake Venom Phospholipases B
The SVPLBs like other snake venom enzymes are secreted as zymogen with the signal peptide, an internal peptide, and a phospholipase domain (Rokyta et al., 2011; Amorim et al., 2017; Figures 4A–D, 5). The zymogen or precursor proteins of SVPLBs contain 547–553 amino acid residues in length (Chatrath et al., 2011; Rokyta et al., 2011; Aird et al., 2017). During the maturation process, the SVPLBs lose the signal peptide. The amino acid sequence analysis by signalP-3.0 (Bendtsen et al., 2004) indicates that this part has 36 amino acid residues (Figure 4A). The signal peptide is removed cotranslationally or by the action of signal peptidases (Paetzel et al., 2002; Figure 5). A second segment (internal peptide) is removed autocatalytically and internally from these enzymes (Oinonen and Rouvinen, 2000). This segment contains 16 amino acid residues (Figure 1). After the removal of the internal peptide, the SVPLBs are divided into two chains like bovine lysosomal phospholipase B-like protein and lysosomal 66.3 kDa proteins from mouse (Lakomek et al., 2009). The two chains are connected by many hydrogen bonds and non-bonded contacts between them (Lakomek et al., 2009). The Kyte–Doolittle plot for hydropathy (Gasteiger et al., 2005) and the temperature B-factor analysis indicate that both the signal and internal peptides are present in the hydrophilic region of the protein (Figures 4B–D).
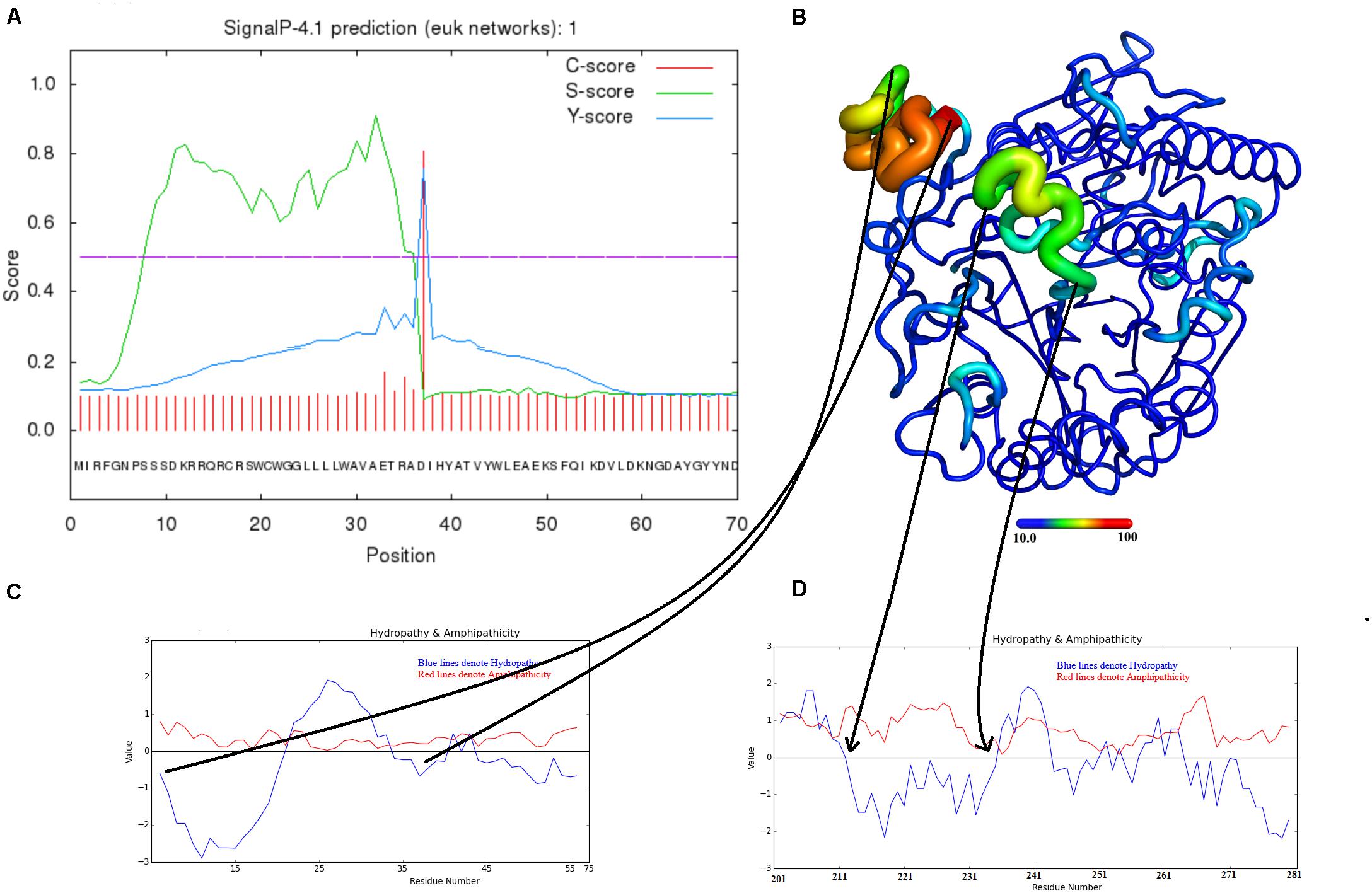
Figure 4. Maturation mechanism for phospholipase B from Bothrops moojeni (PLB_Bm). (A) A signalP-HMM prediction plot, (B,C) a Kyte–Doolittle plot for signal and activation peptides, and (D) ribbon representation of colored B-factor (temperature)

Figure 5. Steps involved in the maturation of phospholipase B from Bothrops moojeni (PLB_Bm). The prepropeptide of PLB_Bm with the signal and internal peptides (colored in black and red, respectively) and the mature protein with chain A (colored in green) and chain B (colored in blue).
Conclusion
• The sequence and structural analysis of PLB_Bm was carried out using several computational biology programs.
• The sequence alignment analysis indicates a high sequence identity (70–97%) among SVPLBs, average sequence identity (63–67%) with cow PLB, and very low sequence identity (34%) with mouse PLB.
• The 3D structural analysis of PLB_Bm indicates that its structure is composed of four-layer αββα sandwich core and has a typical fold of the Ntn-hydrolases.
• Structural comparisons with PLBs from cow and mouse indicated that the surface charge distribution and the average active site cavity volume and depth vary in these enzymes, which may impart variable substrate specificity to these enzymes.
• The maturation process of PLB_Bm involves loss of the signal and internal peptides to convert it into the fully active mature form.
• The structure of PLB_Bm described in this work is solely a predicted structure, and these observations need to be confirmed with experimental evidence like X-rays crystallography (Ullah et al., 2020).
• This work will provide a good starting point for future experimental studies of these enzymes.
Materials and Methods
Sequence Logo Generated From Multiple Sequence Alignment
The Weblogo 3.2 (Schneider and Stephens, 1990; Crooks et al., 2004) was used to generate the sequence logo from multiple sequence alignment using default parameters.
Domain Prediction and Biochemical Properties of the PLB_Bm
The domain organization and biochemical properties of PLB_Bm were predicted using the program ThreaDomEx (Wang et al., 2017) and ProtParam1 (Gasteiger et al., 2005), respectively.
Prediction of Ligand Binding
The ligand-binding sites in PLB_Bm were predicted using the 3DLigandSite (Wass et al., 2010) with parameters set to default.
Prediction of Glycosylation Sites
The ScanProsite tool (De Castro et al., 2006) and NetNGlyc 1.0 Server (Gupta et al., 2004) were used to predict the putative glycosylation sites of PLB_Bm. All the parameters were set to default.
Disulfide Bond Prediction
The disulfide bridges in PLB_Bm were checked using the DiANNA webserver (Ferrè and Clote, 2006) and Dinosolve (Darden et al., 1993; DeLano, 2002; Anandakrishnan et al., 2012; Yaseen and Li, 2013; Maier et al., 2015).
Homology Model Building of PLB_Bm
The 3D structure model of PLB_Bm was produced using various protein modeling programs, like I-TESSER (Roy et al., 2010), the MODELLER 9v19 program (Colovos and Yeates, 1993), and the SWISS Model (Waterhouse et al., 2018). The atomic coordinates of Phospholipase B-like Protein 1 from bovine kidneys (PDB ID: 4BWC) that display 70% amino acid sequence identity with PLB_Bm were used as a template (Repo et al., 2014). The best model was carefully chosen based on the quality and validation reports produced by PROCHECK (Webb and Sali, 2016).
Molecular Dynamics Simulation
The validation of the modeled 3D structure of PLB_Bm was carried out through MD simulation using the programs like GROMACS (Berendsen et al., 1995), MDMoby (Hospital et al., 2012), AMBER16 (Maier et al., 2015), and MDweb (Hospital et al., 2012). The FF14SB force field (Darden et al., 1993) was used for all-atom–protein interaction. The protonation states of the amino acid side chain were determined using the web server H++ (Anandakrishnan et al., 2012) at pH 7.0. The system was neutralized with chloride ions, was placed in a rectangular box of TIP3P water, and extended to at least 15 Å from any protein atom. The bad contact from the modeled structure was removed by energy minimization of the system for 500 conjugate gradients steps using a constant force constraint of 15 kcal/mol.Å2. The gradual heating of the system was carried out from 0 to 300 K for 250 ps with a constant atom number, volume, and temperature (NVT) ensemble. The protein was maintained with a constant force of 10 kcal/mol.Å2. The equilibration step was achieved using the constant atom number, pressure, and temperature (NPT) ensemble for 500 ps. The simulation was carried out for 100 ns with a 4-fs time step. The pressure and temperature were kept constant at 1 atm and 300 K, respectively, by Langevin coupling. The particle-mesh Ewald (PME) method (Darden et al., 1993) was used to compute the long-range electrostatic interactions by keeping the cutoff distance of 10 Å to Van der Waals interactions.
Model Validation
The PROCHECK software (Laskowski et al., 1993, 2001), ERRAT version 2.0 (Colovos and Yeates, 1993), and Verify 3D (Bowie et al., 1991; Lüthy et al., 1992) were used for validation of the built 3D model of PLB_Bm.
Structure Superimposition
The PyMOL molecular graphics visualization program (DeLano, 2002) was used to align the build PLB_Bm model to other homologous proteins from the Protein Data Bank.
Surface Charge Analysis
The PDB2PQR server program (Dolinsky et al., 2004) was used for charge and radius calculations; and the ABPS Tools from PyMOL was used for surface and charge visualization of the protein (DeLano, 2002).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
AU designed the project and reviewed the manuscript. RM drafted and thoroughly checked it. Both authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2020.00175/full#supplementary-material
Footnotes
References
Aird, S. D., da Silva, N. J., Qiu, L., Villar-Briones, A., Saddi, V. A., de Campos Telles, M. P., et al. (2017). Coralsnake venomics: analyses of venom gland transcriptomes and proteomes of six Brazilian taxa. Toxins 9:187. doi: 10.3390/toxins9060187
Aird, S. D., Watanabe, Y., Villar-Briones, A., Roy, M. C., Terada, K., and Mikheyev, A. S. (2013). Quantitative high-throughput profiling of snake venom gland transcriptomes and proteomes (Ovophis okinavensis and Protobothrops flavoviridis). BMC Genomics 14:790. doi: 10.1186/1471-2164-14-790
Amorim, F. G., Morandi-Filho, R., Fujimura, P. T., Ueira-Vieira, C., and Sampaio, S. V. (2017). New findings from the first transcriptome of the Bothrops moojeni snake venom gland. Toxicon 140, 105–117. doi: 10.1016/j.toxicon.2017.10.025
Anandakrishnan, R., Aguilar, B., and Onufriev, A. V. (2012). H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulation. Nucleic Acids Res. 40, W537–W541.
Bendtsen, J. D., Nielsen, H., von Heijne, G., and Brunak, S. (2004). Improved prediction of signal peptides: signalP 3.0. J. Mol. Biol. 340, 783–795. doi: 10.1016/j.jmb.2004.05.028
Berendsen, H. J. C., van der Spoel, D., and van Drunen, R. (1995). GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56. doi: 10.1016/0010-4655(95)00042-e
Bernheimer, A. W., Linder, R., Weinstein, S. A., and Kim, K. S. (1987). Isolation and characterization of a phospholipase B from venom of Collett’s snake, Pseudechis colletti. Toxicon 25, 547–554. doi: 10.1016/0041-0101(87)90290-x
Bernheimer, A. W., Weinstein, S. A., and Linder, R. (1986). Isoelectric analysis of some Australian elapid snake venoms with special reference to phospholipase B and hemolysis. Toxicon 24, 841–849. doi: 10.1016/0041-0101(86)90109-1
Bowie, J., Luthy, R., and Eisenberg, D. (1991). A method to identify protein sequences that fold into a known three-dimensional structure. Science 253, 164–170. doi: 10.1126/science.1853201
Carugo, O. (2018). How large B-factors can be in protein crystal structures. BMC Bioinformatics 19:61. doi: 10.1186/s12859-018-2083-8
Case, D. A., Cheatham, T. E., Darden, T., Gohlke, H., Luo, R., Merz, K. M., et al. (2005). The Amber biomolecular simulation programs. J. Computat. Chem. 26, 1668–1688. doi: 10.1002/jcc.20290
Chapeaurouge, A., Reza, M. A., Mackessy, S. P., Carvalho, P. C., Valente, R. H., Teixeira-Ferreira, A., et al. (2015). Interrogating the venom of the viperid snake sistrurus catenatus edwardsii by a combined approach of electrospray and MALDI mass spectrometry. PLoS One. 10:e0092091. doi: 10.1371/journal.pone.0092091
Chatrath, S. T., Chapeaurouge, A., Lin, Q., Lim, T. K., Dunstan, N., Mirtschin, P., et al. (2011). Identification of novel proteins from the venom of a cryptic snake Drysdalia coronoides by a combined transcriptomics and proteomics approach. J. Proteome Res. 10, 739–750. doi: 10.1021/pr1008916
Colovos, C., and Yeates, T. O. (1993). Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 2, 1511–1519. doi: 10.1002/pro.5560020916
Contardi, A., and Ercoli, A. (1933). Enzymatic cleavage of lecithin and lyso-lecithin. Biochem. Z. 261:275–302.
Crooks, G. E., Hon, G., Chandonia, J. M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
Darden, T., York, D., and Pedersen, L. (1993). Particle mesh Ewald: an N log (N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. doi: 10.1063/1.464397
De Castro, E., Sigrist, C. J. A., Gattiker, A., Bulliard, V., Langendijk-Genevaux, P. S., Gasteiger, E., et al. (2006). ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365.
Doery, H. M., and Pearson, J. E. (1964). Phospholipase B in snake venoms and bee venom. Biochem. J. 92, 599–602. doi: 10.1042/bj0920599
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A., and Baker, N. A. (2004). PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 32, W665–W667.
Fairbairn, D. (1948). The preparation and properties of a lysophospholipase from Penicillium notatum. J. Biol. Chem. 173, 705–714.
Ferrè, F., and Clote, P. (2006). DiANNA 1.1: an extension of the DiANNA web server for ternary cysteine classification. Nucleic Acids Res. 34(Suppl. 2), W182–W185.
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D., et al. (2005). “Protein identification and analysis tools on the ExPASy server,” in The Proteomics Protocols Handbook, ed. J. M. Walker (Totowa, NJ: Humana Press), 571–607. doi: 10.1385/1-59259-890-0:571
Gupta, R., Jung, E., and Brunak, S. (2004). Prediction of N-glycosylation Sites in Human Proteins. Available online: http://www.cbs.dtu.dk/services/NetNGlyc/ (accessed December 2, 2019).
Hospital, A., Andrio, P., Fenollosa, C., Cicin-Sain, D., Orozco, M., and Gelpí, J. L. (2012). MDWeb and MDMoby: an integrated web-based platform for molecular dynamics simulations. Bioinformatics 28, 1278–1279. doi: 10.1093/bioinformatics/bts139
Jiménez-Charris, E., Montealegre-Sanchez, L., Solano-Redondo, L., Mora-Obando, D., Camacho, E., Castro-Herrera, F., et al. (2015). Proteomic and functional analyses of the venom of Porthidium lansbergii lansbergii (Lansberg’s hognose viper) from the atlantic department of Colombia. J. Proteomics 114, 287–299. doi: 10.1016/j.jprot.2014.11.016
Jones, B. K., Saviola, A. J., Reilly, S. B., Stubbs, A. L., Arida, E., Iskandar, D. T., et al. (2019). Venom composition in a phenotypically variable pit viper (Trimeresurus insularis) across the Lesser Sunda Archipelago. J. Proteome. Res. 18, 2206–2220. doi: 10.1021/acs.jproteome.9b00077
Khan, M. A., and Hodgson, E. (1967). Phospholipase activity in Musca domestica L. Comp. Biochem. Physiol. 23, 899–910. doi: 10.1016/0010-406x(67)90350-7
Kovalchuk, S. I., Ziganshin, R. H., Starkov, V. G., Tsetlin, V. I., and Utkin, Y. N. (2016). Quantitative proteomic analysis of venoms from russian vipers of pelias group: phospholipases A2 are the main venom components. Toxins 8:105. doi: 10.3390/toxins8040105
Lakomek, K., Dickmanns, A., Kettwig, M., Urlaub, H., Ficner, R., and Luebke, T. (2009). Initial insight into the function of the lysosomal 66.3 kDa protein from mouse by means of X-ray crystallography. BMC Struct. Biol. 9:56. doi: 10.1186/1472-6807-9-56
Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. doi: 10.1107/s0021889892009944
Laskowski, R. A., MacArthur, M. W., and Thornton, J. M. (2001). “PROCHECK: validation of protein structure coordinates,” in International Tables of Crystallography: Volume F: Crystallography of Biological Macromolecules, eds M. G. Rossmann and E. Arnold (Dordrecht: Kluwer Academic Publishers), 722–725.
Sousa, L. F., Nicolau, C. A., Peixoto, P. S., Bernardoni, J. L., Oliveira, S. S., Portes-Junior, J. A., et al. (2013). Comparison of phylogeny, venom composition and neutralization by antivenom in diverse species of bothrops complex. PLoS Negl. Trop. Dis. 7:e2442. doi: 10.1371/journal.pntd.0002442
Lovell, S. C., Davis, I. W., Arendall, W. B., de Bakker, P. I. W., Word, J. M., Prisant, M. G., et al. (2003). Structure validation by Calpha geometry: phi, psi and C beta deviation. Proteins 50, 437–450.
Lüthy, R., Bowie, J. U., and Eisenberg, D. (1992). Assessment of protein models with three-dimensional profiles. Nature 356, 83–85. doi: 10.1038/356083a0
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., and Simmerling, C. (2015). ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713. doi: 10.1021/acs.jctc.5b00255
Marcon, F., Purtell, L., Santos, J., Hains, P. G., Escoubas, P., Graudins, A., et al. (2013). Characterization of monomeric and multimeric snake neurotoxins and other bioactive proteins from the venom of the lethal Australian common copperhead (Austrelaps superbus). Biochem. Pharmacol. 85, 1555–1573. doi: 10.1016/j.bcp.2013.02.034
Margres, M. J., Aronow, K., Loyacano, J., and Rokyta, D. R. (2013). The venom-gland transcriptome of the eastern coral snake (Micrurus fulvius) reveals high venom complexity in the intragenomic evolution of venoms. BMC Genomics. 14:531. doi: 10.1186/1471-2164-14-531
Matsumoto, Y., Mineta, S., Murayama, K., and Sugimori, D. (2013). A novel phospholipase B from Streptomyces sp. NA684–purification, characterization, gene cloning, extracellular production and prediction of the catalytic residues. FEBS J. 280, 3780–3796. doi: 10.1111/febs.12366
Méndez, R., Bonilla, F., Sasa, M., Dwyer, Q., Fernández, J., and Lomonte, B. (2019). Proteomic profiling, functional characterization, and immunoneutralization of the venom of Porthidium porrasi, a pitviper endemic to Costa Rica. Acta Trop. 193, 113–123. doi: 10.1016/j.actatropica.2019.02.030
Mohamed, A. H., Kamel, A., and Ayobe, M. H. (1969). Studies of phospholipase A and B activities of Egyptian snake venoms and a scorpion toxin. Toxicon 6, 293–298. doi: 10.1016/0041-0101(69)90099-3
Morgan, C. P., Insall, R., Haynes, L., and Cockcroft, S. (2004). Identification of phospholipase B from Dictyostelium discoideum reveals a new lipase family present in mammals, flies and nematodes, but not yeast. Biochem. J. 382(Pt 2), 441–449. doi: 10.1042/bj20040110
Oinonen, C., and Rouvinen, J. (2000). Structural comparison of Ntn-hydrolases. Protein Sci. 9, 2329–2337. doi: 10.1110/ps.9.12.2329
Oliveira, I. S., Cardoso, I. A., Bordon, K. C. F., Carone, S. E. I., Boldrini-França, J., Berto Pucca, M., et al. (2019). Global proteomic and functional analysis of Crotalus durissus collilineatus individual venom variation and its impact on envenoming. J. Proteomics. 191, 153–165. doi: 10.1016/j.jprot.2018.02.020
Paetzel, M., Karla, A., Strynadka, N. C., and Dalbey, R. E. (2002). Signal peptidases. Chem Rev. 102, 4549–4580.
Patra, A., Kalita, B., Chanda, A., and Mukherjee, A. K. (2017). Proteomics and antivenomics of Echis carinatus carinatus venom: correlation with pharmacological properties and pathophysiology of envenomation. Sci. Rep. 7:17119.
Rao, R. H., and Subrahmanyam, D. (1969). Characterization of phospholipase B of Culex pipiens fatigans. J. Lipid Res. 10, 636–641.
Repo, H., Kuokkanen, E., Oksanen, E., Goldman, A., and Heikinheimo, P. (2014). Is the bovine lysosomal phospholipase B-like protein an amidase? Proteins 82, 300–311. doi: 10.1002/prot.24388
Rokyta, D. R., Wray, K. P., Lemmon, A. R., Lemmon, E. M., and Caudle, S. B. (2011). A high-throughput venom-gland transcriptome for the eastern diamondback rattlesnake (Crotalus Adamanteus) and evidence for pervasive positive selection across toxin classes. Toxicon 57, 657–671. doi: 10.1016/j.toxicon.2011.01.008
Roy, A., Kucukural, A., and Zhang, Y. (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738. doi: 10.1038/nprot.2010.5
Saito, K. (2014). Reminiscence of phospholipase B in Penicillium notatum. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 90, 333–346. doi: 10.2183/pjab.90.333
Salomon-Ferrer, R., Case, D. A., and Walker, R. C. (2013). An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 3, 198–210. doi: 10.1002/wcms.1121
Schneider, T. D., and Stephens, R. M. (1990). Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100. doi: 10.1093/nar/18.20.6097
Shiloah, J., Klibansky, C., de Vries, A., and Berger, A. (1973). Phospholipase B activity of a purified phospholipase A from Vipera palestinae venom. J. Lipid. Res. 14, 267–278.
Takasaki, C., and Tamiya, N. (1982). Isolation and properties of lysophospholipases from the venom of an Australian elapid snake, Pseudechis australis. Biochem. J. 203, 269–276. doi: 10.1042/bj2030269
Tan, K. Y., Tan, C. H., Chanhome, L., and Tan, N. H. (2017). Comparative venom gland transcriptomics of Naja kaouthia (monocled cobra) from Malaysia and Thailand: elucidating geographical venom variation and insights into sequence novelty. PeerJ 5:e3142. doi: 10.7717/peerj.3142
Tang, E. L., Tan, C. H., Fung, S. Y., and Tan, N. H. (2016). Venomics of Calloselasma rhodostoma, the Malayan pit viper: a complex toxin arsenal unraveled. J. Proteomics 148, 44–56. doi: 10.1016/j.jprot.2016.07.006
Tang, E. L. H., Tan, N. H., Fung, S. Y., and Tan, C. H. (2019). Comparative proteomes, immunoreactivities and neutralization of procoagulant activities of Calloselasma rhodostoma (Malayan pit viper) venoms from four regions in Southeast Asia. Toxicon 169, 91–102. doi: 10.1016/j.toxicon.2019.08.004
Ullah, A. (2020). Structure-function studies and mechanism of action of snake venom L-Amino acid oxidases. Front. Pharmacol. 11:110. doi: 10.3389/fphar.2020.00110
Ullah, A., Masood, R., Ali, I., Ullah, K., Ali, H., Akbar, H., et al. (2018). Thrombin-like enzymes from snake venom: Structural characterization and mechanism of action. Int. J. Biol. Macromol. 114, 788–811.
Ullah, A., Masood, R., Hayat, Z., and Hafeez, A. (2020). Determining the structures of the snake and spider toxins by x-rays. Methods Mol. Biol. 2068, 163–172. doi: 10.1007/978-1-4939-9845-6_8
Ullah, A., Ullah, K., Ali, H., Betzel, C., and Ur Rehman, S. (2019). The sequence and a three-dimensional structural analysis reveal substrate specificity among snake venom phosphodiesterases. Toxins (Basel). 11:625. doi: 10.3390/toxins11110625
Vanuopadath, M., Sajeev, N., Murali, A. R., Sudish, N., Kangosseri, N., Sebastian, I. R., et al. (2018). Mass spectrometry-assisted venom profiling of Hypnale hypnale found in the Western Ghats of India incorporating de novo sequencing approaches. Int. J. Biol. Macromol. 118(Pt B), 1736–1746. doi: 10.1016/j.ijbiomac.2018.07.016
Verma, S., Dixit, R., and Pandey, K. C. (2016). Cysteine proteases: modes of activation and future prospects as pharmacological targets. Front. Pharmacol. 7:107. doi: 10.3389/fphar.2016.00107
Viala, V. L., Hildebrand, D., Trusch, M., Arni, R. K., Pimenta, D. C., Schlüter, H., et al. (2014). Pseudechis guttatus venom proteome: insights into evolution and toxin clustering. J. Proteomics 110, 32–44. doi: 10.1016/j.jprot.2014.07.030
Wang, Y., Wang, J., Li, R., Shi, Q., Xue, Z., and Zhang, Y. (2017). ThreaDomEx: a unified platform for predicting continuous and discontinuous protein domains by multiple-threading and segment assembly. Nucleic Acids Res. 45, W400–W407.
Wass, M. N., Kelley, L. A., and Sternberg, M. J. (2010). 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res. 38, W469–W473.
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303.
Webb, B., and Sali, A. (2016). Comparative protein structure modeling using modeller. Curr. Protoc. Protein Sci. 86, 2.9.1–2.9.37.
Wiezel, G. A., dos Santos, P. K., Cordeiro, F. A., Bordon, K. C., Selistre-de-Araújo, H. S., Ueberheide, B., et al. (2015). Identification of hyaluronidase and phospholipase B in Lachesis muta rhombeata venom. Toxicon 107(Pt B), 359–368. doi: 10.1016/j.toxicon.2015.08.029
Xue, Z., Xu, D., Wang, Y., and Zhang, Y. (2013). ThreaDom: extracting protein domain boundary information from multiple threading alignments. Bioinformatics 29, i247–i256. doi: 10.1093/bioinformatics/btt209
Yaseen, A., and Li, Y. (2013). Dinosolve: a protein disulfide bonding prediction server using context-based features to enhance prediction accuracy. BMC Bioinform. 14:S9. doi: 10.1186/1471-2105-14-S13-S9
Keywords: snake venom phospholipases B, sequence and three-dimensional structure analysis, glycosylation, structural comparison, structure-based substrate specificity and maturation
Citation: Ullah A and Masood R (2020) The Sequence and Three-Dimensional Structure Characterization of Snake Venom Phospholipases B. Front. Mol. Biosci. 7:175. doi: 10.3389/fmolb.2020.00175
Received: 06 May 2020; Accepted: 06 July 2020;
Published: 05 August 2020.
Edited by:
Annalisa Pastore, King’s College London, United KingdomReviewed by:
Piero Andrea Temussi, University of Naples Federico II, ItalyDelia Picone, University of Naples Federico II, Italy
Copyright © 2020 Ullah and Masood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anwar Ullah, YW53YXJ1bGxhaEBjb21zYXRzLmVkdS5waw==; YW53YXIubXM5MEB5YWhvby5jb20=