 Irene Vanni1,2†
Irene Vanni1,2† Enrica Teresa Tanda3†
Enrica Teresa Tanda3† Bruna Dalmasso1,2
Bruna Dalmasso1,2 Lorenza Pastorino1,2
Lorenza Pastorino1,2 Virginia Andreotti1,2
Virginia Andreotti1,2 William Bruno1,2
William Bruno1,2 Andrea Boutros3
Andrea Boutros3 Francesco Spagnolo3
Francesco Spagnolo3 Paola Ghiorzo1,2*
Paola Ghiorzo1,2*- 1Genetics of Rare Cancers, IRCCS Ospedale Policlinico San Martino, Genova, Italy
- 2Genetics of Rare Cancers, Department of Internal Medicine and Medical Specialties, University of Genoa, Genova, Italy
- 3Medical Oncology, IRCCS Ospedale Policlinico San Martino, Genova, Italy
Melanoma is one of the most aggressive tumors of the skin, and its incidence is growing worldwide. Historically considered a drug resistant disease, since 2011 the therapeutic landscape of melanoma has radically changed. Indeed, the improved knowledge of the immune system and its interactions with the tumor, and the ever more thorough molecular characterization of the disease, has allowed the development of immunotherapy on the one hand, and molecular target therapies on the other. The increased availability of more performing technologies like Next-Generation Sequencing (NGS), and the availability of increasingly large genetic panels, allows the identification of several potential therapeutic targets. In light of this, numerous clinical and preclinical trials are ongoing, to identify new molecular targets. Here, we review the landscape of mutated non-BRAF skin melanoma, in light of recent data deriving from Whole-Exome Sequencing (WES) or Whole-Genome Sequencing (WGS) studies on melanoma cohorts for which information on the mutation rate of each gene was available, for a total of 10 NGS studies and 992 samples, focusing on available, or in experimentation, targeted therapies beyond those targeting mutated BRAF. Namely, we describe 33 established and candidate driver genes altered with frequency greater than 1.5%, and the current status of targeted therapy for each gene. Only 1.1% of the samples showed no coding mutations, whereas 30% showed at least one mutation in the RAS genes (mostly NRAS) and 70% showed mutations outside of the RAS genes, suggesting potential new roads for targeted therapy. Ongoing clinical trials are available for 33.3% of the most frequently altered genes.
Introduction
Cutaneous melanoma is one of the most aggressive malignancies of the skin. Its incidence is globally growing partly due to the increase of early diagnoses, and contextually, the prevalence is also increasing (Bray et al., 2018; Schadendorf et al., 2018). Until 10 years ago, advanced melanoma was associated with poor survival due to the lack of durable responses to conventional chemotherapy and biochemotherapy (Korn et al., 2008), with a median Overall Survival (OS) of about 6 month in patients with stage IV melanoma. Since 2011, however, the rules of the treatment of stage IV melanoma have been completely rewritten, with the introduction of targeted therapies with BRAF and MEK inhibitors (Larkin et al., 2014; Long et al., 2014; Robert et al., 2016), and immunotherapy with the anti CTLA-4 ipilimumab (Hodi et al., 2010) and the anti-PD-1 nivolumab (Robert et al., 2015) and pembrolizumab (Schachter et al., 2017). These new therapeutic approaches improved melanoma prognosis, resulting in a 5-year survival rate of 34–43% (Hamid et al., 2019; Robert et al., 2019). However, mainly because of primary and acquired resistance to treatments, the majority of patients will ultimately relapse, and only patients harboring a BRAF mutation, observed in about 50% of cutaneous melanoma, can receive a targeted treatment with BRAF and MEK inhibitors (Spagnolo et al., 2015). The current state of molecular-target drugs and the current therapeutic scenario for patients with BRAF mutated melanoma, from the introduction of BRAF inhibitors as single agents to modern clinical practice, has been extensively described in a related minireview (Tanda et al., 2020). With the purpose of further improving the prognosis of melanoma patients, several preclinical and clinical trials are studying new actionable mechanisms and/or molecules, to simultaneously tackle multiple resistance mechanisms.
The aim of this review is to describe the landscape of mutated non-BRAF melanoma, in light of recent data deriving from Next-Generation Sequencing (NGS) (or Massive Parallel Sequencing – MPS) analysis, focusing on available, or in experimentation, targeted therapies. The advent of MPS, allowing the simultaneous analysis of several genes, led, in the past two decades, to Whole-Exome Sequencing (WES) and Whole-Genome Sequencing (WGS) studies that found several mutated genes in human cancers. The evolution of molecular testing in melanoma, as well as the main techniques and MPS platforms currently in use for BRAF mutation testing, have been recently reviewed (Vanni et al., 2020).
The first actionable mutation to be targeted by specific drugs in melanoma, BRAF V600, was found in 2002 along several other drivers of human cancers (Davies et al., 2002). Since then, several other genes have been identified as putative drivers of melanomagenesis and/or melanoma progression, and additional candidate drivers are currently being assessed, prompting pharmacogenomics studies on potentially actionable targets (Priestley et al., 2019). However, melanoma is one of the tumors with the highest mutation burden, and results from different studies were frequently not overlapping, possibly due to dissimilar sample size and cohort characteristics (Berger et al., 2012; Hodis et al., 2012; Krauthammer et al., 2012; Snyder et al., 2014; Van Allen et al., 2015). Although this high mutational burden is one of the reason behind the success of immunotherapy in this tumor, it makes it hard to clearly identify novel driver genes that could be used for targeted therapies (Davis et al., 2018).
In 2015, The Cancer Genome Atlas analyzed 333 cutaneous melanoma samples by integrating integrated multi-level genomic analyses, namely WES and low-pass WGS, transcriptome sequencing including miRNA, protein expression, and classified melanoma in four major molecular subtypes: mutant BRAF, mutant RAS, mutant NF1 and triple wild-type (The Cancer Genome Atlas Network, 2015). However, as NF1 mutations can be found in melanomas with concurrent BRAF or NRAS hotspot mutation, a three-group classification of melanoma (mutant BRAF, mutant RAS, non-BRAFmut /non-NRASmut) has been proposed (Palmieri et al., 2018). Although providing an unprecedented insight into the complex mutational spectrum of melanoma, the TCGA study cohort did not include acral and mucosal melanomas. Two years later, this issue was addressed with the molecular characterization of 183 melanoma samples through WGS, including the acral and mucosal subtypes (Hayward et al., 2017). Recently, a joint effort by the TCGA and the ICGC resulted in the description of the molecular spectrum of the largest whole genome dataset of 38 different tumor types, which included a subset of 118 melanoma samples previously described (ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, 2020).
The spectrum of genomic alterations in melanoma involve multiple genes and signaling networks, but the most frequently altered pathways are MAPK, PIK3CA, KIT signaling, and apoptosis/cell senescence pathways (Figure 1). This review focuses on cutaneous melanoma, including the acral melanoma. To obtain an overview of molecular alterations in skin melanoma, we focused our analysis on all WES or WGS studies on melanoma cohorts or pan-cancer cohorts that included melanoma, for which information on the mutation rate of each gene was available. For selecting TCGA melanoma samples we considered the group’s pan-cancer flagship paper (Hoadley et al., 2018), which included the original cohort of the melanoma-only TCGA study, plus an additional set of 60 skin melanoma samples. With these criteria we collected and combined mutational data from 10 studies published from 2012 to 2019, available from either the cBioportal repository (cBioPortal for Cancer Genomics, 2020) or from the tables within the published manuscripts (Hayward et al., 2017; Birkeland et al., 2018; Kontogianni et al., 2018; Wilmott et al., 2019), to obtain cumulative mutational frequencies of all genes assessed by these studies (Table 1) (Berger et al., 2012; Hodis et al., 2012; Krauthammer et al., 2012; Snyder et al., 2014; Van Allen et al., 2015; Hayward et al., 2017; Kontogianni et al., 2018; Wilmott et al., 2019; cBioPortal for Cancer Genomics, 2020). When present, we filtered out uveal and mucosal melanoma samples, as well as synonymous variants. Moreover, only three studies used for this review were performed with WGS (Berger et al., 2012; Hodis et al., 2012; Hayward et al., 2017; Wilmott et al., 2019), while all the others are WES studies. Non-coding regions were not considered for our analysis, except for TERT promoter, whose frequency was calculated on data from a single WGS study (Hayward et al., 2017). Similarly, we used the WGS subset to assess Copy Number Variations (CNVs). An overview of the studies analyzed for this review is found in Table 1.
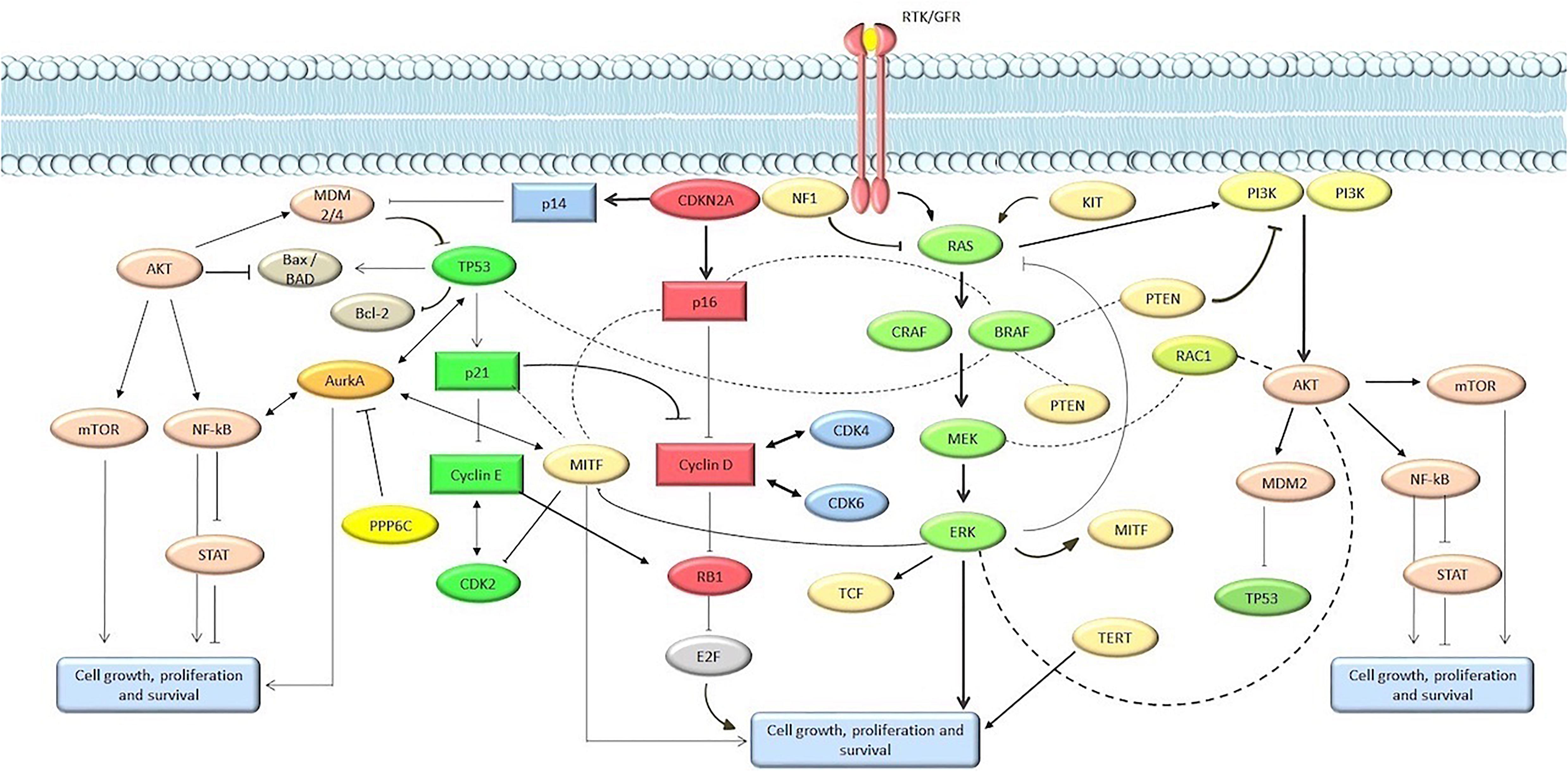
Figure 1. Main pathways involved in melanomagenesis. Genes and proteins are marked in circle and rectangles, respectively.
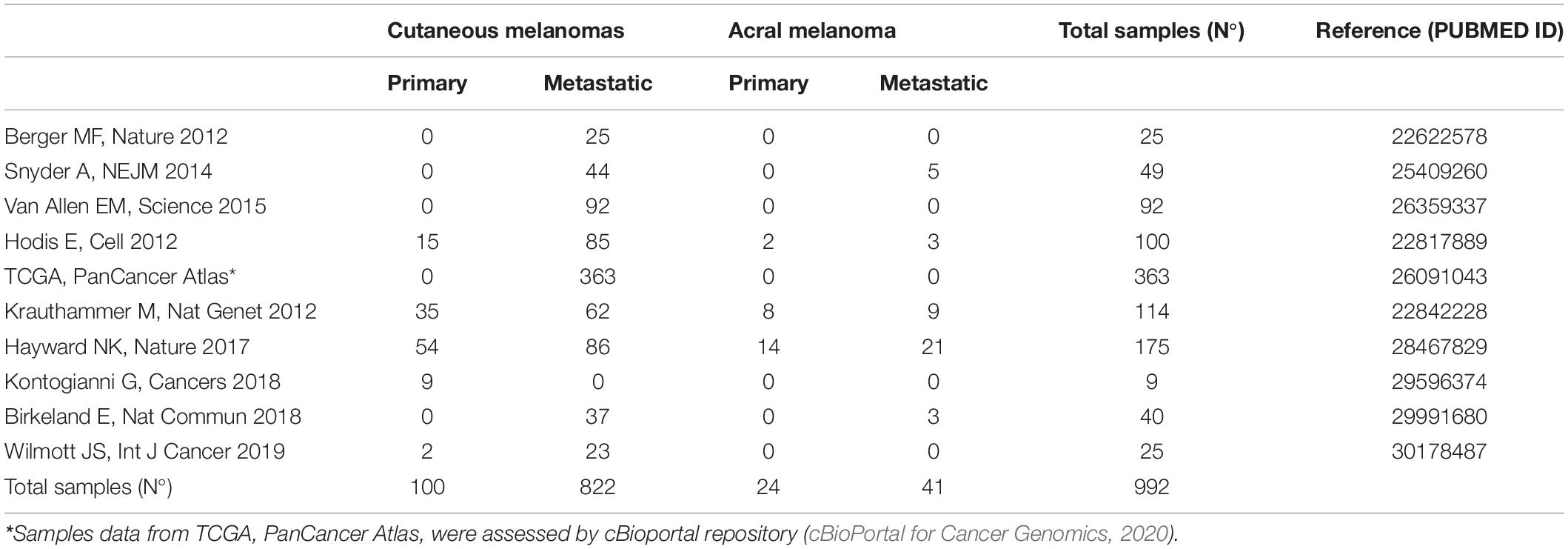
Table 1. Skin melanomas samples from the 10 NGS-based studies evaluated for the mutational and CNV frequency in the present report.
In the following sections, we provide a description of 33 selected established and candidate melanoma driver genes, as well as the mutational and CNV frequency of each gene (Figures 2, 3) in the melanoma samples analyzed, and we describe available, or in experimentation, targeted therapies for each gene/pathway, excluding immunotherapy. Mutational frequency of these genes in melanoma across each study included in the review is displayed in Supplementary Figures S1–S10; ongoing targeted clinical trials are displayed in Table 2.
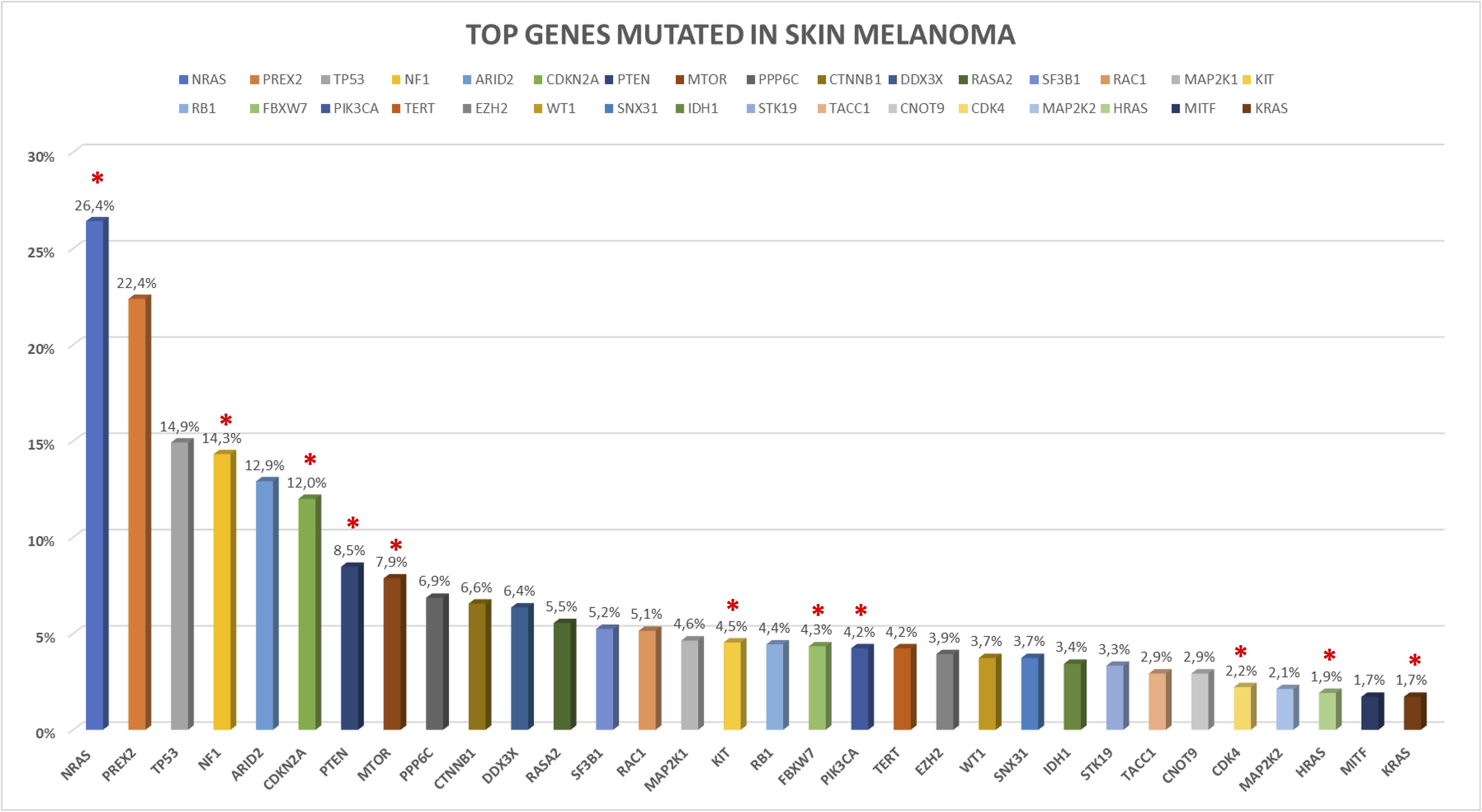
Figure 2. Somatic mutations frequency in top melanoma driver cancer genes based on 992 skin melanoma samples derived by the 10 selected NGS studies. Somatic coding mutations were considered for the analysis excluding synonymous variants. Red asterisks indicate the presence of clinical trials for the altered gene.
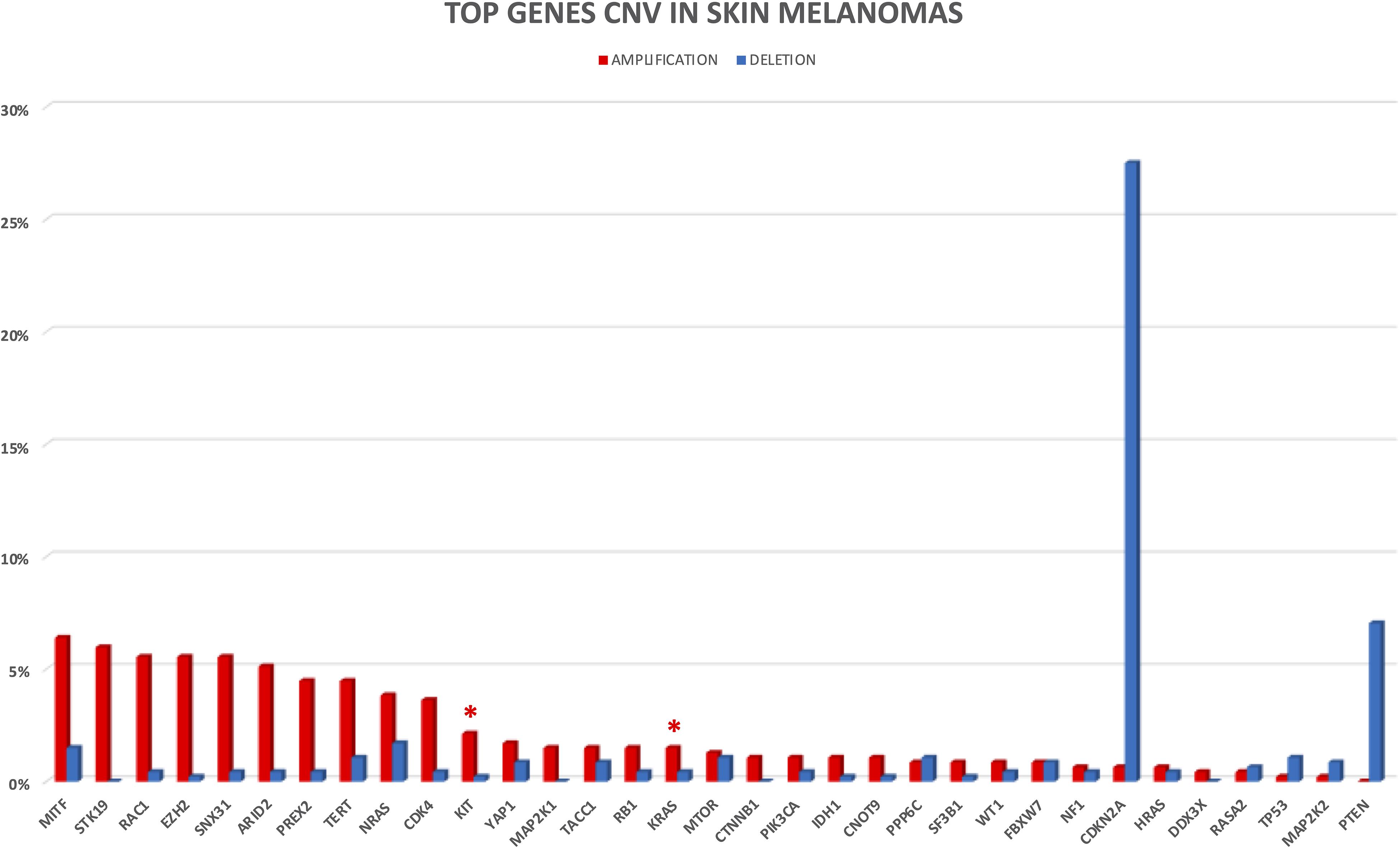
Figure 3. CNV frequency in top melanoma driver cancer genes. Amplifications and deletions are reported in red and blue, respectively. Only four studies with available CNV information were considered for CNV analysis. Red asterisks indicate the presence of clinical trials for the altered gene.
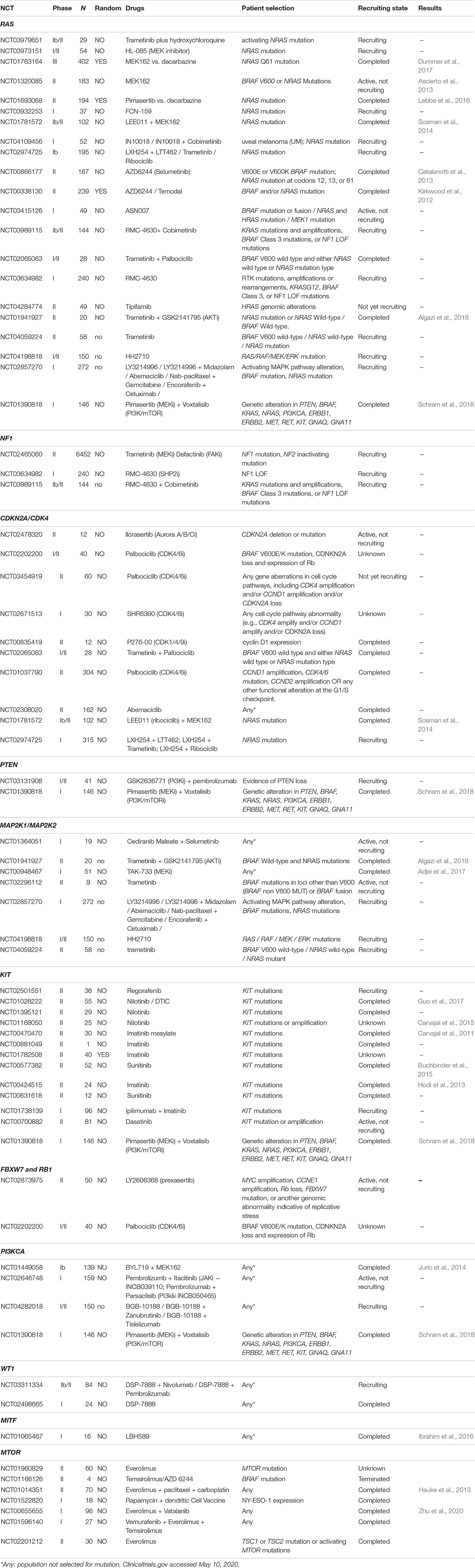
Table 2. Clinical trials.
Moreover, for each of the 33 genes included in this review and listed here by mutational frequency and/or pathway, we also provide molecular function, frequency alterations (mutations plus CNV) in our dataset, and ranked their effect on melanoma according to the DisGeNET Gene–Disease Association (GDA) Score (Piñero et al., 2015) (Figure 4).
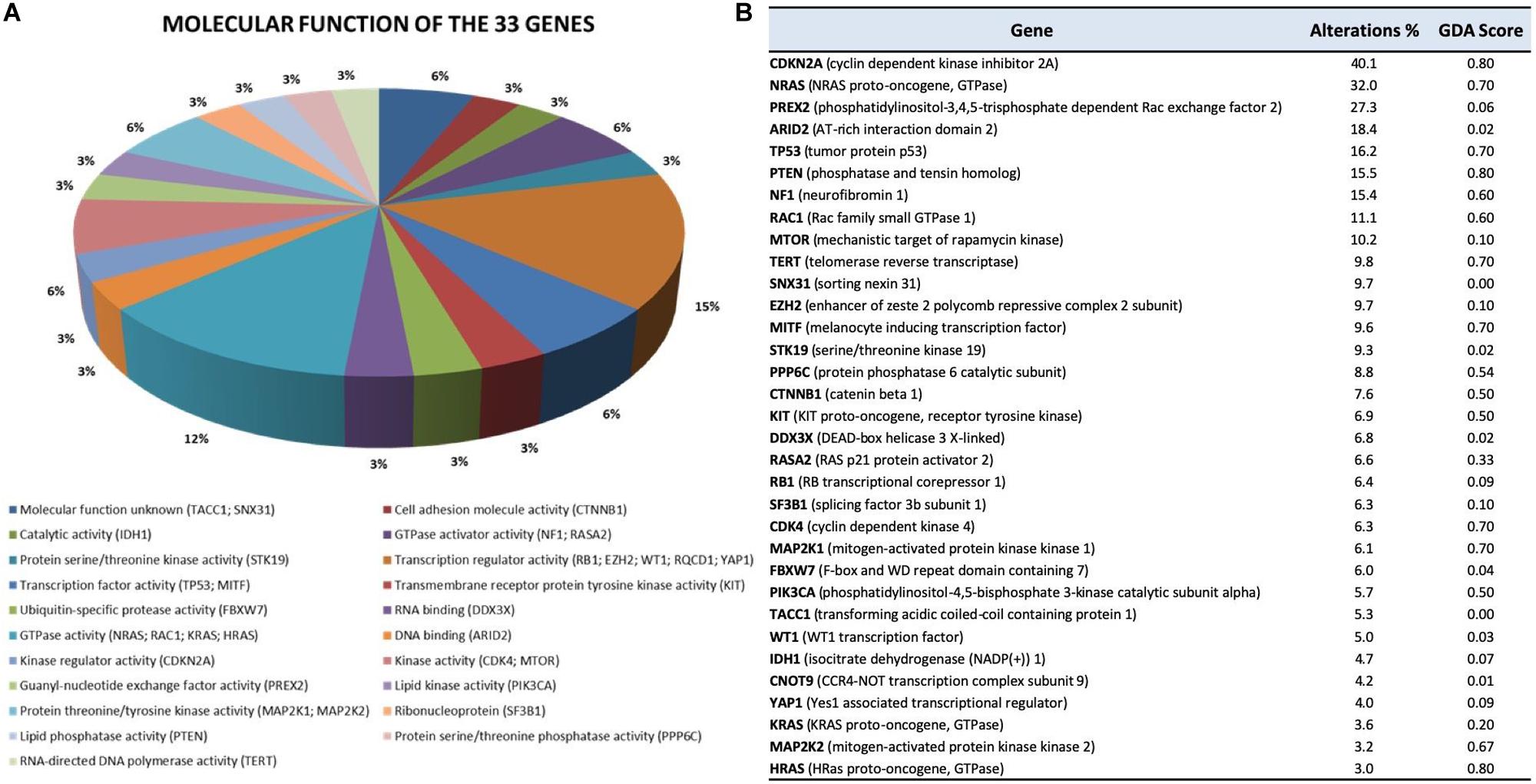
Figure 4. Molecular function (A) and ranking (B) of the 33 genes analyzed. (A) Gene Ontology (GO) molecular function of the 33 genes. GO molecular function was obtained using FunRich v3.1.3, a stand-alone software tool (Pathan et al., 2015). (B) The table reports the frequency of genomic alterations (mutations and CNV) resulting from the 10 selected NGS studies, as well as the Gene–Disease Association (GDA) Score obtained from the DisGeNET (Piñero et al., 2015) database using the keyword “melanoma” (C0025202 result). The GDA Score is computed by integrating evidence from multiple sources. The higher the GDA Score, the more reliable the gene–disease relationship is.
RAS Genes (NRAS/KRAS/HRAS)
Ras is a superfamily of small GTPase proteins that regulate cell growth, survival, differentiation and play a key role in transmitting the signal from Receptor Tyrosine Kinases (RTK) to several downstream signaling pathways, in particularly MAPK (mitogen-activated protein kinase) and PI3K (phosphoinositide 3-kinase). The three RAS tissue specific isoforms (NRAS, neuroblastoma ras viral oncogene homolog; KRAS, Kirsten rat sarcoma viral oncogene homolog; HRAS, Harvey rat sarcoma viral oncogene homolog) are frequently mutated in cancers. In cutaneous melanoma, NRAS is mutated in about 15–30% of cases, while KRAS (∼2%) and HRAS (∼1%) play a minor role (Milagre et al., 2010). NRAS, KRAS, and HRAS, mutations and CNVs in our dataset are reported in Figures 2, 3 and Supplementary Figures S1A–C.
NRAS
The NRAS was the first oncogene identified in melanoma in 1984, and the second most prevalent after BRAF (mutation frequency of 30%) (The Cancer Genome Atlas Network, 2015). NRAS mutations primarily occur at position 61 (Q61R/K/L accounts for about 80%) and, less frequently, at positions 12 and 13 (G12/13 accounts for about 6%) (Bos, 1989; Lee et al., 2011). These mutations cause an altered GTPase activity that keeps NRAS activated: this always-on signal induces a constitutional activation of the whole MAPK pathway with cell proliferation, dysregulation of the cell cycle and activation of other pro-survival pathways (Hodis et al., 2012).
Melanoma patients harboring mutated NRAS display different characteristics compared to those harboring mutated BRAF: they are older (>55 years) and have a story of UV exposure, thicker primary tumors and a higher rate of mitosis. Several studies showed that NRAS mutations may result in an inferior clinical outcome with a shorter Melanoma-Specific Survival (MSS) (Devitt et al., 2011; Ellerhorst et al., 2011), although partially debated (Omholt et al., 2003; Akslen et al., 2005; Edlundh-Rose et al., 2006; Ugurel et al., 2007; Ellerhorst et al., 2011).
The high number of NRAS mutations in cutaneous melanoma did not allow the development of effective drugs: targeting directly NRAS remains a great challenge, and the target therapy for NRAS mutant melanoma is focused on MEK inhibitors.
KRAS
KRAS encodes for two proteins resulting from alternative splicing of exon 4, KRAS4A and KRAS4B. These proteins have different structures in their C-terminal region and use different mechanisms to localize to cellular membranes (Welman et al., 2000). KRAS4B is the most frequent in human cells and differs from KRAS4A for a C-terminal residue which allows to bind calmodulin and induce its phosphorylation by PKC (Villalonga et al., 2001; Sung et al., 2013). Binding between KRAS4B and calmodulin seems to determine drug resistance, facilitate tumorigenesis and express stem-like markers on the cell surface (Wang et al., 2015). KRAS mutations occur most commonly at codon 12 but also at 13 and 61 (Pylayeva-Gupta et al., 2011; Liu et al., 2019). Mutation of G12 interferes with GAP binding and GAP-stimulated GTP hydrolysis and represents ∼12% of all KRAS mutations (Tate et al., 2019; COSMIC, 2020). The mutation in G13 decreases GAP binding and its hydrolysis while mutation in codon 61 has a direct role in inhibiting the hydrolysis reaction (Ostrem and Shokat, 2016). KRAS mutations are found in 15–20% of cancers, mostly in colorectal and pancreatic adenocarcinomas (Chiosea et al., 2011; Hartman et al., 2012; Zhou et al., 2017; Román et al., 2018). In melanoma, KRAS mutations are rare (1.7% of our cases) occurring almost exclusively in codon G12 (Milagre et al., 2010). KRAS mutant remains undruggable.
HRAS
HRAS encodes for a GTPase involved in regulating cell division in response to growth factor stimulation (Wong-Staal et al., 1981). Mutations in HRAS cause cell overgrowth and are implicated in a variety of cancers (Rauen, 2007). HRAS is altered in 0.97% of all cancers and, rarely, in 1.5% of melanoma (AACR Project Genie Consortium, 2017). Moreover, HRAS has been shown to be mutated in Spitz nevi both by CNVs (12/102; 12%) and point mutations (8/12; 67%) (Bastian et al., 1999, 2000). The reason why mutations in HRAS lead to Spitz nevi is unclear but could be related to higher affinity for the PI3K-PKB/AKT pathway which would be able to drive the symmetrical overgrowth of cells with an epithelioid morphology without marked activation of the melanizing pathways (Ross et al., 2011). Spitz nevus should not be regarded as a precursor lesion of melanoma. HRAS mutation analysis seems to be useful in the differential diagnosis between Spitz nevus and Spitzoid melanoma, and the presence of HRAS mutations is a marker of benignity and/or favorable clinical outcome (Dimonitsas et al., 2018).
RAS Genes – A Clinical Approach
To date, therapeutic inhibition of RAS remains an unmet need. Indeed, there are no approved therapies that specifically target NRAS, KRAS or HRAS and RAS-mutated patients are usually treated with immunotherapy. However, several trials are ongoing (Table 2).
Several years ago in vitro studies had already shown that mutated NRAS melanoma cells were sensitive to MEK inhibitors (Solit et al., 2006). However, the efficacy of such drugs on mutated NRAS cells was lower than that observed in mutated BRAF cells, and this lower efficacy could be explained, at least in part, if we consider the complexity of the molecular pathways network involving RAS. From these observations, two hypotheses arose: first, that probably a therapy based on a single drug could be insufficient; second, that the best therapeutic possibility was to target RAS directly (Mandalà et al., 2014). Unfortunately, targeting directly RAS did not give the expected results. Indeed, acting on GTP binding pocket in RAS protein is difficult due to the excessive affinity between RAS and GTP (Baines et al., 2011). In the same way, the inhibition of farnesylation of a cysteine residue, a post translational modification necessary to RAS insertion to the plasma cell membrane, has proven ineffective. Due to these disappointing results, the therapeutic strategies for mutated NRAS melanoma reverted on MEK inhibitors.
Initially, the use of MEK inhibitors led to modest results, with an Overall Response Rate (ORR) of 10% and a high incidence of adverse events (Rinehart et al., 2004; LoRusso et al., 2010). Subsequently, the MEK 1/2 inhibitor selumetinib (AZD-6244) was developed (Yeh et al., 2007). Phase I trial enrolled 11 melanoma patients and showed promising results (Adjei et al., 2008, 142886); on this wave, phase II trials comparing selumetinib and chemotherapy (temozolomide and docetaxel) in BRAF-WT and NRAS-unselected melanoma patients were initiated. The results were unsatisfactory for both trials, with no difference in efficacy outcomes (Kirkwood et al., 2012; Gupta et al., 2014).
Later, other MEK inhibitors were developed. Binimetinib (MEK162) is an allosteric, selective, non-ATP competitive MEK 1/2 inhibitor.
In preclinical studies binimetinib inhibited the growth of NRAS and BRAF mutated melanoma tumor cells (Winski et al., 2010). In the subsequent phase II trial (Ascierto et al., 2013) MEK162 obtained an ORR of 14.5% and a Disease Control Rate (DCR) of 56%. Median Progression Free Survival (PFS) was 3.6 months, underlying a rapid development of resistance, and the median OS 12.2 months (van Herpen et al., 2014). Phase III trial (NEMO) (Dummer et al., 2016) compared the efficacy of MEK162 versus dacarbazine in 402 NRAS mutated, melanoma patients. The ORR was 15% in the binimetinib arm versus 7% in dacarbazine arm. Furthermore, binimetib significantly prolonged median PFS, with 2.8 months [3.9 in patients with normal lactate dehydrogenase (LDH)] versus 1.5 months, respectively [hazard ratio (HR), 0.62]. Interestingly, immunotherapy pretreated patients had a longer median PFS (5.5 months). However, no differences in terms of OS were noted, and binimetinib as single agent did not receive regulatory agencies approval for the treatment of NRAS-mutated melanoma.
Finally, pimasertib (PIM; AS703026) has been evaluated in a phase I trial with encouraging results. Phase II study enrolled 194 patients to be treated with pimasertib or dacarbazine. Results showed a significant benefit for pimasertib, with a median PFS of 13.0 versus 6.9 weeks and a DCR of 37.7% versus 26.6%. Unfortunately, no difference in OS was showed (8.9 vs. 10.6 months) (Lebbe et al., 2016).
Combination of MEK inhibition with other targets is currently being evaluated. Among all, the results of a combination trial performed on 14 patients treated with ribociclib (LEE001) and MEK162 are particularly interesting. Indeed, the combination obtained six partial response and six stable disease, with a DCR of 85% (Sosman et al., 2014).
As we mentioned before, NRAS has to undergo some post-translational modifications, like farnesylation (Konstantinopoulos et al., 2007). Farnesyl Transferase Inhibitors (FTIs), like lonafarnib, was developed in an attempt to exploit this phenomenon. FTIs inhibit the function of RAS and seem to be able to sensitizing melanoma cells to RTK inhibitors like sorafenib (Meier et al., 2009). Unfortunately, FTIs failed in clinical trials showing no efficacy against NRAS and KRAS-driven cancers (Smalley and Eisen, 2003; Konstantinopoulos et al., 2007; Niessner et al., 2011; Gajewski et al., 2012; Margolin et al., 2012; Cox et al., 2015). The reason of this failure seems to be due to the action of geranylgeranyltransferase I (GGTase I) in the alternative prenylation (Whyte et al., 1997). FTIs in combination with GGTase I inhibitors have been tested in clinical trials but turned out to be too toxic (Brock et al., 2016). Other approaches attempting to inhibit some post-translational modifications are currently being evaluated. However, the great limit remains the toxicity of these drugs and challenges in delivering siRNA using nanoparticles (Davis et al., 2010).
PREX2
The PREX2 gene plays oncogenic roles in human cancers, such as melanoma, since it is involved in PIK3CA-PTEN-AKT signaling pathway (Fine et al., 2009; Srijakotre et al., 2017). It has been demonstrated that ectopic expression of mutant PREX2 accelerates tumor formation of immortalized human melanocytes in vivo (Berger et al., 2012). The upregulation of PREX2 and its mRNA increasing expression led to the AKT activation by PTEN phosphorylation and increases tumor proliferation. PREX2 non-synonymous variants have been found in 44.0% of a 25 patients cohort and in 14.0% of a 107 melanoma samples validation cohort (Berger et al., 2012). Notably, the truncating mutation PREX2 E824∗ was further studied to determine its in vivo implications in the context of mutant NRAS (Lissanu Deribe, 2016a). PREX2 truncation E824∗ was found to cooperate with NRAS mutations but not with BRAF V600E mutation, to accelerate melanoma development (Lissanu Deribe, 2016a). In 2017, 100 primary melanoma samples were analyzed by targeted NGS for 35 melanoma-related genes, and PREX2 mutations were reported in 14 samples (de Unamuno Bustos et al., 2017). Interestingly, recently eighty patients with conjunctival melanoma were examined by WES, identifying three PREX2 mutations (frequency of 37%) (Demirci et al., 2019). In our skin melanoma dataset, mutations and CNVs in PREX2 gene are found at frequency of 22.3 and 4.9%, respectively, as shown in Figures 2, 3 and Supplementary Figure S1D.
Although these evidences point to PREX2 as a key player in melanoma, suggesting that PREX2 may be a potential therapeutic target, to date no clinical trials are available.
TP53
The TP53 tumor suppressor gene, which is universally recognized as the “guardian of the genome,” prevents the cell from dividing and promotes apoptosis. Moreover, it is the most frequently mutated gene in human cancer with a significant prevalence of missense mutations (Hainaut and Pfeifer, 2016). The TCGA database analysis identified across 32 different cancer types 3,786/10,225 (36.8%) patients with TP53 somatic mutations (Donehower et al., 2019).
Loss of TP53 was uncommon in thicker, early or in situ melanoma, but is commonly reported as a late event in the development of melanoma (Shain et al., 2015). The Ultra-Violet Radiation (UVR) role on melanoma formation in BRAF V600E mice is often associated with mutations in TP53 (Viros et al., 2014). In our data, mutations and CNVs in the TP53 gene are present at a frequency of 14.9 and 1.3%, respectively (Figures 2, 3 and Supplementary Figure S2A).
Concerning the use and response of immune checkpoint inhibitor therapy, data are controversial. The TP53 mutation had previously been considered a potential negative predictor of metastatic melanoma treated with CTLA-4 blockade (Xiao et al., 2018), while a very recent paper showed that cell cycle regulators, such as TP53 and CDKN2A, do not appear to significantly alter clinical outcomes when immune checkpoint inhibitors are used (DeLeon et al., 2020). At the time of the writing of this manuscript, no clinical trial with TP53-targeting drugs is ongoing.
NF1
NF1 encodes neurofibromin 1, a cytoplasmic protein highly expressed in neurons, Schwann cells, oligodendrocytes, and leukocytes but also involved in RAS pathway as a tumor suppressor through its inhibiting activity as GAP (GTPase-activating protein) that converts the active RAS-GTP to RAS-GDP (Trovó-Marqui and Tajara, 2006). Inactivating variants in NF1, the most frequently mutated gene in melanoma after BRAF/NRAS/TP53, being reported in 10–15% of cases, were described in up to 46% of wild-type BRAF and RAS melanomas, in particular in male, older or chronically sun-exposed patients, and in copresence of mutations in RASopathy genes, e.g., PTPN11 and RASA2, that enhance its role in melanomagenesis, besides the RAS missing inhibition (Krauthammer et al., 2015). A poorer OS for NF1-mutated subtype melanoma has also been described (Cirenajwis et al., 2017). In BRAF V600E melanomas, loss of NF1, frequently co-occurring with BRAF and RAS alterations, allows a high level of activity of RAS-GTP and resistance to BRAF inhibitors (Nissan et al., 2014). Since 2000, a role for NF1 was proposed for the genesis of desmoplastic neurotropic melanoma, an uncommon melanoma with pathologic features in common with schwannomas (Gutmann, 2001; Kiuru and Busam, 2017; Mahalingam, 2017). Moreover, NF1 together with BRAF and NRAS has been found significatively mutated in melanoma (Hayward et al., 2017). In our series of skin melanomas, mutations and CNVs are reported in Figures 2, 3 and Supplementary Figure S2B, with a frequency of 14.3 and 1.1%, respectively.
Regarding therapy, a study revealed Calpain1 (CAPN1), a calcium-dependent neutral cysteine protease, as a novel NF1 binding partner that regulates NF1 degradation in melanoma cells. ShRNA-mediated knockdown of CAPN1 or treatment with a CAPN1 inhibitor showed to stabilize NF1 protein levels, downregulate AKT signaling and melanoma cell growth. Moreover, combination treatment of Calpain inhibitor I with MEK inhibitor Trametinib in different melanoma cells seemed to be more effective in reducing melanoma cell growth compared to treatment with Trametinib alone, suggesting that this combination may have a therapeutic potential in melanoma (Alon et al., 2019, p. 1). This novel mechanism for regulating NF1 in melanoma provides a molecular basis for targeting CAPN1 to suppress Ras activation. Despite these data, this novel approach is waiting to be tested within clinical trials. Currently, there are no ongoing clinical trials that evaluate NF1-targeted drugs, but two experimentations regard specifically NF1-mutated melanoma patients, treated with either a MEK inhibitor plus a FAK inhibitor or with RMC-4630, a potent and selective inhibitor of SHP2. The results of these trials have not been published yet (Table 2).
ARID2
ARID2 encodes a subunit of the SWI/SNF chromatin remodeling complexes (polybromo- and BGR1-associated factor [PBAF]), which facilitates ligand-dependent transcriptional activation by nuclear receptors. The SWI/SNF multiprotein chromatin remodeling complex is involved in regulating cell growth and proliferation. Mutations in nine genes encoding for subunits of the SWI/SNF chromatin remodeling complex are found in 20.0% of human cancers (Shain and Pollack, 2013). ARID2 was found frequently mutated in melanoma, with a frequency ranging from 7.0 to 18.0% (Hodis et al., 2012; Krauthammer et al., 2012; Ticha et al., 2019). In our dataset, mutations and CNVs in ARID2 are found in 12.9 and 5.5%, respectively, as shown in Figures 2, 3 and Supplementary Figure S2C.
To date, studies investigating the biological function of ARID2 in melanocytes and its role as tumor suppressor are missing. In 2018, it has been reported that cancers with inactivating mutations in ARID2 are more sensitive to PD-1 blockade as well as other forms of immunotherapy (Pan et al., 2018). In a very recent study, a higher sensitivity to different DNA-damaging therapies in ARID2-deficient non-small cell lung cancer cells, likely as a result of the ARID2 involvement in DNA repair, was observed (Moreno et al., 2020). In addition, ARID2 deficiency showed synthetic lethality with PARP inhibition using veliparib, an inhibitor that has shown good results in the treatment of breast cancer and is included in several clinical trials on breast, ovarian and, lung cancer (Moreno et al., 2020). Overall, recent data prompt further investigation of the role of ARID2 in melanomagenesis and direct testing of several potential therapeutic compounds already available.
CDKN2A/CDK4 Pathway
CDKN2A
The CDKN2A (Cyclin Dependent Kinase Inhibitor 2A) gene encodes two alternatively spliced variants known to function as inhibitors of G1 progression through different mechanisms, e.g., the tumor suppressor p16INK4A and p14ARF.
p16INK4A blocks the G1 progression, binding CDK4 and CDK6, and preventing them from interaction with D-cyclins and phosphorylation of RB. p14ARF, whose name derives from the presence of an Alternate Reading Frame (ARF) in the transcript, binds MDM2 (Mouse double minute 2 homolog or E3 ubiquitin-protein ligase) whose role is the degradation of p53; therefore, it also plays an indirect role in G1 progression. Furthermore, p14ARF has a role in p53-independent cell functions through the interaction with other proteins (Inoue and Fry, 2018). Loss, e.g., by promoter hypermethylation, or alterations, e.g., homozygous deletions of CDKN2A, are described in several tumors. CDKN2A is altered in 21.4% of cutaneous melanomas (AACR Project Genie Consortium, 2017). The loss of the CDKN2A locus is the most common acquired genetic change in precursor lesions, including in situ melanomas (Shain et al., 2015). Over 75% of cutaneous melanoma metastases have lost one or both alleles of CDKN2A (The Cancer Genome Atlas Network, 2015). Recently, a CRISPR-based study (Zeng et al., 2018) demonstrated that CDKN2A suppresses the initiation of melanoma invasion through inhibition of BRN2, a lineage restricted transcription factor which encodes an established regulator of melanocyte and melanoma invasion (Fane et al., 2017). Then the loss of p16INK4A enhances the motility of melanocytic cells through increased expression of BRN2.
In our dataset, frequencies of CDKN2A CNVs and mutations are 28.1% (27.5 and 0.6% for deletion and amplification, respectively) and 12%, respectively (Figures 2, 3 and Supplementary Figure S2D).
CDK4
CDK4 (cyclin dependent kinase 4) encodes a member of the Ser/Thr protein kinase family responsible for the phosphorylation and regulation of transcription factors, including RB1, SMAD3, MYC, FOXM1, MEP50, able to mediate cell-cycle progression (Sheppard and McArthur, 2013). p16-cyclinD-CDK4/6-retinoblastoma protein pathway, also known as CDK4 pathway, is dysregulated in 90% of melanomas (Curtin et al., 2005).
CDK4 amplifications are common in human cancers (Beroukhim et al., 2010). In our dataset, frequency of CDK4 mutations and CNVs is 2.2 and 4%, respectively (Figures 2, 3 and Supplementary Figure S3A).
CDKN2A and CDK4: A Clinical Perspective
To date, several clinical trials are ongoing, attempting to find a way to modulate this pathway (Spagnolo et al., 2015) (Table 2). With regard to CDKN2A alterations, drugs on study include: ilorasertib (ABT-348), a potent and ATP-competitive multitargeted kinase inhibitor that inhibits Aurora C, Aurora B, and Aurora A and that suppresses RET tyrosine kinase, PDGFRβ and Flt1; palbociclib and SHR6390, two selective inhibitor of the cyclin-dependent kinases CDK4 and CDK6.
Phase I trial with ilorasertib showed two clinical responses among 58 treated patients, and confirmed a good tolerability and safety profile of the drug (Maitland et al., 2018). Palbociclib, together with abemaciclib and ribociclib, have already been approved for the treatment of metastatic breast cancer, after several studies showing their activity in a spectrum of solid tumors including melanoma. Palbociclib is currently under investigation among patients affected by acral melanoma with documented gene aberrations in cell cycle pathways, including CDK4 amplification and/or CCND1 amplification and/or CDKN2A loss. Finally, SHR6390 showed a promising activity in preclinical studies performed on cell lines and human tumor xenograft models.
Considering CDK4/6 alterations, several drugs are being testing.
P276-00 is a novel inhibitor for CDK-1, CDK4 and CDK9 that has been tested in 16 tumor cell lines from different human cancers, showing a significant antiproliferative effect compared to cisplatin. Interestingly, some cancers showed to be particularly sensitive to P279-00, like tumors of central nervous system, NSCLC, breast cancer and melanoma (Joshi et al., 2007).
Ribociclib also showed some activity in melanomas with activating mutations of BRAF or NRAS.
A phase Ib/II study with ribociclib plus MEK162 has been performed in 2013. Among 14 NRAS mutant advanced or metastatic melanoma patients, the combinations of drugs allowed to obtain six partial responses and six stable disease (Sosman et al., 2014).
Abemaciclib, another CDK4/6 inhibitor, structurally different from ribociclib and palbociclib, showed to be effective among several human tumors, including melanoma, in preclinical models (Gelbert et al., 2014; Tate et al., 2014). Of note, abemaciclib demonstrated to be particularly effective in BRAF resistant melanoma cells (Yadav et al., 2014). Subsequently, a phase I trial was conducted to evaluate safety and tolerability of abemaciclib and its antitumor activity. A total of 26 melanoma patients were enrolled: one achieved a partial response and the DCR was 27%. Interestingly, abemaciclib was found in the cerebrospinal fluid demonstrating to pass the blood brain barrier and, on this basis, a trial specifically dedicated to patients with brain metastasis was designed. Unfortunately, no information concerning the melanoma cohort are available to date.
PTEN
The PTEN tumor-suppressor gene encodes for a phosphatidyl-inositol-3,4,5-triphosphate 3-phosphatase which negatively regulate the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) pathway. The PTEN gene, which is also known as MMAC1 (mutated in multiple advanced cancers) and TEP1 (TGF-β regulated and epithelial cell-enriched phosphatase) exhibits both protein and lipid phosphatase activities. Therefore, deletion or inactivation of PTEN results in constitutive AKT activation. Loss of PTEN function through deletion, mutation, and/or decreased expression, has been found in human sporadic cancers, including melanoma (Bonneau and Longy, 2000). PTEN mutations were identified for the first time in 1997 through the analysis of 35 melanoma cell lines (Guldberg et al., 1997). Subsequently, different studies reported that approximately 30% of cutaneous melanoma cell lines harbor PTEN mutations or deletions (Guldberg et al., 1997; Tsao et al., 1998) and in vitro studies were performed attesting the involvement of PTEN LOH in the development of more than 30–40% of melanomas (Robertson et al., 1998). Then, PTEN mutations were identified in 4 of 61 (7.0%) metastatic melanoma tumors (Birck et al., 2000), a lower rate compared to previous studies (Guldberg et al., 1997; Tsao et al., 1998), but the authors explained this disagreement due to technical limitations and the in vitro selection of cells harboring PTEN mutations.
PTEN and NRAS mutations were described as mutually exclusive (Tsao et al., 2000). Moreover, evidence of cooperation between BRAF activating mutations and PTEN loss in melanoma development was found, suggesting that the activation of MAPK and AKT pathways may be required for melanoma progression (Tsao et al., 2004).
In conclusion, PTEN mutations frequently coexist with BRAF mutations, but not with NRAS, which can independently activate the PI3K cascade, with a mutation and deletion rate frequency of 8.5 and 7.0% in our dataset (Figures 2, 3 and Supplementary Figure S3B).
To date, no therapeutic strategy specifically targeting PTEN has been developed. Interestingly, some trial evaluating the safety and efficacy of two Pi3K inhibitor, alone or in combination with pembrolizumab, in patients with loss of PTEN, are ongoing (Table 2).
PPP6C
PPP6C gene encodes the catalytic subunit of the PP6 serine/threonine phosphatase complex and regulates cell cycle progression in response to IL2 receptor stimulation (Bastians et al., 1997; Filali et al., 1999). Loss of PPP6C function has been known to cause the increase of Aurora A activity that, as the Aurora A amplification, leads to chromosome instability (Zeng et al., 2010; Hammond et al., 2013). PPP6C mutations make melanoma cells susceptible to inhibition by Aurora kinase inhibitors (Gold et al., 2014). Moreover, PPP6C mutations in melanoma cells seems to induce an increased autophagy. Indeed, PPP6C mutants bind to the PPP6Rs leading to its rapidly degradation. This increases wild-type PPP6C stability, sensitizing the cells autophagy induction in response to mTORC1 inhibition (Wengrod et al., 2015).
Several NGS studies reported a PPP6C mutations frequency around 7% (Gold et al., 2014).
In our dataset, PPP6C mutations and CNVs are found in 6.9 and 1.9% of cases, respectively (Figures 2, 3 and Supplementary Figure S3C). In conclusion, PPP6C mutations in melanoma may lead to new targeted approaches, such as specific PPP6 inhibitors, but at now, no trials are ongoing.
CTNNB1
CTNNB1 gene encodes β-catenin that is a core component of the adherens junctions that play a key role in maintaining of the epithelial cell layers, and in transmitting the contact inhibition signal, by anchoring the cytoskeleton. CTNNB1 is also part of the Wnt signaling pathway and is involved, e.g., in pre-birth development, the maintenance and renewal of stem cells. Finally, it has a role in the formation of the matrix of hair follicles. Gain-of-function variants of CTNNB1 cause accumulation of the protein in the nucleus, where it promotes cell proliferation by gene-activation, and have been found in several types of cancer. In melanoma cells, growth is promoted by altering expression of MITF (melanocyte inducing transcription factor) (Widlund et al., 2002), although an opposite effect of invasion-blocking MITF mediated has been observed (Arozarena et al., 2011).
The Wnt pathway may be altered in melanomas by different mechanisms, e.g., somatic variants not only in CTNNB1 but also in APC (adenomatous polyposis coli gene) or ICAT (inhibitor of beta-catenin) genes (Reifenberger et al., 2002). However, a major mechanism of promoting melanomagenesis is the cooperation between CTNNB1 and NRAS by suppression of p16INK4a expression, releasing cells from oncogene−induced growth arrest (Curley and Bosenberg, 2008). Furthermore, the CTNNB1 promotes expression, in a positive feedback loop, of Tspan8, which triggers melanoma cell detachment and invasiveness (El Kharbili et al., 2019).
In melanoma, mutations in CTNNB1 (typically missense mutations localized in exon 3) are relatively infrequent (around 7%) (Hodis et al., 2012; Polakis, 2012; Siroy et al., 2015).
Interestingly, CTNNB1 (S33C) mutation was found to confer resistance to imatinib in a metastatic melanoma patients harboring KIT L576P mutation (Cho et al., 2017). In summary, in our dataset frequency of CTNNB1 mutations and CNVs is 6.6 and 1%, respectively (Figures 2, 3 and Supplementary Figure S3D). Currently there are no drugs that target beta-catenin right now, nor ongoing clinical trials.
DDX3X
DDX3X gene encodes an ATP-dependent DEAD-box RNA helicase frequently altered in various human cancers, including melanomas (Stransky et al., 2011; Wang L. et al., 2011; Kandoth et al., 2013; Bol et al., 2015; The Cancer Genome Atlas Network, 2015; Ojha et al., 2015; Hayward et al., 2017).
Several studies reported that DDX3X is involved in double-stranded RNA unwinding, pre-mRNA splicing, RNA export, transcription, and translation (He et al., 2018; Lin, 2019). Despite its important roles in several cytosolic steps of mRNA metabolism, its function in tumorigenesis remains incompletely known. In our dataset, DDX3X mutations and CNVs are found with a frequency of 6.4 and 0.4%, respectively (Figures 2, 3 and Supplementary Figure S4A). Moreover, it has been reported that DDX3X loss decreases MITF mRNA levels, leading to a proliferative-to-metastatic phenotype in vivo, and it is implicated in resistance to BRAF inhibitors (Phung et al., 2019).
These studies reveal a DDX3X role in melanoma cancer providing a new therapeutic target that should be elucidated in the future.
RASA2
RASA2 gene encodes a GTPase-activating protein that suppresses RAS function; therefore mutations or loss of function of RASA2 enhance RAS activation (Yarwood et al., 2006). RASA2 mutations were initially reported in skin melanoma at a frequency ranging from 2 to 8% by three different NGS studies (Supplementary Figure S4B) (Berger et al., 2012; Hodis et al., 2012; Krauthammer et al., 2012, 1). Overall, frequency of RASA2 mutations and CNVs in our skin melanomas dataset is 5.5 and 1%, respectively (Figures 2, 3 and Supplementary Figure S4B).
RASA2 mutations co-occurred in NF1 mutant melanomas that were BRAF-RAS wild type, in a WES study of 213 melanoma samples (62 cell lines and 151 melanomas); 12.2% (26/212) of melanomas were NF1-mutant/BRAF-RAS-wild-type and nine of them harbored co-mutations in RASA2 (2 nonsense and 3 R551C) (Krauthammer et al., 2015).
Recently, RASA2 and NRAS mutations were confirmed to be mutually exclusive, with NF1 mutations significantly co-occurring with RASA2 mutations in BRAF and NRAS wild-type melanomas, since RASA2 inhibits NRAS and NF1 inhibits KRAS and HRAS (Arafeh et al., 2019). Indeed, RASA2- and NF1-mutated genes co-selection in melanoma could be equivalent to oncogenic RAS mutation (Arafeh et al., 2019).
To date, clinical trials direct toward RASA2 are not currently available.
SF3B1
SF3B1 gene encodes subunit 1 of the splicing factor 3b protein complex, a component of the U2 small nuclear ribonucleoprotein complex (snRNP) that participates in the splicing of pre-mRNAs. Mutations in SF3B1 can lead to alternative splicing events for multiple genes and are found in several cancers, including uveal (frequency of 10–20%) (Harbour et al., 2013; Martin et al., 2013, p. 1; Dono et al., 2014, p. 1), mucosal (Newell et al., 2019; Nassar and Tan, 2020), and cutaneous melanoma (Kong et al., 2014). SFRB1 mutations (particularly at codon 625) are rare in cutaneous melanoma despite in uveal, mucosal, leptomeningeal and blue naevi-like cutaneous melanomas are reported (Schilling et al., 2013; Kong et al., 2014). Overall, in our dataset, SF3B1 mutations and CNVs are 5.2 and 1%, respectively, as reported in Figures 2, 3 and Supplementary Figure S4C.
Very interestingly, SF3B1 mutations were found to induce cancer cells to produce an abnormal form of the BRD9 RNA molecule, including noncoding DNA sequences or “junk DNA,” which garbled the genetic message (Inoue et al., 2019). This “junk DNA” originated from a viral element that recently inserted itself into the human genome. The use of CRISPR technology and antisense oligonucleotides to suppresses tumors growth deriving by SF3B1-mutant cells opens for future therapeutic approaches, but to date no clinical trials directed to SF3B1 are available (Inoue et al., 2019).
RAC1
RAC1 encodes for a RHO GTPase that plays a key role in cellular cytoskeleton organization and motility and can induce RAS dependent pathways stimulating cell proliferation (Krauthammer et al., 2012, 2015). WES studies led to the discovery of a hotspot mutation (P29S) in RAC1 gene, defining it as the most frequent driver mutation in sun-exposed melanomas, with a frequency of 5–7% (Hodis et al., 2012; Krauthammer et al., 2012; The Cancer Genome Atlas Network, 2015). The frequency of RAC1 P29S mutation (overall RAC1 P29S mutation of 4.1%) was found to be more prevalent in male patients and similar between primary (9.2%) and metastatic tumors (8.6%) (Krauthammer et al., 2012). Indeed, mutations in RAC1 enhanced melanoma disease progression: RAC1 P29S analysis in a cohort of 814 primary cutaneous melanomas, with known BRAF and NRAS mutation status, revealed an association with increased tumor thickness, increased mitotic rate, ulceration, and presence of lymph node metastases at the time of diagnosis (Mar et al., 2014). In general, frequency of RAC1 mutations and CNVs in our skin melanomas dataset is 5.1 and 5.9%, respectively (Figures 2, 3 and Supplementary Figure S4D).
Activation of RAC1 may be also promoted by mutated PREX2. Indeed, truncating mutations in PREX2 have been shown to activate the GTPase RAC1 abolishing the binding to PTEN and activating the PI3K (phosphatidyl inositol 3 kinase)/Akt signaling pathway (Lissanu Deribe, 2016b; Lissanu Deribe et al., 2016). Concerning treatment, in vitro studies have shown that melanoma cell lines harboring the RAC1 P29S mutation are resistant to BRAF and MEK inhibitors, but its role in conferring this resistance is still to be elucidated (Watson et al., 2014). However, clinical inhibitors of RAC1 are not currently available although SRF/MRTF inhibitors in combination with BRAF inhibitors have been recently demonstrated to have utility in the treatment of BRAF mutant melanoma with an RAC1 P29S mutation (Lionarons et al., 2019).
High levels of PD-L1 in patients with RAC1 P29S mutations compared to wild-type RAC1 melanoma samples from the TCGA cohort were observed (Vu et al., 2015). These findings could open up individualized therapy based on immunological characteristics of patient tumor and the presence of RAC1 P29S mutations with anti-PD-1 or anti-PD-L1 antibodies treatment in melanoma patients with high expression of PD-L1 harboring RAC1 P29S mutation.
MAP2K1/MAP2K2
MAP2K1, mitogen-activated protein kinase 1, also known as MEK1, and MAP2K2 (MEK2), mitogen-activated protein kinase 2, encode for mitogen-activated protein (MAP) kinase involved in many cellular processes, such as proliferation, transcription regulation, differentiation and development, and represent the downstream targets of the RAS-RAF-MAPK cascade. Activating mutations in MAP2K1 and MAP2K2 represent one of the multiple mechanisms of resistance to BRAF and MEK inhibitors (Spagnolo et al., 2015). Overall MAP2K1/MAPK2 mutational and CNV prevalence in our skin melanomas dataset is 6.7 and 2.5%, respectively (Figures 2, 3). In particular, melanoma patients have a higher frequency of MAP2K1 mutations/CNVs compared to MAP2K2 mutations/CNVs (Figures 2, 3). Notably, the coexistence of MAP2K1 mutations and BRAF or NRAS mutations are often observed. In general, MAP2K1/2 mutations in BRAF V600E melanomas are linked to both intrinsic and acquired resistance to BRAF inhibitors.
To date, no clinical trials having MEK1/2-mutated melanoma patients as population of interest are ongoing but, as we mentioned before, MEK inhibitors are being tested in several studies, alone or in combination with other drugs, and their use with BRAF inhibitors has become a standard in the therapy of BRAF mutated metastatic melanoma.
Trametinib is a small molecule and a selective MEK1/2 inhibitor, non-ATP competitive.
Based on the encouraging results obtained in phase I (Infante et al., 2012) and phase II trial (Kim et al., 2013, 1), a phase III study (METRIC) with trametinib monotherapy compared to chemotherapy was performed (Flaherty et al., 2012). A total of 322 patients affected by previously untreated BRAF V600 E/K stage III or IV melanoma were enrolled. Trametinib was associated with a higher PFS (4.8 months vs. 1.5), a greater OS (6-months OS 81% vs. 67%), and a higher rate of responses (22% vs. 8%). Based on these data, trametinib was approved as single agent by the FDA in May 2013. Despite this, currently MEK inhibitors do not play a role in monotherapy in NRAS-mutated or in WT patients but could play a role in combination with immunotherapy or with other target agents.
Finally, TAK-733, a selective MEK1/2 inhibitor, showed a broad antitumor activity in melanoma cell lines and in 10 out of 11 patient-derived xenografts models (Micel et al., 2015). In phase I study, performed among 51 patients, TAK-733 showed a manageable toxicity profile but a limited antitumor activity, with partial responses obtained just in 5% of patients (Table 2) (Adjei et al., 2017).
KIT
KIT encodes for a class III tyrosine kinase receptor that is expressed on several cell types, including melanoblasts and differentiated melanocytes, but also hematopoietic progenitors and mast cells. The binding to the stem cell grow factor causes c-kit homodimerization that leads to the phosphorylation of tyrosine residues by activating the MAPK/ERK and the PI3K/AKT/mTOR pathways (Lennartsson and Rönnstrand, 2012). KIT was initially thought to act as a tumor suppressor gene, because its presence in normal melanocytes and benign nevi, and its loss during progression and in metastatic melanoma was reported (Montone et al., 1997; Shen et al., 2003; Isabel Zhu and Fitzpatrick, 2006). Moreover, loss of c-kit expression was observed in different cultured melanoma cells (Lassam and Bickford, 1992; Natali et al., 1992; Zakut et al., 1993) and related to a higher metastatic potential of melanoma xenografts in nude mice (Gutman et al., 1994). However, KIT also acts as an oncogene. Indeed, it has been found an increase of KIT mutations and/or CNVs in mucosal (39%), acral (36%), and melanomas arose on chronically sun-damaged skin (28%) (Curtin et al., 2006). Interestingly, a recent meta-analysis reported KIT mutations in 497 (9.5%) melanoma patients analyzing 5,224 patients from 32 studies selected (Gong et al., 2018). Moreover, a close association with older age, acral, mucosal, or chronic sun-damage sites, but not with any histological features or tumor stage was found (Gong et al., 2018). Approximately 70% of KIT mutations identified in melanoma and leading to constitutive activation of kinase activity are localized in exon 11 (L576P) or exon 13 (K642E) (Shtivelman et al., 2014). Mutations in KIT are generally mutually exclusive with other driver mutations, including NRAS and BRAF. In our skin melanomas dataset, KIT mutations and CNVs are found in 4.5 and 2.3% of patients, respectively (Figures 2, 3 and Supplementary Figure S5C).
The activity of imatinib mesylate in KIT mutated melanoma patients was explored through three single arm phase II trials (Table 2). Carvajal et al. treated 28 patients affected by KIT mutated advanced or metastatic melanoma with imatinib 400 mg twice daily (Carvajal et al., 2011). Among the 25 evaluable patients, two complete and four partial responses were observed, with a median time to progression of 12 weeks and a median OS of 10.7 months. Best responses were observed among patients with mutations involving recurrent hotspots or with a mutant – WT allelic ratio superior of 1 (40 vs. 0%). Interestingly, particularly good responses were obtained among patients with K642E mutation, that showed an ORR of 50% and a DCR of 100%.
Guo et al. (2011) reported data from 43 treated patients who received imatinib 400 mg daily or, in case of disease progression, 800 mg daily. The median PFS was 3.5 months with a 6-month PFS of 36.6%, while median OS was 12 months. Globally, the (DCR) was 53.3%: 10 patients (23.3%) and 13 patients (3.2%) achieved partial response and stabilization of disease, respectively. Overall, 18 patients (41.9%) achieved tumor regression. Specifically, 9/10 partial responses were observed in patients with mutations in exons 11 or 13. The overall 1-year survival rate was 51%.
Finally, in a third study published by Hodi et al. (2013), 25 patients affected by metastatic melanoma of the mucosa, acral or chronically UV-damaged skin with amplifications or KIT mutations received 400 mg of imatinib daily or, in the absence of a clinical response, twice a day. This study confirmed the clinical activity of imatinib, mainly concerning KIT mutations: indeed, all responses were achieved among patients with KIT mutations while the best response observed in patients with amplifications was stable disease. OS was 12.5 month in the overall cohort, similar to the previously reported trials.
Other KIT inhibitors showed activity in KIT mutated advanced melanomas (Table 2).
Nilotinib is a small molecule that showed activity among KIT mutated advanced or metastatic melanoma patients in four clinical studies. In the first trial published in 2015 (Carvajal et al., 2015), 19 patients, mostly in disease progression after treatment with imatinib, achieved an ORR of 15.8% and a median OS of 9.1 months. In a second trial, 39 patients were treated with nilotinib (Lee S. J et al., 2015): one experienced complete response and six partial response; median PFS was 3.3 months and median OS 11.9 months. The subsequent phase II trial (TEAM), an ORR of 26.2% was achieved among 42 patients naïve for previous KIT inhibition (91% of which in the presence of exon 11 mutations), a median PFS of 4.2 months and a median OS of 18 months. In another phase II study, 25 patients were treated with an ORR of 16% at six months of observation, median PFS of 6 months and median OS of 13.2 months (Guo et al., 2017).
Sunitinib is another molecule tested in 12 patients with advanced or metastatic melanoma, and obtained four clinical responses in a small clinical trial (1 complete response and 3 partial response) (Minor et al., 2012).
Finally, dasatinib was evaluated in 22 patients with advanced or metastatic KIT mutated melanoma. With regard to clinical activity, four partial responses and seven stable diseases were obtained. The median PFS was 4.7 months and median OS 12.3 months (Kalinsky et al., 2017).
In conclusion, to date it is recommended to test KIT mutations (especially exons 11 and 13) in acral, mucosal, and unknown origin melanomas, as well as cutaneous ones arising on chronically damaged skin, to offer an additional therapeutic option.
RB1
RB1 acts as a tumor suppressor gene by regulating cell cycle division: when dephosphorylated, it interacts directly with E2F1 and inhibits its transcriptional activity with cell cycle arrest. Thus, the cell cycle is finely regulated by CDKs with CDKIs varying levels of RB phosphorylation, E2F family and TP53; alterations that disrupt the p16INK4A:cyclinD-CDK4/6:RB functional pathway may the first critical step leading to melanomagenesis (Bartkova et al., 1996; Lee B. et al., 2015). Cells deficient or with low levels of RB go into p53-mediated apoptosis: this phenomenon is particularly important to identify new compounds able to activate p53 (Knudsen and Wang, 2010). In mouse melanoma cells Rb1 cooperates with MITF to activate expression of Tyr and Cdkn1a/p21Cip1 (Carreira et al., 2005). RB1 is also important in maintaining chromatin structure, stabilizing histone methylation (Shao and Robbins, 1995). Summing up, RB is likely a multifunctional protein that binds to at least 100 other proteins (Morris and Dyson, 2001). RB1 is altered in about 4% of all cancers (MCG)1. In our dataset, the most common somatic alterations in RB1 are mutations with a frequency of 4.4% while CNVs are reported in 1.9% of skin melanoma patients (Figures 2, 3 and Supplementary Figure S5D). To date, no clinical trials specifically designed to target RB are ongoing. However, patients with RB1 loss are specifically considered in one clinical trial with LY260636, a checkpoint kinase 1 inhibitor. The trial is active, not recruiting.
FBXW7
FBXW7 is a critical tumor suppressor gene and a member of the F-box protein family, ubiquitin ligase complex, that controls proteasome-mediated degradation of oncoproteins such as cyclin E, c-Myc, Mcl-1, mTOR, Jun, Notch, and AURKA, STAT2 (Minella and Clurman, 2005; Yeh et al., 2018; Lee et al., 2020). Inactivating mutations in FBXW7 have been described in a variety of human tumors and cancer cell lines (Akhoondi et al., 2007). Loss of function of FBXW7 in several human cancers has clinical implications and prognostic value: the use of rapamycin has proven to inhibit breast cancer cells with loss of FBXW7 by mTOR inhibition (Mao et al., 2008; Yeh et al., 2018).
FBXW7 expression was reduced in primary and metastatic melanoma compared with dysplastic and normal nevi and an increase in FBXW7 expression was significantly correlated with a better 5-year patient survival. In vitro studies demonstrated that FBXW7 inhibited human cell migration through MAPK/ERK signaling pathway suggesting its prognostic and potential therapeutic role for melanoma treatment (Cheng et al., 2013). Several WES and WGS studies reported a FBXW7 mutations in around 4% of melanomas (Wei et al., 2011; Berger et al., 2012; Hodis et al., 2012; Krauthammer et al., 2012; The Cancer Genome Atlas Network, 2015; Hayward et al., 2017; ICGC Data Portal, 2020). WES of 103 cutaneous melanomas, including 77 tumor samples and 26 cell lines, found FBXW7 mutations (8.1%) independently of BRAF or NRAS mutation (Aydin et al., 2014). In the same study, the authors discovered that inactivation of FBXW7 gene determines enhanced tumorigenesis by NOTCH1 activation (Aydin et al., 2014). These evidences open up to FBXW7 potential therapeutic targeting through modulating NOTCH1 signaling (Aydin et al., 2014). Moreover, another study revealed FBXW7α deficiency leading to HSF1 (Heat shock factor 1) accumulation and subsequent activation of the invasion-supportive transcriptional program and metastatic potential of human melanoma cells (Kourtis et al., 2015).
In our dataset, frequency of FBXW7 mutations and CNVs is 4.3 and 1.7%, respectively (Figures 2, 3 and Supplementary Figure S6A).
To date, just one clinical trial designed to enroll specifically FBXW7-mutated patients is active (Table 2). LY2606368, the drug on study, performed well in a phase I trial where it was tested in 45 patients affected by solid tumors, who experienced treatment failure with standard therapies. Among 43 evaluable patients, two partial responses and 15 stable diseases were achieved, with a DCR of 37.7% (Hong et al., 2016).
PIK3CA
PIK3CA encodes the protein p110α, the catalytic subunit of phosphatidylinositol 3-kinase (PI3K). PI3K signaling has a role in many cell activities, e.g., cell proliferation, migration, survival.
Alterations in PIK3CA have an oncogenic effect mostly related to activating variants in two hotspots located in the regions of helical and kinase domains. The PI3K-AKT pathway plays a significant role in melanomagenesis, frequently by activating RAS-RAF-MEK-ERK pathway, e.g., for NRAS activating variants or loss of PTEN (Davies, 2012), as demonstrated by studies on resistance to targeted therapies based on BRAF inhibitors (Deng et al., 2012; Penna et al., 2016). Usually, PIK3CA activating mutations are rare in melanoma with a frequency of 5%, despite the ability of activated PIK3CA (H1047R) to cooperate with BRAF V600E promoting melanomagenesis in mouse models (Marsh Durban et al., 2013). PIK3CA mutations frequently co-occurred with either a BRAF or an NRAS mutation (The Cancer Genome Atlas Network, 2015). In our skin melanoma dataset, PIK3CA mutations and CNVs are 4.2 and 1.5%, respectively, as shown in Figures 2, 3 and Supplementary Figure S6B.
Several clinical trials with PI3K inhibitors are ongoing (Table 2).
Alpelisib (BYL-719) is an oral selective inhibitor of PI3K isoform-α that showed to be active against the somatic PI3Kα mutations and wild-type PI3Kα (Fritsch et al., 2014). Based on these data, 58 patients with BRAF or RAS mutated advanced solid tumors were enrolled in a phase Ib trial and were treated with BYL-179 plus MEK162. A total of five NRAS mutated patients experienced a partial response (Juric et al., 2014).
Parsaclisib is a novel Pi3kδ inhibitor that exhibited an excellent profile in xenograft models (Yue et al., 2019, 0504). To date, one clinical trial is ongoing to assess the efficacy and the safety of the combination between parsaclisib plus pembrolizumab and itacitinib, a JAK1 inhibitor. The association parsalisib plus itacitinib showed good results in a phase I/II trial for hematological malignancies, with an ORR of 67–78%, respectively in mantle cell lymphoma and marginal zone lymphoma. Lower results were obtained among patients with diffuse large B-cell lymphoma (ORR 30%) (Forero-Torres et al., 2019).
The combination between pimasertib, a MEK1/2 inhibitor, and voxtalisib, a dual PI3K/mTOR inhibitor among patients affected by solid tumors, including melanoma, was tested in a phase Ib trial in 146 patients. In December 2018, data from this experimentation were published, showing a DCR of 52% with one complete response and five partial responses. Unfortunately, the toxicity profile of this combination was considered not acceptable (Schram et al., 2018).
EZH2
EZH2 encodes a histone methyltransferase that constitutes the catalytic component of the polycomb repressive complex-2 (PRC2) which has a role in epigenetic silencing during cell differentiation, in particular in the development of the hematopoietic and central nervous systems. EZH2 can also induce an epithelial-to-mesenchymal transition in the cancer cells, increases their metastatic potential (Min et al., 2010) and acts as a coactivator for transcription factors including the androgen receptor (Xu et al., 2012).
More recently it has been described the role of EZH2 as a recruitment platform for DNA methyltransferases in epigenetic repression (Viré et al., 2006; Barsotti et al., 2015; Moran et al., 2018).
The EZH2-dependent expression of genes associated with cell motility contributes to early phases of metastasis (Manning et al., 2015) while activating variants promotes melanoma progression inactivating tumor suppressor genes (Tiffen et al., 2015). The reactivation of tumor suppressors was correlated to increased survival confirming that EZH2-mediated epigenetic repression has a major role in advanced melanoma progression (Zingg et al., 2015). A study suggested the possibility that combined immunohistochemical expression of EZH2, H3K4me2, and H3K27me3 might identify cancer cells with potential stem cell properties; another relevant data that many epigenetic changes are pharmacologically reversible (Kampilafkos et al., 2015; Mahmoud et al., 2016).
EZH2 mutations and CNVs in our dataset are detailed in Figures 2, 3 and Supplementary Figure S6C, with an overall mutational rate of 3.9 and 5.7%, respectively.
Of note, coexistence of BRAF V600E mutation and EZH2 amplification is rather prevalent in melanoma. Indeed, in a cohort of 138 patients with BRAF V600E-mutated melanoma, 40 cases (29.0%) showed EZH2 gain. Moreover, a significant difference in overall survival and disease-free survival between no EZH2 copy number gain and gain groups was reported (Yu et al., 2017).
A recent study highlights that both benign melanocytes and cutaneous melanomas frequently harbor amplifications of EZH2 that silence genes correlated to the integrity of the primary cilium (Zingg et al., 2018).
Vemurafenib and trametinib induce senescence consistent with downregulation of c-MYC but an EZH2 variant limits this effect (Hartman et al., 2019). EZH2 has a role in differentiation of CD4+ T-cells and in the function of T regulatory cells. Its activation causes immune suppression and it has been suggested that EZH2 inhibitors may have a role in combination with immunotherapy and targeted therapies to prevent immunosuppression (Tiffen et al., 2016). It has been further demonstrated that EZH2 controls melanoma escaping mechanisms during T cell-targeting immunotherapies (Zingg et al., 2017) and because the upregulation of EZH2 and its histone modification H3K27me3 seems correlated to melanoma progression and resistance to immune checkpoint blockade, clinical trials based on EZH2 inhibitors are strongly suggested (Hoffmann et al., 2020).
WT1
WT1 encodes a transcription factor implied in the prenatal development of kidneys and gonads, mainly known for its action in cell differentiation and apoptosis. Moreover, WT1 has a role in tumorigenesis controlling several other genes [e.g., Pecam-1 (CD31) and c-KIT (CD117)], and modulating vascularity, immune response and metastasis formation (Wagner et al., 2014). High expression of WT1 is described in leukemias and in solid tumors and it seems correlated to the chemoresistance and poor outcome. Interestingly, inhibition of cell proliferation by shRNA-WT1, cisplatin, and gemcitabine in B16F10 cells induces cell death and potentiates the action of anticancer drugs by inducing synergistic effects both in vitro and in vivo (Zapata-Benavides et al., 2019). Although WT1 could be expressed by Spitz naevi and in up to one third of dysplastic naevi, it is considered as a diagnostic tool in melanoma diagnosis (Wilsher and Cheerala, 2007). Indeed, WT1 is expressed in more than 80% of malignant melanoma cells, but not in normal skin or benign melanocytic nevi (Wagner et al., 2008). WT1 protein expression was associated with shorter overall survival in melanoma (Garrido-Ruiz et al., 2010) and deemed as a target antigen for immunotherapy since a novel signaling mechanism mediated by PPARbeta ligands, which led to melanoma cell growth suppression through the direct repression of WT1, was described (Michiels et al., 2010, 1). The silencing of WT1 through shRNAi has a synergistic effect with doxorubicin and cisplatin, sensitizing B16F10 melanoma cells (Zapata-Benavides et al., 2012). WT1 somatic mutations are described in several cancers, with cutaneous melanoma having the greatest prevalence. Two similarly sized WES analysis of 100 and 114 skin melanomas found WT1 somatic mutations in the 3 and 1.8% of skin melanoma patients, respectively (Hodis et al., 2012; Krauthammer et al., 2012) (Supplementary Figure S6D). In summary, frequency of WT1 mutations and CNVs in our skin melanomas dataset are 3.7 and 1.3%, respectively (Figures 2, 3 and Supplementary Figure S6D).
To date, one trial with DSP-7888 plus nivolumab or pembrolizumab in advanced solid tumors including melanoma, is ongoing (Table 2). DSP-7888 is a peptide cancer vaccine that includes WT1 derived peptides: it has shown to induce both a CD8+ and CD4+ mediated immune response against WT1 overexpressing tumor cells.
SNX31
SNX31 encodes for the sorting nexins protein involved in membrane trafficking (Worby and Dixon, 2002; Ghai and Collins, 2011; Ghai et al., 2014). It is upregulated in more than 50% of bladder carcinoma transitional cell, but to date its role in cancer is poorly understood. In melanoma, missense mutations in SNX31 were first reported with a 7% frequency (Hodis et al., 2012). Interestingly, a 9.0% mutation frequency was reported in 46 primary mucosal melanomas (Kim et al., 2017). In our skin melanomas dataset SNX31 mutations and CNVs are 3.7 and 6.0%, respectively (Figures 2, 3 and Supplementary Figure S7A). No clinical trials with SNX31 inhibitors are ongoing.
IDH1
IDH1 encodes for a single, soluble, cytoplasmic isocitrate dehydrogenase 1 enzyme, that converts isocitrate to α-ketoglutarate (also known as 2-oxoglutarate) in an NADP+ dependent manner, protecting cells against reactive oxygen species (ROS) and radiations (Minard and McAlister-Henn, 1999; Jo et al., 2002). First described in glioma (Balss et al., 2008, p. 1; Parsons et al., 2008; Yan et al., 2009, p. 2), mutations in IDH1 and IDH2 (isocitrate dehydrogenase 2 (NADP+), mitochondrial) were described in several cancers, including melanoma (Lopez et al., 2010). IDH1 and IDH2 mutations occur primarily in the catalytic domain at residue R132 in IDH1, and R140 and R172 in IDH2. The mutant IDH1/IDH2 proteins lead to a reduction of α-ketoglutarate (α-KG) with a relative production of oncometabolite 2-hydroxylglutarate (D-2HG) that it is likely to play a major role in the pathophysiology of tumors blocking cellular differentiation by competitively inhibiting αKG-dependent dioxygenases involved in histone and DNA demethylation (Mondesir et al., 2016). The first IDH1 mutation in melanoma was reported in 2010 (Lopez et al., 2010). The authors also revealed BRAF V600E mutation, but no NRAS/TP53/CDKN2A/CDKN2B mutations, in one metastatic sample carrying the R132C mutation, analysing 78 patients. Moreover, IDH2 R172 mutations were not detected in any of these samples (Lopez et al., 2010). The next year, in a cohort of 142 primary non-epithelial tumors, it was shown that about 10.0% of metastatic melanoma lesions (4/39) harbored an IDH1 or IDH2 heterozygous mutation, equally distributed (Shibata et al., 2011). This study confirmed the co-occurrence of BRAF mutation with IDH1 mutation previously observed, and demonstrated that mutant IDH1 conferred in a BRAF V600E melanoma cell line acquired in vivo growth activities and enhanced activation of the MAPK and STAT3 pathways (Lopez et al., 2010; Shibata et al., 2011). Mutant IDH1 also reduced the expression of RASSF1, DHRS1, ADH5, whereas it induced the expression of JUN, MYCN, and ATF3 (Lopez et al., 2010).
In our skin melanoma dataset, mutations of IDH1 are 3.4% (Figure 2 and Supplementary Figure S7B) while CNVs are 1.3% (Figure 3 and Supplementary Figure S7B).
To date, no FDA-approved drugs or clinical trials are available for melanoma patients carrying IDH1/IDH2 mutations. Indeed, only two compounds, Enasidenib (AG-221), a DH2-specific inhibitor, and Ivosidenib (AG-120), a IDH1-specific inhibitor, have been approved by FDA for patients with relapsed or refractory acute myeloid leukemia (Stein et al., 2017; DiNardo et al., 2018).
Thanks to these evidences, future clinical studies could be pursued investigating the efficacy of IDH inhibitors for melanoma cancer.
STK19
STK19, also known as RP1, encodes a nuclear serine/threonine kinase (Shen et al., 1994; Gomez-Escobar et al., 1998). STK19 protein can bind ATP phosphorylated a-casein proteins at Serine/Threonine residues and histone at Serine residues, therefore phosphorylation of STK19 is involved in transcriptional regulation (Gomez-Escobar et al., 1998). STK19 is important for the transcription-related DNA damage response since it is involved in DNA repair during active transcription and in nuclear signal transduction (Boeing et al., 2016). STK19 mutations have been found with a frequency of around 4.1% in melanoma, with D89N mutation present at 3.3%, whereas no SKT19 mutations were detected in nevus associated-melanomas (Hodis et al., 2012; Shitara et al., 2015). In our dataset, STK19 mutations and amplifications are reported in 3.3 and 6.0% of samples selected (Figures 2, 3 and Supplementary Figure S7C).
In a recent study, STK19 was identified as a novel regulator of NRAS function (Yin et al., 2019). STK19 alterations were mutually exclusive with BRAF (Yin et al., 2019). Through in vitro and in vivo experiments STK19 was found to phosphorylate the residue S89 of NRAS activating NRAS signaling via the MEK-ERK and PI3K pathways. In NRAS Q61R transgenic mice the STK19 D89N mutant promoted oncogenic NRAS-driven melanomagenesis. An STK19 inhibitor (ZT-012-037-1) was able to inhibit NRAS activation in NRAS-STK19 mutant mice preventing NRAS-driven melanoma development and growth (Yin et al., 2019), revealing a promising therapeutic strategy for NRAS-mutant melanoma tumors treatment (Yin et al., 2019). Based on these last observations, several natural compound libraries were screened using a phosphorylation assay-based approach and chelidonine was identified as a potent and selective inhibitor of STK19, providing a novel option for targeting NRAS-mutant cancers (Qian et al., 2020). Nevertheless, clinical trials are not available.
MITF
Microphthalmia-associated transcription factor is a basic helix-loop-helix (hHLH)-leucine zipper protein that plays a role in the development of neural crest-derived melanocytes and retinal pigment epithelial cells, and is encoded by MITF gene (Fuse et al., 1999). MITF is directly involved in the expression of genes that encode melanin synthesis enzymes (TYR, TYRP1, and DCT), melanosome proteins (PMEL, MLANA, and RAB27A), proteins involved in the cell cycle (CDKN1A, CDKN2A, TBX2, and CDK2) and in cell survival (e.g., BCL2, BIRC7, HIF1A, and MET) (Levy et al., 2006; Cheli et al., 2010; Vachtenheim and Borovanskı, 2010), DNA replication and repair, cell proliferation, and mitosis (Strub et al., 2011; Webster et al., 2014). This well-known role in melanocyte development led MITF nicknaming as the “master regulator” of melanocytes (Levy et al., 2006). Despite MITF acts together with many transcription factors as SOX10, YY1, TFAP2A, LEF1, RB1, IRF4, and PAX3 (Seberg et al., 2017), its activity level determines the phenotype adopted by melanoma cells, whether invasive, proliferative, or differentiated according to the MITF rheostat model (Goding, 2011). MITF locus is amplified in 7.9% of skin melanomas, while in our dataset, MITF somatic mutations are quite rare, with a frequency of 1.7% (Figures 2, 3 and Supplementary Figure S7D). MITF amplification is more prevalent in metastatic melanomas and correlates with decreased overall patient survival. It is responsible for resistance to conventional chemotherapy and BRAF inhibition (Garraway et al., 2005). Recently, Wang et al. provided evidence that p300/CBP inhibition suppressed the melanoma-driven transcription factor, MITF, and could be utilized as a potential therapy for treating melanoma (Wang R. et al., 2018; Kim et al., 2019). To date, no clinical trials are attempting to target MITF directly; on the other hand, MITF-mediated pathways targeting through histone deacetylase inhibitors (HDACi) has been investigated with disappointing results. Indeed, despite HDACi showed the ability to silence MITF promoter in vitro and in vivo (Yokoyama et al., 2008), this ability was not observed among treated patients in a phase I trial performed between 2010 and 2012. A total of 16 patients affected by unresectable stage III or IV melanoma received panobinostat, a potent HDACi that previously showed promising results among preclinical studies in melanoma samples and hematologic malignancies (Giles et al., 2006). Results from this phase I clinical trial were published in 2016: among 15 evaluable patients, four achieved stable disease, with an ORR of 0% and a disease control rate of 27%. Due to the absence of partial or complete response, and the heavy toxicity of study drug, the trial was stopped (Ibrahim et al., 2016).
Other Genes
TERT
TERT gene needs to be addressed separately from the other genes, since the most common mutations in TERT are localized in TERT promoter and have not been systematically investigated in most of the studies included in our dataset. TERT gene codes for a telomere reverse transcriptase catalytic subunit, the principal mechanism of telomere maintenance in cancer cells.
Most of human cancers re-activate telomerase (Kim et al., 1994). The first studies that identified cancer related TERT promoter mutations were performed in melanoma patients. The most common TERT promoter mutations are −57A/C, −124C/T, −146C/T, upstream the TERT gene ATG (Horn et al., 2013). From TCGA dataset of 9,127 patients and 31 cancer types emerged that 27% of all analyzed samples harbored one of these promoter mutations (Barthel et al., 2017), placing them among the most frequent cancer mutations (Lorbeer and Hockemeyer, 2020). In particular, 73 out 115 SKCM melanoma samples (frequency of 63.4%) harbored TERT promoter mutations (The Cancer Genome Atlas Network, 2015). Interestingly, WGS studies analyzed TERT promoter reporting a frequency of 81.2% in acral and cutaneous melanomas (Supplementary Figure S9) (Hayward et al., 2017).
Then, a discordancy on the mutational status between the primary and metastatic lesion was found in a cohort of 194 primary nodular melanomas matched with 72 loco-regional metastases, while TERT protein expression was found to correlate with reduced patient survival (Hugdahl et al., 2018). A frequent TERT promoter polymorphism at −245 was associated with an increased rate of metastasis in melanoma (Nagore et al., 2019) and inversely correlated with BRAF mutation (Bruno et al., 2018). Moreover, mutations of the TERT promoter are more frequent in fast-growing melanomas rather than slow-growing ones and this characteristic could be used to identify more aggressive tumors that might benefit from adjuvant therapy (Nagore et al., 2016). Recently, CNV was found in 61.5% of acral melanoma which is consistent with a previous study that reported rates from 44.9 to 75% (Liang et al., 2017; Yu et al., 2018, 2020). In our skin melanoma dataset, TERT coding mutations (3.9%) and CNVs (5.5%) are described in Figures 2, 3 and Supplementary Figure S8A. Specifically, the 42 TERT coding variants found in our dataset were all not-pathogenic variants in melanoma, underlying TERT as a polymorphic gene.
No clinical trials for TERT mutated patients or with TERT inhibitors are ongoing.
MTOR
The MTOR gene encodes a protein belonging to the phosphatidylinositol kinase-related kinases family, which regulates cell growth, proliferation, motility and survival, protein synthesis and transcription (Wullschleger et al., 2006). mTOR is regulated and responds to growth factors, energy metabolites and/or levels of nutrients (Vogt, 2001). mTOR is the catalytic subunit of two different multiprotein complexes, mTORC1 (highly sensitive to rapamycin) and mTORC2 (relatively insensitive to rapamycin) with different functions. AKT regulates the mTORC1 complex by phosphorylating and inhibiting the TSC-2 gene (Tuberous Sclerosis 2), which is a GTP-ase activating protein (GAP) that binds to TSC-1 (Tuberin) forming a complex and blocking the G Rheb protein. The inhibition of TSC-2 allows the Rheb protein to accumulate in a GTP-bound state and to activate mTORC1. mTORC1 regulates several key steps of protein synthesis, controlling the expression of proteins that promote cell proliferation and survival. The mechanism controlling mTORC2 is not yet well known; the activation of this complex, however, is linked to the PI3K signaling (Pópulo et al., 2012). mTOR, as key protein of the PI3K/AKT pathway, acts both upstream and downstream of AKT, and aberrant mTOR activation of promotes survival and proliferation of tumor cells in several human cancers (Saxton and Sabatini, 2017; Chamcheu et al., 2019). Nonsynonymous MTOR mutations are present in about 3.61–12% of melanoma patients (The Cancer Genome Atlas Network, 2015; AACR Project Genie Consortium, 2017). In a Chinese cohort of 412 melanoma patients, nonsynonymous MTOR mutations were found by NGS analysis in acral, mucosal, cutaneous melanomas (with and without chronic sun-induced damage), and unknown primary subtypes, with a frequency of 11.0, 14.3, 3.4–6.7, and 11.1%, respectively (Kong et al., 2016). In this study, H1968Y, P2213S and S2215Y were established as gain-of-function mutations sensitive to specific inhibitors (Kong et al., 2016). Similarly to P2213S and S2215Y, the same authors found another gain-of-function mutation (H2189Y) that was sensitive in heterozygous at the AKT inhibitor (AZD5363) and the phosphoinositide 3-kinase inhibitor (LY294002), and in homozygous at the mTOR inhibitor (everolimus) and the AKT inhibitors (AZD5363) and (MK-2206 2HCL), and the phosphoinositide 3-kinase inhibitor (LY294002) (Wu et al., 2018). Interestingly, analyzing 31 candidate genes existing in either the PI3K or MAPK pathway in 105 metastatic melanoma patients, the researchers found two novel nonsynonymous mutations (R2443∗ and L552F) in MTOR gene (Shull et al., 2012).
In our dataset, mutations and CNVs in MTOR are found in 7.9 and 2.3% of the samples, respectively, as shown in Figures 2, 3 and Supplementary Figure S8B. However, among the 78 MTOR coding mutations found in our dataset, only five were pathogenic (6.4%; 2 E1799K, 2 S2215F, and 1 I2500F). Interestingly, these mutations have previously been identified as activating mutations in cancer patients using publicly available databases of cancer genome sequencing data (Grabiner et al., 2014). This study found 33 MTOR mutations that confer pathway activation, not by reducing sensitivity to mTOR inhibitors, but by altering the response of mTORC1 signaling pathway to nutrient deprivation (Grabiner et al., 2014).
Regarding melanoma therapy, MTOR may be targeted by selective inhibitors (Chamcheu et al., 2019). Rapamycin, a specific mTORC1 inhibitor, inhibits the cell growth and proliferation in several melanoma cell lines (Buscà et al., 1996; Molhoek et al., 2005; Karbowniczek et al., 2008). Two other rapamycin analogs, everolimus, and temsirolimus, also showed promising results in preclinical studies but failed the clinical experimentation in melanoma patients (Table 2).
Everolimus, as single agent, was tested in a phase II trial with 53 metastatic melanoma patients: the drug led to obtain just two clinical responses with a heavy toxicity profile and the study was terminated for futility (Vera Aguilera et al., 2018). A combination of everolimus with carboplatin and paclitaxel as I line treatment for metastatic melanoma was also tested in a phase II trial. Among 70 enrolled patients, just 12 showed objective responses, with a median PFS of 4 months and a median OS of 10 months (Hauke et al., 2013). Bevacizumab and everolimus were tested in a phase clinical II trial with 57 metastatic melanoma patients and led to a disease control rate of 70%. Unfortunately, median PFS and OS were just 4 month and 8.6 months, respectively (Hainsworth et al., 2010). Lastly, a recent study evaluated the combination of everolimus, bevacizumab, carboplatin and paclitaxel (CPB) in a randomized phase II trial. A total of 149 patients affected by stage IV melanoma were treated with CPB or with CPB plus everolimus: no differences in terms of PFS or OS were noted among the two groups, but everolimus strongly increased toxicity (McWilliams et al., 2018).
A new attempt to bring attention back to everolimus comes from a recently published phase I study, that evaluated the safety and the efficacy of vatalanib plus everolimus in patients with advanced solid tumors, including melanoma. Among 70 evaluable patients, nine achieved a partial response and 41 a stable disease, with best results obtained in neuroendocrine tumors, renal cancer, melanoma and NSCLC (Zhu et al., 2020).
Regarding the use of everolimus in mTOR mutated patients, only two clinical trials exist to date, to our knowledge. The first one is a phase II clinical trial on everolimus in cancer patients with TSC1 and TSC2 mutation or activating MTOR mutation. The second is a single-arm, open-labeled and single-centered study of everolimus in selective patients with metastatic melanoma and mutations (Kinase domain) of mTOR. Both studies are ongoing, and thus results are not yet available.
Temsirolimus was tested in combination with sorafenib in a phase I (Davies et al., 2012) and in a phase II clinical trials (Margolin et al., 2012), with disappointing results. Indeed, not only this combination failed to achieve any clinical response, but it induced a significant toxicity at higher dose levels. More encouraging results came from a phase II clinical trial with temsirolimus and bevacizumab: among 17 treated patients, three obtained a partial response and nine a stable disease, with best responses obtained in BRAF wild type population (Slingluff et al., 2013).
TACC1
TACC1 gene belongs to the TACC gene family that encodes centrosomal proteins that may play roles in microtubule regulation and spindle function, and, thus, may be an important driver of genomic instability in cancer cells (Anafi et al., 2000; Conte et al., 2003). Moreover, TACC1 proteins play an important role in transcriptional regulation (Gas41, thyroid hormone receptor, and retinoid acid receptor α) and mRNA processing (LSM7 and SmG) (Conte et al., 2002; Guyot et al., 2010). Many studies reported that TACC1 expression was modified in several cancers (Line et al., 2002; Rhodes et al., 2002; Nguyen et al., 2005; Ghayad et al., 2009). TACC1 has been also reported to stimulate the RAS and PI3K pathways playing an oncogenic role in tumor formation in the murine mammary gland (Cully et al., 2005). Finally, FGFR-TACC fusions, including FGFR1-TACC1, were also described in glioblastomas since 2012 (Di Stefano et al., 2015; Lasorella et al., 2017).
TACC1 mutations were first reported in a WES study of 121 melanoma tumor/normal pairs reporting five novel melanoma candidate genes, including TACC1, with a frequency of 7% which was confirmed in a further similar study of 25 metastatic/germline pairs (Berger et al., 2012; Hodis et al., 2012) (Supplementary Figure S8C). Finally, in a recent study several key genes involved in melanomagenesis, including TACC1, in 115 human melanoma cell lines, 248 patient-derived xenografts, 31 cell lines derived from PDX, and 68 patient tumors, an overall frequency rate of 1.47% was reported (Garman et al., 2017). In conclusion, in our dataset TACC1 mutations and CNVs have a frequency of 2.9 and 2.3%, as shown in Figures 2, 3 and Supplementary Figure S8C. No clinical trials are available.
CNOT9
CNOT9, also known as RQCD1, encodes for the CCR4-NOT transcription complex subunit 9 that plays a role as transcription cofactor in multiple biological processes including cellular differentiation and RNA processing (Mathys et al., 2014). Several NGS studies revealed the presence at low frequency of recurrent mutations in RQCD1/CNOT9 (P131L) in melanoma samples (Dutton-Regester et al., 2014). Interestingly, Wong SQ et al. found that melanoma tumors harboring RQCD1/CNOT9 P131L mutation were associated with increased thickness, head and neck and upper limb location, lentigo maligna melanoma subtype, and BRAF V600K but not V600E or NRAS Q61 mutations (Wong et al., 2015). CNOT9 alterations in our dataset are shown in Figures 2, 3 and Supplementary Figure S8D (frequency mutations and CNVs of 2.9 and 1.3%, respectively).
The functional oncogenic role of CNOT9/RQCD1 in melanoma remains currently unknown although limited studies revealed RQCD1 implication in AKT activation and cell proliferation (Ajiro et al., 2009, 2010). In particular, the CNOT9/RQCD1 mutant has demonstrated to stimulate stronger immune responses than the wild type, implicating the formation of a neoantigen that could be a potential therapeutic target although no drugs or clinical trials are available (Wong et al., 2015).
YAP1
YAP1 encodes for transcriptional coactivators belonging to the Hippo pathway. The core of the Hippo pathway in mammals consists of a kinase cascade, MST1/2 and LATS1/2, as well as downstream effectors, transcriptional coactivators YAP and TAZ (Ma et al., 2019). The Hippo pathway plays a crucial role in organ size control by regulation of cell proliferation, apoptosis, cell differentiation, and cell migration. Moreover, the upregulation of the Hippo pathway downstream effectors, such as YAP and TAZ, are common across various cancers since mutations and altered expression of its core components promote the migration, invasion, malignancy of cancer cells (Moroishi et al., 2015; Yu et al., 2015; Han, 2019). Very recently, 9,125 tumor samples were profiled revealing a widespread dysregulation of Hippo pathway components in multiple human cancer types, including melanoma (Sanchez-Vega et al., 2018). In particular, a strong interaction between Hippo pathway and GNAQ and GNA11 oncogenes was observed. Several studies reported the functional role of YAP in uveal melanoma cells carrying GNAQ/11 mutations (Feng et al., 2014; Yu et al., 2014). In uveal melanoma ATCG samples, only one among 80 uveal melanoma patients revealed a missense mutation in YAP (G369R) co-occurring with GNAQ R183Q and GNA11 R183C (Bakhoum and Esmaeli, 2019). Moreover, no YAP CNV was shown in uveal melanoma samples by a comprehensive molecular characterization of the Hippo core genes in 9,125 tumor samples (Wang Y. et al., 2018). In our skin melanoma dataset, YAP1 alterations are reported in Figures 2, 3 and Supplementary Figure S10, with an overall frequency mutation and CNVs of 1.4 and 2.6%, respectively.
Although the central role of YAP in uveal melanomagenesis is deeply investigated, the role of Hippo/YAP signaling in cutaneous melanoma is less understood. Zhang et al. (2019) proved for the first time that YAP hyperactivity is elevated in invasive melanoma cell lines, induce invasion in normally non-invasive melanoma cells, induces spontaneous melanoma metastasis in vivo and promotes melanoma cell invasion by regulating expression of AXL, CYR61 and CRIM1. Moreover, they did not found association between YAP activity and the BRAF/NRAS mutation status. Of note, they found for the first time somatic activating mutations in YAP gene by WES. Indeed, they identified seven independent serine to alanine substitutions in YAP gene (called YAP-7SA) from a primary cutaneous melanoma patient carrying BRAF V600E and RAC1 P29S mutation. In vitro study confirmed that YAP-7SA encodes a hyperactive version of the YAP protein (Zhang et al., 2019). Despite the great interest of scientific community toward the role of Hippo pathway in cancer, no drugs and clinical trials direct to YAP in melanoma are described.
Conclusion and Perspectives
The better definition of the genetic mechanisms underlying melanomagenesis allowed to synthesize new molecular targeted drugs that radically changed the prognosis of melanoma patients.
In this review we analyzed more than 30 genes other than BRAF, that can be or become targets for new molecular target therapies. We collected mutation data on a total of 992 melanoma samples analyzed by WES or WGS to prove the genetic heterogeneity of melanoma, beyond the BRAF mutation. Only 11 of them (1.1%) showed no coding mutations, whereas 298 (30%) showed a mutation in one of the RAS genes (mostly NRAS) and most of the samples studied (698, 70%) showed mutations in non-RAS genes (Figures 2, 3) suggesting potential new roads/candidates for targeted therapy.
We also provide an overview of the related clinical trials, ongoing or completed, to better outline the state of molecular – derived clinical research (Table 2). Among the 33 selected established and candidate melanoma driver genes here described, clinical trials have been performed for 11 genes (33.3%) and this percentage could be increased if we considered ongoing trials with immunotherapy although it was not the topic of our review. Moreover, several preclinical studies are showing that some drugs could have anti-tumor ability toward melanoma driver genes opening future clinical studies and new potential therapeutic perspectives.
In this scenario, we strongly believe that in a near future, the use of NGS could extend the cohort of patients that could benefit by this therapeutic approach. Indeed, at least two clinical trials including melanoma patients (NCT02465060 and NCT02645149) are currently ongoing to prove this concept, enrolling hundreds of patients regardless of their neoplasm, and treating them with the specific drug targeting the specific identified mutation. Hopefully, the results of these experimentations will be a milestone in medical oncology because will allow us to put into practice a real personalized therapy, tailored on what is written in tumor genome.
Author Contributions
IV and ET contributed equally in conceiving the review focus, conducting the literature review and summarizing the manuscript, analyzed data, wrote the first draft, and finalized the manuscript. PG designed and coordinated the review. BD, LP, VA, WB, AB, FS, and PG reviewed the literature, revised and made corrections to the manuscript. All authors have read and agreed to the final version of the manuscript.
Funding
We acknowledge the funds granted by the Italian Ministry of Health RF-2016-02362288, Ricerca Corrente to Ospedale Policlinico San Martino, and University of Genoa, FRA 2017.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2020.00172/full#supplementary-material
FIGURE S1 | NRAS (A), KRAS (B), HRAS (C), and PREX2 (D) mutations and CNV frequency in the 10 NGS selected studies. Mutations are indicated in green and CNV in purple. Only four studies with available CNV information were considered for CNV analysis.
FIGURE S2 | TP53 (A), NF1 (B), ARID2 (C), and CDKN2A (D) mutations and CNV frequency in the 10 NGS selected studies. Mutations are indicated in green and CNV in purple. Only four studies with available CNV information were considered for CNV analysis.
FIGURE S3 | CDK4 (A), PTEN (B), PPP6C (C), and CTNNB1 (D) mutations and CNV frequency in the 10 NGS selected studies. Mutations are indicated in green and CNV in purple. Only four studies with available CNV information were considered for CNV analysis.
FIGURE S4 | DDX3X (A), RASA2 (B), SF3B1 (C), and RAC1 (D) mutations and CNV frequency in the 10 NGS selected studies Mutations are indicated in green and CNV in purple. Only four studies with available CNV information were considered for CNV analysis.
FIGURE S5 | MAP2K1 (A), MAP2K2 (B), KIT (C), and RB1 (D) mutations and CNV frequency in the 10 NGS selected studies. Mutations are indicated in green and CNV in purple. Only four studies with available CNV information were considered for CNV analysis.
FIGURE S6 | FBXW7 (A), PIK3CA (B), EZH2 (C), and WT1 (D) mutations and CNV frequency in the 10 NGS selected studies. Mutations are indicated in green and CNV in purple. Only four studies with available CNV information were considered for CNV analysis.
FIGURE S7 | SNX31 (A), IDH1 (B), STK19 (C), and MITF (D) mutations and CNV frequency in the 10 NGS selected studies. Mutations are indicated in green and CNV in purple. Only four studies with available CNV information were considered for CNV analysis.
FIGURE S8 | TERT (A), MTOR (B), TACC1 (C), and CNOT9 (D) mutations and CNV frequency in the 10 NGS selected studies. Mutations are indicated in green and CNV in purple. Only four studies with available CNV information were considered for CNV analysis.
FIGURE S9 | TERT promoter mutations in 160 skin melanomas. TERT promoter mutations were derived by 129 cutaneous melanoma and 31 acral melanomas (Hayward et al., 2017).
FIGURE S10 | YAP1 mutations and CNV frequency in the 10 NGS selected studies. Mutations are indicated in green and CNV in purple. Only four studies with available CNV information were considered for CNV analysis.
Footnotes
References
AACR Project Genie Consortium (2017). AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. 7, 818–831. doi: 10.1158/2159-8290.CD-17-0151
Adjei, A. A., Cohen, R. B., Franklin, W., Morris, C., Wilson, D., Molina, J. R., et al. (2008). Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J. Clin. Oncol. 26, 2139–2146. doi: 10.1200/JCO.2007.14.4956
Adjei, A. A., LoRusso, P., Ribas, A., Sosman, J. A., Pavlick, A., Dy, G. K., et al. (2017). A phase I dose-escalation study of TAK-733, an investigational oral MEK inhibitor, in patients with advanced solid tumors. Invest. New Drugs 35, 47–58. doi: 10.1007/s10637-016-0391-2
Ajiro, M., Katagiri, T., Ueda, K., Nakagawa, H., Fukukawa, C., Lin, M. L., et al. (2009). Involvement of RQCD1 overexpression, a novel cancer-testis antigen, in the Akt pathway in breast cancer cells. Int. J. Oncol. 35, 673–681.
Ajiro, M., Nishidate, T., Katagiri, T., and Nakamura, Y. (2010). Critical involvement of RQCD1 in the EGFR-Akt pathway in mammary carcinogenesis. Int. J. Oncol. 37, 1085–1093. doi: 10.3892/ijo_00000760
Akhoondi, S., Sun, D., von der Lehr, N., Apostolidou, S., Klotz, K., Maljukova, A., et al. (2007). FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 67, 9006–9012. doi: 10.1158/0008-5472.CAN-07-1320
Akslen, L. A., Angelini, S., Straume, O., Bachmann, I. M., Molven, A., Hemminki, K., et al. (2005). BRAF and NRAS mutations are frequent in nodular melanoma but are not associated with tumor cell proliferation or patient survival. J. Invest. Dermatol. 125, 312–317. doi: 10.1111/j.0022-202X.2005.23788.x
Algazi, A. P., Esteve-Puig, R., Nosrati, A., Hinds, B., Hobbs-Muthukumar, A., Nandoskar, P., et al. (2018). Dual MEK/AKT inhibition with trametinib and GSK2141795 does not yield clinical benefit in metastatic NRAS-mutant and wild-type melanoma. Pigment Cell Melanoma Res. 31, 110–114. doi: 10.1111/pcmr.12644
Alon, M., Arafeh, R., Lee, J. S., Madan, S., Kalaora, S., Nagler, A., et al. (2019). Correction: CAPN1 is a novel binding partner and regulator of the tumor suppressor NF1 in melanoma. Oncotarget 10:1344. doi: 10.18632/oncotarget.26685
Anafi, M., Yang, Y. F., Barlev, N. A., Govindan, M. V., Berger, S. L., Butt, T. R., et al. (2000). GCN5 and ADA adaptor proteins regulate triiodothyronine/GRIP1 and SRC-1 coactivator-dependent gene activation by the human thyroid hormone receptor. Mol. Endocrinol. 14, 718–732. doi: 10.1210/mend.14.5.0457
Arafeh, R., Di Pizio, A., Elkahloun, A. G., Dym, O., Niv, M. Y., and Samuels, Y. (2019). RASA2 and NF1; two-negative regulators of Ras with complementary functions in melanoma. Oncogene 38, 2432–2434. doi: 10.1038/s41388-018-0578-4
Arozarena, I., Bischof, H., Gilby, D., Belloni, B., Dummer, R., and Wellbrock, C. (2011). In melanoma, beta-catenin is a suppressor of invasion. Oncogene 30, 4531–4543. doi: 10.1038/onc.2011.162
Ascierto, P. A., Schadendorf, D., Berking, C., Agarwala, S. S., van Herpen, C. M., Queirolo, P., et al. (2013). MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 14, 249–256. doi: 10.1016/S1470-2045(13)70024-X
Aydin, I. T., Melamed, R. D., Adams, S. J., Castillo-Martin, M., Demir, A., Bryk, D., et al. (2014). FBXW7 mutations in melanoma and a new therapeutic paradigm. J. Natl. Cancer Inst. 106:dju107. doi: 10.1093/jnci/dju107
Baines, A. T., Xu, D., and Der, C. J. (2011). Inhibition of Ras for cancer treatment: the search continues. Future Med. Chem. 3, 1787–1808. doi: 10.4155/fmc.11.121
Bakhoum, M. F., and Esmaeli, B. (2019). Molecular characteristics of uveal melanoma: insights from the cancer genome atlas (TCGA) project. Cancers 11:e081061. doi: 10.3390/cancers11081061
Balss, J., Meyer, J., Mueller, W., Korshunov, A., Hartmann, C., and von Deimling, A. (2008). Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 116, 597–602. doi: 10.1007/s00401-008-0455-2
Barsotti, A. M., Ryskin, M., and Rollins, R. A. (2015). Epigenetic reprogramming in solid tumors: therapeutic implications of EZH2 gain-of-function mutations. Epigenomics 7, 687–690. doi: 10.2217/epi.15.27
Barthel, F. P., Wei, W., Tang, M., Martinez-Ledesma, E., Hu, X., Amin, S. B., et al. (2017). Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 49, 349–357. doi: 10.1038/ng.3781
Bartkova, J., Lukas, J., Guldberg, P., Alsner, J., Kirkin, A. F., Zeuthen, J., et al. (1996). The p16-cyclin D/Cdk4-pRb pathway as a functional unit frequently altered in melanoma pathogenesis. Cancer Res. 56, 5475–5483.
Bastian, B. C., LeBoit, P. E., and Pinkel, D. (2000). Mutations and copy number increase of HRAS in spitz nevi with distinctive histopathological features. Am. J. Pathol. 157, 967–972.
Bastian, B. C., Wesselmann, U., Pinkel, D., and Leboit, P. E. (1999). Molecular cytogenetic analysis of Spitz nevi shows clear differences to melanoma. J. Invest. Dermatol. 113, 1065–1069. doi: 10.1046/j.1523-1747.1999.00787.x
Bastians, H., Krebber, H., Vetrie, D., Hoheisel, J., Lichter, P., Ponstingl, H., et al. (1997). Localization of the novel serine/threonine protein phosphatase 6 gene (PPP6C) to human chromosome Xq22.3. Genomics 41, 296–297. doi: 10.1006/geno.1997.4640
Berger, M. F., Hodis, E., Heffernan, T. P., Deribe, Y. L., Lawrence, M. S., Protopopov, A., et al. (2012). Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 485, 502–506. doi: 10.1038/nature11071
Beroukhim, R., Mermel, C. H., Porter, D., Wei, G., Raychaudhuri, S., Donovan, J., et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. doi: 10.1038/nature08822
Birck, A., Ahrenkiel, V., Zeuthen, J., Hou-Jensen, K., and Guldberg, P. (2000). Mutation and allelic loss of the PTEN/MMAC1 gene in primary and metastatic melanoma biopsies. J. Invest. Dermatol. 114, 277–280. doi: 10.1046/j.1523-1747.2000.00877.x
Birkeland, E., Zhang, S., Poduval, D., Geisler, J., Nakken, S., Vodak, D., et al. (2018). Patterns of genomic evolution in advanced melanoma. Nat. Commun. 9:2665. doi: 10.1038/s41467-018-05063-1
Boeing, S., Williamson, L., Encheva, V., Gori, I., Saunders, R. E., Instrell, R., et al. (2016). Multiomic analysis of the UV-Induced DNA damage response. Cell Rep. 15, 1597–1610. doi: 10.1016/j.celrep.2016.04.047
Bol, G. M., Vesuna, F., Xie, M., Zeng, J., Aziz, K., Gandhi, N., et al. (2015). Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol. Med. 7, 648–669. doi: 10.15252/emmm.201404368
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Brock, E. J., Ji, K., Reiners, J. J., and Mattingly, R. R. (2016). How to target activated ras proteins: direct Inhibition vs. Induced mislocalization. Mini. Rev. Med. Chem. 16, 358–369. doi: 10.2174/1389557515666151001154002
Bruno, W., Martinuzzi, C., Dalmasso, B., Andreotti, V., Pastorino, L., Cabiddu, F., et al. (2018). Correction: combining molecular and immunohistochemical analyses of key drivers in primary melanomas: interplay between germline and somatic variations. Oncotarget 9:28798. doi: 10.18632/oncotarget.25684
Buchbinder, E. I., Sosman, J. A., Lawrence, D. P., McDermott, D. F., Ramaiya, N. H., Van den Abbeele, A. D., et al. (2015). Phase 2 study of sunitinib in patients with metastatic mucosal or acral melanoma. Cancer 121, 4007–4015. doi: 10.1002/cncr.29622
Buscà, R., Bertolotto, C., Ortonne, J. P., and Ballotti, R. (1996). Inhibition of the phosphatidylinositol 3-kinase/p70(S6)-kinase pathway induces B16 melanoma cell differentiation. J. Biol. Chem. 271, 31824–31830. doi: 10.1074/jbc.271.50.31824
Carreira, S., Goodall, J., Aksan, I., La Rocca, S. A., Galibert, M.-D., Denat, L., et al. (2005). Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature 433, 764–769. doi: 10.1038/nature03269
Carvajal, R. D., Antonescu, C. R., Wolchok, J. D., Chapman, P. B., Roman, R.-A., Teitcher, J., et al. (2011). KIT as a therapeutic target in metastatic melanoma. JAMA 305, 2327–2334. doi: 10.1001/jama.2011.746
Carvajal, R. D., Lawrence, D. P., Weber, J. S., Gajewski, T. F., Gonzalez, R., Lutzky, J., et al. (2015). Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin. Cancer Res. 21, 2289–2296. doi: 10.1158/1078-0432.CCR-14-1630
Catalanotti, F., Solit, D. B., Pulitzer, M. P., Berger, M. F., Scott, S. N., Iyriboz, T., et al. (2013). Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K- mutated melanoma. Clin. Cancer Res. 19, 2257–2264. doi: 10.1158/1078-0432.CCR-12-3476
cBioPortal for Cancer Genomics (2020). Available online at: http://www.cbioportal.org/ (accessed April 25, 2020).
Chamcheu, J. C., Roy, T., Uddin, M. B., Banang-Mbeumi, S., Chamcheu, R.-C. N., Walker, A. L., et al. (2019). Role and therapeutic targeting of the PI3K/Akt/mTOR signaling pathway in skin cancer: a review of current status and future trends on natural and synthetic agents therapy. Cells 8:803. doi: 10.3390/cells8080803
Cheli, Y., Ohanna, M., Ballotti, R., and Bertolotto, C. (2010). Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 23, 27–40. doi: 10.1111/j.1755-148X.2009.00653.x
Cheng, Y., Chen, G., Martinka, M., Ho, V., and Li, G. (2013). Prognostic significance of Fbw7 in human melanoma and its role in cell migration. J. Invest. Dermatol. 133, 1794–1802. doi: 10.1038/jid.2013.58
Chiosea, S. I., Sherer, C. K., Jelic, T., and Dacic, S. (2011). KRAS mutant allele-specific imbalance in lung adenocarcinoma. Mod. Pathol. 24, 1571–1577. doi: 10.1038/modpathol.2011.109
Cho, J., Kim, S. Y., Kim, Y. J., Sim, M. H., Kim, S. T., Kim, N. K. D., et al. (2017). Emergence of CTNNB1 mutation at acquired resistance to KIT inhibitor in metastatic melanoma. Clin. Transl. Oncol. 19, 1247–1252. doi: 10.1007/s12094-017-1662-x
Cirenajwis, H., Lauss, M., Ekedahl, H., Törngren, T., Kvist, A., Saal, L. H., et al. (2017). NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol. Oncol. 11, 438–451. doi: 10.1002/1878-0261.12050
Conte, N., Charafe-Jauffret, E., Delaval, B., Adélaïde, J., Ginestier, C., Geneix, J., et al. (2002). Carcinogenesis and translational controls: TACC1 is down-regulated in human cancers and associates with mRNA regulators. Oncogene 21, 5619–5630. doi: 10.1038/sj.onc.1205658
Conte, N., Delaval, B., Ginestier, C., Ferrand, A., Isnardon, D., Larroque, C., et al. (2003). TACC1-chTOG-Aurora A protein complex in breast cancer. Oncogene 22, 8102–8116. doi: 10.1038/sj.onc.1206972
COSMIC (2020). The Catalogue of Somatic Mutations in Cancer (COSMIC). Available online at: https://www.sanger.ac.uk/science/tools/cosmic (accessed April 28, 2020).
Cox, A. D., Der, C. J., and Philips, M. R. (2015). Targeting RAS membrane association: back to the future for Anti-Ras drug discovery? Clin. Cancer Res. 21, 1819–1827. doi: 10.1158/1078-0432.CCR-14-3214
Cully, M., Shiu, J., Piekorz, R. P., Muller, W. J., Done, S. J., and Mak, T. W. (2005). Transforming acidic coiled coil 1 promotes transformation and mammary tumorigenesis. Cancer Res. 65, 10363–10370. doi: 10.1158/0008-5472.CAN-05-1633
Curley, D. P., and Bosenberg, M. W. (2008). A new mechanism of release from senescence: suppression of p16INK4a by beta-catenin. Pigment Cell Melanoma Res. 21, 5–6. doi: 10.1111/j.1755-148X.2007.00434.x
Curtin, J. A., Busam, K., Pinkel, D., and Bastian, B. C. (2006). Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 24, 4340–4346. doi: 10.1200/JCO.2006.06.2984
Curtin, J. A., Fridlyand, J., Kageshita, T., Patel, H. N., Busam, K. J., Kutzner, H., et al. (2005). Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 353, 2135–2147. doi: 10.1056/NEJMoa050092
Davies, H., Bignell, G. R., Cox, C., Stephens, P., Edkins, S., Clegg, S., et al. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949–954. doi: 10.1038/nature00766
Davies, M. A. (2012). The role of the PI3K-AKT pathway in melanoma. Cancer J 18, 142–147. doi: 10.1097/PPO.0b013e31824d448c
Davies, M. A., Fox, P. S., Papadopoulos, N. E., Bedikian, A. Y., Hwu, W.-J., Lazar, A. J., et al. (2012). Phase I study of the combination of sorafenib and temsirolimus in patients with metastatic melanoma. Clin. Cancer Res. 18, 1120–1128. doi: 10.1158/1078-0432.CCR-11-2436
Davis, E. J., Johnson, D. B., Sosman, J. A., and Chandra, S. (2018). Melanoma: what do all the mutations mean? Cancer 124, 3490–3499. doi: 10.1002/cncr.31345
Davis, M. E., Zuckerman, J. E., Choi, C. H. J., Seligson, D., Tolcher, A., Alabi, C. A., et al. (2010). Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 464, 1067–1070. doi: 10.1038/nature08956
de Unamuno Bustos, B., Murria Estal, R., Pérez Simó, G., de Juan Jimenez, I., Escutia Muñoz, B., Rodríguez Serna, M., et al. (2017). Towards personalized medicine in melanoma: implementation of a clinical next-generation sequencing panel. Sci. Rep. 7:495. doi: 10.1038/s41598-017-00606-w
DeLeon, T. T., Almquist, D. R., Kipp, B. R., Langlais, B. T., Mangold, A., Winters, J. L., et al. (2020). Assessment of clinical outcomes with immune checkpoint inhibitor therapy in melanoma patients with CDKN2A and TP53 pathogenic mutations. PLoS One 15:e0230306. doi: 10.1371/journal.pone.0230306
Demirci, H., Demirci, F. Y., Ciftci, S., Elner, V. M., Wu, Y.-M., Ning, Y., et al. (2019). Integrative exome and transcriptome analysis of conjunctival melanoma and its potential application for personalized therapy. JAMA Ophthalmol. 137, 1444–1448. doi: 10.1001/jamaophthalmol.2019.4237
Deng, W., Gopal, Y. N. V., Scott, A., Chen, G., Woodman, S. E., and Davies, M. A. (2012). Role and therapeutic potential of PI3K-mTOR signaling in de novo resistance to BRAF inhibition. Pigment Cell Melanoma Res. 25, 248–258. doi: 10.1111/j.1755-148X.2011.00950.x
Devitt, B., Liu, W., Salemi, R., Wolfe, R., Kelly, J., Tzen, C.-Y., et al. (2011). Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res 24, 666–672. doi: 10.1111/j.1755-148X.2011.00873.x
Di Stefano, A. L., Fucci, A., Frattini, V., Labussiere, M., Mokhtari, K., Zoppoli, P., et al. (2015). Detection, characterization, and inhibition of FGFR-TACC fusions in IDH Wild-type glioma. Clin. Cancer Res. 21, 3307–3317. doi: 10.1158/1078-0432.CCR-14-2199
Dimonitsas, E., Liakea, A., Sakellariou, S., Thymara, I., Giannopoulos, A., Stratigos, A., et al. (2018). An update on molecular alterations in melanocytic tumors with emphasis on Spitzoid lesions. Ann. Transl. Med. 6:249. doi: 10.21037/atm.2018.05.23
DiNardo, C. D., Stein, E. M., de Botton, S., Roboz, G. J., Altman, J. K., Mims, A. S., et al. (2018). Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N. Engl. J. Med. 378, 2386–2398. doi: 10.1056/NEJMoa1716984
Donehower, L. A., Soussi, T., Korkut, A., Liu, Y., Schultz, A., Cardenas, M., et al. (2019). Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. 28:3010. doi: 10.1016/j.celrep.2019.08.061
Dono, M., Angelini, G., Cecconi, M., Amaro, A., Esposito, A. I., Mirisola, V., et al. (2014). Mutation frequencies of GNAQ, GNA11, BAP1, SF3B1, EIF1AX and TERT in uveal melanoma: detection of an activating mutation in the TERT gene promoter in a single case of uveal melanoma. Br. J. Cancer 110, 1058–1065. doi: 10.1038/bjc.2013.804
Dummer, R., Schadendorf, D., Ascierto, P., Fernández, A., Dutriaux, C., Maio, M., et al. (2016). Results of NEMO: a phase III trial of binimetinib (BINI) vs dacarbazine (DTIC) in NRAS-mutant cutaneous melanoma. ASCO Meet. Abstracts 34:9500. doi: 10.1200/JCO.2016.34.15_suppl.9500
Dummer, R., Schadendorf, D., Ascierto, P. A., Arance, A., Dutriaux, C., Di Giacomo, A. M., et al. (2017). Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 18, 435–445. doi: 10.1016/S1470-2045(17)30180-8
Dutton-Regester, K., Gartner, J. J., Emmanuel, R., Qutob, N., Davies, M. A., Gershenwald, J. E., et al. (2014). A highly recurrent RPS27 5’UTR mutation in melanoma. Oncotarget 5, 2912–2917. doi: 10.18632/oncotarget.2048
Edlundh-Rose, E., Egyházi, S., Omholt, K., Månsson-Brahme, E., Platz, A., Hansson, J., et al. (2006). NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Res. 16, 471–478. doi: 10.1097/01.cmr.0000232300.22032.86
El Kharbili, M., Agaësse, G., Barbollat-Boutrand, L., Pommier, R. M., de la Fouchardière, A., Larue, L., et al. (2019). Tspan8-β-catenin positive feedback loop promotes melanoma invasion. Oncogene 38, 3781–3793. doi: 10.1038/s41388-019-0691-z
Ellerhorst, J. A., Greene, V. R., Ekmekcioglu, S., Warneke, C. L., Johnson, M. M., Cooke, C. P., et al. (2011). Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin. Cancer Res. 17, 229–235. doi: 10.1158/1078-0432.CCR-10-2276
Fane, M. E., Chhabra, Y., Hollingsworth, D. E. J., Simmons, J. L., Spoerri, L., Oh, T. G., et al. (2017). NFIB mediates BRN2 driven melanoma cell migration and invasion through regulation of EZH2 and MITF. EBiomedicine 16, 63–75. doi: 10.1016/j.ebiom.2017.01.013
Feng, X., Degese, M. S., Iglesias-Bartolome, R., Vaque, J. P., Molinolo, A. A., Rodrigues, M., et al. (2014). Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 25, 831–845. doi: 10.1016/j.ccr.2014.04.016
Filali, M., Li, S., Kim, H. W., Wadzinski, B., and Kamoun, M. (1999). Identification of a type 6 protein ser/thr phosphatase regulated by interleukin-2 stimulation. J. Cell. Biochem. 73, 153–163.
Fine, B., Hodakoski, C., Koujak, S., Su, T., Saal, L. H., Maurer, M., et al. (2009). Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science 325, 1261–1265. doi: 10.1126/science.1173569
Flaherty, K. T., Robert, C., Hersey, P., Nathan, P., Garbe, C., Milhem, M., et al. (2012). Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 367, 107–114. doi: 10.1056/NEJMoa1203421
Forero-Torres, A., Ramchandren, R., Yacoub, A., Wertheim, M. S., Edenfield, W. J., Caimi, P., et al. (2019). Parsaclisib, a potent and highly selective PI3Kδ inhibitor, in patients with relapsed or refractory B-cell malignancies. Blood 133, 1742–1752. doi: 10.1182/blood-2018-08-867499
Fritsch, C., Huang, A., Chatenay-Rivauday, C., Schnell, C., Reddy, A., Liu, M., et al. (2014). Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther. 13, 1117–1129. doi: 10.1158/1535-7163.MCT-13-0865
Fuse, N., Yasumoto, K., Takeda, K., Amae, S., Yoshizawa, M., Udono, T., et al. (1999). Molecular cloning of cDNA encoding a novel microphthalmia-associated transcription factor isoform with a distinct amino-terminus. J. Biochem. 126, 1043–1051. doi: 10.1093/oxfordjournals.jbchem.a022548
Gajewski, T. F., Salama, A. K., Niedzwiecki, D., Johnson, J., Linette, G., Bucher, C., et al. (2012). Phase II study of the farnesyltransferase inhibitor R115777 in advanced melanoma (CALGB 500104). J. Transl. Med. 10:246. doi: 10.1186/1479-5876-10-246
Garman, B., Anastopoulos, I. N., Krepler, C., Brafford, P., Sproesser, K., Jiang, Y., et al. (2017). Genetic and genomic characterization of 462 melanoma patient-derived xenografts, tumor biopsies, and cell lines. Cell Rep. 21, 1936–1952. doi: 10.1016/j.celrep.2017.10.052
Garraway, L. A., Widlund, H. R., Rubin, M. A., Getz, G., Berger, A. J., Ramaswamy, S., et al. (2005). Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436, 117–122. doi: 10.1038/nature03664
Garrido-Ruiz, M. C., Rodriguez-Pinilla, S. M., Pérez-Gómez, B., and Rodriguez-Peralto, J. L. (2010). WT 1 expression in nevi and melanomas: a marker of melanocytic invasion into the dermis. J. Cutan. Pathol. 37, 542–548. doi: 10.1111/j.1600-0560.2009.01379.x
Gelbert, L. M., Cai, S., Lin, X., Sanchez-Martinez, C., del Prado, M., Lallena, M. J., et al. (2014). Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest. New Drugs 32, 825–837. doi: 10.1007/s10637-014-0120-7
Ghai, R., and Collins, B. M. (2011). PX-FERM proteins: a link between endosomal trafficking and signaling? Small GTPases 2, 259–263. doi: 10.4161/sgtp.2.5.17276
Ghai, R., Mobli, M., and Collins, B. M. (2014). Measuring interactions of FERM domain-containing sorting nexin proteins with endosomal lipids and cargo molecules. Meth. Enzymol. 534, 331–349. doi: 10.1016/B978-0-12-397926-1.00019-6
Ghayad, S. E., Vendrell, J. A., Bieche, I., Spyratos, F., Dumontet, C., Treilleux, I., et al. (2009). Identification of TACC1, NOV, and PTTG1 as new candidate genes associated with endocrine therapy resistance in breast cancer. J. Mol. Endocrinol. 42, 87–103. doi: 10.1677/JME-08-0076
Giles, F., Fischer, T., Cortes, J., Garcia-Manero, G., Beck, J., Ravandi, F., et al. (2006). A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin. Cancer Res. 12, 4628–4635. doi: 10.1158/1078-0432.CCR-06-0511
Goding, C. R. (2011). Commentary. A picture of Mitf in melanoma immortality. Oncogene 30, 2304–2306. doi: 10.1038/onc.2010.641
Gold, H. L., Wengrod, J., de Miera, E. V.-S., Wang, D., Fleming, N., Sikkema, L., et al. (2014). PP6C hotspot mutations in melanoma display sensitivity to Aurora kinase inhibition. Mol. Cancer Res. 12, 433–439. doi: 10.1158/1541-7786.MCR-13-0422
Gomez-Escobar, N., Chou, C. F., Lin, W. W., Hsieh, S. L., and Campbell, R. D. (1998). The G11 gene located in the major histocompatibility complex encodes a novel nuclear serine/threonine protein kinase. J. Biol. Chem. 273, 30954–30960. doi: 10.1074/jbc.273.47.30954
Gong, H. Z., Zheng, H. Y., and Li, J. (2018). The clinical significance of KIT mutations in melanoma: a meta-analysis. Melanoma Res. 28, 259–270. doi: 10.1097/CMR.0000000000000454
Grabiner, B. C., Nardi, V., Birsoy, K., Possemato, R., Shen, K., Sinha, S., et al. (2014). A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 4, 554–563. doi: 10.1158/2159-8290.CD-13-0929
Guldberg, P., Straten, P., Birck, A., Ahrenkiel, V., Kirkin, A. F., and Zeuthen, J. (1997). Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer Res. 57, 3660–3663.
Guo, J., Carvajal, R. D., Dummer, R., Hauschild, A., Daud, A., Bastian, B. C., et al. (2017). Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: final results from the global, single-arm, phase II TEAM trial. Ann. Oncol. 28, 1380–1387. doi: 10.1093/annonc/mdx079
Guo, J., Si, L., Kong, Y., Flaherty, K. T., Xu, X., Zhu, Y., et al. (2011). Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J. Clin. Oncol. 29, 2904–2909. doi: 10.1200/JCO.2010.33.9275
Gupta, A., Love, S., Schuh, A., Shanyinde, M., Larkin, J. M., Plummer, R., et al. (2014). DOC-MEK: a double-blind randomized phase II trial of docetaxel with or without selumetinib in wild-type BRAF advanced melanoma. Ann. Oncol. 25, 968–974. doi: 10.1093/annonc/mdu054
Gutman, M., Singh, R. K., Radinsky, R., and Bar-Eli, M. (1994). Intertumoral heterogeneity of receptor-tyrosine kinases expression in human melanoma cell lines with different metastatic capabilities. Anticancer Res. 14, 1759–1765.
Gutmann, D. H. (2001). The neurofibromatoses: when less is more. Hum. Mol. Genet. 10, 747–755. doi: 10.1093/hmg/10.7.747
Guyot, R., Vincent, S., Bertin, J., Samarut, J., and Ravel-Chapuis, P. (2010). The transforming acidic coiled coil (TACC1) protein modulates the transcriptional activity of the nuclear receptors TR and RAR. BMC Mol. Biol. 11:3. doi: 10.1186/1471-2199-11-3
Hainaut, P., and Pfeifer, G. P. (2016). Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb. Perspect. Med. 6:a026179. doi: 10.1101/cshperspect.a026179
Hainsworth, J. D., Infante, J. R., Spigel, D. R., Peyton, J. D., Thompson, D. S., Lane, C. M., et al. (2010). Bevacizumab and everolimus in the treatment of patients with metastatic melanoma: a phase 2 trial of the sarah cannon oncology research consortium. Cancer 116, 4122–4129. doi: 10.1002/cncr.25320
Hamid, O., Robert, C., Daud, A., Hodi, F. S., Hwu, W. J., Kefford, R., et al. (2019). Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 30, 582–588. doi: 10.1093/annonc/mdz011
Hammond, D., Zeng, K., Espert, A., Bastos, R. N., Baron, R. D., Gruneberg, U., et al. (2013). Melanoma-associated mutations in protein phosphatase 6 cause chromosome instability and DNA damage owing to dysregulated Aurora-A. J. Cell. Sci. 126, 3429–3440. doi: 10.1242/jcs.128397
Han, Y. (2019). Analysis of the role of the hippo pathway in cancer. J. Transl. Med. 17:116. doi: 10.1186/s12967-019-1869-4
Harbour, J. W., Roberson, E. D. O., Anbunathan, H., Onken, M. D., Worley, L. A., and Bowcock, A. M. (2013). Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 45, 133–135. doi: 10.1038/ng.2523
Hartman, D. J., Davison, J. M., Foxwell, T. J., Nikiforova, M. N., and Chiosea, S. I. (2012). Mutant allele-specific imbalance modulates prognostic impact of KRAS mutations in colorectal adenocarcinoma and is associated with worse overall survival. Int. J. Cancer 131, 1810–1817. doi: 10.1002/ijc.27461
Hartman, M. L., Sztiller-Sikorska, M., and Czyz, M. (2019). Whole-exome sequencing reveals novel genetic variants associated with diverse phenotypes of melanoma cells. Mol. Carcinog. 58, 588–602. doi: 10.1002/mc.22953
Hauke, R. J., Infante, J. R., Rubin, M. S., Shih, K. C., Arrowsmith, E. R., and Hainsworth, J. D. (2013). Everolimus in combination with paclitaxel and carboplatin in patients with metastatic melanoma: a phase II trial of the Sarah cannon research institute oncology research consortium. Melanoma Res. 23, 468–473. doi: 10.1097/CMR.0000000000000014
Hayward, N. K., Wilmott, J. S., Waddell, N., Johansson, P. A., Field, M. A., Nones, K., et al. (2017). Whole-genome landscapes of major melanoma subtypes. Nature 545, 175–180. doi: 10.1038/nature22071
He, Y., Zhang, D., Yang, Y., Wang, X., Zhao, X., Zhang, P., et al. (2018). A double-edged function of DDX3, as an oncogene or tumor suppressor, in cancer progression (Review). Oncol. Rep. 39, 883–892. doi: 10.3892/or.2018.6203
Hoadley, K. A., Yau, C., Hinoue, T., Wolf, D. M., Lazar, A. J., Drill, E., et al. (2018). Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304. doi: 10.1016/j.cell.2018.03.022
Hodi, F. S., Corless, C. L., Giobbie-Hurder, A., Fletcher, J. A., Zhu, M., Marino-Enriquez, A., et al. (2013). Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 31, 3182–3190. doi: 10.1200/JCO.2012.47.7836
Hodi, F. S., O’Day, S. J., McDermott, D. F., Weber, R. W., Sosman, J. A., Haanen, J. B., et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723. doi: 10.1056/NEJMoa1003466
Hodis, E., Watson, I. R., Kryukov, G. V., Arold, S. T., Imielinski, M., Theurillat, J.-P., et al. (2012). A landscape of driver mutations in melanoma. Cell 150, 251–263. doi: 10.1016/j.cell.2012.06.024
Hoffmann, F., Niebel, D., Aymans, P., Ferring-Schmitt, S., Dietrich, D., and Landsberg, J. (2020). H3K27me3 and EZH2 expression in melanoma: relevance for melanoma progression and response to immune checkpoint blockade. Clin. Epigenet. 12:24. doi: 10.1186/s13148-020-0818-7
Hong, D., Infante, J., Janku, F., Jones, S., Nguyen, L. M., Burris, H., et al. (2016). Phase I study of LY2606368, a checkpoint kinase 1 inhibitor, in patients with advanced cancer. J. Clin. Oncol. 34, 1764–1771. doi: 10.1200/JCO.2015.64.5788
Horn, S., Figl, A., Rachakonda, P. S., Fischer, C., Sucker, A., Gast, A., et al. (2013). TERT promoter mutations in familial and sporadic melanoma. Science 339, 959–961. doi: 10.1126/science.1230062
Hugdahl, E., Kalvenes, M. B., Mannelqvist, M., Ladstein, R. G., and Akslen, L. A. (2018). Prognostic impact and concordance of TERT promoter mutation and protein expression in matched primary and metastatic cutaneous melanoma. Br. J. Cancer 118, 98–105. doi: 10.1038/bjc.2017.384
Ibrahim, N., Buchbinder, E. I., Granter, S. R., Rodig, S. J., Giobbie-Hurder, A., Becerra, C., et al. (2016). A phase I trial of panobinostat (LBH589) in patients with metastatic melanoma. Cancer Med. 5, 3041–3050. doi: 10.1002/cam4.862
ICGC Data Portal (2020). Available online at: https://dcc.icgc.org/ (accessed April 25, 2020).
ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium (2020). Pan-cancer analysis of whole genomes. Nature 578, 82–93. doi: 10.1038/s41586-020-1969-6
Infante, J. R., Fecher, L. A., Falchook, G. S., Nallapareddy, S., Gordon, M. S., Becerra, C., et al. (2012). Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 13, 773–781. doi: 10.1016/S1470-2045(12)70270-X
Inoue, D., Chew, G.-L., Liu, B., Michel, B. C., Pangallo, J., D’Avino, A. R., et al. (2019). Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 574, 432–436. doi: 10.1038/s41586-019-1646-9
Inoue, K., and Fry, E. A. (2018). Aberrant expression of p14ARF in human cancers: a new biomarker? Tumor Microenviron. 1, 37–44. doi: 10.4103/tme.tme_24_17
Isabel Zhu, Y., and Fitzpatrick, J. E. (2006). Expression of c-kit (CD117) in Spitz nevus and malignant melanoma. J. Cutan. Pathol. 33, 33–37. doi: 10.1111/j.0303-6987.2006.00420.x
Jo, S.-H., Lee, S.-H., Chun, H. S., Lee, S. M., Koh, H.-J., Lee, S.-E., et al. (2002). Cellular defense against UVB-induced phototoxicity by cytosolic NADP(+)-dependent isocitrate dehydrogenase. Biochem. Biophys. Res. Commun. 292, 542–549. doi: 10.1006/bbrc.2002.6667
Joshi, K. S., Rathos, M. J., Mahajan, P., Wagh, V., Shenoy, S., Bhatia, D., et al. (2007). P276-00, a novel cyclin-dependent inhibitor induces G1-G2 arrest, shows antitumor activity on cisplatin-resistant cells and significant in vivo efficacy in tumor models. Mol. Cancer Ther. 6, 926–934. doi: 10.1158/1535-7163.MCT-06-0614
Juric, D., Soria, J.-C., Sharma, S., Banerji, U., Azaro, A., Desai, J., et al. (2014). A phase 1b dose-escalation study of BYL719 plus binimetinib (MEK162) in patients with selected advanced solid tumors. JCO 32:9051. doi: 10.1200/jco.2014.32.15_suppl.9051
Kalinsky, K., Lee, S., Rubin, K. M., Lawrence, D. P., Iafrarte, A. J., Borger, D. R., et al. (2017). A phase 2 trial of dasatinib in patients with locally advanced or stage IV mucosal, acral, or vulvovaginal melanoma: a trial of the ECOG-ACRIN cancer research group (E2607). Cancer 123, 2688–2697. doi: 10.1002/cncr.30663
Kampilafkos, P., Melachrinou, M., Kefalopoulou, Z., Lakoumentas, J., and Sotiropoulou-Bonikou, G. (2015). Epigenetic modifications in cutaneous malignant melanoma: EZH2, H3K4me2, and H3K27me3 immunohistochemical expression is enhanced at the invasion front of the tumor. Am. J. Dermatopathol. 37, 138–144. doi: 10.1097/DAD.0b013e31828a2d54
Kandoth, C., McLellan, M. D., Vandin, F., Ye, K., Niu, B., Lu, C., et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. doi: 10.1038/nature12634
Karbowniczek, M., Spittle, C. S., Morrison, T., Wu, H., and Henske, E. P. (2008). mTOR is activated in the majority of malignant melanomas. J. Invest. Dermatol. 128, 980–987. doi: 10.1038/sj.jid.5701074
Kim, E., Zucconi, B. E., Wu, M., Nocco, S. E., Meyers, D. J., McGee, J. S., et al. (2019). MITF expression predicts therapeutic vulnerability to p300 inhibition in human melanoma. Cancer Res. 79, 2649–2661. doi: 10.1158/0008-5472.CAN-18-2331
Kim, H. S., Jung, M., Kang, H. N., Kim, H., Park, C.-W., Kim, S.-M., et al. (2017). Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene 36, 3334–3345. doi: 10.1038/onc.2016.486
Kim, K. B., Kefford, R., Pavlick, A. C., Infante, J. R., Ribas, A., Sosman, J. A., et al. (2013). Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J. Clin. Oncol. 31, 482–489. doi: 10.1200/JCO.2012.43.5966
Kim, N. W., Piatyszek, M. A., Prowse, K. R., Harley, C. B., West, M. D., Ho, P. L., et al. (1994). Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015. doi: 10.1126/science.7605428
Kirkwood, J. M., Bastholt, L., Robert, C., Sosman, J., Larkin, J., Hersey, P., et al. (2012). Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin. Cancer Res. 18, 555–567. doi: 10.1158/1078-0432.CCR-11-1491
Kiuru, M., and Busam, K. J. (2017). The NF1 gene in tumor syndromes and melanoma. Lab. Invest. 97, 146–157. doi: 10.1038/labinvest.2016.142
Knudsen, E. S., and Wang, J. Y. J. (2010). Targeting the RB-pathway in cancer therapy. Clin. Cancer Res. 16, 1094–1099. doi: 10.1158/1078-0432.CCR-09-0787
Kong, Y., Krauthammer, M., and Halaban, R. (2014). Rare SF3B1 R625 mutations in cutaneous melanoma. Melanoma Res. 24, 332–334. doi: 10.1097/CMR.0000000000000071
Kong, Y., Si, L., Li, Y., Wu, X., Xu, X., Dai, J., et al. (2016). Analysis of mTOR gene aberrations in melanoma patients and evaluation of their sensitivity to PI3K-AKT-mTOR pathway inhibitors. Clin. Cancer Res. 22, 1018–1027. doi: 10.1158/1078-0432.CCR-15-1110
Konstantinopoulos, P. A., Karamouzis, M. V., and Papavassiliou, A. G. (2007). Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 6, 541–555. doi: 10.1038/2221
Kontogianni, G., Piroti, G., Maglogiannis, I., Chatziioannou, A., and Papadodima, O. (2018). Dissecting the mutational landscape of cutaneous melanoma: an omic analysis based on patients from greece. Cancers 10:96. doi: 10.3390/cancers10040096
Korn, E. L., Liu, P.-Y., Lee, S. J., Chapman, J.-A. W., Niedzwiecki, D., Suman, V. J., et al. (2008). Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. JCO 26, 527–534. doi: 10.1200/JCO.2007.12.7837
Kourtis, N., Moubarak, R. S., Aranda-Orgilles, B., Lui, K., Aydin, I. T., Trimarchi, T., et al. (2015). FBXW7 modulates cellular stress response and metastatic potential through ?HSF1 post-translational modification. Nat. Cell Biol. 17, 322–332. doi: 10.1038/ncb3121
Krauthammer, M., Kong, Y., Bacchiocchi, A., Evans, P., Pornputtapong, N., Wu, C., et al. (2015). Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 47, 996–1002. doi: 10.1038/ng.3361
Krauthammer, M., Kong, Y., Ha, B. H., Evans, P., Bacchiocchi, A., McCusker, J. P., et al. (2012). Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 44, 1006–1014. doi: 10.1038/ng.2359
Larkin, J., Ascierto, P. A., Dréno, B., Atkinson, V., Liszkay, G., Maio, M., et al. (2014). Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 371, 1867–1876. doi: 10.1056/NEJMoa1408868
Lasorella, A., Sanson, M., and Iavarone, A. (2017). FGFR-TACC gene fusions in human glioma. Neurooncology 19, 475–483. doi: 10.1093/neuonc/now240
Lassam, N., and Bickford, S. (1992). Loss of c-kit expression in cultured melanoma cells. Oncogene 7, 51–56.
Lebbe, C., Dutriaux, C., Lesimple, T., Schueler, C., Markivskyy, A., and Dreno, B. (2016). Pimasertib (Pim) versus dacarbazine (Dtic) in patients (Pts) With cutaneous nras melanoma: a controlled, open-label phase Ii trial with crossover. Ann. Oncol. 27, 379–400. doi: 10.1093/annonc/mdw379
Lee, B., Sandhu, S., and McArthur, G. (2015). Cell cycle control as a promising target in melanoma. Curr. Opin. Oncol. 27, 141–150. doi: 10.1097/CCO.0000000000000159
Lee, S. J., Kim, T. M., Kim, Y. J., Jang, K.-T., Lee, H. J., Lee, S. N., et al. (2015). Phase II trial of nilotinib in patients with metastatic malignant melanoma harboring KIT gene aberration: a multicenter trial of korean cancer study group (UN10-06). Oncologist 20, 1312–1319. doi: 10.1634/theoncologist.2015-0161
Lee, C.-J., An, H.-J., Kim, S.-M., Yoo, S.-M., Park, J., Lee, G.-E., et al. (2020). FBXW7-mediated stability regulation of signal transducer and activator of transcription 2 in melanoma formation. Proc. Natl. Acad. Sci. U.S.A. 117, 584–594. doi: 10.1073/pnas.1909879116
Lee, J.-H., Choi, J.-W., and Kim, Y.-S. (2011). Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: a meta-analysis. Br. J. Dermatol. 164, 776–784. doi: 10.1111/j.1365-2133.2010.10185.x
Lennartsson, J., and Rönnstrand, L. (2012). Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol. Rev. 92, 1619–1649. doi: 10.1152/physrev.00046.2011
Levy, C., Khaled, M., and Fisher, D. E. (2006). MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 12, 406–414. doi: 10.1016/j.molmed.2006.07.008
Liang, W. S., Hendricks, W., Kiefer, J., Schmidt, J., Sekar, S., Carpten, J., et al. (2017). Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res. 27, 524–532. doi: 10.1101/gr.213348.116
Lin, T.-C. (2019). DDX3X multifunctionally modulates tumor progression and serves as a prognostic indicator to predict cancer outcomes. Int. J. Mol. Sci. 21:281. doi: 10.3390/ijms21010281
Line, A., Slucka, Z., Stengrevics, A., Li, G., and Rees, R. C. (2002). Altered splicing pattern of TACC1 mRNA in gastric cancer. Cancer Genet. Cytogenet. 139, 78–83. doi: 10.1016/s0165-4608(02)00607-6
Lionarons, D. A., Hancock, D. C., Rana, S., East, P., Moore, C., Murillo, M. M., et al. (2019). RAC1P29S induces a mesenchymal phenotypic switch via serum response factor to promote melanoma development and therapy resistance. Cancer Cell 36, 68–83. doi: 10.1016/j.ccell.2019.05.015
Lissanu Deribe, Y. (2016a). Interplay between PREX2 mutations and the PI3K pathway and its effect on epigenetic regulation of gene expression in NRAS-mutant melanoma. Small GTPases 7, 178–185. doi: 10.1080/21541248.2016.1178366
Lissanu Deribe, Y. (2016b). Mechanistic insights into the role of truncating PREX2 mutations in melanoma. Mol. Cell Oncol. 3:e1160174. doi: 10.1080/23723556.2016.1160174
Lissanu Deribe, Y., Shi, Y., Rai, K., Nezi, L., Amin, S. B., Wu, C.-C., et al. (2016). Truncating PREX2 mutations activate its GEF activity and alter gene expression regulation in NRAS-mutant melanoma. Proc. Natl. Acad. Sci. U.S.A. 113, E1296–E1305. doi: 10.1073/pnas.1513801113
Liu, P., Wang, Y., and Li, X. (2019). Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 9, 871–879. doi: 10.1016/j.apsb.2019.03.002
Long, G. V., Stroyakovskiy, D., Gogas, H., Levchenko, E., de Braud, F., Larkin, J., et al. (2014). Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 371, 1877–1888. doi: 10.1056/NEJMoa1406037
Lopez, G. Y., Reitman, Z. J., Solomon, D., Waldman, T., Bigner, D. D., McLendon, R. E., et al. (2010). IDH1(R132) mutation identified in one human melanoma metastasis, but not correlated with metastases to the brain. Biochem. Biophys. Res. Commun. 398, 585–587. doi: 10.1016/j.bbrc.2010.06.125
Lorbeer, F. K., and Hockemeyer, D. (2020). TERT promoter mutations and telomeres during tumorigenesis. Curr. Opin. Genet. Dev. 60, 56–62. doi: 10.1016/j.gde.2020.02.001
LoRusso, P. M., Krishnamurthi, S. S., Rinehart, J. J., Nabell, L. M., Malburg, L., Chapman, P. B., et al. (2010). Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin. Cancer Res. 16, 1924–1937. doi: 10.1158/1078-0432.CCR-09-1883
Ma, S., Meng, Z., Chen, R., and Guan, K.-L. (2019). The hippo pathway: biology and pathophysiology. Annu. Rev. Biochem. 88, 577–604. doi: 10.1146/annurev-biochem-013118-111829
Mahalingam, M. (2017). NF1 and neurofibromin: emerging players in the genetic landscape of desmoplastic melanoma. Adv. Anat. Pathol. 24, 1–14. doi: 10.1097/PAP.0000000000000131
Mahmoud, F., Shields, B., Makhoul, I., Hutchins, L. F., Shalin, S. C., and Tackett, A. J. (2016). Role of EZH2 histone methyltrasferase in melanoma progression and metastasis. Cancer Biol. Ther. 17, 579–591. doi: 10.1080/15384047.2016.1167291
Maitland, M. L., Piha-Paul, S., Falchook, G., Kurzrock, R., Nguyen, L., Janisch, L., et al. (2018). Clinical pharmacodynamic/exposure characterisation of the multikinase inhibitor ilorasertib (ABT-348) in a phase 1 dose-escalation trial. Br. J. Cancer 118, 1042–1050. doi: 10.1038/s41416-018-0020-2
Mandalà, M., Merelli, B., and Massi, D. (2014). Nras in melanoma: targeting the undruggable target. Crit. Rev. Oncol. Hematol. 92, 107–122. doi: 10.1016/j.critrevonc.2014.05.005
Manning, C. S., Hooper, S., and Sahai, E. A. (2015). Intravital imaging of SRF and Notch signalling identifies a key role for EZH2 in invasive melanoma cells. Oncogene 34, 4320–4332. doi: 10.1038/onc.2014.362
Mao, J.-H., Kim, I.-J., Wu, D., Climent, J., Kang, H. C., DelRosario, R., et al. (2008). FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 321, 1499–1502. doi: 10.1126/science.1162981
Mar, V. J., Wong, S. Q., Logan, A., Nguyen, T., Cebon, J., Kelly, J. W., et al. (2014). Clinical and pathological associations of the activating RAC1 P29S mutation in primary cutaneous melanoma. Pigment Cell Melanoma Res. 27, 1117–1125. doi: 10.1111/pcmr.12295
Margolin, K. A., Moon, J., Flaherty, L. E., Lao, C. D., Akerley, W. L., Othus, M., et al. (2012). Randomized phase II trial of sorafenib with temsirolimus or tipifarnib in untreated metastatic melanoma (S0438). Clin. Cancer Res. 18, 1129–1137. doi: 10.1158/1078-0432.CCR-11-2488
Marsh Durban, V., Deuker, M. M., Bosenberg, M. W., Phillips, W., and McMahon, M. (2013). Differential AKT dependency displayed by mouse models of BRAFV600E-initiated melanoma. J. Clin. Invest. 123, 5104–5118. doi: 10.1172/JCI69619
Martin, M., Maßhöfer, L., Temming, P., Rahmann, S., Metz, C., Bornfeld, N., et al. (2013). Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 45, 933–936. doi: 10.1038/ng.2674
Mathys, H., Basquin, J., Ozgur, S., Czarnocki-Cieciura, M., Bonneau, F., Aartse, A., et al. (2014). Structural and biochemical insights to the role of the CCR4-NOT complex and DDX6 ATPase in microRNA repression. Mol. Cell 54, 751–765. doi: 10.1016/j.molcel.2014.03.036
McWilliams, R. R., Allred, J. B., Slostad, J. A., Katipamula, R., Dronca, R. S., Rumilla, K. M., et al. (2018). NCCTG N0879 (Alliance): a randomized phase 2 cooperative group trial of carboplatin, paclitaxel, and bevacizumab?±?everolimus for metastatic melanoma. Cancer 124, 537–545. doi: 10.1002/cncr.31072
Meier, F. E., Niessner, H., Flaherty, K., Schadendorf, D., Sinnberg, T., Schittek, B., et al. (2009). Effect of the farnesyl transferase inhibitor lonafarnib on sensitivity of melanoma cells to the multikinase inhibitor sorafenib and on Rheb farnesylation and mTOR signaling. JCO 27:9077. doi: 10.1200/jco.2009.27.15_suppl.9077
Micel, L. N., Tentler, J. J., Tan, A.-C., Selby, H. M., Brunkow, K. L., Robertson, K. M., et al. (2015). Antitumor activity of the MEK inhibitor TAK-733 against melanoma cell lines and patient-derived tumor explants. Mol. Cancer Ther. 14, 317–325. doi: 10.1158/1535-7163.MCT-13-1012
Michiels, J.-F., Perrin, C., Leccia, N., Massi, D., Grimaldi, P., and Wagner, N. (2010). PPARbeta activation inhibits melanoma cell proliferation involving repression of the Wilms’ tumour suppressor WT1. Pflugers Arch. 459, 689–703. doi: 10.1007/s00424-009-0776-6
Milagre, C., Dhomen, N., Geyer, F. C., Hayward, R., Lambros, M., Reis-Filho, J. S., et al. (2010). A mouse model of melanoma driven by oncogenic KRAS. Cancer Res. 70, 5549–5557. doi: 10.1158/0008-5472.CAN-09-4254
Min, J., Zaslavsky, A., Fedele, G., McLaughlin, S. K., Reczek, E. E., De Raedt, T., et al. (2010). An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat. Med. 16, 286–294. doi: 10.1038/nm.2100
Minard, K. I., and McAlister-Henn, L. (1999). Dependence of peroxisomal beta-oxidation on cytosolic sources of NADPH. J. Biol. Chem. 274, 3402–3406. doi: 10.1074/jbc.274.6.3402
Minella, A. C., and Clurman, B. E. (2005). Mechanisms of tumor suppression by the SCF(Fbw7). Cell Cycle 4, 1356–1359. doi: 10.4161/cc.4.10.2058
Minor, D. R., Kashani-Sabet, M., Garrido, M., O’Day, S. J., Hamid, O., and Bastian, B. C. (2012). Sunitinib therapy for melanoma patients with KIT mutations. Clin. Cancer Res. 18, 1457–1463. doi: 10.1158/1078-0432.CCR-11-1987
Molhoek, K. R., Brautigan, D. L., and Slingluff, C. L. (2005). Synergistic inhibition of human melanoma proliferation by combination treatment with B-Raf inhibitor BAY43-9006 and mTOR inhibitor Rapamycin. J. Transl. Med. 3:39. doi: 10.1186/1479-5876-3-39
Mondesir, J., Willekens, C., Touat, M., and de Botton, S. (2016). IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. J. Blood Med. 7, 171–180. doi: 10.2147/JBM.S70716
Montone, K. T., van Belle, P., Elenitsas, R., and Elder, D. E. (1997). Proto-oncogene c-kit expression in malignant melanoma: protein loss with tumor progression. Mod. Pathol. 10, 939–944.
Moran, B., Silva, R., Perry, A. S., and Gallagher, W. M. (2018). Epigenetics of malignant melanoma. Semin. Cancer Biol. 51, 80–88. doi: 10.1016/j.semcancer.2017.10.006
Moreno, T., González-Silva, L., Monterde, B., Betancor-Fernández, I., Revilla, C., Agraz-Doblas, A., et al. (2020). ARID2 deficiency promotes tumor progression and is associated with higher sensitivity to PARP inhibition in lung cancer. bioRxiv [Preprint], doi: 10.1101/2020.01.10.898726
Moroishi, T., Hansen, C. G., and Guan, K.-L. (2015). The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 15, 73–79. doi: 10.1038/nrc3876
Morris, E. J., and Dyson, N. J. (2001). Retinoblastoma protein partners. Adv. Cancer Res. 82, 1–54. doi: 10.1016/s0065-230x(01)82001-7
Nagore, E., Heidenreich, B., Requena, C., García-Casado, Z., Martorell-Calatayud, A., Pont-Sanjuan, V., et al. (2016). TERT promoter mutations associate with fast-growing melanoma. Pigment Cell Melanom. Res. 29, 236–238. doi: 10.1111/pcmr.12441
Nagore, E., Rachakonda, S., and Kumar, R. (2019). TERT promoter mutations in melanoma survival. Oncotarget 10, 1546–1548. doi: 10.18632/oncotarget.26688
Nassar, K. W., and Tan, A. C. (2020). The mutational landscape of mucosal melanoma. Semin. Cancer Biol. 61, 139–148. doi: 10.1016/j.semcancer.2019.09.013
Natali, P. G., Nicotra, M. R., Winkler, A. B., Cavaliere, R., Bigotti, A., and Ullrich, A. (1992). Progression of human cutaneous melanoma is associated with loss of expression of c-kit proto-oncogene receptor. Int. J. Cancer 52, 197–201. doi: 10.1002/ijc.2910520207
Newell, F., Kong, Y., Wilmott, J. S., Johansson, P. A., Ferguson, P. M., Cui, C., et al. (2019). Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat. Commun. 10:3163. doi: 10.1038/s41467-019-11107-x
Nguyen, H. G., Chinnappan, D., Urano, T., and Ravid, K. (2005). Mechanism of Aurora-B degradation and its dependency on intact KEN and A-boxes: identification of an aneuploidy-promoting property. Mol. Cell. Biol. 25, 4977–4992. doi: 10.1128/MCB.25.12.4977-4992.2005
Niessner, H., Beck, D., Sinnberg, T., Lasithiotakis, K., Maczey, E., Gogel, J., et al. (2011). The farnesyl transferase inhibitor lonafarnib inhibits mTOR signaling and enforces sorafenib-induced apoptosis in melanoma cells. J. Invest. Dermatol. 131, 468–479. doi: 10.1038/jid.2010.297
Nissan, M. H., Pratilas, C. A., Jones, A. M., Ramirez, R., Won, H., Liu, C., et al. (2014). Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 74, 2340–2350. doi: 10.1158/0008-5472.CAN-13-2625
Ojha, J., Secreto, C. R., Rabe, K. G., Van Dyke, D. L., Kortum, K. M., Slager, S. L., et al. (2015). Identification of recurrent truncated DDX3X mutations in chronic lymphocytic leukaemia. Br. J. Haematol. 169, 445–448. doi: 10.1111/bjh.13211
Omholt, K., Platz, A., Kanter, L., Ringborg, U., and Hansson, J. (2003). NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin. Cancer Res. 9, 6483–6488.
Ostrem, J. M. L., and Shokat, K. M. (2016). Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discov. 15, 771–785. doi: 10.1038/nrd.2016.139
Palmieri, G., Colombino, M., Casula, M., Manca, A., Mandalà, M., Cossu, A., et al. (2018). Molecular pathways in melanomagenesis: what we learned from next-generation sequencing approaches. Curr. Oncol. Rep. 20:86. doi: 10.1007/s11912-018-0733-7
Pan, D., Kobayashi, A., Jiang, P., Ferrari de Andrade, L., Tay, R. E., Luoma, A. M., et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. doi: 10.1126/science.aao1710
Parsons, D. W., Jones, S., Zhang, X., Lin, J. C.-H., Leary, R. J., Angenendt, P., et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812. doi: 10.1126/science.1164382
Pathan, M., Keerthikumar, S., Ang, C. S., Gangoda, L., Quek, C. Y., Williamson, N. A., et al. (2015). FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics 15, 2597–2601.
Penna, I., Molla, A., Grazia, G., Cleris, L., Nicolini, G., Perrone, F., et al. (2016). Primary cross-resistance to BRAFV600E-, MEK1/2- and PI3K/mTOR-specific inhibitors in BRAF-mutant melanoma cells counteracted by dual pathway blockade. Oncotarget 7, 3947–3965. doi: 10.18632/oncotarget.6600
Phung, B., Cieśla, M., Sanna, A., Guzzi, N., Beneventi, G., Cao Thi Ngoc, P., et al. (2019). The X-linked DDX3X RNA helicase dictates translation reprogramming and metastasis in melanoma. Cell Rep. 27, 3573–3586. doi: 10.1016/j.celrep.2019.05.069
Piñero, J., Queralt-Rosinach, N., Bravo, A., Deu-Pons, J., Bauer-Mehren, A., Baron, M., et al. (2015). DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database 2015:bav028. doi: 10.1093/database/bav028
Polakis, P. (2012). Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 4:8052. doi: 10.1101/cshperspect.a008052
Pópulo, H., Lopes, J. M., and Soares, P. (2012). The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 13, 1886–1918. doi: 10.3390/ijms13021886
Priestley, P., Baber, J., Lolkema, M. P., Steeghs, N., de Bruijn, E., Shale, C., et al. (2019). Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216. doi: 10.1038/s41586-019-1689-y
Pylayeva-Gupta, Y., Grabocka, E., and Bar-Sagi, D. (2011). RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774. doi: 10.1038/nrc3106
Qian, L., Chen, K., Wang, C., Chen, Z., Meng, Z., and Wang, P. (2020). Targeting NRAS-mutant cancers with the selective STK19 kinase inhibitor chelidonine. Clin. Cancer Res. 26, 2604. doi: 10.1158/1078-0432.CCR-19-2604
Rauen, K. A. (2007). HRAS and the costello syndrome. Clin. Genet. 71, 101–108. doi: 10.1111/j.1399-0004.2007.00743.x
Reifenberger, J., Knobbe, C. B., Wolter, M., Blaschke, B., Schulte, K. W., Pietsch, T., et al. (2002). Molecular genetic analysis of malignant melanomas for aberrations of the WNT signaling pathway genes CTNNB1, APC, ICAT and BTRC. Int. J. Cancer 100, 549–556. doi: 10.1002/ijc.10512
Rhodes, D. R., Barrette, T. R., Rubin, M. A., Ghosh, D., and Chinnaiyan, A. M. (2002). Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res. 62, 4427–4433.
Rinehart, J., Adjei, A. A., Lorusso, P. M., Waterhouse, D., Hecht, J. R., Natale, R. B., et al. (2004). Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 22, 4456–4462. doi: 10.1200/JCO.2004.01.185
Robert, C., Grob, J. J., Stroyakovskiy, D., Karaszewska, B., Hauschild, A., Levchenko, E., et al. (2019). Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. N. Engl. J. Med. 381, 626–636. doi: 10.1056/NEJMoa1904059
Robert, C., Karaszewska, B., Schachter, J., Rutkowski, P., Mackiewicz, A., Stroyakovskiy, D., et al. (2016). Three-year estimate of overall survival in COMBI-v, a randomized phase 3 study evaluating first-line dabrafenib (D) + trametinib (T) in patients (pts) with unresectable or metastatic BRAF V600E/K-mutant cutaneous melanoma. Ann. Oncol. 27:37. doi: 10.1093/annonc/mdw435.37
Robert, C., Long, G. V., Brady, B., Dutriaux, C., Maio, M., Mortier, L., et al. (2015). Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 372, 320–330. doi: 10.1056/NEJMoa1412082
Robertson, G. P., Furnari, F. B., Miele, M. E., Glendening, M. J., Welch, D. R., Fountain, J. W., et al. (1998). In vitro loss of heterozygosity targets the PTEN/MMAC1 gene in melanoma. Proc. Natl. Acad. Sci. U.S.A. 95, 9418–9423.
Román, M., Baraibar, I., López, I., Nadal, E., Rolfo, C., Vicent, S., et al. (2018). KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Mol. Cancer 17:33. doi: 10.1186/s12943-018-0789-x
Ross, A. L., Sanchez, M. I., and Grichnik, J. M. (2011). Molecular nevogenesis. Dermatol. Res. Pract. 2011:463184. doi: 10.1155/2011/463184
Sanchez-Vega, F., Mina, M., Armenia, J., Chatila, W. K., Luna, A., La, K. C., et al. (2018). Oncogenic signaling pathways in the cancer genome atlas. Cell 173, 321–337. doi: 10.1016/j.cell.2018.03.035
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371. doi: 10.1016/j.cell.2017.03.035
Schachter, J., Ribas, A., Long, G. V., Arance, A., Grob, J.-J., Mortier, L., et al. (2017). Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 390, 1853–1862. doi: 10.1016/S0140-6736(17)31601-X
Schadendorf, D., van Akkooi, A. C. J., Berking, C., Griewank, K. G., Gutzmer, R., Hauschild, A., et al. (2018). Melanoma. Lancet 392, 971–984. doi: 10.1016/S0140-6736(18)31559-9
Schilling, B., Bielefeld, N., Sucker, A., Hillen, U., Zimmer, L., Schadendorf, D., et al. (2013). Lack of SF3B1 R625 mutations in cutaneous melanoma. Diagn. Pathol. 8:87. doi: 10.1186/1746-1596-8-87
Schram, A. M., Gandhi, L., Mita, M. M., Damstrup, L., Campana, F., Hidalgo, M., et al. (2018). A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br. J. Cancer 119, 1471–1476. doi: 10.1038/s41416-018-0322-4
Seberg, H. E., Van Otterloo, E., and Cornell, R. A. (2017). Beyond MITF: multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment Cell Melanom. Res. 30, 454–466. doi: 10.1111/pcmr.12611
Shain, A. H., and Pollack, J. R. (2013). The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One 8:e55119. doi: 10.1371/journal.pone.0055119
Shain, A. H., Yeh, I., Kovalyshyn, I., Sriharan, A., Talevich, E., Gagnon, A., et al. (2015). The genetic evolution of melanoma from precursor lesions. N. Engl. J. Med. 373, 1926–1936. doi: 10.1056/NEJMoa1502583
Shao, Z., and Robbins, P. D. (1995). Differential regulation of E2F and Sp1-mediated transcription by G1 cyclins. Oncogene 10, 221–228.
Shen, L., Wu, L. C., Sanlioglu, S., Chen, R., Mendoza, A. R., Dangel, A. W., et al. (1994). Structure and genetics of the partially duplicated gene RP located immediately upstream of the complement C4A and the C4B genes in the HLA class III region. Molecular cloning, exon-intron structure, composite retroposon, and breakpoint of gene duplication. J. Biol. Chem. 269, 8466–8476.
Shen, S. S., Zhang, P. S., Eton, O., and Prieto, V. G. (2003). Analysis of protein tyrosine kinase expression in melanocytic lesions by tissue array. J. Cutan. Pathol. 30, 539–547. doi: 10.1034/j.1600-0560.2003.00090.x
Sheppard, K. E., and McArthur, G. A. (2013). The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin. Cancer Res. 19, 5320–5328. doi: 10.1158/1078-0432.CCR-13-0259
Shibata, T., Kokubu, A., Miyamoto, M., Sasajima, Y., and Yamazaki, N. (2011). Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am. J. Pathol. 178, 1395–1402. doi: 10.1016/j.ajpath.2010.12.011
Shitara, D., Tell-Martí, G., Badenas, C., Enokihara, M. M. S. S., Alós, L., Larque, A. B., et al. (2015). Mutational status of naevus-associated melanomas. Br. J. Dermatol. 173, 671–680. doi: 10.1111/bjd.13829
Shtivelman, E., Davies, M. Q. A., Hwu, P., Yang, J., Lotem, M., Oren, M., et al. (2014). Pathways and therapeutic targets in melanoma. Oncotarget 5, 1701–1752. doi: 10.18632/oncotarget.1892
Shull, A. Y., Latham-Schwark, A., Ramasamy, P., Leskoske, K., Oroian, D., Birtwistle, M. R., et al. (2012). Novel somatic mutations to PI3K pathway genes in metastatic melanoma. PLoS One 7:e43369. doi: 10.1371/journal.pone.0043369
Siroy, A. E., Boland, G. M., Milton, D. R., Roszik, J., Frankian, S., Malke, J., et al. (2015). Beyond BRAF(V600): clinical mutation panel testing by next-generation sequencing in advanced melanoma. J. Invest. Dermatol. 135, 508–515. doi: 10.1038/jid.2014.366
Slingluff, C. L., Petroni, G. R., Molhoek, K. R., Brautigan, D. L., Chianese-Bullock, K. A., Shada, A. L., et al. (2013). Clinical activity and safety of combination therapy with temsirolimus and bevacizumab for advanced melanoma: a phase II trial (CTEP 7190/Mel47). Clin. Cancer Res. 19, 3611–3620. doi: 10.1158/1078-0432.CCR-12-3919
Smalley, K. S. M., and Eisen, T. G. (2003). Farnesyl transferase inhibitor SCH66336 is cytostatic, pro-apoptotic and enhances chemosensitivity to cisplatin in melanoma cells. Int. J. Cancer 105, 165–175. doi: 10.1002/ijc.11064
Snyder, A., Makarov, V., Merghoub, T., Yuan, J., Zaretsky, J. M., Desrichard, A., et al. (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199. doi: 10.1056/NEJMoa1406498
Solit, D. B., Garraway, L. A., Pratilas, C. A., Sawai, A., Getz, G., Basso, A., et al. (2006). BRAF mutation predicts sensitivity to MEK inhibition. Nature 439, 358–362. doi: 10.1038/nature04304
Sosman, J. A., Kittaneh, M., Lolkema, M. P. J. K., Postow, M. A., Schwartz, G., Franklin, C., et al. (2014). A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: early encouraging clinical activity. JCO 32:9009. doi: 10.1200/jco.2014.32.15_suppl.9009
Spagnolo, F., Ghiorzo, P., Orgiano, L., Pastorino, L., Picasso, V., Tornari, E., et al. (2015). BRAF-mutant melanoma: treatment approaches, resistance mechanisms, and diagnostic strategies. Oncol. Targets Ther. 8, 157–168. doi: 10.2147/OTT.S39096
Srijakotre, N., Man, J., Ooms, L. M., Lucato, C. M., Ellisdon, A. M., and Mitchell, C. A. (2017). P-Rex1 and P-Rex2 RacGEFs and cancer. Biochem. Soc. Trans. 45, 963–977. doi: 10.1042/BST20160269
Stein, E. M., DiNardo, C. D., Pollyea, D. A., Fathi, A. T., Roboz, G. J., Altman, J. K., et al. (2017). Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130, 722–731. doi: 10.1182/blood-2017-04-779405
Stransky, N., Egloff, A. M., Tward, A. D., Kostic, A. D., Cibulskis, K., Sivachenko, A., et al. (2011). The mutational landscape of head and neck squamous cell carcinoma. Science 333, 1157–1160. doi: 10.1126/science.1208130
Strub, T., Giuliano, S., Ye, T., Bonet, C., Keime, C., Kobi, D., et al. (2011). Essential role of microphthalmia transcription factor for DNA replication, mitosis and genomic stability in melanoma. Oncogene 30, 2319–2332. doi: 10.1038/onc.2010.612
Sung, P. J., Tsai, F. D., Vais, H., Court, H., Yang, J., Fehrenbacher, N., et al. (2013). Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc. Natl. Acad. Sci. U.S.A. 110, 20593–20598. doi: 10.1073/pnas.1306431110
Tanda, E. T., Vanni, I., Boutros, A., Andreotti, V., Bruno, W., Ghiorzo, P., et al. (2020). Current state of target treatment in BRAF mutated melanoma. Front. Mol. Biosci. 7:154. doi: 10.3389/fmolb.2020.00154
Tate, J. G., Bamford, S., Jubb, H. C., Sondka, Z., Beare, D. M., Bindal, N., et al. (2019). COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947. doi: 10.1093/nar/gky1015
Tate, S. C., Cai, S., Ajamie, R. T., Burke, T., Beckmann, R. P., Chan, E. M., et al. (2014). Semi-mechanistic pharmacokinetic/pharmacodynamic modeling of the antitumor activity of LY2835219, a new cyclin-dependent kinase 4/6 inhibitor, in mice bearing human tumor xenografts. Clin. Cancer Res. 20, 3763–3774. doi: 10.1158/1078-0432.CCR-13-2846
The Cancer Genome Atlas Network (2015). Genomic classification of cutaneous melanoma. Cell 161, 1681–1696. doi: 10.1016/j.cell.2015.05.044
Ticha, I., Hojny, J., Michalkova, R., Kodet, O., Krkavcova, E., Hajkova, N., et al. (2019). A comprehensive evaluation of pathogenic mutations in primary cutaneous melanomas, including the identification of novel loss-of-function variants. Sci. Rep. 9:17050. doi: 10.1038/s41598-019-53636-x
Tiffen, J. C., Gallagher, S. J., Tseng, H.-Y., Filipp, F. V., and Hersey, P. (2016). EZH2 as a mediator of treatment resistance in melanoma. Pigment Cell Melanom. Res. 29, 500–507. doi: 10.1111/pcmr.12481
Tiffen, J. C., Gunatilake, D., Gallagher, S. J., Gowrishankar, K., Heinemann, A., Cullinane, C., et al. (2015). Targeting activating mutations of EZH2 leads to potent cell growth inhibition in human melanoma by derepression of tumor suppressor genes. Oncotarget 6, 27023–27036. doi: 10.18632/oncotarget.4809
Trovó-Marqui, A. B., and Tajara, E. H. (2006). Neurofibromin: a general outlook. Clin. Genet. 70, 1–13. doi: 10.1111/j.1399-0004.2006.00639.x
Tsao, H., Goel, V., Wu, H., Yang, G., and Haluska, F. G. (2004). Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J. Invest. Dermatol. 122, 337–341. doi: 10.1046/j.0022-202X.2004.22243.x
Tsao, H., Zhang, X., Benoit, E., and Haluska, F. G. (1998). Identification of PTEN/MMAC1 alterations in uncultured melanomas and melanoma cell lines. Oncogene 16, 3397–3402. doi: 10.1038/sj.onc.1201881
Tsao, H., Zhang, X., Fowlkes, K., and Haluska, F. G. (2000). Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res. 60, 1800–1804.
Ugurel, S., Thirumaran, R. K., Bloethner, S., Gast, A., Sucker, A., Mueller-Berghaus, J., et al. (2007). B-RAF and N-RAS mutations are preserved during short time in vitro propagation and differentially impact prognosis. PLoS One 2:e236. doi: 10.1371/journal.pone.0000236
Vachtenheim, J., and Borovanskı, J. (2010). “Transcription physiology” of pigment formation in melanocytes: central role of MITF. Exp. Dermatol. 19, 617–627. doi: 10.1111/j.1600-0625.2009.01053.x
Van Allen, E. M., Miao, D., Schilling, B., Shukla, S. A., Blank, C., Zimmer, L., et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. doi: 10.1126/science.aad0095
van Herpen, C., Agarwala, S. S., Hauschild, A., Dummer, R., Berking, C., Beck, J. T., et al. (2014). LBA35 - overall survival and biomarker results from a phase 2 study of mek1/2 inhibitor binimetinib (Mek162) in patients with advanced NRAS-mutant melanoma. Ann. Oncol. 25:43. doi: 10.1093/annonc/mdu438.43
Vanni, I., Tanda, E. T., Spagnolo, F., Andreotti, V., Bruno, W., and Ghiorzo, P. (2020). The current state of molecular testing in the BRAF-mutated melanoma landscape. Front. Mol. Biosci. 7:113. doi: 10.3389/fmolb.2020.00113
Vera Aguilera, J., Rao, R. D., Allred, J. B., Suman, V. J., Windschitl, H. E., Kaur, J. S., et al. (2018). Phase II study of everolimus in metastatic malignant melanoma (NCCTG-N0377, Alliance). Oncologist 23:887. doi: 10.1634/theoncologist.2018-0100
Villalonga, P., López-Alcalá, C., Bosch, M., Chiloeches, A., Rocamora, N., Gil, J., et al. (2001). Calmodulin binds to K-Ras, but Not to H- or N-Ras, and modulates its downstream signaling. Mol. Cell Biol. 21, 7345–7354. doi: 10.1128/MCB.21.21.7345-7354.2001
Viré, E., Brenner, C., Deplus, R., Blanchon, L., Fraga, M., Didelot, C., et al. (2006). The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439, 871–874. doi: 10.1038/nature04431
Viros, A., Sanchez-Laorden, B., Pedersen, M., Furney, S. J., Rae, J., Hogan, K., et al. (2014). Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature 511, 478–482. doi: 10.1038/nature13298
Vogt, P. K. (2001). PI 3-kinase, mTOR, protein synthesis and cancer. Trends Mol. Med. 7, 482–484. doi: 10.1016/s1471-4914(01)02161-x
Vu, H. L., Rosenbaum, S., Purwin, T. J., Davies, M. A., and Aplin, A. E. (2015). RAC1 P29S regulates PD-L1 expression in melanoma. Pigment Cell Melanom. Res. 28, 590–598. doi: 10.1111/pcmr.12392
Wagner, K.-D., Cherfils-Vicini, J., Hosen, N., Hohenstein, P., Gilson, E., Hastie, N. D., et al. (2014). The Wilms’ tumour suppressor Wt1 is a major regulator of tumour angiogenesis and progression. Nat. Commun. 5:5852. doi: 10.1038/ncomms6852
Wagner, N., Panelos, J., Massi, D., and Wagner, K.-D. (2008). The Wilms’ tumor suppressor WT1 is associated with melanoma proliferation. Pflugers Arch. 455, 839–847. doi: 10.1007/s00424-007-0340-1
Wang, L., Lawrence, M. S., Wan, Y., Stojanov, P., Sougnez, C., Stevenson, K., et al. (2011). SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 365, 2497–2506. doi: 10.1056/NEJMoa1109016
Wang, M.-T., Holderfield, M., Galeas, J., Delrosario, R., To, M. D., Balmain, A., et al. (2015). K-Ras promotes tumorigenicity through suppression of non-canonical Wnt signaling. Cell 163, 1237–1251. doi: 10.1016/j.cell.2015.10.041
Wang, R., He, Y., Robinson, V., Yang, Z., Hessler, P., Lasko, L. M., et al. (2018). Targeting lineage-specific MITF pathway in human melanoma cell lines by A-485, the selective small-molecule inhibitor of p300/CBP. Mol. Cancer Ther. 17, 2543–2550. doi: 10.1158/1535-7163.MCT-18-0511
Wang, Y., Xu, X., Maglic, D., Dill, M. T., Mojumdar, K., Ng, P. K.-S., et al. (2018). Comprehensive molecular characterization of the hippo signaling pathway in cancer. Cell Rep. 25, 1304–1317. doi: 10.1016/j.celrep.2018.10.001
Watson, I. R., Li, L., Cabeceiras, P. K., Mahdavi, M., Gutschner, T., Genovese, G., et al. (2014). The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res. 74, 4845–4852. doi: 10.1158/0008-5472.CAN-14-1232-T
Webster, D. E., Barajas, B., Bussat, R. T., Yan, K. J., Neela, P. H., Flockhart, R. J., et al. (2014). Enhancer-targeted genome editing selectively blocks innate resistance to oncokinase inhibition. Genome Res. 24, 751–760. doi: 10.1101/gr.166231.113
Wei, X., Walia, V., Lin, J. C., Teer, J. K., Prickett, T. D., Gartner, J., et al. (2011). Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat. Genet. 43, 442–446. doi: 10.1038/ng.810
Welman, A., Burger, M. M., and Hagmann, J. (2000). Structure and function of the C-terminal hypervariable region of K-Ras4B in plasma membrane targetting and transformation. Oncogene 19, 4582–4591. doi: 10.1038/sj.onc.1203818
Wengrod, J., Wang, D., Weiss, S., Zhong, H., Osman, I., and Gardner, L. B. (2015). Phosphorylation of eIF2α triggered by mTORC1 inhibition and PP6C activation is required for autophagy and is aberrant in PP6C-mutated melanoma. Sci. Signal 8:ra27. doi: 10.1126/scisignal.aaa0899
Whyte, D. B., Kirschmeier, P., Hockenberry, T. N., Nunez-Oliva, I., James, L., Catino, J. J., et al. (1997). K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 272, 14459–14464. doi: 10.1074/jbc.272.22.14459
Widlund, H. R., Horstmann, M. A., Price, E. R., Cui, J., Lessnick, S. L., Wu, M., et al. (2002). Beta-catenin-induced melanoma growth requires the downstream target Microphthalmia-associated transcription factor. J. Cell Biol. 158, 1079–1087. doi: 10.1083/jcb.200202049
Wilmott, J. S., Johansson, P. A., Newell, F., Waddell, N., Ferguson, P., Quek, C., et al. (2019). Whole genome sequencing of melanomas in adolescent and young adults reveals distinct mutation landscapes and the potential role of germline variants in disease susceptibility. Int. J. Cancer 144, 1049–1060. doi: 10.1002/ijc.31791
Wilsher, M., and Cheerala, B. (2007). WT1 as a complementary marker of malignant melanoma: an immunohistochemical study of whole sections. Histopathology 51, 605–610. doi: 10.1111/j.1365-2559.2007.02843.x
Winski, L., Anderson, D., and Bouhana, K. (2010). MEK162 (ARRY-162), a Novel MEK 1/2 Inhibitor, Inhibits Tumor Growth Regardless of KRas/Raf Pathway Mutations. EORTC - NCI - AACR, Berlin, 2010. Available online at: https://www.cureus.com/posters/103-mek162-arry-162-a-novel-mek-12-inhibitor-inhibits-tumor-growth-regardless-of-krasraf-pathway-mutations (accessed April 27, 2020).
Wong, S. Q., Behren, A., Mar, V. J., Woods, K., Li, J., Martin, C., et al. (2015). Whole exome sequencing identifies a recurrent RQCD1 P131L mutation in cutaneous melanoma. Oncotarget 6, 1115–1127. doi: 10.18632/oncotarget.2747
Wong-Staal, F., Dalla-Favera, R., Franchini, G., Gelmann, E. P., and Gallo, R. C. (1981). Three distinct genes in human DNA related to the transforming genes of mammalian sarcoma retroviruses. Science 213, 226–228. doi: 10.1126/science.6264598
Worby, C. A., and Dixon, J. E. (2002). Sorting out the cellular functions of sorting nexins. Nat. Rev. Mol. Cell Biol. 3, 919–931. doi: 10.1038/nrm974
Wu, X., Yu, J., Yan, J., Dai, J., Si, L., Chi, Z., et al. (2018). PI3K/AKT/mTOR pathway inhibitors inhibit the growth of melanoma cells with mTOR H2189Y mutations in vitro. Cancer Biol. Ther. 19, 584–589. doi: 10.1080/15384047.2018.1435221
Wullschleger, S., Loewith, R., and Hall, M. N. (2006). TOR signaling in growth and metabolism. Cell 124, 471–484. doi: 10.1016/j.cell.2006.01.016
Xiao, W., Du, N., Huang, T., Guo, J., Mo, X., Yuan, T., et al. (2018). TP53 mutation as potential negative predictor for response of anti-CTLA-4 therapy in metastatic melanoma. EBiomedicine 32, 119–124. doi: 10.1016/j.ebiom.2018.05.019
Xu, K., Wu, Z. J., Groner, A. C., He, H. H., Cai, C., Lis, R. T., et al. (2012). EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 338, 1465–1469. doi: 10.1126/science.1227604
Yadav, V., Burke, T. F., Huber, L., Van Horn, R. D., Zhang, Y., Buchanan, S. G., et al. (2014). The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol. Cancer Ther. 13, 2253–2263. doi: 10.1158/1535-7163.MCT-14-0257
Yan, H., Parsons, D. W., Jin, G., McLendon, R., Rasheed, B. A., Yuan, W., et al. (2009). IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773. doi: 10.1056/NEJMoa0808710
Yarwood, S., Bouyoucef-Cherchalli, D., Cullen, P. J., and Kupzig, S. (2006). The GAP1 family of GTPase-activating proteins: spatial and temporal regulators of small GTPase signalling. Biochem. Soc. Trans. 34, 846–850. doi: 10.1042/BST0340846
Yeh, C.-H., Bellon, M., and Nicot, C. (2018). FBXW7: a critical tumor suppressor of human cancers. Mol. Cancer 17:115. doi: 10.1186/s12943-018-0857-2
Yeh, T. C., Marsh, V., Bernat, B. A., Ballard, J., Colwell, H., Evans, R. J., et al. (2007). Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin. Cancer Res. 13, 1576–1583. doi: 10.1158/1078-0432.CCR-06-1150
Yin, C., Zhu, B., Zhang, T., Liu, T., Chen, S., Liu, Y., et al. (2019). Pharmacological targeting of STK19 inhibits oncogenic NRAS-driven melanomagenesis. Cell 176, 1113–1127. doi: 10.1016/j.cell.2019.01.002
Yokoyama, S., Feige, E., Poling, L. L., Levy, C., Widlund, H. R., Khaled, M., et al. (2008). Pharmacologic suppression of MITF expression via HDAC inhibitors in the melanocyte lineage. Pigment Cell Melanom. Res. 21, 457–463. doi: 10.1111/j.1755-148X.2008.00480.x
Yu, F.-X., Luo, J., Mo, J.-S., Liu, G., Kim, Y. C., Meng, Z., et al. (2014). Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 25, 822–830. doi: 10.1016/j.ccr.2014.04.017
Yu, F.-X., Zhao, B., and Guan, K.-L. (2015). Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163, 811–828. doi: 10.1016/j.cell.2015.10.044
Yu, H., Ma, M., Yan, J., Xu, L., Yu, J., Dai, J., et al. (2017). Identification of coexistence of BRAF V600E mutation and EZH2 gain specifically in melanoma as a promising target for combination therapy. J. Transl. Med. 15:243. doi: 10.1186/s12967-017-1344-z
Yu, J., Yu, J., Wu, X., Guo, Q., Yin, T., Cheng, Z., et al. (2020). The TERT copy number gain is sensitive to telomerase inhibitors in human melanoma. Clin. Sci. 134, 193–205. doi: 10.1042/CS20190890
Yu, S., Xu, T., Dai, J., Ma, M., Tang, H., Chi, Z., et al. (2018). TERT copy gain predicts the outcome of high-dose interferon α-2b therapy in acral melanoma. Oncol. Targets Ther. 11, 4097–4104. doi: 10.2147/OTT.S158239
Yue, E. W., Li, Y.-L., Douty, B., He, C., Mei, S., Wayland, B., et al. (2019). INCB050465 (Parsaclisib), a novel next-generation inhibitor of phosphoinositide 3-kinase delta (PI3Kδ). ACS Med. Chem. Lett. 10, 1554–1560. doi: 10.1021/acsmedchemlett.9b00334
Zakut, R., Perlis, R., Eliyahu, S., Yarden, Y., Givol, D., Lyman, S. D., et al. (1993). KIT ligand (mast cell growth factor) inhibits the growth of KIT-expressing melanoma cells. Oncogene 8, 2221–2229.
Zapata-Benavides, P., Manilla-Muñoz, E., Zamora-Avila, D. E., Saavedra-Alonso, S., Franco-Molina, M. A., Trejo-Avila, L. M., et al. (2012). WT1 silencing by RNAi synergizes with chemotherapeutic agents and induces chemosensitization to doxorubicin and cisplatin in B16F10 murine melanoma cells. Oncol. Lett. 3, 751–755. doi: 10.3892/ol.2012.578
Zapata-Benavides, P., Thompson-Armendariz, F. G., Arellano-Rodríguez, M., Franco-Molina, M. A., Mendoza-Gamboa, E., Saavedra-Alonso, S., et al. (2019). shRNA-WT1 potentiates anticancer effects of gemcitabine and cisplatin against B16F10 lung metastases in vitro and in vivo. Vivo 33, 777–785. doi: 10.21873/invivo.11539
Zeng, H., Jorapur, A., Shain, A. H., Lang, U. E., Torres, R., Zhang, Y., et al. (2018). Bi-allelic Loss of CDKN2A initiates melanoma invasion via BRN2 activation. Cancer Cell 34, 56–68. doi: 10.1016/j.ccell.2018.05.014
Zeng, K., Bastos, R. N., Barr, F. A., and Gruneberg, U. (2010). Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J. Cell Biol. 191, 1315–1332. doi: 10.1083/jcb.201008106
Zhang, X., Tang, J. Z., Vergara, I. A., Zhang, Y., Szeto, P., Yang, L., et al. (2019). Somatic hypermutation of the YAP oncogene in a human cutaneous melanoma. Mol. Cancer Res. 17, 1435–1449. doi: 10.1158/1541-7786.MCR-18-0407
Zhou, J.-D., Yao, D.-M., Li, X.-X., Zhang, T.-J., Zhang, W., Ma, J.-C., et al. (2017). KRAS overexpression independent of RAS mutations confers an adverse prognosis in cytogenetically normal acute myeloid leukemia. Oncotarget 8, 66087–66097. doi: 10.18632/oncotarget.19798
Zhu, M., Molina, J. R., Dy, G. K., Croghan, G. A., Qi, Y., Glockner, J., et al. (2020). A phase I study of the VEGFR kinase inhibitor vatalanib in combination with the mTOR inhibitor, everolimus, in patients with advanced solid tumors. Invest. New Drugs. doi: 10.1007/s10637-020-00936-z [Epub ahead of print].
Zingg, D., Arenas-Ramirez, N., Sahin, D., Rosalia, R. A., Antunes, A. T., Haeusel, J., et al. (2017). The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. 20, 854–867. doi: 10.1016/j.celrep.2017.07.007
Zingg, D., Debbache, J., Peña-Hernández, R., Antunes, A. T., Schaefer, S. M., Cheng, P. F., et al. (2018). EZH2-mediated primary cilium deconstruction drives metastatic melanoma formation. Cancer Cell 34, 69–84. doi: 10.1016/j.ccell.2018.06.001
Keywords: melanoma, non-BRAF mutation, targeted therapy, driver mutations, genetic, heterogeneity, WES, WGS
Citation: Vanni I, Tanda ET, Dalmasso B, Pastorino L, Andreotti V, Bruno W, Boutros A, Spagnolo F and Ghiorzo P (2020) Non-BRAF Mutant Melanoma: Molecular Features and Therapeutical Implications. Front. Mol. Biosci. 7:172. doi: 10.3389/fmolb.2020.00172
Received: 14 May 2020; Accepted: 03 July 2020;
Published: 24 July 2020.
Edited by:
Cristina Pellegrini, University of L’Aquila, ItalyReviewed by:
Lixia Gao, Chongqing University of Arts and Sciences, ChinaAustin Shull, Presbyterian College, United States
Copyright © 2020 Vanni, Tanda, Dalmasso, Pastorino, Andreotti, Bruno, Boutros, Spagnolo and Ghiorzo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Ghiorzo, cGFvbGEuZ2hpb3J6b0B1bmlnZS5pdA==
†These authors have contributed equally to this work