 Jacob M. Favret
Jacob M. Favret Nadav I. Weinstock
Nadav I. Weinstock M. Laura Feltri
M. Laura Feltri Daesung Shin
Daesung Shin- Hunter James Kelly Research Institute, Department of Biochemistry and Neurology, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo, NY, United States
There are over 50 lysosomal hydrolase deficiencies, many of which cause neurodegeneration, cognitive decline and death. In recent years, a number of broad innovative therapies have been proposed and investigated for lysosomal storage diseases (LSDs), such as enzyme replacement, substrate reduction, pharmacologic chaperones, stem cell transplantation, and various forms of gene therapy. Murine models that accurately reflect the phenotypes observed in human LSDs are critical for the development, assessment and implementation of novel translational therapies. The goal of this review is to summarize the neurodegenerative murine LSD models available that recapitulate human disease, and the pre-clinical studies previously conducted. We also describe some limitations and difficulties in working with mouse models of neurodegenerative LSDs.
Introduction
The lysosome orchestrates a number of cellular homeostatic processes, primarily focused on the catabolism of diverse macromolecules. Lysosomes contain more than 50 unique acid hydrolases, each facilitating the degradation of specific metabolites including glycosides, sulfates, phosphates, various lipids, phospholipids, proteins and nucleic acids. Lysosomal hydrolases are synthesized in the ER, tagged with a mannose-6-phosphate (M6P) residue in the Golgi apparatus, and properly trafficked to the lysosome via M6P receptors. These receptors are also expressed at the plasma membrane of cells, thus allowing for the internalization of secreted lysosomal enzymes from the environment (Platt and Walkley, 2004).
Lysosomal storage diseases (LSDs) are a heterogeneous group of inherited diseases, caused by mutations leading to a deficiency of lysosomal hydrolases or transporters. LSDs lead to the accumulation of specific substrates within the lysosomal compartment, consequentially triggering a number of secondary cellular responses that result in cellular dysfunction, death and tissue damage. Substrate storage causes a wide range of perturbed cellular functions including the loss-of-function in housekeeping processes and pathways, modulation of signal transduction cascades, aberrant activation of inflammatory responses, impaired intracellular trafficking of vesicles and membrane-bound proteins, and disequilibrium of autophagic flux (Walkley, 2009). Although the nature of substrate accumulation and generalized lysosomal dysfunction is seemingly intuitive, there are a number of intriguing unanswered questions in the field of LSDs. For example, though lysosomal hydrolases are generally ubiquitously expressed, the lysosomal storage often varies even among neighboring cells (Marques and Saftig, 2019). This may be explained by a number of factors including the ability of cells to upregulate the lysosomal-autophagy pathway (Schultz et al., 2011) or whether cells have alternate strategies to dispose of stored material (Schultz et al., 2011; Ferraz et al., 2016).
Due to the heterogeneous accumulation of enzymatic substrates among multiple tissues and organs, the phenotypes among LSDs vary widely and often include visceral, ocular, hematologic, skeletal and neurological manifestations. In particular, those related to the involvement of the central nervous system (CNS) may cause progressive neurodegeneration and severe cognitive impairment. Approximately two thirds of LSD patients display CNS imparment to some extent, resulting in progressive neurodegeneration (Parenti et al., 2013). Post-mitotic cells such as neurons rely heavily on the endolysosomal and autophagic systems to prevent accumulation of debris that would otherwise become toxic. Furthermore, the extreme anatomical architecture of neurons makes the actual sequestration and degradation of substrates very challenging, as lysosomes have to travel very long distances from the cell soma to distal axons and dendrites. On top of all of this, lysosomal and autophagic functions decrease in aging (Cuervo and Dice, 2000; Lynch and Bi, 2003; Kurz et al., 2008), further pressuring the system’s efficiency. Therefore, it is not surprising that many LSDs have nervous system involvement and that various aging-related neurodegenerative diseases are caused, at least in part, by endolysosomal dysfunction. These include Parkinson’s (Anglade et al., 1997), Alzheimer’s (Nixon et al., 2005), Huntington’s diseases (Rudnicki et al., 2008), and amyotrophic lateral sclerosis (Sasaki, 2011) caused by the accumulation of aberrant or misfolded proteins. Due to the common underlying mechanisms between LSDs and neurodegenerative diseases, the development of novel treatments for LSDs may have supplemental benefits for a larger spectrum of neurodegenerative conditions. Here, we review currently available mouse models for neurodegenerative LSDs and discuss how those models have been used for pre-clinical trials and have helped move therapies forward.
Main Text
Therapeutic Approaches for LSDs
During the past three decades, research in the field of LSDs has made marked progress. Innovation of novel therapeutic approaches has given hope to many where historically the outlook has been bleak. Some LSDs are now treatable, though most cannot be treated after symptoms begin. The major strategy implemented in the treatment of LSDs is to restore or replace the defective enzyme’s activity. These modalities include hematopoietic stem cell transplantation (HSCT), enzyme replacement therapy (ERT), pharmacological chaperone therapy (PCT) and gene therapy (GT). Alternative approaches include substrate reduction therapy (SRT), based on reducing the synthesis of the substrates stored in the lysosomes (Figure 1; Eisenstein, 2016).
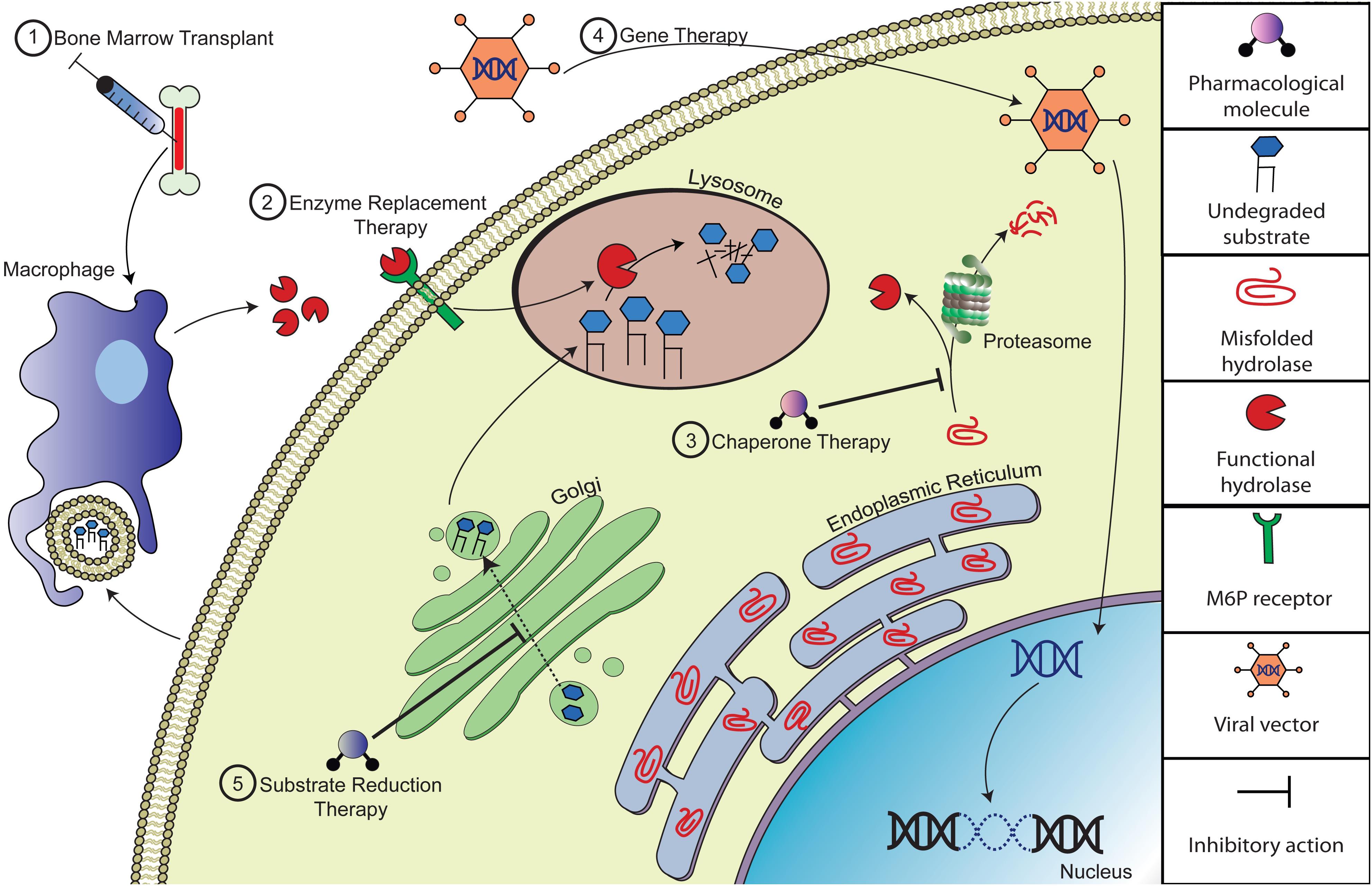
Figure 1. Depiction of potential therapeutic approaches to correct substrate accumulation of lysosomal storage diseases. (1) Bone marrow transplantation (BMT) alleviates storage by introducing normal donor-derived macrophages that will cross-correct with viable lysosomal hydrolases and/or phagocytize excess substrate. (2) Enzyme replacement therapy (ERT) compensates for the loss of endogenous hydrolase activity by providing recombinant enzyme, which could be taken up by mannose/M6P receptors on the cell surface. (3) Pharmacologic chaperone therapy (PCT) can improve the catalytic activity of misfolded lysosomal enzyme by promoting folding and acquisition of functional conformation of the nascent mutant peptide and thus evading pre-mature degradation. (4) Gene therapy (GT) is a therapeutic approach designed to deliver recombinant DNA to enzyme deficient cells, often via viral vector therapy. (5) Substrate reduction therapy (SRT) involves delivery of small molecule inhibitors that reduce biosynthesis of the specific accumulating substrate.
Hematopoietic Stem Cell Transplantation Therapy
Hematopoietic stem cell therapy (HSCT) is one of the more common treatments of LSDs. HSCT is the main therapy for mucopolysaccharidosis (MPS) I and Krabbe disease, and has been used in other LSDs including MPS II, MPS IVA, MPS VII, metachromatic leukodystrophy and fucosidosis (Poswar et al., 2019). HSCT allows for the delivery and engraftment of donor derived stem cells in patients with LSDs. The healthy cells repopulate in specific tissues and secrete functional lysosomal hydrolases into the extracellular space and into the blood circulation. The secreted normal enzyme may be taken up by the endogenous cells to cross-correct the enzyme deficiency of the mutated cells (Prasad and Kurtzberg, 2009). Additional benefits of HSCT have also been speculated to be immunomodulatory. HSCT-derived macrophages, which have functional lysosomal enzymes, may be able to better phagocytize dying cells/debris. Generally, earlier HSCT leads to improved outcomes but is only efficacious if delivered to pre-symptomatic patients, presumably before the occurrence of irreversible cell damage. This is particularly true for the neurologic symptoms of LSDs. Recently, genetically modified HSCT, termed HSCGT (Hematopoietic Stem Cell - Gene Therapy) has been used successfully to treat metachromatic leukodystrophy patients (MLD) (Biffi et al., 2013; Lorioli and Biffi, 2015) who re-introduced the patients’ own CD34+cells with lentivirus-transfected cells, overexpressing arylsulfatase A (ARSA). Similarly, MPS IIIA phase I/II clinical trials of SGSH are currently underway based on an HSCGT pre-clinical study in MPSIIIA mice (Bhaumik et al., 1999; Ellison et al., 2019). Alternatively, gene editing approaches have also been implemented to modify HSCT or induced pluripotent stem cells (iPSCs) (Mandal et al., 2014; Christensen and Choy, 2017). Other non-LSDs like cerebral adrenoleukodystrophy (Eichler et al., 2017), have had success treating patients with HSCGT which has garnered significant attention and excitement. Furthermore, alternative stem cell transplantation, like oligodendrocyte progenitor cell implantation (Scaravilli and Jacobs, 1981, 1982; Scaravilli and Suzuki, 1983; Givogri et al., 2006) has also been explored in MLD.
Enzyme Replacement Therapy
Enzyme replacement therapy (ERT) consists of periodic intravenous infusions of recombinant lysosomal enzyme in patients with LSDs. The first use of ERT was the use of glucocerebrosidase for Gaucher disease in 1991 (Barton et al., 1991). ERT has since been used for Farber’s disease, Pompe disease, MPS types I, II, IVA, VI, and VII and lysosomal acid lipase deficiency, and is currently being developed for others (Poswar et al., 2019). It is now possible to mass produce purified enzyme due to advances in recombinant DNA techniques. Once injected, the normal recombinant enzymes are distributed to tissues, internalized by endocytosis and targeted to the lysosomal compartment, where they replace the defective enzyme. Receptor-mediated endocytosis underlies the cellular uptake of lysosomal enzymes with mannose residues that bind to mannose receptors on the cell surface, as well as M6P residues that bind to M6P receptors (Platt et al., 2018). A major limitation of ERT is that not all organs are freely accessible to the administered enzyme. Recombinant enzymes are large molecules that do not passively diffuse across membranes. Consequently, the enzyme is unable to reach therapeutic concentrations in some of the key target tissues. Most notably, there is poor efficacy of recombinant enzyme in reaching the CNS, due in part to restricted diffusion across the blood brain barrier (BBB). To overcome the challenges presented by the BBB, it has been tried to make modified enzymes to be transported through existing systems, such as the insulin or transferrin receptors. Moreover, direct administration of recombinant enzyme into the CNS has proven an effective method of distribution (Platt et al., 2018; Kohlschütter et al., 2019). Another major limitation of ERT is that the exogenous recombinant enzyme can elicit an immune reaction. These responses include hypersensitivity reactions, neutralizing antibodies to the recombinant enzyme and altered enzyme turnover and uptake (Brooks, 1999).
Pharmacological Chaperone Therapy
Pharmacological chaperone therapy (PCT) attempts to rescue reduced or absent function of mutant lysosomal protein which is misfolded or mis-trafficked. The approach uses small molecule ligands which bind and stabilize mutant enzyme. The binding of chaperone to mutant enzyme facilitates increased cellular enzyme concentrations, improved enzyme trafficking and increased lysosomal activity (Parenti et al., 2015). For example, sub-inhibitory concentrations of active site inhibitors can stabilize the mutant enzyme, which extends half-life (Platt et al., 2018). Competitive enzyme inhibitors are expected to be effective as active site specific chaperones, because of their high affinity to the catalytic domain (Fan, 2008). As a result, the enzymatic activity of the mutant protein is partially rescued. Minor increases in enzymatic activity have a favorable impact on the clearance of storage material and thus patient status and rate of disease progression. PCTs have several advantages, as compared to other therapies, as they can be administered orally, allowing for a non-invasive treatment and are non-immunogenic. Furthermore, pharmaceutical chaperones are generally small enough to diffuse passively across cell membranes and reach therapeutic concentrations in different tissues and systems, including the CNS. A major limitation of PCT is that it cannot be used for stop-codon mutations because they result in premature termination or nonsense mRNA decay. For this type of nonsense mutant, other type of small molecules that can override or read-through the stop-codons are now under development (Platt et al., 2018). The development of nonsense mutation LSD mouse models has better allows researchers to test these nonsense-suppression therapies (NST). This is of importance as many LSDs harbor nonsensense mutations. For example, greater than 50% of CLN1 disease is caused by nonsense mutations (Miller et al., 2015).
Gene Therapy
Gene therapy (GT) for LSDs is a rapidly advancing field of treatment. The major approach in GT is the direct transfer of the defective gene into the cells of the patient. The normal gene product is generally delivered to the patient via a viral vector (Ohashi, 2019). The first GT for LSDs was for Gaucher disease in 1998, which resulted in transient enzymatic expression in the patient (Dunbar et al., 1998). However, retroviruses generally only infect mitotically active cell types. This property seriously limits the usefulness of retroviruses as the vector of GT for CNS disease, in which most cells are post-mitotic. To overcome this limitation, other viral vectors have been studied and implemented, including adeno-associated virus (AAV) and lentivirus (LV) (Suzuki, 2004). AAV has been especially effective in correcting genetic diseases, quickly becoming one of the most promising viral vectors for the treatment of LSDs. AAVs are capable of infecting cells that are not going through mitosis and persist primarily as non-integrative episomal units. Therefore, various AAV serotypes have been developed with particular tropism for cells and tissues of interest, including neurons and glia, and tested in pre-clinical mouse models of LSDs (Bailey et al., 2018). AAV-mediated GT successfully improved the phenotypes of GM1 gangliosidosis, MPS I & IIIB, Sandhoff disease, metachromatic leukodystrophy, and Krabbe disease (Gonzalez and Baldo, 2017). Based on the positive results from these animal models, multiple phase I/II clinical trials are currently being conducted, many of which have promising results. Of note, multiple technical approaches exist regarding the site of injection, particularly in regards to treating the CNS in neurodegenerative LSDs. Some approaches have implemented systemic intravenous GT administration (Fu et al., 2016), while others attempted more localized intracerebral (Winner et al., 2016) or intrathecal administration of the viral vector (Karumuthil-Melethil et al., 2016; Bey et al., 2017), or an approach across multiple sites (Marshall et al., 2018). Furthermore, major advances in the realm of GT in recent years has been centered on the bourgeoning field of gene editing via CRISPR/Cas9 and zinc finger nuclease (ZFN) technologies. These approaches either introduce exogenous genetic material or repair the defunct endogenous locus. Alternatively, non-viral methods of introducing the gene of interest such as electro gene therapy, the phiC31, minicircle and Sleeping Beauty gene transfer systems have been implemented pre-clinically to test the efficacy of non-viral gene therapy (Aronovich et al., 2007, 2009; Osborn et al., 2008; Stilhano et al., 2015). General advantages of GT over other approaches are the stable and long-term production of therapeutic protein. GT therefore has advantages compared to ERT and small molecule therapies, which require life-long treatment. Concerns of GT do remain, including the risk that the modification of genomic DNA in the patient increases the risk of carcinogenesis (Chandler et al., 2017a). Furthermore, the expression of supra-physiologic levels of enzyme may cause unintended side effects. Finally, one challenge remaining in certain cases is the broad distribution of the viral vector to all the tissues involved (Cearley and Wolfe, 2007).
Substrate Reduction Therapy
Substrate reduction therapy (SRT) often uses small-molecule inhibitors to partially inhibit specific steps of the biosynthetic pathways of substrates that accumulate in LSDs (Radin, 1996). Two SRT drugs for Gaucher disease (miglustat and eliglustat tartrate) have been approved and others are undergoing clinical trials (Coutinho et al., 2016). Since SRT drugs are orally administered, SRT does not involve an invasive delivery. Due to their general low molecular mass, SRT drugs are non-immunogenic, and mostly can cross the BBB. Although in principle SRT could be useful for all LSDs, it is restricted to the one whose specific upstream biosynthetic pathways have been identified. An intriguing expansion of this approach can be thought of as genetic SRT, in which small interfering RNAs could theoretically be used to silence enzymes responsible for the production of LSD accumulated substrates (Coutinho et al., 2016). Other forms of SRT include dietary modification and restriction of substrate intake (Denny et al., 2010; Soga et al., 2015).
Combination Therapy
Since LSDs are multisystem disorders, multiple therapies may be required to effectively treat different components of disease pathology. Due to the partial effect of different modalities of therapies, combining different forms of therapy can be more effective than a single approach. Therefore, combination therapies to address the diverse symptoms of LSDs may be required (Platt et al., 2018). In animal models, the efficacy of combination therapy is actively being investigated, and in some cases the synergy that is reported is striking. For example, combining different forms of therapy has had profound improvements of survival on the twitcher mouse model of Krabbe disease (KD). In particular, the use of HSCT has been particularly efficacious in synergizing with viral-directed gene therapy. The combination of HSCT and GT has been replicated by many different labs with different viral vectors and regiments of delivery (Hawkins-Salsbury et al., 2015; Rafi et al., 2015b).
Autophagy Modulators
Additional strategies for treating LSDs involve targeting common mechanisms involved in LSD and neurodegenerative pathophysiology. The benefit of these approaches, which are downstream of the specific lysosomal hydrolase mutation, is that they can theoretically be applied broadly. For example, defects in autophagy have been associated with a large number of LSDs (Settembre et al., 2008; Seranova et al., 2017). Recent attempts to increase autophagy in LSDs has been successfully employed in a variety of pre-clinical models including overexpression of the MiTF transcription factors TFEB and TFE3 (Spampanato et al., 2013; Rega et al., 2016) or by manipulation of the mTOR pathway (Bartolomeo et al., 2014). These approaches also have the added benefit of increasing lysosomal exocytosis, which is thought to decrease the lysosomal storage burden and associated pathology. Alternatively, for neurodegenerative LSDs, attempts to decrease the accumulation of misfolded proteins and restore proper autophagy have been employed (Monaco et al., 2020). While most of these trials remain in the pre-clinical stage, their clinical applications seem promising.
Other Therapies Under Development
A large number of alternative therapeutic strategies relating to inflammation and neurodegeneration have also been tested in various pre-clinical trials. Prolonged neuroinflammation contributes to neuronal degeneration and can exacerbate the LSD phenotype and pre-clinical trials have exhibited the beneficial effects of non-steroidal anti-inflammatory drugs (NSAIDs) and other anti-inflammatory (AI) agents (Stein et al., 2015; Dannhausen et al., 2018). Some pre-clinical trials target proteins involved in modulating reactions to symptoms of the particular LSD; for example abolishing macrophage-inflammatory protein (MIP)-1 activity (Wu and Proia, 2004) to attenuate inflammation. LSDs are also often accompanied by oxidative stress, which can be treated with pharmacologic or dietary antioxidants (Wei et al., 2011; Hawkins-Salsbury et al., 2012; Saha et al., 2012; Donida et al., 2017). Akin to other neurodegenerative diseases such as Alzheimer’s disease and Huntington’s disease, LSDs can elicit aberrant neuronal signaling leading to neurotransmitter mediated excitotoxicity. Pre-clinical trials evaluating receptor antagonism (RA), AMPA-RA (Kovacs and Pearce, 2008) and NMDA-RA (Finn et al., 2013) have improved the neurobehavioral phenotypes associated with LSDs. Furthermore, a number of pharmacological agents have been explored eliciting various effects from modulating membrane fluidity (Schultz et al., 2018), modulating Ca2+ (Chang et al., 2007) or cholesterol levels (Erickson et al., 2000; Pelled et al., 2003; Kim et al., 2007; Repa et al., 2007; Abi-Mosleh et al., 2009; Liu et al., 2010; Taylor et al., 2012; Hovakimyan et al., 2013; Nusca et al., 2014; Tanaka et al., 2014; Soga et al., 2015; Demais et al., 2016; Liou et al., 2016), and enhancing enzyme activity (Arroyo et al., 2014) via the use of neurosteroids (NS) (Griffin et al., 2004; Liao et al., 2009). There is also interest in facilitating sphingolipid degradation via the upregulation of heat shock proteins (HSP) (Chung et al., 2016; Kirkegaard et al., 2016).
Mouse Models of Neurodegenerative LSDs
The majority of LSDs are associated with neurodegenerative features, that are often progressive over the course of the disease. Therapies are available only for a small subset of LSDs and have not been much effective on neurological symptoms. Mouse models have played a major role in the development and improvement of novel therapeutic modalities. Pre-clinical animal models are particularly useful as they can aid in elucidating key molecular changes involved with disease pathogenesis. Various LSD mouse models have been characterized and developed and are extensively being used for the design of novel therapeutics. The use of authentic pre-clinical animal models has been shown to be predictive of therapeutic outcomes in LSD human clinical trials, and therefore reduces the time and cost of drug development. In addition, due to the small patient numbers available when studying LSDs and other rare diseases, it is difficult to standardize endpoint measurements and generate the statistical power necessary for accurate interpretation and study design (Augustine et al., 2013). Therefore, the use of pre-clinical models is important for acquiring as much information as possible about the safety and efficacy of new therapies. Here, we categorize currently available models for neurodegenerative LSDs and elaborate on how those models have been used for pre-clinical purposes. We explore the critical role of animal models in developing novel therapies and discuss broadly the advantages and caveats of existing animal models. We restrict our focus to LSDs which include neurological disease, and specifically review the mucopolysaccharidoses (MPSs), glycoproteinoses, sphingolipidoses, lysosomal transport disorders, multiple enzyme deficiency, glycogen storage diseases, and neuronal ceroid lipofuscinosis (Table 1).
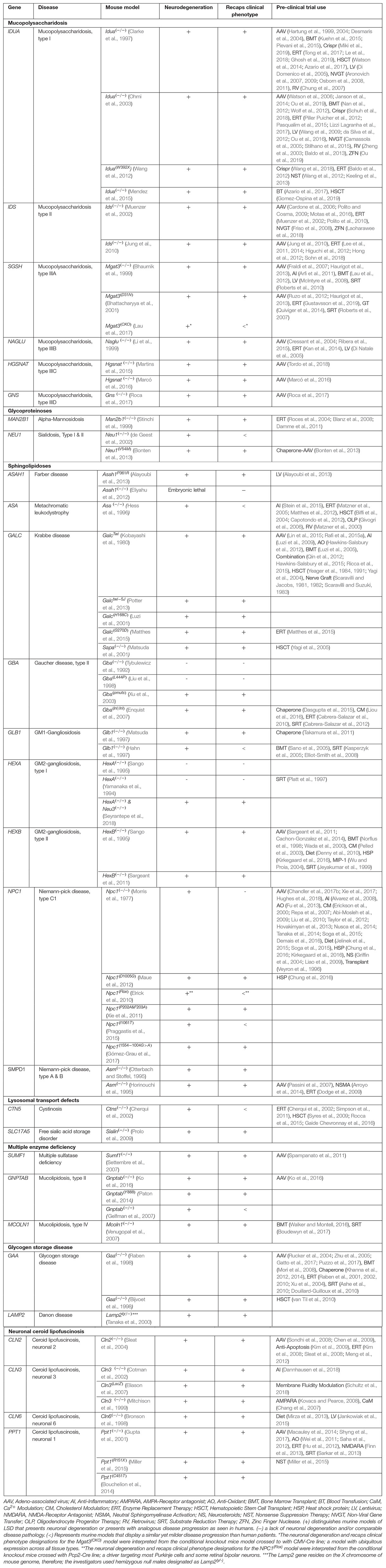
Table 1. List of LSDs having neurodegeneration and currently available pre-clinical mouse models.
Mucopolysaccharidoses
The mucopolysaccharidoses (MPS) are a family of lysosomal storage diseases wherein patients have an inability to properly metabolize glycosaminoglycan’s (GAGs) resulting in toxic accumulation of undigested dermatan sulfate (DS), heparan sulfate (HS) and/or keratin sulfate (KS) in the lysosome. GAGs are complex polymers comprised of alternating sulfated or amino disaccharides attached to protein cores, and are distributed in a wide variety of tissues, including bones, cartilage and the nervous system. There are eleven enzymes involved in the degradation of GAGs and mutations in any of these enzymes can elicit one of the seven characterized MPS. MPS are classified into 7 subtypes, with varied clinical phenotypes. The incidence for all types of MPS is estimated at 1 in 20,000 live births. The degree of CNS dysfunction, if any, varies widely among the 7 subtypes, but it seems to correlate with the degree of storage of HS, a major component of the extracellular matrix of the CNS. For example, MPS III A-D (Sanfilippo A-D) manifest as primarily CNS disorders, and often present with aggressive behavior and subsequent neurologic decline. On the other end of the spectrum, MPS VI (Maroteaux-Lamy), MPS IV A-B (Morquio) and MPS IX (Natowicz syndrome) present primarily as soft tissue or skeletal disease without neurological involvement. Other MPS can have variable degrees of neurologic involvement, in addition to soft tissue and skeletal disease, including MPS I (Hurler, Hurler-Scheie, Sheie), MPS II (Hunter) and MPS VII (Sly) (Coutinho et al., 2012).
1. MPS, Type I (OMIM [Online Mendelian Inheritance in Man] #252800) is caused by mutations in IDUA, which encodes a glycosidase involved in degrading HS and DS. MPS I is a multisystem disorder that ranges over a continuum of severity from severe (Hurler; MPS I-H) to attenuated (Scheie; MPS I-S) symptoms. Intellectual disability and developmental delay is common in MPS I-H, and neurological involvement and learning disabilities can be present in MPS I-S (Coutinho et al., 2012). Standard of care treatment includes HSCT, which can increase survival and peripheral symptoms, and may slow the development of mild, but not of severe, cognitive impairment. ERT with laronidase (Aldurazyme) is approved for non-CNS symptoms of MPS I (Al-Sannaa et al., 2015). Two MPS I-H mouse models were generated using targeted knockout cassettes in Idua, as well as a knock-in nonsense mutations (Idua-W392X) (Clarke et al., 1997; Ohmi et al., 2003; Wang et al., 2012). Knockout MPS I mice (Idua -/-) exhibit cognitive and motor defects with storage of GAGs in Purkinje cells (Baldo et al., 2012). Systemic delivery of apoE-fused IDUA protein produced from erythroid/megakaryocytic cells via LV-mediated HSCGT successfully corrected metabolic and behavioral deficits in MPS I mice (El-Amouri et al., 2014). Furthermore, efforts to directly edit the genome and correct mutations are currently underway. Several groups have undertaken attempts using genome editing tools such as the CRISPR/Cas9 and ZFN systems to directly modify the endogenous Idua locus or introduce an exogenous Idua gene (Schuh et al., 2018; Wang et al., 2018; Gomez-Ospina et al., 2019). These techniques have shown preliminarily success at promoting functional IDUA and reducing GAG storage. A ZFN mediated approach of delivering Idua to hepatocytes under control of the albumin promoter was particularly effective in distributing functional IDUA throughout secondary tissues via cross-correction, consequently reducing storage pathology (Ou et al., 2019). The therapy is currently in phase I/II of clinical trials.
2. MPS, Type II (OMIM #309900) is caused by mutations in the X-linked gene IDS (iduronate 2-sulfatase), which encodes the enzyme that catabolizes DS and HS. Infants with MPS II experience neurologic symptoms in the first decade of life and often experience developmental regression after 5 years of age (Scarpa, 2018). Weekly ERT infusions of idursulfase (Elaprase) can treat somatic manifestations and improves survival but does not treat the neurological disease (Burrow and Leslie, 2008). An Ids(–/–) mouse model of MPS II accumulate GAGs diffusely and shows neuronal necrosis in the brainstem and spinal cord by 60 weeks of age (Muenzer et al., 2002; Jung et al., 2010). AAV2/8-mediated GT restored DS activity in plasma and tissue of this null mice and cleared the accumulated GAGs in all the tissues (Motas et al., 2016). Akin to MPS I, genome editing via ZFN has been implemented as therapeutic option for MPS II, which elicited a vast improvement in neurocognition following reduction of accumulated GAGs by active re-introduction of Ids to hepatocytes and passive cross-correction through all tissues (Laoharawee et al., 2018). The pre-clinical success of the ZFN mediated genome editing has led to phase I/II clinical trials assessing the drugs efficacy in humans and optimal dosage as administered by intravenous injection.
3. MPS, Type III is characterized by progressive CNS degeneration and manifests clinically as developmental regression, severe intellectual disability and psychiatric manifestations. There are four subtypes of MPS III, distinguished as types ‘A-D,’ which are caused by mutations in one of four genes, all required for the proper degradation of HS (Wagner and Northrup, 2019). Unfortunately, there is no effective therapy available for any form of MPS III, aside from clinical management of neurological symptoms. Therefore, the role of animal models for the various forms of MPS III are especially important in the design of emerging and future therapies (Fedele, 2015).
a. MPS, Type III A (OMIM # 252900) is caused by a mutation in SGSH (N-sulfoglucosamine sulfohydrolase), resulting in intellectual disability, seizures and hyperactivity (Wagner and Northrup, 2019). The spontaneous mutant Mgat3(–/–) represents an authentic animal model of the disease manifesting hyperactivity and shortened lifespan akin to the human phenotype (Bhaumik et al., 1999). This animal has been used for GT and SRT studies which have successfully extended mutant lifespan and reduced associated symptomology (McIntyre et al., 2008; Roberts et al., 2010; Haurigot et al., 2013).
b. MPS, Type III B (OMIM # 252920) is caused by a mutation in NAGLU (N-alpha-acetylglucosaminidase) and presents with progressive neurological deterioration and seizures (Wagner and Northrup, 2019). The KO mouse model Naglu(–/–) had a comparatively mild phenotype (Li et al., 1999). In this knockout mouse, cellular inclusions in neurons were observed but the resulting behavioral changes were less prominent, although hypoactive behavior is apparent. This model has been used to test GT and ERT, which improved the behavior and neuropathology of Naglu(–/–) mice (Cressant et al., 2004; Kan et al., 2014; Ribera et al., 2015).
c. MPS, Type III C (OMIM #252930) is caused by a mutation in HGSNAT (Heparan acetyl-CoA:α-glucosaminide N-acetyltransferase), causing mental retardation and hyperactivity. Prominent atrophy occurs in the parieto-occipital region with a significant thinning of the corpus callosum white matter density (Wagner and Northrup, 2019). Two different Hgsnat(–/–) mice were generated. Both of which recapitulate human disease pathology including altered locomotor capabilities, hyperactivity, decline in cognitive memory ability, and shortened lifespan (Martins et al., 2015; Marcó et al., 2016). An AAV2 variant, AAV-TT, was used in a GT study in the Hgsnat(–/–) mouse model, and was able to correct the neurological phenotype (Tordo et al., 2018).
d. MPS, Type III D (OMIM #252940) is caused by mutations in GNS (N-acetylglucosamine-6-sulfatase) resulting in hyperactivity and mental retardation. White matter lesions are particularly present in the periventricular subcortical white matter regions (Wagner and Northrup, 2019). A Gns(–/–) mouse was generated exhibiting similar symptomology to the human disease, such as widespread neuroinflammation, reduced locomotion, and a decrease in lifespan. AAV-mediated GT of GNS to the cerebrospinal fluid in this mouse model ameliorated disease pathology, resolving lysosomal storage, neuroinflammation and behavioral phenotypes (Roca et al., 2017).
Glycoproteinoses
The glycoproteinoses are a group of LSDs caused by defects in the catabolism of glycoproteins containing N-linked or O-linked oligosaccharides. The degradation of glycan moieties occurs in a stepwise fashion thus the failure of one enzyme causes a complete blockade of the cycle. Therefore, most patients with glycoproteinoses present with very similar clinical findings (Michalski and Klein, 1999).
1. α-Mannosidosis (OMIM #248500) is caused by mutations in MAN2B1 (alpha-D-mannosidase), an enzyme involved in glycoprotein metabolism that results in accumulation of undigested oligosaccharides (Beck et al., 2013). A clinical spectrum of disease exists, spanning from mild (type 1), moderate (type 2) and severe (type 3). Severe patients have primary CNS disease, primarily involving cerebellar dysfunction and ataxia, as well as severe intellectual disability and developmental regression (Michalski and Klein, 1999). ERT with recombinant human α-mannosidase (Velmanase alfa) is regarded as the standard-of-care treatment for α-mannosidosis, which has been shown to cause improvements in both biochemical and functional endpoints of disease progression (Borgwardt et al., 2018; Lund et al., 2018). Man2b1(–/–) mice were generated, and resemble a mild form of the disease (Stinchi et al., 1999). Glycoproteins accumulate in Purkinje cells of the cerebellum as well as cortical neurons and pyramidal neurons of the hippocampus. The cerebellar pathology of Man2b1(–/–) mice had a partial rescue in a pre-clinical trial using recombinant MAN2B1 ERT (Blanz et al., 2008; Damme et al., 2011).
2. Sialidosis, Type II (OMIM #256550) is caused by a mutation of NEU1 (Neuraminidase-1), leading to the lysosomal accumulation of sialylated glycopeptides and oligosaccharides, manifesting in gait disturbances, corneal clouding and psychomotor retardation. Patients present with decreased cerebellum volume, as well as cortical and occipital lobe atrophy (Gultekin et al., 2018). There is no specific treatment for Sialidosis, Type II. Neu1(–/–) and Neu1(V54M) models of the disease were established. The phenotypes of Neu1(–/–) mice are not similar to those found in the human disease, because there was a lack of early degeneration in cerebellar Purkinje neurons (de Geest et al., 2002). The Neu1(V54M) models a non-neuronopathic form of the disease. Therefore, there is currently a lack of neuronopathic representative models of Sialidosis, Type II (Bonten et al., 2013).
Sphingolipidoses
The sphingolipidoses are characterized by the intracellular accumulation of sphingolipids. Sphingolipids are lipids that contain an aliphatic amino alcohol head group, or a structurally similar element. There are three categories of sphingolipids: ceramides, phosphosphingolipids and glycosphingolipids. These biologically active lipids form microdomains in the plasma membrane and facilitate signal transduction, cell recognition and physical protection. Accumulation of specific sphingolipids leads to the unique clinical manifestation of each sphingolipidosis. Despite their various phenotypes, all sphingolipidoses present at least some form of neuronopathic pathophysiology (Platt et al., 2018).
1. Farber lipogranulomatosis (OMIM #228000) is caused by mutations in ASAH1 (Acid ceramidase), leads to the accumulation of lysosomal ceramide, causing neonatal joint deformities and a characteristic hoarse voice/cry, with life expectancy of approximately 2 years. Neurologic symptoms are difficult to fully characterize due to the severity of the phenotype which includes joint immobility and early death. Older patients often develop motor defects and intellectual disabilities. A more mild spectrum of disease caused by ASAH1 mutations is categorized as spinal muscular atrophy with progressive myoclonic epilepsy, which includes progressive lower motor neuron disease in young children, accompanied by myoclonic and atonic seizures. The tissues of afflicted patients contain granulomatous and lipid-laden macrophages. The liver, spleen, lungs and heart are particularly affected with progressive CNS degeneration and impairments in psychomotor development (Bongarzone et al., 2012). There is no specific treatment for Farber disease. While a complete ‘knockout’ of ASAH1 (Asah1-/-) mice resulted in early embryonic lethality due to oocyte apoptosis, a ‘knock-in’ allele in Asah1, harboring a classical Farber missense mutation (P361R), accurately models most of the characteristics of human disease. This may also indicate a specific role for ASAH1 in murine development. Interestingly, intravenous lentiviral gene transfer expressing human Acid ceramidase in Asah1P361R/P361R reduced the symptoms, though the mice still succumbed to disease. Conditional Asah1 floxed mouse have also been generated, which will be helpful in testing cell specificity and efficacy of various therapies (Eliyahu et al., 2012; Alayoubi et al., 2013).
2. Metachromatic leukodystrophy (MLD, OMIM #250100) is due to mutations in ARSA (Arylsulfatase A), necessary for the metabolism of sulfatide, causing hypotonia, mental deterioration and cognitive regression. The accumulation of cerebroside sulfate causes demyelination of the frontal and parietal periventricular and central zones (Eichler et al., 2009). Currently, no effective treatment is available to reverse the deterioration and loss of function that MLD causes. An Arsa(–/–) mouse was generated which displays a milder neuropathological phenotype than human cases. The model has been useful in showing that attempts to rescue the mild phenotype via various therapies including ERT, HSCT and GT were successful in ameliorating the pathology (Hess et al., 1996). The lack of a complete recapitulation of the clinical and pathological MLD phenotype led to the generation of the Arsa(–/–) mice with the addition of neural cells overexpressing the sulfatide synthesizing enzymes, including UDP-galactose: ceramide galactosyltransferase (CGT) and cerebroside sulfotransferase (CST). These CGT/Arsa(–/–) and CST/Arsa(–/–) mice had increased sulfatide storage in myelin-forming cells, resulting in axonal degeneration leading to the development of neurological symptoms similar to MLD (Patil and Maegawa, 2013).
3. Krabbe disease (OMIM #245200) is caused by a deficiency of GALC (Galactosylceramidase), which leads to the accumulation of psychosine (galactosylsphingosine), a metabolite of galactosylceramide (Li et al., 2019). Clinical symptoms often present in the 1st year of life including irritability, spasticity and developmental delay. Underlying pathology includes widespread demyelination and neurodegeneration (Li et al., 2019). HSCT appears to be of some benefit in cases of later onset or in infantile patients who have been diagnosed before symptoms begin (Wright et al., 2017). Several Galc mutant mice have been reported, including two spontaneous mutants twitcher (Duchen et al., 1980) and twi-5J (Potter et al., 2013), humanized GALC transgenic (Gasperi et al., 2004), GALC-Gly270Asp (Matthes et al., 2015), Galc-His168Cys knock-in mice (Luzi et al., 2001) and Saposin A knockout mice (Matsuda et al., 2001). The twitcher mouse (GalcW339X/W339X) is an authentic murine model of Krabbe disease, presenting a near identical neurological phenotype to the human disease (Kobayashi et al., 1980). Due to the highly authentic nature of the twitcher model of KD, an incredible number of therapeutic strategies have been tested on twitcher mice. These include HSCT (Yeager et al., 1984), neural and mesenchymal stem cell transplantation (Taylor and Snyder, 1997; Strazza et al., 2009; Neri et al., 2011; Ripoll et al., 2011), SRT (LeVine et al., 2000), anti-oxidant therapy (Pannuzzo et al., 2010), PCT (Berardi et al., 2014), ERT (Lee et al., 2005) and GT alone (Shen et al., 2001; Lin et al., 2005; Lee et al., 2007; Rafi et al., 2012, 2015a; Pan et al., 2019). Although viral gene therapy should theoretically be curative, the pre-clinical efficacy in vivo has had major limitations. Viral-mediated GT trials in the twitcher tends to only modestly improve survival, typically by 1.5-2 fold. Therefore, multiple approaches have been made to improve delivery and transducing ability. These include diffuse and multiple injection points by employing multiple intracerebral points of injection or a combination of intracerebral, intrathecal and intravenous point. Interestingly a recent paper had far better success in the twitcher mouse by using much higher doses of AAV9 (Marshall et al., 2018). Additionally, due to the partial effect among different modalities of therapies, a number of groups have approached the concept of combining different forms of therapy. The effect of combining different forms of therapies has had profound improvements of survival of both the twitcher and canine models of KD. In particular, a robust synergistic effect of HSCT and viral-directed GT has been noted and replicated by multiple labs (Reddy et al., 2011; Rafi et al., 2015a; Ricca et al., 2015). Furthermore, triple combined therapy of HSCT, GT and SRT with L-cycloserine resulted in an unprecedented increase in lifespan with improved motor function, persistent GALC expression, nearly normal psychosine levels, and decreased neuroinflammation in the twitcher (Hawkins-Salsbury et al., 2015), suggesting that simultaneous-treatment of multiple pathogenic aspects of KD may be necessary for synergistic increases in therapeutic efficacy of KD patients. The twi-5J (Galc E130K/E130K) mice manifest a more severe phenotype with shorter lifespan than the twitcher (Potter et al., 2013), indicating the possible existence of a toxic gain-of function mutation from misfolded GALC protein.
4. Gaucher Disease (OMIM #230900) is caused by mutations of GBA (Acid β-glucosidase), which leads to the accumulation of glucosylceramide and glucosylsphingosine. Gaucher disease is characterized clinically as types 1-3, with types 2 and 3 having primary CNS disease. Infantile Gaucher disease (type 2) often presents with signs of bulbar and pyramidal neuronal atrophy, as well as cognitive impairment and progressive neurological deterioration and is usually fatal by age 2 (Pastores and Hughes, 2018). Treatment for types 1 and 3 include SRT, and ERT including miglustat (Zavesca) and eligustat tartrate (Cerdelga). PCT with ambroxol, a drug that breaks up phlegm of respiratory diseases, has been evaluated for pre-clinical testing for Gaucher disease (Bendikov-Bar et al., 2013). Unfortunately, there is no effective treatment for the neurologic damage of GD types 2 and 3. HSCT can reverse the non-neurological effects of the disease, but the procedure carries a high risk and is rarely performed in individuals with Gaucher disease (Riboldi and Fonzo, 2019). Interestingly, patients with type 1 are at increased risk for Parkinson’s disease and Lewy Body Dementia. It has been suggested that accumulated glucosylceramide may directly influence amyloid formation of α-synuclein by stabilizing soluble oligomeric intermediates in the lysosome of dopaminergic neurons of GBA mutant, that is one of hallmarks of Parkinson’s disease (Mazzulli et al., 2011). Numerous murine models have been created, including Gba(–/–),Gba(L444),Gba(pmuts), and Gba(lnl/lnl). The full KO and L444P models both die within 24–48 h of birth due to a compromised endothelial layer (Tybulewicz et al., 1992; Liu et al., 1998). The latter two models were able to circumvent the endothelial lethality barrier by either using cre-driven expression to negate endothelial expression or by using point mutations causing a less severe phenotype (Xu et al., 2003; Enquist et al., 2007). The lnl model has been useful for pre-clinical trials with research using PCT, ERT and SRT related therapies being tested on the model. Some of these studies have highlighted limitations also seen in clinical ERT trials, namely the poor distribution of recombinant enzyme penetrating the CNS. Conditional Gba floxed mice are also available that have shown cell-specific toxicity in which neuroinflammation is triggered by molecules released from dying neurons, astrocytes and oligodendrocytes (Farfel-Becker et al., 2011; Platt et al., 2018).
5. Type 1 GM1-Gangliosidosis (OMIM #230500) is caused by mutations in GLB1 (beta-galactosidase-1) leading to the accumulation of GM1 gangliosides and presenting with prompt psychomotor dysfunction and general CNS degeneration within the first 6 months of life. Severe cerebral atrophy leads to death, often within the first 2 years of life (Platt et al., 2018). While there is no effective medical treatment, anti-convulsants may control seizures secondary to GM1 Gangliosidosis. Thus far, two Glb1(–/–) mutants have been generated that express disease symptomology consistent with the human disease such as paralysis and premature death (Hahn et al., 1997; Matsuda et al., 1997). These models have been used to study PCT, SRT and HSCT as potential therapeutics which have all shown promise in reversing the neurological phenotype (Kasperzyk et al., 2005; Sano et al., 2005; Elliot-Smith et al., 2008; Takamura et al., 2011).
6. GM2-gangliosidosis, Type I (OMIM #272800; also called as Tay-Sachs Disease) is caused by mutations in HEXA (β-hexosaminidase A), which normally degrades GM2 gangliosides, and typically manifests within 6 months of life. Tay-Sachs is characterized by progressive hypotonia, weakness, neurodegeneration and death by 4 years of age. Neuropathology includes less pronounced fissures and enlarged sulci with a great loss of neuronal density in the cerebral cortex along with demyelination of cerebral white matter (Kaback and Desnick, 2011). There is no effective treatment for Tay-Sachs beyond palliative care. Two HexA(–/–) murine models have been produced, though none of them seem to recapitulate human disease. Instead, the mouse models are asymptomatic due to a rescue pathway involving Sialidase, not present in humans (Yamanaka et al., 1994; Sango et al., 1995). Recently, a dual KO model HexA(–/–) and Neu3(–/–) has since been generated that nullifies the bypass pathway via Sialidase (Seyrantepe et al., 2018) and more closely resembles the human disease.
7. GM2-gangliosidosis, Type II (OMIM #268800; also called as Sandhoff disease) is due to mutations in HEXB (β-hexosaminidase B), and results in deficiencies of both HexA and HexB enzymatic activities. Sandhoff disease is therefore often indistinguishable from the Tay-Sachs disease phenotype (Platt et al., 2018). Although there is no available specific treatment, supportive treatment such as proper nutrition and hydration, as well as use of anti-convulsants for patients with Sandhoff disease. HexB(–/–) mice were developed that recapitulates the pathology seen in human patients. Various approaches including GT and HSCT in the pre-clinical trials in this model showed slow disease progression and prolong lifespan (Sargeant et al., 2011).
8. Niemann-Pick Disease Type A (NPD-A) & B (OMIM #257200) are caused by mutations in ASM (Acid sphingomyelinase), which cleaves the phosphorylcholine group from sphingomyelin. NPD-A is categorized as the neuronopathic form of disease, often manifesting in early childhood, while NPD-B does not involve CNS manifestations. Neurologic symptoms of NPD-A include progressive hypotonia, psychomotor developmental regression and relentless neurologic deterioration. There is significant atrophy in the cerebellar and cortical neurons with presentation of foam cells (Vanier, 2013). Two Asm(–/–) mouse models have been generated that reproduce human disease symptomology. They have been used to study GT and ERT effects on Niemann-Pick disease (Horinouchi et al., 1995; Otterbach and Stoffel, 1995). These studies have shown limitations in therapeutic techniques such as viral vector-mediated GT as intracerebral injections were insufficient to deliver vectors throughout the CNS, instead requiring intracerebroventricular injections to alleviate motor abnormities.
9. Niemann-Pick disease, Type C1 (OMIM # 257220) and Type C2 (OMIM #607625) are due to mutations in NPC1 and NPC2, respectively. These proteins are involved with the trafficking of cholesterol and lipids within lysosomes and endosomes. Specifically NPC1 is a multipass lysosomal membrane protein that transports sphingosine out of lysosomes, whereas NPC2 is a soluble cholesterol-binding protein. NPC causes a secondary reduction of ASM, thus producing overlapping symptoms with NPD-A and B. Patients with NPC presents with mental degeneration, dementia and dystonia due to neuronal atrophy, particularly present in the Purkinje cells of the cerebellum (Vanier, 2013; Platt et al., 2018). Supportive care is essential and substantially improves the quality of life of NPC patients. Numerous murine models of NPC have been generated including Npc1(–/–), as well as multiple missense knockin models (D1005G, P202A, F203A, I1061T, and 1554-1004 G > A) and a conditional floxed model. The latter has been used for the cell-specific knockout of Npc1 in Purkinje cells (Morris et al., 1977; Elrick et al., 2010; Xie et al., 2011; Maue et al., 2012; Praggastis et al., 2015; Gómez-Grau et al., 2017). Depending on the point mutation induced, many of the models fully recapitulate the human disease phenotype including neurodegeneration, shortened lifespan and Purkinje cell atrophy. The I1061T mutant, which harbors the most common human NPC1 mutation, was generated to specifically study the effects of proteostatic modulation on disease progression. Various preclinical trials including GT, ERT, and transplant in these models have shown different but promising levels of success in prolonging mutant lifespan, reducing Purkinje cell atrophy and the storage phenotype (Veyron et al., 1996; Soga et al., 2015; Chandler et al., 2017b; Xie et al., 2017; Hughes et al., 2018).
Lysosomal Transport Defects
Whereas a majority of LSDs are the result of a defunct catabolic enzyme unable to metabolize its substrate, lysosomal transport defects are the results of mutations to intracellular membrane transporters. The absence of specific transmembrane transporters can lead to substrate being trapped in the lysosome, barred from cellular recycling. The two most common forms of lysosomal transport defects are Cystinosis and Sialic Acid storage disorders (Mancini et al., 2000).
1. Cystinosis (OMIM #219800) is due to mutations in CTNS (Cystinosin), which normally allows for the exit of cystine from the lysosome. Cystinosis has primarily nephropathic sequelae including renal tubular Fanconi syndrome (generalized proximal tubular dysfunction) and progressive glomerular failure. Advances in therapy including cystine-depleting agents and renal transplantation have allowed patients to survive longer and revealed some neurologic dysfunction and cerebellar calcification (Nesterova and Gahl, 2017). A Ctns(–/–) mouse model was developed that displays some of the secondary symptoms of Cystinosis, though it lacks the primary ailment in renal failure. Nonetheless, the murine model has still been useful in testing potential therapeutics such as HSCT and medicinal treatments such as Cysteamine, the first line of defense drug against cysteine accumulation (Cherqui et al., 2002; Syres et al., 2009; Simpson et al., 2011; Rocca et al., 2015; Gaide Chevronnay et al., 2016).
2. Sialic acid storage disorders (infantile free sialic acid storage disease [ISSD]; OMIM #269920, Salla disease; OMIM #604369) are a spectrum of disorders due to mutations in SLC17A5 (Sialin), which are autosomal recessive neurodegenerative disorders that present as a severe infantile form (ISSD) or as a slowly progressive adult form (Salla disease). General symptoms include developmental delay, low muscle tone, abnormal movements, and seizures. They are progressive, and symptoms get worse over time. Sialin is a sialic acid lysosomal membrane transport protein (Adams and Gahl, 2013). Since there is no cure, supportive treatments including anti-convulsants and physical therapy are recommended for the patients. Recently developed Slc17a5(-/-) mice successfully show early neurobehavioral milestones including hypomyelination and leukoencephalopathy, but exhibit progressive delay of later-stage sensory and motor milestones such as grasping, twitching and locomotion development (Stroobants et al., 2017).
Multiple Enzyme Deficiency
Multiple enzyme deficiency LSDs refer to those LSDs where more than one enzyme involved in the catabolism of lipids may be indirectly affected, thus eliciting a pathology associated with multiple LSDs due to a single mutation (Schlotawa et al., 2019).
1. Multiple sulfatase deficiency (MSD, OMIM #272200) is due to a mutation in SUMF1 (sulfatase modifying factor 1) resulting in a dysfunctional or complete absence of formylglycine-generating enzyme (Fge). Fge is responsible for post-translationally activating sulfatase enzymes that could affect the activity of all 13 sulfatase enzymes. While symptoms of MSD patients can be highly variable, many may display characteristics of the mucopolysaccharidoses, including developmental regression and neurologic deterioration (Schlotawa et al., 2019). There is no cure for MSD. Similarly to MSD patients, Sumf1(-/-) mice display early mortality, congenital growth retardation, skeletal abnormalities, and neurological defects including widespread neurodegeneration and neuroinflammation (Settembre et al., 2007). Combined brain and systemic AAV mediated GT in this mouse model resulted in significant improvement in both growth rate and lifespan (Surace et al., 2007; Spampanato et al., 2011).
2. Mucolipidosis (ML), Types II (I-Cell disease, OMIM #252500), III α/β (Pseudo-Hurler polydystrophy, OMIM #252600) and III gamma (OMIM #252605) are caused by mutations in GNPTAB (for ML II, ML III α/β) and GNPTG (ML III γ). GNPTAB and GNPTG give rise to the enzyme N-acetylglucosamine-1-phosphotransferase, a phosphotransferase which normally phosphorylates mannose residues for proper enzymatic trafficking of hydrolases to the lysosome. In addition to widespread musculoskeletal and cardiac phenotypes, patients with ML II have delays in motor milestones and expressive language (Leroy et al., 2019). There is no specific therapy to cure ML II and III. Speech and physical therapies can improve motor and speech delays. The Gnptab(-/-) mouse showed impaired growth, retinal degeneration, lesions in secretory epithelial cells of exocrine glands, and elevated levels of serum acid hydrolases. However, this mutant presented with a relatively normal lifespan and did not develop characteristic disease features, such as skeletal and facial abnormalities (Vogel et al., 2009). Another mouse model Nymphe (nym/nym), which was recovered from an N-ethyl-N-nitrosourea screen, and carries the patient mutation Y888X, recapitulates the major features of the human disease including motor dysfunction and psychomotor retardation with progressive neurodegeneration of Purkinje cells. Treatment with 2-hydroxypropyl-β-cyclodextrin delayed Purkinje cell loss in a NPC model, but had no effect on Purkinje cell loss in the Nymphe mouse. This finding suggested that the loss of Npc2 (Niemann-Pick, Type C2) expression in the Nymphe mouse brain is not a primary molecular mechanism causing Purkinje cell degeneration (Paton et al., 2014).
3. Mucolipidosis, Type IV (MLIV) is caused by mutations in MCOLN1 (Mucolipin 1), an endo-lysosomal pH sensitive channel that facilitates diffusion of monovalent and divalent ions (Dong et al., 2008) and facilitates lysosomal and autophagic regulation via its interaction with TFEB and mTOR signaling (Zhang et al., 2016). Clinical findings often include developmental delay, gross psychomotor impairments and failure to reach developmental milestones. There is no specific treatment to this disorder. Neurological impairments are attributed to developmental dysregulation of the corpus callosum in addition to atrophy and axonal spheroids in the cerebellum (Schiffmann et al., 2014). An authentic murine model of MLIV, Mcoln1(–/–), has been generated and closely resembles the pathology seen in human patients including hind limb paralysis and reduced lifespan (Venugopal et al., 2007). The model has been used to study the effects of HSCT and drug administration to improve the neuropathology associated with the disease with promising results (Walker and Montell, 2016).
Glycogen Storage Disease
Glycogen storage diseases are caused by impaired glycogen degradation or synthesis. Of the 13 known glycogen storage diseases, only one fits the criteria of classification as an LSD, namely Glycogen storage disease (GSD) II. GSD II is typically referred as Pompe disease, though the variant GSD-Type IIb (Danon disease) also exists and shares a similar phenotype. There is debate as to whether Danon disease should be classified as a glycogen storage disease as the mutation occurs in a lysosomal membrane protein and glycogen accumulation is not always present (Nishino et al., 2000).
1. Pompe disease (GSD II, OMIM #232300) is caused by mutations in GAA (Acid α-Glucosidase), which encodes an enzyme that degrades the α-1,4 and α-1,6 linkages required for the degradation of glycogen. The infantile-onset form of disease presents with hypotonia, feeding difficulties and is fatal due to cardiorespiratory failure. The effects on the CNS are still obscure due to the early age of death, however, research has shown a delay in reaching myelination milestones in patients (Chien et al., 2006). ERT of Myozyme (alglucosidase alfa) clearly prolongs overall survival for infantile-onset Pompe disease. Early diagnosis and early treatment leads to much better outcomes (Capelle et al., 2018). ERT with Lumizyme (alglucosidase alfa) is approved for patients with late-onset Pompe disease without evidence of cardiac hypertrophy, which improves survival and ambulation maintained over time. Lumizyme and Myozyme have the same generic ingredient (alglucosidase alfa; recombinant human GAA) and manufacturer (Genzyme Co.), but have the difference in the manufacturing process (Schoser et al., 2017). Two authentic murine Gaa(-/-) models have been established, and both of them have analogous pathology to the human disease with hallmarks such as muscle weakening and gait abnormalities (Bijvoet et al., 1998; Raben et al., 1998). The models have proven useful in pre-clinical trials with a variety of treatments including gene therapy, BMT, PCT, ERT and glycogen accumulation suppression (Raben et al., 2001, 2002, 2010; Rucker et al., 2004; Xu et al., 2004; Zhu et al., 2005; Mori et al., 2008; Khanna et al., 2014; Gatto et al., 2017; Puzzo et al., 2017).
2. Danon disease (GSD IIb, OMIM #300257) is an X-linked disease caused by mutations in LAMP2 and has overlapping symptoms with Pompe disease, including diffuse hypotonia and hypertrophic cardiomyopathy. However, unlike Pompe disease, most male patients with Danon disease have mild intellectual disability (Cenacchi et al., 2019). There is no approved treatment. A mouse model of Lamp2 knockouts exist and have inflammation, motor deficits and impaired learning (Rothaug et al., 2015). AAV-mediated GT increased survival rate of this mutant (Tanaka et al., 2000; Adler et al., 2019).
Neuronal Ceroid-Lipofuscinoses
The final category of inherited, neurodegenerative, LSDs include the neuronal ceroid-lipofuscinoses (NCLs). They are generally characterized clinically by neurologic deterioration, seizures and early death. While the NCL phenotypes resemble each other, there exists broad genetic heterogeneity and multiple mechanisms of pathogenesis. To date, thirteen genes have been identified to cause NCLs including PPT1, TPP1, CLN3, CLN5, CLN6, MFSD8, CLN8, CTSD, DNAJC5, CTSF, ATP13A2, GRN, and KCTD7 (Mole and Williams, 2013). While various therapeutic strategies are being explored for NCLs, there is only one clinically approved drug, cerliponase alfa, that effectively attenuates the progression of a specific form of NCLs (CLN2) (Kohlschütter et al., 2019).
1. CLN1 (OMIM #256730; infantile NCL, Santavuori-Haltia) is caused by mutations in PPT1, which encodes Palmitoyl-protein thioesterase-1, responsible for the catabolism of thioester-linked fatty acyl groups from cysteine residues. CLN1 presents with infantile developmental delays, myoclonic jerks and seizures. The symptoms are due to neuronal atrophy, particularly in the cortex and cerebellum (Mole et al., 2005). Three murine models have been established including a full null Ppt1(–/–) and two common missense mutations Ppt1(R151X) and Ppt1(C451T) (Gupta et al., 2001). All of them have been shown to recapitulate many of the pathological features observed in CLN1 patients, such as accumulation of autofluorescent granular osmiophilic deposits in neural and visceral tissues, rapidly progressing neurodegeneration in the brain, motor abnormalities, seizures and premature death. These models have been used in the development of therapies such as GT, dietary supplements, pharmacological and ERT. The combination of these therapies has additionally been shown to prolong the diseased animals’ lifespan. Ppt1(R151X) mutant mice treated with the read-through drug ataluren (PTC124) have been shown to increase CLN1 enzyme activity (Miller et al., 2015).
2. CLN2 (OMIM #204500, late-infantile NCL, Jansky–Bielschowsky) is caused by mutations in TPP1, which encodes tripeptidyl peptidase 1, involved in the cleavage of N-terminal tripeptides. Patients with CLN2 develop symptoms between the age of 2 to 4 years, including ataxia, seizures and mental deterioration. The formation of curvilinear body, that is an intermingled twisted microtubular substructure, is a hallmark of the disease and is often accompanied by cortical thinning (Mole and Williams, 2013). Intrathecal ERT delivery of cerliponase alfa (a recombinant human proenzyme of TPP1) is clinically approved for affected children (Kohlschütter et al., 2019). The Cln2(–/–) mouse recapitulated the clinical course of CLN2 with axonal degeneration, Purkinje cell atrophy and a reduced lifespan. The model has been used to study GT, ERT and anti-apoptosis manipulation all with varying degrees of success (Kohlschütter and Schulz, 2016).
3. CLN3 (OMIM #204200, juvenile NCL, Spielmeyer-Vogt) is caused by mutations in the CLN3 gene, which encodes a lysosomal transmembrane protein. Symptoms are progressive, and include visual impairments between 2 to 4 years of age and epilepsy with generalized tonic-clonic seizures after 9 years of age, as well as ataxia and motor dysfunction (Mole et al., 2005). Three full KO models of Cln3 have thus far been established all displaying hallmarks of CLN3 such as progressive neurodegeneration (Mitchison et al., 1999; Cotman et al., 2002; Eliason et al., 2007). To date there has been research using these models in alleviating CLN3 pathology with administration of various anti-inflammatory and neuroprotective compounds such as ibuprofen and lamotrigine to modulate intracellular inflammatory conditions (Mirza et al., 2019; Tarczyluk-Wells et al., 2019).
4. CLN6 (OMIM #601780, adult NCL) is caused by mutations in CLN6, a transmembrane protein of the endoplasmic reticulum. Patients with CLN6 generally begin to exhibit symptoms before the age of 40 (mean age 28). Clinical manifestations of disease first present as myoclonus epilepsy, followed by ataxia and dementia due to cerebral and cerebellar atrophy (Berkovic et al., 2019). A neuronal ceroid lipofuscinosis (Nclf) murine model was established by a one base pair insertion in the orthologous mouse Cln6 gene resulting in a frame shift defect that recapitulates many aspects of the disease such as Wallerian degeneration and paralysis (Bronson et al., 1998). This model has been used to research the effects of GT and diet intervention on disease progression (Kohlschütter et al., 2019).
Discussion
Limitations of Using Mouse Models to Study Human LSDs
Mouse models have been used for biomedical research since the beginning of the 20th century (Ericsson et al., 2013). These models have been used widely for a number of reasons including their relative low cost, high level of reproducibility, and short lifespans. Furthermore, the similarity of the mouse and human genome have facilitated the study of human disease in murine models. Early advances in modern mouse models began with the development of transgenic mice and knockout mice, which progressed to the advance of conditional mutagenesis (Orban et al., 1992), inducible mutagenesis (Doetschman and Azhar, 2012) and fluorescent reporters (Ikawa et al., 1995). Consequentially, the use of mouse models has drastically outpaced the use of rats and other mammals for modeling of human disease (Ericsson et al., 2013). Furthermore, modern techniques and services have rapidly increased the speed and decreased the cost of producing mouse models of interest.
It is crucial to have highly predictive animal models as pre-clinical test tools for the development of LSD therapies, where small patient sample sizes and variable ages of presentation make it difficult to standardize endpoint evaluation in clinical tests. While many mouse models of LSDs accurately reflect the human symptomatology of the disease they model, there are a number of examples in which generated models fall short. For example, Arsa(–/–) MLD mice show mild neurological symptoms that are only observed by the end of a normal lifespan, and fail to show sulfatide accumulation and demyelination (Patil and Maegawa, 2013). These differences may be explained by the fundamental differences of mice and human development, particularly in regards to neurodevelopment (Lin et al., 2014). Despite the homology of genes between different species, it is obvious that there are many variables that are not well replicated by simply introducing the same genetic mutation responsible for a human disease into a mouse model. Attempts to generate a more ‘authentic’ human model may consequentially produce a mouse model similar to the human disease, but nonetheless artificial. For example, Arsa(–/–) mice with neural cells overexpressing the sulfatide synthesizing enzyme CGT or CST have more similar pathology to human MLD patients (Patil and Maegawa, 2013). Furthermore, the artificial nature of ‘knockout’ systems sometimes trigger compensatory mechanisms not seen in human disease (El-Brolosy and Stainier, 2017).
Since naturally occurring spontaneous mutant mice are rare and, the vast majority of LSD mice are knockout or knock-in animals. While these models have provided us with significant advances in our understanding of pathogenesis and therapy, these artificial disease models have limitations. Firstly, complete knockouts have limited relevance to the human phenotype, as most patients expressing a low level of functioning or malfunctioning protein. These complete null genotypes occasionally make it hard to reintroduce target proteins, such as gene− or enzyme−replacement therapy, as the novel protein could trigger a neutralizing immune response. This may result in confusion in predicting the efficacy of such treatments in patients. Moreover, not all LSDs are associated with loss−of−function mutations, several are associated with toxic gain−of−function mutations which are often overlooked.
Particularly disconcerting is the high number of ‘successful’ pre-clinical trials that are not successful when replicated in human clinical trials. An analysis of the reported causes of drug candidate attrition during 2013–2015 revealed that past failures in phase II clinical trials were primarily due to insufficient efficacy (48%) and safety (25%). Similarly, insufficient efficacy was the primary reason for failure in phase III (55%), followed by safety (14%). When examining these failures by therapeutic areas, 13% and 17% of the failures were in metabolic and CNS diseases, respectively (Harrison, 2016). There are a number of potential attributable factors to this problem. One explanation for this gap in translation is the lack of robustness in the conduction of the pre-clinical trials. The efficiency of the new treatment under the different conditions indicates the treatment’s robustness (Carlson and Doyle, 2002; Xu et al., 2016). In many cases, pre-clinical trials are not being designed to influence human clinical trials and lack crucial features, such as statistical power, blinding, and appropriately sized control groups (Freise et al., 2011). In addition, design of pre-clinical animal experiments do not reflect all aspects of the challenge originated from the varied age and complex genetic makeup of patients (Lowenstein and Castro, 2009). For example, fixed genetic strains of inbred mice minimize the complexity of some disease phenotypes. Genetic backgrounds of mice have been shown to directly influence disease pathology and survival in models of LSDs (Tominaga et al., 2004), and can therefore present a confounding variable if not properly taken into account. If it is shown that a particular treatment works not only in certain mouse strain but also in additional mouse genetic backgrounds, that will increase the robustness of the treatment. Furthermore, the small size of mice, and in particular mice brains, compared to humans, potentially allows for easier treatment of mice. Alternative approaches include more rigorous testing of pre-clinical trials and the use of large animal models (Techiryan et al., 2018). Additional technologies such as induced pluripotent stem cells and organoids are particularly appealing as using cells from humans patients can more accurately model certain aspects of disease.
Perspectives
The past three decades have given rise to significant progress in the treatment of LSDs, and thus different therapeutic options are now available and are being actively pursued. Unfortunately, many LSDs remain untreatable and most of the therapeutics offered have important limitations related to a number of issues including bioavailability, toxicity and limited efficacy. Nonetheless, the success of a number of innovative and promising studies is suggestive that many of these LSDs will have novel treatments in the coming years. The possibility of combining different approaches in order to maximize therapeutic efficacy, as well as the ability to personalize treatment options for each individual patient are exciting realities. The use of mouse models continues to be an invaluable tool in the advancement of therapeutic interventions for LSDs.
Author Contributions
JF, NW, and DS collected all information of LSD animal models and JF, NW, MF, and DS wrote the manuscript.
Funding
This work was supported by grants from the National Institutes of Health (R01-NS112327 and R56-NS106023 to DS), (R01-NS111715 to MF), and (F30-NS090835 to NW).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank L. Wrabetz and other members of the Hunter James Kelly Research Institute for constructive discussions over the years.
References
Abi-Mosleh, L., Infante, R. E., Radhakrishnan, A., Goldstein, J. L., and Brown, M. S. (2009). Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc. Natl. Acad. Sci. U.S.A. 106, 19316–19321. doi: 10.1073/pnas.0910916106
Adams, D., and Gahl, W. A. (2013). Free Sialic Acid Storage Disorders. Available online at: https://rarediseases.org/rare-diseases/lysosomal-free-sialic-acid-storage-disorders/.
Adler, E., Gault, E., Villarruel, E., Hashem, S., Nelson, B., and Manso, A. M. (2019). AAV9.LAMP-2B improves metabolic and physiologic function in murine and human in-vitro models of Danon disease. J. Am. Coll. Cardiol. 73:687. doi: 10.1016/S0735-1097(19)31295-1
Alayoubi, A. M., Wang, J. C., Au, B. C., Carpentier, S., Garcia, V., Dworski, S., et al. (2013). Systemic ceramide accumulation leads to severe and varied pathological consequences. EMBO Mol. Med. 5, 827–842. doi: 10.1002/emmm.201202301
Al-Sannaa, N. A., Bay, L., Barbouth, D. S., Benhayoun, Y., Goizet, C., Guelbert, N., et al. (2015). Early treatment with laronidase improves clinical outcomes in patients with attenuated MPS I: a retrospective case series analysis of nine sibships. Orphanet. J. Rare Dis. 10, 131. doi: 10.1186/s13023-015-0344-4
Alvarez, A. R., Klein, A., Castro, J., Cancino, G. I., Amigo, J., Mosqueira, M., et al. (2008). Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB J. 22, 3617–3627. doi: 10.1096/fj.07-102715
Anglade, P., Vyas, S., Javoy-Agid, F., Herrero, M., Michel, P., Marquez, J., et al. (1997). Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 12, 25–32.
Arfi, A., Richard, M., Gandolphe, C., Bonnefont-Rousselot, D., Thérond, P., and Scherman, D. (2011). Neuroinflammatory and oxidative stress phenomena in MPS IIIA mouse model: the positive effect of long-term aspirin treatment. Mol. Genet. Metab. 103, 18–25. doi: 10.1016/j.ymgme.2011.01.015
Aronovich, E. L., Bell, J. B., Belur, L. R., Gunther, R., Koniar, B., Erickson, D. C. C., et al. (2007). Prolonged expression of a lysosomal enzyme in mouse liver after Sleeping Beauty transposon-mediated gene delivery: implications for non-viral gene therapy of mucopolysaccharidoses. J. Gene Med. 9, 403–415. doi: 10.1002/jgm.1028
Aronovich, E. L., Bell, J. B., Khan, S. A., Belur, L. R., Gunther, R., Koniar, B., et al. (2009). Systemic correction of storage disease in MPS I NOD/SCID mice using the sleeping beauty transposon system. Mol. Ther. 17, 1136–1144. doi: 10.1038/mt.2009.87
Arroyo, A. I., Camoletto, P. G., Morando, L., Sassoe-Pognetto, M., Giustetto, M., Van Veldhoven, P. P., et al. (2014). Pharmacological reversion of sphingomyelin-induced dendritic spine anomalies in a Niemann Pick disease type A mouse model. EMBO Mol. Med. 6, 398–413. doi: 10.1002/emmm.201302649
Ashe, K. M., Taylor, K. M., Chu, Q., Meyers, E., Ellis, A., Jingozyan, V., et al. (2010). Inhibition of glycogen biosynthesis via mTORC1 suppression as an adjunct therapy for Pompe disease. Mol. Genet. Metab. 100, 309–315. doi: 10.1016/j.ymgme.2010.05.001
Augustine, E. F., Adams, H. R., and Mink, J. W. (2013). Clinical trials in rare disease: challenges and opportunities. J. Child. Neurol. 28, 1142–1150. doi: 10.1177/0883073813495959
Azario, I., Pievani, A., Del Priore, F., Antolini, L., Santi, L., Corsi, A., et al. (2017). Neonatal umbilical cord blood transplantation halts skeletal disease progression in the murine model of MPS-I. Sci. Rep. 7:9473. doi: 10.1038/s41598-017-09958-9
Bailey, R. M., Armao, D., Kalburgi, S. N., and Gray, S. J. (2018). Development of intrathecal AAV9 gene therapy for giant axonal neuropathy. Mol. Ther. Methods Clin. Dev. 9, 160–171. doi: 10.1016/j.omtm.2018.02.005
Baldo, G., Mayer, F. Q., Martinelli, B., Meyer, F. S., Burin, M., Meurer, L., et al. (2012). Intraperitoneal implant of recombinant encapsulated cells overexpressing alpha-L-iduronidase partially corrects visceral pathology in mucopolysaccharidosis type I mice. Cytotherapy 14, 860–867. doi: 10.3109/14653249.2012.672730
Baldo, G., Wozniak, D. F., Ohlemiller, K. K., Zhang, Y., Giugliani, R., and Ponder, K. P. (2013). Retroviral-vector-mediated gene therapy to mucopolysaccharidosis I mice improves sensorimotor impairments and other behavioral deficits. J. Inherit. Metab. Dis. 36, 499–512. doi: 10.1007/s10545-012-9530-x
Bartolomeo, R., Cinque, L., Leonibus, C. D., Forrester, A., Salzano, A. C., Monfregola, J., et al. (2014). mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J. Clin. Invest. 127, 3717–3729. doi: 10.1172/JCI94130
Barton, N. W., Brady, R. O., Dambrosia, J. M., Bisceglie, A. M. D., Doppelt, S. H., Hill, S. C., et al. (1991). Replacement therapy for inherited enzyme de?ciency: macrophage-targeted glucocerebrosidase for Gaucher’s disease. N. Engl. J. Med. 324, 1464–1470. doi: 10.1056/NEJM199105233242104
Beck, M., Olsen, K. J., Wraith, J. E., Zeman, J., Michalski, J.-C., Saftig, P., et al. (2013). Natural history of alpha mannosidosis a longitudinal study. Orphanet. J. Rare Dis. 8:88. doi: 10.1186/1750-1172-8-88
Bendikov-Bar, I., Maor, G., Filocamo, M., and Horowitz, M. (2013). Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 50, 141–145. doi: 10.1016/j.bcmd.2012.10.007
Berardi, A. S., Pannuzzo, G., Graziano, A., Costantino-Ceccarini, E., Piomboni, P., and Luddi, A. (2014). Pharmacological chaperones increase residual β-galactocerebrosidase activity in fibroblasts from Krabbe patients. Molecular genetics and metabolism 112, 294–301. doi: 10.1016/j.ymgme.2014.05.009
Berkovic, S. F., Oliver, K. L., Canafoglia, L., Krieger, P., Damiano, J. A., Hildebrand, M. S., et al. (2019). Kufs disease due to mutation of CLN6: clinical, pathological and molecular genetic features. Brain 142, 59–69. doi: 10.1093/brain/awy297
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Efficient CNS targeting in adult mice by intrathecal infusion of single-stranded AAV9-GFP for gene therapy of neurological disorders. Gene Ther. 24, 325–332. doi: 10.1038/gt.2017.18
Bhattacharyya, R., Gliddon, B., Beccari, T., Hopwood, J. J., and Stanley, P. (2001). A novel missense mutation in lysosomal sulfamidase is the basis of MPS III A in a spontaneous mouse mutant. Glycobiology 11, 99–103. doi: 10.1093/glycob/11.1.99
Bhaumik, M., Muller, V. J., Rozaklis, T., Johnson, L., Dobrenis, K., Bhattacharyya, R., et al. (1999). A mouse model for mucopolysaccharidosis type III A (Sanfilippo syndrome). Glycobiology 9, 1389–1396. doi: 10.1093/glycob/9.12.1389
Biffi, A., De Palma, M., Quattrini, A., Del Carro, U., Amadio, S., Visigalli, I., et al. (2004). Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Invest. 113, 1118–1129. doi: 10.1172/JCI200419205
Biffi, A., Montini, E., Lorioli, L., Cesani, M., Fumagalli, F., Plati, T., et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341:1233158. doi: 10.1126/science.1233158
Bijvoet, A. G., Van De Kamp, E. H., Kroos, M. A., Ding, J. H., Yang, B. Z., Visser, P., et al. (1998). Generalized glycogen storage and cardiomegaly in a knockout mouse model of Pompe disease. Hum. Mol. Genet. 7, 53–62. doi: 10.1093/hmg/7.1.53
Blanz, J., Stroobants, S., Lullmann-Rauch, R., Morelle, W., Ludemann, M., D’hooge, R., et al. (2008). Reversal of peripheral and central neural storage and ataxia after recombinant enzyme replacement therapy in alpha-mannosidosis mice. Hum. Mol. Genet. 17, 3437–3445. doi: 10.1093/hmg/ddn23
Bongarzone, E. R., Givogri, M. I., De Vivo, D. C., and Dimauro, S. (2012). “Chapter 43 – inborn metabolic defects of lysosomes, peroxisomes, carbohydrates, fatty acids and mitochondria,” in Basic Neurochemistry Principles of Molecular, Cellular, and Medical Neurobiology, 18th Edn. 755–782. doi: 10.1016/B978-0-12-374947-5.00043-2
Bonten, E. J., Yogalingam, G., Hu, H., Gomero, E., Van De Vlekkert, D., and D’azzo, A. (2013). Chaperone-mediated gene therapy with recombinant AAV-PPCA in a new mouse model of type I sialidosis. Biochim. Et Biophys. Acta 1832, 1784–1792. doi: 10.1016/j.bbadis.2013.06.002
Borgwardt, L., Guffon, N., Amraoui, Y., Dali, C. I., Meirleir, L. D., Gil-Campos, M., et al. (2018). Efficacy and safety of Velmanase alfa in the treatment of patients with alpha-mannosidosis: results from the core and extension phase analysis of a phase III multicentre, double-blind, randomised, placebo-controlled trial. J. Inherit. Metab. Dis. 41, 1215–1223. doi: 10.1007/s10545-018-0185-0
Bouchelion, A., Zhang, Z., Li, Y., Qian, H., and Mukherjee, A. B. (2014). Mice homozygous for c.451C>T mutation in Cln1 gene recapitulate INCL phenotype. Ann. Clin. Transl. Neurol. 1, 1006–1023. doi: 10.1002/acn3.144
Boudewyn, L. C., Sikora, J., Kuchar, L., Ledvinova, J., Grishchuk, Y., Wang, S. L., et al. (2017). N-butyldeoxynojirimycin delays motor deficits, cerebellar microgliosis, and Purkinje cell loss in a mouse model of mucolipidosis type IV. Neurobiol. Dis. 105, 257–270. doi: 10.1016/j.nbd.2017.06.003
Bronson, R. T., Donahue, L. R., Johnson, K. R., Tanner, A., Lane, P. W., and Faust, J. R. (1998). Neuronal ceroid lipofuscinosis (nclf), a new disorder of the mouse linked to chromosome 9. Am. J. Med. Genet. 77, 289–297. doi: 10.1002/(SICI)1096-8628(19980526)77:4<289::AID-AJMG8>3.0.CO;2-I
Brooks, D. A. (1999). Immune response to enzyme replacement therapy in lysosomal storage disorder patients and animal models. Mol. Genet. Metab. 68, 268–275. doi: 10.1006/mgme.1999.2894
Burrow, T. A., and Leslie, N. D. (2008). Review of the use of idursulfase in the treatment of mucopolysaccharidosis II. Biologics 2, 311–320. doi: 10.1517/14656566.9.2.311
Cabrera-Salazar, M. A., Bercury, S. D., Ziegler, R. J., Marshall, J., Hodges, B. L., Chuang, W. L., et al. (2010). Intracerebroventricular delivery of glucocerebrosidase reduces substrates and increases lifespan in a mouse model of neuronopathic Gaucher disease. Exp. Neurol. 225, 436–444. doi: 10.1016/j.expneurol.2010.07.023
Cabrera-Salazar, M. A., Deriso, M., Bercury, S. D., Li, L., Lydon, J. T., Weber, W., et al. (2012). Systemic delivery of a glucosylceramide synthase inhibitor reduces CNS substrates and increases lifespan in a mouse model of type 2 Gaucher disease. PLoS ONE 7:e43310. doi: 10.1371/journal.pone.0043310
Cachon-Gonzalez, M. B., Wang, S. Z., Ziegler, R., Cheng, S. H., and Cox, T. M. (2014). Reversibility of neuropathology in Tay-Sachs-related diseases. Hum. Mol. Genet. 23, 730–748. doi: 10.1093/hmg/ddt459
Camassola, M., Braga, L. M., Delgado-Cañedo, A., Dalberto, T. P., Matte, U., Burin, M., et al. (2005). Nonviral in vivo gene transfer in the mucopolysaccharidosis I murine model. J. Inherit. Metab. Dis. 28, 1035–1043. doi: 10.1007/s10545-005-0070-5
Capelle, C. I. V., Poelman, E., Frohn-Mulder, I. M., Koopman, L. P., Hout, J. M. P. V. D., Régal, L., et al. (2018). Cardiac outcome in classic infantile Pompe disease after 13?years of treatment with recombinant human acid alpha-glucosidase. Int. J. Cardiol. 269, 104–110. doi: 10.1016/j.ijcard.2018.07.091
Capotondo, A., Milazzo, R., Politi, L. S., Quattrini, A., Palini, A., Plati, T., et al. (2012). Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc. Natl. Acad. Sci. U.S.A. 109, 15018–15023. doi: 10.1073/pnas.1205858109
Cardone, M., Polito, V. A., Pepe, S., Mann, L., D’azzo, A., Auricchio, A., et al. (2006). Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery. Hum. Mol. Genet. 15, 1225–1236. doi: 10.1093/hmg/ddl038
Carlson, J. M., and Doyle, J. (2002). Complexity and robustness. Proc. Natl. Acad. Sci. U.S.A. 99, 2538–2545. doi: 10.1073/pnas.012582499
Cearley, C. N., and Wolfe, J. H. (2007). A single injection of an adeno-associated virus vector into nuclei with divergent connections results in widespread vector distribution in the brain and global correction of a neurogenetic disease. J. Neurosci. 27, 9928–9940. doi: 10.1523/JNEUROSCI.2185-07.2007
Cenacchi, G., Papa, V., Pegoraro, V., Marozzo, R., Fanin, M., and Angelini, C. (2019). Review: danon disease: review of natural history and recent advances. Neuropathol. Appl. Neurobiol. [Epub ahead of print]. doi: 10.1111/nan.12587
Chandler, R. J., Sands, M. S., and Venditti, C. P. (2017a). Recombinant adeno-associated viral integration and genotoxicity: insights from animal models. Hum. Gene Ther. 28, 314–322. doi: 10.1089/hum.2017.009
Chandler, R. J., Williams, I. M., Gibson, A. L., Davidson, C. D., Incao, A. A., Hubbard, B. T., et al. (2017b). Systemic AAV9 gene therapy improves the lifespan of mice with Niemann-Pick disease, type C1. Hum. Mol. Genet. 26, 52–64. doi: 10.1093/hmg/ddw367
Chang, J. W., Choi, H., Kim, H. J., Jo, D. G., Jeon, Y. J., Noh, J. Y., et al. (2007). Neuronal vulnerability of CLN3 deletion to calcium-induced cytotoxicity is mediated by calsenilin. Hum. Mol. Genet. 16, 317–326. doi: 10.1093/hmg/ddl466
Chen, Y. H., Chang, M., and Davidson, B. L. (2009). Molecular signatures of disease brain endothelia provide new sites for CNS-directed enzyme therapy. Nat. Med. 15, 1215–1218. doi: 10.1038/nm.2025
Cherqui, S., Sevin, C., Hamard, G., Kalatzis, V., Sich, M., Pequignot, M. O., et al. (2002). Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol. Cell Biol. 22, 7622–7632. doi: 10.1128/MCB.22.21.7622-7632.2002
Chien, Y. H., Lee, N. C., Peng, S. F., and Hwu, W. L. (2006). Brain development in infantile-onset Pompe disease treated by enzyme replacement therapy. Pediatr. Res. 60, 349–352. doi: 10.1203/01.pdr.0000233014.84318.4e
Christensen, C. L., and Choy, F. Y. M. (2017). A prospective treatment option for lysosomal storage diseases: CRISPR/Cas9 gene editing technology for mutation correction in induced pluripotent stem cells. Diseases 5:6. doi: 10.3390/diseases5010006
Chung, C., Elrick, M. J., Dell’orco, J. M., Qin, Z. S., Kalyana-Sundaram, S., Chinnaiyan, A. M., et al. (2016). Heat shock protein beta-1 modifies anterior to posterior purkinje cell vulnerability in a mouse model of niemann-pick type C disease. PLoS Genet. 12:e1006042. doi: 10.1371/journal.pgen.1006042
Chung, S., Ma, X., Liu, Y., Lee, D., Tittiger, M., and Ponder, K. P. (2007). Effect of neonatal administration of a retroviral vector expressing alpha-L-iduronidase upon lysosomal storage in brain and other organs in mucopolysaccharidosis I mice. Mol. Genet. Metabol. 90, 181–192. doi: 10.1016/j.ymgme.2006.08.001
Clarke, L. A., Russell, C. S., Pownall, S., Warrington, C. L., Borowski, A., Dimmick, J. E., et al. (1997). Murine mucopolysaccharidosis type I: targeted disruption of the murine alpha-L-iduronidase gene. Hum. Mol. Genet. 6, 503–511. doi: 10.1093/hmg/6.4.503
Cotman, S. L., Vrbanac, V., Lebel, L. A., Lee, R. L., Johnson, K. A., Donahue, L. R., et al. (2002). Cln3(Deltaex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum. Mol. Genet. 11, 2709–2721. doi: 10.1093/hmg/11.22.2709
Coutinho, M. F., Lacerda, L., and Alves, S. (2012). Glycosaminoglycan storage disorders: a review. Biochem. Res. Int. 2012:471325. doi: 10.1155/2012/471325
Coutinho, M. F., Santos, J. I., and Alves, S. (2016). Less is more: substrate reduction therapy for lysosomal storage disorders. Int. J. Mol. Sci. 17:1065. doi: 10.3390/ijms17071065
Cressant, A., Desmaris, N., Verot, L., Brejot, T., Froissart, R., Vanier, M. T., et al. (2004). Improved behavior and neuropathology in the mouse model of Sanfilippo type IIIB disease after adeno-associated virus-mediated gene transfer in the striatum. J. Neurosci. 24, 10229–10239. doi: 10.1523/JNEUROSCI.3558-04.2004
Cuervo, A. M., and Dice, J. F. (2000). When lysosomes get old?. Exp. Gerontol. 35, 119–131. doi: 10.1016/S0531-5565(00)00075-9
da Silva, F. H., Pereira, V. G., Yasumura, E. G., Tenório, L. Z., De Carvalho, L. P., Lisboa, B. C. G., et al. (2012). Treatment of adult MPSI mouse brains with IDUA-expressing mesenchymal stem cells decreases GAG deposition and improves exploratory behavior. Genet. Vaccines Ther. 10:2. doi: 10.1186/1479-0556-10-2
Damme, M., Stroobants, S., Walkley, S. U., Lullmann-Rauch, R., D’hooge, R., Fogh, J., et al. (2011). Cerebellar alterations and gait defects as therapeutic outcome measures for enzyme replacement therapy in alpha-mannosidosis. J. Neuropathol. Exp. Neurol. 70, 83–94. doi: 10.1097/NEN.0b013e31820428fa
Dannhausen, K., Mohle, C., and Langmann, T. (2018). Immunomodulation with minocycline rescues retinal degeneration in juvenile neuronal ceroid lipofuscinosis mice highly susceptible to light damage. Dis. Model Mech. 11:dmm033597. doi: 10.1242/dmm.033597
Dasgupta, N., Xu, Y. H., Li, R., Peng, Y., Pandey, M. K., Tinch, S. L., et al. (2015). Neuronopathic Gaucher disease: dysregulated mRNAs and miRNAs in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Hum. Mol. Genet. 24, 7031–7048. doi: 10.1093/hmg/ddv404
de Geest, N., Bonten, E., Mann, L., De Sousa-Hitzler, J., Hahn, C., and D’azzo, A. (2002). Systemic and neurologic abnormalities distinguish the lysosomal disorders sialidosis and galactosialidosis in mice. Hum. Mol. Genet. 11, 1455–1464. doi: 10.1093/hmg/11.12.1455
Demais, V., Barthelemy, A., Perraut, M., Ungerer, N., Keime, C., Reibel, S., et al. (2016). Reversal of pathologic lipid accumulation in NPC1-deficient neurons by drug-promoted release of LAMP1-coated lamellar inclusions. J. Neurosci. 36, 8012–8025. doi: 10.1523/JNEUROSCI.0900-16.2016
Denny, C. A., Heinecke, K. A., Kim, Y. P., Baek, R. C., Loh, K. S., Butters, T. D., et al. (2010). Restricted ketogenic diet enhances the therapeutic action of N-butyldeoxynojirimycin towards brain GM2 accumulation in adult Sandhoff disease mice. J. Neurochem. 113, 1525–1535. doi: 10.1111/j.1471-4159.2010.06733.x
Desmaris, N., Verot, L., Puech, J. P., Caillaud, C., Vanier, M. T., and Heard, J. M. (2004). Prevention of neuropathology in the mouse model of Hurler syndrome. Ann. Neurol. 56, 68–76. doi: 10.1002/ana.20150
Di Domenico, C., Villani, G. R. D., Di Napoli, D., Reyero, E. G. Y., Lombardo, A., Naldini, L., et al. (2005). Gene therapy for a mucopolysaccharidosis type I murine model with lentiviral-IDUA vector. Hum. Gene Ther. 16, 81–90. doi: 10.1089/hum.2005.16.81
Di Natale, P., Di Domenico, C., Gargiulo, N., Castaldo, S., Gonzalez, Y. R. E., Mithbaokar, P., et al. (2005). Treatment of the mouse model of mucopolysaccharidosis type IIIB with lentiviral-NAGLU vector. Biochem. J. 388, 639–646. doi: 10.1042/BJ20041702
Dodge, J. C., Clarke, J., Treleaven, C. M., Taksir, T. V., Griffiths, D. A., Yang, W., et al. (2009). Intracerebroventricular infusion of acid sphingomyelinase corrects CNS manifestations in a mouse model of niemann-Pick A disease. Exp. Neurol. 215, 349–357. doi: 10.1016/j.expneurol.2008.10.021
Doetschman, T., and Azhar, M. (2012). Cardiac-specific inducible and conditional gene targeting in mice. Cir. Res. 110, 1498–1512. doi: 10.1161/CIRCRESAHA.112.265066
Dong, X.-P., Cheng, X., Mills, E., Delling, M., Wang, F., Kurz, T., et al. (2008). The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455, 992–996. doi: 10.1038/nature07311
Donida, B., Jacques, C. E. D., Mescka, C. P., Rodrigues, D. G. B., Marchetti, D. P., Ribas, G., et al. (2017). Oxidative damage and redox in lysosomal storage disorders: biochemical markers. Clin. Chim. Acta 466, 46–53. doi: 10.1016/j.cca.2017.01.007
Douillard-Guilloux, G., Raben, N., Takikita, S., Ferry, A., Vignaud, A., Guillet-Deniau, I., et al. (2010). Restoration of muscle functionality by genetic suppression of glycogen synthesis in a murine model of Pompe disease. Hum. Mol. Genet. 19, 684–696. doi: 10.1093/hmg/ddp535
Duchen, L. W., Eicher, E. M., Jacobs, J. M., Scaravilli, F., and Teixeira, F. (1980). Hereditary leucodystrophy in the mouse: the new mutant twitcher. Brain 103, 695–710. doi: 10.1093/brain/103.3.695
Dunbar, C. E., Kohn, D. B., Schiffmann, R., Barton, N. W., Nolta, J. A., Esplin, J. A., et al. (1998). Retroviral transfer of the glucocerebrosidase gene into CD34+ cells from patients with gaucher disease: in vivo detection of transduced cells without myeloablation. Hum. Gene Ther. 9, 2629–2640. doi: 10.1089/10430349850019463
Eichler, F., Duncan, C., Musolino, P. L., Orchard, P. J., Oliveira, S. D., Thrasher, A. J., et al. (2017). Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N. Engl. J. Med. 377, 1630–1638. doi: 10.1056/NEJMoa1700554
Eichler, F., Grodd, W., Grant, E., Sessa, M., Biffi, A., Bley, A., et al. (2009). Metachromatic leukodystrophy: a scoring system for brain MR imaging observations. AJNR Am. J. Neuroradiol. 30, 1893–1897. doi: 10.3174/ajnr.A1739
El-Amouri, S. S., Dai, M., Han, J.-F., Brady, R. O., and Pan, D. (2014). Normalization and improvement of CNS deficits in mice with hurler syndrome after long-term peripheral delivery of BBB-targeted iduronidase. Mol. Ther. 22, 2028–2037. doi: 10.1038/mt.2014.152
El-Brolosy, M. A., and Stainier, D. Y. R. (2017). Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet. 13:e1006780. doi: 10.1371/journal.pgen.1006780
Eliason, S. L., Stein, C. S., Mao, Q., Tecedor, L., Ding, S. L., Gaines, D. M., et al. (2007). A knock-in reporter model of Batten disease. J. Neurosci. 27, 9826–9834. doi: 10.1523/JNEUROSCI.1710-07.2007
Eliyahu, E., Shtraizent, N., Shalgi, R., and Schuchman, E. H. (2012). Construction of conditional acid ceramidase knockout mice and in vivo effects on oocyte development and fertility. Cell Physiol. Biochem. 30, 735–748. doi: 10.1159/000341453
Elliot-Smith, E., Speak, A. O., Lloyd-Evans, E., Smith, D. A., Van Der Spoel, A. C., Jeyakumar, M., et al. (2008). Beneficial effects of substrate reduction therapy in a mouse model of GM1 gangliosidosis. Mol. Genet. Metab. 94, 204–211. doi: 10.1016/j.ymgme.2008.02.005
Ellison, S. M., Liao, A., Wood, S., Taylor, J., Youshani, A. S., Rowlston, S., et al. (2019). Pre-clinical safety and efficacy of lentiviral vector-mediated ex vivo stem cell gene therapy for the treatment of mucopolysaccharidosis IIIA. Mol. Ther. Methods Clin. Dev. 13, 399–413. doi: 10.1016/j.omtm.2019.04.001
Elrick, M. J., Pacheco, C. D., Yu, T., Dadgar, N., Shakkottai, V. G., Ware, C., et al. (2010). Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum. Mol. Genet. 19, 837–847. doi: 10.1093/hmg/ddp552
Enquist, I. B., Lo Bianco, C., Ooka, A., Nilsson, E., Månsson, J.-E., Ehinger, M., et al. (2007). Murine models of acute neuronopathic Gaucher disease. Proc. Natl. Acad. Sci. U.S.A. 104, 17483–17488. doi: 10.1073/pnas.0708086104
Erickson, R. P., Garver, W. S., Camargo, F., Hossain, G. S., and Heidenreich, R. A. (2000). Pharmacological and genetic modifications of somatic cholesterol do not substantially alter the course of CNS disease in Niemann-Pick C mice. J. Inherit. Metab. Dis. 23, 54–62. doi: 10.1023/A:1005650930330
Ericsson, A. C., Crim, M. J., and Franklin, C. L. (2013). A brief history of animal modeling. Mol. Med. 110, 201–205.
Fan, J.-Q. (2008). A counterintuitive approach to treat enzyme deficiencies: useof enzyme inhibitors for restoring mutant enzyme activity. Biol. Chem. 389, 1–11. doi: 10.1515/BC.2008.009
Farfel-Becker, T., Vitner, E. B., and Futerman, A. H. (2011). Animal models for Gaucher disease research. Dis. Model Mech. 4, 746–752. doi: 10.1242/dmm.008185
Fedele, A. O. (2015). Sanfilippo syndrome: causes, consequences, and treatments. Appl. Clin. Genet. 8, 269–281. doi: 10.2147/TACG.S57672
Ferraz, M. J., Marques, A. R. A., Appelman, M. D., Verhoek, M., Strijland, A., Mirzaian, M., et al. (2016). Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 590, 716–725. doi: 10.1002/1873-3468.12104
Finn, R., Kovács, A. D., and Pearce, D. A. (2013). Treatment of the Ppt1(-/-) mouse model of infantile neuronal ceroid lipofuscinosis with the N-methyl-D-aspartate (n.d.) receptor antagonist memantine. J. Child Neurol. 28, 1159–1168. doi: 10.1177/0883073813494480
Fraldi, A., Biffi, A., Lombardi, A., Visigalli, I., Pepe, S., Settembre, C., et al. (2007). SUMF1 enhances sulfatase activities in vivo in five sulfatase deficiencies. Biochem. J. 403, 305–312. doi: 10.1042/BJ20061783
Freise, K. J., Lin, T. L., Fan, T. M., Recta, V., and Clark, T. P. (2011). Evidence-based medicine: the design and interpretation of noninferiority clinical trials in veterinary medicine. J. Vet. Int. Med. 27, 1305–1317. doi: 10.1111/jvim.12211
Friso, A., Tomanin, R., Zanetti, A., Mennuni, C., Calvaruso, F., La Monica, N., et al. (2008). Gene therapy of Hunter syndrome: evaluation of the efficiency of muscle electro gene transfer for the production and release of recombinant iduronate-2-sulfatase (IDS). Biochim. Et Biophys. Acta 1782, 574–580. doi: 10.1016/j.bbadis.2008.07.001
Fu, H., Cataldi, M. P., Ware, T. A., Zaraspe, K., Meadows, A. S., Murrey, D. A., et al. (2016). Functional correction of neurological and somatic disorders at later stages of disease in MPS IIIA mice by systemic scAAV9-hSGSH gene delivery. Mol. Ther. Methods Clin. Dev. 3:16036. doi: 10.1038/mtm.2016.36
Fu, R., Wassif, C. A., Yanjanin, N. M., Watkins-Chow, D. E., Baxter, L. L., Incao, A., et al. (2013). Efficacy of N-acetylcysteine in phenotypic suppression of mouse models of Niemann-Pick disease, type C1. Hum. Mol. Genet. 22, 3508–3523. doi: 10.1093/hmg/ddt206
Gaide Chevronnay, H. P., Janssens, V., Van Der Smissen, P., Rocca, C. J., Liao, X. H., Refetoff, S., et al. (2016). Hematopoietic stem cells transplantation can normalize thyroid function in a cystinosis mouse model. Endocrinology 157, 1363–1371. doi: 10.1210/en.2015-1762
Gasperi, R. D., Friedrich, V. L., Perez, G. M., Senturk, E., Wen, P. H., Kelley, K., et al. (2004). Transgenic rescue of Krabbe disease in the twitcher mouse. Gene Ther. 11, 1188–1194. doi: 10.1038/sj.gt.3302282
Gatto, F., Rossi, B., Tarallo, A., Polishchuk, E., Polishchuk, R., Carrella, A., et al. (2017). AAV-mediated transcription factor EB (TFEB) gene delivery ameliorates muscle pathology and function in the murine model of Pompe Disease. Sci. Rep. 7:15089. doi: 10.1038/s41598-017-15352-2
Gelfman, C. M., Vogel, P., Issa, T. M., Turner, C. A., Lee, W.-S., Kornfeld, S., et al. (2007). Mice lacking alpha/beta subunits of GlcNAc-1-phosphotransferase exhibit growth retardation, retinal degeneration, and secretory cell lesions. Investig. Ophthalmol. Vis. Sci. 48, 5221–5228. doi: 10.1167/iovs.07-0452
Ghosh, A., Liao, A., O’leary, C., Mercer, J., Tylee, K., Goenka, A., et al. (2019). Strategies for the induction of immune tolerance to enzyme replacement therapy in mucopolysaccharidosis type I. Molecular therapy. Methods Clin. Dev. 13, 321–333. doi: 10.1016/j.omtm.2019.02.007
Givogri, M. I., Galbiati, F., Fasano, S., Amadio, S., Perani, L., Superchi, D., et al. (2006). Oligodendroglial progenitor cell therapy limits central neurological deficits in mice with metachromatic leukodystrophy. J. Neurosci. 26, 3109–3119. doi: 10.1523/JNEUROSCI.4366-05.2006
Gómez-Grau, M., Albaigès, J., Casas, J., Auladell, C., Dierssen, M., Vilageliu, L., et al. (2017). New murine niemann-pick type C models bearing a pseudoexon-generating mutation recapitulate the main neurobehavioural and molecular features of the disease. Sci. Rep. 7:41931. doi: 10.1038/srep41931
Gomez-Ospina, N., Scharenberg, S. G., Mostrel, N., Bak, R. O., Mantri, S., Quadros, R. M., et al. (2019). Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 10:4045. doi: 10.1038/s41467-019-11962-8
Gonzalez, E. A., and Baldo, G. (2017). Gene therapy for lysosomal storage disorders: recent advances and limitations. J. Inborn Errors Metab. Screen. 5, 1–6. doi: 10.1177/2326409816689786
Griffin, L. D., Gong, W., Verot, L., and Mellon, S. H. (2004). Niemann-pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat. Med. 10, 704–711. doi: 10.1038/nm1073
Gultekin, M., Bayramov, R., Karaca, C., and Acer, N. (2018). Sialidosis type I presenting with a novel mutation and advanced neuroimaging features. Neurosciences (Riyadh) 23, 57–61. doi: 10.17712/nsj.2018.1.20170328
Gupta, P., Soyombo, A. A., Atashband, A., Wisniewski, K. E., Shelton, J. M., Richardson, J. A., et al. (2001). Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc. Natl. Acad. Sci. U.S.A. 98, 13566–13571. doi: 10.1073/pnas.251485198
Gustavsson, S., Sjöström, E. O., Tjernberg, A., Janson, J., Westermark, U., Andersson, T., et al. (2019). Intravenous delivery of a chemically modified sulfamidase efficiently reduces heparan sulfate storage and brain pathology in mucopolysaccharidosis IIIA mice. Mol. Genet. Metab. 21:100510. doi: 10.1016/j.ymgmr.2019.100510
Hahn, C. N., Del Pilar, Martin, M., Schröder, M., Vanier, M. T., Hara, Y., et al. (1997). Generalized CNS disease and massive GM1-ganglioside accumulation in mice defective in lysosomal acid beta-galactosidase. Hum. Mol. Genet. 6, 205–211. doi: 10.1093/hmg/6.2.205
Harrison, R. K. (2016). Phase II and Phase III Failures: 2013-2015 Nat. Rev. Drug Discov. 15, 817–818. doi: 10.1038/nrd.2016.184
Hartung, S. D., Frandsen, J. L., Pan, D., Koniar, B. L., Graupman, P., Gunther, R., et al. (2004). Correction of metabolic, craniofacial, and neurologic abnormalities in MPS I mice treated at birth with adeno-associated virus vector transducing the human alpha-L-iduronidase gene. Mol. Ther. J. Am. Soc. Gene Ther. 9, 866–875. doi: 10.1016/j.ymthe.2004.03.011
Hartung, S. D., Reddy, R. G., Whitley, C. B., and Mcivor, R. S. (1999). Enzymatic correction and cross-correction of mucopolysaccharidosis type I fibroblasts by adeno-associated virus-mediated transduction of the alpha-L-iduronidase gene. Hum. Gene Ther. 10, 2163–2172. doi: 10.1089/10430349950017158
Haurigot, V., Marco, S., Ribera, A., Garcia, M., Ruzo, A., Villacampa, P., et al. (2013). Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. J. Clin. Invest. 123, 3254–3271. doi: 10.1172/JCI66778
Hawkins-Salsbury, J. A., Qin, E. Y., Reddy, A. S., Vogler, C. A., and Sands, M. S. (2012). Oxidative stress as a therapeutic target in globoid cell leukodystrophy. Exp. Neurol. 237, 444–452. doi: 10.1016/j.expneurol.2012.07.013
Hawkins-Salsbury, J. A., Shea, L., Jiang, X., Hunter, D. A., Guzman, A. M., Reddy, A. S., et al. (2015). Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. J. Neurosci. 35, 6495–6505. doi: 10.1523/JNEUROSCI.4199-14.2015
Hess, B., Saftig, P., Hartmann, D., Coenen, R., Lullmann-Rauch, R., Goebel, H. H., et al. (1996). Phenotype of arylsulfatase a-deficient mice: relationship to human metachromatic leukodystrophy. Proc. Natl. Acad. Sci. U.S.A. 93, 14821–14826. doi: 10.1073/pnas.93.25.14821
Higuchi, T., Shimizu, H., Fukuda, T., Kawagoe, S., Matsumoto, J., Shimada, Y., et al. (2012). Enzyme replacement therapy (ERT) procedure for mucopolysaccharidosis type II (MPS II) by intraventricular administration (IVA) in murine MPS II. Mol. Genet. Metab. 107, 122–128. doi: 10.1016/j.ymgme.2012.05.005
Hong, S. H., Chu, H., Kim, K. R., Ko, M. H., Kwon, S. Y., Moon, I. J., et al. (2012). Auditory characteristics and therapeutic effects of enzyme replacement in mouse model of the mucopolysaccharidosis (MPS) II. Am. J. Med. Genet. Part A 158A, 2131–2138. doi: 10.1002/ajmg.a.35498
Horinouchi, K., Erlich, S., Perl, D. P., Ferlinz, K., Bisgaier, C. L., Sandhoff, K., et al. (1995). Acid sphingomyelinase deficient mice: a model of types A and B niemann-pick disease. Nat. Genet. 10, 288–293. doi: 10.1038/ng0795-288
Hovakimyan, M., Maass, F., Petersen, J., Holzmann, C., Witt, M., Lukas, J., et al. (2013). Combined therapy with cyclodextrin/allopregnanolone and miglustat improves motor but not cognitive functions in niemann-pick Type C1 mice. Neuroscience 252, 201–211. doi: 10.1016/j.neuroscience.2013.08.001
Hu, J., Lu, J. Y., Wong, A. M., Hynan, L. S., Birnbaum, S. G., Yilmaz, D. S., et al. (2012). Intravenous high-dose enzyme replacement therapy with recombinant palmitoyl-protein thioesterase reduces visceral lysosomal storage and modestly prolongs survival in a preclinical mouse model of infantile neuronal ceroid lipofuscinosis. Mol. Genet. Metab. 107, 213–221. doi: 10.1016/j.ymgme.2012.05.009
Hughes, M. P., Smith, D. A., Morris, L., Fletcher, C., Colaco, A., Huebecker, M., et al. (2018). AAV9 intracerebroventricular gene therapy improves lifespan, locomotor function and pathology in a mouse model of Niemann-Pick type C1 disease. Hum. Mol. Genet. 27, 3079–3098. doi: 10.1093/hmg/ddy212
Ikawa, M., Kominami, K., Yoshimura, Y., Tanaka, K., Nishimune, Y., and Okabe, M. (1995). Green fluorescent protein as a marker in transgenic mice. Dev. Growth Differ. 37, 455–459. doi: 10.1046/j.1440-169X.1995.t01-2-00012.x
Jankowiak, W., Kruszewski, K., Flachsbarth, K., Skevas, C., Richard, G., Ruther, K., et al. (2015). Sustained neural stem cell-based intraocular delivery of CNTF attenuates photoreceptor loss in the nclf mouse model of neuronal ceroid lipofuscinosis. PLoS ONE 10:e0127204. doi: 10.1371/journal.pone.0127204
Janson, C. G., Romanova, L. G., Leone, P., Nan, Z., Belur, L., Mcivor, R. S., et al. (2014). Comparison of endovascular and intraventricular gene therapy with adeno-associated virus-α-L-iduronidase for hurler disease. Neurosurgery 74, 99–111. doi: 10.1227/NEU.0000000000000157
Jelinek, D., Castillo, J. J., Heidenreich, R. A., and Garver, W. S. (2015). The C57BL/6J niemann-Pick C1 mouse model with decreased gene dosage is susceptible to increased weight gain when fed a high-fat diet: confirmation of a gene-diet interaction. Gene 568, 112–113. doi: 10.1016/j.gene.2015.05.025
Jeyakumar, M., Butters, T. D., Cortina-Borja, M., Hunnam, V., Proia, R. L., Perry, V. H., et al. (1999). Delayed symptom onset and increased life expectancy in Sandhoff disease mice treated with N-butyldeoxynojirimycin. Proc. Natl. Acad. Sci. U.S.A. 96, 6388–6393. doi: 10.1073/pnas.96.11.6388
Jung, S.-C., Park, E.-S., Choi, E. N., Kim, C. H., Kim, S. J., and Jin, D.-K. (2010). Characterization of a novel mucopolysaccharidosis type II mouse model and recombinant AAV2/8 vector-mediated gene therapy. Mol. Cells 30, 13–18. doi: 10.1007/s10059-010-0083-2
Kaback, M. M., and Desnick, R. J. (2011). “Hexosaminidase A deficiency,” in SourceGeneReviews§[Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington), 1993–2020.
Kan, S. H., Aoyagi-Scharber, M., Le, S. Q., Vincelette, J., Ohmi, K., Bullens, S., et al. (2014). Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc. Natl. Acad. Sci. U.S.A. 111, 14870–14875. doi: 10.1073/pnas.1416660111
Karumuthil-Melethil, S., Marshall, M. S., Heindel, C., Jakubauskas, B., Bongarzone, E. R., and Gray, S. J. (2016). Intrathecal administration of AAV/GALC Vectors in 10-11-day-old twitcher mice improves survival and is enhanced by bone marrow transplant. J. Neurosci. Res. 94, 1138–1151. doi: 10.1002/jnr.23882
Kasperzyk, J. L., D’azzo, A., Platt, F. M., Alroy, J., and Seyfried, T. N. (2005). Substrate reduction reduces gangliosides in postnatal cerebrum-brainstem and cerebellum in GM1 gangliosidosis mice. J. Lipid Res. 46, 744–751. doi: 10.1194/jlr.M400411-JLR200
Keeling, K. M., Wang, D., Dai, Y., Murugesan, S., Chenna, B., Clark, J., et al. (2013). Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PLoS ONE 8:e60478. doi: 10.1371/journal.pone.0060478
Khanna, R., Flanagan, J. J., Feng, J., Soska, R., Frascella, M., Pellegrino, L. J., et al. (2012). The pharmacological chaperone AT2220 increases recombinant human acid alpha-glucosidase uptake and glycogen reduction in a mouse model of Pompe disease. PLoS ONE 7:e40776. doi: 10.1371/journal.pone.0040776
Khanna, R., Powe, A. C. Jr., Lun, Y., Soska, R., Feng, J., et al. (2014). The pharmacological chaperone AT2220 increases the specific activity and lysosomal delivery of mutant acid alpha-glucosidase, and promotes glycogen reduction in a transgenic mouse model of Pompe disease. PLoS ONE 9:e102092. doi: 10.1371/journal.pone.0102092
Kim, K. H., Pham, C. T., Sleat, D. E., and Lobel, P. (2008). Dipeptidyl-peptidase I does not functionally compensate for the loss of tripeptidyl-peptidase I in the neurodegenerative disease late-infantile neuronal ceroid lipofuscinosis. Biochem. J. 415, 225–232. doi: 10.1042/BJ20080411
Kim, K. H., Sleat, D. E., Bernard, O., and Lobel, P. (2009). Genetic modulation of apoptotic pathways fails to alter disease course in tripeptidyl-peptidase 1 deficient mice. Neurosci. Lett. 453, 27–30. doi: 10.1016/j.neulet.2009.01.072
Kim, S.-J., Lee, B.-H., Lee, Y.-S., and Kang, K.-S. (2007). Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochem. Biophys. Res. Commun. 360, 593–599. doi: 10.1016/j.bbrc.2007.06.116
Kirkegaard, T., Gray, J., Priestman, D. A., Wallom, K. L., Atkins, J., Olsen, O. D., et al. (2016). Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med. 8, 355ra118. doi: 10.1126/scitranslmed.aad9823
Ko, A.-R., Jin, D.-K., Cho, S. Y., Park, S. W., Przybylska, M., Yew, N. S., et al. (2016). AAV8-mediated expression of N-acetylglucosamine-1-phosphate transferase attenuates bone loss in a mouse model of mucolipidosis II. Mol. Genet. Metab. 117, 447–455. doi: 10.1016/j.ymgme.2016.02.001
Kobayashi, T., Yamanaka, T., Jacobs, J. M., Teixeira, F., and Suzuki, K. (1980). The Twitcher mouse: an enzymatically authentic model of human globoid cell leukodystrophy (Krabbe disease). Brain Res. 202, 479–483. doi: 10.1016/0006-8993(80)90159-6
Kohlschütter, A., and Schulz, A. (2016). CLN2 disease (Classic Late Infantile Neuronal Ceroid Lipofuscinosis). Pediatr. Endocrinol. Rev. 13(Suppl. 1), 682–688.
Kohlschütter, A., Schulz, A., Bartsch, U., and Storch, S. (2019). Current and emerging treatment strategies for neuronal ceroid lipofuscinoses. CNS Drugs 33, 315–325. doi: 10.1007/s40263-019-00620-8
Kovacs, A. D., and Pearce, D. A. (2008). Attenuation of AMPA receptor activity improves motor skills in a mouse model of juvenile Batten disease. Exp. Neurol. 209, 288–291. doi: 10.1016/j.expneurol.2007.09.012
Kuehn, S. C., Koehne, T., Cornils, K., Markmann, S., Riedel, C., Pestka, J. M., et al. (2015). Impaired bone remodeling and its correction by combination therapy in a mouse model of mucopolysaccharidosis-I. Hum. Mol. Genet. 24, 7075–7086. doi: 10.1093/hmg/ddv407
Kurz, T., Terman, A., Gustafsson, B., and Brunk, U. T. (2008). Lysosomes and oxidative stress in aging and apoptosis. Biochim. Biophys. Acta (BBA) 1780, 1291–1303. doi: 10.1016/j.bbagen.2008.01.009
Laoharawee, K., Dekelver, R. C., Podetz-Pedersen, K. M., Rohde, M., Sproul, S., Nguyen, H.-O., et al. (2018). Dose-dependent prevention of metabolic and neurologic disease in murine MPS II by ZFN-mediated in vivo genome editing. Mol. Ther. 26, 1127–1136. doi: 10.1016/j.ymthe.2018.03.002
Lau, A. A., King, B. M., Thorsen, C. L., Hassiotis, S., Beard, H., Trim, P. J., et al. (2017). A novel conditional sgsh knockout mouse model recapitulates phenotypic and neuropathic deficits of sanfilippo syndrome. J. Inherit. Metab. Dis. 40, 715–724. doi: 10.1007/s10545-017-0044-4
Lau, A. A., Shamsani, N. J., Winner, L. K., Hassiotis, S., King, B. M., Hopwood, J. J., et al. (2012). Neonatal bone marrow transplantation in MPS IIIA mice. JIMD Rep. 8, 121–132. doi: 10.1007/8904_2012_169
Le, S. Q., Kan, S.-H., Clarke, D., Sanghez, V., Egeland, M., Vondrak, K. N., et al. (2018). A humoral immune response alters the distribution of enzyme replacement therapy in murine mucopolysaccharidosis type I. Molecular Therapy. Methods Clin. Dev. 8, 42–51. doi: 10.1016/j.omtm.2017.09.008
Lee, J. H., Choe, Y. H., Kim, S. J., Paik, K. H., and Jin, D.-K. (2011). Changes in glycogen and glycosaminoglycan levels in hepatocytes of iduronate-2-sulfatase knockout mice before and after recombinant iduronate-2-sulfatase supplementation. Yonsei Med. J. 52, 263–267. doi: 10.3349/ymj.2011.52.2.263
Lee, S.-C., Lee, J., Jin, D.-K., Kim, J.-S., Jeon, E.-S., Kwun, Y. H., et al. (2014). Improvement of cardiac function by short-term enzyme replacement therapy in a murine model of cardiomyopathy associated with Hunter syndrome evaluated by serial echocardiography with speckle tracking 2-D strain analysis. Mol. Genet. Metab. 112, 218–223. doi: 10.1016/j.ymgme.2014.04.005
Lee, W. C., Courtenay, A., Troendle, F. J., Stallings-Mann, M. L., Dickey, C. A., Delucia, M. W., et al. (2005). Enzyme replacement therapy results in substantial improvements in early clinical phenotype in a mouse model of globoid cell leukodystrophy. FASEB J. 19, 1549–1551. doi: 10.1096/fj.05-3826fje
Lee, W. C., Tsoi, Y. K., Troendle, F. J., Delucia, M. W., Ahmed, Z., Dicky, C. A., et al. (2007). Single-dose intracerebroventricular administration of galactocerebrosidase improves survival in a mouse model of globoid cell leukodystrophy. FASEB J. 21, 2520–2527. doi: 10.1096/fj.06-6169com
Leroy, J. G., Cathey, S. S., and Friez, M. J. (2019). “GNPTAB-related disorders,” in SourceGeneReviews§[Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington), 1993–2020.
LeVine, S. M., Pedchenko, T. V., Bronshteyn, I. G., and Pinson, D. M. (2000). L-cycloserine slows the clinical and pathological course in mice with globoid cell leukodystrophy (twitcher mice). J. Neurosci. Res. 60, 231–236. doi: 10.1002/(SICI)1097-4547(20000415)60:2<231::AID-JNR12>3.0.CO;2-E
Li, H. H., Yu, W. H., Rozengurt, N., Zhao, H. Z., Lyons, K. M., Anagnostaras, S., et al. (1999). Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proc. Natl. Acad. Sci. U.S.A. 96, 14505–14510. doi: 10.1073/pnas.96.25.14505
Li, Y., Xu, Y., Benitez, B. A., Nagree, M. S., Dearborn, J. T., Jiang, X., et al. (2019). Genetic ablation of acid ceramidase in Krabbe disease confirms the psychosine hypothesis and identifies a new therapeutic target. Proc. Natl. Acad. Sci. U.S.A. 116, 20097–20103. doi: 10.1073/pnas.1912108116
Liao, G., Cheung, S., Galeano, J., Ji, A. X., Qin, Q., and Bi, X. (2009). Allopregnanolone treatment delays cholesterol accumulation and reduces autophagic/lysosomal dysfunction and inflammation in Npc1-/- mouse brain. Brain Res. 1270, 140–151. doi: 10.1016/j.brainres.2009.03.027
Lin, D., Fantz, C. R., Levy, B., Rafi, M. A., Vogler, C., Wenger, D. A., et al. (2005). AAV2/5 vector expressing galactocerebrosidase ameliorates CNS disease in the murine model of globoid-cell leukodystrophy more efficiently than AAV2. Mol. Ther. 12, 422–430. doi: 10.1016/j.ymthe.2005.04.019
Lin, D. S., Hsiao, C. D., Lee, A. Y., Ho, C. S., Liu, H. L., Wang, T. J., et al. (2015). Mitigation of cerebellar neuropathy in globoid cell leukodystrophy mice by AAV-mediated gene therapy. Gene 571, 81–90. doi: 10.1016/j.gene.2015.06.049
Lin, S., Lin, Y., Nery, J. R., Urich, M. A., Breschi, A., Davis, C. A., et al. (2014). Comparison of the transcriptional landscapes between human and mouse tissues. Proc. Natl. Acad. Sci. U.S.A. 111, 17224–17229. doi: 10.1073/pnas.1413624111
Liou, B., Peng, Y., Li, R., Inskeep, V., Zhang, W., Quinn, B., et al. (2016). Modulating ryanodine receptors with dantrolene attenuates neuronopathic phenotype in Gaucher disease mice. Hum. Mol. Genet. 25, 5126–5141. doi: 10.1093/hmg/ddw322
Liu, B., Ramirez, C. M., Miller, A. M., Repa, J. J., Turley, S. D., and Dietschy, J. M. (2010). Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J. Lipid Res. 51, 933–944. doi: 10.1194/jlr.M000257
Liu, Y., Suzuki, K., Reed, J. D., Grinberg, A., Westphal, H., Hoffmann, A., et al. (1998). Mice with type 2 and 3 Gaucher disease point mutations generated by a single insertion mutagenesis procedure. Proc. Natl. Acad. Sci. U.S.A. 95, 2503–2508. doi: 10.1073/pnas.95.5.2503
Lizzi Lagranha, V., Zambiasi Martinelli, B., Baldo, G., Ávila Testa, G., Giacomet de Carvalho, T., Giugliani, R., et al. (2017). Subcutaneous implantation of microencapsulated cells overexpressing α-L-iduronidase for mucopolysaccharidosis type I treatment. J. Mater. Sci. Mater. Med. 28:43. doi: 10.1007/s10856-017-5844-4
Lorioli, L., and Biffi, A. (2015). Hematopoietic stem cell transplantation for metachromatic leukodystrophy. Expert Opin. Orphan. Drugs 3, 911–919. doi: 10.1517/21678707.2015.1062754
Lowenstein, P. R., and Castro, M. G. (2009). Uncertainty in the translation of preclinical experiments to clinical trials. Why do Most Phase III Clinical Trials Fail? Curr. Gene Ther. 9, 368–374. doi: 10.2174/156652309789753392
Lund, A. M., Borgwardt, L., Cattaneo, F., Ardigò, D., Geraci, S., Gil-Campos, M., et al. (2018). Comprehensive long-term efficacy and safety of recombinant human alpha-mannosidase (velmanase alfa) treatment in patients with alpha-mannosidosis. J. Inherit. Metab. Dis. 41, 1225–1233. doi: 10.1007/s10545-018-0175-2
Luzi, P., Abraham, R. M., Rafi, M. A., Curtis, M., Hooper, D. C., and Wenger, D. A. (2009). Effects of treatments on inflammatory and apoptotic markers in the CNS of mice with globoid cell leukodystrophy. Brain Res. 1300, 146–158. doi: 10.1016/j.brainres.2009.09.017
Luzi, P., Rafi, M. A., Zaka, M., Curtis, M., Vanier, M. T., and Wenger, D. A. (2001). Generation of a mouse with low galactocerebrosidase activity by gene targeting: a new model of globoid cell leukodystrophy (Krabbe disease). Mol. Genet. Metab. 73, 211–223. doi: 10.1006/mgme.2001.3194
Luzi, P., Rafi, M. A., Zaka, M., Rao, H. Z., Curtis, M., Vanier, M. T., et al. (2005). Biochemical and pathological evaluation of long-lived mice with globoid cell leukodystrophy after bone marrow transplantation. Mol. Genet. Metab. 86, 150–159. doi: 10.1016/j.ymgme.2005.06.023
Lynch, G., and Bi, X. (2003). Lysosomes and brain aging in mammals. Neurochem. Res. 28, 1725–1734. doi: 10.1023/A:1026069223763
Macauley, S. L., Wong, A. M., Shyng, C., Augner, D. P., Dearborn, J. T., Pearse, Y., et al. (2014). An anti-neuroinflammatory that targets dysregulated glia enhances the efficacy of CNS-directed gene therapy in murine infantile neuronal ceroid lipofuscinosis. J. Neurosci. 34, 13077–13082. doi: 10.1523/JNEUROSCI.2518-14.2014
Mancini, G. M., Havelaar, A. C., and Verheijen, F. W. (2000). Lysosomal transport disorders. J. Inherit. Metab. Dis. 23, 278–292. doi: 10.1023/A:1005640214408
Mandal, P. K., Ferreira, L. M. R., Collins, R., Meissner, T. B., Boutwell, C. L., Friesen, M., et al. (2014). Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 15, 643–652. doi: 10.1016/j.stem.2014.10.004
Marcó, S., Pujol, A., Roca, C., Motas, S., Ribera, A., Garcia, M., et al. (2016). Progressive neurologic and somatic disease in a novel mouse model of human mucopolysaccharidosis type IIIC. Dis. Model Mech. 9, 999–1013. doi: 10.1242/dmm.025171
Marques, A. R. A., and Saftig, P. (2019). Lysosomal storage disorders – challenges, concepts and avenues for therapy: beyond rare diseases. J. Cell Sci. 132, jcs221739. doi: 10.1242/jcs.221739
Marshall, M. S., Issa, Y., Jakubauskas, B., Stoskute, M., Elackattu, V., Marshall, J. N., et al. (2018). Long-term improvement of neurological signs and metabolic dysfunction in a mouse model of Krabbe’s disease after global gene therapy. Mol. Ther. 26, 874–889. doi: 10.1016/j.ymthe.2018.01.009
Martins, C., Hulkova, H., Dridi, L., Dormoy-Raclet, V., Grigoryeva, L., Choi, Y., et al. (2015). Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 138, 336–355. doi: 10.1093/brain/awu355
Matsuda, J., Suzuki, O., Oshima, A., Ogura, A., Noguchi, Y., Yamamoto, Y., et al. (1997). Beta-galactosidase-deficient mouse as an animal model for GM1-gangliosidosis. Glycoconj J. 14, 729–736. doi: 10.1023/A:1018573518127
Matsuda, J., Vanier, M. T., Saito, Y., Tohyama, J., Suzuki, K., and Suzuki, K. (2001). A mutation in the saposin A domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Hum. Mol. Genet. 10, 1191–1199. doi: 10.1093/hmg/10.11.1191
Matthes, F., Andersson, C., Stein, A., Eistrup, C., Fogh, J., Gieselmann, V., et al. (2015). Enzyme replacement therapy of a novel humanized mouse model of globoid cell leukodystrophy. Exp. Neurol. 271, 36–45. doi: 10.1016/j.expneurol.2015.04.020
Matthes, F., Stroobants, S., Gerlach, D., Wohlenberg, C., Wessig, C., Fogh, J., et al. (2012). Efficacy of enzyme replacement therapy in an aggravated mouse model of metachromatic leukodystrophy declines with age. Hum. Mol. Genet. 21, 2599–2609. doi: 10.1093/hmg/dds086
Matzner, U., Habetha, M., and Gieselmann, V. (2000). Retrovirally expressed human arylsulfatase A corrects the metabolic defect of arylsulfatase A-deficient mouse cells. Gene Ther. 7, 805–812. doi: 10.1038/sj.gt.3301150
Matzner, U., Herbst, E., Hedayati, K. K., Lullmann-Rauch, R., Wessig, C., Schroder, S., et al. (2005). Enzyme replacement improves nervous system pathology and function in a mouse model for metachromatic leukodystrophy. Hum. Mol. Genet. 14, 1139–1152. doi: 10.1093/hmg/ddi126
Maue, R. A., Burgess, R. W., Wang, B., Wooley, C. M., Seburn, K. L., Vanier, M. T., et al. (2012). A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum. Mol. Genet. 21, 730–750. doi: 10.1093/hmg/ddr505
Mazzulli, J. R., Xu, Y.-H., Sun, Y., Knight, A. L., Mclean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
McIntyre, C., Derrick Roberts, A. L., Ranieri, E., Clements, P. R., Byers, S., and Anson, D. S. (2008). Lentiviral-mediated gene therapy for murine mucopolysaccharidosis type IIIA. Mol. Genet. Metab. 93, 411–418. doi: 10.1016/j.ymgme.2007.11.008
Mendez, D. C., Stover, A. E., Rangel, A. D., Brick, D. J., Nethercott, H. E., Torres, M. A., et al. (2015). A novel, long-lived, and highly engraftable immunodeficient mouse model of mucopolysaccharidosis type I. Molecular Therapy. Methods Clin. Dev. 2:14068. doi: 10.1038/mtm.2014.68
Meng, Y., Sohar, I., Wang, L., Sleat, D. E., and Lobel, P. (2012). Systemic administration of tripeptidyl peptidase I in a mouse model of late infantile neuronal ceroid lipofuscinosis: effect of glycan modification. PLoS ONE 7:e40509. doi: 10.1371/journal.pone.0040509
Michalski, J. C., and Klein, A. (1999). Glycoprotein lysosomal storage disorders: alpha- and beta-mannosidosis, fucosidosis and alpha-N-acetylgalactosaminidase deficiency. Biochim. Biophys. Acta 1455, 69–84. doi: 10.1016/S0925-4439(99)00077-0
Miki, T., Vazquez, L., Yanuaria, L., Lopez, O., Garcia, I. M., Ohashi, K., et al. (2019). Induced pluripotent stem cell derivation and ex vivo gene correction using a mucopolysaccharidosis type 1 disease mouse model. Stem Cells Int. 2019, 6978303. doi: 10.1155/2019/6978303
Miller, J. N., Kovacs, A. D., and Pearce, D. A. (2015). The novel Cln1(R151X) mouse model of infantile neuronal ceroid lipofuscinosis (INCL) for testing nonsense suppression therapy. Hum. Mol. Genet. 24, 185–196. doi: 10.1093/hmg/ddu428
Mirza, M., Vainshtein, A., Dironza, A., Chandrachud, U., Haslett, L. J., Palmieri, M., et al. (2019). The CLN3 gene and protein: what we know. Mol. Genet. Genomic Med. 7:e859. doi: 10.1002/mgg3.859
Mirza, M., Volz, C., Karlstetter, M., Langiu, M., Somogyi, A., Ruonala, M. O., et al. (2013). Progressive retinal degeneration and glial activation in the CLN6 (nclf) mouse model of neuronal ceroid lipofuscinosis: a beneficial effect of DHA and curcumin supplementation. PLoS ONE 8:e75963. doi: 10.1371/journal.pone.0075963
Mitchison, H. M., Bernard, D. J., Greene, N. D., Cooper, J. D., Junaid, M. A., Pullarkat, R. K., et al. (1999). Targeted disruption of the Cln3 gene provides a mouse model for Batten disease. The Batten Mouse Model Consortium [corrected]. Neurobiol. Dis. 6, 321–334. doi: 10.1006/nbdi.1999.0267
Mole, S. E., and Williams, R. E. (2013). “Neuronal ceroid-lipofuscinoses,” in SourceGeneReviews§[Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington). doi: 10.1093/med/9780199590018.001.0001
Mole, S. E., Williams, R. E., and Goebel, H. H. (2005). Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 6, 107–126. doi: 10.1007/s10048-005-0218-3
Monaco, A., Maffia, V., Sorrentino, N. C., Sambri, I., Ezhova, Y., Giuliano, T., et al. (2020). The amyloid inhibitor CLR01 relieves autophagy and ameliorates neuropathology in a severe lysosomal storage disease. Mol. Ther. [Epub ahead of print]. doi: 10.1016/j.ymthe.2020.02.005
Mori, J., Ishihara, Y., Matsuo, K., Nakajima, H., Terada, N., Kosaka, K., et al. (2008). Hematopoietic contribution to skeletal muscle regeneration in acid alpha-glucosidase knockout mice. J. Histochem. Cytochem. 56, 811–817. doi: 10.1369/jhc.2008.951244
Morris, M. D., Bhuvanswaran, C., and Boothe, A. D. (1977). Tissue cholesterol storage disorder in balb/c mice. Abstr. Subject Strain Bibliogr. 1977:2723.
Motas, S., Haurigot, V., Garcia, M., Marcó, S., Ribera, A., Roca, C., et al. (2016). CNS-directed gene therapy for the treatment of neurologic and somatic mucopolysaccharidosis type II (Hunter syndrome). JCI Insight 1:e86696. doi: 10.1172/jci.insight.86696
Muenzer, J., Lamsa, J. C., Garcia, A., Dacosta, J., Garcia, J., and Treco, D. A. (2002). Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): a preliminary report. Acta Paediatr. (Oslo, Norway: 1992). Suppl. 91, 98–99. doi: 10.1111/j.1651-2227.2002.tb03118.x
Nan, Z., Shekels, L., Ryabinin, O., Evavold, C., Nelson, M. S., Khan, S. A., et al. (2012). Intracerebroventricular transplantation of human bone marrow-derived multipotent progenitor cells in an immunodeficient mouse model of mucopolysaccharidosis type I (MPS-I). Cell Transpl. 21, 1577–1593. doi: 10.3727/096368912X636894
Neri, M., Ricca, A., Di Girolamo, I., Cavazzin, C., Orlacchio, A., Martino, S., et al. (2011). Neural stem cell gene therapy ameliorates pathology and function in a mouse model of globoid cell leukodystrophy. Stem Cells 29, 1559–1571. doi: 10.1002/stem.701
Nesterova, G., and Gahl, W. A. (2017). “Cystinosis,” in SourceGeneReviews§[Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Nishino, I., Fu, J., Tanji, K., Yamada, T., Shimojo, S., Koori, T., et al. (2000). Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406, 906–910. doi: 10.1038/35022604
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., et al. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122. doi: 10.1093/jnen/64.2.113
Norflus, F., Tifft, C. J., Mcdonald, M. P., Goldstein, G., Crawley, J. N., Hoffmann, A., et al. (1998). Bone marrow transplantation prolongs life span and ameliorates neurologic manifestations in Sandhoff disease mice. J. Clin. Invest. 101, 1881–1888. doi: 10.1172/JCI2127
Nusca, S., Canterini, S., Palladino, G., Bruno, F., Mangia, F., Erickson, R. P., et al. (2014). A marked paucity of granule cells in the developing cerebellum of the Npc1(–/–) mouse is corrected by a single injection of hydroxypropyl-beta-cyclodextrin. Neurobiol. Dis. 70, 117–126. doi: 10.1016/j.nbd.2014.06.012
Ohashi, T. (2019). Gene therapy for lysosomal storage diseases and peroxisomal diseases. J. Hum. Genet. 64, 139–143. doi: 10.1038/s10038-018-0537-5
Ohmi, K., Greenberg, D. S., Rajavel, K. S., Ryazantsev, S., Li, H. H., and Neufeld, E. F. (2003). Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. U.S.A. 100, 1902–1907. doi: 10.1073/pnas.252784899
Orban, P. C., Chui, D., and Marth, J. D. (1992). Tissue- and site-specific DNA recombination in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 89, 6861–6865. doi: 10.1073/pnas.89.15.6861
Osborn, M. J., Mcelmurry, R. T., Lees, C. J., Defeo, A. P., Chen, Z.-Y., Kay, M. A., et al. (2011). Minicircle DNA-based gene therapy coupled with immune modulation permits long-term expression of α-L-iduronidase in mice with mucopolysaccharidosis type I. Mol. Ther. 19, 450–460. doi: 10.1038/mt.2010.249
Osborn, M. J., Mcelmurry, R. T., Peacock, B., Tolar, J., and Blazar, B. R. (2008). Targeting of the CNS in MPS-IH using a nonviral transferrin-alpha-L-iduronidase fusion gene product. Mol. Ther. 16, 1459–1466. doi: 10.1038/mt.2008.119
Otterbach, B., and Stoffel, W. (1995). Acid sphingomyelinase-deficient mice mimic the neurovisceral form of human lysosomal storage disease (Niemann-Pick disease). Cell 81, 1053–1061. doi: 10.1016/S0092-8674(05)80010-8
Ou, L., Dekelver, R. C., Rohde, M., Tom, S., Radeke, R., St Martin, S. J., et al. (2019). ZFN-Mediated In Vivo Genome Editing Corrects Murine Hurler Syndrome. Mol. Ther. 27, 178–187. doi: 10.1016/j.ymthe.2018.10.018
Ou, L., Przybilla, M. J., Koniar, B. L., and Whitley, C. B. (2016). Elements of lentiviral vector design toward gene therapy for treating mucopolysaccharidosis I. Mol. Genet. Metab. Rep. 8, 87–93. doi: 10.1016/j.ymgmr.2015.11.004
Pan, X., Sands, S. A., Yue, Y., Zhang, K., Levine, S. M., and Duan, D. (2019). An engineered galactosylceramidase construct improves AAV gene therapy for krabbe disease in twitcher mice. Hum. Gene Ther. 30, 1039–1051. doi: 10.1089/hum.2019.008
Pannuzzo, G., Cardile, V., Costantino-Ceccarini, E., Alvares, E., Mazzone, D., and Perciavalle, V. (2010). A galactose-free diet enriched in soy isoflavones and antioxidants results in delayed onset of symptoms of Krabbe disease in twitcher mice. Mol. Genet. Metab. 100, 234–240. doi: 10.1016/j.ymgme.2010.03.021
Parenti, G., Andria, G., and Valenzano, K. J. (2015). Pharmacological chaperone therapy: preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol. Ther. 23, 1138–1148. doi: 10.1038/mt.2015.62
Parenti, G., Pignata, C., Vajro, P., and Salerno, M. (2013). New strategies for the treatment of lysosomal storage diseases (Review). Int. J. Mol. Med. 31, 11–20. doi: 10.3892/ijmm.2012.1187
Pasqualim, G., Baldo, G., De Carvalho, T. G., Tavares, A. M. V., Giugliani, R., and Matte, U. (2015). Effects of enzyme replacement therapy started late in a murine model of mucopolysaccharidosis type I. PLoS ONE 10:e0117271. doi: 10.1371/journal.pone.0117271
Passini, M. A., Bu, J., Fidler, J. A., Ziegler, R. J., Foley, J. W., Dodge, J. C., et al. (2007). Combination brain and systemic injections of AAV provide maximal functional and survival benefits in the Niemann-Pick mouse. Proc. Natl. Acad. Sci. U.S.A. 104, 9505–9510. doi: 10.1073/pnas.0703509104
Pastores, G. M., and Hughes, D. A. (2018). “Gaucher disease,” in SourceGeneReviews§[Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Patil, S. A., and Maegawa, G. H. (2013). Developing therapeutic approaches for metachromatic leukodystrophy. Drug Des. Dev. Ther. 7, 729–745. doi: 10.2147/DDDT.S15467
Paton, L., Bitoun, E., Kenyon, J., Priestman, D. A., Oliver, P. L., Edwards, B., et al. (2014). A novel mouse model of a patient mucolipidosis II mutation recapitulates disease pathology. J. Biol. Chem. 289, 26709–26721. doi: 10.1074/jbc.M114.586156
Pelled, D., Lloyd-Evans, E., Riebeling, C., Jeyakumar, M., Platt, F. M., and Futerman, A. H. (2003). Inhibition of calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase in a mouse model of Sandhoff disease and prevention by treatment with N-butyldeoxynojirimycin. J. Biol. Chem. 278, 29496–29501. doi: 10.1074/jbc.M302964200
Pievani, A., Azario, I., Antolini, L., Shimada, T., Patel, P., Remoli, C., et al. (2015). Neonatal bone marrow transplantation prevents bone pathology in a mouse model of mucopolysaccharidosis type I. Blood 125, 1662–1671. doi: 10.1182/blood-2014-06-581207
Piller Puicher, E., Tomanin, R., Salvalaio, M., Friso, A., Hortelano, G., Marin, O., et al. (2012). Encapsulated engineered myoblasts can cure Hurler syndrome: preclinical experiments in the mouse model. Gene Ther. 19, 355–364. doi: 10.1038/gt.2011.94
Platt, F., and Walkley, S. U. (2004). Lysosomal Defects and Storage. Oxford: Oxford University Press. doi: 10.1093/acprof:oso/9780198508786.003.0002
Platt, F. M., D’azzo, A., Davidson, B. L., Neufeld, E. F., and Tifft, C. J. (2018). Lysosomal storage diseases. Nat. Rev. Dis Primers 4:27. doi: 10.1038/s41572-018-0025-4
Platt, F. M., Neises, G. R., Reinkensmeier, G., Townsend, M. J., Perry, V. H., Proia, R. L., et al. (1997). Prevention of lysosomal storage in Tay-Sachs mice treated with N-butyldeoxynojirimycin. Science 276, 428–431. doi: 10.1126/science.276.5311.428
Polito, V. A., Abbondante, S., Polishchuk, R. S., Nusco, E., Salvia, R., and Cosma, M. P. (2010). Correction of CNS defects in the MPSII mouse model via systemic enzyme replacement therapy. Hum. Mol. Genet. 19, 4871–4885. doi: 10.1093/hmg/ddq420
Polito, V. A., and Cosma, M. P. (2009). IDS crossing of the blood-brain barrier corrects CNS defects in MPSII mice. Am. J. Hum. Genet. 85, 296–301. doi: 10.1016/j.ajhg.2009.07.011
Poswar, F. D. O., Vairo, F., Burin, M., Michelin-Tirelli, K., Brusius-Facchin, A. C., Kubaski, F., et al. (2019). Lysosomal diseases: overview on current diagnosis and treatment. Genet. Mol. Biol. 42, 165–177. doi: 10.1590/1678-4685-gmb-2018-0159
Potter, G. B., Santos, M., Davisson, M. T., Rowitch, D. H., Marks, D. L., Bongarzone, E. R., et al. (2013). Missense mutation in mouse GALC mimics human gene defect and offers new insights into Krabbe disease. Hum. Mol. Genet. 22, 3397–3414. doi: 10.1093/hmg/ddt190
Praggastis, M., Tortelli, B., Zhang, J., Fujiwara, H., Sidhu, R., Chacko, A., et al. (2015). A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci. 35, 8091–8106. doi: 10.1523/JNEUROSCI.4173-14.2015
Prasad, V. K., and Kurtzberg, J. (2009). Cord blood and bone marrow transplantation in inherited metabolic diseases: scienti?c basis, current status and future directions. Br. J. Haematol. 148, 356–372. doi: 10.1111/j.1365-2141.2009.07974.x
Prolo, L. M., Vogel, H., and Reimer, R. J. (2009). The lysosomal sialic acid transporter sialin is required for normal CNS myelination. J. Neurosci. 29, 15355–15365. doi: 10.1523/JNEUROSCI.3005-09.2009
Puzzo, F., Colella, P., Biferi, M. G., Bali, D., Paulk, N. K., Vidal, P., et al. (2017). Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid alpha-glucosidase. Sci. Transl. Med. 9:eaam6375. doi: 10.1126/scitranslmed.aam6375
Qin, E. Y., Hawkins-Salsbury, J. A., Jiang, X., Reddy, A. S., Farber, N. B., Ory, D. S., et al. (2012). Bone marrow transplantation increases efficacy of central nervous system-directed enzyme replacement therapy in the murine model of globoid cell leukodystrophy. Mol. Genet. Metab. 107, 186–196. doi: 10.1016/j.ymgme.2012.05.021
Quiviger, M., Arfi, A., Mansard, D., Delacotte, L., Pastor, M., Scherman, D., et al. (2014). High and prolonged sulfamidase secretion by the liver of MPS-IIIA mice following hydrodynamic tail vein delivery of antibiotic-Free pFAR4 plasmid vector. Gene Ther. 21, 1001–1007. doi: 10.1038/gt.2014.75
Raben, N., Jatkar, T., Lee, A., Lu, N., Dwivedi, S., Nagaraju, K., et al. (2002). Glycogen stored in skeletal but not in cardiac muscle in acid alpha-glucosidase mutant (Pompe) mice is highly resistant to transgene-encoded human enzyme. Mol. Ther. 6, 601–608. doi: 10.1016/S1525-0016(02)90716-1
Raben, N., Lu, N., Nagaraju, K., Rivera, Y., Lee, A., Yan, B., et al. (2001). Conditional tissue-specific expression of the acid alpha-glucosidase (GAA) gene in the GAA knockout mice: implications for therapy. Hum. Mol. Genet. 10, 2039–2047. doi: 10.1093/hmg/10.19.2039
Raben, N., Nagaraju, K., Lee, E., Kessler, P., Byrne, B., Lee, L., et al. (1998). Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J. Biol. Chem. 273, 19086–19092. doi: 10.1074/jbc.273.30.19086
Raben, N., Schreiner, C., Baum, R., Takikita, S., Xu, S., Xie, T., et al. (2010). Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder–murine Pompe disease. Autophagy 6, 1078–1089. doi: 10.4161/auto.6.8.13378
Radin, N. S. (1996). Treatment of Gaucher disease with an enzyme inhibitor. Glycoconj J. 13, 153–157. doi: 10.1007/BF00731489
Rafi, M. A., Rao, H. Z., Luzi, P., Curtis, M. T., and Wenger, D. A. (2012). Extended normal life after AAVrh10-mediated gene therapy in the mouse model of krabbe disease. Mol. Ther. 20, 2031–2042. doi: 10.1038/mt.2012.153
Rafi, M. A., Rao, H. Z., Luzi, P., Luddi, A., Curtis, M. T., and Wenger, D. A. (2015a). Intravenous injection of AAVrh10-GALC after the neonatal period in twitcher mice results in significant expression in the central and peripheral nervous systems and improvement of clinical features. Mol. Genet. Metab. 114, 459–466. doi: 10.1016/j.ymgme.2014.12.300
Rafi, M. A., Rao, H. Z., Luzi, P., and Wenger, D. A. (2015b). Long-term improvements in lifespan and pathology in CNS and PNS after BMT plus one intravenous injection of AAVrh10-GALC in twitcher mice. Mol. Ther. 23, 1681–1690. doi: 10.1038/mt.2015.145
Reddy, A. S., Kim, J. H., Hawkins-Salsbury, J. A., Macauley, S. L., Tracy, E. T., Vogler, C. A., et al. (2011). Bone marrow transplantation augments the effect of brain- and spinal cord-directed adeno-associated virus 2/5 gene therapy by altering inflammation in the murine model of globoid-cell leukodystrophy. J. Neurosci. 31, 9945–9957. doi: 10.1523/JNEUROSCI.1802-11.2011
Rega, L. R., Polishchuk, E., Montefusco, S., Napolitano, G., Tozzi, G., Zhang, J., et al. (2016). Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kid. Int. 89, 862–873. doi: 10.1016/j.kint.2015.12.045
Repa, J. J., Li, H., Frank-Cannon, T. C., Valasek, M. A., Turley, S. D., Tansey, M. G., et al. (2007). Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. J. Neurosci. 27, 14470–14480. doi: 10.1523/JNEUROSCI.4823-07.2007
Ribera, A., Haurigot, V., Garcia, M., Marco, S., Motas, S., Villacampa, P., et al. (2015). Biochemical, histological and functional correction of mucopolysaccharidosis type IIIB by intra-cerebrospinal fluid gene therapy. Hum. Mol. Genet. 24, 2078–2095. doi: 10.1093/hmg/ddu727
Riboldi, G. M., and Fonzo, A. B. D. (2019). GBA, gaucher disease, and Parkinson’s disease: from genetic to clinic to new therapeutic approaches. Cells 8:364. doi: 10.3390/cells8040364
Ricca, A., Rufo, N., Ungari, S., Morena, F., Martino, S., Kulik, W., et al. (2015). Combined gene/cell therapies provide long-term and pervasive rescue of multiple pathological symptoms in a murine model of globoid cell leukodystrophy. Hum. Mol. Genet. 24, 3372–3389. doi: 10.1093/hmg/ddv086
Ripoll, C. B., Flaat, M., Klopf-Eiermann, J., Fisher-Perkins, J. M., Trygg, C. B., Scruggs, B. A., et al. (2011). Mesenchymal lineage stem cells have pronounced anti-inflammatory effects in the twitcher mouse model of Krabbe’s disease. Stem Cells 29, 67–77. doi: 10.1002/stem.555
Roberts, A. L., Fletcher, J. M., Moore, L., and Byers, S. (2010). Trans-generational exposure to low levels of rhodamine B does not adversely affect litter size or liver function in murine mucopolysaccharidosis type IIIA. Mol. Genet. Metab. 101, 208–213. doi: 10.1016/j.ymgme.2010.06.008
Roberts, A. L. K., Rees, M. H., Klebe, S., Fletcher, J. M., and Byers, S. (2007). Improvement in behaviour after substrate deprivation therapy with rhodamine b in a mouse model of MPS IIIA. Mol. Genet. Metab. 92, 115–121. doi: 10.1016/j.ymgme.2007.06.016
Roca, C., Motas, S., Marco, S., Ribera, A., Sanchez, V., Sanchez, X., et al. (2017). Disease correction by AAV-mediated gene therapy in a new mouse model of mucopolysaccharidosis type IIID. Hum. Mol. Genet. 26, 1535–1551. doi: 10.1093/hmg/ddx058
Rocca, C. J., Kreymerman, A., Ur, S. N., Frizzi, K. E., Naphade, S., Lau, A., et al. (2015). Treatment of inherited eye defects by systemic hematopoietic stem cell transplantation. Invest. Ophthalmol. Vis. Sci. 56, 7214–7223. doi: 10.1167/iovs.15-17107
Roces, D. P., Lullmann-Rauch, R., Peng, J., Balducci, C., Andersson, C., Tollersrud, O., et al. (2004). Efficacy of enzyme replacement therapy in alpha-mannosidosis mice: a preclinical animal study. Hum. Mol. Genet. 13, 1979–1988. doi: 10.1093/hmg/ddh220
Rothaug, M., Stroobants, S., Schweizer, M., Peters, J., Zunke, F., Allerding, M., et al. (2015). LAMP-2 deficiency leads to hippocampal dysfunction but normal clearance of neuronal substrates of chaperone-mediated autophagy in a mouse model for Danon disease. Acta Neuropathol. Commun. 3:6. doi: 10.1186/s40478-014-0182-y
Rucker, M., Fraites, T. J. Jr., Porvasnik, S. L., Lewis, M. A., Zolotukhin, I., et al. (2004). Rescue of enzyme deficiency in embryonic diaphragm in a mouse model of metabolic myopathy: Pompe disease. Development 131, 3007–3019. doi: 10.1242/dev.01169
Rudnicki, D. D., Pletnikova, O., Vonsattel, J.-P. G., Ross, C. A., and Margolis, R. L. (2008). A comparison of huntington disease and huntington disease-like 2 neuropathology. J. Neuropathol. Exp. Neurol. 67, 366–374. doi: 10.1097/NEN.0b013e31816b4aee
Ruzo, A., Marcó, S., García, M., Villacampa, P., Ribera, A., Ayuso, E., et al. (2012). Correction of pathological accumulation of glycosaminoglycans in central nervous system and peripheral tissues of MPSIIIA mice through systemic AAV9 gene transfer. Hum. Gene Ther. 23, 1237–1246. doi: 10.1089/hum.2012.029
Saha, A., Sarkar, C., Singh, S. P., Zhang, Z., Munasinghe, J., Peng, S., et al. (2012). The blood-brain barrier is disrupted in a mouse model of infantile neuronal ceroid lipofuscinosis: amelioration by resveratrol. Hum. Mol. Genet. 21, 2233–2244. doi: 10.1093/hmg/dds038
Sango, K., Yamanaka, S., Hoffmann, A., Okuda, Y., Grinberg, A., Westphal, H., et al. (1995). Mouse models of tay-sachs and sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat. Genet. 11, 170–176. doi: 10.1038/ng1095-170
Sano, R., Tessitore, A., Ingrassia, A., and D’azzo, A. (2005). Chemokine-induced recruitment of genetically modified bone marrow cells into the CNS of GM1-gangliosidosis mice corrects neuronal pathology. Blood 106, 2259–2268. doi: 10.1182/blood-2005-03-1189
Sargeant, T. J., Wang, S., Bradley, J., Smith, N. J., Raha, A. A., Mcnair, R., et al. (2011). Adeno-associated virus-mediated expression of beta-hexosaminidase prevents neuronal loss in the Sandhoff mouse brain. Hum. Mol. Genet. 20, 4371–4380. doi: 10.1093/hmg/ddr364
Sarkar, C., Chandra, G., Peng, S., Zhang, Z., Liu, A., and Mukherjee, A. B. (2013). Neuroprotection and lifespan extension in Ppt1(-/-) mice by NtBuHA: therapeutic implications for INCL. Nat. Neurosci. 16, 1608–1617. doi: 10.1038/nn.3526
Sasaki, S. (2011). Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Ex. Neurol. 70, 349–359. doi: 10.1097/NEN.0b013e3182160690
Scaravilli, F., and Jacobs, J. M. (1981). Peripheral nerve grafts in hereditary leukodystrophic mutant mice (twitcher). Nature 290, 56–58. doi: 10.1038/290056a0
Scaravilli, F., and Jacobs, J. M. (1982). Improved myelination in nerve grafts from the leucodystrophic twitcher into trembler mice: evidence for enzyme replacement. Brain Res. 237, 163–172. doi: 10.1016/0006-8993(82)90564-9
Scaravilli, F., and Suzuki, K. (1983). Enzyme replacement in grafted nerve of twitcher mouse. Nature 305, 713–715. doi: 10.1038/305713a0
Scarpa, M. (2018). “Mucopolysaccharidosis type II, synonyms: hunter syndrome, iduronate-2-sulfatase deficiency, MPS II,” in GeneReviews§[Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, et al. (Seattle, WA: University of Washington, Seattle).
Schiffmann, R., Mayfield, J., Swift, C., and Nestrasil, I. (2014). Quantitative neuroimaging in mucolipidosis type IV. Mol. Genet. Metab. 111, 147–151. doi: 10.1016/j.ymgme.2013.11.007
Schlotawa, L., Adang, L., Castro, M. D., and Ahrens-Nicklas, R. (2019). “Multiple sulfatase deficiency,” in GeneReviews, eds M. P. Adam, H. H. Arranger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Schoser, B., Stewart, A., Kanters, S., Hamed, A., Jansen, J., Chan, K., et al. (2017). Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J. Neurol. 264, 621–630. doi: 10.1007/s00415-016-8219-8
Schuh, R. S., Poletto, É, Pasqualim, G., Tavares, A. M. V., Meyer, F. S., Gonzalez, E. A., et al. (2018). In vivo genome editing of mucopolysaccharidosis I mice using the CRISPR/Cas9 system. J. Control Release 288, 23–33. doi: 10.1016/j.jconrel.2018.08.031
Schultz, M. L., Tecedor, L., Chang, M., and Davidson, B. L. (2011). Clarifying lysosomal storage diseases. Trends Neurosci. 34, 401–410. doi: 10.1016/j.tins.2011.05.006
Schultz, M. L., Tecedor, L., Lysenko, E., Ramachandran, S., Stein, C. S., and Davidson, B. L. (2018). Modulating membrane fluidity corrects Batten disease phenotypes in vitro and in vivo. Neurobiol. Dis. 115, 182–193. doi: 10.1016/j.nbd.2018.04.010
Seranova, E., Connolly, K. J., Zatyka, M., Rosenstock, T. R., Barrett, T., Tuxworth, R. I., et al. (2017). Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 61, 733–749. doi: 10.1042/EBC20170055
Settembre, C., Annunziata, I., Spampanato, C., Zarcone, D., Cobellis, G., Nusco, E., et al. (2007). Systemic inflammation and neurodegeneration in a mouse model of multiple sulfatase deficiency. Proc. Natl. Acad. Sci. U.S.A. 104, 4506–4511. doi: 10.1073/pnas.0700382104
Settembre, C., Fraldi, A., Jahreiss, L., Spampanato, C., Venturi, C., Medina, D., et al. (2008). A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 17, 119–129. doi: 10.1093/hmg/ddm289
Seyrantepe, V., Demir, S. A., Timur, Z. K., Von Gerichten, J., Marsching, C., Erdemli, E., et al. (2018). Murine sialidase Neu3 facilitates GM2 degradation and bypass in mouse model of tay-sachs disease. Exp. Neurol. 299, 26–41. doi: 10.1016/j.expneurol.2017.09.012
Shen, J. S., Watabe, K., Ohashi, T., and Eto, Y. (2001). Intraventricular administration of recombinant adenovirus to neonatal twitcher mouse leads to clinicopathological improvements. Gene Ther. 8, 1081–1087. doi: 10.1038/sj.gt.3301495
Shyng, C., Nelvagal, H. R., Dearborn, J. T., Tyynela, J., Schmidt, R. E., Sands, M. S., et al. (2017). Synergistic effects of treating the spinal cord and brain in CLN1 disease. Proc. Natl. Acad. Sci. U.S.A. 114, E5920–E5929. doi: 10.1073/pnas.1701832114
Simpson, J. L., Nien, C. J., Flynn, K. J., and Jester, J. V. (2011). Evaluation of topical cysteamine therapy in the CTNS(-/-) knockout mouse using in vivo confocal microscopy. Mol. Vis. 17, 2649–2654.
Sleat, D. E., El-Banna, M., Sohar, I., Kim, K. H., Dobrenis, K., Walkley, S. U., et al. (2008). Residual levels of tripeptidyl-peptidase I activity dramatically ameliorate disease in late-infantile neuronal ceroid lipofuscinosis. Mol. Genet. Metab. 94, 222–233. doi: 10.1016/j.ymgme.2008.01.014
Sleat, D. E., Wiseman, J. A., El-Banna, M., Kim, K. H., Mao, Q., Price, S., et al. (2004). A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. J. Neurosci. 24, 9117–9126. doi: 10.1523/JNEUROSCI.2729-04.2004
Soga, M., Ishitsuka, Y., Hamasaki, M., Yoneda, K., Furuya, H., Matsuo, M., et al. (2015). HPGCD outperforms HPBCD as a potential treatment for niemann-Pick disease type C during disease modeling with iPS cells. Stem Cells 33, 1075–1088. doi: 10.1002/stem.1917
Sohn, Y. B., Ko, A.-R., Seong, M.-R., Lee, S., Kim, M. R., Cho, S. Y., et al. (2018). The efficacy of intracerebroventricular idursulfase-beta enzyme replacement therapy in mucopolysaccharidosis II murine model: heparan sulfate in cerebrospinal fluid as a clinical biomarker of neuropathology. J. Inherit.Metab. Dis. 41, 1235–1246. doi: 10.1007/s10545-018-0221-0
Sondhi, D., Peterson, D. A., Edelstein, A. M., Del Fierro, K., Hackett, N. R., and Crystal, R. G. (2008). Survival advantage of neonatal CNS gene transfer for late infantile neuronal ceroid lipofuscinosis. Exp. Neurol. 213, 18–27. doi: 10.1016/j.expneurol.2008.04.022
Spampanato, C., Feeney, E., Li, L., Cardone, M., Lim, J.-A., Annunziata, F., et al. (2013). Transcription factor EB (TFEB) is a new therapeutic target for pompe disease. EMBO Mol. Med. 5, 691–706. doi: 10.1002/emmm.201202176
Spampanato, C., Leonibus, E. D., Dama, P., Gargiulo, A., Fraldi, A., Sorrentino, N. C., et al. (2011). Efficacy of a combined intracerebral and systemic gene delivery approach for the treatment of a severe lysosomal storage disorder. Mol. Ther. 19, 860–869. doi: 10.1038/mt.2010.299
Stein, A., Stroobants, S., Gieselmann, V., D’hooge, R., and Matzner, U. (2015). Anti-inflammatory therapy with simvastatin improves neuroinflammation and CNS function in a mouse model of metachromatic leukodystrophy. Mol. Ther. 23, 1160–1168. doi: 10.1038/mt.2015.69
Stilhano, R. S., Martin, P. K. M., De Melo, S. M., Samoto, V. Y., Peres, G. B., and Da Silva, et al. (2015). α- L-iduronidase gene-based therapy using the phiC31 system to treat mucopolysaccharidose type I mice. J. Gene Med. 17, 1–13. doi: 10.1002/jgm.2818
Stinchi, S., Lullmann-Rauch, R., Hartmann, D., Coenen, R., Beccari, T., Orlacchio, A., et al. (1999). Targeted disruption of the lysosomal alpha-mannosidase gene results in mice resembling a mild form of human alpha-mannosidosis. Hum. Mol. Genet. 8, 1365–1372. doi: 10.1093/hmg/8.8.1365
Strazza, M., Luddi, A., Carbone, M., Rafi, M. A., Costantino-Ceccarini, E., and Wenger, D. A. (2009). Significant correction of pathology in brains of twitcher mice following injection of genetically modified mouse neural progenitor cells. Mol. Genet. Metab. 97, 27–34. doi: 10.1016/j.ymgme.2009.01.005
Stroobants, S., Acker, N. G. G. V., Verheijen, F. W., Goris, I., Daneels, G. F. T., Schot, R., et al. (2017). Progressive leukoencephalopathy impairs neurobehavioral development in sialin-deficient mice. Exp. Neurol. 291, 106–119. doi: 10.1016/j.expneurol.2017.02.009
Surace, E. M., Spampanato, C., Settembre, C., and Ballabio, A. (2007). Systemic administration of AAV9-SUMF1 gene results in re-activation of sulfatases in a mouse model of multiple sulfatase deficiency (MSD). Mol. Ther. 15:S37. doi: 10.1016/S1525-0016(16)44300-5
Suzuki, K. (2004). Krabbe Disease: Myelin Biology and Disorders. Amsterdam: Elsevier. doi: 10.1016/B978-012439510-7/50088-7
Syres, K., Harrison, F., Tadlock, M., Jester, J. V., Simpson, J., Roy, S., et al. (2009). Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood 114, 2542–2552. doi: 10.1182/blood-2009-03-213934
Takamura, A., Higaki, K., Ninomiya, H., Takai, T., Matsuda, J., Iida, M., et al. (2011). Lysosomal accumulation of Trk protein in brain of GM(1) -gangliosidosis mouse and its restoration by chemical chaperone. J. Neurochem. 118, 399–406. doi: 10.1111/j.1471-4159.2011.07310.x
Tanaka, Y., Guhde, G., Suter, A., Eskelinen, E. L., Hartmann, D., Lüllmann-Rauch, R., et al. (2000). Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406, 902–906. doi: 10.1038/35022595
Tanaka, Y., Ishitsuka, Y., Yamada, Y., Kondo, Y., Takeo, T., Nakagata, N., et al. (2014). Influence of Npc1 genotype on the toxicity of hydroxypropyl-β-cyclodextrin, a potentially therapeutic agent, in Niemann-Pick Type C disease models. Mol. Genet. Metab. Rep. 1, 19–30. doi: 10.1016/j.ymgmr.2013.12.003
Tarczyluk-Wells, M. A., Salzlechner, C., Najafi, A. R., Lim, M. J., Smith, D., Platt, F. M., et al. (2019). Combined anti-inflammatory and neuroprotective treatments have the potential to impact disease phenotypes in Cln3-/- Mice. Front. Neurol. 10:963. doi: 10.3389/fneur.2019.00963
Taylor, A. M., Liu, B., Mari, Y., Liu, B., and Repa, J. J. (2012). Cyclodextrin mediates rapid changes in lipid balance in Npc1-/- mice without carrying cholesterol through the bloodstream. J. Lipid Res. 53, 2331–2342. doi: 10.1194/jlr.M028241
Taylor, R., and Snyder, E. (1997). “Widespread engraftment of neural progenitor and stem-like cells throughout the(mouse)brain,” in Transplantation Proceedings, ed. B. D. Kahan (Amsterdam: Elsevier), 845–847. doi: 10.1016/S0041-1345(96)00163-7
Techiryan, G., Weil, B. R., Palka, B. A., and Jr, J. M. C. (2018). Effect of intracoronary metformin on myocardial infarct size in swine. Circ. Res. 123, 986–995. doi: 10.1161/CIRCRESAHA.118.313341
Tominaga, K., Matsuda, J., Kido, M., Naito, E., Yokota, I., Toida, K., et al. (2004). Genetic background markedly influences vulnerability of the hippocampal neuronal organization in the “twitcher” mouse model of globoid cell leukodystrophy. J. Neurosci. Res. 77, 507–516. doi: 10.1002/jnr.20190
Tong, W., Dwyer, C. A., Thacker, B. E., Glass, C. A., Brown, J. R., Hamill, K., et al. (2017). Guanidinylated neomycin conjugation enhances intranasal enzyme replacement in the brain. Mol. Ther. 25, 2743–2752. doi: 10.1016/j.ymthe.2017.08.007
Tordo, J., O’leary, C., Antunes, A., Palomar, N., Aldrin-Kirk, P., Basche, M., et al. (2018). A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain 141, 2014–2031. doi: 10.1093/brain/awy126
Tybulewicz, V. L., Tremblay, M. L., Lamarca, M. E., Willemsen, R., Stubblefield, B. K., Winfield, S., et al. (1992). Animal model of Gaucher’s disease from targeted disruption of the mouse glucocerebrosidase gene. Nature 357, 407–410. doi: 10.1038/357407a0
van Til, N. P., Stok, M., Aerts Kaya, F. S., De Waard, M. C., Farahbakhshian, E., Visser, T. P., et al. (2010). Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype. Blood 115, 5329–5337. doi: 10.1182/blood-2009-11-252874
Vanier, M. T. (2013). Niemann–Pick diseases. Handb. Clin. Neurol. 113, 1717–1721. doi: 10.1016/B978-0-444-59565-2.00041-1
Venugopal, B., Browning, M. F., Curcio-Morelli, C., Varro, A., Michaud, N., Nanthakumar, N., et al. (2007). Neurologic, gastric, and opthalmologic pathologies in a murine model of mucolipidosis type IV. Am. J. Hum. Genet. 81, 1070–1083. doi: 10.1086/521954
Veyron, P., Mutin, M., and Touraine, J. L. (1996). Transplantation of fetal liver cells corrects accumulation of lipids in tissues and prevents fatal neuropathy in cholesterol-storage disease BALB/c mice. Transplantation 62, 1039–1045. doi: 10.1097/00007890-199610270-00001
Vogel, P., Payne, B. J., Read, R., Lee, W.-S., Gelfman, C. M., and Kornfeld, S. (2009). Comparative pathology of murine mucolipidosis types II and IIIC. Vet. Pathol. 46, 313–324. doi: 10.1354/vp.46-2-313
Wada, R., Tifft, C. J., and Proia, R. L. (2000). Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. U.S.A. 97, 10954–10959. doi: 10.1073/pnas.97.20.10954
Wagner, V. F., and Northrup, H. (2019). “Mucopolysaccharidosis type III,” in SourceGeneReviews§[Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Walker, M. T., and Montell, C. (2016). Suppression of the motor deficit in a mucolipidosis type IV mouse model by bone marrow transplantation. Hum. Mol. Genet. 25, 2752–2761. doi: 10.1093/hmg/ddw132
Walkley, S. U. (2009). Pathogenic cascades in lysosomal disease – Why so complex? J. Inherit. Metab. Dis. 32, 181–189. doi: 10.1007/s10545-008-1040-5
Wang, D., Belakhov, V., Kandasamy, J., Baasov, T., Li, S.-C., Li, Y.-T., et al. (2012). The designer aminoglycoside NB84 significantly reduces glycosaminoglycan accumulation associated with MPS I-H in the Idua-W392X mouse. Mol. Genet. Metab. 105, 116–125. doi: 10.1016/j.ymgme.2011.10.005
Wang, D., Li, J., Song, C.-Q., Tran, K., Mou, H., Wu, P.-H., et al. (2018). Cas9-mediated allelic exchange repairs compound heterozygous recessive mutations in mice. Nat. Biotechnol. 36, 839–842. doi: 10.1038/nbt.4219
Wang, D., Zhang, W., Kalfa, T. A., Grabowski, G., Davies, S., Malik, P., et al. (2009). Reprogramming erythroid cells for lysosomal enzyme production leads to visceral and CNS cross-correction in mice with Hurler syndrome. Proc. Natl. Acad. Sci. U.S.A. 106, 19958–19963. doi: 10.1073/pnas.0908528106
Watson, G., Bastacky, J., Belichenko, P., Buddhikot, M., Jungles, S., Vellard, M., et al. (2006). Intrathecal administration of AAV vectors for the treatment of lysosomal storage in the brains of MPS I mice. Gene Ther. 13, 917–925. doi: 10.1038/sj.gt.3302735
Watson, H. A., Holley, R. J., Langford-Smith, K. J., Wilkinson, F. L., Van Kuppevelt, T. H., Wynn, R. F., et al. (2014). Heparan sulfate inhibits hematopoietic stem and progenitor cell migration and engraftment in mucopolysaccharidosis I. J. Biol. Chem. 289, 36194–36203. doi: 10.1074/jbc.M114.599944
Wei, H., Zhang, Z., Saha, A., Peng, S., Chandra, G., Quezado, Z., et al. (2011). Disruption of adaptive energy metabolism and elevated ribosomal p-S6K1 levels contribute to INCL pathogenesis: partial rescue by resveratrol. Hum. Mol. Genet. 20, 1111–1121. doi: 10.1093/hmg/ddq555
Winner, L. K., Beard, H., Hassiotis, S., Lau, A. A., Luck, A. J., Hopwood, J. J., et al. (2016). A preclinical study evaluating AAVrh10-based gene therapy for sanfilippo syndrome. Hum. Gene Ther. 27, 363–375. doi: 10.1089/hum.2015.170
Wolf, D. A., Hanson, L. R., Aronovich, E. L., Nan, Z., Low, W. C., Frey, W. H., et al. (2012). Lysosomal enzyme can bypass the blood-brain barrier and reach the CNS following intranasal administration. Mol. Genet. Metab. 106, 131–134. doi: 10.1016/j.ymgme.2012.02.006
Wright, M. D., Poe, M. D., Derenzo, A., Haldal, S., and Escolar, M. L. (2017). Developmental outcomes of cord blood transplantation for Krabbe disease – A 15-year study. Neurology 89, 1365–1372. doi: 10.1212/WNL.0000000000004418
Wu, Y. P., and Proia, R. L. (2004). Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in Sandhoff disease mice. Proc. Natl. Acad. Sci. U.S.A. 101, 8425–8430. doi: 10.1073/pnas.0400625101
Xie, C., Gong, X. M., Luo, J., Li, B. L., and Song, B. L. (2017). AAV9-NPC1 significantly ameliorates Purkinje cell death and behavioral abnormalities in mouse NPC disease. J. Lipid Res. 58, 512–518. doi: 10.1194/jlr.M071274
Xie, X., Brown, M. S., Shelton, J. M., Richardson, J. A., Goldstein, J. L., and Liang, G. (2011). Amino acid substitution in NPC1 that abolishes cholesterol binding reproduces phenotype of complete NPC1 deficiency in mice. Proc. Natl. Acad. Sci. U.S.A. 108, 15330–15335. doi: 10.1073/pnas.1112751108
Xu, F., Ding, E., Liao, S. X., Migone, F., Dai, J., Schneider, A., et al. (2004). Improved efficacy of gene therapy approaches for Pompe disease using a new, immune-deficient GSD-II mouse model. Gene Ther. 11, 1590–1598. doi: 10.1038/sj.gt.3302314
Xu, M., Motabar, O., Ferrer, M., Marugan, J. J., Zheng, W., and Ottinger, E. A. (2016). Disease models for the development of therapies for lysosomal storage diseases. Ann. N. Y. Acad. Sci. 1371, 15–29. doi: 10.1111/nyas.13052
Xu, Y. H., Quinn, B., Witte, D., and Grabowski, G. A. (2003). Viable mouse models of acid beta-glucosidase deficiency: the defect in Gaucher disease. Am. J. Pathol. 163, 2093–2101. doi: 10.1016/S0002-9440(10)63566-3
Yagi, T., Matsuda, J., Tominaga, K., Suzuki, K., and Suzuki, K. (2005). Hematopoietic cell transplantation ameliorates clinical phenotype and progression of the CNS pathology in the mouse model of late onset Krabbe disease. J. Neuropathol. Exp. Neurol. 64, 565–575. doi: 10.1097/01.jnen.0000171646.01966.0c
Yagi, T., Mcmahon, E. J., Takikita, S., Mohri, I., Matsushima, G. K., and Suzuki, K. (2004). Fate of donor hematopoietic cells in demyelinating mutant mouse, twitcher, following transplantation of GFP+ bone marrow cells. Neurobiol. Dis. 16, 98–109. doi: 10.1016/j.nbd.2004.01.002
Yamanaka, S., Johnson, M. D., Grinberg, A., Westphal, H., Crawley, J. N., Taniike, M., et al. (1994). Targeted disruption of the Hexa gene results in mice with biochemical and pathologic features of Tay-Sachs disease. Proc. Natl. Acad. Sci. U.S.A. 91, 9975–9979. doi: 10.1073/pnas.91.21.9975
Yeager, A. M., Brennan, S., Tiffany, C., Moser, H. W., and Santos, G. W. (1984). Prolonged survival and remyelination after hematopoietic cell transplantation in the twitcher mouse. Science 225, 1052–1054. doi: 10.1126/science.6382609
Yeager, A. M., Shinohara, M., and Shinn, C. (1991). Hematopoietic cell transplantation after administration of high-dose busulfan in murine globoid cell leukodystrophy (the twitcher mouse). Pediatr. Res. 29, 302–305. doi: 10.1203/00006450-199103000-00016
Zhang, X., Cheng, X., Yu, L., Yang, J., Calvo, R., Patnaik, S., et al. (2016). MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 7:12109. doi: 10.1038/ncomms12109
Zheng, Y., Rozengurt, N., Ryazantsev, S., Kohn, D. B., Satake, N., and Neufeld, E. F. (2003). Treatment of the mouse model of mucopolysaccharidosis I with retrovirally transduced bone marrow. Mol. Genet. Metab. 79, 233–244. doi: 10.1016/S1096-7192(03)00116-1
Zhu, Y., Li, X., Mcvie-Wylie, A., Jiang, C., Thurberg, B. L., Raben, N., et al. (2005). Carbohydrate-remodelled acid alpha-glucosidase with higher affinity for the cation-independent mannose 6-phosphate receptor demonstrates improved delivery to muscles of Pompe mice. Biochem. J. 389, 619–628. doi: 10.1042/BJ20050364
Keywords: lysosomal diseases, preclinical mouse models, HSCT, enzyme replacement therapy, gene therapy, chaperone therapy, substrate reduction therapy
Citation: Favret JM, Weinstock NI, Feltri ML and Shin D (2020) Pre-clinical Mouse Models of Neurodegenerative Lysosomal Storage Diseases. Front. Mol. Biosci. 7:57. doi: 10.3389/fmolb.2020.00057
Received: 28 January 2020; Accepted: 20 March 2020;
Published: 15 April 2020.
Edited by:
Angela Gritti, San Raffaele Scientific Institute (IRCCS), ItalyReviewed by:
Alessandro Fraldi, Telethon Institute of Genetics and Medicine, ItalyMaria Irene Givogri, The University of Illinois at Chicago, United States
Copyright © 2020 Favret, Weinstock, Feltri and Shin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daesung Shin, ZGFlc3VuZ3NAYnVmZmFsby5lZHU=
†These authors have contributed equally to this work