
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Mol. Biosci., 05 July 2018
Sec. RNA
Volume 5 - 2018 | https://doi.org/10.3389/fmolb.2018.00062
 Markus Haak1
Markus Haak1 Svenja Vinke1Willy Keller1,2Julian Droste1,2
Svenja Vinke1Willy Keller1,2Julian Droste1,2 Christian Rückert1,2,3
Christian Rückert1,2,3 Jörn Kalinowski1,2
Jörn Kalinowski1,2 Boas Pucker1,2,4*
Boas Pucker1,2,4*Croton tiglium is one of more than 1,200 different species in the large genus Croton, belonging to the family Euphorbiaceae (Kalwij, 2012; The Plant List, 2014). C. tiglium can be found in subtropical and tropical regions on both hemispheres (Salatino et al., 2007). This plant was first mentioned in the medical literature over 2,200 years ago in China. The medical relevance is probably due to a huge variety of different secondary metabolites (Pope, 1824). Traditionally utilized as a purgative to treat gastrointestinal and intestinal disorders, as an abortifacient and counterirritant, the commercially available seed oil of C. tiglium is nowadays applied in homeopathy and acupuncture (Gläser et al., 1988). The pharmacologic mechanism of the laxative properties of ethanol extracts of C. tiglium has been studied on rat intestinal epithelium (Tsai et al., 2004). C. tiglium produces various phorbol esters, including substances that were reported to be tumor-promoting (Van Duuren et al., 1963), antileukemic and antimycobacterial (Goel et al., 2007; Salatino et al., 2007), and even candidates for the treatment of HIV (El-Mekkawy et al., 2000). Beside the tumor-promoting factors, some cytotoxic phorbol esters were isolated from plant extracts and evaluated in cell culture assays (Zhang et al., 2013). In contrast to the co-carcinogenic substances, C. tiglium was shown to produce a ribonucleoside analog of guanosine with antitumor activity (Kim et al., 1994).
In this work, we present a comprehensive de novo transcriptome assembly of C. tiglium based on a normalized library to cover a huge variety of transcripts. In addition, tissue-specific transcript libraries were generated to enable differential gene expression analysis between tissues. This will facilitate the identification of candidate genes involved in growth, development, and metabolism of this plant species.
Tissue samples from Croton tiglium L. were kindly provided by the botanic garden of the Phillipps-Universität Marburg (https://www.uni-marburg.de/botgart). The plants were cultivated in individual buckets in a greenhouse at 18–20°C, a relative humidity of 60–80% and daylight. The substrate consisted of potting soil mixed with sand, lava and clay, fertilized every 2 weeks with WUXAL Top N and WUXAL Super liquid fertilizer. The collected samples were frozen in liquid nitrogen immediately and kept on dry ice until RNA extraction.
Total RNA was extracted from frozen tissue samples of young leaves, stem, inflorescence, seeds, and roots. Mortar and pestle were used to grind the material in liquid nitrogen. The Spectrum Plant Total RNA Kit (Sigma Aldrich) was used for RNA extraction according to the supplier's instructions. A DropSense16 (Trinean) was used for quantification and quality control. RNA samples with insufficient purity were purified with the RNA Clean & Concentrator Kit (Zymo Research) prior to library preparation.
A pooled total RNA sample from all five tissues was used for the construction of a normalized, stranded library (vertis Biotechnologie AG). In parallel, tissue-specific libraries with an average insert size of 400 bp were prepared according to the Illumina TrueSeq Stranded mRNA Sample Preparation Guide. These libraries represent young leaves, stem, inflorescence, seed, and root.
Sequencing of the normalized library was performed on two lanes of an Illumina HiSeq1500 in Rapid Run mode generating about 47.4 million 2 × 250 nt paired-end reads. Sequencing of the tissue-specific libraries was performed on the same machine generating between 20 and 44 million 2 × 75 nt paired-end reads per tissue-specific library (Supplementary Table 1).
FastQC (Andrews, 2010) was applied to check the quality of all sequencing data. Low quality regions and adapter fragments were removed from the reads via Trimmomatic 0.36 (Bolger et al., 2014). Removal of adapters was performed based on all known Illumina adapter sequences with the options 2:30:10. A sliding window of the length 4 was used to clip reads once the average PHRED score dropped below 15. Reads below the length cutoff of 100 nt were discarded. Pairs with only one surviving read were dropped after trimming.
Trinity v2.4.0 (Grabherr et al., 2011; Haas et al., 2013) was applied with different k-mer sizes and the stranded flag for the de novo transcriptome assembly based on all 2 × 250 nt paired-end reads of the normalized library (SRR6239853). Finally, 24 was identified as the best k-mer size based on comparison of the results by assembly size, number of contigs, assembly continuity, and recovered benchmarking genes. The minimal length of contigs to report was set to 200 nt.
Assembly completeness was investigated by computing assemblies for subsets of the data. Basic statistics like assembly size, number of contigs, and N50 were compared via customized python scripts. Benchmarking Universal Single-Copy Orthologs (BUSCO) v2.0 (Simão et al., 2015) was run on all subset assemblies in transcriptome mode to quantify the completeness. A dedicated python script was deployed to identify remaining adapter sequences in contigs via BLASTn (Altschul et al., 1990) (e-value < 0.01 and word_size = 4) and to clip them afterwards. Assembled sequences were kept if the surviving part still exceeds the minimal length cutoff. A dedicated python script was deployed to distinguish true plant contigs from bacterial and other contaminations. First, BLASTn (Altschul et al., 1990) with stringent parameters (e-value < 0.00001, alignment length >100, similarity >80%) against the Jatropha curcas GZQX0401 genome sequence (GCA_000696525.1; Zhang et al., 2014) was applied. Second, all contigs without a significant hit were subjected to a subsequent BLASTn (Altschul et al., 1990) against nt. All sequences with best hits against bacterial genomes were removed, while sequences without hits were kept in this step. Finally, sequences shorter than 400 nt were removed from the final assembly. Since the assembly is based on 2 × 250 nt PE reads, smaller contigs are probably assembly artifacts.
Peptide-encoding contigs were identified by a python script, based on results from Transdecoder (Haas et al., 2013), ORFfinder (Wheeler et al., 2003), and ORFpredictor (Min et al., 2005). Local alignments for all predicted peptide sequences against Swiss-Prot (Bairoch and Apweiler, 2000; The UniProt Consortium, 2017) were computed via DIAMOND (Buchfink et al., 2014). The longest continuous peptide sequence per contig starting with Methionine was selected unless similarity to a Swiss-Prot sequence pointed toward another predicted peptide sequence on the same contig. In case of multiple predicted sequences with similarity to Swiss-Prot sequences, the selection was based on the alignment score. Finally, only predicted sequences longer than 100 amino acids or with a significant sequence similarity to a Swiss-Prot entry (e-value < e-10) were kept.
Reciprocal best BLAST hits (RBHs) against Arabidopsis thaliana Araport11 representative peptides (Cheng et al., 2017) were identified as previously described (Pucker et al., 2016) to transfer the functional annotation. In addition, RBHs against Beta vulgaris BeetSet-2 (Minoche et al., 2015), J. curcas JatCur_1.0 (Zhang et al., 2014), and Vitis vinifera PN40024 with the annotation V2.1 provided by CRIBI (Vitulo et al., 2014) were identified to cover a broad range of phylogenetically diverse plant species. All predicted peptide sequences were screened by InterProScan5 (Finn et al., 2017) to assign GO terms.
Reads from tissue-specific data sets were mapped to the final transcriptome assembly via STAR (Dobin et al., 2013) requiring at least 90% of the read sequence to match with at least 95% identity. featureCounts (Liao et al., 2014) was applied to quantify the abundance of all sequences in the assembly. Since most transcripts were represented by multiple contigs probably due to different splice variants and alleles, we decided to include multi-mapped reads. VENN diagram generation was performed at http://bioinformatics.psb.ugent.be/webtools/Venn/ to illustrate tissue-specific transcript abundance.
In total, 45.5 million 2 × 250 nt paired-end reads were assembled into the 391.5 Mbp transcriptome comprising 379,585 contigs. Through all filter steps the size was reduced to 345.7 Mbp comprising only 224,425 contigs. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GGDV00000000. The version described in this paper is the first version, GGDV01000000. The high continuity of the assembled contigs can be described by the E90N50 of 3,115 nt and the E90N90 of 1,645 nt. These statistics are Nx values of an assembly subset, which accounts for 90% of the expression. The completeness check indicated a sufficient amount of sequencing data was generated. An assembly with only 20% of the sequencing data almost reached the final assembly in terms of size and contained genes. The selected k-mer size of 24 for the final assembly resulted in the best assembly based on size, continuity and recovered benchmarking genes (Figure 1). Benchmarking Universal Single-Copy Orthologs (BUSCO) revealed the presence of 95.1% complete BUSCO genes in the initial assembly. In addition, 2.6% of all BUSCO genes are present in fragmented form and only 2.3% are missing in this de novo transcriptome assembly.
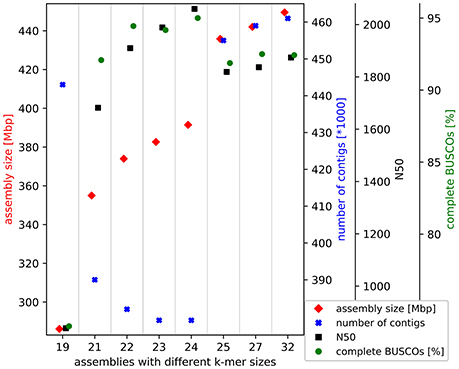
Figure 1. Croton tiglium de novo transcriptome assembly benchmarking. Different k-mer sizes were tested for this de novo transcriptome assembly via Trinity (Grabherr et al., 2011). Results are evaluated based on assembly size, continuity and recovered benchmarking genes (BUSCOs).
A set of 122,206 representative peptide sequences (Supplementary File 1) was inferred by selecting only the best predicted peptide sequence per contig (see methods for details). The position of the corresponding protein coding sequence was identified (Supplementary File 2). Comparison of these predicted peptide sequence to peptide sequence sets of well annotated plant species like A. thaliana resulted in 8,858 RBHs. These hits enabled the transfer of functional annotation information in addition to 113,097 assigned GOs (Supplementary Table 2). Comparison to additional plant genomes revealed 8,623 RBHs against B. vulgaris, 10,822 against J. curcas, and 9,687 against Vitis vinifera.
Analysis of the tissue-specific abundance of the corresponding transcripts (Supplementary Table 3) revealed a high number of shared transcripts (Figure 2). The number of tissue-specific transcripts ranges from 1,049 (stem) to 28,330 (root).
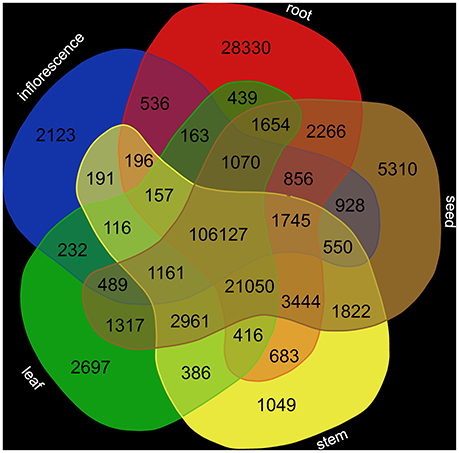
Figure 2. Transcript abundance in different tissues. Transcript abundance was quantified based on RNA-Seq read mapping via STAR (Dobin et al., 2013) and following summarization via featureCounts (Liao et al., 2014).
The transcriptome sequences of C. tiglium were assembled de novo and tissue-specific abundance of transcripts was quantified. High quality of this de novo transcriptome assembly is indicated by the high percentage of completely recovered BUSCO genes, since other recent de novo transcriptome assemblies of plants reached slightly lower values of up to 93% (Babineau et al., 2017; Blande et al., 2017). Sufficient sequencing depth is indicated by the number of detected complete BUSCO genes in subset assemblies. Probably, most genes would be represented in a transcriptome assembly, based on only 20% of the provided data set. A high number of assembled alternative transcripts per gene could explain some of the reported duplicated BUSCOs. A strong heterozygosity of C. tiglium would be another explanation, because both alleles could be represented. Not all assembled transcript sequences were identified in samples of single tissues. One explanation is the nature of the normalized library used for the generation of the de novo assembly leading to an enrichment of rare transcripts to enable a comprehensive representation of the transcriptome. In contrast, tissue-specific data sets are dominated by abundant transcripts and thus do not recover all transcript in this assembly.
This de novo transcriptome assembly can serve as a reference for the investigation of over 1,200 species in the large genus Croton. The availability of comprehensive sequence information for C. tiglium is the first step toward the development of medical applications. Enzymes for the synthesis of various secondary metabolites described in this species may be identified from the provided set of predicted peptides, assembled transcripts, or even based on the raw sequencing reads.
Efforts were already made toward the identification of genes putatively involved in the biosynthesis of the guanosine analog isoguanosine. The transcriptome resource of C. tiglium revealed an additional isoform of the GMPS gene involved in GMP synthesis (Supplementary File 3). Heterologous expression and in vitro enzyme assays of the purified enzymes followed by HPLC/MS analysis revealed substantial differences in the reaction products of the two isoforms that differ from a GMP standard, supporting the postulated involvement of this additional isoform in the isoguanosine biosynthesis (Karsten et al., 2017).
All raw sequencing reads were submitted to the SRA https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?study=SRP123217. The de novo assembled transcript sequences are available at DDBJ/EMBL/GenBank through the BioProject https://www.ncbi.nlm.nih.gov/bioproject/416498.
MH, SV, and WK harvested the samples and extracted the RNA. BP performed bioinformatic analysis of the sequencing data. MH, JD, JK, CR, and BP wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Anika Winkler and Katharina Hanuschka for library preparation and sequencing as well as the entire iGEM team Bielefeld-CeBiTec 2017 and the Chair of Genome Research for excellent support. Furthermore, we would like to thank the botanical garden of the Phillipps-Universität Marburg for providing tissue samples from Croton tiglium L. We acknowledge support for the Article Processing Charge by the Deutsche Forschungsgemeinschaft and the Open Access Publication Fund of Bielefeld University.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmolb.2018.00062/full#supplementary-material
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andrews, S. (2010). FastQC A Quality Control tool for High Throughput Sequence Data. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (Accessed December 14, 2017).
Babineau, M., Mahmood, K., Mathiassen, S. K., Kudsk, P., and Kristensen, M. (2017). De novo transcriptome assembly analysis of weed Apera spica-venti from seven tissues and growth stages. BMC Genomics 18:128. doi: 10.1186/s12864-017-3538-4
Bairoch, A., and Apweiler, R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. doi: 10.1093/nar/28.1.45
Blande, D., Halimaa, P., Tervahauta, A. I., Aarts, M. G., and Kärenlampi, S. O. (2017). De novo transcriptome assemblies of four accessions of the metal hyperaccumulator plant Noccaea caerulescens. Sci. Data 4:160131. doi: 10.1038/sdata.2016.131
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Cheng, C. Y., Krishnakumar, V., Chan, A. P., Thibaud-Nissen, F., Schobel, S., and Town, C. D. (2017). Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 89, 789–804. doi: 10.1111/tpj.13415
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
El-Mekkawy, S., Meselhy, M. R., Nakamura, N., Hattori, M., Kawahata, T., and Otake, T. (2000). Anti-HIV-1 phorbol esters from the seeds of Croton tiglium. Phytochemistry 53, 457–464. doi: 10.1016/S0031-9422(99)00556-7
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199. doi: 10.1093/nar/gkw1107
Gläser, S., Sorg, B., and Hecker, E. (1988). A method for quantitative determination of polyfunctional diterpene esters of the tigliane type in Croton tiglium. Planta Med. 54:580. doi: 10.1055/s-2006-962595
Goel, G., Makkar, H. P. S., Francis, G., and Becker, K. (2007). Phorbol esters: structure, biological activity, and toxicity in animals. Int. J. Toxicol. 26, 279–288. doi: 10.1080/10915810701464641
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Kalwij, J. M. (2012). Review of ‘The Plant List, a working list of all plant species’. J. Veg. Sci. 23, 998–1002. doi: 10.1111/j.1654-1103.2012.01407.x
Karsten, L., Bergen, D., Drake, C., Dymek, S., Edich, M., Haak, M., et al. (2017). Expanding the Genetic Code. Available online at: https://www.researchgate.net/publication/321586905_Expanding_The_Genetic_Code (Accessed June 25, 2018).
Kim, J. H., Lee, S. J., Han, Y. B., Moon, J. J., and Kim, J. B. (1994). Isolation of isoguanosine from Croton tiglium and its antitumor activity. Arch. Pharm. Res. 17, 115–118. doi: 10.1007/BF02974234
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Min, X. J., Butler, G., Storms, R., and Tsang, A. (2005). OrfPredictor: predicting protein-coding regions in EST-derived sequences. Nucleic Acids Res. 33, W677–W680. doi: 10.1093/nar/gki394
Minoche, A. E., Dohm, J. C., Schneider, J., Holtgräwe, D., Viehöver, P., Montfort, M., et al. (2015). Exploiting single-molecule transcript sequencing for eukaryotic gene prediction. Genome Biol. 16:184. doi: 10.1186/s13059-015-0729-7
Pucker, B., Holtgräwe, D., Rosleff Sörensen, T., Stracke, R., Viehöver, P., and Weisshaar, B. (2016). A de novo genome sequence assembly of the Arabidopsis thaliana accession niederzenz-1 displays presence/absence variation and strong synteny. PLoS ONE 11:e0164321. doi: 10.1371/journal.pone.0164321
Salatino, A., Salatino, M. L. F., and Negri, G. (2007). Traditional uses, chemistry and pharmacology of Croton species (Euphorbiaceae). J. Braz. Chem. Soc. 18, 11–33. doi: 10.1590/S0103-50532007000100002
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
The Plant List (2014). The Plant List. Available online at: http://www.theplantlist.org/ (Accessed December 15, 2017).
The UniProt Consortium (2017). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169. doi: 10.1093/nar/gkh131
Tsai, J. C., Tsai, S., and Chang, W. C. (2004). Effect of ethanol extracts of three Chinese medicinal plants with laxative properties on ion transport of the rat intestinal epithelia. Biol. Pharm. Bull. 27, 162–165. doi: 10.1248/bpb.27.162
Van Duuren, B. L., Orris, L., and Arroyo, E. (1963). Tumour-enhancing activity of the active principles of Croton tiglium L. Nature 200, 1115–1116. doi: 10.1038/2001115a0
Vitulo, N., Forcato, C., Carpinelli, E., Telatin, A., Campagna, D., D'Angelo, M., et al. (2014). A deep survey of alternative splicing in grape reveals changes in the splicing machinery related to tissue, stress condition and genotype. BMC Plant Biol. 14:99. doi: 10.1186/1471-2229-14-99
Wheeler, D. L., Church, D. M., Federhen, S., Lash, A. E., Madden, T. L., Pontius, J. U., et al. (2003). Database resources of the national center for biotechnology. Nucleic Acids Res. 31, 28–33. doi: 10.1093/nar/gkg033
Zhang, L., Zhang, C., Wu, P., Chen, Y., Li, M., Jiang, H., et al. (2014). Global analysis of gene expression profiles in physic nut (Jatropha curcas L.) seedlings exposed to salt stress. PLoS ONE 9:e97878. doi: 10.1371/journal.pone.0097878
Keywords: Croton tiglium, RNA-Seq, Trinity, reciprocal best BLAST hit, annotation transfer, gene expression, tissue-specific, anti-cancer
Citation: Haak M, Vinke S, Keller W, Droste J, Rückert C, Kalinowski J and Pucker B (2018) High Quality de Novo Transcriptome Assembly of Croton tiglium. Front. Mol. Biosci. 5:62. doi: 10.3389/fmolb.2018.00062
Received: 03 January 2018; Accepted: 18 June 2018;
Published: 05 July 2018.
Edited by:
Stefano Volinia, University of Ferrara, ItalyReviewed by:
David Arnosti, Michigan State University, United StatesCopyright © 2018 Haak, Vinke, Keller, Droste, Rückert, Kalinowski and Pucker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Boas Pucker, YnB1Y2tlckBjZWJpdGVjLnVuaS1iaWVsZWZlbGQuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.