 Maia Lanfranco
Maia Lanfranco Neville Vassallo
Neville Vassallo Ruben J. Cauchi
Ruben J. Cauchi- 1Department of Physiology and Biochemistry, Faculty of Medicine and Surgery, University of Malta, Msida, Malta
- 2Center for Molecular Medicine and Biobanking, University of Malta, Msida, Malta
- 3Institut de Génétique Moléculaire de Montpellier, Center National de la Recherche Scientifique-UMR 5535, Université de Montpellier, Montpellier, France
Spinal Muscular Atrophy (SMA) is a neuromuscular disorder that results from decreased levels of the survival motor neuron (SMN) protein. SMN is part of a multiprotein complex that also includes Gemins 2–8 and Unrip. The SMN-Gemins complex cooperates with the protein arginine methyltransferase 5 (PRMT5) complex, whose constituents include WD45, PRMT5 and pICln. Both complexes function as molecular chaperones, interacting with and assisting in the assembly of an Sm protein core onto small nuclear RNAs (snRNAs) to generate small nuclear ribonucleoproteins (snRNPs), which are the operating components of the spliceosome. Molecular and structural studies have refined our knowledge of the key events taking place within the crowded environment of cells and the numerous precautions undertaken to ensure the faithful assembly of snRNPs. Nonetheless, it remains unclear whether a loss of chaperoning in snRNP assembly, considered as a “housekeeping” activity, is responsible for the selective neuromuscular phenotype in SMA. This review thus shines light on in vivo studies that point toward disturbances in snRNP assembly and the consequential transcriptome abnormalities as the primary drivers of the progressive neuromuscular degeneration underpinning the disease. Disruption of U1 snRNP or snRNP assembly factors other than SMN induces phenotypes that mirror aspects of SMN deficiency, and splicing defects, described in numerous SMA models, can lead to a DNA damage and stress response that compromises the survival of the motor system. Restoring the correct chaperoning of snRNP assembly is therefore predicted to enhance the benefit of SMA therapeutic modalities based on augmenting SMN expression.
Introduction
Spinal Muscular Atrophy (SMA) is a neuromuscular disorder that can afflict both infants and adults. Patients present with loss of lower motor neurons and profound muscle weakness leading to immobility and, in severe cases, respiratory failure and death (Kolb and Kissel, 2011). The recent availability of an effective therapy is the culmination of more than two decades of research aimed at characterizing the molecular genetics underlying the disease following the discovery that SMA is caused by mutation or homozygous deletion of the survival motor neuron 1 (SMN1), a gene encoding the SMN protein. Due to a quirk in human evolution, SMN is also encoded by the highly homologous SMN2 gene. Nonetheless, a single nucleotide substitution (C/T) in exon 7, converts an exon splicing enhancer to a silencer, hence inducing the omission of exon 7 from most of the SMN2-derived mRNA transcripts. This alteration leads to the production of an unstable truncated protein isoform (SMNΔ7) that is rapidly degraded, although in the absence of complete penetrance, full-length, functional SMN is still encoded by a small portion of SMN2 transcripts that evade exon 7 skipping (reviewed in Burghes and Beattie, 2009). In the context of SMA, the levels of SMN are sufficient to prevent lethality yet not enough to fully compensate for the loss of SMN1. The inverse correlation between SMN2 copy number and SMA severity elevated SMN2 to the leading disease genetic modifier (Wirth et al., 2013). The antisense oligonucleotide (ASO) nusinersen (marketed as Spinraza), recently approved for a broad patient population, is the first successful output of a campaign aimed at identifying therapeutics that promote exon 7 inclusion in SMN2 transcripts. The backdrop is a string of favorable results from animal models up to clinical trials, all showing that nusinersen enhanced SMN protein levels adequately enough to improve disease phenotypes (Faravelli et al., 2015; Farrar et al., 2016).
Interestingly, ASOs and other therapeutic approaches that treat SMA by augmenting SMN expression, including chief amongst others viral-mediated SMN gene delivery, and orally bioavailable small molecules that correct SMN2 splicing, are not the obvious sequel to basic knowledge gained on SMN function. SMN, a ubiquitously expressed protein, is known to partner with Gemins 2–8 and Unrip to form a complex that is indispensable for chaperoning the assembly of small nuclear ribonucleoproteins (snRNPs), core elements of the spliceosome. In this review, we present a refined view of the key snRNP assembly events taking place within the crowded environment of cells, and evaluate the evidence favoring the classification of SMA as a chaperonopathy or a disorder arising from a disturbance in the chaperoning of snRNP assembly with the consequential transcriptome abnormalities as the primary drivers of neuromuscular degeneration in SMA patients. A better understanding of the mechanisms underpinning the disease can open up novel therapeutic routes that complement or accentuate the effect of the mainstream approach.
Anatomy of the SMN-Gemins Complex Chaperone Machine
In its simplest version, the SMN-Gemins complex is composed of only SMN (Yab8p) and Gemin2 (Yip1p), a situation that is typical in the fission yeast Schizosaccharomyces pombe (Hannus et al., 2000). Complexity was gained in evolution through the incorporation of the remaining constituents (Kroiss et al., 2008; Cauchi, 2010). The fruit fly Drosophila melanogaster possesses a minimalistic complex that, in addition to SMN and Gemin2, also includes Gemin3 and Gemin5 (Cauchi et al., 2010). Besides physical associations (Cauchi et al., 2008; Kroiss et al., 2008; Shpargel et al., 2009; Guruharsha et al., 2011), genetic interactions between members of the Drosophila SMN-Gemins complex indicate that SMN and its Gemin associates were conserved during evolution not as independent entities but rather as a genetic network (Borg et al., 2015) (Figure 1A). Vertebrates, including humans, have the most elaborate SMN-Gemins complex counting SMN, seven Gemin proteins (Gemin2-Gemin8), and Unrip as its members. Comprehensive biochemical studies revealed a modular composition with the SMN-Gemin8-Gemin7 module placed at its center, thereby allowing the recruitment of the Gemin2-Gemin5 and Gemin6-Unrip subunits mainly via SMN and Gemin7, respectively. The Gemin3-Gemin4 block latches to the complex via both SMN and Gemin8 (Otter et al., 2007). Additional interactions are thought to further stabilize the complex (Otter et al., 2007; Ogawa et al., 2009) (Figure 1A). SMN, Gemin2, Gemin4, and Gemin8 can self-associate (Lorson et al., 1998; Young et al., 2000; Ogawa et al., 2007; Otter et al., 2007), hence their oligomerization propensity means that SMN-Gemins complexes can reach large macromolecular sizes, at least in vertebrates. Cell biology studies have confirmed the clustering of SMN-Gemins complex members to form membrane-less structures named Gems if nuclear (Liu and Dreyfuss, 1996; Cauchi, 2011) or U bodies if cytoplasmic (Liu and Gall, 2007; Cauchi et al., 2010).
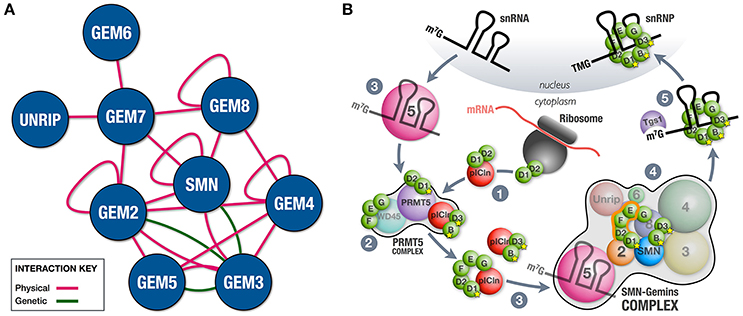
Figure 1. SMN-Gemins complex and its role in chaperoning snRNP assembly. (A) Integration of physical and genetic interactions between the constituent members of the SMN-Gemins complex. Physical interactions were identified through various biochemical techniques including two-hybrid, pulldown and co-immunoprecipitation assays in multiple organisms (Lorson et al., 1998; Young et al., 2000; Ogawa et al., 2007, 2009; Otter et al., 2007; Cauchi et al., 2008; Kroiss et al., 2008; Shpargel et al., 2009; Guruharsha et al., 2011). Changes in the levels of SMN or Gemins enhanced viability and motor phenotypes of a hypomorphic Gemin3 mutant in Drosophila, thereby allowing the identification of genetic interactions (Borg et al., 2015). Interaction network was composed using the esyN tool. (B) Cooperation between the PRMT5 and SMN-Gemins complexes in the cytoplasm ensures the faithful assembly of snRNPs. (1) On translation, Sm D2 protein remains attached to the ribosome. Its release occurs subsequent to the formation of the Sm D2/D1 heterodimer and association with pICln. (2) Select Sm proteins are then post-translationally modified by the PRMT5 complex. (3) The SMN-Gemins complex receives pre-organized Sm subsets and snRNAs from pICln and Gemin5, respectively. (4) Within the SMN-Gemins complex, the majority of Sm proteins are handled by Gemin2 until their uploading onto the delivered snRNAs. (5) Tgs1 ensures cap hypermethylation of assembled snRNPs prior to their nuclear import.
It has long been known that loss of SMN is incompatible with life (reviewed in Burghes and Beattie, 2009). The same outcome applies to additional SMN-Gemins complex members investigated thus far. To this end, knockout or RNAi-mediated knockdown of Gemin2, Gemin3 or Gemin5 leads to lethality in various organisms (reviewed in Borg and Cauchi, 2014). This might indicate that the constituents of the SMN-Gemins complex are not redundant, hence, the function of one component cannot be covered by another. Alternatively, or additionally, an imbalance in the protein levels of its members can destabilize the SMN-Gemins complex. Experiments inducing a gain-of-function were revelatory in this regard. Indeed, overexpression of Gemin2 is deleterious in both yeast and flies (Borg et al., 2015). Furthermore, the upregulation of SMN or Gemin5 can have a negative impact on fly viability only when either perturbation is combined with a Gemin3 hypomorphic mutant (Borg et al., 2015). These findings are in line with studies that underscore the interdependence of constituent levels within the SMN-Gemins complex. Hence, cells with low amounts of SMN, including those derived from SMA patients, were found to have reduced protein levels of select Gemins (Jablonka et al., 2002; Helmken et al., 2003; Feng et al., 2005; Shpargel and Matera, 2005; Carissimi et al., 2006; Gabanella et al., 2007; Hao le et al., 2007). In agreement, severe SMA mice were also shown to have a significant reduction in the levels of a subset of Gemin proteins in the spinal cord (Gabanella et al., 2007; Zhang et al., 2008). A similar effect can be achieved on knockdown of select Gemins (Shpargel and Matera, 2005; Ogawa et al., 2007). Furthermore, the half-life of SMN was decreased by mutations interfering with its incorporation within the SMN-Gemins complex (Burnett et al., 2009).
A Refined View of Chaperoning Activities During snRNP Biogenesis
In addition to being an essential step in gene expression, splicing of pre-mRNA transcripts is also crucial for the generation of diverse proteomes in eukaryotes. United in the major spliceosome, U1, U2, U4/U6, and U5 snRNPs catalyze the removal of the majority of pre-mRNA introns. The less abundant minor spliceosome, which processes a rare non-canonical group of introns is however composed of U11, U12, U4atac/U6atac and U5 snRNPs. Not considering the varying number of specific protein components, spliceosomal snRNPs are in essence composed of a short noncoding RNA (snRNA) bound to a heptameric Sm/Lsm protein ring (reviewed in Matera and Wang, 2014). Cells take numerous precautions to ensure the faithful assembly of snRNPs. Hence, key events of the snRNP production cycle take place in the cytoplasm to limit contact of partially assembled snRNPs with their nuclear substrates. Importantly, the process involves cooperation between the SMN-Gemins complex, and the protein arginine methyltransferase 5 (PRMT5) complex, whose constituents include WD45, PRMT5 and pICln. This brings the number of trans-acting assembly factors to at least twelve, a count far greater than the parts to be assembled (reviewed in Fischer et al., 2011). The mis-assembly evading measures put forward by cells most probably address the low intrinsic selectively of Sm proteins for snRNAs. As we discuss below, recent molecular and structural studies mostly focusing on Sm-class snRNPs have started to unravel the chaperoning activities of each factor during the uploading of the Sm ring onto a conserved short uridine-rich sequence motif, known as the Sm site, within snRNAs.
snRNP assembly is thought to occur during two phases, the early one dominated by the PRMT5 complex, whereas in the late one, the SMN-Gemins complex is central (Figure 1B). In the early assembly phase, the newly translated Sm D2 protein is thought to remain attached to the ribosome. Formation of the Sm D2/D1 dimer and its association with pICln ensures their release and the subsequent delivery to the PRMT5 complex (Paknia et al., 2016). Here, designated arginine residues of a bound Sm protein subset (B/B', D1, and D3) are symmetrically dimethylated by PRMT5 and, possibly PRMT7, a modification thought to enhance their affinity for the SMN-Gemins complex (reviewed in Fischer et al., 2011). The conclusion of this phase is marked by the formation of the Sm D1/D2/F/E/G and Sm B/D3 sub-complexes, each bound by pICln to prevent premature RNA interactions. Whereas pICln is dismissed, the pre-organized Sm proteins are handed over to the SMN-Gemins complex, a step that signals the initiation of the late assembly phase (Chari et al., 2008; Grimm et al., 2013). Gemin2 is the SMN-Gemins complex subunit that handles the majority of Sm proteins by hugging the crescent-shaped Sm D1/D2/F/E/G pentamer and blocking RNA binding capacity until delivery of snRNAs (Zhang et al., 2011; Grimm et al., 2013). The factor that channels snRNAs to the SMN-Gemins complex is Gemin5 (Battle et al., 2006; Yong et al., 2010), though U1-70K, a component of U1 snRNP, can substitute Gemin5 in a U1-exclusive snRNP assembly pathway (So et al., 2016). Gemin5 is capable of recognizing the Sm site and 7-methylguanosine (m7G) cap of nuclear-exported snRNAs via the first and second WD40 repeat domains, respectively (Lau et al., 2009; Jin et al., 2016; Tang et al., 2016; Xu et al., 2016). Although binding to both structures cannot be done simultaneously, this dual recognition tactic is thought to enhance stringency of snRNP assembly. The mechanism ensuring Sm ring closure remains unclear though Unrip and the Gemin6/Gemin7 dimer might have a leading role with the latter thought to act as a temporary substitute for the Sm B/D3 dimer (Ma et al., 2005; Ogawa et al., 2009).
The exact role of other SMN-Gemins complex members in snRNP assembly will probably be unraveled by future mechanistic and structure-based studies. Nevertheless, in vitro studies using purified reconstituted systems have recently shown that Gemins 3, 4 and even Gemin5 were dispensable for the assembly and proofreading of snRNAs (Neuenkirchen et al., 2015). This goes against findings by earlier reports demonstrating that snRNP assembly was disrupted on RNAi-mediated knockdown of Gemin3-8 and Unrip in cell culture (Feng et al., 2005; Grimmler et al., 2005; Shpargel and Matera, 2005; Carissimi et al., 2006). It could very well be argued that this outcome is an indirect effect brought about by the destabilization of the SMN-Gemins complex. In support, changes in the levels of its components are known to disrupt the function of the SMN-Gemins complex in vivo (Borg et al., 2015). However, it is highly likely that all components of the SMN-Gemins complex have key roles, at least in vivo, and their participation in chaperoning snRNP assembly drives the reaction forward in addition to increasing its efficiency. Hence, in an in vivo setting, ATP breakdown and RNA or RNP remodeling during snRNP biogenesis is a job most probably attributed to Gemin3, a well-known DEAD-box RNA helicase (Charroux et al., 1999; Yan et al., 2003). In a final step during the late assembly phase, the SMN-Gemins complex recruits trimethylguanosine synthase 1 (Tgs1), an enzyme that hypermethylates the 7-methylguanosine (m7G) cap of assembled snRNPs to a 2,2,7-trimethylguanosine (TMG) cap (Mouaikel et al., 2002, 2003). Both the TMG cap and the Sm ring act as a localization signal for their import to the nucleus where they are expected to operate subsequent to a maturation stage in the Cajal body (reviewed in Stanek, 2016).
In vivo Studies Linking snRNP Assembly Defects to Neuromuscular Dysfunction
Several key studies making use of animal models strongly support the possibility that altered snRNP production due to defective chaperoning downstream to SMN deficiency can lead to the neuromuscular defects that are typical in SMA (Table 1). It has long been known that SMN levels strongly stipulate the snRNP assembly capacity of cell extracts (Wan et al., 2005; Boulisfane et al., 2011). In the spinal cord, maximal snRNP assembly chaperoning activity overlaps with the highest demands for SMN during the development of the neuromuscular system (Gabanella et al., 2005; Foust et al., 2010; Le et al., 2011; Lutz et al., 2011; Kariya et al., 2014). Importantly, snRNP assembly capacity in the spinal cord of SMA mouse models was found to determine disease severity, hence, the greatest perturbation was observed in severe SMA mice (Gabanella et al., 2007). In a reciprocal experiment, the disease phenotype was rescued in mice following the introduction of the SMNA111G allele, which is capable of chaperoning snRNP assembly. Correction of SMA was dependent on snRNP assembly activity in the spinal cord (Workman et al., 2009). This study is in agreement with an earlier report demonstrating that motor neuron degeneration was rescued when purified snRNPs were injected in SMN-deficient zebrafish embryos (Winkler et al., 2005). Interestingly, in line with earlier findings demonstrating that knockdown of pICln or U1 snRNP leads to SMA-like defects in zebrafish (Winkler et al., 2005; Yu et al., 2015) (Table 1), disruption of either pICln or Tgs1 was found to result in motor defects that mirror those described on loss of SMN or Gemins in Drosophila (Borg et al., 2016). pICln and Tgs1 are two factors that are known to have a leading role in the early and late phase of snRNP assembly, respectively. Importantly, unlike the Gemins (reviewed in Cauchi, 2010), they have never been directly linked to the assembly and transport of messenger ribonucleoprotein (mRNP) complexes along axons, which is often considered as the primary non-canonical activity of the SMN-Gemins complex (reviewed in Donlin-Asp et al., 2016).
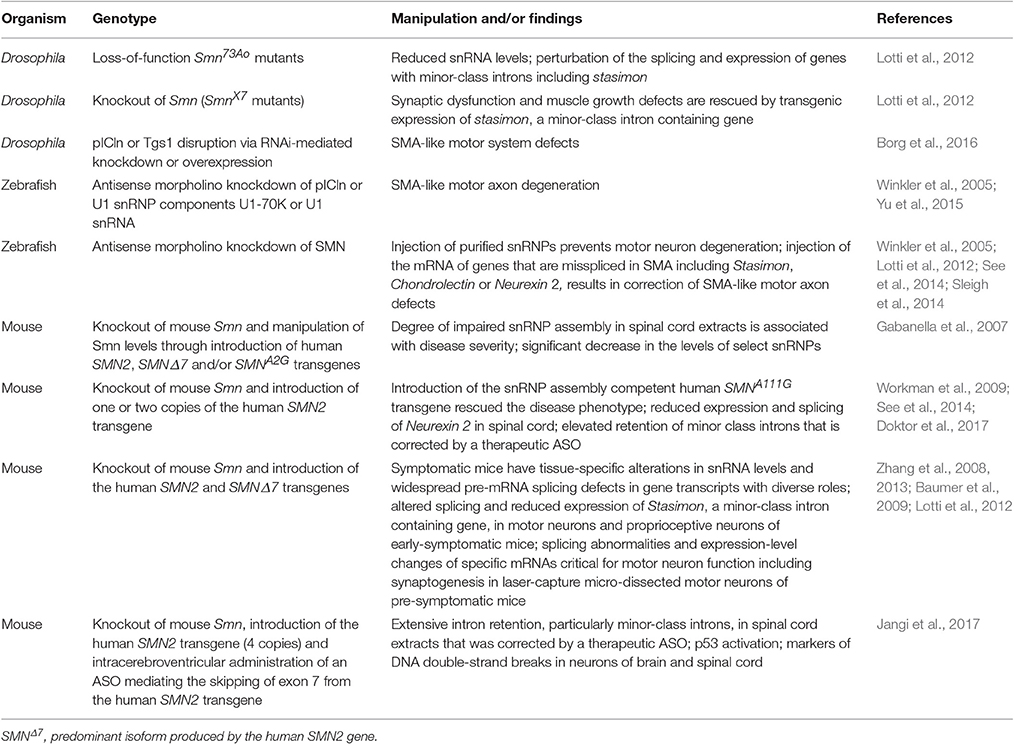
Table 1. Key studies in animal models linking motor dysfunction to perturbation in snRNP biogenesis.
Consistent with a fundamental role for SMN in chaperoning snRNP assembly, several studies were successful in identifying splicing defects as a consequence of SMN loss and, importantly, explain how missplicing of specific transcripts leads to motor dysfunction in SMA. Whereas symptomatic SMA mice were shown to have widespread pre-mRNA splicing defects in numerous transcripts of diverse genes (Zhang et al., 2008; Baumer et al., 2009), at a pre-symptomatic stage, they exhibit dysregulation of genes that are critical for the function of the motor neuron and may thus contribute to SMA's signature pathology (Zhang et al., 2013). Similarly, in Drosophila, SMN deficiency perturbed the splicing and expression of several genes including that of stasimon, whose correct splicing is dependent on the minor spliceosome. Stasimon mRNA expression and splicing was also found perturbed in the constituent neurons of the sensory-motor circuit in SMA mice. Restoration of stasimon expression in the motor circuit was found to correct in part the motor system defects in Drosophila Smn mutants and Smn-deficient zebrafish, thus establishing stasimon as an SMN target gene (Lotti et al., 2012). Other studies have since focused on additional genes that are misspliced in SMA models including Neurexin2 (See et al., 2014) and Chondrolectin (Sleigh et al., 2014), both of which are important for motor neuron axon outgrowth. Further still, following up on a previous study in a genetic model (Doktor et al., 2017), an ASO-inducible model of SMA was recently found to have widespread intron retention, particularly those spliced by the minor spliceosome, in spinal cord extracts. Importantly, these changes were rescued by a therapeutic ASO, thereby indicating that intron removal is directly correlated with SMN levels. Interestingly, intron retention was associated with a strong induction of the p53 pathway, and markers of DNA double-strand breaks were apparent in the neurons of the spinal cord and brain of SMA mice (Jangi et al., 2017). Thus, it is highly likely that instead of single gene effects, inefficiencies in pre-mRNA processing consequent to severe SMN deficiency lead to a DNA damage and stress response that compromises the survival of the motor neuron. Nonetheless, the reasons why the neuromuscular system remains highly vulnerable to damage warrants further investigation.
Conclusion
The ample evidence linking defective chaperoning of snRNP assembly to neuromuscular dysfunction is not only consistent with SMA being a chaperonopathy but also sets the scene for the discovery of therapies that target this pathway. Inhibition of RNA decay pathways to correct snRNP levels (Shukla and Parker, 2014) or suppression of genome instability induced by intron retention (Jangi et al., 2017) are two treatment routes that are successful, at least in vitro. Although the effectiveness of these and other approaches on animal models or humans, remains to be investigated, the suppression of snRNP hypo-assembly and splicing defects may provide benefits to SMA patients beyond the benefit of SMN restoration alone. Considering that distrubances in snRNP assembly are also a component of the pathogenesis of the adult-onset amyotrophic lateral sclerosis (ALS) (Cauchi, 2014; Sun et al., 2015; Yu et al., 2015), such therapeutic strategies are expected to have broad implications in motor neuron disease.
Author Contributions
RC conceived the review focus; ML, NV, and RC conducted the literature review, wrote and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Dr. Rémy Bordonné for his valuable collaboration and comments on the manuscript. Work in the authors' laboratory is supported by the University of Malta, the ALS Malta Foundation, and the Malta Council for Science and Technology. ML is supported by the Endeavor Scholarship Scheme (Malta), part-financed by the EU—European Social Fund under Operational Programme II—Cohesion Policy 2014-2020, “Investing in human capital to create more opportunities and promote the well-being of society.”
References
Battle, D. J., Lau, C. K., Wan, L., Deng, H., Lotti, F., and Dreyfuss, G. (2006). The Gemin5 protein of the SMN complex identifies snRNAs. Mol. Cell 23, 273–279. doi: 10.1016/j.molcel.2006.05.036
Baumer, D., Lee, S., Nicholson, G., Davies, J. L., Parkinson, N. J., Murray, L. M., et al. (2009). Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 5:e1000773. doi: 10.1371/journal.pgen.1000773
Borg, R., and Cauchi, R. J. (2014). GEMINs: potential therapeutic targets for spinal muscular atrophy? Front. Neurosci. 8:325. doi: 10.3389/fnins.2014.00325
Borg, R. M., Bordonne, R., Vassallo, N., and Cauchi, R. J. (2015). Genetic interactions between the members of the SMN-gemins complex in Drosophila. PLoS ONE 10:e0130974. doi: 10.1371/journal.pone.0130974
Borg, R. M., Fenech Salerno, B., Vassallo, N., Bordonne, R., and Cauchi, R. J. (2016). Disruption of snRNP biogenesis factors Tgs1 and pICln induces phenotypes that mirror aspects of SMN-Gemins complex perturbation in Drosophila, providing new insights into spinal muscular atrophy. Neurobiol. Dis. 94, 245–258. doi: 10.1016/j.nbd.2016.06.015
Boulisfane, N., Choleza, M., Rage, F., Neel, H., Soret, J., and Bordonne, R. (2011). Impaired minor tri-snRNP assembly generates differential splicing defects of U12-type introns in lymphoblasts derived from a type I SMA patient. Hum. Mol. Genet. 20, 641–648. doi: 10.1093/hmg/ddq508
Burghes, A. H., and Beattie, C. E. (2009). Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 10, 597–609. doi: 10.1038/nrn2670
Burnett, B. G., Munoz, E., Tandon, A., Kwon, D. Y., Sumner, C. J., and Fischbeck, K. H. (2009). Regulation of SMN protein stability. Mol. Cell. Biol. 29, 1107–1115. doi: 10.1128/MCB.01262-08
Carissimi, C., Saieva, L., Baccon, J., Chiarella, P., Maiolica, A., Sawyer, A., et al. (2006). Gemin8 is a novel component of the survival motor neuron complex and functions in small nuclear ribonucleoprotein assembly. J. Biol. Chem. 281, 8126–8134. doi: 10.1074/jbc.M512243200
Cauchi, R. J. (2010). SMN and Gemins: ‘we are family’…or are we? Insights into the partnership between Gemins and the spinal muscular atrophy disease protein SMN. Bioessays 32, 1077–1089. doi: 10.1002/bies.201000088
Cauchi, R. J. (2011). Gem formation upon constitutive Gemin3 overexpression in Drosophila. Cell Biol. Int. 35, 1233–1238. doi: 10.1042/CBI20110147
Cauchi, R. J. (2014). Gem depletion: amyotrophic lateral sclerosis and spinal muscular atrophy crossover. CNS Neurosci. Ther. 20, 574–581. doi: 10.1111/cns.12242
Cauchi, R. J., Davies, K. E., and Liu, J. L. (2008). A motor function for the DEAD-box RNA helicase, Gemin3, in Drosophila. PLoS Genet. 4:e1000265. doi: 10.1371/journal.pgen.1000265
Cauchi, R. J., Sanchez-Pulido, L., and Liu, J. L. (2010). Drosophila SMN complex proteins Gemin2, Gemin3, and Gemin5 are components of U bodies. Exp. Cell Res. 316, 2354–2364. doi: 10.1016/j.yexcr.2010.05.001
Chari, A., Golas, M. M., Klingenhager, M., Neuenkirchen, N., Sander, B., Englbrecht, C., et al. (2008). An assembly chaperone collaborates with the SMN complex to generate spliceosomal SnRNPs. Cell 135, 497–509. doi: 10.1016/j.cell.2008.09.020
Charroux, B., Pellizzoni, L., Perkinson, R. A., Shevchenko, A., Mann, M., and Dreyfuss, G. (1999). Gemin3: a novel DEAD box protein that interacts with SMN, the spinal muscular atrophy gene product, and is a component of Gems. J. Cell Biol. 147, 1181–1193. doi: 10.1083/jcb.147.6.1181
Doktor, T. K., Hua, Y., Andersen, H. S., Broner, S., Liu, Y. H., Wieckowska, A., et al. (2017). RNA-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of U12-dependent introns. Nucleic Acids Res. 45, 395–416. doi: 10.1093/nar/gkw731
Donlin-Asp, P. G., Bassell, G. J., and Rossoll, W. (2016). A role for the survival of motor neuron protein in mRNP assembly and transport. Curr. Opin. Neurobiol. 39, 53–61. doi: 10.1016/j.conb.2016.04.004
Faravelli, I., Nizzardo, M., Comi, G. P., and Corti, S. (2015). Spinal muscular atrophy–recent therapeutic advances for an old challenge. Nat. Rev. Neurol. 11, 351–359. doi: 10.1038/nrneurol.2015.77
Farrar, M. A., Park, S. B., Vucic, S., Carey, K. A., Turner, B. J., Gillingwater, T. H., et al. (2016). Emerging therapies and challenges in Spinal Muscular Atrophy. Ann. Neurol. 81, 355–368. doi: 10.1002/ana.24864
Feng, W., Gubitz, A. K., Wan, L., Battle, D. J., Dostie, J., Golembe, T. J., et al. (2005). Gemins modulate the expression and activity of the SMN complex. Hum. Mol. Genet. 14, 1605–1611. doi: 10.1093/hmg/ddi168
Fischer, U., Englbrecht, C., and Chari, A. (2011). Biogenesis of spliceosomal small nuclear ribonucleoproteins. Wiley Interdiscip. Rev. 2, 718–731. doi: 10.1002/wrna.87
Foust, K. D., Wang, X., McGovern, V. L., Braun, L., Bevan, A. K., Haidet, A. M., et al. (2010). Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 28, 271–274. doi: 10.1038/nbt.1610
Gabanella, F., Butchbach, M. E., Saieva, L., Carissimi, C., Burghes, A. H., and Pellizzoni, L. (2007). Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE 2:e921. doi: 10.1371/journal.pone.0000921
Gabanella, F., Carissimi, C., Usiello, A., and Pellizzoni, L. (2005). The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum. Mol. Genet. 14, 3629–3642. doi: 10.1093/hmg/ddi390
Grimm, C., Chari, A., Pelz, J. P., Kuper, J., Kisker, C., Diederichs, K., et al. (2013). Structural basis of assembly chaperone-mediated snRNP formation. Mol. Cell 49, 692–703. doi: 10.1016/j.molcel.2012.12.009
Grimmler, M., Otter, S., Peter, C., Muller, F., Chari, A., and Fischer, U. (2005). Unrip, a factor implicated in cap-independent translation, associates with the cytosolic SMN complex and influences its intracellular localization. Hum. Mol. Genet. 14, 3099–3111. doi: 10.1093/hmg/ddi343
Guruharsha, K. G., Rual, J. F., Zhai, B., Mintseris, J., Vaidya, P., Vaidya, N., et al. (2011). A protein complex network of Drosophila melanogaster. Cell 147, 690–703. doi: 10.1016/j.cell.2011.08.047
Hannus, S., Buhler, D., Romano, M., Seraphin, B., and Fischer, U. (2000). The Schizosaccharomyces pombe protein Yab8p and a novel factor, Yip1p, share structural and functional similarity with the spinal muscular atrophy-associated proteins SMN and SIP1. Hum. Mol. Genet. 9, 663–674. doi: 10.1093/hmg/9.5.663
Hao le, T., Fuller, H. R., Lam le, T., Le, T. T., Burghes, A. H., and Morris, G. E. (2007). Absence of gemin5 from SMN complexes in nuclear Cajal bodies. BMC Cell Biol. 8:28. doi: 10.1186/1471-2121-8-28
Helmken, C., Hofmann, Y., Schoenen, F., Oprea, G., Raschke, H., Rudnik-Schoneborn, S., et al. (2003). Evidence for a modifying pathway in SMA discordant families: reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum. Genet. 114, 11–21. doi: 10.1007/s00439-003-1025-2
Jablonka, S., Holtmann, B., Meister, G., Bandilla, M., Rossoll, W., Fischer, U., et al. (2002). Gene targeting of Gemin2 in mice reveals a correlation between defects in the biogenesis of U snRNPs and motoneuron cell death. Proc. Natl. Acad. U.S.A. 99, 10126–10131. doi: 10.1073/pnas.152318699
Jangi, M., Fleet, C., Cullen, P., Gupta, S. V., Mekhoubad, S., Chiao, E., et al. (2017). SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc. Natl. Acad. Sci. U.S.A. 114, E2347–E2356. doi: 10.1073/pnas.1613181114
Jin, W., Wang, Y., Liu, C. P., Yang, N., Jin, M., Cong, Y., et al. (2016). Structural basis for snRNA recognition by the double-WD40 repeat domain of Gemin5. Genes Dev. 30, 2391–2403. doi: 10.1101/gad.291377.116
Kariya, S., Obis, T., Garone, C., Akay, T., Sera, F., Iwata, S., et al. (2014). Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J. Clin. Invest. 124, 785–800. doi: 10.1172/JCI72017
Kolb, S. J., and Kissel, J. T. (2011). Spinal muscular atrophy: a timely review. Arch. Neurol. 68, 979–984. doi: 10.1001/archneurol.2011.74
Kroiss, M., Schultz, J., Wiesner, J., Chari, A., Sickmann, A., and Fischer, U. (2008). Evolution of an RNP assembly system: a minimal SMN complex facilitates formation of UsnRNPs in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 105, 10045–10050. doi: 10.1073/pnas.0802287105
Lau, C. K., Bachorik, J. L., and Dreyfuss, G. (2009). Gemin5-snRNA interaction reveals an RNA binding function for WD repeat domains. Nat. Struct. Mol. Biol. 16, 486–491. doi: 10.1038/nsmb.1584
Le, T. T., McGovern, V. L., Alwine, I. E., Wang, X., Massoni-Laporte, A., Rich, M. M., et al. (2011). Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet. 20, 3578–3591. doi: 10.1093/hmg/ddr275
Liu, J. L., and Gall, J. G. (2007). U bodies are cytoplasmic structures that contain uridine-rich small nuclear ribonucleoproteins and associate with P bodies. Proc. Natl. Acad. Sci. U.S.A. 104, 11655–11659. doi: 10.1073/pnas.0704977104
Liu, Q., and Dreyfuss, G. (1996). A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 15, 3555–3565.
Lorson, C. L., Strasswimmer, J., Yao, J. M., Baleja, J. D., Hahnen, E., Wirth, B., et al. (1998). SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 19, 63–66. doi: 10.1038/ng0598-63
Lotti, F., Imlach, W. L., Saieva, L., Beck, E. S., Hao le, T., Li, D. K., et al. (2012). An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151, 440–454. doi: 10.1016/j.cell.2012.09.012
Lutz, C. M., Kariya, S., Patruni, S., Osborne, M. A., Liu, D., Henderson, C. E., et al. (2011). Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J. Clin. Invest. 121, 3029–3041. doi: 10.1172/JCI57291
Ma, Y., Dostie, J., Dreyfuss, G., and Van Duyne, G. D. (2005). The Gemin6-Gemin7 heterodimer from the survival of motor neurons complex has an Sm protein-like structure. Structure 13, 883–892. doi: 10.1016/j.str.2005.03.014
Matera, A. G., and Wang, Z. (2014). A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 15, 108–121. doi: 10.1038/nrm3742
Mouaikel, J., Narayanan, U., Verheggen, C., Matera, A. G., Bertrand, E., Tazi, J., et al. (2003). Interaction between the small-nuclear-RNA cap hypermethylase and the spinal muscular atrophy protein, survival of motor neuron. EMBO Rep. 4, 616–622. doi: 10.1038/sj.embor.embor863
Mouaikel, J., Verheggen, C., Bertrand, E., Tazi, J., and Bordonne, R. (2002). Hypermethylation of the cap structure of both yeast snRNAs and snoRNAs requires a conserved methyltransferase that is localized to the nucleolus. Mol. Cell 9, 891–901. doi: 10.1016/S1097-2765(02)00484-7
Neuenkirchen, N., Englbrecht, C., Ohmer, J., Ziegenhals, T., Chari, A., and Fischer, U. (2015). Reconstitution of the human U snRNP assembly machinery reveals stepwise Sm protein organization. EMBO J. 34, 1925–1941. doi: 10.15252/embj.201490350
Ogawa, C., Usui, K., Aoki, M., Ito, F., Itoh, M., Kai, C., et al. (2007). Gemin2 plays an important role in stabilizing the survival of motor neuron complex. J. Biol. Chem. 282, 11122–11134. doi: 10.1074/jbc.M609297200
Ogawa, C., Usui, K., Ito, F., Itoh, M., Hayashizaki, Y., and Suzuki, H. (2009). Role of survival motor neuron complex components in small nuclear ribonucleoprotein assembly. J. Biol. Chem. 284, 14609–14617. doi: 10.1074/jbc.M809031200
Otter, S., Grimmler, M., Neuenkirchen, N., Chari, A., Sickmann, A., and Fischer, U. (2007). A comprehensive interaction map of the human survival of motor neuron (SMN) complex. J. Biol. Chem. 282, 5825–5833. doi: 10.1074/jbc.M608528200
Paknia, E., Chari, A., Stark, H., and Fischer, U. (2016). The ribosome cooperates with the assembly chaperone pICln to initiate formation of snRNPs. Cell Rep. 16, 3103–3112. doi: 10.1016/j.celrep.2016.08.047
See, K., Yadav, P., Giegerich, M., Cheong, P. S., Graf, M., Vyas, H., et al. (2014). SMN deficiency alters Nrxn2 expression and splicing in zebrafish and mouse models of spinal muscular atrophy. Hum. Mol. Genet. 23, 1754–1770. doi: 10.1093/hmg/ddt567
Shpargel, K. B., and Matera, G. (2005). Gemin proteins are required for efficient assembly of Sm-class ribonucleoproteins. Proc. Natl. Acad. U.S.A. 102, 17372–17377. doi: 10.1073/pnas.0508947102
Shpargel, K. B., Praveen, K., Rajendra, T. K., and Matera, A. G. (2009). Gemin3 is an essential gene required for larval motor function and pupation in Drosophila. Mol. Biol. Cell 20, 90–101. doi: 10.1091/mbc.E08-01-0024
Shukla, S., and Parker, R. (2014). Quality control of assembly-defective U1 snRNAs by decapping and 5′-to-3′ exonucleolytic digestion. Proc. Natl. Acad. Sci. U.S.A. 111, E3277–E3286. doi: 10.1073/pnas.1412614111
Sleigh, J. N., Barreiro-Iglesias, A., Oliver, P. L., Biba, A., Becker, T., Davies, K. E., et al. (2014). Chondrolectin affects cell survival and neuronal outgrowth in in vitro and in vivo models of spinal muscular atrophy. Hum. Mol. Genet. 23, 855–869. doi: 10.1093/hmg/ddt477
So, B. R., Wan, L., Zhang, Z., Li, P., Babiash, E., Duan, J., et al. (2016). A U1 snRNP-specific assembly pathway reveals the SMN complex as a versatile hub for RNP exchange. Nat. Struct. Mol. Biol. 23, 225–230. doi: 10.1038/nsmb.3167
Stanek, D. (2016). Cajal bodies and snRNPs - friends with benefits. RNA Biol. 14, 1–9. doi: 10.1080/15476286.2016.1231359
Sun, S., Ling, S. C., Qiu, J., Albuquerque, C. P., Zhou, Y., Tokunaga, S., et al. (2015). ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat. Commun. 6:6171. doi: 10.1038/ncomms7171
Tang, X., Bharath, S. R., Piao, S., Tan, V. Q., Bowler, M. W., and Song, H. (2016). Structural basis for specific recognition of pre-snRNA by Gemin5. Cell Res. 26, 1353–1356. doi: 10.1038/cr.2016.133
Wan, L., Battle, D. J., Yong, J., Gubitz, A. K., Kolb, S. J., Wang, J., et al. (2005). The survival of motor neurons protein determines the capacity for snRNP assembly: biochemical deficiency in spinal muscular atrophy. Mol. Cell. Biol. 25, 5543–5551. doi: 10.1128/MCB.25.13.5543-5551.2005
Winkler, C., Eggert, C., Gradl, D., Meister, G., Giegerich, M., Wedlich, D., et al. (2005). Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev. 19, 2320–2330. doi: 10.1101/gad.342005
Wirth, B., Garbes, L., and Riessland, M. (2013). How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr. Opin. Genet. Dev. 23, 330–338. doi: 10.1016/j.gde.2013.03.003
Workman, E., Saieva, L., Carrel, T. L., Crawford, T. O., Liu, D., Lutz, C., et al. (2009). A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum. Mol. Genet. 18, 2215–2229. doi: 10.1093/hmg/ddp157
Xu, C., Ishikawa, H., Izumikawa, K., Li, L., He, H., Nobe, Y., et al. (2016). Structural insights into Gemin5-guided selection of pre-snRNAs for snRNP assembly. Genes Dev. 30, 2376–2390. doi: 10.1101/gad.288340.116
Yan, X., Mouillet, J. F., Ou, Q., and Sadovsky, Y. (2003). A novel domain within the DEAD-box protein DP103 is essential for transcriptional repression and helicase activity. Mol. Cell. Biol. 23, 414–423. doi: 10.1128/MCB.23.1.414-423.2003
Yong, J., Kasim, M., Bachorik, J. L., Wan, L., and Dreyfuss, G. (2010). Gemin5 delivers snRNA precursors to the SMN complex for snRNP biogenesis. Mol. Cell 38, 551–562. doi: 10.1016/j.molcel.2010.03.014
Young, P. J., Man, N. T., Lorson, C. L., Le, T. T., Androphy, E. J., Burghes, A. H., et al. (2000). The exon 2b region of the spinal muscular atrophy protein, SMN, is involved in self-association and SIP1 binding. Hum. Mol. Genet. 9, 2869–2877. doi: 10.1093/hmg/9.19.2869
Yu, Y., Chi, B., Xia, W., Gangopadhyay, J., Yamazaki, T., Winkelbauer-Hurt, M. E., et al. (2015). U1 snRNP is mislocalized in ALS patient fibroblasts bearing NLS mutations in FUS and is required for motor neuron outgrowth in zebrafish. Nucleic Acids Res. 43, 3208–3218. doi: 10.1093/nar/gkv157
Zhang, R., So, B. R., Li, P., Yong, J., Glisovic, T., Wan, L., et al. (2011). Structure of a key intermediate of the SMN complex reveals Gemin2's crucial function in snRNP assembly. Cell 146, 384–395. doi: 10.1016/j.cell.2011.06.043
Zhang, Z., Lotti, F., Dittmar, K., Younis, I., Wan, L., Kasim, M., et al. (2008). SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133, 585–600. doi: 10.1016/j.cell.2008.03.031
Keywords: survival motor neuron, SMN-Gemins complex, snRNP assembly, missplicing, motor neuron disease (MND), amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), spliceosome
Citation: Lanfranco M, Vassallo N and Cauchi RJ (2017) Spinal Muscular Atrophy: From Defective Chaperoning of snRNP Assembly to Neuromuscular Dysfunction. Front. Mol. Biosci. 4:41. doi: 10.3389/fmolb.2017.00041
Received: 05 April 2017; Accepted: 26 May 2017;
Published: 08 June 2017.
Edited by:
Alberto J. L. Macario, University of Maryland at Baltimore and Institute of Marine and Environmental Technology, United States; Istituto Euro-Mediterraneo di Scienza e Tecnologia, ItalyReviewed by:
Umesh K. Jinwal, University of South Florida, United StatesLeonid Breydo, University of South Florida, United States
Copyright © 2017 Lanfranco, Vassallo and Cauchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruben J. Cauchi, cnViZW4uY2F1Y2hpQHVtLmVkdS5tdA==