 Andrew R. Castle
Andrew R. Castle Andrew C. Gill
Andrew C. Gill
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Biosci., 06 April 2017
Sec. Protein Folding, Misfolding and Degradation
Volume 4 - 2017 | https://doi.org/10.3389/fmolb.2017.00019
The prion protein, PrPC, is a small, cell-surface glycoprotein notable primarily for its critical role in pathogenesis of the neurodegenerative disorders known as prion diseases. A hallmark of prion diseases is the conversion of PrPC into an abnormally folded isoform, which provides a template for further pathogenic conversion of PrPC, allowing disease to spread from cell to cell and, in some circumstances, to transfer to a new host. In addition to the putative neurotoxicity caused by the misfolded form(s), loss of normal PrPC function could be an integral part of the neurodegenerative processes and, consequently, significant research efforts have been directed toward determining the physiological functions of PrPC. In this review, we first summarise important aspects of the biochemistry of PrPC before moving on to address the current understanding of the various proposed functions of the protein, including details of the underlying molecular mechanisms potentially involved in these functions. Over years of study, PrPC has been associated with a wide array of different cellular processes and many interacting partners have been suggested. However, recent studies have cast doubt on the previously well-established links between PrPC and processes such as stress-protection, copper homeostasis and neuronal excitability. Instead, the functions best-supported by the current literature include regulation of myelin maintenance and of processes linked to cellular differentiation, including proliferation, adhesion, and control of cell morphology. Intriguing connections have also been made between PrPC and the modulation of circadian rhythm, glucose homeostasis, immune function and cellular iron uptake, all of which warrant further investigation.
The cell surface glycoprotein known as the cellular prion protein (PrPC) has been the subject of intensive study since it was first proposed that misfolding of PrPC plays a key role in the pathogenesis of the neurodegenerative disorders referred to as either the transmissible spongiform encephalopathies (TSEs) or prion diseases (Prusiner, 1982). Rather than focusing on the involvement of prion protein in disease, this review provides an overview of the physiological function of PrPC as it is currently understood. However, before addressing PrPC function, we first introduce some important aspects of the biochemistry/molecular biology of PrPC and its gene (PRNP) that are of functional relevance, starting with an exploration of the evolutionary history of PRNP.
PrPC is encoded by the PRNP gene located on chromosome 20 (in humans—in mice this is chromosome 2) and is remarkably conserved throughout vertebrates. In addition to PRNP, the mammalian prion gene family includes SPRN, which encodes a protein referred to as shadoo (Sho), and PRND, which encodes a protein known as doppel (Dpl). Sho and Dpl share structural similarities to PrPC (see below and Figure 2) and several theories have been put forward to explain the development of this gene family over evolutionary time. Three such theories are outlined in diagrammatic form in Figure 1. Figure 1A shows an explanation proposed by Schmitt-Ulms et al. (2009) and developed in other, more recent publications (Ehsani et al., 2011, 2012). The authors of these papers provided evidence that the prion gene family is evolutionarily descended from the LIV-1 branch of the ZIP (Zrt-, Irt-like) metal ion transporter family. Specifically, Ehsani et al. (2011) suggested that the ancestral PRNP gene could have been created as a result of reverse transcription of the mRNA of a ZIP gene, followed by insertion into the genome at a new position, a process known as retroposition. This event is proposed to have occurred around the time of the first vertebrates, which would explain the presence of PRNP homologs in all vertebrate lineages. In this model, a subsequent gene duplication event, in addition to some form of genomic rearrangement, led to modern-day SPRN, which is situated on chromosome 10 in the human genome. A later, local duplication would have then given rise to modern-day PRNP and PRND, which are directly adjacent to one another in the genome (Westaway et al., 2011). Figure 1B displays an alternative scenario also outlined by Schmitt-Ulms et al. (2009) but incorporating earlier research suggesting that SPRN may be evolutionarily more ancient than PRNP (Premzl et al., 2004). As per the first theory, the genetic material encoding the PrPC C-terminal domain could have originated from an ancestral ZIP gene. Contrastingly, the sequences encoding the N-terminal domain may have arisen from a pre-existing SPRN-like gene (SPRNB1) that was created by earlier duplications of an SPRN founder gene. The generation of intergenic mRNA transcripts or an alternative form of local rearrangement could have caused these sequences to merge to form the ancestral PRNP (Westaway et al., 2011). A third model (Figure 1C) is that ancestral PRNP and SRPN genes could have evolved out of ZIP genes in separate events (Westaway et al., 2011).
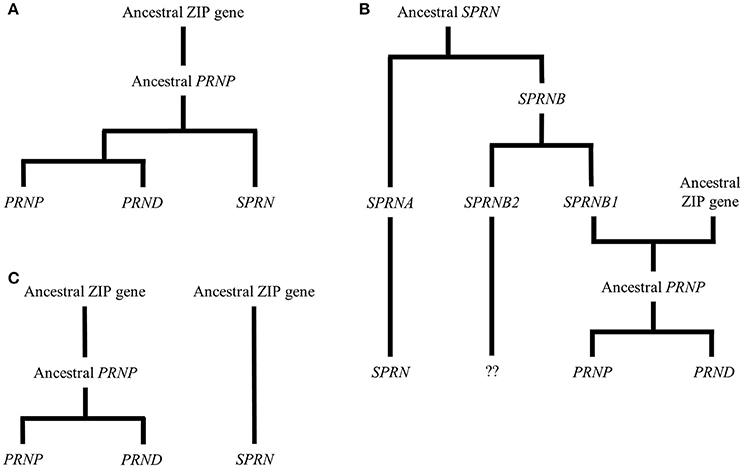
Figure 1. Theories of the evolutionary history of the prion gene family. The figure shows three possibilities for the evolution of the mammalian prion gene family. (A) Schmitt-Ulms et al. (2009) proposed that an ancestral PRNP gene evolved from a member of the ZIP metal ion transporter family. Subsequently, this PRNP precursor gave rise to the modern-day prion gene family through local duplications and other genomic rearrangements. (B) This alternative version, also put forward by Schmitt-Ulms et al. (2009), incorporates additional research by Premzl et al. (2004) suggesting that SPRN existed before PRNP and that the genetic material encoding the N-terminal domain of an ancestral PrPC evolved from a gene called SPRNB1 that, itself, had emerged from the original SPRN. The genetic material encoding the C-terminal domain of the ancestral PrPC is proposed to have derived from a ZIP gene and a later, local duplication would have then created modern-day PRNP and PRND. Although, descendants of SPRNB2 are found in fish, this gene is thought either to have been deleted or to have evolved beyond detectability in the mammalian lineage (Premzl et al., 2004). (C) A further possibility is that ancestral PRNP and SRPN genes could have evolved out of ZIP genes in separate events (Westaway et al., 2011).
Although PRNP has a short GC-rich region immediately upstream of its transcription start site, as well as other features common to housekeeping genes (Puckett et al., 1991; Sakudo et al., 2010), intron 1 and the sequences upstream of the transcription start site also contain evolutionarily conserved, putative binding sites for numerous transcription factors, including Sp1 (Basler et al., 1986), activator proteins 1 and 2 (Mahal et al., 2001), forkhead box protein O3 (Liu et al., 2013), regulatory factor X1, heat shock factor 2, GATA-binding factor 3, thyrotrophic embryonic factor, myocyte enhancer factor 2, ecotropic viral integration site 1, E4 promoter-binding protein, 4 and nuclear matrix protein 4/cas-interacting zinc finger protein (Kim et al., 2008). These regulatory sequences presumably enable dynamic control of PrPC expression in response to various stimuli, for example, treatment of cultured cells with nerve growth factor, insulin or insulin-like growth factor induces PrPC expression (Kuwahara et al., 2000; Zawlik et al., 2006; Liu et al., 2013). Additionally, endoplasmic reticulum (ER) stress, oxidative stress and genotoxic stress are all reported to cause upregulation of PrPC expression (Dery et al., 2013; Cichon and Brown, 2014; Bravard et al., 2015).
PrPC is synthesised as a precursor protein of 253 amino acids (human numbering) with an N-terminal signal peptide that codes for entry into the ER. However, seemingly because of an inefficiency in this ER-targeting signal (Rane et al., 2004), a small percentage of precursor molecules may fail to translocate fully into the ER lumen. Some of this PrPC is consequently retained in the cytoplasm. Interestingly, the levels of this immature, non-translocated PrPC may be upregulated by ER stress (Orsi et al., 2006). Another form, known as CtmPrP, partially enters the ER—the hydrophobic domain of PrPC (see below) acts as a transmembrane domain, leaving the C-terminal region within the ER lumen and the N-terminal region in the cytoplasm. Reports suggest that CtmPrP is retained either in the ER or the Golgi apparatus before eventual degradation by the proteasome (Stewart et al., 2001, 2005). A second transmembrane form of PrPC called NtmPrP can be produced if the molecule inserts into the ER membrane in the opposite orientation to CtmPrP. However, NtmPrP has rarely been studied in any detail (Chakrabarti et al., 2009). These forms appear more related to disease than physiological PrPC function and are not considered further here.
The vast majority of immature PrPC molecules translocate properly into the ER, enabling cleavage of a C-terminal signal peptide and addition of a glycophosphatidylinositol (GPI) anchor. Removal of both signal sequences results in a mature protein of 208 amino acids (residues 23–230 of the precursor protein), the structure of which is shown in Figure 2. The N-terminal domain is traditionally viewed as intrinsically disordered, but may possess elements of stable structure (Gill et al., 2000; Blanch et al., 2004; Taubner et al., 2010) that could enable PrPC to interact with multiple partners (Bakkebo et al., 2015). The N-terminal domain also contains four tandem repeats of a sequence of eight amino acids, a region of PrPC that is missing in Dpl. These octapeptide repeats may be functionally significant since analysis of PrPC sequences from 53 species showed excellent conservation (Kim et al., 2008). A hydrophobic section (approximately residues 112–133) spans the divide between the N- and C-terminal domains and may be involved in PrPC dimerisation (Rambold et al., 2008; Beland and Roucou, 2013). The PrPC C-terminal domain has a globular structure, consisting of three α-helices, two β-strands and interconnecting loops (Riek et al., 1996; Haire et al., 2004). There is also a disulphide bond between residues 179 and 214 (Zahn et al., 2000) and N-linked glycans can be added at residues 181 and 197—the vast majority of PrPC is generally thought to be di-glycosylated, although this may not be the case in all tissue/cell types (Williams et al., 2004). Notably, the entire globular domain is missing in Sho, but this protein does possess degenerate peptide repeats in the N-terminal region.
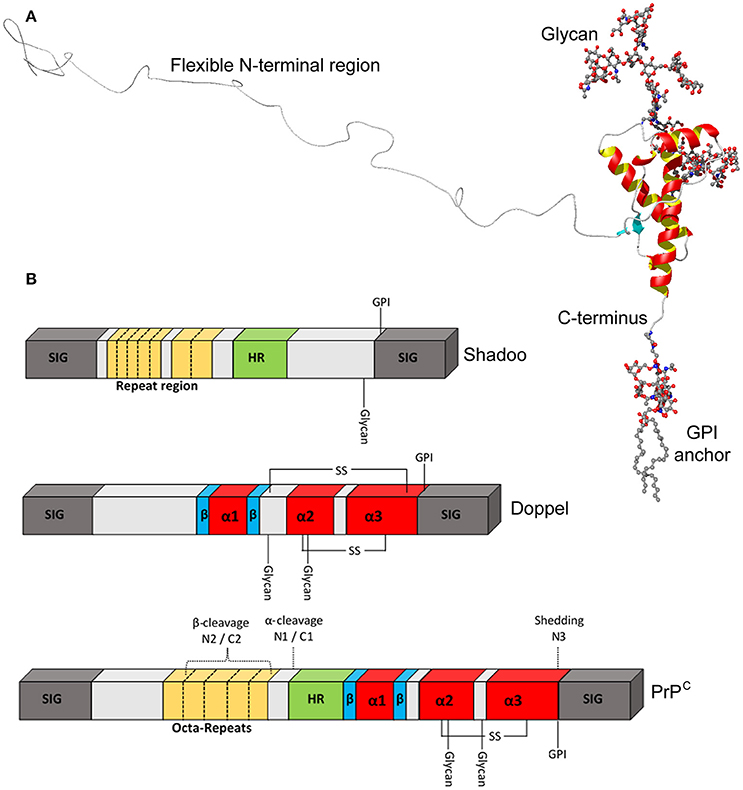
Figure 2. Structural features of PrPC. (A) Ribbon diagram of the PrPC molecule. The C-terminal domain contains three α-helices, shown in red and yellow, and two β-strands shown in turquoise, whereas the N-terminal domain has been added on in a “random” configuration. (B) Schematic representation of PrPC, Sho, and Dpl to highlight key structural features in greater detail. PrPC possesses octa-peptide repeats in the N-terminal region, whilst shadoo has two different, imperfect repeat stretches. SIG, N- and C-terminal signal peptides; HR, hydrophobic region.
After passing through the ER, PrPC moves to the Golgi apparatus where the N-linked glycans mature and the protein is sorted for trafficking to the cell surface. Once there, the GPI anchor attaches PrPC to the extra-cytoplasmic face of the cell membrane, specifically in microdomains called lipid rafts (Vey et al., 1996). Dpl and Sho also both have GPI anchors and are therefore similarly targeted to the cell surface (Silverman et al., 2000; Watts et al., 2007). Interestingly, not all PrPC is found at the cell surface. The protein seems to be subject to cycles of internalisation followed by trafficking back to the cell membrane via recycling endosomes (Shyng et al., 1993; Magalhaes et al., 2002; Sunyach et al., 2003), a process that presumably exists to enable tight control of the cell surface pool of PrPC. Studies using a green fluorescent protein reporter system have suggested that this recycling process may also result in PrPC expression within the Golgi apparatus (Lee et al., 2001; Magalhaes et al., 2002; Nikles et al., 2008). Furthermore, there are reports that PrPC can be present in the nucleus (Gu et al., 2003; Morel et al., 2008; Besnier et al., 2015; Bravard et al., 2015) and in mitochondria (Hachiya et al., 2005; Satoh et al., 2005; Sorice et al., 2012; Faris et al., 2017). Regardless of the different subcellular locations that PrPC can reside, the functional form is believed to be that present on the cell surface.
PrPC is most highly expressed in the central nervous system (CNS) although PRNP transcripts can also be found in many other tissue/cell types, albeit at somewhat lower levels. Publically available PRNP expression data from microarray analyses of various human tissues and cell types (Su et al., 2004) are shown in Figure 3 in the form of a bar chart for purposes of comparison. By contrast, in the same microarray dataset SPRN transcripts were found to be restricted to CNS and peripheral nervous system (PNS) tissues, whilst PRND expression was confined largely to the testes. At the protein level, PrPC expression in the brain appears to increase throughout development, reaching a peak in early life before reducing somewhat toward adulthood (Sales et al., 2002; Adle-Biassette et al., 2006). It is unclear how PrPC levels in the brain are affected by the ageing process—one study found increased PrPC expression in the brains of aged mice (Williams et al., 2004), whilst analysis of post-mortem human brain tissues showed that PrPC expression in the hippocampus was significantly reduced in older individuals (Whitehouse et al., 2010).
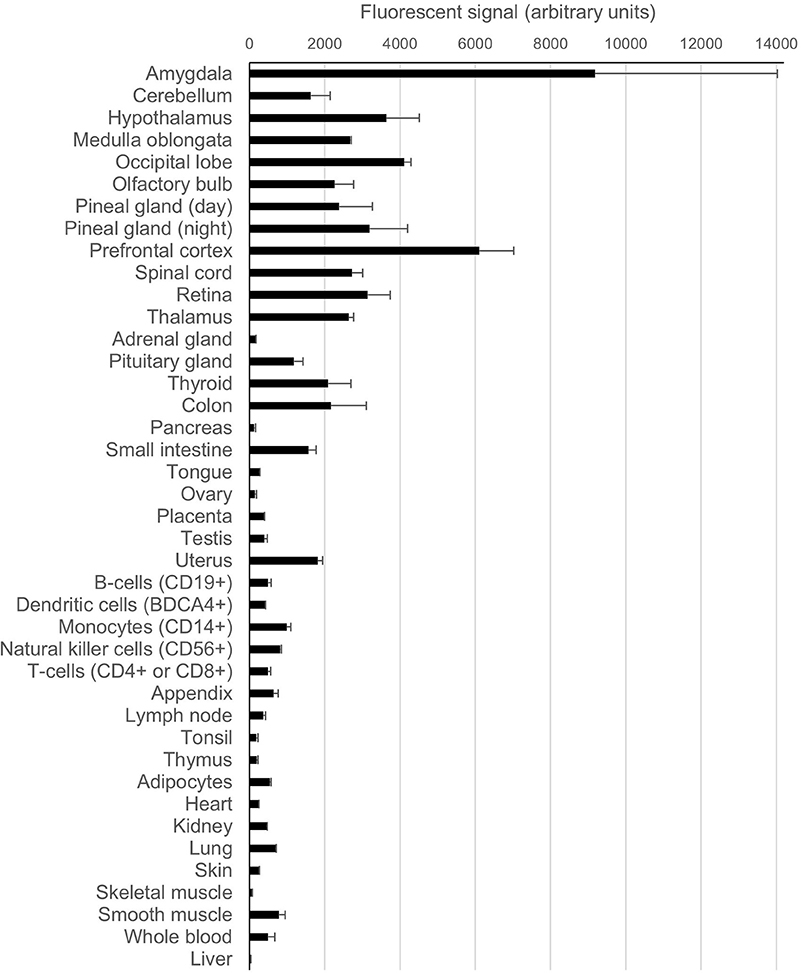
Figure 3. Expression levels of PRNP in various human tissues and cell types. The data for this chart originate from a publically available microarray dataset (GeneAtlas U133A, probeset 201300_s) originally published by Su et al. (2004) that was accessed through the BioGPS gene annotation portal (Wu et al., 2016). The bars on the chart indicate the arithmetic mean fluorescence signals from the replicate analytes; the error bars show standard error of the mean.
Most reports indicate that PrPC is expressed not only in neurons but also in astrocytes (Lima et al., 2007; Hartmann et al., 2013), oligodendrocytes (Moser et al., 1995; Bribian et al., 2012), and microglia (Adle-Biassette et al., 2006), although one study found that PrPC expression was inhibited both in vitro and in vivo following differentiation of neural precursors into glial cell types (Steele et al., 2006). Non-neural cells of the forebrain also stain for PrPC, including the endothelial cells in blood vessel walls (Adle-Biassette et al., 2006). Furthermore, PrPC expression has been reported in the PNS, including the dorsal and ventral root ganglia of the spinal cord (Tremblay et al., 2007; Peralta et al., 2012; Ganley et al., 2015), sensory and motor axons (Manson et al., 1992) and Schwann cells (Follet et al., 2002). Outside of the nervous system, PrPC expression has been detected in immune cells, including T-lymphocytes, natural killer cells and mast cells (Durig et al., 2000; Haddon et al., 2009), and also in multiple organs, including the heart, pancreas, intestine, spleen, liver, and kidneys (Peralta and Eyestone, 2009).
So far in this review, PrPC has been referred to as a single entity. However, as summarised in Figure 4, PrPC can be proteolytically processed in several ways. Firstly, PrPC can be enzymatically cleaved at the peptidyl bond between residues 110 and 111 (human numbering) that lies just outside the hydrophobic domain of the protein (Chen et al., 1995; Watt et al., 2005). A recent study proposed that this “α” form of cleavage may also occur at alternative sites within the hydrophobic domain itself (McDonald et al., 2014). Members of the disintegrin and metalloproteinase domain-containing protein (ADAM) family may be responsible for α-cleavage (Vincent et al., 2001; Cisse et al., 2008; McDonald et al., 2014), although not all studies support this idea (Beland et al., 2012; Wik et al., 2012). α-Cleavage is thought to occur either in an acidic endosomal compartment (Shyng et al., 1993) or within the Golgi apparatus (Walmsley et al., 2009) and results in the production of the N-terminal fragment N1, which is released from the cell, and the C-terminal fragment C1, which seems to be trafficked to the cell membrane as per the full length protein (Harris et al., 1993; Vincent et al., 2000; Laffont-Proust et al., 2006). A large proportion of the cellular pool of PrP molecules may be in the form of C1—around 50% on average in sheep cerebral cortex, for example (Campbell et al., 2013). In addition to the α form of processing, PrPC can be subject to cleavage within its octapeptide repeat region (McMahon et al., 2001). Recent findings from cell-free experiments using recombinant PrP suggest that this “β” cleavage can occur between the adjacent His and Gly residues of each octapeptide sequence (residues 61/62, 69/70, 77/78, and 85/86 for human PrPC) (McDonald et al., 2014). Whilst cleavage was equally likely at each site, the authors of this study caution that the presence of PrPC binding partners may cause a particular site to be favoured in vivo. β-Cleavage seems to be dependent upon the combined presence of Cu2+ and reactive oxygen species (ROS) (McMahon et al., 2001), although enzymatic processing by ADAM8 is also a possibility (McDonald et al., 2014). The apparent role for ROS in β-cleavage suggests that it might be a response to oxidative stress (Watt et al., 2005), although β-cleavage also seems to occur physiologically, since the resulting C-terminal fragment, C2, is found in healthy brain tissues from various species, albeit in small amounts (Mange et al., 2004; Campbell et al., 2013). β-Cleavage is thought to take place at the cell surface, leading to retainment of C2 on the cell membrane and release of the N-terminal fragment N2 (Mange et al., 2004; Watt et al., 2005). In a third form of PrPC processing, referred to as “shedding,” the protein can be cleaved at the 227/228 peptidyl bond (mouse numbering) by ADAM10, which removes the GPI anchor and the three adjacent amino acid residues, allowing the remaining protein fragment, sometimes known as N3, to be released into the extracellular medium (Taylor et al., 2009; McDonald et al., 2014).
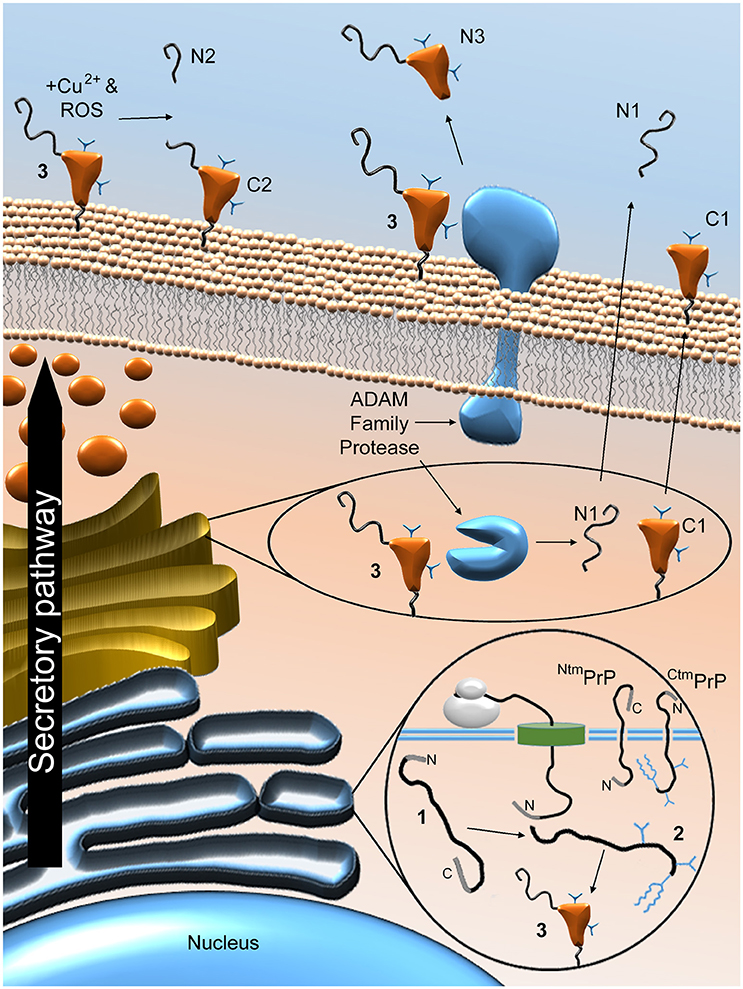
Figure 4. Proteolytic processing of PrPC. Post-translational and proteolytic processing events create multiple distinct PrP fragments. Ribosomal expression of PrPC is concomitant with ER translocation. Imperfect translocation can result in NtmPrP or CtmPrP. Once in the ER, the immature protein (1) is N- and C-terminally truncated, glycosylated, the membrane anchor is added and the single disulphide bond is formed to produce the mature protein (2), before (potentially chaperone-mediated) folding to produce the folded form (3). Enzymatic α-cleavage, possibly mediated by ADAM family proteases, results in the production of N1 and C1 and is thought to occur either in an acidic endosomal compartment or within the Golgi apparatus. These fragments and the remaining, uncleaved PrPC molecules are trafficked to the cell surface. Once there, PrPC can be subject to β-cleavage, possibly stimulated by the combined presence of ROS and Cu2+, leading to the production of N2 and C2. ADAM protease-mediated shedding may also occur, which results in cleavage of PrPC near its GPI anchor, thereby producing the N3 fragment. The sites of proteolytic cleavage are shown schematically in Figure 2B.
Before turning to PrPC function, some introductory comments on the diseases that made the prion protein notorious are warranted, since the role that this protein plays in these diseases drives the abundance of studies into its function. Prion diseases are infectious diseases characterised by long, pre-clinical incubation periods followed by rapidly progressing neurodegeneration. Although rare, prion diseases are uniformly fatal and many mammalian species are susceptible (Fernandez-Borges et al., 2012). Animal prion diseases include scrapie in sheep and goats, bovine spongiform encephalopathy in cattle and goats, and chronic wasting disease in deer, whilst sporadic Creutzfeldt-Jakob disease is the most common human prion disease (Head, 2013). Early studies of the infectious agent suggested that it was too small and too resistant to UV irradiation to contain any living organism, even a virus (Alper et al., 1966, 1978; Alper, 1985). Subsequently, a protein was isolated from prion-infected hamster brains and its concentration appeared to be proportional to prion infectivity (Prusiner et al., 1982; McKinley et al., 1983). This discovery led to the prion hypothesis, which proposes that the novel protein is the major component of the infectious agent of all prion diseases, the term “prion” deriving from proteinaceous infectious particle (Prusiner, 1982). The infectious protein was later shown to be a host genome-encoded protein, now known as PrPC, that can undergo conversion to a partially protease-resistant, misfolded form referred to as PrPSc (McKinley et al., 1983; Prusiner et al., 1985; Horwich and Weissman, 1997). The misfolding process leads to a reduction in the amount of α-helical structure and an increase in β-sheet conformation (Pan et al., 1993), which makes PrPSc prone to aggregation. PrPSc is also thought to act as a template for further pathogenic conversion of the normally folded form (Horwich and Weissman, 1997), enabling disease to spread from cell to cell, from individual to individual and, in rarer instances, to transfer between species.
In addition to its role in prion disease transmission, PrPSc is thought to be involved in the disease processes that cause neuronal cell death, especially when aggregated into soluble oligomers (Ugalde et al., 2016). The loss of normal PrPC function may also play a role in the pathogenesis of prion disease, since data from various animal models indicate that PrPC levels fall as disease progresses (Mays et al., 2014). Given the absence of disease-modifying treatments for these diseases, therapeutic interventions that further reduce PrPC expression or block its ability to interact with PrPSc are being investigated; better knowledge of PrPC function would aid risk assessment of these approaches. Additionally, knowing which proteins interact with PrPC physiologically might enable the development of other treatment strategies for prion diseases. Finally, as described later in this review, some beneficial functions of PrPC could be exploited independently for therapeutic purposes. For these reasons, the physiological function of PrPC has been investigated intensively over recent years and the knowledge gained from such research will be summarised in the following sections.
One way to assess protein function is to analyse the consequences of ablating expression in animals. As such, several groups have independently generated lines of PrPC-knockout mice, as summarised in Table 1. The first PrPC-knockouts were created in the early 1990s by gene targeting methods; these lines are referred to as Zurich I (Bueler et al., 1992) and Npu (Manson et al., 1994). Whilst the lack of PrPC expression completely prevented scrapie transmission to these mice, thereby providing strong evidence for the prion hypothesis (Bueler et al., 1993; Prusiner et al., 1993), no other striking phenotypes were identified in initial analyses (Bueler et al., 1992; Manson et al., 1994). These results were surprising given that the structure and amino acid sequence of PrPC are both well-conserved among mammalian species, which hints at an important function for the protein (Pastore and Zagari, 2007). However, more recent studies have identified several abnormalities of these PrPC-null mice and these findings will be covered in later sections.
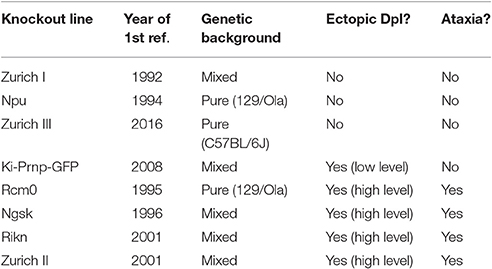
Table 1. PrPC-knockout mouse lines.
In the years following the generation of the Zurich I and Npu PrPC-null mice, several groups independently produced additional knockout lines, known as Rcm0 (Moore et al., 1995), Ngsk (Sakaguchi et al., 1996), Rikn (Yokoyama et al., 2001), and Zurich II (Rossi et al., 2001). Unlike the Zurich I and Npu knockout mice, all the newer lines were shown to develop a late-onset ataxia due to death of cerebellar Purkinje neurons (Sakaguchi et al., 1996; Moore et al., 1999; Rossi et al., 2001; Yokoyama et al., 2001). The reintroduction of PrPC into the Ngsk line rescued the ataxic phenotype, which seemed to confirm that it resulted from ablation of PrPC expression (Nishida et al., 1999). However, the particular gene targeting methods used for production of the Rcm0, Ngsk, Rikn and Zurich II knockouts led to the generation of intergenic mRNA transcripts from the undisrupted, non-protein-coding exons of Prnp and the exons of the neighbouring Prnd. Therefore, Dpl was being expressed under the control of the Prnp regulatory sequences, leading to ectopic Dpl expression in the brains of the PrPC-null mice, but not the wild type controls (Moore et al., 1999). The presence of the ataxic phenotype only in knockout mice with ectopic Dpl expression suggested that the cerebellar Purkinje neurons were dying from Dpl-mediated neurotoxicity rather than a lack of PrPC. Further evidence obtained in vitro confirmed that Dpl was toxic to neuronal cells, but only in the absence of PrPC expression (Sakudo et al., 2005; Qin et al., 2006). Although, the ability of PrPC to block the neurotoxic effects of Dpl may derive from a physical interaction between the proteins (Qin et al., 2006), this is unlikely to be a genuine physiological function of PrPC in the brain given that PrPC and Dpl are not typically co-expressed.
The ectopic Dpl expression makes it difficult to interpret the phenotypes of RcmO, Ngsk, Rikn, and Zurich II PrPC-knockout mice. A further knockout line (Ki-Prnp-GFP), in which the protein-coding exon of Prnp was replaced with genetic material encoding enhanced green fluorescent protein (Heikenwalder et al., 2008), also exhibit aberrant expression of Prnd transcripts in the brain but the levels seem low enough not to cause any overt abnormalities (Jackson et al., 2014). However, one issue that does affect the Ki-Prnp-GFP line as well as the much more frequently studied Zurich I line is their mixed genetic backgrounds. As outlined in a review by Steele et al. (2007), the result is that the genomes of these PrPC-null mice and their wild type counterparts may differ at sites other than the Prnp locus. Importantly, even extensive backcrossing to one of the parental strains or to a different inbred strain is unlikely to eliminate this issue completely. This is because alleles of genes linked to Prnp that derive from the embryonic stem cells used for gene targeting will tend to persist in the PrPC-knockout line due to the low probability of recombination between neighbouring genes (Gerlai, 1996; Nuvolone et al., 2013). Therefore, rather than resulting from a lack of PrPC expression, some of the phenotypes of the Zurich I PrPC-null mice may be due instead to the presence of different alleles of Prnp-linked genes compared with the wild type controls. Indeed, a specific phenotype associated with Zurich I (and Ngsk) PrPC-null mice—increased phagocytosis of apoptotic cells by macrophages—was traced to polymorphisms in the nearby gene that encodes signal-regulatory protein α-1 (Nuvolone et al., 2013). Due to their pure genetic backgrounds, the Npu line and a recently derived knockout line called Zurich III, which has been created using transcription activator-like effector nuclease genome editing technology (Nuvolone et al., 2016), are free from the problems caused by interfering linked genes.
Although, mice have been used in the great majority of studies to investigate PrPC function in vivo, PrPC expression has also been knocked out in goats and in cattle, both natural hosts of prion disease. Whilst no in-depth phenotypic description of the PrPC-null goats has been published, the animals in question were reported to be healthy up to at least 5 months of age (Yu et al., 2009; Zhu et al., 2009). Similarly, detailed clinical and histopathological examinations in addition to analyses of blood samples and isolated peripheral blood lymphocytes uncovered no overt abnormalities of PrPC-null cattle (Richt et al., 2007). However, these observations were made only in relatively young individuals (up to 20 months of age) and the samples sizes were small, meaning that subtle phenotypes would have been difficult to detect.
PrPC function has also been interrogated using zebrafish, in which three homologs of mammalian PRNP have been identified. These homologs are known as prp1 and prp2, encoding the proteins PrP1 and PrP2, respectively, and a more divergent PRNP-like gene known as prp3, which codes for PrP3 (Cotto et al., 2005). Conflicting data on the spatiotemporal expression pattern of PrP1 during embryonic development have been published (Cotto et al., 2005; Malaga-Trillo et al., 2009). The evidence is clearer for PrP2 expression, which is reportedly absent in the early embryo but reaches high levels in the nervous system during later developmental stages and is maintained following hatching (Malaga-Trillo et al., 2009; Fleisch et al., 2013). Knockdown of PrP1 or PrP2 expression using morpholinos has identified putative roles for both proteins in regulating cell adhesion (Malaga-Trillo et al., 2009; Huc-Brandt et al., 2014; Sempou et al., 2016). Furthermore, complete knockout of PrP2 expression using zinc finger nuclease technology increased susceptibility to seizures (Fleisch et al., 2013), suggesting that PrP2 might modulate neuronal excitability. There is some evidence that mammalian PrPC has similar functions to the zebrafish PrPs and this will be described in more detail in subsequent sections.
Recently, the first non-experimental animals without PrPC expression were identified. Naturally occurring PrPC-null animals represent a useful resource for studying PrPC function since they are free from the confounding factors that can be introduced by genetic manipulation. The animals in question were goats of the Norwegian Dairy Goat breed that were homozygous for a Prnp allele with a premature stop codon at position 32, just a few amino acids after the end of the N-terminal signal peptide (Benestad et al., 2012). These PrPC-null goats appeared to reproduce and behave normally (Benestad et al., 2012), although it has been reported that they may have increased red blood cell and neutrophil counts (Reiten et al., 2015). One would expect novel insights to arise from these animals in due course.
Significant research efforts have been directed toward investigating the physiological function of PrPC in various cellular and animal models. The results from these studies have suggested roles for PrPC in numerous processes, as will be explained in the following sections. In order to simplify our description of PrPC function, we have aimed to group together putative functions that may be related, including some that may seem unconnected at first glance but could, in fact, result from PrPC regulating the same underlying biochemical pathways.
The most extensive body of work relating to PrPC function addresses the putative stress-protective properties of the protein. Initial evidence came from PrPC protecting neuronal “HpL” cells from serum withdrawal (Kuwahara et al., 1999)—the loss of growth and survival factors present in serum results in activation of mitochondria-dependent apoptotic signalling driven by a protein called Bax (Deckwerth et al., 1996). Further studies using HpL cell lines appeared to confirm that PrPC expression confers protection against serum deprivation-induced apoptosis (Kim et al., 2004; Wu et al., 2008). However, as described by Kuwahara et al. (1999), the HpL cell lines were generated by immortalisation of primary hippocampal neurons derived from embryonic Rikn PrPC-null mice. As explained previously, Dpl is ectopically expressed in the brains of these mice; consequently, the HpL cell lines also express Dpl (Sakudo et al., 2005). Therefore, the greater susceptibility of HpL cells to serum deprivation compared with control cell lines derived from wild type mice may have been caused by the toxic effects of Dpl rather than the absence of PrPC expression. Additionally, the increased resistance to serum withdrawal following reintroduction of PrPC into the HpL cells (Kim et al., 2004; Wu et al., 2008) may merely have been caused by PrPC interacting with Dpl and inhibiting its neurotoxicity (Moore et al., 1999; Sakudo et al., 2005; Qin et al., 2006), which, as previously mentioned, is unlikely to be a physiologically relevant function of PrPC. More recently, the Zurich I line of PrPC-knockout mice, which do not have ectopic Dpl expression in the CNS, was used for the production of additional immortalised hippocampal cell lines, although there have been conflicting reports over whether or not PrPC expression confers significant protection against serum deprivation in these cells (Nishimura et al., 2007; Oh et al., 2008). Nonetheless, PrPC expression has been shown to protect human primary neurons and MCF-7 breast cancer cells from microinjection or transfection with Bax-expressing constructs (Bounhar et al., 2001; Roucou et al., 2005). In addition, viability of MDA-MB-435 breast cancer cells following serum deprivation was reduced by RNA interference-mediated knockdown of PrPC expression (Yu et al., 2012).
A number of studies have assessed how PrPC affects the cellular response to staurosporine, a potent but relatively non-selective adenosine triphosphate-competitive kinase inhibitor (Ruegg and Burgess, 1989) that seems to induce mitochondria-independent apoptotic signalling as well as affecting the same apoptotic pathways activated by serum deprivation (Zhang et al., 2004). PrPC expression has been reported to protect primary hippocampal neurons from staurosporine-mediated cell death, possibly through an interaction with stress-induced phosphoprotein 1 (STI1) (Lopes et al., 2005; Beraldo et al., 2010; Ostapchenko et al., 2013). STI1 is a secreted protein thought to interact with PrPC, leading to activation of the pro-survival protein kinase A (PKA) signalling pathway (Zanata et al., 2002; Lopes et al., 2005). Interestingly, studies using primary cortical neurons derived from Zurich I PrPC-knockout mice found that transfection with a vector encoding full length PrPC actually increased susceptibility to staurosporine (Paitel et al., 2004), whereas the N1 fragment produced by α-cleavage was neuroprotective (Guillot-Sestier et al., 2009). There are several additional reports of full length PrPC expression conferring reduced viability in response to staurosporine exposure in a number of cell lines (Paitel et al., 2002, 2003; Sunyach et al., 2007; Guillot-Sestier et al., 2009).
In addition to direct effects on apoptosis, PrPC reportedly protects cells from oxidative stress. For example, basal levels of ROS and lipid peroxidation were lower in PrPC-transfected neuroblastoma and epithelial cell lines compared with untransfected controls (Rachidi et al., 2003; Zeng et al., 2003). Moreover, PrPC expression by primary neurons, astrocytes and cell lines has been associated with lower levels of damage following exposure to various oxidative toxins (Brown et al., 1997b, 2002; Anantharam et al., 2008; Dupiereux et al., 2008; Bertuchi et al., 2012). A possible mechanism is that PrPC modulates the activities of the antioxidant enzymes that convert ROS into less toxic products—several studies have shown lower superoxide dismutase and glutathione peroxidase activities in the absence of PrPC expression (Brown et al., 1997b; Miele et al., 2002; Rachidi et al., 2003; Sakudo et al., 2003; Paterson et al., 2008). Since ROS may induce β-cleavage of PrPC in the presence of Cu2+, the C2 or N2 fragments could be responsible for the putative antioxidant properties of PrPC. Indeed, it has been reported that N2 lowers ROS production in response to serum deprivation in neuronal cell lines and neural stem cells (NSCs) (Haigh et al., 2015a,b). As well as potentially increasing the activities of antioxidant enzymes, a recent study proposed that PrPC translocates to the nucleus in response to oxidative stress-induced DNA damage and directly activates the base excision repair pathway by interacting with AP endonuclease and enhancing its activity (Bravard et al., 2015). Additionally, PrPC-mediated activation of the tyrosine-protein kinase Fyn might lead to a stress-protective release of calcium ions from stores in the ER (Krebs et al., 2007). However, not all the evidence agrees with an antioxidant role for PrPC. Other studies have found no differences in superoxide dismutase activity between PrPC-null and wild type mice in the spinal cord, spleen or brain (Hutter et al., 2003; Steinacker et al., 2010). Additionally, although PrPC protected neuroblastoma cells from the oxidative toxin 3-morpholinosynonimine hydrochloride through activation of the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)-RACα serine/threonine-protein kinase (Akt) signalling pathway, PrPC transfection actually increased susceptibility to hydrogen peroxide treatment (Vassallo et al., 2005). Incidentally, PI3K-Akt signalling can also activate the mammalian target of rapamycin (mTOR) pathway, leading to reduced levels of autophagy, thereby rationalising the reported regulation of autophagic flux by PrPC (Nah et al., 2013; Shin et al., 2014).
Finally, PrPC has been implicated in the response to ER stress, which is caused by accumulation of unfolded/misfolded proteins within the ER. A cell responds to ER stress by triggering the unfolded protein response, which consists of: (1) increased expression of chaperones to improve protein folding; (2) global inhibition of protein synthesis; (3) an increase in ER volume; and (4) activation of the ER-associated protein degradation pathway, a key pathway in neurodegenerative disorders (Halliday and Mallucci, 2014). Interestingly, ER stress-response elements have been found within the human PRNP promoter and they appear active, since PrPC expression was induced following treatment of breast carcinoma cells with toxins that cause ER stress (Dery et al., 2013). In the same study, knockdown of PrPC expression in several cancer cell lines resulted in increased cell death in response to these toxins. Higher PrPC expression in spite of the global, ER stress-induced reduction in protein synthesis suggests a potential role for PrPC in the unfolded protein response, although results from other studies argue against a protective role for PrPC during ER stress (Roucou et al., 2005; Anantharam et al., 2008).
In conclusion, the idea that PrPC has a direct stress-protective function remains unproven. For example, there is no clear data supporting a protective role for PrPC expression in the response to ER stress and, depending on the cellular context, PrPC expression is seemingly either pro-survival or pro-apoptotic following exposure to staurosporine. There is evidence that PrPC expression protects cells from serum deprivation, although this is weakened by the use of the Dpl-expressing HpL cells in several of the relevant studies. Arguably, the evidence that PrPC can protect cells from oxidative stress is more convincing, although, as far as we are aware, this putative function has yet to be confirmed in the genetically pure Npu or Zurich III lines of PrPC-knockout mice.
In addition to its reported role in neuroprotection, several studies have shown that PrPC promotes neurite outgrowth, and potential explanations include interactions of PrPC with STI1 (Lopes et al., 2005), neural cell adhesion molecule 1 (NCAM1) (Santuccione et al., 2005), epidermal growth factor receptors (Llorens et al., 2013), integrins (Loubet et al., 2012), laminin (Graner et al., 2000a), or metabotropic glutamate receptors (mGluRs) (Beraldo et al., 2011). The downstream signalling responsible may include inhibition of the ras homolog gene family, member A (RhoA)-Rho-associated protein kinase (ROCK) pathway (Loubet et al., 2012). When activated, this signalling pathway stabilises the actin cytoskeleton, thereby inhibiting the development of filopodia—dynamic protrusions from the neurite growth cone that respond to the extracellular environment to guide migration of the developing neurite (O'Connor et al., 1990). Activation of the extracellular signal-regulated kinases 1 and 2 (ERK1/2), PI3K-Akt, and protein kinase c (PKC) signalling pathways may also be involved in mediating PrPC-dependent neurite outgrowth (Lopes et al., 2005; Caetano et al., 2008; Beraldo et al., 2011; Llorens et al., 2013). It should be cautioned that signal-regulatory protein α-1 reportedly modulates neurite outgrowth (Wang and Pfenninger, 2006) and, as previously mentioned, polymorphisms between Zurich I PrPC-null mice and their wild type counterparts in the gene encoding signal-regulatory protein α-1 can be present even after extensive backcrossing to an inbred strain (Nuvolone et al., 2016). This confounding factor affects the interpretation of several studies that used cells derived from these mice to show apparent regulation of neurite outgrowth by PrPC (Lopes et al., 2005; Santuccione et al., 2005; Beraldo et al., 2011). However, investigations of other cell lines are free from this problem and a connection between PrPC expression and neurite outgrowth has been demonstrated repeatedly in such models (Graner et al., 2000a,b; Loubet et al., 2012; Llorens et al., 2013).
Neurite outgrowth is a feature of neuronal differentiation, which raises the possibility that the effects of PrPC expression on neurite outgrowth may be consequences of PrPC regulating differentiation. Indeed, such a role may not be restricted to the nervous system, since there is evidence that PrPC can influence some of the earliest differentiation processes that occur during embryogenesis. For example, PrPC may indirectly modulate NCAM1 polysialylation in order to regulate the epithelial-to-mesenchymal transition (Mehrabian et al., 2015, 2016), a vital developmental process that alters cell adhesion and enables cell migration. Ectodermal cells in the developing embryo must undergo epithelial-to-mesenchymal transition as part of gastrulation, which may explain why knockdown of PrP1 expression in zebrafish embryos has been shown to prevent this process (Malaga-Trillo et al., 2009). This finding suggests that PrPC may have an evolutionarily-conserved role in regulating cell adhesion. Indeed, zebrafish PrP1 and PrP2 are reported to regulate the connections between adherens junctions and the actin cytoskeleton, mainly by affecting the localisation of E-cadherin and β-catenin to these junctions (Malaga-Trillo et al., 2009; Sempou et al., 2016). This process seems to rely upon signalling through members of the Src family of tyrosine-protein kinases (Sempou et al., 2016) that could be mediated by PrP1/2 interacting with the zebrafish NCAM1 ortholog (Santuccione et al., 2005). PrPC has been reported to regulate other differentiation processes. For example, knocking down PrPC expression in cultured human embryonic stem cells delayed spontaneous differentiation into the three germ layers (Lee and Baskakov, 2013). In a similar manner, PrPC expression has been shown to promote guided differentiation of cultured human embryonic stem cells and neural precursors into neurons, astrocytes and oligodendrocytes (Steele et al., 2006; Lee and Baskakov, 2014). Furthermore, a role in regulating cell adhesion suggests that PrPC expression in cancer may affect the tendency to metastasise. Indeed, Chieng and Say (2015) found that overexpression of PrPC in a colon adenocarcinoma cell line increased invasiveness, whilst a study of pancreatic ductal adenocarcinoma patients reported that PrPC-positive tumours were associated with reduced survival time from diagnosis (Sy et al., 2011). PrPC expression was also shown to be upregulated in cancerous tissue from human gastric cancer patients compared with adjacent non-cancerous tissue from the same individuals (Zhou et al., 2014), although a recent study of a larger patient group found the opposite correlation (Tang et al., 2016).
Differentiation often involves a change in cell morphology, such as neurite extension, and this will likely require altered expression of cytoskeletal proteins. Several proteomic studies have identified such changes in cell lines in which PrPC was knocked down or overexpressed (Provansal et al., 2010; Weiss et al., 2010; Mehrabian et al., 2014) and also in PrPC-null liver tissues compared with wild type controls (Arora et al., 2013). Analyses of brain tissues have been less successful at detecting differential expression of cytoskeletal proteins (or of any proteins for that matter), although this could be because cell type-specific effects of PrPC are averaged out over an entire tissue (Crecelius et al., 2008; Mehrabian et al., 2016). Another common feature of differentiation is a change of cell cycle progression. For example, neural precursors withdraw from the cell cycle as they differentiate into post-mitotic neurons. In this regard, PrPC has been shown to inhibit proliferation of oligodendrocyte precursors (Bribian et al., 2012), neuronal cells (Kim et al., 2005) and cells derived from the intestinal epithelium (Morel et al., 2008). In the case of NSCs, one study found that expression of full length PrPC promoted proliferation, whilst the N1 and N2 fragments appeared to be inhibitory (Haigh and Collins, 2014). Interestingly, proliferation and self-renewal of NSCs and their differentiation into neurons seem to rely, in part, upon signalling mediated by ROS (Le Belle et al., 2011) and the effects of the different N-terminal PrP fragments on NSC proliferation apparently arose from their modulation of intracellular ROS levels (Haigh and Collins, 2014). Therefore, regulation of NSC proliferation by PrPC may be a consequence of its putative antioxidant properties or, conversely, protection from oxidative stress by PrPC may result from a role in regulating physiological production of ROS for signalling purposes. PrPC has also been observed to promote proliferation of colon adenocarcinoma cells (Chieng and Say, 2015), neuroblastoma cells (Llorens et al., 2013), cancer stem cells that give rise to glioblastoma (Corsaro et al., 2016), and precursor cells in intestinal organoid cultures (Besnier et al., 2015). In neuroblastoma cells, PrPC was proposed to interact with the epidermal growth factor receptor to promote activation of the PI3K-Akt pathway (Llorens et al., 2013). PI3K-Akt-dependent mTOR activation might also drive the increased protein synthesis required for differentiation processes, such as changes to morphology (Roffe et al., 2010).
Although, PrPC-dependent regulation of neurite outgrowth, cell adhesion, expression of cytoskeletal proteins and proliferation could have been addressed separately, we argue that these putative functions can be understood in the context of PrPC modulating different types of cellular differentiation. This explanation is attractive because, rather than affecting various cellular processes via independent mechanisms, PrPC may, instead, regulate the function of a single interacting partner that underpins differentiation—given that altered regulation of cell adhesion would have an impact on many differentiation processes the interacting partner could be NCAM1, but there are many other possibilities and further research will be required to obtain a definitive answer.
As described in the previous section, interactions between PrPC and mGLuRs have been linked to regulation of neurite outgrowth (Beraldo et al., 2011). There are also reports of PrPC interacting with other neurotransmitter receptors, including α7 nicotinic acetylcholine (Beraldo et al., 2010), kainate (Carulla et al., 2011), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (Kleene et al., 2007), and N-methyl-D-aspartate receptors (NMDARs) (Khosravani et al., 2008). NMDARs are a subclass of ionotropic glutamate receptors and are particularly implicated in excitotoxic neuronal cell death, which occurs when overactivation of NMDARs causes dysregulation of intracellular calcium homeostasis, leading to disruption of various physiological processes (Bondy and Lee, 1993). NMDAR-mediated excitotoxicity contributes to neuronal death in prion diseases and other neurodegenerative disorders (Muller et al., 1993; Gorman, 2008). Intriguingly, PrPC has been reported to inhibit activity of NMDARs containing the GluN2D subunit, which suggests that PrPC may protect neurons from excitotoxic death (Khosravani et al., 2008). There is evidence for this in vivo, for example, Zurich I PrPC-knockout mice displayed increased excitotoxicity in the retina in response to damaging light intensities (Frigg et al., 2006) and in the hippocampus following injection with N-methyl-D-aspartate (Khosravani et al., 2008), which selectively activates NMDARs. Given that excitotoxicity is the major cause of neuronal death following ischaemic stroke (Lai et al., 2014), it is also interesting to note that experimental stroke has been shown to induce PrPC expression in the affected brain region in both rats and mice (Weise et al., 2004; Shyu et al., 2005). Furthermore, Zurich I PrPC-null mice displayed greater infarct volumes than wild type controls after transient or permanent middle cerebral artery occlusion (Spudich et al., 2005; Weise et al., 2006), whilst injection of a PrPC-expressing adenovirus construct into rat brains reduced infarct volume in a similar experimental model (Shyu et al., 2005). In addition to potentially modulating NMDAR activity, involvement of PrPC in the cellular response to oxidative stress could explain the putative protective effect of PrPC expression following stroke, since oxidative stress is a feature of excitotoxicity.
Increased sensitivity to seizures induced by high-dose kainate treatment has been observed in studies of Zurich I PrPC-null mice (Rangel et al., 2009; Carulla et al., 2011). However, this finding has been called into question by Striebel et al. (2013b), who presented evidence that Prnp flanking genes may be responsible for the seizure phenotype rather than Prnp itself. Further adding to the complexity is a recent report suggesting that, whilst Prnp flanking genes may affect sensitivity to kainate treatment, PrPC expression also has a neuroprotective effect (Carulla et al., 2015). Additionally, one study found that hippocampal slices from Zurich I PrPC-null mice were actually more resistant than wild type controls to three different seizure-inducing protocols, including removal of Mg2+ from the culture medium, which leads to hyperexcitability of NMDARs, and treatment with gamma-aminobutyric acid (GABA) receptor antagonists (Ratte et al., 2011).
If PrPC can modulate neuronal excitability then one might expect a lack of PrPC expression to affect synaptic plasticity. Indeed, Collinge et al. (1994) showed that Zurich I PrPC-knockout mice had disrupted hippocampal long-term potentiation (LTP), a form of synaptic plasticity involved in memory formation. These mice displayed abnormal behaviour in nest building and novel environment exploration tasks and more pronounced age-related decline in short-term memory compared to wild type controls (Schmitz et al., 2014), effects that are possibly underpinned by impaired LTP. Intriguingly, it has been reported that the effect of ageing on memory can be blocked by infusing wild type mice with a peptide containing the putative PrPC-binding site of STI1 (Rial et al., 2009). Mice overexpressing PrPC were similarly protected from age-related memory decline in the same study. NMDAR activity is required for LTP in the area of the hippocampus that was used by Collinge et al. (1994), suggesting that the disrupted LTP observed by the authors in PrPC-null hippocampal slices could have been caused by PrPC ablation affecting NMDAR activity, as has been described previously (Khosravani et al., 2008). However, Collinge et al. (1994) found that the contribution of NMDAR-mediated currents to excitatory postsynaptic potentials was unaffected by PrPC knockout. Instead, the lack of PrPC expression resulted in weaker fast inhibitory postsynaptic potentials generated through GABA receptors. In contrast, a different study identified no effects of PrPC knockout on inhibitory currents generated by GABA receptors nor, indeed, on LTP itself (Lledo et al., 1996).
Together, the findings reported in this section paint a confusing picture, indicating that further work is required to determine conclusively how PrPC affects neuronal excitability. There is increasing recognition of the contribution of Prnp flanking genes to phenotypes displayed by PrPC-knockout mice that were generated on mixed genetic backgrounds, an issue that confounds interpretation of many studies connecting PrPC to the regulation of neuronal excitability—this problem is documented in more detail in a couple of recent review articles (Striebel et al., 2013a; del Rio and Gavin, 2016). Nevertheless, there are data from animal models other than mice that support the involvement of PrPC in the modulation of neuronal excitability. For example, knocking out PrP2 expression in zebrafish using zinc finger nuclease technology affected NMDAR currents and increased susceptibility to seizures induced by the drug pentylenetetrazol (Fleisch et al., 2013). Furthermore, in a study of the Drosophila neuromuscular junction, researchers found that murine PrPC expression resulted in enlarged synaptic vesicles and increased the probability of neurotransmitter release (Robinson et al., 2014).
One phenotype of PrPC-knockout mice that was discovered relatively recently is a widespread, adult-onset demyelination of the PNS. This phenotype has now been observed in Zurich I and Npu knockout mice (Bremer et al., 2010) as well as the newly generated Zurich III line (Nuvolone et al., 2016), which provides strong evidence that PrPC is involved in myelin maintenance. Intriguingly, Bremer et al. (2010) showed that neuron-specific PrPC expression was sufficient to rescue the demyelination phenotype, whereas PrPC expressed only in Schwann cells had little effect. More recent work has shown that PrPC may promote myelin maintenance through an interaction between its extreme N-terminal region (residues 23–33) and G-protein coupled receptor 126 (GPR126) on the surface of Schwann cells (Kuffer et al., 2016). There is some evidence that α-cleavage of neuronal PrPC is needed to prevent PNS demyelination (Bremer et al., 2010), perhaps indicating that the released N1 fragment, which contains the 23–33 region, is responsible for the interaction with GPR126. Binding of the PrPC N-terminus seems to promote receptor activation, initiating signalling through the cyclic adenosine monophosphate (cAMP)-PKA pathway (Kuffer et al., 2016). In zebrafish, PKA acting downstream of GPR126 was shown to induce myelination during embryonic development by upregulating expression of E3 SUMO-protein ligase EGR2 (Glenn and Talbot, 2013), a transcription factor that also seems to regulate myelin maintenance (Decker et al., 2006). In spite of the importance of the GPR126-PKA signalling axis in the initial myelination of PNS axons, PrPC-null mice appear to develop morphologically-normal myelin before onset of the demyelinating polyneuropathy (Bremer et al., 2010). Kuffer et al. (2016) suggested that developmental myelination may progress normally in the PrPC-knockout mice because the lack of PrPC expression is compensated for by other GPR126 ligands such as type IV collagen and laminin-211, which have both been reported to regulate GPR126 activity (Paavola et al., 2014; Petersen et al., 2015). However, it has also been reported that GPR126-PKA signalling is not required for myelin maintenance. For example, E3 SUMO-protein ligase EGR2 expression by zebrafish Schwann cells was shown to be independent of GPR126 signalling following completion of initial myelination (Glenn and Talbot, 2013). Thus, the molecular mechanisms by which PrPC affects myelin integrity are still to be determined conclusively.
No myelin-related abnormalities have been identified within the CNS of PrPC-knockout mice (Bremer et al., 2010), perhaps because CNS myelination reportedly does not depend upon GPR126 activity (Monk et al., 2011). However, it is of note that spinal cord expression of PrPC was found to be reduced in human patients with multiple sclerosis, a demyelinating disease of the CNS (Scalabrino et al., 2015).
The distinctive clinical feature of a human prion disease known as fatal familial insomnia is severe disruption of the sleep-wake cycle (Collins et al., 2001). Strikingly, PrPC-null mice also exhibit alterations to their sleep structure, including faster cycling through the different stages of sleep and more brief awakenings than wild type mice (Tobler et al., 1996, 1997; Sanchez-Alavez et al., 2007). These issues may be caused by changes to the regulation of melatonin production—melatonin is synthesised by the pineal gland and regulates sleep timing as well as other processes. Melatonin production is under the control of the suprachiasmatic nucleus of the hypothalamus, which is the master regulator of the circadian rhythms that drive the sleep-wake cycle. Thus, melatonin levels vary according to a 24-h cycle—for nocturnal animals such as mice, levels are highest in the hours of darkness and lower levels are required for sleep initiation. One study found that serum melatonin levels of Zurich I and Npu PrPC-knockout mice were considerably lower than in wild type mice during the dark phase of the cycle but were higher during the light phase (Brown et al., 2002), which probably contributed to the sleep disruption. A network of proteins called the circadian clock work together to generate circadian rhythms and, strikingly, Prnp mRNA levels seem to be subject to circadian oscillations in the suprachiasmatic nucleus in addition to other areas of the rat forebrain (Cagampang et al., 1999). Together, these lines of evidence suggest that PrPC might have a regulatory role in the sleep-wake cycle, raising the possibility that a loss of this function is involved in the pathogenesis of fatal familial insomnia.
Given that metabolic processes are also subject to circadian regulation (Dibner et al., 2010), one might expect PrPC-knockout mice to show signs of altered metabolism. Interestingly, recent data suggest that PrPC knockout does have an effect on glucose homeostasis. For example, Strom et al. (2011) demonstrated that, compared with wild type controls, blood glucose levels of PrPC-null mice were slower to return to normal after intraperitoneal injection of glucose. In the same study, pancreatic insulin secretion and the effects of intraperitoneal insulin injection on blood glucose levels were shown to be similar irrespective of PrPC expression, however, it has been reported that insulin resistance develops more quickly in PrPC-null mice in response to a high fat diet (Brito et al., 2013). More work is required to unpick the mechanisms behind these phenotypes, although evidence obtained in vitro suggests that PrPC-mediated Fyn kinase signalling can lead to activation of hypoxia-inducible factor-2α and subsequent upregulation of glucose transporter 1 expression, resulting in increased glucose uptake by cells (Li et al., 2011). Additionally, glucose homeostasis is known to be regulated by the PI3K-Akt pathway (Schultze et al., 2012), which PrPC has also been reported to modulate (Vassallo et al., 2005; Roffe et al., 2010; Llorens et al., 2013).
A number of early studies of PrPC function focused on an apparent ability to bind Cu2+ ions at the cell membrane through the octapeptide repeat region (Hornshaw et al., 1995a,b; Brown et al., 1997a). Subsequently, PrPC interactions with Cu2+ have been implicated in the regulation of NMDAR activity (Gasperini et al., 2015), astrocytic glutamate uptake (Brown and Mohn, 1999), protection against oxidative stress (Rachidi et al., 2003; Watt et al., 2007) and maintenance of Cu2+ homeostasis in the placenta (Alfaidy et al., 2013). In addition, binding of Cu2+ by PrPC in the presence of ROS appears to promote β-cleavage of PrPC, leading to production of C2 and N2 (McMahon et al., 2001; Watt et al., 2005; McDonald et al., 2014). However, whilst it is clear that PrPC can bind Cu2+ in vitro, the relevance of this interaction in vivo has been questioned. For example, it was reported that PrPC-dependent internalisation of Cu2+ by cells in culture occurred only when the extracellular Cu2+ concentration exceeded physiologically relevant levels (Rachidi et al., 2003). Furthermore, although some studies have shown that PrPC expression can protect neuronal and non-neuronal cell lines from oxidative stress caused by excessive Cu2+ (Rachidi et al., 2003; Watt et al., 2007), Cingaram et al. (2015) found that transfecting PrPC into the Zpl hippocampal cell line, originally derived from Zurich I PrPC-knockout mice, did not result in protection from Cu2+-mediated toxicity.
Experiments in neuronal cell lines showed that PrPC transfection did not confer protection against the oxidative DNA damage and subsequent cell death caused by treatment with high levels of Mn2+, Co2+, or Zn2+ (Watt et al., 2007; Cingaram et al., 2015). However, PrPC expression did protect neuroblastoma cells from the toxic effects of Fe2+ treatment (Watt et al., 2007). Furthermore, PrPC appears to be involved in the physiological uptake of iron by cells; this was first shown in neuroblastoma cells, which became more efficient at sequestering iron from the culture medium following overexpression of PrPC (Singh et al., 2009b). Singh et al. (2009a) also showed that PrPC transfection increased expression of the iron-storage protein ferritin and reduced expression of both the iron-transport protein transferrin and the transferrin receptor. These alterations to the expression levels of iron-binding proteins are part of the homeostatic response that ensures that the amount of labile iron within cells remains constant when iron uptake is enhanced (Hare et al., 2013). Further evidence supporting a role for PrPC in iron homeostasis has been obtained in vivo, where the liver, spleen, brain and kidneys of Zurich I PrPC-null mice have all been shown to have lower total levels of iron than wild type controls (Singh et al., 2009a; Haldar et al., 2015). Consequently, PrPC-knockout also reduced ferritin expression and increased expression of transferrin and its receptor in various tissues (Singh et al., 2009a). Most iron circulating in the bloodstream is in the form of Fe3+ bound to transferrin (Hare et al., 2013); however, Fe3+ must be reduced to Fe2+ by a ferrireductase to enable uptake by cells. PrPC itself may have ferrireductase activity (Singh et al., 2013; Haldar et al., 2015) and may act to enhance iron uptake through divalent-metal transporter 1 and ZIP14 (Tripathi et al., 2015), although an alternative explanation is that PrPC binds through its octapeptide repeat region to a ferrireductase in order to modulate its activity (Tripathi et al., 2015). One final point to make is that PrPC-null mice are only mildly anaemic, probably because, in spite of the low levels of stored iron, sufficient quantities of labile iron were present to maintain normal cellular function (Singh et al., 2009a).
In conclusion, more than two decades after the discovery that PrPC can bind Cu2+, the functional significance of this interaction is still unclear. However, the recent reports proposing that PrPC is involved in iron homeostasis are intriguing. Further research is necessary to clarify the mechanisms responsible and to determine whether other PrPC-knockout mouse lines also display signs of anaemia.
Because prion diseases are neurodegenerative disorders, much of the research into PrPC function has focused on its role in the nervous system. However, PrPC is also expressed in immune cells, including T-lymphocytes, natural killer cells, macrophages and mast cells (Durig et al., 2000; Haddon et al., 2009). Mast cells reportedly express high levels of PrPC, some of which is shed from the cell membrane as the N3 fragment (Haddon et al., 2009). When activated, mast cells release various inflammatory mediators, including histamine, prostaglandins and cytokines (Stempelj and Ferjan, 2005). Interestingly, treatment of cultured mast cells with compounds that induce activation resulted in rapid shedding of a large proportion of the cellular pool of PrPC, suggesting that PrPC could be involved in the inflammatory response (Haddon et al., 2009). Mast cells are also involved in angiogenesis (Maltby et al., 2009), a process that PrPC seems to modulate within the placenta (Alfaidy et al., 2013), although this function may be more linked to the putative antioxidant properties of PrPC. Moreover, mast cells release pain mediators upon activation (Chatterjea and Martinov, 2015) and ablation of PrPC expression in mice is associated with altered sensitivity to pain. However, whilst one study found that PrPC-null mice showed a hypersensitivity to pain (Gadotti and Zamponi, 2011), another demonstrated that PrPC-null mice were more resistant than wild type controls (Meotti et al., 2007). It is conceivable that the apparent influence of PrPC expression on pain sensitivity stems from a role in regulating mast cell function, although Gadotti and Zamponi (2011) provided evidence that loss of PrPC-dependent NMDAR inhibition was responsible for the hypersensitivity to pain displayed by the PrPC-knockout mice in their study.
PrPC expression by T-cells may be involved in their differentiation, since knockdown of PrPC resulted in an increased tendency for T-cells to develop a pro-inflammatory phenotype (Hu et al., 2010). Additionally, in a model of autoimmune encephalomyelitis, PrPC-knockout mice experienced a more severe disease phenotype that was linked to increased T-cell-dependent neuroinflammation (Tsutsui et al., 2008), possibly because PrPC is involved in the signalling complexes that regulate T-cell activation (Mabbott et al., 1997; Mattei et al., 2004). In contrast, another study employing the autoimmune encephalomyelitis model reported no evidence that T-cells were driven toward a more inflammatory phenotype by PrPC knockout and, instead, the increased disease severity seemed to result from loss of PrPC expression within the CNS, not the immune system (Gourdain et al., 2012).
As explained previously, the first non-laboratory animals lacking PrPC expression were recently identified (Benestad et al., 2012). No overt abnormalities were detected in these PrPC-null goats (Benestad et al., 2012), although increased neutrophil numbers compared with PrPC-expressing controls were reported recently (Reiten et al., 2015). Further study of these PrPC-null goats may help to improve understanding of how PrPC expression influences the immune system, knowledge that is currently rather limited. Of particular interest is the putative function of PrPC expression in mast cells, given the multiple lines of circumstantial evidence, detailed in this section, that hint at an important role for PrPC in these cells.
Another function sometimes ascribed to PrPC is maintenance of mitochondrial homeostasis. For instance, whilst mouse studies have shown that PrPC-knockout does not affect the membrane potential or baseline respiration rate of mitochondria, nor the activities of the individual complexes of the electron transport chain (Brown et al., 2002; Miele et al., 2002; Lobao-Soares et al., 2005), transcriptomic, and proteomic analyses of brain tissue and cultured cells have identified specific subunits of each of the five electron transport chain complexes that vary in their expression between PrPC-null and wild type mice (Miele et al., 2002; Ramljak et al., 2008; Stella et al., 2012). Furthermore, the Npu line of PrPC-null mice were found to have reduced numbers of mitochondria in the brain and myocardium, although the mitochondria that were present were larger and had increased maximal respiratory capacities, presumably to compensate for the lower numbers (Miele et al., 2002; Paterson et al., 2008). The enhanced maximal respiratory capacity of individual PrPC-null mitochondria seemed to be enabled by increased activity of electron transport chain complex I, which was, in turn, associated with an increase in superoxide production (Paterson et al., 2008). In line with these findings, superoxide dismutase 2, which is found within mitochondria and converts superoxide into either hydrogen peroxide or molecular oxygen, was reportedly more active in PrPC-null mice (Brown et al., 1997b; Miele et al., 2002; Paterson et al., 2008). Furthermore, it is conceivable that the reported antioxidant properties of PrPC could derive from a role in maintaining mitochondrial homeostasis and, consequently, preventing excessive production of ROS. It should be emphasised, however, that the evidence suggesting that PrPC may regulate mitochondrial function comes from a very limited number of studies and, as far as we aware, the only published examples of mitochondrial phenotypes in PrPC-null mice are from studies of the Npu line.
β-Cleavage of the amyloid precursor protein (APP) by beta-secretase 1 produces a fragment called sAPPβ that is subsequently cleaved by gamma-secretase to produce amyloid beta (Aβ) peptides. Aβ accumulates during pathogenesis of Alzheimer's disease (AD) and PrPC reportedly acts as an inhibitor of β-secretase 1 (Parkin et al., 2007; Whitehouse et al., 2013), thereby reducing the amount of Aβ produced and suggesting that PrPC expression should protect against the development of AD. A feedback loop has also been proposed that consists of the APP intracellular domain (the other fragment produced by cleavage of sAPPβ) activating cellular tumour antigen p53, which subsequently upregulates PrPC expression, leading to further inhibition of β-secretase 1 activity (Vincent et al., 2009). However, recent data have questioned whether the APP intracellular domain is able to induce PrPC expression (Lewis et al., 2012), whilst another study found that PrPC expression actually led to higher rates of APP β-cleavage (McHugh et al., 2012). In addition, removing PrPC from human APP-expressing mice has no effect on processing or Aβ levels (Whitehouse et al., 2016).
In addition to potentially affecting APP processing, it has been suggested that PrPC downregulates transcription of microtubule-associated protein tau by activating Fyn kinase signalling (Chen et al., 2013). This putative function would also be protective against AD, since accumulation of neurofibrillary tangles consisting of hyperphosphorylated tau is another major molecular feature of the disease. Conversely, oligomeric forms of Aβ are reported to bind to PrPC at either its extreme N-terminal region (Chen et al., 2010; Dohler et al., 2014) or between residues 92 and 110 (Lauren et al., 2009), potentially eliciting toxic effects through hyperactivation of Fyn kinase (Vergara et al., 2015). The C1 fragment, created by α-cleavage of PrPC, lacks both the putative Aβ binding sites. Therefore, the reduced levels of hippocampal PrPC and the increased rates of PrPC α-cleavage in the cortex that have been reported in the brains of sporadic AD patients could be part of a protective response (Whitehouse et al., 2010; Beland et al., 2014). On the other hand, one study found that higher levels of PrPC in serum were correlated with poorer cognitive function in an elderly human population, although the increase in serum PrPC could be a consequence of loss of PrPC from neuronal membranes (Breitling et al., 2012). Overall, the conflicting nature of the evidence obtained to date makes it difficult to determine whether the net effect of PrPC expression on AD pathogenesis is positive or negative.
The preceding sections of this review have addressed many of the diverse functions ascribed to PrPC. Putative interacting partners of PrPC that may be involved in these functions have also been mentioned and Table 2 provides information about some of the interactions best supported by the literature. Given that the GPI-anchored PrPC molecule lacks an intracellular domain, it is likely that the protein interacts with one or more co-receptors at the cell surface to mediate downstream signalling. Several potential co-receptors are included in Table 2, such as NCAM1 and mGluRs, which may be involved in PrPC-dependent neurite outgrowth (Santuccione et al., 2005; Beraldo et al., 2011). Furthermore, the extracellular matrix protein laminin may act as a PrPC ligand (Graner et al., 2000a) to activate neuritogenic signalling downstream of mGluRs (Beraldo et al., 2011). Likewise, STI1 released by astrocytes (Lima et al., 2007) may form a complex with nicotinic acetylcholine receptors and PrPC on the neuronal surface, leading to activation of the acetylcholine receptors and consequent pro-survival and neuritogenic signalling (Beraldo et al., 2010). Interactions between PrPC and neurotransmitter receptors are also implicated in the regulation of neuronal excitability, although some of these findings have been disputed, as explained previously. Further PrPC binding partners listed in Table 2 include proteins that are important at cell-cell junctions, such as desmoplakin and junction plakoglobin (Zafar et al., 2011; Besnier et al., 2015), and may be linked to the role PrPC is thought to play in the regulation of cell adhesion (Malaga-Trillo et al., 2009; Sempou et al., 2016). Additionally, PrPC reportedly binds to cytoskeletal proteins, such as tubulin and vimentin (Nieznanski et al., 2005; Zafar et al., 2011), although, given that PrPC is usually expressed at the cell surface, these findings could be artefactual. For example, cell or tissue lysis carried out prior to analysis of protein-protein interactions can result in association of proteins that do not interact physiologically because of their differing subcellular localisation. An alternative explanation is that PrPC does interact with cytoskeletal proteins but does so indirectly—not all techniques used to identify binding partners of a protein can distinguish between direct and indirect interactions. Desmoplakin, for example, could provide the link between PrPC and tubulin or vimentin, since desmoplakin is reported to interact with the microtubule-binding protein end-binding 1 (Patel et al., 2014) and with vimentin intermediate filaments (Stappenbeck and Green, 1992). In fact, many of the proteins that appear to interact with PrPC may merely be part of the same multiprotein complex or complexes rather than being direct binding partners. Alternatively, the relatively flexible structure of the PrPC N-terminal domain may allow the protein to bind directly to multiple partners (Bakkebo et al., 2015), potentially enabling PrPC to act as a scaffolding protein that mediates the formation of a number of different multiprotein complexes at the cell surface, as has been proposed previously (Linden et al., 2008).
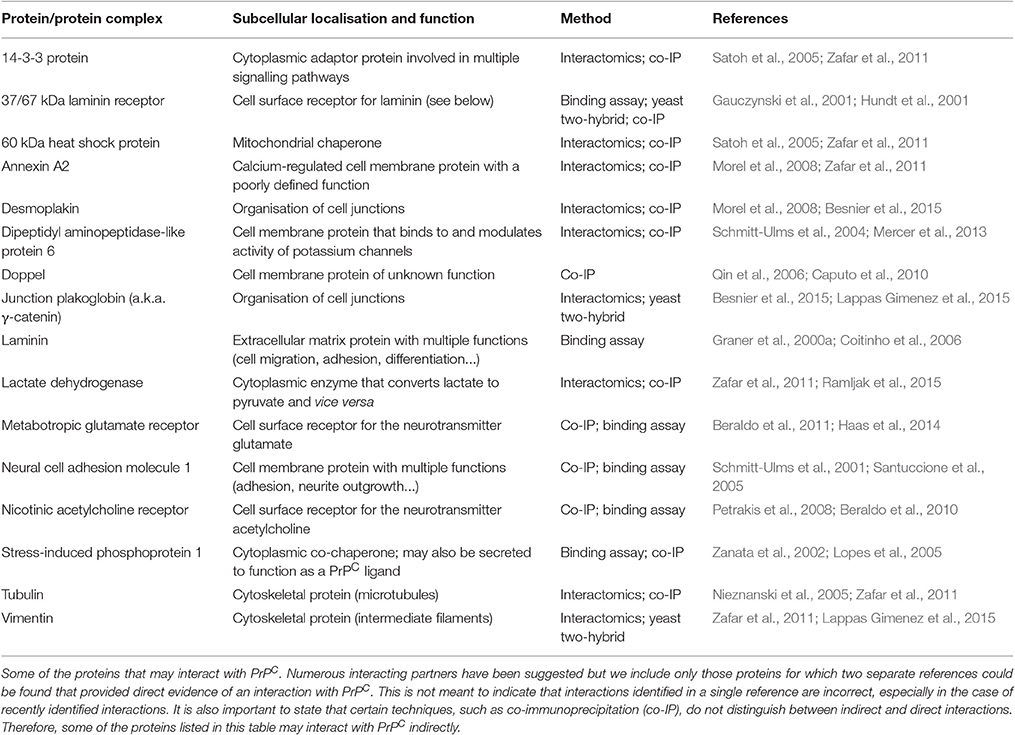
Table 2. Reported interacting partners of PrPC.
Interactions between PrPC and its co-receptor(s) initiate downstream signalling and Figure 5 provides an overview of the signalling pathways that seem to be affected by PrPC expression as well as some of the cellular processes that may be regulated in a PrPC-dependent manner through these pathways. Further information on these signal transduction pathways was retrieved from several review articles (Dhillon et al., 2007; Newton, 2010; Rosse et al., 2010; Zhang and Yu, 2012; Martini et al., 2014), enabling some of the crosstalk between pathways to be included in the figure. In brief, evidence suggests that PrPC expression affects the activities of the ERK1/2 (Lopes et al., 2005; Caetano et al., 2008; Beraldo et al., 2011) and PI3K-Akt signalling pathways (Vassallo et al., 2005; Roffe et al., 2010; Llorens et al., 2013), both of which are involved in functions ascribed to PrPC such as regulation of protein synthesis (Roffe et al., 2010) and autophagy (Nah et al., 2013; Shin et al., 2014). In addition, differential activation of the PI3K-Akt pathway may explain the effects of PrPC expression on proliferation (Llorens et al., 2013). Furthermore, PrPC appears to modulate cAMP-PKA signalling to stimulate the maintenance of PNS myelin (Kuffer et al., 2016) and to promote cell survival (Lopes et al., 2005), although it should be stressed that the literature as a whole does not provide strong support for PrPC having a direct pro-survival role, as explained previously. PrPC expression also seems to activate signalling through Src family kinases, such as Fyn kinase (Krebs et al., 2007; Chen et al., 2013), leading to changes in cell adhesion properties (Sempou et al., 2016) as well as potentially affecting the cellular uptake of glucose (Li et al., 2011), a process that can additionally be regulated by the PI3K-Akt pathway (Schultze et al., 2012). Finally, PrPC-dependent regulation of several pathways, including RhoA-ROCK (Loubet et al., 2012) and PKC (Beraldo et al., 2011) signalling, reportedly results in changes to cell morphology, such as neuritogenesis.
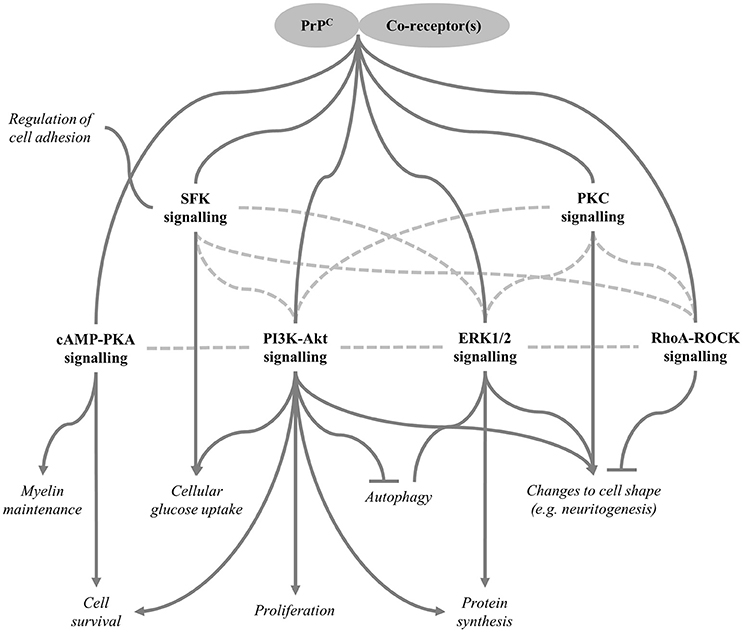
Figure 5. Signalling pathways regulated by PrPC. Various downstream signalling pathways are reportedly modulated as a result of PrPC interacting with specific co-receptors (some candidate co-receptors are included in Table 2). Cellular functions regulated by these pathways are shown in italics. Arrows indicate positive regulation, inhibition is shown by the flat-ended lines, and connectors without arrows indicate that the direction of regulation may be context-specific. Dotted lines represent crosstalk between pathways; therefore, some of the pathways shown may be regulated indirectly through other pathways rather than modulated directly by PrPC. SFK, Src family kinase, which includes Fyn kinase.
The extensive nature of the crosstalk between pathways affected by PrPC expression raises the possibility that they may not all be regulated by PrPC directly; instead, the altered activation states of some of the pathways may be indirect consequences of effects on other signalling cascades. Moreover, although PrPC expression has an activating effect on most of the pathways shown in Figure 5, it is highly unlikely that this is the case for all tissue/cell types. For example, the PI3K-Akt pathway is thought to promote proliferation (Martini et al., 2014) and yet PrPC expression has been shown to have opposing effects on proliferation in different experimental models. Context-dependent regulation of cell signalling processes by PrPC could result from co-receptors and/or downstream components of the relevant signalling pathways being differentially expressed in different tissue/cell types.
Since it was first recognised that prion protein misfolding plays an important role in the pathogenesis of prion diseases, significant attention has been directed toward understanding the physiological function of PrPC. One goal of such research has been to determine whether the disease processes of prion diseases are driven mainly by toxic gain of function caused by conversion of PrPC into PrPSc or by a loss of normal PrPC function. However, this question has yet to be answered conclusively, because a clear description of PrPC function remains elusive. This is not to say that no progress has been made; in fact, PrPC has been linked to roles in a wide variety of cellular processes. Furthermore, numerous putative interacting partners of PrPC have been identified and PrPC expression has been shown to affect a number of downstream signalling pathways. These findings have led to proposals that PrPC is a scaffolding protein that regulates the formation of various multiprotein complexes at the cell surface. However, given the potential for experimental artefacts when studying protein-protein interactions in addition to the possible detection of indirect rather than direct PrPC binding partners, the actual number of direct PrPC interactors may be considerably lower than it appears. It is also possible that some of the signalling processes reportedly affected by PrPC expression are differentially activated due to crosstalk with other pathways rather than as a result of direct regulation by PrPC. Further adding to the complexity of understanding PrPC function is recent evidence that has weakened support for the involvement of PrPC in processes such as stress-protection, copper homeostasis and the modulation of neuronal excitability.
Arguably, the best-supported phenotype of PrPC-knockout mice is adult-onset demyelination of the PNS, suggesting that PrPC expression is important for myelin maintenance. Additionally, there is accumulating evidence that PrPC expression modulates proliferation, differentiation, adhesion properties and cell shape in multiple cell types. Given that proliferation rate, adhesion properties and cell shape are all likely to be affected when cells differentiate, we have suggested in this review that PrPC may regulate a single underlying mechanism connected to differentiation and further work will be required to determine whether or not this is the case. Another intriguing phenotype of PrPC-null mice is altered sleep structure, which may result from disruption of the normal circadian variation in melatonin levels. However, there have been few publications on this topic, which is surprising since a role of PrPC in sleep regulation would suggest that loss or subversion of normal PrPC function could be involved in the development of fatal familial insomnia. Other areas of study that could be explored further include the link between PrPC and iron homeostasis as well as the putative involvement of PrPC in immune function, especially in the context of mast cells, which reportedly shed large quantities of the N3 fragment from the cell surface when activated.
Aside from the involvement of prion protein in prion diseases, there are wider reasons for pursuing investigations to clarify the function of PrPC. Firstly, given that PrPC has been shown to affect processes such as proliferation, cellular differentiation and adhesion, the expression levels of PrPC are likely to have an influence on cancer progression. Although, some attention has been focused on this topic in recent years, further research would help to determine whether treatments that modulate PrPC activity or expression levels are beneficial in cancer. Secondly, the putative connection between PrPC and glucose homeostasis makes the protein a potential target for treating metabolic disorders such as diabetes. Thirdly, given that dementia is one of the most pressing medical issues of our time, the apparent role of PrPC in the cell biology of AD warrants further attention, especially since the relevant data obtained to date are rather contradictory. Together, these areas provide further impetus for continued research into the cell biological mechanisms regulated by PrPC, raising the possibility that a clear understanding of PrPC function will be obtained in the near future.
AC conducted the literature review and wrote the first draft of the manuscript. AG supervised the work, prepared publication-ready figures and corrected the manuscript ready for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AC was supported by a Doctoral Training Programme grant (BB/JO1446X/1) from the BBSRC, UK. AG is supported, in part, by an Institute Strategic Programme Grant (BB/J004332/1) from the BBSRC, UK.
Adle-Biassette, H., Verney, C., Peoc'h, K., Dauge, M. C., Razavi, F., Choudat, L., et al. (2006). Immunohistochemical expression of prion protein (PrPC) in the human forebrain during development. J. Neuropathol. Exp. Neurol. 65, 698–706.
Alfaidy, N., Chauvet, S., Donadio-Andrei, S., Salomon, A., Saoudi, Y., Richaud, P., et al. (2013). Prion protein expression and functional importance in developmental angiogenesis: role in oxidative stress and copper homeostasis. Antioxid. Redox Signal. 18, 400–411. doi: 10.1089/ars.2012.4637
Alper, T. (1985). Scrapie agent unlike viruses in size and susceptibility to inactivation by ionizing or ultraviolet-radiation. Nature 317, 750–750. doi: 10.1038/317750a0
Alper, T., Haig, D. A., and Clarke, M. C. (1966). Exceptionally small size of scrapie agent. Biochem. Biophys. Res. Commun. 22, 278–284. doi: 10.1016/0006-291x(66)90478-5
Alper, T., Haig, D. A., and Clarke, M. C. (1978). Scrapie agent - evidence against its dependence for replication on intrinsic nucleic-acid. J. Gen. Virol. 41, 503–516. doi: 10.1099/0022-1317-41-3-503
Anantharam, V., Kanthasamy, A., Choi, C. J., Martin, D. P., Latchoumycandane, C., Richt, J. A., et al. (2008). Opposing roles of prion protein in oxidative stress- and ER stress-induced apoptotic signaling. Free Radic. Biol. Med. 45, 1530–1541. doi: 10.1016/j.freeradbiomed.2008.08.028
Arora, A. S., Zafar, S., Schmitz, M., and Zerr, I. (2013). Differential proteome analysis of cytoskeleton associated proteins in the liver of PrP knockout mice. Prion 7, 69. doi: 10.4161/pri.24865
Bakkebo, M. K., Mouillet-Richard, S., Espenes, A., Goldmann, W., Tatzelt, J., and Tranulis, M. A. (2015). The cellular prion protein: a player in immunological quiescence. Front. Immunol. 6:450. doi: 10.3389/fimmu.2015.00450
Basler, K., Oesch, B., Scott, M., Westaway, D., Walchli, M., Groth, D. F., et al. (1986). Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 46, 417–428. doi: 10.1016/0092-8674(86)90662-8
Beland, M., Bedard, M., Tremblay, G., Lavigne, P., and Roucou, X. (2014). A beta induces its own prion protein N-terminal fragment (PrPN1)-mediated neutralization in amorphous aggregates. Neurobiol. Aging 35, 1537–1548. doi: 10.1016/j.neurobiolaging.2014.02.001
Beland, M., Motard, J., Barbarin, A., and Roucou, X. (2012). PrPC homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J. Neurosci. 32, 13255–13263. doi: 10.1523/jneurosci.2236-12.2012
Beland, M., and Roucou, X. (2013). Homodimerization as a molecular switch between low and high efficiency PrPC cell surface delivery and neuroprotective activity. Prion 7, 170–174. doi: 10.1523/jneurosci.2236-12.2012
Benestad, S. L., Austbo, L., Tranulis, M. A., Espenes, A., and Olsaker, I. (2012). Healthy goats naturally devoid of prion protein. Vet. Res. 43:87. doi: 10.1186/1297-9716-43-87
Beraldo, F. H., Arantes, C. P., Santos, T. G., Machado, C. F., Roffe, M., Hajj, G. N., et al. (2011). Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma 1 chain. FASEB J. 25, 265–279. doi: 10.1096/fj.10-161653
Beraldo, F. H., Arantes, C. P., Santos, T. G., Queiroz, N. G. T., Young, K., Rylett, R. J., et al. (2010). Role of α7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem. 285, 36542–36550. doi: 10.1074/jbc.M110.157263
Bertuchi, F. R., Bourgeon, D. M. G., Landemberger, M. C., Martins, V. R., and Cerchiaro, G. (2012). PrPC displays an essential protective role from oxidative stress in an astrocyte cell line derived from PrPC knockout mice. Biochem. Biophys. Res. Commun. 418, 27–32. doi: 10.1016/j.bbrc.2011.12.098
Besnier, L. S., Cardot, P., Da Rocha, B., Simon, A., Loew, D., Klein, C., et al. (2015). The cellular prion protein PrPc is a partner of the Wnt pathway in intestinal epithelial cells. Mol. Biol. Cell 26, 3313–3328. doi: 10.1091/mbc.E14-11-1534
Blanch, E. W., Gill, A. C., Rhie, A. G. O., Hope, J., Hecht, L., Nielsen, K., et al. (2004). Raman optical activity demonstrates poly(L-proline) II helix in the N-terminal region of the ovine prion protein: implications for function and misfunction. J. Mol. Biol. 343, 467–476. doi: 10.1016/j.jmb.2004.08.058
Bondy, S. C., and Lee, D. K. (1993). Oxidative stress-induced by glutamate receptor agonists. Brain Res. 610, 229–233. doi: 10.1016/0006-8993(93)91405-h
Bounhar, Y., Zhang, Y., Goodyer, C. G., and LeBlanc, A. (2001). Prion protein protects human neurons against Bax-mediated apoptosis. J. Biol. Chem. 276, 39145–39149. doi: 10.1074/jbc.C100443200
Bravard, A., Auvre, F., Fantini, D., Bernardino-Sgherri, J., Sissoeff, L., Daynac, M., et al. (2015). The prion protein is critical for DNA repair and cell survival after genotoxic stress. Nucleic Acids Res. 43, 904–916. doi: 10.1093/nar/gku1342
Breitling, L. P., Muller, H., Stegmaier, C., Kliegel, M., and Brenner, H. (2012). Association of prion protein with cognitive functioning in humans. Exp. Gerontol. 47, 919–924. doi: 10.1016/j.exger.2012.08.001
Bremer, J., Baumann, F., Tiberi, C., Wessig, C., Fischer, H., Schwarz, P., et al. (2010). Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 13, U310–U319. doi: 10.1038/nn.2483
Bribian, A., Fontana, X., Llorens, F., Gavin, R., Reina, M., Garcia-Verdugo, J. M., et al. (2012). Role of the cellular prion protein in oligodendrocyte precursor cell proliferation and differentiation in the developing and adult mouse CNS. PLoS ONE 7:e33872. doi: 10.1371/journal.pone.0033872
Brito, G., Roffe, M., Lupinacci, F., Santos, T., Beraldo, F., Martins, V. R., et al. (2013). The role of cellular prion protein in the regulation of insulin signaling. Mol. Biol. Cell 24, 3775. doi: 10.1091/mbc.E13-10-0584
Brown, D. R., and Mohn, C. M. (1999). Astrocytic glutamate uptake and prion protein expression. Glia 25, 282–292. doi: 10.1002/(sici)1098-1136(19990201)25:3<282::aid-glia8>3.0.co;2-n
Brown, D. R., Nicholas, R. S. J., and Canevari, L. (2002). Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J. Neurosci. Res. 67, 211–224. doi: 10.1002/jnr.10118
Brown, D. R., Qin, K. F., Herms, J. W., Madlung, A., Manson, J., Strome, R., et al. (1997a). The cellular prion protein binds copper in vivo. Nature 390, 684–687
Brown, D. R., SchulzSchaeffer, W. J., Schmidt, B., and Kretzschmar, H. A. (1997b). Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp. Neurol. 146, 104–112. doi: 10.1006/exnr.1997.6505
Bueler, H., Aguzzi, A., Sailer, A., Greiner, R. A., Autenried, P., Aguet, M., et al. (1993). Mice devoid of prp are resistant to scrapie. Cell 73, 1339–1347. doi: 10.1016/0092-8674(93)90360-3
Bueler, H., Fischer, M., Lang, Y., Bluethmann, H., Lipp, H. P., Dearmond, S. J., et al. (1992). Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature 356, 577–582. doi: 10.1038/356577a0
Caetano, F. A., Lopes, M. H., Hajj, G. N. M., Machado, C. F., Arantes, C. P., Magalhaes, A. C., et al. (2008). Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J. Neurosci. 28, 6691–6702. doi: 10.1523/jneurosci.1701-08.2008
Cagampang, F. R. A., Whatley, S. A., Mitchell, A. L., Powell, J. F., Campbell, I. C., and Coen, C. W. (1999). Circadian regulation of prion protein messenger RNA in the rat forebrain: a widespread and synchronous rhythm. Neuroscience 91, 1201–1204. doi: 10.1016/s0306-4522(99)00092-5
Campbell, L., Gill, A. C., McGovern, G., Jalland, C. M. O., Hopkins, J., Tranulis, M. A., et al. (2013). The PrPC Cl fragment derived from the ovine A(136)R(154)R(171) PRNP allele is highly abundant in sheep brain and inhibits fibrillisation of full-length PrPC protein in vitro. Biochim. Biophys. Acta Mol. Basis Dis. 1832, 826–836. doi: 10.1016/j.bbadis.2013.02.020
Caputo, A., Sarnataro, D., Campana, V., Costanzo, M., Negro, A., Sorgato, M. C., et al. (2010). Doppel and PrPC co-immunoprecipitate in detergent-resistant membrane domains of epithelial FRT cells. Biochem. J. 425, 341–351. doi: 10.1042/bj20091050
Carulla, P., Bribian, A., Rangel, A., Gavin, R., Ferrer, I., Caelles, C., et al. (2011). Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol. Biol. Cell 22, 3041–3054. doi: 10.1091/mbc.E11-04-0321
Carulla, P., Llorens, F., Matamoros-Angles, A., Aguilar-Calvo, P., Carlos Espinosa, J., Gavin, R., et al. (2015). Involvement of PrPC in kainate-induced excitotoxicity in several mouse strains. Sci. Rep. 5:11971. doi: 10.1038/srep11971
Chakrabarti, O., Ashok, A., and Hegde, R. S. (2009). Prion protein biosynthesis and its emerging role in neurodegeneration. Trends Biochem. Sci. 34, 287–295. doi: 10.1016/j.tibs.2009.03.001
Chatterjea, D., and Martinov, T. (2015). Mast cells: versatile gatekeepers of pain. Mol. Immunol. 63, 38–44. doi: 10.1016/j.molimm.2014.03.001
Chen, R. J., Chang, W. W., Lin, Y. C., Cheng, P. L., and Chen, Y. R. (2013). Alzheimer's Amyloid-β oligomers rescue cellular prion protein induced Tau reduction via the Fyn pathway. ACS Chem. Neurosci. 4, 1287–1296. doi: 10.1021/cn400085q
Chen, S. G., Teplow, D. B., Parchi, P., Teller, J. K., Gambetti, P., and Autiliogambetti, L. (1995). Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 270, 19173–19180.
Chen, S. G., Yadav, S. P., and Surewicz, W. K. (2010). Interaction between human prion protein and Amyloid-β (Aβ) oligomers role Of N-terminal residues. J. Biol. Chem. 285, 26377–26383. doi: 10.1074/jbc.M110.145516
Chieng, C. K. L., and Say, Y. H. (2015). Cellular prion protein contributes to LS 174T colon cancer cell carcinogenesis by increasing invasiveness and resistance against doxorubicin-induced apoptosis. Tumor Biol. 36, 8107–8120. doi: 10.1007/s13277-015-3530-z
Cichon, A.-C., and Brown, D. R. (2014). Nrf-2 regulation of prion protein expression is independent of oxidative stress. Mol. Cell. Neurosci. 63, 31–37. doi: 10.1016/j.mcn.2014.09.001
Cingaram, P. K. R., Nyeste, A., Dondapati, D. T., Fodor, E., and Welker, E. (2015). Prion protein does not confer resistance to hippocampus-derived Zpl cells against the toxic effects of Cu2+, Mn2+, Zn2+ and Co2+ not supporting a general protective role for PrP in transition metal induced toxicity. PLoS ONE 10:e0139219. doi: 10.1371/journal.pone.0139219
Cisse, M. A., Louis, K., Braun, U., Mari, B., Leitges, M., Slack, B. E., et al. (2008). Isoform-specific contribution of protein kinase C to prion processing. Mol. Cell. Neurosci. 39, 400–410. doi: 10.1016/j.mcn.2008.07.013
Coitinho, A. S., Freitas, A. R. O., Lopes, M. H., Hajj, G. N. M., Roesler, R., Walz, R., et al. (2006). The interaction between prion protein and laminin modulates memory consolidation. Eur. J. Neurosci. 24, 3255–3264. doi: 10.1111/j.1460-9568.2006.05156.x
Collinge, J., Whittington, M. A., Sidle, K. C. L., Smith, C. J., Palmer, M. S., Clarke, A. R., et al. (1994). Prion protein IS necessary for normal synaptic function. Nature 370, 295–297. doi: 10.1038/370295a0
Collins, S., McLean, C. A., and Masters, C. L. (2001). Gerstmann-Straussier-Scheinker syndrome, fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J. Clin. Neurosci. 8, 387–397. doi: 10.1054/jocn.2001.0919
Corsaro, A., Bajetto, A., Thellung, S., Begani, G., Villa, V., Nizzari, M., et al. (2016). Cellular prion protein controls stem cell-like properties of human glioblastoma tumor-initiating cells. Oncotarget 7, 38638–38657. doi: 10.18632/oncotarget.9575
Cotto, E., Andre, M., Forgue, J., Fleury, H. J., and Babin, P. J. (2005). Molecular characterization, phylogenetic relationships, and developmental expression patterns of prion genes in zebrafish (Danio rerio). FEBS J. 272, 500–513. doi: 10.1111/j.1742-4658.2004.04492.x
Crecelius, A. C., Helmstetter, D., Strangmann, J., Mitteregger, G., Frohlich, T., Arnold, G. J., et al. (2008). The brain proteome profile is highly conserved between Prnp(-/-) and Prnp(+/+) mice. Neuroreport 19, 1027–1031. doi: 10.1097/WNR.0b013e3283046157.
Decker, L., Desmarquet-Trin-Dinh, C., Taillebourg, E., Ghislain, J., Vallat, J. M., and Charnay, P. (2006). Peripheral myelin maintenance is a dynamic process requiring constant Krox20 expression. J. Neurosci. 26, 9771–9779. doi: 10.1523/jneurosci.0716-06.2006
Deckwerth, T. L., Elliott, J. L., Knudson, C. M., Johnson, E. M., Snider, W. D., and Korsmeyer, S. J. (1996). BAX is required for neuronal death after trophic factor deprivation and during development. Neuron 17, 401–411. doi: 10.1016/s0896-6273(00)80173-7
del Rio, J. A., and Gavin, R. (2016). Functions of the cellular prion protein, the end of Moore's law, and Ockham's razor theory. Prion 10, 25–40. doi: 10.1080/19336896.2015.1126038
Dery, M. A., Jodoin, J., Ursini-Siegel, J., Aleynikova, O., Ferrario, C., Hassan, S., et al. (2013). Endoplasmic reticulum stress induces PRNP prion protein gene expression in breast cancer. Breast Cancer Res. 15:R22. doi: 10.1186/bcr3398
Dhillon, A. S., Hagan, S., Rath, O., and Kolch, W. (2007). MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290. doi: 10.1038/sj.onc.1210421
Dibner, C., Schibler, U., and Albrecht, U. (2010). The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Ann. Rev. Physiol. 72, 517–549. doi: 10.1146/annurev-physiol-021909-135821
Dohler, F., Sepulveda-Falla, D., Krasemann, S., Altmeppen, H., Schluter, H., Hildebrand, D., et al. (2014). High molecular mass assemblies of amyloid-beta oligomers bind prion protein in patients with Alzheimer's disease. Brain 137, 873–886. doi: 10.1093/brain/awt375
Dupiereux, I., Falisse-Poirrier, N., Zorzi, W., Watt, N. T., Thellin, O., Zorzi, D., et al. (2008). Protective effect of prion protein via the N-terminal region in mediating a protective effect on paraquat-induced oxidative injury in neuronal cells. J. Neurosci. Res. 86, 653–659. doi: 10.1002/jnr.21506
Durig, J., Giese, A., Schulz-Schaeffer, W., Rosenthal, C., Schmucker, U., Bieschke, J., et al. (2000). Differential constitutive and activation-dependent expression of prion protein in human peripheral blood leucocytes. Br. J. Haematol. 108, 488–496. doi: 10.1046/j.1365-2141.2000.01881.x
Ehsani, S., Huo, H. R., Salehzadeh, A., Pocanschi, C. L., Watts, J. C., Wille, H., et al. (2011). Family reunion - The ZIP/prion gene family. Prog. Neurobiol. 93, 405–420. doi: 10.1016/j.pneurobio.2010.12.001
Ehsani, S., Mehrabian, M., Pocanschi, C. L., and Schmitt-Ulms, G. (2012). The ZIP-prion connection. Prion 6, 317–321. doi: 10.1371/journal.pone.e0026800
Faris, R., Moore, R. A., Ward, A., Race, B., Dorward, D. W., Hollister, J. R., et al. (2017). Cellular prion protein is present in mitochondria of healthy mice. Sci. Rep. 7:41556. doi: 10.1038/srep41556
Fernandez-Borges, N., Chianini, F., Erana, H., Vidal, E., Eaton, S. L., Pintado, B., et al. (2012). Naturally prion resistant mammals A utopia? Prion 6, 425–429. doi: 10.4161/pri.22057
Fleisch, V. C., Leighton, P. L. A., Wang, H., Pillay, L. M., Ritz, R. G., Bhinder, G., et al. (2013). Targeted mutation of the gene encoding prion protein in zebrafish reveals a conserved role in neuron excitability. Neurobiol. Dis. 55, 11–25. doi: 10.1016/j.nbd.2013.03.007
Follet, J., Lemaire-Vieille, C., Blanquet-Grossard, F., Podevin-Dimster, V., Lehmann, S., Chauvin, J. P., et al. (2002). PrP expression and replication by Schwann cells: implications in prion spreading. J. Virol. 76, 2434–2439. doi: 10.1128/jvi.76.5.2434-2439.2002
Frigg, R., Wenzel, A., Samardzija, M., Oesch, B., Wariwoda, H., Navarini, A. A., et al. (2006). The prion protein is neuroprotective against retinal degeneration in vivo. Exp. Eye Res. 83, 1350–1358. doi: 10.1016/j.exer.2006.07.010
Gadotti, V. M., and Zamponi, G. W. (2011). Cellular prion protein protects from inflammatory and neuropathic pain. Mol. Pain 7:59. doi: 10.1186/1744-8069-7-59
Ganley, R. P., Iwagaki, N., del Rio, P., Baseer, N., Dickie, A. C., Boyle, K. A., et al. (2015). Inhibitory interneurons that express GFP in the PrP-GFP mouse spinal cord are morphologically heterogeneous, innervated by several classes of primary afferent and include lamina i projection neurons among their postsynaptic targets. J. Neurosci. 35, 7626–7642. doi: 10.1523/jneurosci.0406-15.2015
Gasperini, L., Meneghetti, E., Pastore, B., Benetti, F., and Legname, G. (2015). Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid. Redox Signal. 22, 772–784. doi: 10.1089/ars.2014.6032
Gauczynski, S., Peyrin, J. M., Haik, S., Leucht, C., Hundt, C., Rieger, R., et al. (2001). The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 20, 5863–5875. doi: 10.1093/emboj/20.21.5863
Gerlai, R. (1996). Gene-targeting studies of mammalian behavior: Is it the mutation or the background genotype? Trends Neurosci. 19, 177–181. doi: 10.1016/s0166-2236(96)20020-7
Gill, A. C., Ritchie, M. A., Hunt, L. G., Steane, S. E., Davies, K. G., Bocking, S. P., et al. (2000). Post-translational hydroxylation at the N-terminus of the prion protein reveals presence of PPII structure in vivo. EMBO J. 19, 5324–5331. doi: 10.1093/emboj/19.20.5324
Glenn, T. D., and Talbot, W. S. (2013). Analysis of Gpr126 function defines distinct mechanisms controlling the initiation and maturation of myelin. Development 140, 3167–3175. doi: 10.1242/dev.093401
Gorman, A. M. (2008). Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. J. Cell. Mol. Med. 12, 2263–2280. doi: 10.1111/j.1582-4934.2008.00402.x
Gourdain, P., Ballerini, C., Nicot, A. B., and Carnaud, C. (2012). Exacerbation of experimental autoimmune encephalomyelitis in prion protein (PrPc)-null mice: evidence for a critical role of the central nervous system. J. Neuroinflammation 9:25. doi: 10.1186/1742-2094-9-25
Graner, E., Mercadante, A. F., Zanata, S. M., Forlenza, O. V., Cabral, A. L. B., Veiga, S. S., et al. (2000a). Cellular prion protein binds laminin and mediates neuritogenesis. Mol. Brain Res. 76, 85–92. doi: 10.1016/s0169-328x(99)00334-4
Graner, E., Mercadante, A. F., Zanata, S. M., Martins, V. R., Jay, D. G., and Brentani, R. R. (2000b). Laminin-induced PC-12 cell differentiation is inhibited following laser inactivation of cellular prion protein. FEBS Lett. 482, 257–260. doi: 10.1016/s0014-5793(00)02070-6
Gu, Y. P., Hinnerwisch, J., Fredricks, R., Kalepu, S., Mishra, R. S., and Singh, N. (2003). Identification of cryptic nuclear localization signals in the prion protein. Neurobiol. Dis. 12, 133–149. doi: 10.1016/s0969-9961(02)00014-1
Guillot-Sestier, M. V., Sunyach, C., Druon, C., Scarzello, S., and Checler, F. (2009). The alpha-Secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J. Biol. Chem. 284, 35973–35986. doi: 10.1074/jbc.M109.051086
Haas, L. T., Kostylev, M. A., and Strittmatter, S. M. (2014). Therapeutic molecules and endogenous ligands regulate the interaction between brain cellular prion protein (PrPC) and Metabotropic Glutamate Receptor 5 (mGluR5). J. Biol. Chem. 289, 28460–28477. doi: 10.1074/jbc.M114.584342
Hachiya, N. S., Yamada, M., Watanabe, K., Jozuka, A., Ohkubo, T., Sano, K., et al. (2005). Mitochondrial localization of cellular prion protein (PrPC) invokes neuronal apoptosis in aged transgenic mice overexpressing PrPC. Neurosci. Lett. 374, 98–103. doi: 10.1016/j.neulet.2004.10.044
Haddon, D. J., Hughes, M. R., Antignano, F., Westaway, D., Cashman, N. R., and McNagny, K. M. (2009). Prion protein expression and release by mast cells after activation. J. Infect. Dis. 200, 827–831. doi: 10.1086/605022
Haigh, C., and Collins, S. (2014). Oxidative modulation of neural stem cell growth by prion protein cleavage fragments. Free Radic. Biol. Med. 76, S24–S24. doi: 10.1016/j.freeradbiomed.2014.10.476
Haigh, C. L., McGlade, A. R., and Collins, S. J. (2015a). MEK1 transduces the prion protein N2 fragment antioxidant effects. Cell. Mol. Life Sci. 72, 1613–1629. doi: 10.1007/s00018-014-1777-y
Haigh, C. L., Tumpach, C., Drew, S. C., and Collins, S. J. (2015b). The prion protein N1 and N2 cleavage fragments bind to phosphatidylserine and phosphatidic acid; relevance to stress-protection responses. PLoS ONE 10:e0134680. doi: 10.1371/journal.pone.0134680
Haire, L. F., Whyte, S. M., Vasisht, N., Gill, A. C., Verma, C., Dodson, E. J., et al. (2004). The crystal structure of the globular domain of sheep prion protein. J. Mol. Biol. 336, 1175–1183. doi: 10.1016/j.jmb.2003.12.059
Haldar, S., Tripathi, A., Qian, J., Beserra, A., Suda, S., McElwee, M., et al. (2015). Prion protein promotes kidney iron uptake via its ferrireductase activity. J. Biol. Chem. 290, 5512–5522. doi: 10.1074/jbc.M114.607507
Halliday, M., and Mallucci, G. R. (2014). Targeting the unfolded protein response in neurodegeneration: a new approach to therapy. Neuropharmacology 76, 169–174. doi: 10.1016/j.neuropharm.2013.08.034
Hare, D., Ayton, S., Bush, A., and Lei, P. (2013). A delicate balance: iron metabolism and diseases of the brain. Front. Aging Neurosci. 5:34. doi: 10.3389/fnagi.2013.00034
Harris, D. A., Huber, M. T., Vandijken, P., Shyng, S. L., Chait, B. T., and Wang, R. (1993). Processing of a cellular prion protein - identification of N-terminal and c-terminal cleavage sites. Biochemistry 32, 1009–1016. doi: 10.1021/bi00055a003
Hartmann, C. A., Martins, V. R., and Lima, F. R. S. (2013). High levels of cellular prion protein improve astrocyte development. FEBS Lett. 587, 238–244. doi: 10.1016/j.febslet.2012.11.032
Head, M. W. (2013). Human prion diseases: molecular, cellular and population biology. Neuropathology 33, 221–236. doi: 10.1111/neup.12016
Heikenwalder, M., Kurrer, M. O., Margalith, I., Kranich, J., Zeller, N., Haybaeck, J., et al. (2008). Lymphotoxin-dependent prion replication in inflammatory stromal cells of granulomas. Immunity 29, 998–1008. doi: 10.1016/j.immuni.2008.10.014
Hornshaw, M. P., McDermott, J. R., and Candy, J. M. (1995a). Copper-binding to the N-terminal tandem repeat regions of mammalian and avian prion protein. Biochem. Biophys. Res. Commun. 207, 621–629. doi: 10.1006/bbrc.1995.1233
Hornshaw, M. P., McDermott, J. R., Candy, J. M., and Lakey, J. H. (1995b). Copper-binding to the N-terminal tandem repeat region of mammalian and avian prion protein - structural studies using synthetic peptides. Biochem. Biophys. Res. Commun. 214, 993–999. doi: 10.1006/bbrc.1995.2384
Horwich, A. L., and Weissman, J. S. (1997). Deadly conformations - Protein misfolding in prion disease. Cell 89, 499–510. doi: 10.1016/s0092-8674(00)80232-9
Hu, W., Nessler, S., Hemmer, B., Eagar, T. N., Kane, L. P., Leliveld, S. R., et al. (2010). Pharmacological prion protein silencing accelerates central nervous system autoimmune disease via T cell receptor signalling. Brain 133, 375–388. doi: 10.1093/brain/awp298
Huc-Brandt, S., Hieu, N., Imberdis, T., Cubedo, N., Silhol, M., Leighton, P. L. A., et al. (2014). Zebrafish prion protein PrP2 controls collective migration process during lateral line sensory system development. PLoS ONE 9:e113331. doi: 10.1371/journal.pone.0113331
Hundt, C., Peyrin, J. M., Haik, S., Gauczynski, S., Leucht, C., Rieger, R., et al. (2001). Identification of interaction domains of the prion protein with its 37-kDa/67-kDa laminin receptor. EMBO J. 20, 5876–5886. doi: 10.1093/emboj/20.21.5876
Hutter, G., Heppner, F. L., and Aguzzi, A. (2003). No superoxide dismutase activity of cellular prion protein in vivo. Biol. Chem. 384, 1279–1285. doi: 10.1515/bc.2003.142
Jackson, W. S., Krost, C., Borkowski, A. W., and Kaczmarczyk, L. (2014). Translation of the prion protein mRNA Is robust in astrocytes but does not amplify during reactive astrocytosis in the mouse brain. PLoS ONE 9:e95958. doi: 10.1371/journal.pone.0095958
Khosravani, H., Zhang, Y. F., Tsutsui, S., Hameed, S., Altier, C., Hamid, J., et al. (2008). Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol. 181, 551–565. doi: 10.1083/jcb.200711002
Kim, B. H., Kim, J. I., Choi, E. K., Carp, R. I., and Kim, Y. S. (2005). A neuronal cell line that does not express either prion or doppel proteins. Neuroreport 16, 425–429. doi: 10.1097/00001756-200504040-00002
Kim, B. H., Lee, H. G., Choi, J. K., Kim, J. I., Choi, E. K., Carp, R. I., et al. (2004). The cellular prion protein (PrPC) prevents apoptotic neuronal cell death and mitochondrial dysfunction induced by serum deprivation. Mol. Brain Res. 124, 40–50. doi: 10.1016/j.molbrainres.2004.02.005
Kim, Y., Lee, J., and Lee, C. (2008). In silico, comparative analysis of DNA and amino acid sequences for prion protein gene. Transbound. Emerg. Dis. 55, 105–114. doi: 10.1111/j.1865-1682.2007.00997.x
Kleene, R., Loers, G., Langer, J., Frobert, Y., Buck, F., and Schachner, M. (2007). Prion protein regulates glutamate-dependent lactate transport of Astrocytes. J. Neurosci. 27, 12331–12340. doi: 10.1523/jneurosci.1358-07.2007
Krebs, B., Wiebelitz, A., Balitzki-Korte, B., Vassallo, N., Paluch, S., Mitteregger, G., et al. (2007). Cellular prion protein modulates the intracellular calcium response to hydrogen peroxide. J. Neurochem. 100, 358–367. doi: 10.1111/j.1471-4159.2006.04256.x
Kuffer, A., Lakkaraju, A. K., Mogha, A., Petersen, S. C., Airich, K., Doucerain, C., et al. (2016). The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 536, 464–468. doi: 10.1038/nature19312
Kuwahara, C., Kubosaki, A., Nishimura, T., Nasu, Y., Nakamura, Y., Saeki, K., et al. (2000). Enhanced expression of cellular prion protein gene by insulin or nerve growth factor in immortalized mouse neuronal precursor cell lines. Biochem. Biophys. Res. Commun. 268, 763–766. doi: 10.1006/bbrc.2000.2152
Kuwahara, C., Takeuchi, A. M., Nishimura, T., Haraguchi, K., Kubosaki, A., Matsumoto, Y., et al. (1999). Prions prevent neuronal cell-line death. Nature 400, 225–226. doi: 10.1038/22241
Laffont-Proust, I., Hassig, R., Haik, S., Simon, S., Grassi, J., Fonta, C., et al. (2006). Truncated PrPc in mammalian brain: interspecies variation and location in membrane rafts. Biol. Chem. 387, 297–300. doi: 10.1515/bc.2006.039
Lai, T. W., Zhang, S., and Wang, Y. T. (2014). Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 115, 157–188. doi: 10.1016/j.pneurobio.2013.11.006
Lappas Gimenez, A. P., Luciani Richter, L. M., Atherino, M. C., Branco Beirao, B. C., Favaro, C. Jr., Moura Costa, M. D., et al. (2015). Identification of novel putative-binding proteins for cellular prion protein and a specific interaction with the STIP1 homology and U-Box-containing protein 1. Prion 9, 355–366. doi: 10.1080/19336896.2015.1075347
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, U1128–U1184. doi: 10.1038/nature07761
Le Belle, J. E., Orozco, N. M., Paucar, A. A., Saxe, J. P., Mottahedeh, J., Pyle, A. D., et al. (2011). Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 8, 59–71. doi: 10.1016/j.stem.2010.11.028
Lee, K. S., Magalhaes, A. C., Zanata, S. M., Brentani, R. R., Martins, V. R., and Prado, M. A. M. (2001). Internalization of mammalian fluorescent cellular prion protein and N-terminal deletion mutants in living cells. J. Neurochem. 79, 79–87. doi: 10.1046/j.1471-4159.2001.00529.x
Lee, Y. J., and Baskakov, I. V. (2013). The cellular form of the prion protein is involved in controlling cell cycle dynamics, self-renewal, and the fate of human embryonic stem cell differentiation. J. Neurochem. 124, 310–322. doi: 10.1111/j.1471-4159.2012.07913.x
Lee, Y. J., and Baskakov, I. V. (2014). The cellular form of the prion protein guides the differentiation of human embryonic stem cells into neuron-, oligodendrocyte-, and astrocyte-committed lineages. Prion 8, 266–275. doi: 10.4161/pri.32079
Lewis, V., Whitehouse, I. J., Baybutt, H., Manson, J. C., Collins, S. J., and Hooper, N. M. (2012). Cellular prion protein expression is not regulated by the Alzheimer's amyloid precursor protein intracellular domain. PLoS ONE 7:e31754. doi: 10.1371/journal.pone.0031754
Li, Q. Q., Sun, Y. P., Ruan, C. P., Xu, X. Y., Ge, J. H., He, J., et al. (2011). Cellular prion protein promotes glucose uptake through the Fyn-HIF-2 alpha-Glut1 pathway to support colorectal cancer cell survival. Cancer Sci. 102, 400–406. doi: 10.1111/j.1349-7006.2010.01811.x
Lima, F. R. S., Arantes, C. P., Muras, A. G., Nomizo, R., Brentani, R. R., and Martins, V. R. (2007). Cellular prion protein expression in astrocytes modulates neuronal survival and differentiation. J. Neurochem. 103, 2164–2176. doi: 10.1111/j.1471-4159.2007.04904.x
Linden, R., Martins, V. R., Prado, M. A. M., Cammarota, M., Izquierdo, I., and Brentani, R. R. (2008). Physiology of the prion protein. Physiol. Rev. 88, 673–728. doi: 10.1152/physrev.00007.2007
Liu, T., Yi, W. J., Feng, B. Y., Zhou, Z., and Xiao, G. F. (2013). IGF-1-induced enhancement of PRNP expression depends on the negative regulation of transcription factor FOXO3a. PLoS ONE 8:e71896. doi: 10.1371/journal.pone.0071896
Lledo, P. M., Tremblay, P., Dearmond, S. J., Prusiner, S. B., and Nicoll, R. A. (1996). Mice deficient for prion protein exhibit normal neuronal excitability and synaptic transmission in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 93, 2403–2407. doi: 10.1073/pnas.93.6.2403
Llorens, F., Carulla, P., Villa, A., Torres, J. M., Fortes, P., Ferrer, I., et al. (2013). PrPC regulates epidermal growth factor receptor function and cell shape dynamics in Neuro2a cells. J. Neurochem. 127, 124–138. doi: 10.1111/jnc.12283
Lobao-Soares, B., Bianchin, M. M., Linhares, M. N., Carqueja, C. L., Tasca, C. I., Souza, M., et al. (2005). Normal brain mitochondrial respiration in adult mice lacking cellular prion protein. Neurosci. Lett. 375, 203–206. doi: 10.1016/j.neulet.2004.11.012
Lopes, M. H., Hajj, G. N. M., Muras, A. G., Mancini, G. L., Castro, R., Ribeiro, K. C. B., et al. (2005). Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 25, 11330–11339. doi: 10.1523/jneurosci.2313-05.2005
Loubet, D., Dakowski, C., Pietri, M., Pradines, E., Bernard, S., Callebert, J., et al. (2012). Neuritogenesis: the prion protein controls beta 1 integrin signaling activity. FASEB J. 26, 678–690. doi: 10.1096/fj.11-185579
Mabbott, N. A., Brown, K. L., Manson, J., and Bruce, M. E. (1997). T-lymphocyte activation and the cellular form of the prion protein. Immunology 92, 161–165. doi: 10.1046/j.1365-2567.1997.00331.x
Magalhaes, A. C., Silva, J. A., Lee, K. S., Martins, V. R., Prado, V. F., Ferguson, S. S. G., et al. (2002). Endocytic intermediates involved with the intracellular trafficking of a fluorescent cellular prion protein. J. Biol. Chem. 277, 33311–33318. doi: 10.1074/jbc.M203661200
Mahal, S. P., Asante, E. A., Antoniou, M., and Collinge, J. (2001). Isolation and functional characterisation of the promoter region of the human prion protein gene. Gene 268, 105–114. doi: 10.1016/s0378-1119(01)00424-3
Malaga-Trillo, E., Solis, G. P., Schrock, Y., Geiss, C., Luncz, L., Thomanetz, V., et al. (2009). Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 7:e55. doi: 10.1371/journal.pbio.1000055
Maltby, S., Khazaie, K., and McNagny, K. M. (2009). Mast cells in tumor growth: angiogenesis, tissue remodelling and immune-modulation. Biochim. Biophys. Acta 1796, 19–26. doi: 10.1016/j.bbcan.2009.02.001
Mange, A., Beranger, F., Poec'h, K., Onodera, T., Frobert, Y., and Lehmann, S. (2004). Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol. Cell 96, 125–132. doi: 10.1016/j.biolcel.2003.11.007
Manson, J. C., Clarke, A. R., Hooper, M. L., Aitchison, L., McConnell, I., and Hope, J. (1994). 129/Ola mice carrying a null mutation in PrP that abolishes messenger-RNA production are developmentally normal. Mol. Neurobiol. 8, 121–127. doi: 10.1007/bf02780662
Manson, J., West, J. D., Thomson, V., McBride, P., Kaufman, M. H., and Hope, J. (1992). The prion protein gene - a role in mouse embryogenesis. Development 115, 117–122
Martini, M., De Santis, M. C., Braccini, L., Gulluni, F., and Hirsch, E. (2014). PI3K/AKT signaling pathway and cancer: an updated review. Ann. Med. 46, 372–383. doi: 10.3109/07853890.2014.912836
Mattei, V., Garofalo, T., Misasi, R., Circella, A., Manganelli, V., Lucania, G., et al. (2004). Prion protein is a component of the multimolecular signaling complex involved in T cell activation. FEBS Lett. 560, 14–18. doi: 10.1016/s0014-5793(04)00029-8
Mays, C. E., Kim, C., Haldiman, T., van der Merwe, J., Lau, A., Yang, J., et al. (2014). Prion disease tempo determined by host-dependent substrate reduction. J. Clin. Invest. 124, 847–858. doi: 10.1172/jci72241
McDonald, A. J., Dibble, J. P., Evans, E. G. B., and Millhauser, G. L. (2014). A new paradigm for enzymatic control of α-cleavage and β-cleavage of the prion protein. J. Biol. Chem. 289, 803–813. doi: 10.1074/jbc.M113.502351
McHugh, P. C., Wright, J. A., Williams, R. J., and Brown, D. R. (2012). Prion protein expression alters APP cleavage without interaction with BACE-1. Neurochem. Int. 61, 672–680. doi: 10.1016/j.neuint.2012.07.002
McKinley, M. P., Bolton, D. C., and Prusiner, S. B. (1983). A protease-resistant protein is a structural component of the scrapie prion. Cell 35, 57–62. doi: 10.1016/0092-8674(83)90207-6
McMahon, H. E. M., Mange, A., Nishida, N., Creminon, C., Casanova, D., and Lehmann, S. (2001). Cleavage of the amino terminus of the prion protein by reactive oxygen species. J. Biol. Chem. 276, 2286–2291. doi: 10.1074/jbc.M007243200
Mehrabian, M., Brethour, D., MacIsaac, S., Kim, J. K., Gunawardana, C. G., Wang, H., et al. (2014). CRISPR-Cas9-based knockout of the prion protein and its effect on the proteome. PLoS ONE 9:e114594. doi: 10.1371/journal.pone.0114594
Mehrabian, M., Brethour, D., Wang, H., Xi, Z., Rogaeva, E., and Schmitt-Ulms, G. (2015). The prion protein controls polysialylation of neural cell adhesion molecule 1 during cellular morphogenesis. PLoS ONE 10:e0133741. doi: 10.1371/journal.pone.0133741
Mehrabian, M., Brethour, D., Williams, D., Wang, H. S., Arnould, H., Schneider, B., et al. (2016). Prion protein deficiency causes diverse proteome shifts in cell models that escape detection in brain tissue. PLoS ONE 11:e0156779. doi: 10.1371/journal.pone.0156779
Meotti, F. C., Carqueja, C. L., Gadotti, V. D., Tasca, C. I., Walz, R., and Santos, A. R. S. (2007). Involvement of cellular prion protein in the nociceptive response in mice. Brain Res. 1151, 84–90. doi: 10.1016/j.brainres.2007.03.024
Mercer, R. C. C., Ma, L., Watts, J. C., Strome, R., Wohlgemuth, S., Yang, J., et al. (2013). The prion protein modulates A-type K+ currents mediated by Kv4.2 complexes through dipeptidyl aminopeptidase-like protein 6. J. Biol. Chem. 288, 37241–37255. doi: 10.1074/jbc.M113.488650
Miele, G., Jeffrey, M., Turnbull, D., Manson, J., and Clinton, M. (2002). Ablation of cellular prion protein expression affects mitochondrial numbers and morphology. Biochem. Biophys. Res. Commun. 291, 372–377. doi: 10.1006/bbrc.2002.6460
Monk, K. R., Oshima, K., Jors, S., Heller, S., and Talbot, W. S. (2011). Gpr126 is essential for peripheral nerve development and myelination in mammals. Development 138, 2673–2680. doi: 10.1242/dev.062224
Moore, R. C., Lee, I. Y., Silverman, G. L., Harrison, P. M., Strome, R., Heinrich, C., et al. (1999). Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein Doppel. J. Mol. Biol. 292, 797–817. doi: 10.1006/jmbi.1999.3108
Moore, R. C., Redhead, N. J., Selfridge, J., Hope, J., Manson, J. C., and Melton, D. W. (1995). Double replacement gene targeting for the production of A series of mouse strains with different prion protein gene alterations. Nat. BioTechnol. 13, 999–1004. doi: 10.1038/nbt0995-999
Morel, E., Fouquet, S., Strup-Perrot, C., Thievend, C. P., Petit, C., Loew, D., et al. (2008). The cellular prion protein PrPc is involved in the proliferation of epithelial cells and in the distribution of junction-associated proteins. PLoS ONE 3:e3000. doi: 10.1371/journal.pone.0003000
Moser, M., Colello, R. J., Pott, U., and Oesch, B. (1995). Developmental expression of the prion protein gene in glial-cells. Neuron 14, 509–517. doi: 10.1016/0896-6273(95)90307-0
Muller, W. E. G., Ushijima, H., Schroder, H. C., Forrest, J. M. S., Schatton, W. F. H., Rytik, P. G., et al. (1993). Cytoprotective effect of NMDA receptor antagonists on prion protein (PRION(SC))-induced toxicity in rat cortical cell-cultures. Eur. J. Pharmacol. Mol. Pharmacol. Sect. 246, 261–267. doi: 10.1016/0922-4106(93)90040-g
Nah, J., Pyo, J. O., Jung, S., Yoo, S. M., Kam, T. I., Chang, J., et al. (2013). BECN1/Beclin 1 is recruited into lipid rafts by prion to activate autophagy in response to amyloid β 42. Autophagy 9, 2009–2021. doi: 10.4161/auto.26118
Newton, A. C. (2010). Protein kinase C: poised to signal. Am. J. Physiol. Endocrinol. Metab. 298, E395–E402. doi: 10.1152/ajpendo.00477.2009
Nieznanski, K., Nieznanska, H., Skowronek, K. J., Osiecka, K. M., and Stepkowski, D. (2005). Direct interaction between prion protein and tubulin. Biochem. Biophys. Res. Commun. 334, 403–411. doi: 10.1016/j.bbrc.2005.06.092
Nikles, D., Vana, K., Gauczynski, S., Knetsch, H., Ludewigs, H., and Weiss, S. (2008). Subcellular localization of prion proteins and the 37 kDa/67 kDa laminin receptor fused to fluorescent proteins. Biochim. Biophys. Acta 1782, 335–340. doi: 10.1016/j.bbadis.2008.02.003
Nishida, N., Tremblay, P., Sugimoto, T., Shigematsu, K., Shirabe, S., Petromilli, C., et al. (1999). A mouse prion protein transgene rescues mice deficient for the prion protein gene from Purkinje cell degeneration and demyelination. Lab. Investi. 79, 689–697
Nishimura, T., Sakudo, A., Hashiyama, Y., Yachi, A., Saeki, K., Matsumoto, Y., et al. (2007). Serum withdrawal-induced apoptosis in Zrchl prion protein (PrP) gene-deficient neuronal cell line is suppressed by PrP, independent of Doppel. Microbiol. Immunol. 51, 457–466. doi: 10.1111/j.1348-0421.2007.tb03920.x
Nuvolone, M., Hermann, M., Sorce, S., Russo, G., Tiberi, C., Schwarz, P., et al. (2016). Strictly co-isogenic C57BL/6J-Prnp(-/-) mice: A rigorous resource for prion science. J. Exp. Med. 213, 313–327. doi: 10.1084/jem.20151610
Nuvolone, M., Kana, V., Hutter, G., Sakata, D., Mortin-Toth, S. M., Russo, G., et al. (2013). SIRP α polymorphisms, but not the prion protein, control phagocytosis of apoptotic cells. J. Exp. Med. 210, 2539–2552. doi: 10.1084/jem.20131274
O'Connor, T. P., Duerr, J. S., and Bentley, D. (1990). Pioneer growth cone steering decisions mediated by single filopodial contacts in situ. J. Neurosci. 10, 3935–3946.
Oh, J. M., Shin, H. Y., Park, S. J., Kim, B. H., Choi, J. K., Choi, E. K., et al. (2008). The involvement of cellular prion protein in the autophagy pathway in neuronal cells. Mol. Cell. Neurosci. 39, 238–247. doi: 10.1016/j.mcn.2008.07.003
Orsi, A., Fioriti, L., Chiesa, R., and Sitia, R. (2006). Conditions of endoplasmic reticulum stress favor the accumulation of cytosolic prion protein. J. Biol. Chem. 281, 30431–30438. doi: 10.1074/jbc.M605320200
Ostapchenko, V. G., Beraldo, F. H., Mohammad, A. H., Xie, Y. F., Hirata, P. H. F., Magalhaes, A. C., et al. (2013). The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-β oligomer toxicity. J. Neurosci. 33, 16552–16564. doi: 10.1523/jneurosci.3214-13.2013
Paavola, K. J., Sidik, H., Zuchero, J. B., Eckart, M., and Talbot, W. S. (2014). Type IV collagen is an activating ligand for the adhesion G protein-coupled receptor GPR126. Sci. Signal. 7:ra76. doi: 10.1126/scisignal.2005347
Paitel, E., da Costa, C. A., Vilette, D., Grassi, J., and Checler, F. (2002). Overexpression of PrPc triggers caspase 3 activation: potentiation by proteasome inhibitors and blockade by anti-PrP antibodies. J. Neurochem. 83, 1208–1214. doi: 10.1046/j.1471-4159.2002.01234.x
Paitel, E., Fahraeus, R., and Checler, F. (2003). Cellular prion protein sensitizes neurons to apoptotic stimuli through Mdm2-regulated and p53-dependent caspase 3-like activation. J. Biol. Chem. 278, 10061–10066. doi: 10.1074/jbc.M211580200
Paitel, E., Sunyach, C., da Costa, C. A., Bourdon, J. C., Vincent, B., and Checler, F. (2004). Primary cultured neurons devoid of cellular prion display lower responsiveness to Staurosporine through the control of p53 at both transcriptional and post-transcriptional levels. J. Biol. Chem. 279, 612–618. doi: 10.1074/jbc.M310453200
Pan, K. M., Baldwin, M., Nguyen, J., Gasset, M., Serban, A., Groth, D., et al. (1993). Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. U.S.A. 90, 10962–10966. doi: 10.1073/pnas.90.23.10962
Parkin, E. T., Watt, N. T., Hussain, I., Eckman, E. A., Eckman, C. B., Manson, J. C., et al. (2007). Cellular prion protein regulates β-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 104, 11062–11067. doi: 10.1073/pnas.0609621104
Pastore, A., and Zagari, A. (2007). A structural overview of the vertebrate prion proteins. Prion 1, 185–197. doi: 10.4161/pri.1.3.5281
Patel, D. M., Dubash, A. D., Kreitzer, G., and Green, K. J. (2014). Disease mutations in desmoplakin inhibit Cx43 membrane targeting mediated by desmoplakin-EB1 interactions. J. Cell Biol. 206, 779–797. doi: 10.1083/jcb.201312110
Paterson, A. W. J., Curtis, J. C., and MacLeod, N. K. (2008). Complex I specific increase in superoxide formation and respiration rate by PrP-null mouse brain mitochondria. J. Neurochem. 105, 177–191. doi: 10.1111/j.1471-4159.2007.5123.x
Peralta, O. A., and Eyestone, W. H. (2009). Quantitative and qualitative analysis of cellular prion protein (PrPC) expression in bovine somatic tissues. Prion 3, 161–170. doi: 10.4161/pri.3.3.9772
Peralta, O. A., Huckle, W. R., and Eyestone, W. H. (2012). Developmental expression of the cellular prion protein (PrPC) in bovine embryos. Mol. Reprod. Dev. 79, 488–498. doi: 10.1002/mrd.22057
Petersen, S. C., Luo, R., Liebscher, I., Giera, S., Jeong, S. J., Mogha, A., et al. (2015). The adhesion GPCR GPR126 has distinct, domain-dependent functions in schwann cell development mediated by interaction with laminin-211. Neuron 85, 755–769. doi: 10.1016/j.neuron.2014.12.057
Petrakis, S., Irinopoulou, T., Panagiotidis, C. H., Engelstein, R., Lindstrom, J., Orr-Urtreger, A., et al. (2008). Cellular prion protein co-localizes with nAChR β4 subunit in brain and gastrointestinal tract. Eur. J. Neurosci. 27, 612–620. doi: 10.1111/j.1460-9568.2008.06037.x
Premzl, M., Gready, J. E., Jermiin, L. S., Simonic, T., and Graves, J. A. M. (2004). Evolution of vertebrate genes related to prion and Shadoo proteins - Clues from comparative genomic analysis. Mol. Biol. Evol. 21, 2210–2231. doi: 10.1093/molbev/msh245
Provansal, M., Roche, S., Pastore, M., Casanova, D., Belondrade, M., Alais, S., et al. (2010). Proteomic consequences of expression and pathological conversion of the prion protein in inducible neuroblastoma N2a cells. Prion 4, 292–301. doi: 10.4161/pri.4.4.13435
Prusiner, S. B. (1982). Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144. doi: 10.1126/science.6801762
Prusiner, S. B., Bolton, D. C., Groth, D. F., Bowman, K. A., Cochran, S. P., and McKinley, M. P. (1982). Further purification and characterization of scrapie prions. Biochemistry 21, 6942–6950. doi: 10.1021/bi00269a050
Prusiner, S. B., Groth, D., Serban, A., Koehler, R., Foster, D., Torchia, M., et al. (1993). Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc. Natl. Acad. Sci. U.S.A. 90, 10608–10612. doi: 10.1073/pnas.90.22.10608
Prusiner, S., Oesch, B., Walchli, M., Westaway, D., McKinley, M., Teplow, D., et al. (1985). Molecular-cloning studies demonstrate that cellular genomic DNA encodes the scrapie prion protein. Clin. Res. 33, A565–A565.
Puckett, C., Concannon, P., Casey, C., and Hood, L. (1991). Genomic structure of the human prion protein gene. Am. J. Hum. Genet. 49, 320–329.
Qin, K., Zhao, L., Tang, Y., Bhatta, S., Simard, J. M., and Zhao, R. Y. (2006). Doppel-induced apoptosis and counteraction by cellular prion protein in neuroblastoma and astrocytes. Neuroscience 141, 1375–1388. doi: 10.1016/j.neuroscience.2006.04.068
Rachidi, W., Vilette, D., Guiraud, P., Arlotto, M., Riondel, J., Laude, H., et al. (2003). Expression of prion protein increases cellular copper binding and antioxidant enzyme activities but not copper delivery. J. Biol. Chem. 278, 9064–9072. doi: 10.1074/jbc.M211830200
Rambold, A. S., Muller, V., Ron, U., Ben-Tal, N., Winklhofer, K. F., and Tatzelt, J. (2008). Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J. 27, 1974–1984. doi: 10.1038/emboj.2008.122
Ramljak, S., Asif, A. R., Armstrong, V. W., Wrede, A., Groschup, M. H., Buschman, A., et al. (2008). Physiological role of the cellular prion protein (PrPc): protein profiling study in two cell culture systems. J. Proteome Res. 7, 2681–2695. doi: 10.1021/pr7007187
Ramljak, S., Schmitz, M., Zafar, S., Wrede, A., Schenkel, S., Asif, A. R., et al. (2015). Cellular prion protein directly interacts with and enhances lactate dehydrogenase expression under hypoxic conditions. Exp. Neurol. 271, 155–167. doi: 10.1016/j.expneurol.2015.04.025
Rane, N. S., Yonkovich, J. L., and Hegde, R. S. (2004). Protection from cytosolic prion protein toxicity by modulation of protein translocation. EMBO J. 23, 4550–4559. doi: 10.1038/sj.emboj.7600462
Rangel, A., Madronal, N., Masso, A. G. I., Gavin, R., Llorens, F., Sumoy, L., et al. (2009). Regulation of GABA(A) and glutamate receptor expression, synaptic facilitation and long-term potentiation in the hippocampus of prion mutant mice. PLoS ONE 4:e7592. doi: 10.1371/journal.pone.0007592
Ratte, S., Vreugdenhil, M., Boult, J. K. R., Patel, A., Asante, E. A., Collinge, J., et al. (2011). Threshold for epileptiform activity is elevated in prion knockout mice. Neuroscience 179, 56–61. doi: 10.1016/j.neuroscience.2011.01.053
Reiten, M. R., Bakkebo, M. K., Tranulis, M. A., Espenes, A., and Boysen, P. (2015). Hematological shift but no evidence of immunological impairment in goat kids naturally devoid of the cellular prion protein (PrPC). Prion 9, S81–S81. doi: 10.1080/19336896.2015.1033248
Rial, D., Duarte, F. S., Xikota, J. C., Schmitz, A. E., Dafre, A. L., Figueiredo, C. P., et al. (2009). Cellular prion protein modulates age-related behavioral and neurochemical alterations in mice. Neuroscience 164, 896–907. doi: 10.1016/j.neuroscience.2009.09.005
Richt, J. A., Kasinathan, P., Hamir, A. N., Castilla, J., Sathiyaseelan, T., Vargas, F., et al. (2007). Production of cattle lacking prion protein. Nat. Biotechnol. 25, 132–138. doi: 10.1038/nbt1271
Riek, R., Hornemann, S., Wider, G., Billeter, M., Glockshuber, R., and Wuthrich, K. (1996). NMR structure of the mouse prion protein domain PrP(121-231). Nature 382, 180–182. doi: 10.1038/382180a0
Robinson, S. W., Nugent, M. L., Dinsdale, D., and Steinert, J. R. (2014). Prion protein facilitates synaptic vesicle release by enhancing release probability. Hum. Mol. Genet. 23, 4581–4596. doi: 10.1093/hmg/ddu171
Roffe, M., Beraldo, F. H., Bester, R., Nunziante, M., Bach, C., Mancini, G., et al. (2010). Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. Natl. Acad. Sci. U.S.A. 107, 13147–13152. doi: 10.1073/pnas.1000784107
Rosse, C., Linch, M., Kermorgant, S., Cameron, A. J. M., Boeckeler, K., and Parker, P. J. (2010). PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 11, 103–112. doi: 10.1038/nrm2847
Rossi, D., Cozzio, A., Flechsig, E., Klein, M. A., Rulicke, T., Aguzzi, A., et al. (2001). Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 20, 694–702. doi: 10.1093/emboj/20.4.694
Roucou, X., Giannopoulos, P. N., Zhang, Y., Jodoin, J., Goodyer, C. G., and LeBlanc, A. (2005). Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 12, 783–795. doi: 10.1038/sj.cdd.4401629
Ruegg, U. T., and Burgess, G. M. (1989). Staurosporine, K-252 and UCN-01 - potent but nonspecific inhibitors of protein-kinases. Trends Pharmacol. Sci. 10, 218–220. doi: 10.1016/0165-6147(89)90263-0
Sakaguchi, S., Katamine, S., Nishida, N., Moriuchi, R., Shigematsu, K., Sugimoto, T., et al. (1996). Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted Prp gene. Nature 380, 528–531. doi: 10.1038/380528a0
Sakudo, A., Lee, D. C., Nakamura, I., Taniuchi, Y., Saeki, K., Matsumoto, Y., et al. (2005). Cell-autonomous PrP-Doppel interaction regulates apoptosis in PrP gene-deficient neuronal cells. Biochem. Biophys. Res. Commun. 333, 448–454. doi: 10.1016/j.bbrc.2005.05.128
Sakudo, A., Lee, D. C., Saeki, K., Nakamura, Y., Inoue, K., Matsumoto, Y., et al. (2003). Impairment of superoxide dismutase activation by N-terminally truncated prion protein (PrP) in PrP-deficient neuronal cell line. Biochem. Biophys. Res. Commun. 308, 660–667. doi: 10.1016/s0006-29ix(03)01459-1
Sakudo, A., Xue, G. A., Kawashita, N., Ano, Y., Takagi, T., Shintani, H., et al. (2010). Structure of the prion protein and its gene: an analysis using bioinformatics and computer simulation. Curr. Protein Pept. Sci. 11, 166–179. doi: 10.2174/138920310790848386
Sales, N., Hassig, R., Rodolfo, K., Di Giamberardino, L., Traiffort, E., Ruat, M., et al. (2002). Developmental expression of the cellular prion protein in elongating axons. Eur. J. Neurosci. 15, 1163–1177. doi: 10.1046/j.1460-9568.2002.01953.x
Sanchez-Alavez, M., Conti, B., Moroncini, G., and Criado, J. R. (2007). Contributions of neuronal prion protein on sleep recovery and stress response following sleep deprivation. Brain Res. 1158, 71–80. doi: 10.1016/j.brainres.2007.05.010
Santuccione, A., Sytnyk, V., Leshchyns'ka, I., and Schachner, M. (2005). Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59(fyn) and to enhance neurite outgrowth. J. Cell Biol. 169, 341–354. doi: 10.1083/jcb.200409127
Satoh, J., Onoue, H., Arima, K., and Yamamura, T. (2005). The 14-3-3 protein forms a molecular complex with heat shock protein Hsp60 and cellular prion protein. J. Neuropathol. Exp. Neurol. 64, 858–868. doi: 10.1097/01.jnen.0000182979.56612.08
Scalabrino, G., Veber, D., De Giuseppe, R., and Roncaroli, F. (2015). Low levels of cobalamin, epidermal growth factor, and normal prions in multiple sclerosis spinal cord. Neuroscience 298, 293–301. doi: 10.1016/j.neuroscience.2015.04.020
Schmitt-Ulms, G., Ehsani, S., Watts, J. C., Westaway, D., and Wille, H. (2009). Evolutionary descent of prion genes from the ZIP family of metal ion transporters. PLoS ONE 4:e7208. doi: 10.1371/journal.pone.0007208
Schmitt-Ulms, G., Hansen, K., Liu, J. L., Cowdrey, C., Yang, J., DeArmond, S. J., et al. (2004). Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat. Biotechnol. 22, 724–731. doi: 10.1038/nbt969
Schmitt-Ulms, G., Legname, G., Baldwin, M. A., Ball, H. L., Bradon, N., Bosque, P. J., et al. (2001). Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 314, 1209–1225. doi: 10.1006/jmbi.2001.5183
Schmitz, M., Greis, C., Ottis, P., Silva, C. J., Schulz-Schaeffer, W. J., Wrede, A., et al. (2014). Loss of prion protein leads to age-dependent behavioral abnormalities and changes in cytoskeletal protein expression. Mol. Neurobiol. 50, 923–936. doi: 10.1007/s12035-014-8655-3
Schultze, S. M., Hemmings, B. A., Niessen, M., and Tschopp, O. (2012). PI3K/AKT, MAPK and AMPK signalling: protein kinases in glucose homeostasis. Expert Rev. Mol. Med. 14:1. doi: 10.1017/s1462399411002109
Sempou, E., Biasini, E., Pinzon-Olejua, A., Harris, D. A., and Malaga-Trillo, E. (2016). Activation of zebrafish Src family kinases by the prion protein is an amyloid-β-sensitive signal that prevents the endocytosis and degradation of E-cadherin/β-catenin complexes in vivo. Mol. Neurodegener. 11:5. doi: 10.1186/s13024-016-0076-5
Shin, H. Y., Park, J. H., Carp, R. I., Choi, E. K., and Kim, Y. S. (2014). Deficiency of prion protein induces impaired autophagic flux in neurons. Front. Aging Neurosci. 6:207. doi: 10.3389/fnagi.2014.00207
Shyng, S. L., Huber, M. T., and Harris, D. A. (1993). A prion protein cycles between the cell-surface and an endocytic compartment in cultured neuroblastoma-cells. J. Biol. Chem. 268, 15922–15928.
Shyu, W. C., Lin, S. Z., Chiang, M. F., Ding, D. C., Li, K. W., Chen, S. F., et al. (2005). Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J. Neurosci. 25, 8967–8977. doi: 10.1523/jneurosci.1115-05.2005
Silverman, G. L., Qin, K. F., Moore, R. C., Yang, Y., Mastrangelo, P., Tremblay, P., et al. (2000). Doppel is an N-glycosylated, glycosylphosphatidylinositol-anchored protein - Expression in testis and ectopic production in the brains of Prnp(o/o) mice predisposed to Purkinje cell loss. J. Biol. Chem. 275, 26834–26841. doi: 10.1074/jbc.M003888200
Singh, A., Haldar, S., Horback, K., Tom, C., Zhou, L., Meyerson, H., et al. (2013). Prion protein regulates iron transport by functioning as a ferrireductase. J. Alzheimers Dis. 35, 541–552. doi: 10.3233/jad-130218
Singh, A., Kong, Q. Z., Luo, X., Petersen, R. B., Meyerson, H., and Singh, N. (2009a). Prion Protein (PrP) knock-out mice show altered iron metabolism: a functional role for prp in iron uptake and transport. PLoS ONE 4:e6115. doi: 10.1371/journal.pone.0006115
Singh, A., Mohan, M. L., Isaac, A. O., Luo, X., Petrak, J., Vyoral, D., et al. (2009b). Prion protein modulates cellular iron uptake: a novel function with implications for prion disease pathogenesis. PLoS ONE 4:e4468. doi: 10.1371/journal.pone.0004468
Sorice, M., Mattei, V., Tasciotti, V., Manganelli, V., Garofalo, T., and Misasi, R. (2012). Trafficking of PrPC to mitochondrial raft-like microdomains during cell apoptosis. Prion 6, 354–358. doi: 10.4161/pri.20479
Spudich, A., Frigg, R., Kilic, E., Kilic, U., Oesch, B., Raeber, A., et al. (2005). Aggravation of ischemic brain injury by prion protein deficiency: role of ERK-1/-2 and STAT-1. Neurobiol. Dis. 20, 442–449. doi: 10.1016/j.nbd.2005.04.002
Stappenbeck, T. S., and Green, K. J. (1992). The desmoplakin carboxyl terminus coaligns with and specifically disrupts intermediate filament networks when expressed in cultured-cells. J. Cell Biol. 116, 1197–1209. doi: 10.1083/jcb.116.5.1197
Steele, A. D., Emsley, J. G., Ozdinler, P. H., Lindquist, S., and Macklis, J. D. (2006). Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc. Natl. Acad. Sci. U.S.A. 103, 3416–3421. doi: 10.1073/pnas.0511290103
Steele, A. D., Lindquist, S., and Aguzzi, A. (2007). The prion protein knockout mouse a phenotype under challenge. Prion 1, 83–93.
Steinacker, P., Hawlik, A., Lehnert, S., Jahn, O., Meier, S., Gorz, E., et al. (2010). Neuroprotective function of cellular prion protein in a mouse model of amyotrophic lateral sclerosis. Am. J. Pathol. 176, 1409–1420. doi: 10.2353/ajpath.2010.090355
Stella, R., Cifani, P., Peggion, C., Hansson, K., Lazzari, C., Bendz, M., et al. (2012). Relative quantification of membrane proteins in wild-type and prion protein (PrP)-knockout cerebellar granule neurons. J. Proteome Res. 11, 523–536. doi: 10.1021/pr200759m
Stempelj, M., and Ferjan, I. (2005). Signaling pathway in nerve growth factor induced histamine release from rat mast cells. Inflamm. Res. 54, 344–349. doi: 10.1007/s00011-005-1364-7
Stewart, R. S., Drisaldi, B., and Harris, D. A. (2001). A transmembrane form of the prion protein contains an uncleaved signal peptide and is retained in the endoplasmic reticululm. Mol. Biol. Cell 12, 881–889. doi: 10.1091/mbc.12.4.881
Stewart, R. S., Piccardo, P., Ghetti, B., and Harris, D. A. (2005). Neurodegenerative illness in transgenic mice expressing a transmembrane form of the prion protein. J. Neurosci. 25, 3469–3477. doi: 10.1523/jneurosci.0105-05.2005
Striebel, J. F., Race, B., and Chesebro, B. (2013a). Prion protein and susceptibility to kainate-induced seizures Genetic pitfalls in the use of PrP knockout mice. Prion 7, 280–285. doi: 10.4161/pri.25738
Striebel, J. F., Race, B., Pathmajeyan, M., Rangel, A., and Chesebro, B. (2013b). Lack of influence of prion protein gene expression on kainate-induced seizures in mice: studies using congenic, coisogenic and transgenic strains. Neuroscience 238, 11–18. doi: 10.1016/j.neuroscience.2013.02.004
Strom, A., Wang, G. S., and Scott, F. W. (2011). Impaired glucose tolerance in mice lacking cellular prion protein. Pancreas 40, 229–232. doi: 10.1097/MPA.0b013e3181f7e547
Su, A. I., Wiltshire, T., Batalov, S., Lapp, H., Ching, K. A., Block, D., et al. (2004). A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 101, 6062–6067. doi: 10.1073/pnas.0400782101
Sunyach, C., Cisse, M. A., da Costa, C. A., and Checler, F. (2007). The C-terminal products of cellular prion protein processing, C1 and C2, exert distinct influence on p53-dependent staurosporine-induced caspase-3 activation. J. Biol. Chem. 282, 1956–1963. doi: 10.1074/jbc.M609663200
Sunyach, C., Jen, A., Deng, J., Fitzgerald, K. T., Frobert, Y., Grassi, J., et al. (2003). The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 22, 3591–3601. doi: 10.1093/emboj/cdg344
Sy, M. S., Altekruse, S. F., Li, C. Y., Lynch, C. F., Goodman, M. T., Hernandez, B. Y., et al. (2011). Association of prion protein expression with pancreatic adenocarcinoma survival in the SEER residual tissue repository. Cancer Biomark. 10, 251–258. doi: 10.3233/cbm-2012-0256
Tang, Z. Q., Ma, J., Zhang, W., Gong, C. G., He, J., Wang, Y., et al. (2016). The role of prion protein expression in predicting gastric cancer prognosis. J. Cancer 7, 984–990. doi: 10.7150/jca.14237
Taubner, L. M., Bienkiewicz, E. A., Copie, V., and Caughey, B. (2010). Structure of the flexible amino-terminal domain of prion protein bound to a sulfated glycan. J. Mol. Biol. 395, 475–490. doi: 10.1016/j.jmb.2009.10.075
Taylor, D. R., Parkin, E. T., Cocklin, S. L., Ault, J. R., Ashcroft, A. E., Turner, A. J., et al. (2009). Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J. Biol. Chem. 284, 22590–22600. doi: 10.1074/jbc.M109.032599
Tobler, I., Deboer, T., and Fischer, M. (1997). Sleep and sleep regulation in normal and prion protein-deficient mice. J. Neurosci. 17, 1869–1879.
Tobler, I., Gaus, S. E., Deboer, T., Achermann, P., Fischer, M., Rulicke, T., et al. (1996). Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 380, 639–642. doi: 10.1038/380639a0
Tremblay, P., Bouzamondo-Bernstein, E., Heinrich, C., Prusiner, S. B., and DeArmond, S. J. (2007). Developmental expression of PrP in the post-implantation embryo. Brain Res. 1139, 60–67. doi: 10.1016/j.brainres.2006.12.055
Tripathi, A. K., Haldar, S., Qian, J., Beserra, A., Suda, S., Singh, A., et al. (2015). Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radic. Biol. Med. 84, 322–330. doi: 10.1016/j.freeradbiomed.2015.03.037
Tsutsui, S., Hahn, J. N., Johnson, T. A., Ali, Z., and Jirik, F. R. (2008). Absence of the cellular prion protein exacerbates and prolongs neuroinflammation in experimental autoimmune encephalomyelitis. Am. J. Pathol. 173, 1029–1041. doi: 10.2353/ajpath.2008.071062
Ugalde, C. L., Finkelstein, D. I., Lawson, V. A., and Hill, A. F. (2016). Pathogenic mechanisms of prion protein, amyloid-beta and alpha-synuclein misfolding: the prion concept and neurotoxicity of protein oligomers. J. Neurochem. 139, 162–180. doi: 10.1111/jnc.13772
Vassallo, N., Herms, J., Behrens, C., Krebs, B., Saeki, K., Onodera, T., et al. (2005). Activation of phosphatidylinositol in 3-kinase by cellular prion protein and its role cell survival. Biochem. Biophys. Res. Commun. 332, 75–82. doi: 10.1016/j.bbrc.2005.04.099
Vergara, C., Ordonez-Gutierrez, L., Wandosell, F., Ferrer, I., del Rio, J. A., and Gavin, R. (2015). Role of PrPC expression in tau protein levels and phosphorylation in Alzheimer's disease evolution. Mol. Neurobiol. 51, 1206–1220. doi: 10.1007/s12035-014-8793-7
Vey, M., Pilkuhn, S., Wille, H., Nixon, R., Dearmond, S. J., Smart, E. J., et al. (1996). Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc. Natl. Acad. Sci. U.S.A. 93, 14945–14949. doi: 10.1073/pnas.93.25.14945
Vincent, B., Paitel, E., Frobert, Y., Lehmann, S., Grassi, J., and Checler, F. (2000). Phorbol ester-regulated cleavage of normal prion protein in HEK293 human cells and murine neurons. J. Biol. Chem. 275, 35612–35616. doi: 10.1074/jbc.M004628200
Vincent, B., Paitel, E., Saftig, P., Frobert, Y., Hartmann, D., De Strooper, B., et al. (2001). The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J. Biol. Chem. 276, 37743–37746. doi: 10.1074/jbc.M105677200
Vincent, B., Sunyach, C., Orzechowski, H. D., St George-Hyslop, P., and Checler, F. (2009). P53-dependent transcriptional control of cellular prion by presenilins. J. Neurosci. 29, 6752–6760. doi: 10.1523/jneurosci.0789-09.2009
Walmsley, A. R., Watt, N. T., Taylor, D. R., Perera, W. S. S., and Hooper, N. M. (2009). α-cleavage of the prion protein occurs in a late compartment of the secretory pathway and is independent of lipid rafts. Mol. Cell. Neurosci. 40, 242–248. doi: 10.1016/j.mcn.2008.10.012
Wang, X. X. X., and Pfenninger, K. H. (2006). Functional analysis of SIRPα in the growth cone. J. Cell Sci. 119, 172–183. doi: 10.1242/jcs.02710
Watt, N. T., Routledge, M. N., Wild, C. P., and Hooper, N. M. (2007). Cellular prion protein protects against reactive-oxygen-species-induced DNA damage. Free Radic. Biol. Med. 43, 959–967. doi: 10.1016/j.freeradbiomed.2007.06.004
Watt, N. T., Taylor, D. R., Gillott, A., Thomas, D. A., Perera, W. S. S., and Hooper, N. M. (2005). Reactive oxygen species-mediated β-cleavage of the prion protein in the cellular response to oxidative stress. J. Biol. Chem. 280, 35914–35921. doi: 10.1074/jbc.M507327200
Watts, J. C., Drisaldi, B., Ng, V., Yang, J., Strome, B., Horne, P., et al. (2007). The CNS glycoprotein Shadoo has PrPC-like protective properties and displays reduced levels in prion infections. EMBO J. 26, 4038–4050. doi: 10.1038/sj.emboj.7601830
Weise, J., Crome, O., Sandau, R., Schulz-Schaeffer, W., Bahr, M., and Zerr, I. (2004). Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci. Lett. 372, 146–150. doi: 10.1016/j.neulet.2004.09.030
Weise, J., Sandau, R., Schwarting, S., Crome, O., Wrede, A., Schulz-Schaeffer, W., et al. (2006). Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 37, 1296–1300. doi: 10.1161/01.STR.0000217262.03192.d4
Weiss, E., Ramljak, S., Asif, A. R., Ciesielczyk, B., Schmitz, M., Gawinecka, J., et al. (2010). Cellular prion protein overexpression disturbs cellular homeostasis in SH-Sy5y neuroblastoma cells but does not alter p53 expression: a proteomic study. Neuroscience 169, 1640–1650. doi: 10.1016/j.neuroscience.2010.06.013
Westaway, D., Daude, N., Wohlgemuth, S., and Harrison, P. (2011). “The PrP-like proteins Shadoo and Doppel,” in Prion Proteins, ed J. Tatzelt (Berlin; Heidelberg: Springer), 225–256.
Whitehouse, I. J., Brown, D., Baybutt, H., Diack, A. B., Kellett, K. A. B., Piccardo, P., et al. (2016). Ablation of prion protein in wild type human amyloid precursor protein (app) transgenic mice does not alter the proteolysis of app, levels of amyloid-beta or pathologic phenotype. PLoS ONE 11:e0159119. doi: 10.1371/journal.pone.0159119
Whitehouse, I. J., Jackson, C., Turner, A. J., and Hooper, N. M. (2010). Prion protein is reduced in aging and in sporadic but not in familial Alzheimer's disease. J. Alzheimers Dis. 22, 1023–1031. doi: 10.3233/jad-2010-101071
Whitehouse, I. J., Miners, J. S., Glennon, E. B. C., Kehoe, P. G., Love, S., Kellett, K. A. B., et al. (2013). Prion protein is decreased in Alzheimer's brain and inversely correlates with BACE1 activity, amyloid-β levels and braak stage. PLoS ONE 8:e59554. doi: 10.1371/journal.pone.0059554
Wik, L., Klingeborn, M., Willander, H., and Linne, T. (2012). Separate mechanisms act concurrently to shed and release the prion protein from the cell. Prion 6, 498–509. doi: 10.4161/pri.22588
Williams, W. M., Stadtman, E. R., and Moskovitz, J. (2004). Ageing and exposure to oxidative stress in vivo differentially affect cellular levels of PrPc in mouse cerebral microvessels and brain parenchyma. Neuropathol. Appl. Neurobiol. 30, 161–168. doi: 10.1111/j.1365-2990.2004.00523.x
Wu, C. L., Jin, X. F., Tsueng, G., Afrasiabi, C., and Su, A. I. (2016). BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 44, D313–D316. doi: 10.1093/nar/gkv1104
Wu, G., Nakajima, K., Takeyama, N., Yukawa, M., Taniuchi, Y., Sakudo, A., et al. (2008). Species-specific anti-apoptotic activity of cellular prion protein in a mouse PrP-deficient neuronal cell line transfected with mouse, hamster, and bovine Prnp. Neurosci. Lett. 446, 11–15. doi: 10.1016/j.neulet.2008.09.020
Yokoyama, T., Kimura, K. M., Ushiki, Y., Yamada, S., Morooka, A., Nakashiba, T., et al. (2001). In vivo conversion of cellular prion protein to pathogenic isoforms, as monitored by conformation-specific antibodies. J. Biol. Chem. 276, 11265–11271. doi: 10.1074/jbc.M008734200
Yu, G. H., Chen, J. Q., Xu, Y. Y., Zhu, C. H., Yu, H. Q., Liu, S. G., et al. (2009). Generation of Goats Lacking Prion Protein. Mol. Reprod. Dev. 76, 3–3. doi: 10.1002/mrd.20960
Yu, G. H., Jiang, L. M., Xu, Y. Y., Guo, H. W., Liu, H. Y., Zhang, Y., et al. (2012). Silencing prion protein in MDA-MB-435 breast cancer cells leads to pleiotropic cellular responses to cytotoxic stimuli. PLoS ONE 7:e48146. doi: 10.1371/journal.pone.0048146
Zafar, S., von Ahsen, N., Oellerich, M., Zerr, I., Schulz-Schaeffer, W. J., Armstrong, V. W., et al. (2011). Proteomics approach to identify the interacting partners of cellular prion protein and characterization of Rab7a interaction in neuronal cells. J. Proteome Res. 10, 3123–3135. doi: 10.1021/pr2001989
Zahn, R., Liu, A. Z., Luhrs, T., Riek, R., von Schroetter, C., Garcia, F. L., et al. (2000). NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. U.S.A. 97, 145–150. doi: 10.1073/pnas.97.1.145
Zanata, S. M., Lopes, M. H., Mercadante, A. F., Hajj, G. N. M., Chiarini, L. B., Nomizo, R., et al. (2002). Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 21, 3307–3316. doi: 10.1093/emboj/cdf325
Zawlik, I., Witusik, M., Hulas-Bigoszewska, K., Piaskowski, S., Szybka, M., Golanska, E., et al. (2006). Regulation of PrPc expression: Nerve Growth Factor (NGF) activates the prion gene promoter through the MEK1 pathway in PC12 cells. Neurosci. Lett. 400, 58–62. doi: 10.1016/j.neulet.2006.02.021
Zeng, F. N., Watt, N. T., Walmsley, A. R., and Hooper, N. M. (2003). Tethering the N-terminus of the prion protein compromises the cellular response to oxidative stress. J. Neurochem. 84, 480–490. doi: 10.1046/j.1471-4159.2003.01529.x
Zhang, S. Y., and Yu, D. H. (2012). Targeting Src family kinases in anti-cancer therapies: turning promise into triumph. Trends Pharmacol. Sci. 33, 122–128. doi: 10.1016/j.tips.2011.11.002
Zhang, X. D., Gillespie, S. K., and Hersey, P. (2004). Staurosporine induces apoptosis of melanoma by both caspase-dependent and -independent apoptotic pathways. Mol. Cancer Ther. 3, 187–197.
Zhou, L., Shang, Y. L., Liu, C. H., Li, J. G., Hu, H., Liang, C., et al. (2014). Overexpression of PrPc, combined with MGr1-Ag/37LRP, is predictive of poor prognosis in gastric cancer. Int. J. Cancer 135, 2329–2337. doi: 10.1002/ijc.28883
Keywords: prion, PrPC, transmissible spongiform encephalopathies, differentiation, adhesion, proliferation, myelin maintenance, stress protection
Citation: Castle AR and Gill AC (2017) Physiological Functions of the Cellular Prion Protein. Front. Mol. Biosci. 4:19. doi: 10.3389/fmolb.2017.00019
Received: 20 February 2017; Accepted: 22 March 2017;
Published: 06 April 2017.
Edited by:
James Shorter, University of Pennsylvania, USAReviewed by:
Jiyan Ma, Van Andel Institute, USACopyright © 2017 Castle and Gill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew C. Gill, YW5kcmV3LmdpbGxAcm9zbGluLmVkLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.