 Peter Bross
Peter Bross Paula Fernandez-Guerra
Paula Fernandez-Guerra
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Biosci. , 31 August 2016
Sec. Protein Folding, Misfolding and Degradation
Volume 3 - 2016 | https://doi.org/10.3389/fmolb.2016.00049
This article is part of the Research Topic Pathologic conditions of the human nervous and muscular systems associated with mutant chaperones: molecular and mechanistic aspects View all 12 articles
Heat shock protein 60 (HSP60) forms together with heat shock protein 10 (HSP10) double-barrel chaperonin complexes that are essential for folding to the native state of proteins in the mitochondrial matrix space. Two extremely rare monogenic disorders have been described that are caused by missense mutations in the HSPD1 gene that encodes the HSP60 subunit of the HSP60/HSP10 chaperonin complex. Investigations of the molecular mechanisms underlying these disorders have revealed that different degrees of reduced HSP60 function produce distinct neurological phenotypes. While mutations with deleterious or strong dominant negative effects are not compatible with life, HSPD1 gene variations found in the human population impair HSP60 function and depending on the mechanism and degree of HSP60 dys- and mal-function cause different phenotypes. We here summarize the knowledge on the effects of disturbances of the function of the HSP60/HSP10 chaperonin complex by disease-associated mutations.
The type I chaperonins, a subclass of the molecular chaperone family of proteins, assist folding of proteins in the bacterial cytosol, the mitochondrial matrix space, and the chloroplast stroma. Like its bacterial and chloroplast homologs the mitochondrial HSP60/HSP10 complex is composed of two seven-meric rings of the large subunit (HSP60) stacked back to back (Nisemblat et al., 2015; Figure 1A). The HSP60 ring structures enclose an inner cavity that is sealed by lids formed by seven-meric rings of the small subunit (HSP10). With the exception of a few endosymbionts, homologs of these proteins are abundantly expressed in mitochondria, chloroplasts and bacteria. The functional folding cycle of the mammalian HSP60/HSP10 complex (Nielsen and Cowan, 1998; Levy-Rimler et al., 2001, 2002) has to a large degree been elucidated in analogy to detailed studies of the homologous GroEL/GroES complex of E. coli bacteria (Horwich, 2013; Hayer-Hartl et al., 2015). Cycles including binding of proteins undergoing folding to the HSP60 rings, their encapsulation by association of HSP10 rings and dissociation of both the HSP10 ring and the enclosed protein are orchestrated by ATP binding, hydrolysis and release of ADP by the HSP60 subunits. These cycles promote folding of proteins to the native state, but not every cycle results in successful folding. Some proteins may require several rounds. Knock-out experiments have shown that the genes encoding homologs of the HSP60/HSP10 complex are essential in organisms from bacteria to mice (Cheng et al., 1989; Fayet et al., 1989; Perezgasga et al., 1999; Christensen et al., 2010). In humans only few gene variations altering the amino acid sequence of HSP60 and HSP10 have been described (Table 1). However, very rare disease-associated missense mutations in HSP60 have been associated with a dominant form of hereditary spastic paraplegia (HSP; Hansen et al., 2002) and a recessively inherited white matter disorder called MitCHAP60 disease (Magen et al., 2008). In another article published under this research topic we describe the first potentially disease associated mutation in HSP10 that has been identified in a patient with a neurological and developmental disorder (Bie et al., submitted).
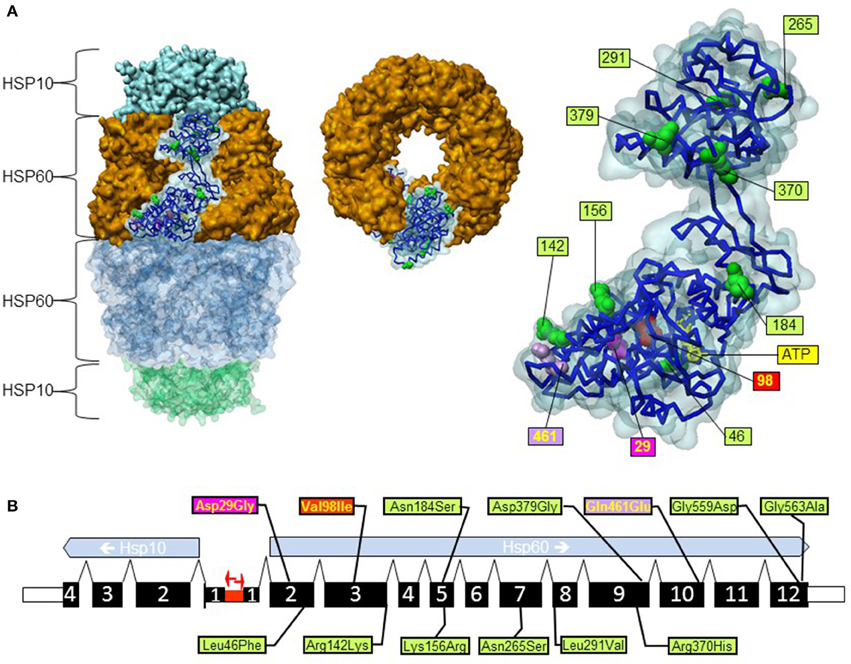
Figure 1. Position of gene variations in the HSPD1 gene and the HSP60 protein structure. (A) Structure of the HSP60/HSP10 complex and position of mutations. 3d structure representations were created based on PDB coordinates 4pj1 (Nisemblat et al., 2015) using the software Discovery Studio 4.5.0.15071 (Biovia). Left: surface representation of the structure of the HSP60/HSP10 complex; HSP60 and HSP10 rings are indicated. The surface representation of one subunit of the upper HSP60 ring is shown transparent and the carbon backbone and space filling representation of amino acid positions where missense variations have been found are shown. Middle: upside view of the upper HSP60 ring. Right: enlarged view of the highlighted subunit from the complex shown on the left with numbering of positions with missense variations. The mutations p.Gly559Asp and p.Gly563Ala are not shown because the C-terminal part of the HSP60 protein is not contained in the crystal structure. The bound ATP molecule is shown as yellow sticks. (B) Exon structure of the human HSPD1 and HSPE1 genes encoding HSP60 and HSP10. The two genes are situated in a head to head configuration on chromosome 2 with a common bidirectional promoter (red arrows). Exons are numbered and their coding parts are given as broad bars. Positions of missense variations are shown; color coding is in relation to disease-association (see text).
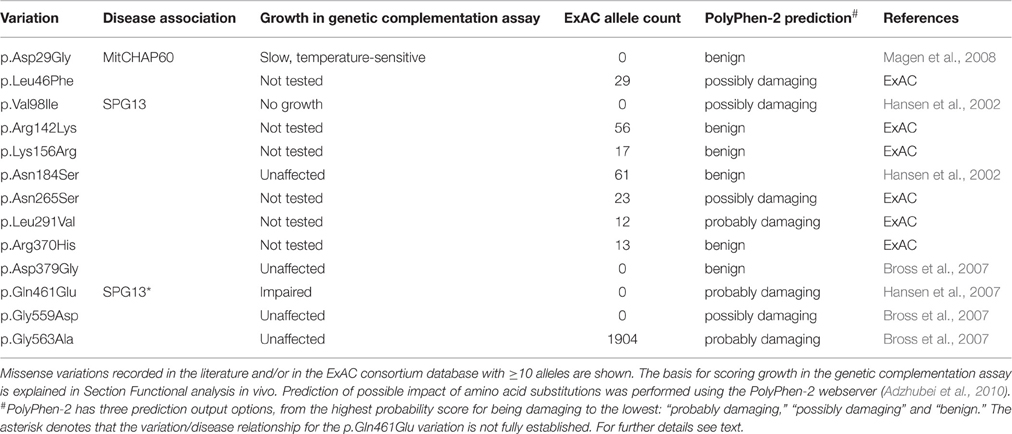
Table 1. Missense variations in the HSPD1 gene encoding HSP60.
The genomic structure for the human HSPD1 and HSPE1 genes encoding the proteins HSP60 and HSP10, respectively, has been characterized experimentally (Hansen et al., 2003). The HSPD1 and HSPE1 genes are localized at chromosome locus 2q33.1 in a head to head arrangement with a bidirectional common promoter (Figure 1). Such a head to head arrangement is evolutionary conserved from C. elegans to humans (Bross, 2015). The two genes comprise ~17 kb and consist of 12 and 4 exons, respectively (Figure 1B). The first exon of Hsp60 is non-coding and the first exon of Hsp10 contains only the first codon (Hansen et al., 2003). At present there are two amino acid variations in HSP60 known for which a clear disease-association has been established: the HSP60-p.Val98Ile mutation associated with a dominantly inherited form of HSP (SPG13; OMIM #605280; Hansen et al., 2002) and the HSP60-p.Asp29Gly mutation causing a recessively inherited white matter disease called MitCHAP60 disease (OMIM #612233; Magen et al., 2008). Besides these two, a number of other variations have been described (Figure 1 and Table 1; Hansen et al., 2002, 2007; Bross et al., 2007). The Exome Aggregation Consortium webserver (ExAC; Cambridge, MA (URL: http://exac.broadinstitute.org) [June, 2016 accessed]) lists 95 missense variations for the protein product of the canonical HSPD1 transcript. This website provides high quality exome sequencing data from more than 60,000 unrelated adults without severe pediatric disease. Of the 95 HSPD1 missense variations 60 have only been seen in one single allele and only 9 have been observed in more than 10 of the ~120,000 alleles recorded (Table 1).
Table 1 lists both the HSPD1 missense variations described in the literature and those present in ≥10 alleles in the ExAC Consortium Website. The functional consequences for some of these variations have been investigated previously and the results are summarized in this review.
Expression in E. coli had indicated that the two disease-associated variant proteins HSP60-p.Val98Ile and HSP60-p.Asp29Gly displayed similar stability as wild type HSP60 suggesting that the amino acid replacements caused functional impairment (Hansen et al., 2002; Magen et al., 2008). Indeed, the purified HSP60-p.Val98Ile protein assembled into native ring complexes in the same way and with similar efficiency as the wild type HSP60 protein but displayed a reduced ATPase hydrolysis rate and a severely decreased in vitro refolding activity with malate dehydrogenase as substrate (Bross et al., 2008). The ATPase function and refolding activity of the purified HSP60-p.Asp29Gly variant protein associated with MitCHAP60 disease was found decreased in a similar way (Parnas et al., 2009). This study also indicated impaired stability of HSP60-p.Asp29Gly oligomers causing their disassembly at low protein concentrations.
A sophisticated genetic complementation assay for analyzing the function of variants of the human HSP60/HSP10 chaperonin complex has been developed in the lab of Costa Georgopoulos (Richardson et al., 2001). This assay is based on knocking out the chromosomal groESgroEL operon and testing whether cell viability and growth properties can be maintained by providing the bacterial cells with a plasmid encoding the chaperonin gene variants to be investigated. Knock-out cells are not viable, but providing the cells with plasmids with cDNAs encoding the wild type human HSP60 and HSP10 genes maintains cell viability demonstrating that the human HSP60/HSP10 complex can functionally replace the bacterial GroEL/GroES complex (Richardson et al., 2001).
Seven human HSP60 missense variants have so-far been investigated using this functional assay (Table 1). These studies showed that the SPG13-associated mutant HSP60-p.Val98Ile was unable to functionally replace the bacterial chaperonin. However, cells expressing the HSP60-p.Asp29Gly variant displayed slow and temperature-sensitive growth. Cells with the HSP60-p.Gln461Glu variation found in a sporadic spastic paraplegia patient also displayed impaired growth. The four other HSP60 variants studied: p.Asn184Ser, p.Asp379Gly, p.Gly559Asp, and p.Gly563Ala, behaved like wild type HSP60 in the genetic complementation assay suggesting that they have no significant effect.
Bioinformatics prediction of the effects of the variations using the PolyPhen-2 tool (Adzhubei et al., 2010) predicts damaging effects for the HSP60-p.Val98Ile and HSP60-Gln461Glu variations, but benign for the HSP60-p.Asp29Gly for which a clear disease relationship has been established. PolyPhen-2 also predicts “damaging” for the two carboxy-terminal variations and four of the six missense variants found in more than 10 alleles in the ExAC dataset. Given the discrepancy between experimental and prediction results this prediction results must be taken with caution. Inspection of the position of the variations in the crystal structure of the HSP60/HSP10 complex (Nisemblat et al., 2015) shows that the variations with an established disease-association (HSP60-p.Asp29Gly and HSP60-p.Val98Ile) are localized in the core of the equatorial domain of the HSP60 protein, whereas most of the other variation sites are localized on the surface of HSP60 subunits (Figure 1A).
The SPG13-associated mutation HSP60-p.Val98Ile is dominantly inherited, i.e., the patient cells express both a wild type and a mutant allele and these two variant proteins are likely on equal terms incorporated into HSP60 ring structures resulting in heteromeric rings with stochastically distributed content of the two variants. To test whether incorporation of mutant HSP60-p.Val98Ile subunits together with wild type HSP60 subunits into HSP60 ring complexes would cause a dominant negative effect, the complementation assay was further engineered. E. coli cells with the deletion of the endogenous groESgroEL operon and containing a plasmid with an IPTG-inducible operons comprising HSP10 and wild type HSP60 were transformed with a second plasmid comprising an arabinose-inducible operon with the respective mutant HSP60 variant and HSP10 (Bross et al., 2008). There was no effect on growth in cells expressing both wild type HSP60 and HSP60-p.Val98Ile. As a control, expression of an artificially constructed ATPase-deficient HSP60 variant together with wild type HSP60 blocked growth. These experiments strongly indicated that the HSP60-p.Val98Ile mutation exerts no significant dominant negative effect on co-expressed wild type HSP60 protein.
Notwithstanding the rarity, studies of the very large index family that led to the discovery of the association of the HSP60-p.Val98Ile mutation with HSP has given a firm basis for the mutation/disease relationship (Hansen et al., 2002). SPG13 is dominantly inherited with a pure spastic paraplegia phenotype with high penetrance (Fontaine et al., 2000). Hereditary spastic paraplegia is a complex disease both genetically and clinically with mutations in more than 60 different genes established as causal and with all inheritance modes (Kumar et al., 2015; Tesson et al., 2015; Di Fabio et al., 2016). The characteristic spastic gait has given the acronym SPG and subsequent numerals distinguish the different genetic forms of HSP. Spastic gait and spasticity in the lower limbs are also observed as a side phenotype in many other neurological diseases. The disease is only classified as HSP if the paraplegia is the major clinical characteristic. Clinical analysis of the 10 MitChap60 patients homozygous for the HSP60-p.Asp29Gly mutation revealed also spastic paraplegia in all them (Magen et al., 2008). However, this “side-phenotype” was overshadowed by the much more severe presentation characterized by early-onset, profound cerebral involvement and lethality of these patients. A number of other SPG genes are also disease genes in other neurological diseases illustrating that maintaining the classical distinctions becomes more and more difficult (Tesson et al., 2015). Rather, the phenotype in a given patient depends on multiple factors, like the mutated gene, the nature of the mutation and its location in the protein, the zygosity of the mutation, and influences of modifier variants and the environment.
In spite of widely spread genetic screening, so far only one single additional spastic paraplegia patient heterozygous for another mutation in HSP60 (HSP60-p.Gln461Glu) has been reported (Hansen et al., 2007). The causative nature of this mutation is uncertain because two siblings carrying this mutation were asymptomatic. However, as the genetic complementation test showed a mild functional impairment (See Section Functional analysis in vivo), this variation may be disease-associated with reduced penetrance. The most frequently observed amino acid variation in HSP60, HSP60-p.Gly563Ala, has been found in homozygous form in one sporadic Danish spastic paraplegia patient (Svenstrup et al., 2009). Frequency analysis of this polymorphisms in Danish controls showed an allele frequency of 1.3% and similar allele frequencies were observed in all ethnic groups in the ExAC database. In addition, the number of homozygotes in the ExAC database is consistent with Hardy Weinberg distribution and the genetic complementation assay did not indicate impaired function of the HSP60-p.Gly563Ala protein. Taken together this suggests that this variation has no significant effect on function (Bross et al., 2007).
Effects of expressing the SPG13- and MitCHap60-associated mutant proteins on mitochondrial morphology have been assessed in Cos-7 cells transfected with cDNAs encoding the disease-associated mutant proteins (Miyamoto et al., 2015, 2016). Cos-7 cells expressing either the mutant variants HSP60-p.Asp29Gly, HSP60-p.Val98Ile or HSP60-p.Gln461Glu displayed increased mitochondrial fission and decreased mitochondrial membrane potential whereas Cos-7 cells transfected with wild type HSP60 cDNA did not. This indicates that the studied mutant proteins interfere with the function of the endogenous wild type protein.
ShRNA-mediated knock-down of HSP60 in human HEK293 cells decreased the steady state levels of the mitochondrial medium-chain acyl-CoA dehydrogenase (Corydon et al., 2005), an enzyme whose subunits transiently interact with HSP60 before assembling into functional tetramers (Yokota et al., 1992; Saijo et al., 1994). Folding of ectopically expressed mitochondria-targeted green fluorescence protein (GFP) was decreased both in HEK293 cells in which HSP60 was knocked down and in HEK293 expressing a dominant negative ATPase-deficient HSP60 variant (Corydon et al., 2005; Bie et al., 2011). Furthermore, a series of assays of HSP60 knock-down in the mouse hypothalamic cell line N25/2 revealed decreases in mitochondrial respiration, levels of respiratory chain subunits, mitochondrial DNA levels and an increase in mitochondrial volume and mitochondrial superoxide (Kleinridders et al., 2013). These cellular models can be used for further studies elucidating the multiple effects of deficiency of the HSP60/HSP10 chaperonin complex.
Knock-out of both HSP60-encoding alleles in mice is not compatible with life. Such embryos died early during development (Christensen et al., 2010). However, mice which are heterozygous for one HSP60 knock-out allele, and which express half levels of HSP60 protein, developed normally and were borne in the expected Hardy-Weinberg frequency. More thorough long-term analysis of these mice revealed a late onset and slowly progressive deficit in motor functions recapitulating features of HSP SPG13 in humans (Magnoni et al., 2013). The phenotype was accompanied by morphological changes of mitochondria in spinal cord axons. Furthermore, decreased ATP synthesis was observed in mitochondria isolated from brain cortex and spinal cord. The respiratory chain defect could be narrowed down to impaired activity of respiratory chain complex III. Proteomic analysis of mitochondria from mutant mouse tissues consistently revealed decreased levels of the UQCRC1 subunit of complex III in these tissues. As UQCRC1 transcript levels were even increased and an effect on translation is improbable, this suggested that deficiency of the HSP60/HSP10 chaperonin complex resulted in impaired folding of the UQCRC1 protein entailing premature degradation of the UQCRC1 protein. Based on the same criteria and supported by direct interaction with HSP60, the manganese-dependent superoxide dismutase SOD2 was identified as another protein that is highly dependent on appropriate HSP60/HSP10 chaperone complex function (Magnoni et al., 2014).
Proteins like UQCRC1 or SOD2 thus appear to depend more than others proteins on folding assistance by the HSP60/HSP10 complex. For E. coli 85 proteins that display obligate dependence on folding assistance by the bacterial chaperonin complex have been characterized (Kerner et al., 2005). Identification of those proteins whose folding obligatorily requires the human HSP60/HSP0 complex is still lacking. Such knowledge would be very helpful to identify further mitochondrial functions affected by deficiencies of the HSP60/HSP10 complex.
Different mutations in HSP60 or its partner protein HSP10 lead to distinct phenotypes of neurological disorders with a clear mitochondrial dysfunction pattern. These diseases are very rare as deleterious effects of mutations in these essential genes are not compatible with normal embryonal development (Christensen et al., 2010). The broader use of exome sequencing will likely reveal more cases also including de novo mutations in the HSPD1 and HSPE1 genes in sporadic diseases. Clinical geneticists should therefore be aware of this and it will be important to collect the knowledge of these rare cases to be able study genotype/phenotype relationships and to assess the disease-causing nature of variations.
Besides being affected by mutations, the activity and function of the HSP60/HSP10 complex can also be regulated by its expression levels. The regulation of the transcription levels of both proteins occurs via different elements in the bidirectional promoter. SP1 elements provide robust house-keeping levels of expression and on top of that heat-shock elements, mitochondrial unfolding protein response elements, and STAT3 elements further modulate expression adapting it to specific situations (Zhao et al., 2002; Hansen et al., 2003; Horibe and Hoogenraad, 2007; Kim and Lee, 2007; Kim et al., 2007; Kleinridders et al., 2013). One study also shows that Hsp60 mRNA is a direct target of miR-1 and miR-206 in cardiomyocytes (Shan et al., 2010).
Indeed, dysregulation of HSP60 expression in hypothalamus has been implicated with type 2 diabetes mellitus (Kleinridders et al., 2013) and changes in chaperonin expression and activity have been observed in several diseases such as cardiomyopathies, autoimmune disorders, and cancer (Cappello et al., 2014).
Finally, posttranslational modifications of the HSP60/HSP10 complex may regulate the activity of the complex. Like other molecular chaperones HSP60 is a highly modified protein with a long list of PTMs recorded in the UniProt database (Consortium, 2015): phosphorylation, acetylation, succinylation, malonylation, nitrosylation, sumoylation, ubiquitination, N-glycosylation, and O-GlcNAcylation. Acetylation of the co-chaperonin HSP10 has been indicated to affect activity of the chaperonin complex (Lu et al., 2014). One of the HSP60 missense variations shown in Table 1, Lysine-156, has been described before as modified by acetylation both in the human acute myeloid leukemia cell line MV4-11 (Choudhary et al., 2009) and in mouse liver HSP60 (Rardin et al., 2013). The p.Lys156Arg missense variation is classified as benign by the bioinformatics tools due to conservation of the positive charge, but elimination of this acetylation site may affect regulation of HSP60 function. Regulation of the activity of the HSP60/HSP10 complex at different levels may thus be a crucial hub for development of mitochondrial dysfunction, a hallmark in many disease processes.
PF and PB have both contributed to writing the draft and producing the final version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The research of PB has been supported by the Ludvig and Sara Elsass Foundation. The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Bie, A. S., Palmfeldt, J., Hansen, J., Christensen, R., Gregersen, N., Corydon, T. J., et al. (2011). A cell model to study different degrees of Hsp60 deficiency in HEK293 cells. Cell Stress Chaperones 16, 633–640. doi: 10.1007/s12192-011-0275-5
Bross, P., Li, Z., Hansen, J., Hansen, J. J., Nielsen, M. N., Corydon, T. J., et al. (2007). Single-nucleotide variations in the genes encoding the mitochondrial Hsp60/Hsp10 chaperone system and their disease-causing potential. J. Hum. Genet. 52, 56–65. doi: 10.1007/s10038-006-0080-7
Bross, P., Naundrup, S., Hansen, J., Nielsen, M. N., Christensen, J. H., Kruhoffer, M., et al. (2008). The HSP60-(P.val98ile) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both in vitro and in vivo. J. Biol. Chem. 283, 15694–15700. doi: 10.1074/jbc.M800548200
Cappello, F., Marino Gammazza, A., Palumbo Piccionello, A., Campanella, C., Pace, A., Conway de Macario, E., et al. (2014). Hsp60 chaperonopathies and chaperonotherapy: targets and agents. Expert Opin. Ther. Targets 18, 185–208. doi: 10.1517/14728222.2014.856417
Cheng, M. Y., Hartl, F. U., Martin, J., Pollock, R. A., Kalousek, F., Neupert, W., et al. (1989). Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature 337, 620–625.
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. doi: 10.1126/science.1175371
Christensen, J. H., Nielsen, M. N., Hansen, J., Füchtbauer, A., Füchtbauer, E. M., West, M., et al. (2010). Inactivation of the hereditary spastic paraplegia-associated Hspd1 gene encoding the Hsp60 chaperone results in early embryonic lethality in mice. Cell Stress Chaperones 15, 851–863. doi: 10.1007/s12192-010-0194-x
Consortium, T. U. (2015). UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212. doi: 10.1093/nar/gku989
Corydon, T. J., Hansen, J., Bross, P., and Jensen, T. G. (2005). Down-regulation of Hsp60 expression by RNAi impairs folding of medium-chain acyl-CoA dehydogenase wild-type and disease-associated proteins. Mol. Genet. Metab. 85, 260–270. doi: 10.1016/j.ymgme.2005.04.003
Di Fabio, R., Storti, E., Tessa, A., Pierelli, F., Morani, F., and Santorelli, F. M. (2016). Hereditary spastic paraplegia: pathology, genetics and therapeutic prospects. Expert Opin. Orphan Drugs 4, 429–442. doi: 10.1517/21678707.2016.1153964
Fayet, O., Ziegelhoffer, T., and Georgopoulos, C. (1989). The groES and groEL heat shock gene products of Escherichia coli are essential for bacterial growth at all temperatures. J. Bacteriol. 171, 1379–1385.
Fontaine, B., Davoine, C. S., Dürr, A., Paternotte, C., Feki, I., Weissenbach, J., et al. (2000). A new locus for autosomal dominant pure spastic paraplegia, on chromosome 2q24-q34. Am. J. Hum. Genet. 66, 702–707. doi: 10.1086/302776
Hansen, J. J., Bross, P., Westergaard, M., Nielsen, M. N., Eiberg, H., Børglum, A. D., et al. (2003). Genomic structure of the human mitochondrial chaperonin genes: HSP60 and HSP10 are localised head to head on chromosome 2 separated by a bidirectional promoter. Hum. Genet. 112, 71–77. doi: 10.1007/s00439-002-0837-9
Hansen, J. J., Dürr, A., Cournu-Rebeix, I., Georgopoulos, C., Ang, D., Nielsen, M. N., et al. (2002). Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am. J. Hum. Genet. 70, 1328–1332. doi: 10.1086/339935
Hansen, J., Svenstrup, K., Ang, D., Nielsen, M. N., Christensen, J. H., Gregersen, N., et al. (2007). A novel mutation in the HSPD1 gene in a patient with hereditary spastic paraplegia. J. Neurol. 254, 897–900. doi: 10.1007/s00415-006-0470-y
Hayer-Hartl, M., Bracher, A., and Hartl, F. U. (2015). The GroEL-GroES chaperonin machine: a nano-cage for protein folding. Trends Biochem. Sci. 441, 62–76. doi: 10.1016/j.tibs.2015.07.009
Horibe, T., and Hoogenraad, N. J. (2007). The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2:e835. doi: 10.1371/journal.pone.0000835
Horwich, A. L. (2013). Chaperonin-mediated protein folding. J. Biol. Chem. 288, 23622–23632. doi: 10.1074/jbc.X113.497321
Kerner, M. J., Naylor, D. J., Ishihama, Y., Maier, T., Chang, H. C., Stines, A. P., et al. (2005). Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell 122, 209–220. doi: 10.1016/j.cell.2005.05.028
Kim, S. W., Kim, J. B., Kim, J. H., and Lee, J. K. (2007). Interferon-gamma-induced expressions of heat shock protein 60 and heat shock protein 10 in C6 astroglioma cells: identification of the signal transducers and activators of transcription 3-binding site in bidirectional promoter. Neuroreport 18, 385–389. doi: 10.1097/WNR.0b013e32801299cc
Kim, S. W., and Lee, J. K. (2007). NO-induced downregulation of HSP10 and HSP60 expression in the postischemic brain. J. Neurosci. Res. 85, 1252–1259. doi: 10.1002/jnr.21236
Kleinridders, A., Lauritzen, H. P., Ussar, S., Christensen, J. H., Mori, M. A., Bross, P., et al. (2013). Leptin regulation of Hsp60 impacts hypothalamic insulin signaling. J. Clin. Invest. 123, 4667–4680. doi: 10.1172/JCI67615
Kumar, K. R., Blair, N. F., and Sue, C. M. (2015). An update on the hereditary spastic paraplegias: new genes and new disease models. Mov. Disord. Clin. Pract. 2, 213–223. doi: 10.1002/mdc3.12184
Levy-Rimler, G., Bell, R. E., Ben-Tal, N., and Azem, A. (2002). Type I chaperonins: not all are created equal. FEBS Lett. 529, 1. doi: 10.1016/S0014-5793(02)03178-2
Levy-Rimler, G., Viitanen, P., Weiss, C., Sharkia, R., Greenberg, A., Niv, A., et al. (2001). The effect of nucleotides and mitochondrial chaperonin 10 on the structure and chaperone activity of mitochondrial chaperonin 60. Eur. J. Biochem. 268, 3465–3472. doi: 10.1046/j.1432-1327.2001.02243.x
Lu, Z., Chen, Y., Aponte, A. M., Battaglia, V., Gucek, M., and Sack, M. N. (2014). Prolonged fasting identifies heat shock protein 10 as a sirtuin 3 substrate: elucidating a new mechanism linking mitochondrial protein acetylation to fatty acid oxidation enzyme folding and function. J. Biol. Chem. 290, 2466–2476. doi: 10.1074/jbc.M114.606228
Magen, D., Georgopoulos, C., Bross, P., Ang, D., Segev, Y., Goldsher, D., et al. (2008). Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am. J. Hum. Genet. 83, 30–42. doi: 10.1016/j.ajhg.2008.05.016
Magnoni, R., Palmfeldt, J., Christensen, J. H., Sand, M., Maltecca, F., Corydon, T. J., et al. (2013). Late onset motoneuron disorder caused by mitochondrial Hsp60 chaperone deficiency in mice. Neurobiol. Dis. 54, 12–23. doi: 10.1016/j.nbd.2013.02.012
Magnoni, R., Palmfeldt, J., Hansen, J., Christensen, J. H., Corydon, T. J., and Bross, P. (2014). The Hsp60 folding machinery is crucial for manganese superoxide dismutase folding and function. Free Radic. Res. 48, 168–179. doi: 10.3109/10715762.2013.858147
Miyamoto, Y., Eguchi, T., Kawahara, K., Hasegawa, N., Nakamura, K., Funakoshi-Tago, M., et al. (2015). Hypomyelinating leukodystrophy-associated missense mutation in HSPD1 blunts mitochondrial dynamics. Biochem. Biophys. Res. Commun. 462, 275–281. doi: 10.1016/j.bbrc.2015.04.132
Miyamoto, Y., Megumi, F.-T., Hasegawa, N., Eguchi, T., Tanoue, A., Tamura, H., et al. (2016). Data supporting mitochondrial morphological changes by SPG13-associated HSPD1 mutants. Data Brief 6, 482–488. doi: 10.1016/j.dib.2015.12.038
Nielsen, K. L., and Cowan, N. J. (1998). A single ring is sufficient for productive chaperonin-mediated folding in vivo. Mol. Cell 2, 93–99. doi: 10.1016/S1097-2765(00)80117-3
Nisemblat, S., Yaniv, O., Parnas, A., Frolow, F., and Azem, A. (2015). Crystal structure of the human mitochondrial chaperonin symmetrical football complex. Proc. Natl. Acad. Sci. U.S.A. 112, 6044–6049. doi: 10.1073/pnas.1411718112
Parnas, A., Nadler, M., Nisemblat, S., Horovitz, A., Mandel, H., and Azem, A. (2009). The MitCHAP-60 disease is due to entropic destabilization of the human mitochondrial Hsp60 oligomer. J. Biol. Chem. 284, 28198–28203. doi: 10.1074/jbc.M109.031997
Perezgasga, L., Segovia, L., and Zurita, M. (1999). Molecular characterization of the 5′ control region and of two lethal alleles affecting the hsp60 gene in Drosophila melanogaster. FEBS Lett. 456, 269–273. doi: 10.1016/S0014-5793(99)00963-1
Rardin, M. J., Newman, J. C., Held, J. M., Cusack, M. P., Sorensen, D. J., Li, B., et al. (2013). Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. U.S.A. 110, 6601–6606. doi: 10.1073/pnas.1302961110
Richardson, A., Schwager, F., Landry, S. J., and Georgopoulos, C. (2001). The importance of a mobile loop in regulating chaperonin/ co-chaperonin interaction: humans versus Escherichia coli. J. Biol. Chem. 276, 4981–4987. doi: 10.1074/jbc.M008628200
Saijo, T., Welch, W. J., and Tanaka, K. (1994). Intramitochondrial folding and assembly of medium-chain acyl-CoA dehydrogenase (MCAD) - Demonstration of impaired transfer of K304E-variant MCAD from its complex with Hsp60 to the native tetramer. J. Biol. Chem. 269, 4401–4408.
Shan, Z. X., Lin, Q. X., Deng, C. Y., Zhu, J. N., Mai, L. P., Liu, J. L., et al. (2010). miR-1/miR-206 regulate Hsp60 expression contributing to glucose-mediated apoptosis in cardiomyocytes. FEBS Lett. 584, 3592–3600. doi: 10.1016/j.febslet.2010.07.027
Svenstrup, K., Bross, P., Koefoed, P., Hjermind, L. E., Eiberg, H., Born, A. P., et al. (2009). Sequence variants in SPAST, SPG3A and HSPD1 in hereditary spastic paraplegia. J. Neurol. Sci. 284, 90–95. doi: 10.1016/j.jns.2009.04.024
Tesson, C., Koht, J., and Stevanin, G. (2015). Delving into the complexity of hereditary spastic paraplegias: how unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum. Genet. 134, 511–538. doi: 10.1007/s00439-015-1536-7
Yokota, I., Saijo, T., Vockley, J., and Tanaka, K. (1992). Impaired tetramer assembly of variant medium-chain acyl-coenzyme A dehydrogenase with a glutamate or aspartate substitution for lysine 304 causing instability of the protein. J. Biol. Chem. 267, 26004–26010.
Keywords: HSP60, chaperonin, neurological disease, protein folding problem, mitochondria, gene variation
Citation: Bross P and Fernandez-Guerra P (2016) Disease-Associated Mutations in the HSPD1 Gene Encoding the Large Subunit of the Mitochondrial HSP60/HSP10 Chaperonin Complex. Front. Mol. Biosci. 3:49. doi: 10.3389/fmolb.2016.00049
Received: 15 July 2016; Accepted: 22 August 2016;
Published: 31 August 2016.
Edited by:
Alberto J. L. Macario, University of Maryland at Baltimore, USAReviewed by:
Carlos H. Ramos, State University of Campinas, BrazilCopyright © 2016 Bross and Fernandez-Guerra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Bross, cGV0ZXIuYnJvc3NAY2xpbi5hdS5kaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.