 Marten Szibor
Marten Szibor Marie Mühlon1
Marie Mühlon1- 1Department of Cardiothoracic Surgery, Center for Sepsis Control and Care (CSCC), Jena University Hospital, Friedrich-Schiller University of Jena, Jena, Germany
- 2BioMediTech and Tampere University Hospital, Faculty of Medicine and Health Technology, Tampere University, Tampere, Finland
- 3Department of Environmental and Biological Sciences, University of Eastern Finland, Joensuu, Finland
Cardiomyocytes depend on mitochondrial oxidative phosphorylation (OXPHOS) for energy metabolism, which is facilitated by the mitochondrial electron transfer system (ETS). In a series of thermogenic redox reactions, electrons are shuttled through the ETS to oxygen as the final electron acceptor. This electron transfer is coupled to proton translocation across the inner mitochondrial membrane, which itself is the main driving force for ATP production. Oxygen availability is thus a prerequisite for ATP production and consequently contractility. Notably, cardiomyocytes are exceptionally large cells and densely packed with contractile structures, which constrains intracellular oxygen distribution. Moreover, oxygen must pass through layers of actively respiring mitochondria to reach the ones located in the innermost contractile compartment. Indeed, uneven oxygen distribution was observed in cardiomyocytes, suggesting that local ATP supply may also vary according to oxygen availability. Here, we discuss how spatial adjustment of bioenergetics to intracellular oxygen fluctuations may underlie cardiac contractile adaptation and how this adaptation may pose a risk for the development of contractile failure.
1 Introduction
The adult human heart turns over the staggering amount of 6 kg of ATP per day to maintain contractility and organ homeostasis (Ingwall, 2002). The amount of ATP present in the organ, however, is comparatively low at about 0.7 g ATP per 250 g of cardiac tissue. This suffices to support organ function for about 10 heartbeats or, in terms of time, approximately 10 seconds (Ingwall, 2002). Continuous replenishment of ATP is thus a vital necessity.
The heart relies on the highly efficient oxidative phosphorylation (OXPHOS) system for adequate ATP production and enters contractile dysfunction when this system is faulty (Sabbah, 2020; Schwartz et al., 2022). The main building block of OXPHOS is the mitochondrial respiratory chain or electron transfer system (ETS), facilitating a series of thermogenic redox reactions for which oxygen serves as the terminal electron acceptor. Electron transfer through the ETS is coupled to proton translocation across the inner mitochondrial membrane, which in turn generates the main driving force for ATP production (Mitchell, 1961). Oxygen abundance is therefore a prerequisite for cardiac energy metabolism (bioenergetics) (Figure 1A). This way, OXPHOS provides the mammalian heart with an impressive reserve capacity (Cooke et al., 1998) without a need for anaerobic metabolism as seen in skeletal muscle (Hargreaves and Spriet, 2020).
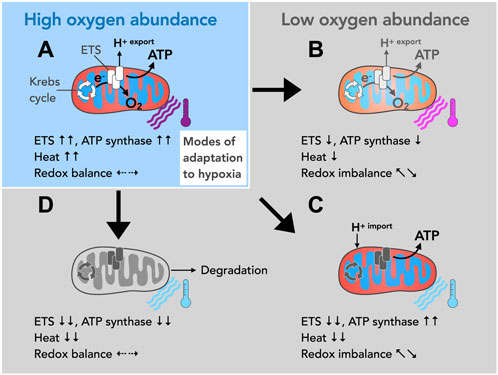
FIGURE 1. Possible modes of mitochondrial adaption to different oxygen levels. (A) Oxygen is the final electron acceptor for a series of thermogenic redox reactions facilitated by the ETS. These redox reactions are coupled to proton translocation (H+ export) across the inner mitochondrial membrane making the ETS the main building block for oxidative phosphorylation (OXPHOS), i.e., mitochondrial ATP generation. ETS activity also keeps upstream metabolic circuits operational such as the tricarboxylic acid cycle (TCA, also known as the Krebs cycle), and produces heat and maintains cellular redox balance. (B–D) Due to their exceptional size, their contractile structures and their respiratory activity, radial oxygen gradients emerge within cardiomyocytes, which make adaptation mechanisms necessary. (B) Oxygen deprivation impairs ETS activity and thereby restricts proton translocation across the inner membrane. This decreases the driving force for ATP production and affects heat production and redox balance. More importantly, since well-oxygenated and less oxygenated mitochondrial populations may exist within one cardiomyocyte, differentially energized zones may build up and affect cellular functions. (C) According to the theory of proton conduction, hypoxic mitochondria may import the H+ gradient from well-oxygenated mitochondria. In principle, this mechanism would allow mitochondrial ATP production in the absence of oxygen. However, upstream metabolic cycles such as the TCA cycle, heat production and the redox balance would inevitably be disturbed. (D) The downregulation of ETS and ATP synthase activity makes the affected mitochondria susceptible to degradation (autophagy) but more importantly renders them metabolically quiescent. A partial downregulation of activity (as described in B) or a reversal in the mode of direction of the ATP synthase (as described in the text) may precede such state of metabolic hibernation.
Notably, across species cardiomyocytes are exceptionally large cells and densely packed with mitochondria and contractile structures (Schaper et al., 1985; Barth et al., 1992). Both, their overall cell size, and intracellular structures constitute a significant hurdle for oxygen distribution. Moreover, oxygen must pass through different layers of actively respiring mitochondria to reach the innermost contractile compartments to satisfy their need for cellular respiration. It stands to reason that adequate oxygen distribution is additionally compromised under conditions of increased workload such as pressure overload and hypertrophy (Seiden et al., 1988; Sabbah, 2017), potentially rendering oxygen delivery a limiting determinant of ETS-driven redox responses. This would inevitably affect a number of processes intrinsically linked to ATP production including cellular redox balance, mitochondrial heat production and would eventually impair organ homeostasis and function (Suomalainen and Battersby, 2017; Bertero and Maack, 2018) (Figures 1B–D).
2 The emergence of oxygen gradients within cardiomyocytes
Given their large size, complex intracellular structures, and high metabolic activity, radial gradients of substrates, waste products, and ions conceivably occur within cardiomyocytes. The visualization of pronounced radial oxygen gradients in cultured rat cardiomyocytes was nevertheless a remarkable observation (Takahashi and Doi, 1996; Takahashi et al., 1998; Takahashi et al., 1999; Takahashi and Asano, 2002). Oxygen gradients emerged under conditions when cardiomyocytes were paced or treated with respiratory uncouplers. Although both interventions may appear as nonphysiological forms of stress, the observed oxygen gradients are likely to occur also in vivo in particular when oxygen becomes scarce or diffusion distances increase (Seiden et al., 1988; Sabbah, 2017). A major limitation to date in validating the biological significance of such gradients in vivo is owed to a lack of suitable oxygen biosensors and imaging systems. Yet, the possibility that oxygen may be unevenly distributed within cardiomyocytes raises several vital questions such as the nature of subcellular OXPHOS control mechanisms, and how organ homeostasis and function can be maintained despite presumably diverging metabolic signals. Interestingly, both uncoupling and pacing caused similar steep radial oxygen gradients. However, the corresponding NADH fluorescence showed divergent signals (Takahashi and Asano, 2002). In the case of uncoupling (maximal rate of respiration without ATP production), there was an increase in the NADH signal, indicative of hypoxia-induced respiratory inhibition. In the case of pacing, however, the NADH signal did not increase, which was interpreted as the presence of mechanisms that allow mitochondria with intact respiratory control to maintain electron transfer through the ETS even when oxygen becomes scarce (Takahashi and Asano, 2002). The nature of subcellular control mechanism remained elusive but several modes of spatial adaptation of bioenergetics to oxygen availability are conceivable, with each of which having a different effect on cardiac function.
3 Potential modes of spatial OXPHOS adjustment
3.1 Radial adjustment of OXPHOS activity may follow oxygen availability
Oxygen has long been proposed as the dominant regulator of both ATP-producing and ATP-consuming processes (Hochachka, 2003). Such regulatory control, however, implies that mitochondria are able to monitor the amount of available oxygen in order to adjust their energy metabolism accordingly. Indeed, recent evidence suggests that a key determinant of oxygen sensing may lie within the mitochondrial ETS itself (Sommer et al., 2017; Moreno-Domínguez et al., 2020; Sommer et al., 2020). Using such a mechanism, mitochondria may decrease the activity and adapt the composition of the OXPHOS system when oxygen levels are low. Conversely, OXPHOS activity could be gradually ramped up and reorganized during reoxygenation using the same mechanism (Figure 1B). In metabolically active cells of the size of cardiomyocytes with intracellular radial oxygen gradients (Takahashi and Doi, 1996; Takahashi et al., 1998; Takahashi et al., 1999; Takahashi and Asano, 2002), however, this would result in subcellular compartments being differentially energized, provided ATP content strictly follows oxygen availability. This type of spatial adaptation of bioenergetics harbors the risk of creating intracellular zones with different contractility, which may eventually lead to organ dysfunction.
Apart from limiting OXPHOS activity, a drop of oxygen below a critical threshold stabilizes hypoxia inducible factors (HIF), in particular HIF1α, thereby activating signaling cascades that rule cellular metabolism, mitochondrial ATP production, and overall signal transduction in a retrograde manner (Huang et al., 2004; Löfstedt et al., 2007; Krishnan et al., 2009; Liang et al., 2023; Sato and Takeda, 2023). In addition, the heart continuously adapts its metabolism dynamically, for example, through the glucose-fatty acid cycle or Randle cycle, where a decrease in the rate of fatty acid-β oxidation leads to a corresponding increase in the rate of glucose oxidation (Randle, 1998; Hue and Taegtmeyer, 2009). This is advantageous, e.g., in the ischemic heart, since glucose oxidation produces more ATP per oxygen molecule consumed thus increasing cardiac efficiency under conditions of oxygen deprivation. However, if such shift in substrate usage occurs differentially in metabolically divergent zones within 1 cell and how this may be orchestrated remains elusive.
Hypoxic signaling could be avoided if oxygen levels are kept above the threshold critical for HIF1α stabilization (Hagen et al., 2003; Sato and Takeda, 2023). The observed stable NADH signal in paced cardiomyocytes (Takahashi and Asano, 2002) may equally be interpreted by a decrease of mitochondrial respiration in an attempt to maintain oxygen levels, provided the Krebs cycle activity closely follows ETS activity and therefore regenerates less NADH. The apparently stable NADH levels may thus indicate a lower turnover. It would be interesting to know whether local lactate levels increased as a result of compensatory stimulation of glycolysis during pacing and/or uncoupling. In addition to an oxygen sensor within the ETS (Sommer et al., 2017; Moreno-Domínguez et al., 2020; Sommer et al., 2020), extramitochondrial cues could prompt mitochondria to cease respiration, e.g., when oxygen levels fall in relation to endogenously produced respiratory inhibitors such as nitric oxide. Disruption of mitochondrial respiration as a means of redistributing oxygen to non-respiratory oxygen-dependent targets including prolyl hydroxylases has previously been suggested (Hagen et al., 2003). In any case, a gradual adjustment of ETS-mediated redox responses likely leads to unevenly distributed ATP levels causing depolarization deficits and asynchronous contractile forces.
3.2 Proton-conducting mitochondrial fibers may render oxygen gradients irrelevant
The dilemma of unevenly distributed ATP levels due to potentially hypoxic regions was seemingly solved by an ingenious concept. It stressed the fact that striated muscles, skeletal muscle and heart, contain at least two spatially distinct mitochondrial subpopulations, i.e., subsarcolemmal (SSM) and interfibrillar mitochondria (IFM) (Palmer et al., 1977). IFM located deep between the contractile structures should experience lower oxygen availability compared to SSM underneath the sarcolemma. Since mitochondria dynamically undergo fusion and fission, it was proposed that well-oxygenated SSM may support ATP production in supposedly less oxygenated IFM by forming proton-conducting fibers between the two compartments (Skulachev, 2001). Indeed, the existence of mitochondrial networks has been demonstrated in different cell types including fibroblasts and cardiomyocytes, in the latter called streptio mitochondriale (Amchenkova et al., 1988; Skulachev, 2001). In principle, the formation of streptio mitochondriale would allow ATP production in IFM even in the absence of local oxygen (Figure 1C). This elegant concept, however, poses a metabolic problem as the activity of upstream metabolic circuits such as the Krebs cycle are mandatorily dependent on redox reactions executed by the ETS. The Krebs cycle certainly shows tremendous plasticity in adapting to different metabolic conditions and may, for example, during hypoxia, operate in opposite directions (Chinopoulos, 2013). Indeed, an unusual accumulation of the Krebs cycle intermediate succinate was observed during cardiac ischemia (Chouchani et al., 2014; Chouchani et al., 2016), which may be beneficial during ischemia itself as it is thought to improve ischemic bioenergetics (Zhang et al., 2018). However, succinate accumulated during ischemia is rapidly oxidized upon reoxygenation, partially by reverse electron transport (RET), which produces excessive amounts of reactive oxygen species (ROS), a critical determinant of reperfusion injury (Chouchani et al., 2014; Chouchani et al., 2016). Fluctuating oxygen levels in cardiomyocytes could therefore lead to repetitive ROS-mediated injuries and altered Krebs cycle activities and/or accumulation of its intermediates. A pathologic accumulation of Krebs cycle intermediates, in turn, has consequences beyond bioenergetics as such metabolites fuel different stress response pathways, alter the metabolic flux and cause epigenetic alterations within the host cell and neighboring cells (Tretter et al., 2016; Chinopoulos, 2019; Martínez-Reyes and Chandel, 2020). Taken together, ATP level may be maintained in hypoxic cell segments by proton-conducting mitochondrial fibers, but it seems questionable how local restoration of ATP levels alone can exert beneficial effects if a disrupted ETS simultaneously causes a greater metabolic crisis.
3.3 Potential segmental adaption by mitochondrial hibernation
Equally possible is a radical third mode of adaptation, namely, the spatial downregulation of under-oxygenated mitochondria to complete inactivity, in particular the IFM compartment (Figure 1D). While a decrease in ETS function following radial oxygen gradients is readily conceivable, the obligatory dependence of the heart on oxidative metabolism appears to impose the maintenance of ATP-synthesizing activity by all means. The latter has led to the concept of proton conduction to maintain ATP synthesis in the absence of oxygen. Quite to the contrary, however, mitochondrial ATP synthase can dynamically shift between two opposing activity states: an ATP-synthesizing and an ATP-hydrolyzing state (Acin-Perez et al., 2023). The transition from ATP-synthesizing to ATP-hydrolyzing is triggered by a loss of membrane potential and thus correlates reciprocally with ETS function and presumably oxygen availability. At the molecular level, this switch to ATP hydrolysis is achieved by reversing the direction of the enzymatic reaction, whereby the ATP synthase mutates into an ATPase that pumps protons against a chemical gradient. The reversal of activity has long been recognized as a protective response to acute stress, such as myocardial infarction and oxygen starvation (St-Pierre et al., 2000). Surprisingly, it was recently described that the level of membrane potential may be heterogeneous along the inner membrane (Wolf et al., 2019). From this it follows that sections with high and low potential can alternate suggesting that ATP-synthesizing and ATP-hydrolyzing enzymes may coexist even within one mitochondrion. Therefore, a locally restricted shift in hypoxic cell segments towards ATP-hydrolyzing activity is possible. Of note, long-term activation of ATP hydrolysis in greater cellular regions, e.g., the IFM compartment, inevitably would lead to ATP depletion. In support of this notion, overexpression of a mitochondrial protein that inhibits ATP hydrolysis (ATPase inhibiting factor 1, ATPIF1) and/or pharmacological inhibition of ATP hydrolysis conferred cardioprotection (Rouslin and Broge, 1989; 1993a; 1993b; Rouslin et al., 1995) and improved the phenotype of a mouse model of Duchenne muscular dystrophy (Acin-Perez et al., 2023). Caution is warranted, however, as the molecular mechanisms and biological effects of ATPIF1 expression appear to be multifaceted. While ATPIF1 may thus control a local reserve of ATP synthase that can be switched on or off upon demand (Romero-Carramiñana et al., 2023), it can also promote pathological cardiac remodeling and mitochondrial dysfunction when overexpressed (Pavez-Giani et al., 2021).
The inhibition of ATP-hydrolyzing activity of the mitochondrial ATP synthase emerged as particularly beneficial for the human heart. Unlike in rodents (fast heart rate species), in which ATP hydrolysis appears to be the predominant reaction when respiration is disrupted, the human heart mitochondrial ATP synthase (slow heart rate species) becomes catalytically inactive, presumably to preserve ATP levels (Rouslin and Broge, 1989; Rouslin and Broge, 1993a; Rouslin and Broge, 1993b; Rouslin et al., 1995). Together with impaired ETS-linked redox reactions, however, such inhibition of ATP hydrolysis (and proton translocation) leads to a decrease in mitochondrial membrane potential and makes mitochondria susceptible to degradation. More importantly, it impairs mitochondrial functions beyond ATP production, including ion homeostasis and protein import inducing a state of mitochondrial hibernation (metabolic quiescence) (Figure 1D). Mitochondrial hibernation may therefore also explain another unusual phenomenon, the metabolic stability of the mammalian heart. The canine heart, for instance, shows remarkably stable levels of energy metabolites despite large fluctuations in workload and oxygen consumption (Balaban et al., 1986), a phenomenon coined the “stability paradox” (Hochachka, 1999; Hochachka, 2003). The actual physiological role of streptio mitochondriale might thus be the delineation of metabolic segments and adaptation of contractile function as contractility in metabolically quiescent segments of cardiomyocytes is not conceivable. Taken together, an oxygen-controlled switch between activity and hibernation of mitochondria may allow the heart to adapt to different demands.
3.4 Could impaired substrate supply rule spatial OXPHOS adjustment?
To always maintain their contractile function, cardiomyocytes are metabolic omnivores (Aubert et al., 2013), they can flexibly switch between different substrates, and mitochondria are essential players in the maintenance of this flexibility (Muoio, 2014). Yet, local starvation of substrates may account for spatial adjustment of bioenergetics. Under physiologic conditions, fatty acid oxidation generates 60%–90% of ATP whilst oxidation of carbohydrates, and to a lesser degree ketones and amino acids provide the remaining 10%–40% (Bertero and Maack, 2018; Popoiu et al., 2023). The Randle cycle (Randle et al., 1963; Hue and Taegtmeyer, 2009) may thus become metabolically relevant when fatty acid oxidation outpaces intracellular transport mechanisms. Previous work, however, makes the concept of a spatial fatty acid deficiency seem unlikely. It has been demonstrated that the cytoplasmic transporter protein, fatty acid-binding protein (FABP), is present in abundance throughout cardiomyocytes suggesting that local fatty acid supply is not a rate-limiting condition (Luiken et al., 2003). Conversely, it was previously shown that high glucose levels can impair the activity of ETS complex I (Cannino et al., 2012). It is unclear, however, if a shift in the glucose to fatty acid ratio has the same effect. The metabolic flexibility may also force the heart to replace fatty acids by other substrates, e.g., ketones (Ho et al., 2019; Ho et al., 2021). Ketone metabolism generally follows its availability and is thus independent from the mechanisms ruling the substrate switch described by the Randle cycle. However, ketones render the heart energetically less efficient (Ho et al., 2019; Ho et al., 2021) arguing against spatial substrate starvation as OXPHOS control mechanism but instead suggesting that a loss of spatial bioenergetic efficiency causes the heart to gradually become energy depleted.
Support for the idea of spatial energy starvation also comes from another observation. In pressure-overloaded human hearts, a significant correlation was observed between a decrease in ejection fraction and myocyte degeneration concomitant with an increase in autophagy (Hein et al., 2003). Mitochondrial hibernation (Figure 1D) decreases the membrane potential, which eventually predisposes mitochondria to degradation explaining why autophagy and OXPHOS activity are reciprocally regulated (Gustafsson and Gottlieb, 2009; Gottlieb and Mentzer, 2010; Rambold and Lippincott-Schwartz, 2011). Induction of mitochondrial hibernation due to insufficient oxygen availability may thus be the first step in a series of events that trigger autophagy and loss of cellular structures including mitochondria and myofibrils. Initially, this adaptive mechanism should lead to a decrease in contractility but eventually push the heart toward a tipping point at which the transition from compensated hypertrophy to contractile failure occurs. In support of this notion, we observed in a mouse model of inflammatory cardiomyopathy that enhanced tissue oxygenation alone was sufficient to restore cardiac contractility and reverse spatial dedifferentiation (Dhandapani et al., 2019). Furthermore, treatment with hyperbaric oxygen has previously shown beneficial effects, e.g., it conferred cardioprotective effects under conditions such as carbon monoxide poisoning and ischemia (Swift et al., 1992; Sterling et al., 1993; Lund et al., 2000; Prockop and Chichkova, 2007; Jørgensen et al., 2013). Taken together, this suggests that oxygen is an important determinant of contractile adaptation and the development of contractile failure possibly through its spatial effect on bioenergetics.
4 Conclusion
Oxygen abundance is a prerequisite for mitochondrial bioenergetics and thus for cardiac contractile function. Therefore, oxygen sensing is vital for metabolic adaptation and is achieved by various means such as oxygen sensing within the ETS (Sommer et al., 2017; 2020; Moreno-Domínguez et al., 2020), reversal in the mode of direction of the ATP synthase or inhibition of its hydrolytic activity (Rouslin and Broge, 1989; Rouslin and Broge, 1993a; Rouslin and Broge, 1993b; Rouslin et al., 1995; Acin-Perez et al., 2023) and altered substrate selection (Randle, 1998; Hue and Taegtmeyer, 2009). Many of these processes are controlled by HIF proteins, which are stabilized or degraded in an oxygen-sensitive manner (Huang et al., 2004; Löfstedt et al., 2007; Krishnan et al., 2009; Liang et al., 2023; Sato and Takeda, 2023). Previously, critical attention has been paid to actual oxygen contents in tissues (Carreau et al., 2011) and the pitfalls arising from cell culture experiments carried out at ambient air (Ast and Mootha, 2019; Keeley and Mann, 2019). Relatively little is known about consequences of radial oxygen gradients that can build up within cells. A better understanding of this phenomenon, however, is important as it implies that different metabolic segments coexist within 1 cell.
In cultured rat cardiomyocytes, for instance, the formation of radial oxygen gradients was visualized (Takahashi and Doi, 1996; Takahashi et al., 1998; Takahashi et al., 1999; Takahashi and Asano, 2002). Since cardiomyocytes rely on OXPHOS for energy metabolism, the heterogeneity of oxygen distribution predicts the subsequent formation of ATP gradients that may cause depolarization deficits and asynchronous contractility. To avoid ATP gradients, it has been hypothesized that proton conduction along mitochondrial fibers to oxygen-depleted compartments may drive local ATP synthase activity (Skulachev, 2001). This concept, however, suffers from another shortcoming, i.e., a stalling of metabolic circuits such as the Krebs cycle upstream of non-respiring mitochondria. Such a metabolic disruption also inevitably leads to an aberrant accumulation of metabolic intermediates such as succinate, which has consequences beyond energy metabolism (Chouchani et al., 2014; Martínez-Reyes and Chandel, 2020; Murphy and Chouchani, 2022).
Considering the various complications arising from different metabolic zones within one cardiomyocyte, we propose that spatial OXPHOS adaptation to a state of hibernation-like inactivity is the most likely response to local oxygen deprivation. In such a state, both the ETS-linked redox reactions and mitochondrial ATP synthase activity must be downregulated, including the hydrolyzing state of ATP synthase, which is thought to limit ATP exhaustion when oxygen is scarce (Rouslin and Broge, 1989; Rouslin and Broge, 1993a; Rouslin and Broge, 1993b; Rouslin et al., 1995). ATP levels in the cell center may nevertheless drop to a level impairing cardiac contractility. A hibernating-like state also causes a loss of membrane potential, which renders mitochondria susceptible to degradation. It stands to reason that the loss of mitochondrial mass due to degradation beyond a critical threshold defines the transition from cardiac contractile adaptation to contractile dysfunction.
Taken together, spatial regulation of bioenergetics may be the key mechanism for contractile adaptation to fluctuating tissue oxygen levels. If oxygen deprivation persists, loss of mitochondrial membrane potential and subsequently activation of autophagic activity may shift the heart towards the development of contractile failure. This puts spatial regulation of bioenergetics at the center of contractile adaptation, with oxygen being its chief determinant.
Author contributions
MS: Conceptualization, Supervision, Visualization, Writing–original draft, Writing–review and editing. MM: Writing–review and editing. TD: Writing–review and editing. JLOP: Conceptualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. MS was supported by the German Federal Ministry of Education and Research, BMBF (FKZ 10/44).
Acknowledgments
The authors thank Jürgen Holtz and Adam R. Wende for critical reading of the manuscript and valuable discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Acin-Perez, R., Benincá, C., Rio, L. F. D., Shu, C., Baghdasarian, S., Zanette, V., et al. (2023). Inhibition of ATP synthase reverse activity restores energy homeostasis in mitochondrial pathologies. EMBO J. 42, e111699. doi:10.15252/embj.2022111699
Amchenkova, A. A., Bakeeva, L. E., Chentsov, Y. S., Skulachev, V. P., and Zorov, D. B. (1988). Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J. Cell Biol. 107, 481–495. doi:10.1083/jcb.107.2.481
Ast, T., and Mootha, V. K. (2019). Oxygen and mammalian cell culture: are we repeating the experiment of Dr. Ox? Nat. Metab. 1, 858–860. doi:10.1038/s42255-019-0105-0
Aubert, G., Vega, R. B., and Kelly, D. P. (2013). Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart. Biochimica Biophysica Acta Bba - Mol Cell Res 1833, 840–847. doi:10.1016/j.bbamcr.2012.08.015
Balaban, R., Kantor, H., Katz, L., and Briggs, R. (1986). Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science 232, 1121–1123. doi:10.1126/science.3704638
Barth, E., Stämmler, G., Speiser, B., and Schaper, J. (1992). Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell Cardiol. 24, 669–681. doi:10.1016/0022-2828(92)93381-s
Bertero, E., and Maack, C. (2018). Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 15, 457–470. doi:10.1038/s41569-018-0044-6
Cannino, G., El-Khoury, R., Pirinen, M., Hutz, B., Rustin, P., Jacobs, H. T., et al. (2012). Glucose modulates respiratory complex I activity in response to acute mitochondrial dysfunction. J. Biol. Chem. 287, 38729–38740. doi:10.1074/jbc.m112.386060
Carreau, A., Hafny-Rahbi, B. E., Matejuk, A., Grillon, C., and Kieda, C. (2011). Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 15, 1239–1253. doi:10.1111/j.1582-4934.2011.01258.x
Chinopoulos, C. (2013). Which way does the citric acid cycle turn during hypoxia? The critical role of α-ketoglutarate dehydrogenase complex. J. Neurosci. Res. 91, 1030–1043. doi:10.1002/jnr.23196
Chinopoulos, C. (2019). Succinate in ischemia: where does it come from? Int. J. Biochem. Cell Biol. 115, 105580. doi:10.1016/j.biocel.2019.105580
Chouchani, E. T., Pell, V. R., Gaude, E., Aksentijević, D., Sundier, S. Y., Robb, E. L., et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. doi:10.1038/nature13909
Chouchani, E. T., Pell, V. R., James, A. M., Work, L. M., Saeb-Parsy, K., Frezza, C., et al. (2016). A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 23, 254–263. doi:10.1016/j.cmet.2015.12.009
Cooke, G. A., Marshall, P., Al-Timman, J. K., Wright, D. J., Riley, R., Hainsworth, R., et al. (1998). Physiological cardiac reserve: development of a non-invasive method and first estimates in man. Heart 79, 289–294. doi:10.1136/hrt.79.3.289
Dhandapani, P. K., Begines-Moreno, I. M., Brea-Calvo, G., Gärtner, U., Graeber, T. G., Sanchez, G. J., et al. (2019). Hyperoxia but not AOX expression mitigates pathological cardiac remodeling in a mouse model of inflammatory cardiomyopathy. Sci. Rep. 9, 12741. doi:10.1038/s41598-019-49231-9
Gottlieb, R. A., and Mentzer, R. M. (2010). Autophagy during cardiac stress: joys and frustrations of autophagy. Annu. Rev. Physiol. 72, 45–59. doi:10.1146/annurev-physiol-021909-135757
Gustafsson, A. B., and Gottlieb, R. A. (2009). Autophagy in ischemic heart disease. Circ. Res. 104, 150–158. doi:10.1161/circresaha.108.187427
Hagen, T., Taylor, C. T., Lam, F., and Moncada, S. (2003). Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science 302, 1975–1978. doi:10.1126/science.1088805
Hargreaves, M., and Spriet, L. L. (2020). Skeletal muscle energy metabolism during exercise. Nat. Metab. 2, 817–828. doi:10.1038/s42255-020-0251-4
Hein, S., Arnon, E., Kostin, S., Schonburg, M., Elsasser, A., Polyakova, V., et al. (2003). Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation 107, 984–991. doi:10.1161/01.cir.0000051865.66123.b7
Ho, K. L., Karwi, Q. G., Wagg, C., Zhang, L., Vo, K., Altamimi, T., et al. (2021). Ketones can become the major fuel source for the heart but do not increase cardiac efficiency. Cardiovasc Res. 117, 1178–1187. doi:10.1093/cvr/cvaa143
Ho, K. L., Zhang, L., Wagg, C., Batran, R. A., Gopal, K., Levasseur, J., et al. (2019). Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc Res. 115, 1606–1616. doi:10.1093/cvr/cvz045
Hochachka, P. W. (1999). The metabolic implications of intracellular circulation. Proc. Natl. Acad. Sci. 96, 12233–12239. doi:10.1073/pnas.96.22.12233
Hochachka, P. W. (2003). Intracellular convection, homeostasis and metabolic regulation. J. Exp. Biol. 206, 2001–2009. doi:10.1242/jeb.00402
Huang, Y., Hickey, R. P., Yeh, J. L., Liu, D., Dadak, A., Young, L. H., et al. (2004). Cardiac myocyte-specific HIF-1alpha deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. Faseb J. 18, 1138–1140. doi:10.1096/fj.04-1510fje
Hue, L., and Taegtmeyer, H. (2009). The Randle cycle revisited: a new head for an old hat. Am. J. Physiol-endoc M. 297, E578–E591. doi:10.1152/ajpendo.00093.2009
Jørgensen, A., Foster, P. P., Brubakk, A. O., and Eftedal, I. (2013). Effects of hyperbaric oxygen preconditioning on cardiac stress markers after simulated diving. Physiol. Rep. 1, e00169. doi:10.1002/phy2.169
Keeley, T. P., and Mann, G. E. (2019). Defining physiological normoxia for improved translation of cell physiology to animal models and humans. Physiol. Rev. 99, 161–234. doi:10.1152/physrev.00041.2017
Krishnan, J., Suter, M., Windak, R., Krebs, T., Felley, A., Montessuit, C., et al. (2009). Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 9, 512–524. doi:10.1016/j.cmet.2009.05.005
Liang, Y., Ruan, W., Jiang, Y., Smalling, R., Yuan, X., and Eltzschig, H. K. (2023). Interplay of hypoxia-inducible factors and oxygen therapy in cardiovascular medicine. Nat. Rev. Cardiol. 20, 723–737. doi:10.1038/s41569-023-00886-y
Löfstedt, T., Fredlund, E., Holmquist-Mengelbier, L., Pietras, A., Ovenberger, M., Poellinger, L., et al. (2007). Hypoxia inducible factor-2alpha in cancer. Cell Cycle 6, 919–926. doi:10.4161/cc.6.8.4133
Luiken, J. J. F. P., Koonen, D. P. Y., Coumans, W. A., Pelsers, M. M. A. L., Binas, B., Bonen, A., et al. (2003). Long-chain fatty acid uptake by skeletal muscle is impaired in homozygous, but not heterozygous, heart-type-FABP null mice. Lipids 38, 491–496. doi:10.1007/s11745-003-1089-6
Lund, V., Klossner, J., Sariola-Heinonen, K., Jalonen, J., and Sariola-Heinonen, K. (2000). Hyperbaric oxygen increases parasympathetic activity in professional divers. Acta Physiol. Scand. 170, 39–44. doi:10.1046/j.1365-201x.2000.00761.x
Martínez-Reyes, I., and Chandel, N. S. (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102. doi:10.1038/s41467-019-13668-3
Mitchell, P. (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191, 144–148. doi:10.1038/191144a0
Moreno-Domínguez, A., Ortega-Sáenz, P., Gao, L., Colinas, O., García-Flores, P., Bonilla-Henao, V., et al. (2020). Acute O2 sensing through HIF2α-dependent expression of atypical cytochrome oxidase subunits in arterial chemoreceptors. Sci. Signal 13, eaay9452. doi:10.1126/scisignal.aay9452
Muoio, D. M. (2014). Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell 159, 1253–1262. doi:10.1016/j.cell.2014.11.034
Murphy, M. P., and Chouchani, E. T. (2022). Why succinate? Physiological regulation by a mitochondrial coenzyme Q sentinel. Nat. Chem. Biol. 18, 461–469. doi:10.1038/s41589-022-01004-8
Palmer, J. W., Tandler, B., and Hoppel, C. L. (1977). Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 252, 8731–8739. doi:10.1016/s0021-9258(19)75283-1
Pavez-Giani, M. G., Sánchez-Aguilera, P. I., Bomer, N., Miyamoto, S., Booij, H. G., Giraldo, P., et al. (2021). ATPase inhibitory factor-1 disrupts mitochondrial Ca2+ handling and promotes pathological cardiac hypertrophy through CaMKIIδ. Int. J. Mol. Sci. 22, 4427. doi:10.3390/ijms22094427
Popoiu, T.-A., Dudek, J., Maack, C., and Bertero, E. (2023). Cardiac involvement in mitochondrial disorders. Curr. Hear Fail Rep. 20, 76–87. doi:10.1007/s11897-023-00592-3
Prockop, L. D., and Chichkova, R. I. (2007). Carbon monoxide intoxication: an updated review. J. Neurol. Sci. 262, 122–130. doi:10.1016/j.jns.2007.06.037
Rambold, A. S., and Lippincott-Schwartz, J. (2011). Mechanisms of mitochondria and autophagy crosstalk. Cell Cycle 10, 4032–4038. doi:10.4161/cc.10.23.18384
Randle, P. J. (1998). Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab. Rev. 14, 263–283. doi:10.1002/(sici)1099-0895(199812)14:4<263::aid-dmr233>3.0.co;2-c
Randle, P. J., Garland, P. B., Hales, C. N., and Newsholme, E. A. (1963). The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 281, 785–789. doi:10.1016/s0140-6736(63)91500-9
Romero-Carramiñana, I., Esparza-Moltó, P. B., Domínguez-Zorita, S., Nuevo-Tapioles, C., and Cuezva, J. M. (2023). IF1 promotes oligomeric assemblies of sluggish ATP synthase and outlines the heterogeneity of the mitochondrial membrane potential. Commun. Biol. 6, 836. doi:10.1038/s42003-023-05214-1
Rouslin, W., and Broge, C. W. (1989). Regulation of mitochondrial matrix pH and adenosine 5′-triphosphatase activity during ischemia in slow heart-rate hearts. J. Biol. Chem. 264, 15224–15229. doi:10.1016/s0021-9258(19)84813-5
Rouslin, W., and Broge, C. W. (1993a). Factors affecting the species-homologous and species-heterologous binding of mitochondrial ATPase inhibitor, IF1, to the mitochondrial ATPase of slow and fast heart-rate hearts. Arch. Biochem. Biophys. 303, 443–450. doi:10.1006/abbi.1993.1307
Rouslin, W., and Broge, C. W. (1993b). Mechanisms of ATP conservation during ischemia in slow and fast heart rate hearts. Am. J. Physiol-Cell Physiol. 264, C209–C216. doi:10.1152/ajpcell.1993.264.1.c209
Rouslin, W., Frank, G. D., and Broge, C. W. (1995). Content and binding characteristics of the mitochondrial ATPase inhibitor, IF1 in the tissues of several slow and fast heart-rate homeothermic species and in two poikilotherms. J. Bioenerg. Biomembr. 27, 117–125. doi:10.1007/bf02110339
Sabbah, H. N. (2017). Silent disease progression in clinically stable heart failure. Eur. J. Heart Fail 19, 469–478. doi:10.1002/ejhf.705
Sabbah, H. N. (2020). Targeting the mitochondria in heart failure A translational perspective. Jacc Basic Transl. Sci. 5, 88–106. doi:10.1016/j.jacbts.2019.07.009
Sato, T., and Takeda, N. (2023). The roles of HIF-1α signaling in cardiovascular diseases. J. Cardiol. 81, 202–208. doi:10.1016/j.jjcc.2022.09.002
Schaper, J., Meiser, E., and Stämmler, G. (1985). Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 56, 377–391. doi:10.1161/01.res.56.3.377
Schwartz, B., Gjini, P., Gopal, D. M., and Fetterman, J. L. (2022). Inefficient batteries in heart failure metabolic bottlenecks disrupting the mitochondrial ecosystem. JACC Basic Transl. Sci. 7, 1161–1179. doi:10.1016/j.jacbts.2022.03.017
Seiden, D., Navidad, P., and Weiss, H. R. (1988). Oxygen diffusion distance in thyroxine-induced hypertrophic rabbit myocardium. J. Mol. Cell Cardiol. 20, 917–930. doi:10.1016/s0022-2828(88)80146-9
Skulachev, V. P. (2001). Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem. Sci. 26, 23–29. doi:10.1016/s0968-0004(00)01735-7
Sommer, N., Alebrahimdehkordi, N., Pak, O., Knoepp, F., Strielkov, I., Scheibe, S., et al. (2020). Bypassing mitochondrial complex III using alternative oxidase inhibits acute pulmonary oxygen sensing. Sci. Adv. 6, eaba0694. doi:10.1126/sciadv.aba0694
Sommer, N., Hüttemann, M., Pak, O., Scheibe, S., Knoepp, F., Sinkler, C., et al. (2017). Mitochondrial complex IV subunit 4 isoform 2 is essential for acute pulmonary oxygen sensing. Circ. Res. 121, 424–438. doi:10.1161/circresaha.116.310482
Sterling, D. L., Thornton, J. D., Swafford, A., Gottlieb, S. F., Bishop, S. P., Stanley, A. W., et al. (1993). Hyperbaric oxygen limits infarct size in ischemic rabbit myocardium in vivo. Circulation 88, 1931–1936. doi:10.1161/01.cir.88.4.1931
St-Pierre, J., Brand, M. D., and Boutilier, R. G. (2000). Mitochondria as ATP consumers: cellular treason in anoxia. Proc. Natl. Acad. Sci. 97, 8670–8674. doi:10.1073/pnas.140093597
Suomalainen, A., and Battersby, B. J. (2017). Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 19, 77–92. doi:10.1038/nrm.2017.66
Swift, P. C., Turner, J. H., Oxer, H. F., O’Shea, J. P., Lane, G. K., and Woollard, K. V. (1992). Myocardial hibernation identified by hyperbaric oxygen treatment and echocardiography in postinfarction patients: comparison with exercise thallium scintigraphy. Am. Heart J. 124, 1151–1158. doi:10.1016/0002-8703(92)90394-b
Takahashi, E., and Asano, K. (2002). Mitochondrial respiratory control can compensate for intracellular O2 gradients in cardiomyocytes at low PO2. Am. J. Physiol-Heart Circ. Physiol. 283, H871–H878. doi:10.1152/ajpheart.00162.2002
Takahashi, E., and Doi, K. (1996). Regulation of oxygen diffusion in hypoxic isolated cardiac myocytes. Am. J. Physiol-Heart Circ. Physiol. 271, H1734–H1738. doi:10.1152/ajpheart.1996.271.5.h1734
Takahashi, E., Endoh, H., and Doi, K. (1999). Intracellular gradients of O2supply to mitochondria in actively respiring single cardiomyocyte of rats. Am. J. Physiol-Heart Circ. Physiol. 276, H718–H724. doi:10.1152/ajpheart.1999.276.2.h718
Takahashi, E., Sato, K., Endoh, H., Xu, Z.-L., and Doi, K. (1998). Direct observation of radial intracellular P O 2 gradients in a single cardiomyocyte of the rat. Am. J. Physiol-Heart Circ. Physiol. 275, H225–H233. doi:10.1152/ajpheart.1998.275.1.h225
Tretter, L., Patocs, A., and Chinopoulos, C. (2016). Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochimica Biophysica Acta Bba - Bioenergetics 1857, 1086–1101. doi:10.1016/j.bbabio.2016.03.012
Wolf, D. M., Segawa, M., Kondadi, A. K., Anand, R., Bailey, S. T., Reichert, A. S., et al. (2019). Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 38, e101056. doi:10.15252/embj.2018101056
Keywords: mitochondria, spatial bioenergetics, mammalian heart, contractility, heart failure
Citation: Szibor M, Mühlon M, Doenst T and Pohjoismäki JLO (2023) Spatial adjustment of bioenergetics, a possible determinant of contractile adaptation and development of contractile failure. Front. Mol. Med. 3:1305960. doi: 10.3389/fmmed.2023.1305960
Received: 02 October 2023; Accepted: 23 November 2023;
Published: 06 December 2023.
Edited by:
Jan Dudek, University of Würzburg, GermanyReviewed by:
Mario Pavez-Giani, University of Göttingen, GermanyCopyright © 2023 Szibor, Mühlon, Doenst and Pohjoismäki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marten Szibor, bWFydGVuLnN6aWJvckB0dW5pLmZp; Jaakko L. O. Pohjoismäki, amFha2tvLnBvaGpvaXNtYWtpQHVlZi5maQ==