
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Med, 08 January 2024
Sec. Molecular Pathology
Volume 3 - 2023 | https://doi.org/10.3389/fmmed.2023.1283170
 Claudia Theys1*
Claudia Theys1* Tineke Vanderhaeghen2,3
Tineke Vanderhaeghen2,3 Evelien Van Dijck4Cedric Peleman5,6Anne Scheepers4
Evelien Van Dijck4Cedric Peleman5,6Anne Scheepers4 Joe Ibrahim4Ligia Mateiu4Steven Timmermans2,3Tom Vanden Berghe2,3,6
Joe Ibrahim4Ligia Mateiu4Steven Timmermans2,3Tom Vanden Berghe2,3,6 Sven M. Francque5,7
Sven M. Francque5,7 Wim Van Hul4
Wim Van Hul4 Claude Libert2,3
Claude Libert2,3 Wim Vanden Berghe1*
Wim Vanden Berghe1*Metabolic Dysfunction Associated Steatotic Liver Disease (MASLD) is a growing epidemic with an estimated prevalence of 20%–30% in Europe and the most common cause of chronic liver disease worldwide. The onset and progression of MASLD are orchestrated by an interplay of the metabolic environment with genetic and epigenetic factors. Emerging evidence suggests altered DNA methylation pattern as a major determinant of MASLD pathogenesis coinciding with progressive DNA hypermethylation and gene silencing of the liver-specific nuclear receptor PPARα, a key regulator of lipid metabolism. To investigate how PPARα loss of function contributes to epigenetic dysregulation in MASLD pathology, we studied DNA methylation changes in liver biopsies of WT and hepatocyte-specific PPARα KO mice, following a 6-week CDAHFD (choline-deficient, L-amino acid-defined, high-fat diet) or chow diet. Interestingly, genetic loss of PPARα function in hepatocyte-specific KO mice could be phenocopied by a 6-week CDAHFD diet in WT mice which promotes epigenetic silencing of PPARα function via DNA hypermethylation, similar to MASLD pathology. Remarkably, genetic and lipid diet-induced loss of PPARα function triggers compensatory activation of multiple lipid sensing transcription factors and epigenetic writer-eraser-reader proteins, which promotes the epigenetic transition from lipid metabolic stress towards ferroptosis and pyroptosis lipid hepatoxicity pathways associated with advanced MASLD. In conclusion, we show that PPARα function is essential to support lipid homeostasis and to suppress the epigenetic progression of ferroptosis-pyroptosis lipid damage associated pathways towards MASLD fibrosis.
Non-alcoholic fatty liver disease (NAFLD), recently re-named and re-defined as Metabolic Dysfunction Associated Steatotic Liver Disease (MASLD) (Rinella et al., 2023), is a growing epidemic, paralleling the increase of obesity in western diet consuming countries. MASLD shares, in part, the common pathogenesis of metabolic syndrome including obesity, hyperlipidaemia, insulin resistance, mitochondrial damage, oxidative stress response, and the release of inflammatory cytokines. It has an estimated prevalence of 20%–30% in Europe and is the most common cause of chronic liver disease worldwide (Almeda-Valdés et al., 2009; Loomba and Sanyal, 2013; Younossi et al., 2016). MASLD consists of a spectrum of liver disorders ranging from isolated steatosis to Metabolic Dysfunction Associated Steatohepatitis (MASH) which predisposes patients to progressive fibrosis, cirrhosis and hepatocarcinoma but also extrahepatic diseases, especially cardiovascular diseases (Stepan et al., 2012; Chalasani et al., 2018). Dysregulation of insulin secretion and dyslipidaemia due to obesity and other lifestyle variables are the primary contributors to the establishment of MASLD. Although the prevalence keeps growing, there is still no FDA-approved treatment for MASLD. Therefore, with no drugs available, the mainstay of MASLD management remains lifestyle changes with exercise and dietary modifications (Vilar-Gomez et al., 2015; Romero-Gómez et al., 2017).
The onset and progression of MASLD are orchestrated by an interplay of metabolic environment with genetic and epigenetic (lifestyle, environment) factors (Tiffon, 2018; Juanola et al., 2021). An accumulating body of studies revealed progressive DNA methylation changes across different stages of MASLD pathogenesis, although the underlying mechanisms remain poorly understood (Ahrens et al., 2013; Murphy et al., 2013; Kitamoto et al., 2015; Zeybel et al., 2015; Hardy et al., 2017; Mwinyi et al., 2017; Wegermann et al., 2018; Lai et al., 2020). DNA methylation signatures that can affect gene expression are influenced by environmental and lifestyle experiences such as diet, obesity and physical activity and are reversible (Berger et al., 2009; Alegría-Torres et al., 2011; Tiffon, 2018). Hence, DNA methylation signatures and modifiers in MASLD may provide the basis for developing biomarkers indicating the onset and progression of MASLD and therapeutics for MASLD. More specifically, MASLD patients show global hepatic DNA hypomethylation in parallel with increasing hepatic inflammation grade, disease progression and increased hypermethylation of the promotor sequence of the nuclear receptor peroxisome proliferator-activated receptor-α (PPARα) gene (Francque et al., 2015; Li et al., 2018; Lai et al., 2020). Whether loss of PPARα function is a cause or consequence of epigenetic dysregulation in MASLD pathology requires further investigation.
PPARα is part of the PPAR nuclear receptor family that consists of three isoforms: PPARα, PPARβ and PPARγ. All three isoforms are involved in lipid metabolism, but are most abundantly expressed in liver, skeletal muscle and adipocytes respectively (Bugge and Mandrup, 2010; Francque et al., 2021). Since PPARα is abundantly expressed in the liver, known as a key regulator of lipid metabolism, and downregulated in MASLD patients correlating with the disease stage, several agonists have been therapeutically evaluated over the years (Francque et al., 2015; Kersten and Stienstra, 2017). However, PPARα agonists that only target PPARα failed to show convincingly positive results in clinical trials (Lange et al., 2022). Therefore, research is currently more shifting towards drugs targeting multiple therapeutic targets, i.e., pan-PPAR agonists (e.g., Lanifibranor), but also epigenetic modulators (e.g., vitamine E) (Remely et al., 2017; Boubia et al., 2018). Both have already shown promising results in clinical trials of MASLD patients, suggesting a crucial role for PPAR interplay with epigenetic control mechanisms in the development of MASLD (Remely et al., 2017; Boubia et al., 2018; Sumida and Yoneda, 2018). Indeed, recent papers demonstrate significant demethylation of PPARα target metabolic genes upon activation of PPARα (Ehara et al., 2015; Hashimot et al., 2018; Yuan et al., 2018; Moody et al., 2019). Besides, PPARα interactions with epigenetic enzymes have already been identified in different tissues including liver and colon (Hervouet et al., 2010; Luo et al., 2019; Aibara et al., 2022). To further characterize epigenetic “driver” or “passenger” functions of PPARα in MASLD, we compared genome-wide DNA methylation and transcriptome changes in livers of wild type (WT) and hepatocyte-specific PPARα knock out (KO) mice, receiving control chow diet versus MASLD promoting high fat diet (CDAHFD). Characterisation of genome-wide DNA methylation and gene expression changes might provide new insights in PPARα-dependent (epigenetic driver) versus independent (epigenetic passenger) functions, with potential clinical relevance in precision medicine for disease management and staging of MASLD progression.
PPARα KO C57BL/6J (PPARafl/fl AlbuminCreTg/+) mice and WT C57BL/6J (PPARafl/fl AlbuminCre+/+) mice (IRC-VIB, UGent) were housed in a temperature-controlled, specific pathogen free (SPF) air-conditioned animal house with 14/10h light/dark cycles and received food and water ad libitum. 7-week old male hepatocyte-specific PPARα KO and WT mice were fed either a chow diet (normal standard diet, containing 9% energy from fat, 58% from carbohydrates, and 33% from protein) or a CDAHFD (choline-deficient L-amino acid defined high-fat diet, A06071302, New Brunswick, NJ United States, containing 62% energy from fat, 20% from carbohydrates, and 18% from protein) for 6 weeks ad libitum creating 4 different treatment groups. At 13 weeks, the mice were sacrificed by cervical dislocation after anaesthesia with ketamine and xylazine diluted in phosphate-buffered saline (PBS) (2:2:6). Liver samples were immediately snap frozen and stored at −80°C for further analysis. The animal experiments were approved by the institutional ethics committee for animal welfare of the Faculty of Sciences, Ghent University, Belgium (EC2021-071).
The liver was excised from euthanized mice and washed in PBS. Excised liver was fixed in 4% paraformaldehyde overnight at 4°C, dehydrated and embedded in paraffin. The excised tissue sections of 5 μm were cut and stained with haematoxylin-eosin (H&E) using standard protocols. Liver sections were also stained using Masson’s Trichrome staining kit (HT15, Sigma-Aldrich), following the manufacturer’s protocol. In short, liver slices were deparaffinized and hydrated using BIDI. Next, the liver slices were treated with the mordant preheated Bouin’s solution for 30 min at 60°C. Then, the slices were washed and stained with Weigert’s iron haematoxylin at room temperature. After washing and rinsing, the liver slices were stained with Biebrich Scarlet-Acid Fuchsin. After rinsing the tissue slices, the slices were put in a Phosphotungstic/phosphomolybdic Acid solution followed by an Aniline Blue solution and 1% acetic acid. Finally, the liver slices were dipped once in 70% ethanol and 90% ethanol followed by washing the liver slices with 100% ethanol and xylene before mounting the liver slices.
Total RNA was extracted from the livers of the mice after tissue disruption with the TissueRuptor (Qiagen) with the RNeasy kit (Qiagen, 75162), according to the manufacturer’s protocol. Afterwards RNA quantity was determined using QubitTM RNA Broad Range Assay kit with the aid of the Invitrogen QubitTM Fluorometer (Thermo Fisher Scientific, United States). The extracted RNA was stored at −80°C until further analysis.
Total isolated RNA of the livers of 3 mice of each treatment group were sent to Novogene Leading Edge Genomic Services & Solutions for RNA sequencing analysis on the Novaseq6000 platform. In brief, messenger RNA was purified from total RNA using poly-T oligo-attached magnetic beads. After fragmentation, the first strand cDNA was synthesized using random hexamer primers, followed by the second strand cDNA synthesis and library construction. The library was checked with Qubit and real-time PCR for quantification and bioanalyzer for size distribution detection. Quantified libraries were pooled and sequenced on Illumina platforms, according to effective library concentration and data amount. The quality of the raw sequencing reads was evaluated using FastQC (v0.11.5) (Andrews, 2020) and subsequent alignment to genome reference consortium mouse build 38 (GRCm38) was performed with the STAR (v.2.7.3a) tool (Dobin et al., 2013). Differential gene expression and pathway analysis were performed using DESeq2 R package software (Love et al., 2014) and the Omics Playground tool (v2.8.12) platform, which was also used for further visualisation. RNA sequencing was validated by qPCR and deposited in the NCBI GEO database with accession number GSE238201.
After RNA extraction, total RNA was converted into cDNA with the iScriptTM cDNA Synthesis Kit (BioRad, 1708890) according to the manufacturer’s protocol. Next, qPCR analysis was performed using the PowerUp SYBRTM green PCR master mix (Thermo Fisher Scientific, United States) according to the manufacturer’s instructions. In brief, a 20 µL reaction volume mix per sample was prepared containing 10 µL PowerUp SYBR Green Master Mix, 0.4 µM forward and reverse primer (Supplementary Table S1), and nuclease-free water. The following PCR program was applied on the Rotor-Gene Q qPCR machine of Qiagen: 50°C for 2 min, 95°C for 2 min, 40 cycles denaturation (95°C, 15 s) and annealing/extension (60°C, 1 min), and dissociation (60°C–95°C). Each sample was run in triplicate. The mean value of the triplicates was taken to calculate the ΔΔCt-values using GAPDH and YWHAZ as the normalisation genes. PPARα and DNMT1 primer sequences (Supplementary Table S1) were designed by Primer3 and synthesized by Integrated DNA Technologies (IDT, United States). Statistical analysis was carried out using a One-Way ANOVA test with Tukey’s correction for multiple comparisons. p-value < 0.05 was considered statistically significant.
For western blot analysis, liver tissue was disrupted with the TissueRuptor (Qiagen). Next, cells were lysed in 0.5 mL 1x RIPA lysis buffer (150 mM NaCl, 0.1% Triton x-100, 0.1% SDS, 50 mM Tris-HCl pH 8 supplemented with protease inhibitor cocktail (Sigma-Aldrich, Germany)) on ice for 15 min. Afterwards cells were briefly sonicated and centrifugated at 13,000 rpm for 15 min at 4°C. Next, supernatant with soluble protein extract was transferred to new Eppendorf tubes and used for protein quantification with Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, United States). After protein extraction, SDS-PAGE was performed to separate proteins on a 6%–12% gradient Bis-Tris gel. First, samples were mixed with Laemmli buffer (Biorad, United States) and 50 mM 1,4- dithiothreitol (DTT) and then heated at 70°C for 10 min to denaturate the protein. Afterwards, both the samples and protein ladder (BenchMark™ Protein Ladder, Thermo Fisher Scientific, United States) were loaded on the Bis-Tris gel at a protein concentration of 10 µg/well (PPARα, DNMT1, NRF2), 20µg/well (ACSM2A, SLC27A2, CYP7B1, CPT1A) or 100µg/well (Caspase-1 and NLRP3). Electrophoresis was performed in a Mini-PROTEAN Tetra Cell System (Biorad, United States) using a high molecular weight buffer (100 mM MOPS, 100 mM Tris, 0.2% SDS, 2 mM EDTA, 5 mM sodium bisulphite). Afterwards, the proteins were transferred to pre-wet nitrocellulose membranes (Cytiva, United States) for 1 h at 4°C on 250 mA. After blocking the membranes in 5% milk/TBST blocking buffer for 1 h at room temperature, the primary antibodies anti-PPARα (Abcam, #ab126285), anti-DNMT1 (Imgenex, #60B1220.2), anti-NLRP3 (Bio-connect, #AG-20B-0014-C100), anti-Caspase-1 (Bio-connect, AG-20B-0048-C100), anti-NRF2 (Proteintech, #16396-1-AP), anti-CYP7B1 (ProteinTech, #24889-1-AP) and anti-ACSM2A (ProteinTech, #22862-1-AP) were diluted 1:1000; anti-SLC27A2 (ProteinTech, #14048-1-AP) was diluted 1:2000; and anti-CPT1A (ProteinTech, #15184-1-AP) was diltuded 1:4000 in the blocking buffer and incubated overnight at 4°C. The next day, membranes were washed three times with TBST and incubated with HRP-conjugated anti-rabbit secondary antibody (PPARα, DNMT1 and NRF2) or HRP-conjugated anti-mouse secondary antibody (NLRP3 and Caspase-1) diluted in blocking buffer (1:2000) for 1 h at room temperature. Anti-GAPDH antibody (Bioké #5174S, diluted 1:1000) in blocking buffer was used as loading control. Protein detection was performed on the Amarshan imager 680 (Cytiva, United States) using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, 34577) and quantified using ImageJ software. Statistical analysis was carried out using a One-Way ANOVA test with Tukey’s correction for multiple comparisons. p-value < 0.05 was considered statistically significant.
Liver tissue was disrupted with the TissueLyser II (Qiagen) on 20Hz for 5 min at 4°C in 1 mL PBS. Afterwards 100 µL of the tissue lysate was pipetted in a 96 well plate for MDA quantification and the remaining sample was used for further protein quantification with the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, United States) according to the manufacturer’s protocol. At the same time, a 1:2 serial dilution of 1,1,3,3-tetramethoxypropane (0–20 µM) in MiliQ was made to form a standard curve of MDA under acidic conditions. Subsequently, a working solutions consisting of 0.5 mg N-methyl phenyl indol (NMPI), 0.2 mL Acetonnitrile and 0.08 mL Methanol was added per 100 µL of sample or standard. Afterwards, 75 µL of 37% chloric acid was added to the reaction and the samples were incubated for 45min at 70°C. Next, the reaction was stopped by a centrifugation for 10min at 15000rpm at 4°C and the amount of carbocyanine dye formed during this reaction of MDA with NMPI, was measured at 595 nm using the 2103 EnVision™ Multilabel Plate Reader (Perkin Elmer, United States). The final concentration of MDA was further corrected for protein concentration. Statistical analysis was carried out using a One-Way ANOVA test with Dunnett’s correction for multiple comparisons. p-value < 0.05 was considered statistically significant.
Whole-genome methylation profiling was performed on the livers of 3 mice of each treatment group using the Infinium Mouse Methylation BeadChip array (Illumina, San Diego, CA, United States) at the Centre for Medical Genetics (Antwerp University Hospital (UZA), University of Antwerp). Genomic DNA (gDNA) was extracted from the livers using the Dneasy Blood & Tissue Kit (Qiagen, 69504, Courtaboeuf, France) according to the manufacturer’s protocol. DNA concentration and purity were determined by the Qubit 4 Fluorometer (Thermo Fisher Scientific, Q33238). Next, 750 ng DNA was bisulphite converted with the EZ DNA Methylation Kit (Zymo Research, D5001/D5002, Irvine, CA, United States) according to the manufacturer’s instructions. Successful bisulphite conversion was confirmed by PCR with the PyroMark PCR kit (Qiagen) in a region of the Line1 gene (Supplementary Table S2). The resulting PCR products were run on a 2% agarose gel. This converted DNA was then further hybridized with the Illumina Infinium Mouse Methylation BeadChip (Illumina, San Diego, CA, United States) according to the manufacturer’s instructions. In brief, converted DNA was amplified overnight and fragmented enzymatically. Precipitated DNA was resuspended in hybridisation buffer and dispended onto the BeadChips. The hybridisation procedure was performed at 48°C overnight using an Illumina Hybridisation oven. After hybridisation, free DNA was washed away, and single nucleotide extension followed by fluorescent readout was performed. The BeadChips were imaged using an Illumina iScan (Illumina, San Diego, CA, United States). The platform interrogates more than 285,000 methylation sites per sample at single-nucleotide resolution. Annotations for the interrogated sites were taken from Illumina’s BeadChip array manifest based on genome build mm10. Raw intensity data from IDAT files was read and processed in R (v. 4.2.0) via the Enmix package and beta values were normalised with the Enmix D method (Xu et al., 2016). Data pre-processing consisted of masking probes with poor design, control probes, and non-cg and non-ch probes. Detection p-values were inferred using SeSAMe’s pOOBAH (p-value with out-of-band array hybridization) algorithm. Probes with detection p-values > 0.01 or more than 10% of NA values were filtered out. No samples had more than 10% missing values, thus all were considered for further analysis. Further probe-type bias adjustment was applied with the Regression on Correlated Probes method (Niu et al., 2016). The difference in signal intensity between the two-colour channels (dye bias correction) was corrected for using a flexible exponential-normal mixture distribution model. Background correction was done using the Out-Of-Band algorithm. To identify significantly differentially methylated CpGs between the different groups of mice, the Wilcoxon rank-sum test with a Bonferroni correction (p < 0.01) for the total amount of CpGs in the Mouse Methylation BeadChip was used. Further Metascape pathway analysis of genes with a delta beta (DB) > |0.1| and FDR <0.05 was performed with the online Metascape Web tool (Zhou et al., 2019). Methylation data was deposited in the NCBI GEO database with accession number GSE238173.
Pyrosequencing was used to validate methylation of the RETSAT and Eci1 promotor identified by BeadChip analysis. The sequences of the promotor region of the RETSAT and Eci1 gene were retrieved from the Ensemble website (http://genome.ucsc.edu/). Primers were designed based on this sequence and the PyroMark Assay Design Software 2.0.2. (Qiagen) (Supplementary Table S1). Genomic DNA (gDNA) was extracted from the livers using the Dneasy Blood & Tissue Kit (Qiagen, 69504, Courtaboeuf, France) according to the manufacturer’s protocol. DNA concentration and purity was determined by the Qubit 4 Fluorometer (Thermo Fisher Scientific, Q33238). Next, 750 ng DNA was bisulphite converted with the EZ DNA Methylation Kit (Zymo Research, D5001/D5002, Irvine, CA, United States) according to the manufacturer’s instructions. Successful bisulphite conversion was confirmed by PCR with the PyroMark PCR kit (Qiagen) in a region of the Line1 gene (Supplementary Table S2). The resulting PCR products were run on a 2% agarose gel. After successful bisulphite conversion, a PCR was performed with the PyroMark PCR kit (Qiagen) and a forward and biotinylated reverse primer specific to a cytosine in the promotor region of the RETSAT and Eci1 gene. Afterwards 20 µL of this biotinylated product was further used for pyrosequencing using the PyroMark Q24 system (Qiagen) and PyroMark Q24 advanced reagent kit (Qiagen) in combination with a sequencing primer covering 1 CpG according to the manufacturer’s protocol. Analysis was performed using the Pyromark Q24 Advanced software (version 3.0) for detection and quantification of methylation patterns in the target regions. The only values that were reported to be technically reliable by the PyroMark Q24 Software 2.0.8 (Qiagen) were used for statistical analysis. The One-Way ANOVA was performed to assess differential methylation of RETSAT and Eci1 gene between different treatment groups.
PPARα, a key player of lipid metabolism and energy homeostasis, is typically downregulated in the livers of MASLD patients (Francque et al., 2015). To characterize the functional role of PPARα in MASLD development, we studied a hepatocyte-specific PPARα KO mouse model following a 6-week CDAHFD (HFD) to simulate liver pathological properties of a prolonged western diet. First, lack of PPARα expression in PPARα KO liver samples was confirmed at the RNA and protein level by qPCR and western blot analysis, respectively (Figures 1A, B). QPCR and western analysis clearly confirm lack of significant PPARα expression in PPARα KO liver samples as expected (some background Ppara mRNA residual transcription may originate from traces of non-hepatocytes (mainly from stellate cells) present in the liver biopsies (Gonzalez-Sanchez et al., 2017)). Interestingly, RNAseq transcriptome profiling revealed high similarities in transcription profiles of PPARα KO mice on chow diet versus WT mice following 6-week CDAHFD diet. This suggests that a CDAHFD partially phenocopies loss of hepatocyte function of PPARα, closely resembling a genetic PPARα KO approach (Figure 1C; Supplementary Figure S1). Along the same line, GO gene set enrichment analysis confirms mitochondrial dysfunctions due to multiple changes in bile and fatty acid metabolism, amino acid catabolism and inflammation, in response to CDAHFD and upon genetic PPARα KO or combinations thereof (Figure 1D).

FIGURE 1. (A) Relative Ppara mRNA expression in PPARα WT and KO mice after a 6-week chow or CDAHFD. (B) Western blot detection and quantification of PPARα and GAPDH expression levels after a 6-week chow or CDAHFD in WT and KO mice. Data are plotted as the mean ± s.d., n = 3 biologically independent replicates. (*p < 0.05, **p < 0.01 ***p < 0.001) (C) UMAP representation of gene clustering based on geneset co-expression in PPARα WT and KO mice after a 6-week chow or CDAHFD (D) GO activation matrix representation of pathway enrichment analysis of significantly up- or downregulated pathways in both comparisons of KO mice versus WT mice on a chow or CDAHFD respectively. The size of the circles in the GO activation matrix corresponds to their relative activation, and are colored according to their upregulation (red) or downregulation (blue) in the contrast profile (meta.q < 0.05).
In line with reduced PPARα expression reported in MASLD/MASH patients, both the qPCR and western blot also reveal decreased PPARα expression in the WT mice following 6-week CDAHFD diet (Francque et al., 2015). The latter suggests that a high fat diet may gradually decrease PPARα expression and as such progressively phenocopies the transcriptome signature of a genetic hepatocyte-specific PPARα KO model. Furthermore, liver sections were scored based on the Clinical Research Network and Steatosis-Activity-Fibrosis NASH scoring systems to assess the disease stage of the mice (Figure 2A; Supplementary Table S2) (Kleiner et al., 2005; Bedossa et al., 2012). These results show that 6-week CDAHFD in WT and PPAR KO mice, both result in MASH features including steatosis, ballooning, lobular inflammation and fibrosis, similar to liver histopathology in MASH patients. Interestingly, PPARα KO mice on a normal chow diet already reveal MASLD features such as accumulation of lipid droplets.
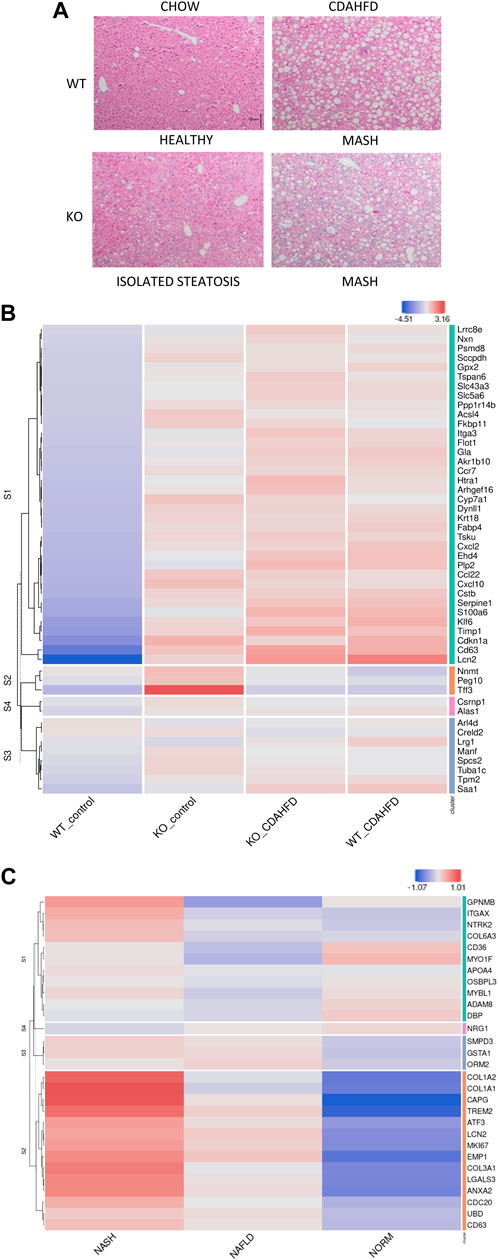
FIGURE 2. (A) H&E staining of liver sections of WT and hepatocyte PPARα KO mice after following 6-week chow or CDAHFD (scaling bar: 100μm) (B) Heatmap representation of MASLD signature in KO or WT mice after 6-week chow or CDAHFD (n = 3). (C) Heatmap representation of PPARα KO signature in MASLD (NAFLD) and MASH (NASH) patients of the GEO dataset GSE126848.
To further characterize whether these histopathological changes also correlate with a MASLD/MASH disease signature in patients, we cross compared our gene expression profiles with the publicly available MASLD/MASH transcriptome signature of liver biopsies of a patient cohort with varying degrees of MASLD (GSE126848) (Supplementary Table S3). Interestingly, the hepatocyte-specific PPARα KO mouse model on both a chow and 6-week CDAHFD diet as well as WT mice after a 6-week CDAHFD diet, reveal a similar hyperactivated MASLD/MASH transcriptome signature, which confirms the involvement of a PPARα loss of function and disrupted fatty acid metabolism in the MASLD/MASH disease aetiology (Figure 2B). Accordingly, the MASLD/MASH heatmap of GSE126848 also phenocopies the PPARα loss of function transcription signature of the hepatocyte-specific PPARα KO mouse model (Figure 2C).
Besides similarities in lipid metabolic gene expression changes between CDAHFD diet WT and chow/CDAHFD PPARα KO mice, genetically and diet-induced PPARα loss of function also regulate overlapping bile and fatty acid responsive transcription factors, nuclear receptors and epigenetic writer-reader-eraser proteins, including multiple DNA (hydroxy)methylating enzymes and DNA Methyl-binding factors (Figures 3A, B). For example, weakly increased RNA and protein expression levels of DNMT1 can be observed in PPARα KO and CDAHFD diet conditions (Figures 3C, D). Remarkably, Homer motif analysis revealed that several of these differentially expressed transcription factors and DNA Methyl-binding proteins themselves contain PPARα binding motifs (PPRE) motifs. Since these proteins can directly regulate epigenetic enzymes, these results suggest that epigenetic enzyme expression and activity too might be under lipidomic PPARα control (Supplementary Table S5).
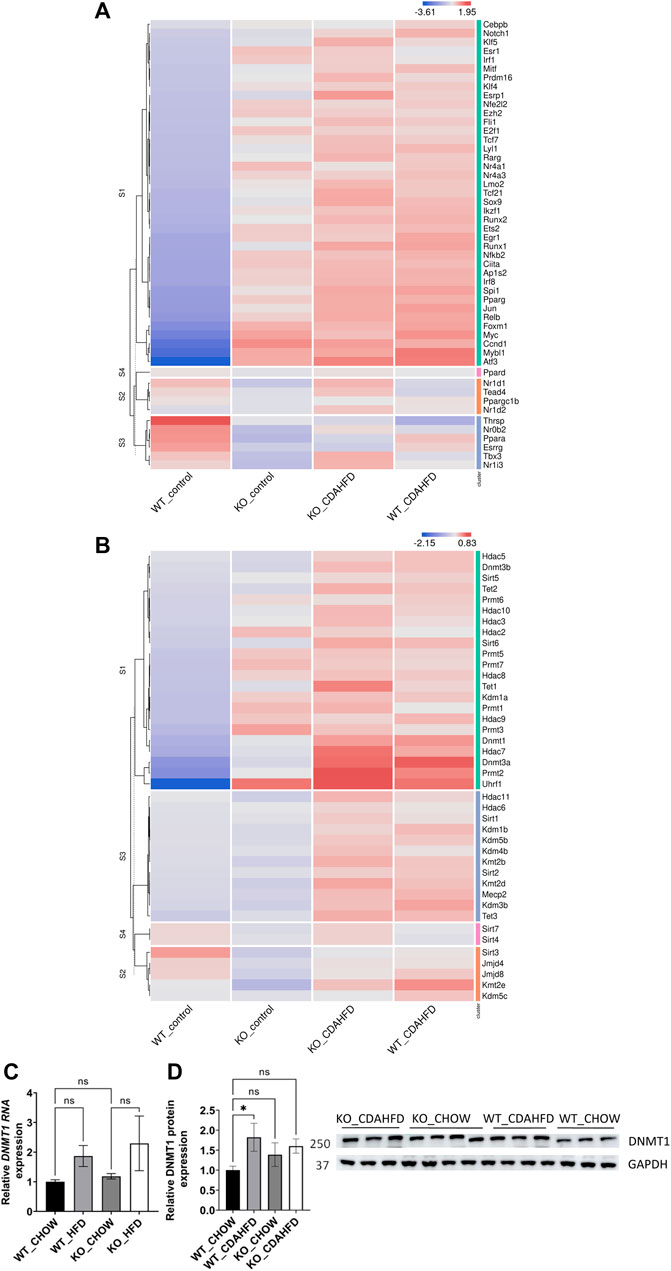
FIGURE 3. Heatmap representation of differentially expressed (A) transcription factors and nuclear receptors or (B) epigenetic writers-readers-eraser proteins in KO or WT mice after 6-week chow or CDAHFD (n = 3). (C) qPCR and (D) western blot detection and quantification of DNMT1 and GAPDH expression levels after 6-week chow or CDAHFD in WT and KO mice. Data are plotted as the mean ± s.d., n = 3 biologically independent replicates. (ns p > 0.05, *p < 0.05, **p < 0.01 ***p < 0.001)
Since altered DNA methylation has been identified as a key determinant of MASLD pathogenesis (Li et al., 2018; Hyun and Jung, 2020; Lai et al., 2020; Bu et al., 2022; Kuramoto et al., 2022; Melton et al., 2023), we next applied Infinium mouse methylation Beadchip array studies to map genome-wide DNA methylation changes in liver biopsies of WT and hepatocyte-specific PPARα KO mice, following 6-week chow or CDAHFD diet. As shown in Figure 4, the WT mice on a CDAHFD show predominant hypermethylation of the promotor region of PPARα compared to the WT mice on a chow diet, which could explain the gradual silencing of PPARα expression following a 6-week CDAHFD diet. Besides, both genetically and diet-induced PPARα loss of function trigger massive -partially redundant- DNA methylation changes in genes involved in fatty acid and bile acid metabolism, nuclear hormone (steroid) receptor and inflammatory cytokine pathways, according to Metascape enrichment analysis (Zhou et al., 2019) (Figures 5A, B). Remarkably, TRRUST motif analysis of hypermethylated genes in PPARα KO mice shows highly significant enrichment of PPRE (Figure 5C), which is still significantly enriched in WT CDAHFD mice which only partially express PPARα protein (Figure 5D). Along the same line, the cross-comparison of a list of PPARα target genes (Supplementary Table S4) with our lists of differentially methylated genes, identified various common hypermethylated target genes in the PPARα KO mice, following chow or CDAHFD diet, whereas CDAHFD diet in WT liver cells with partially decreased PPARα expression shows a mixed hypo/hypermethylation pattern (Supplementary Figure S2). Although most of the selected hypermethylated genes involved in fatty acid or bile acid metabolism, including PPARα metabolic target genes, are downregulated in the hepatocyte-specific PPARα KO mice and WT mice on a CDAHFD with a partial expression of PPARα, few genes are upregulated (Figure 5E). Remarkably, further validation on protein level showed that proteins involved in mitochondrial lipid uptake (CPT1A) are significantly upregulated by the loss of PPARα function, while lipid catabolic proteins involved in lipid or bile acid catabolism are downregulated (CYP7B1 and ACSM2). The latter emphasizes the dual epigenetic regulation of lipid metabolic genes via PPARα (Supplementary Figure S3). Of special note, bisulfite converted DNA assay does not allow discrimination between DNA methylation and hydroxymethylation changes that have been associated with gene silencing and gene activation responses respectively. Indeed, PPARα regulatory functions have recently been described for both DNA methylation as well as hydroxymethylation and may need a more detailed in-depth molecular investigation (Theys et al., 2022).
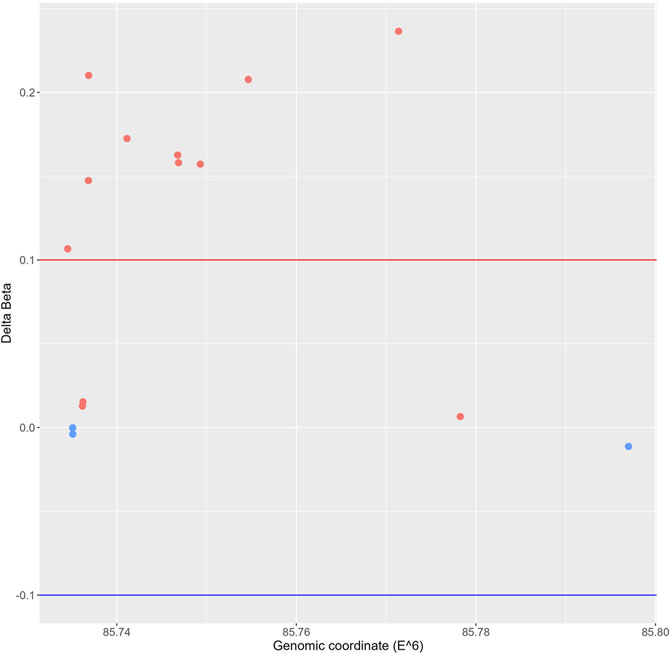
FIGURE 4. Genomic scatterplot of delta beta values of differently methylated probes in the promotor region of the PPARα gene in the WT group on a 6-week CDAHFD compared to the WT mice on 6-week chow diet.
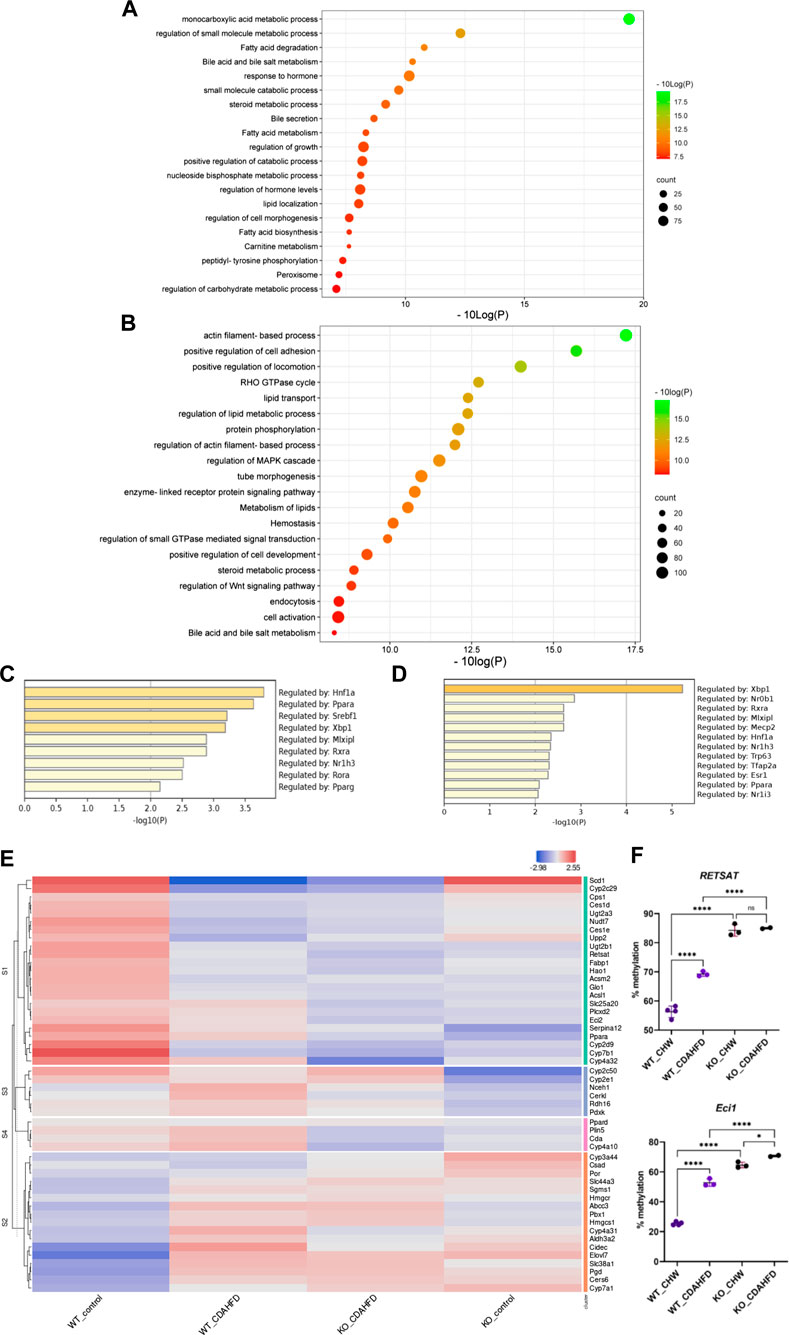
FIGURE 5. Metascape pathway analysis of differently methylated genes in (A) KO mice compared to WT mice on a chow diet (FDR<0.05; DB > 0.15) and (B) WT mice on a CDAHFD compared to a chow diet (FDR<0.05; DB > 0.25). TRRUST transcription factor analysis of differently hypermethylated genes in (C) KO mice compared to WT mice on a chow diet (FDR<0.05; DB > 0.15) (D) WT mice on a CDAHFD compared to a chow diet (FDR<0.05; DB > 0.25). (E) Heatmap representation of the expression of genes involved in lipid or bile acid metabolic pathways that are hypermethylated in WT mice on a CDAHFD and KO mice on a control chow diet compared to WT mice on a control chow diet. (F) Pyrosequencing validation of two PPARα target genes (ns p > 0.05, *p < 0.05, ****p < 0.0001).
To further validate our epic array hypermethylation data, we applied bisulfite pyrosequencing of PPARα target genes RETSAT and Eci1, which are according to the EPIC data both hypermethylated by a diet-induced or genetic KO of PPARα (delta beta KO_chow vs. WT_chow: 0,48-0,29; KO_CDAHFD vs. WT_CDAHFD: 0,17-0,14; WT_CDAHFD vs. WT_chow: 0,31 -0,18). Moreover, these genes are involved in retinol metabolism and beta oxidation respectively and thereby control lipid metabolism (Pang et al., 2017; Bramhecha et al., 2022). As shown in Figure 5F relative DNA methylation is strongly increased when PPARα is knocked out, or modestly increased upon CDAHFD diet in WT mice with partially decreased PPARα expression. These results suggest that PPARα targeting of lipid metabolic genes may be essential to protect against epigenetic DNA methylation modifications.
Furthermore, these results could also be confirmed in Epic Beadchip DNA methylation array data of MASLD liver biopsies of MASH patients (GSE241366) with advanced fibrosis in comparison to healthy tissue controls, which reveal similar enrichment of differentially methylated genes involved in lipid metabolism (fatty acid, bile acid) with PPARα as one of the top enriched TF motifs in general (Figure 6A) and especially of the hypermethylated genes (p-value < 0.05) (Figure 6B). Altogether this suggests that PPARα protects against epigenetic DNA (hyper)methylation of lipid metabolic genes involved in the progression of MASLD.
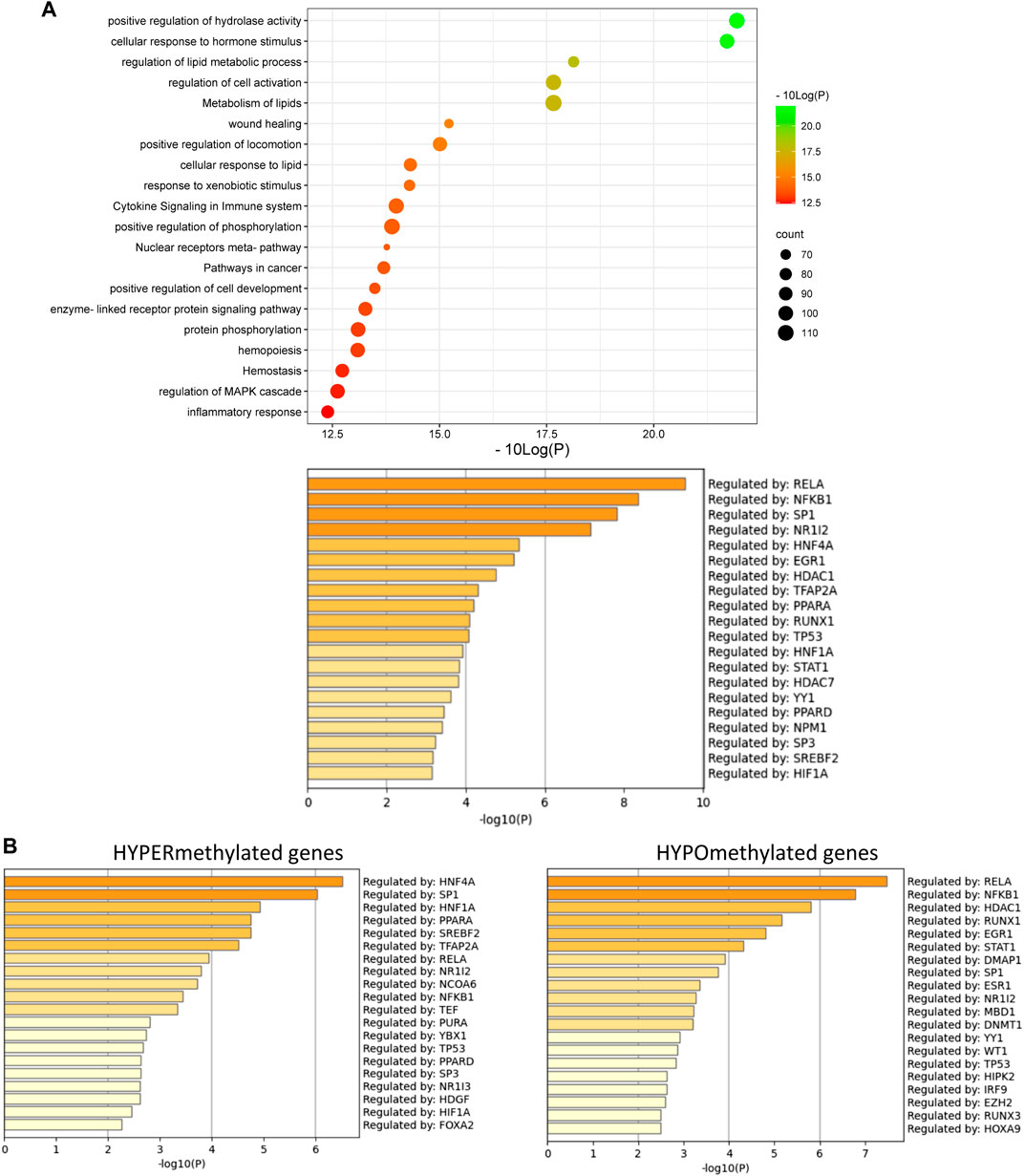
FIGURE 6. (A) Metascape pathway analysis (top) and TRRUST transcription factor analysis (bottom) of significant differently methylated genes in MASH patients with advanced fibrosis versus healthy individuals (B) TRRUST transcription factor analysis of significant hypermethylated and hypomethylated genes in MASH patients with advanced fibrosis versus healthy individuals. (p-value < 0.05; DB < |0.3|).
Of special note, in line with the fact that PPARα can also indirectly regulate genes via transrepression of other bound transcription factors such as NFκB (Delerive et al., 1999; Zúñiga et al., 2011; Tayyeb et al., 2020), we also observe multiple epigenetic changes of NFκB-driven (NFκB1, RelA, RelB) inflammatory target genes in the mouse/patient samples, which may further contribute to lipid-inflammation tissue damage in MASLD.
Lipotoxic hepatocyte injury is a primary event in MASH, characterized by excess triglyceride accumulation stored as lipid droplets in the cytosol of hepatocytes, which is deemed the first stage of MASLD. Hepatic steatosis may further develop into MASH, fibrosis, cirrhosis and eventually hepatocellular carcinoma without timely interventions. Recent evidence suggests that hepatic ferroptosis and pyroptosis play an important role in this lipotoxic pathological progression of MASLD (Tsurusaki et al., 2019; Gaul et al., 2021; Zhang et al., 2021; Knorr et al., 2022).
Ferroptosis, a recently recognized nonapoptotic form of regulated cell death that is characterized by iron-dependent lipid peroxidation, was recently confirmed to be the initial cell death process that triggers MASH (Dixon et al., 2012; Tsurusaki et al., 2019; Zhang et al., 2021). Besides, new results identify hepatocyte pyroptosis and release of NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome components as an additional mechanism to propagate liver injury and liver fibrosis development in MASH progression (Csak et al., 2011; Wree et al., 2014a; Beie et al., 2018; Wu et al., 2019). As the liver is a “first pass” organ, continually challenged with diverse microbial particles from the intestine as well as endogenous metabolic stress signals (fatty acid, bile acid), hepatocytes are capable of undergoing NLRP3-mediated pyroptotic cell death and release extracellular NLRP3 inflammasome complexes into the extracellular space. These extracellular inflammasomes can be internalized by hepatic stellate cells leading to their activation and subsequent liver fibrogenesis (Kisseleva and Brenner, 2007; Watanabe et al., 2009; Wree et al., 2014b; Wu et al., 2019; Knorr et al., 2022).
To evaluate whether PPARα loss may impact ferroptosis/pyroptosis pathways in MASLD/MASH, we next performed a cross comparison of our gene expression mouse data with publicly available ferroptosis/pyroptosis RNAseq based transcriptome datasets (Hassannia et al., 2018; Ye et al., 2021; De Backer et al., 2022; Deng et al., 2022; Wang et al., 2022; Zuo et al., 2022; Zhou et al., 2023). Remarkably, both genetic and diet-induced PPARα loss reveal strong hyperactivation of lipotoxic ferroptosis/pyroptosis signatures (Figure 7). Indeed, further protein level analysis confirmed a significant upregulation of the nuclear factor E2 related factor 2 (Nrf2/NFE2L2), a key regulator of the ferroptosis (Hassannia et al., 2018; Pan et al., 2021) and pyroptosis (Hurtado-Navarro et al., 2022) pathways, in PPARα KO mice on chow diet and WT or KO mice on CDAHFD diet. Moreover, a significant upregulation of malondialdehyde (MDA), which represents increased lipid peroxidation, was found under a CDAHFD in both the KO and WT mice, in line with observations in MASLD/MASH patient samples (Zhang et al., 2021). Furthermore, protein validation of Caspase 1 and NLRP3 showed that a genetic and diet-induced PPARα loss induce an upregulation of NLRP3. However Caspase 1 is not further cleaved in its catalytic domains p10 and p20, indicating that PPARα loss increases sensitivity for pyroptosis without inducing further pyroptotic cell death. Of special note, when cross comparing differentially methylated target genes of advanced MASH (versus healthy liver biopsies), with both ferroptosis/pyroptosis genesets, we could identify various novel epigenetic biomarkers of ferroptosis/pyroptosis lipotoxicity (Figure 9; Supplementary Table S6).
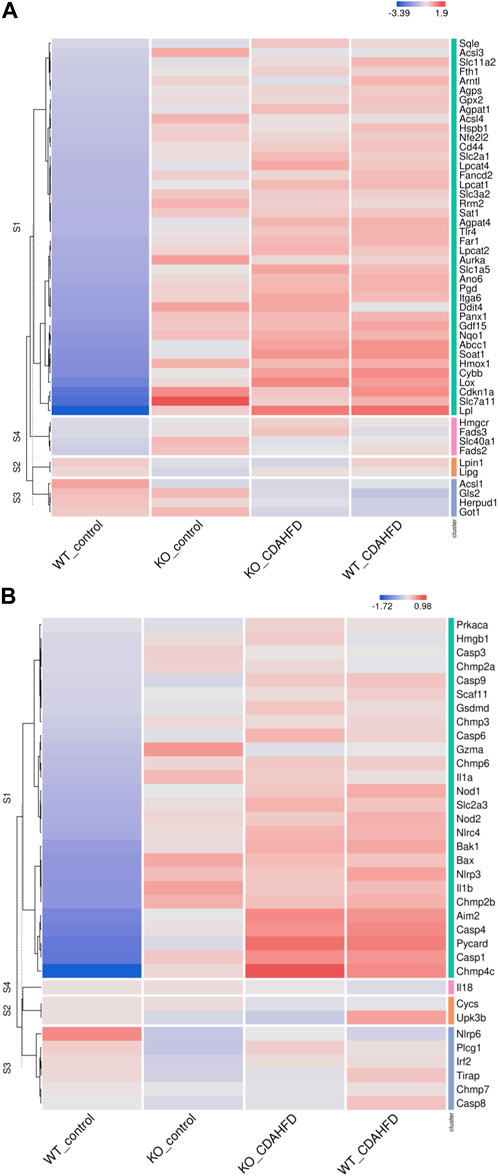
FIGURE 7. Heatmap representation of (A) ferroptosis and (B) pyroptosis signature in WT or PPARα KO mice on a 6-week chow or CDAHFD.
Altogether, these results suggest that PPARα function is essential to prevent the epigenetic transition from lipid homeostasis to MASH/MASLD lipotoxicity. In addition, epigenetic ferroptosis/pyroptosis biomarkers might hold promise as new precision medicine tools in MASLD/MASH disease management and patient stratification of lipotoxic liver damage.
The nuclear receptor PPARα is a critical regulator of lipid metabolism and MASLD progression. Surprisingly, PPARα ligands have only shown limited therapeutic benefits against MASLD in (pre)clinical trial studies (Lange et al., 2022). Of special note, recent evidence suggests a possible involvement of epigenetic silencing mechanisms in PPARα functions in MASLD progression which may counteract therapeutic actions of PPAR ligands (Theys et al., 2022). In this respect, to further characterise reciprocal crosstalk of epigenetic regulatory mechanisms with PPARα functions in lipid metabolism and MASLD progression, we have cross compared DNA methylation and gene expression patterns of chow or CDAHFD hepatocyte-specific PPARα KO mice versus liver biopsies of MASLD/MASH patient samples.
Upon comparing gene expression changes of liver biopsies of hepatocyte-specific PPARα KO mice versus WT mice following 6-week chow diet, or WT mice following 6-week CDAHFD, we observed strong similarities in transcriptome signatures with the CDAHFD WT mice. This reveals that CDAHFD phenocopies to some extent a genetic KO of PPARα liver functions. QPCR and western blot analysis of PPARα expression indeed confirmed a lack of PPARα protein expression, whereas CDAHFD revealed significantly decreased PPARα expression as compared to chow diet fed WT mice. Moreover, in line with the gene expression profiles in mice, publicly available gene expression datasets of liver biopsies of MASLD/MASH patients show high similarities with the PPARα KO transcriptome signature, which reveals loss of PPARα function in these patients. This is in line with reduced PPARα expression levels which have been observed in MASH/MASLD patients (Francque et al., 2015). Reciprocally, we observed that transcriptome profiles of CDAHFD fed WT and PPARα KO mice also show high similarity to a MASLD/MASH patient signature, which suggests that CDAHFD fed WT and PPARα KO mice are clinically relevant mouse models for molecular biochemical investigation of MASH/MASLD disease. In line with these results, histological staining of liver biopsies confirmed increased frequency of lipid droplets in chow fed PPARα KO mice (stage 2, isolated steatosis), as well as inflammatory ballooning and fibrosis properties in CDAHFD fed PPARα KO/WT mice (stage 3, MASH). These results also confirm the data of Matsumoto et al. (2013) who has previously shown that a CDAHFD can induce MASH with fibrosis in mice in 6-week time, which is relatively fast compared to classical HFD used to induce MASLD in mice. By studying this mouse model, we were able to functionally characterize epigenetic driver and passenger functions of PPARα in lipid metabolism in relation to MASLD/MASH, which has not been addressed before.
Remarkably, diet and genetic PPARα knockout mice elicit similar transcriptional activation of multiple transcription factors, steroid hormone receptors and epigenetic factors involved in metabolic stress responses in the liver (Figures 3A, B). This is not completely unexpected, since loss of hepatocyte PPARα function results in loss of lipid metabolism homeostasis due to impaired fatty acid, bile acid and amino acid catabolic processes (Figure 1D) which results in major changes in the lipidome composition (Régnier et al., 2020). Accordingly, multiple compensation mechanisms of lipid sensing transcription factors and nuclear hormone receptors (Myc, NR4A1, NR4A3, PPARδ/γ, E2F1, PPARGC1B, Nrf2/NFE2L2, TCF21) are activated to mitigate lipidomic stress and to alleviate mitochondrial metabolic stress (Walczak and Tontonoz, 2002; Zirath et al., 2013; Denechaud et al., 2015; Sharma et al., 2018; Yan et al., 2018; Li et al., 2020; Cariello et al., 2021; Mohs et al., 2021; Ni et al., 2023). Similarly, expression of various epigenetic factors (DNMT, TET, SIRT, HDAC, Uhrf1) changes upon lipid metabolic inflammatory stress (Kemper et al., 2013; Ding et al., 2017; Keleher et al., 2018; Wang et al., 2020; Byrnes et al., 2022; Hou et al., 2022; Dong, 2023; Sukur et al., 2023), some of which contain PPRE motifs in their gene promoters (Supplementary Table S5). Although the full mechanism has not been resolved, it appears that there is reciprocal regulation between PPARα protein levels versus expression of DNMT, TET and other DNA-methyl binding proteins such as Uhrf1 (Unoki et al., 2004; Ashraf et al., 2017; Ren, 2022). Moreover, besides transcriptional control mechanisms, PPARα-dependent β-oxidation also promotes (mitochondrial) protein hyperacetylation via increased acetyl-coA production, which can change protein function, localisation, interaction and/or stability (Pougovkina et al., 2014).
Not surprisingly, diet and genetic loss of PPARα hepatocyte function trigger massive DNA methylation changes of multiple genes associated with fatty acid, bile acid and steroid hormone receptor pathways, including PPARα, which changes the expression of multiple lipid metabolic genes (Figure 5). Since bisulfite sequencing does not discriminate between DNA methylation and hydroxymethylation changes, epigenetic changes trigger mixed metabolic gene silencing-activation effects involved in lipid metabolism and MASLD (RETSAT, FABP1, Eci1/2, Cyp7a1) (Lyall et al., 2016; Chiang, 2017; Heidenreich et al., 2017; Mukai et al., 2017; Lyall et al., 2020; Rom et al., 2020). Moreover, epigenetic changes following loss of PPARα functions seem to fail to mitigate liver metabolic dysfunctions (autophagy, mitophagy, lipophagy), since downstream gene expression profiles and key regulatory proteins of ferroptosis and pyroptosis lipotoxicity pathways are highly enriched (Figures 7, 8). Indeed, liver overload of fatty acid and bile acid metabolites promotes lipid peroxidation ferroptosis damage and sensitizes for inflammation-induced steatosis-pyroptosis (Figure 8), which can finally trigger liver fibrosis and cirrhosis or hepatocellular carcinoma (Tsurusaki et al., 2019; Wu et al., 2019; Gaul et al., 2021; Wu et al., 2021; Zhang et al., 2021; Chen et al., 2022; Knorr et al., 2022). Accordingly, we identified various epigenetic changes in ferroptosis-pyroptosis target genes in liver biopsies of late-stage MASLD/MASH patients, which could hold promise as novel stochastic biomarkers of lipid-related inflammatory liver damage (Figure 9; Supplementary Table S6), besides fibrosis stage (Sokolowska et al., 2022; Sun et al., 2022) or epigenetic clock age (Loomba et al., 2018; Li et al., 2022). Interestingly, PPARα was recently shown to protect against liver ferroptosis and might further suppress pathological progression into liver pyroptosis-fibrosis-cirrhosis (Grabacka et al., 2021; Xing et al., 2022; Qiu et al., 2023; Xiao et al., 2023). Of special note, PPARα DNA-binding was demonstrated to the Gpx4 promoter by ChIP experiments (Xing et al., 2022). These findings suggest that ferroptosis inhibitors and epigenetic drug combination therapies with PPAR ligands could hold promise to treat MASLD/MASH.
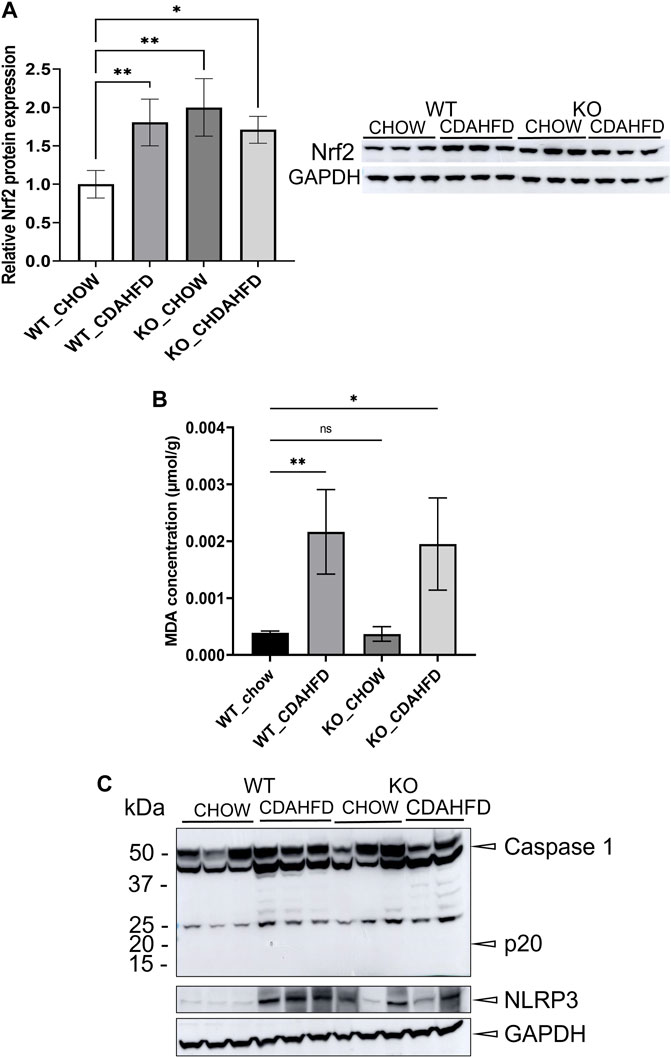
FIGURE 8. (A) Western blot detection and quantification of NRF2 and GAPDH protein expression levels and (B) MDA concentration in liver samples of WT and KO mice after a 6-week chow or CDAHFD (C) Western blot detection of Caspase 1, NLRP3 and GAPDH in WT and KO mice after a 6-week chow or CDAHFD. Data are plotted as the mean ± s.d (ns p > 0.05, *p < 0.05, **p < 0.01 ***p < 0.001).
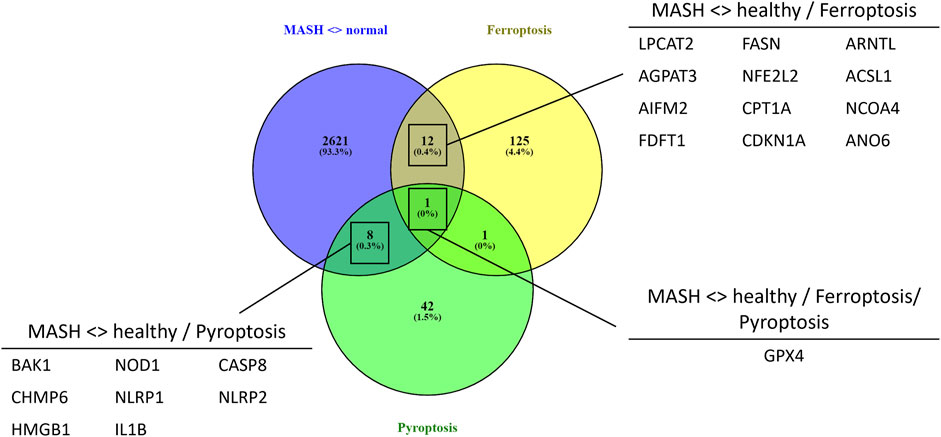
FIGURE 9. Venn diagram showing the overlap between significant differentially methylated genes in MASH patients compared to normal liver controls and a ferroptosis or pyroptosis signature (p-value <0.05, DB>|0.3|).
In conclusion, we demonstrate that loss of PPARα function promotes epigenetic dysregulation of lipid homeostasis, driving ferroptosis and pyroptosis lipotoxicity in MASLD. Of special note, loss of function of a single lipid metabolic PPARα hub seems to cause a lipidomic shockwave of gene expression changes of lipid sensing transcription factors and epigenetic enzymes, which fail to mitigate lipid metabolic stress and trigger epigenetic transition towards lipid hepatotoxicity driving fibrosis. This may explain why monotargeted therapeutic strategies in MASLD/MASH may not be effective to “cure” the multi-factorial nature of MASLD involving genetic predisposition, environmental factors (lifestyle, diet), insulin resistance, disordered lipid metabolism, mitochondrial dysfunction, lipotoxicity, hyperinflammation, oxidative stress, etc. This urges for applying integrative multi-omics systems biology approaches (incorporating data on genetic variants, epigenetic phenomena (i.e., DNA methylation, histone modifications and long non-coding RNA affecting gene expression), gut microbiota dysbiosis, and metabolomics/lipidomic fingerprints) to gain a deeper understanding of the molecular and physiological processes underlying MASLD pathogenesis and phenotype heterogeneity, as well as facilitating the further identification of lipidome-associated epigenetic biomarkers of disease progression and therapeutic targets for the implementation of tailored nutritional strategies (Buzzetti et al., 2016; Soltis et al., 2017; Fang et al., 2018; Ore and Akinloye, 2021; Minami et al., 2023). In this respect, besides pharmaceutical combination therapies, diet interventions and herbal phytomedicinal therapies may also have a role to play in the treatment of MASLD, due to their numerous bioactive constituents and the multiple pharmacological actions they exhibit (Gao et al., 2020; Ore and Akinloye, 2021; Cheng et al., 2022). Finally, to capture a full understanding of adverse MASLD epigenome dysregulation, it will be mandatory to also integrate the complex epi-lipidomic post-translational modification landscape of transcription factors, histones and epigenetic modifiers which control the lipid metabolic network signalling activities in MASLD progression (Amado et al., 2014; Kebede et al., 2017; Sabari et al., 2017; Nieborak and Schneider, 2018; Bartke and Schneider, 2020; Dai et al., 2020; Arima et al., 2021; Guerra et al., 2022).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, GSE238173 https://www.ncbi.nlm.nih.gov/, GSE238201.
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements. The animal study was approved by the institutional ethics committee for animal welfare of the Faculty of Sciences, Ghent University, Belgium (EC2021-071). The study was conducted in accordance with the local legislation and institutional requirements.
CT: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Validation, Visualization, Writing–original draft, Writing–review and editing. TiV: Investigation, Methodology, Resources, Writing–review and editing. EV: Investigation, Writing–review and editing. CP: Formal Analysis, Writing–review and editing. AS: Investigation, Methodology, Writing–review and editing. JI: Formal Analysis, Methodology, Software, Writing–review and editing. LM: Formal Analysis, Methodology, Software, Writing–review and editing. ST: Formal Analysis, Software, Writing–review and editing. ToV: Supervision, Writing–review and editing. SF: Funding acquisition, Supervision, Writing–review and editing. WVn: Funding acquisition, Project administration, Supervision, Writing–review and editing. CL: Methodology, Project administration, Supervision, Writing–review and editing. WVd: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by a GOA-grant (FFB180348/36572) and Methusalem-OEC grant—“GENOMED” (Grant Number: FFB190208).
We would like to thank EV, supported by a GOA-grant (FFB180348/36572) and a FWO-grant (Fonds Wetenschappelijk Onderzoek) (Grant Number: 11E6921N), for letting us use the human Beadchip data as validation for our findings.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmmed.2023.1283170/full#supplementary-material
SUPPLEMENTARY TABLE S1 | Overview of qPCR, PCR and pyrosequencing primers used in this study. (b = biotin tag)
SUPPLEMENTARY TABLE S2 | Full CRN and SAF scoring of the mice used for RNA sequencing and Beadchip analysis.
Ahrens, M., Ammerpohl, O., von Schönfels, W., Kolarova, J., Bens, S., Itzel, T., et al. (2013). DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 18 (2), 296–302. doi:10.1016/j.cmet.2013.07.004
Aibara, D., Takahashi, S., Yagai, T., Kim, D., Brocker, C. N., Levi, M., et al. (2022). Gene repression through epigenetic modulation by PPARA enhances hepatocellular proliferation. iScience 25 (5), 104196. doi:10.1016/j.isci.2022.104196
Alegría-Torres, J. A., Baccarelli, A., and Bollati, V. (2011). Epigenetics and lifestyle. Epigenomics 3 (3), 267–277. doi:10.2217/epi.11.22
Almeda-Valdés, P., Cuevas-Ramos, D., and Alberto Aguilar-Salinas, C. (2009). Metabolic syndrome and non-alcoholic fatty liver disease. Ann. Hepatol. 8, S18–S24. doi:10.1016/s1665-2681(19)31822-8
Amado, F. M., Barros, A., Azevedo, A. L., Vitorino, R., and Ferreira, R. (2014). An integrated perspective and functional impact of the mitochondrial acetylome. Expert Rev. Proteomics 11 (3), 383–394. doi:10.1586/14789450.2014.899470
Andrews, S. (2020). Babraham bioinformatics - FastQC A quality control tool for high throughput sequence data. Vol. 5, Soil.
Arima, Y., Nakagawa, Y., Takeo, T., Ishida, T., Yamada, T., Hino, S., et al. (2021). Murine neonatal ketogenesis preserves mitochondrial energetics by preventing protein hyperacetylation. Nat. Metab. 3 (2), 196–210. doi:10.1038/s42255-021-00342-6
Ashraf, W., Ibrahim, A., Alhosin, M., Zaayter, L., Ouararhni, K., Papin, C., et al. (2017). The epigenetic integrator UHRF1: on the road to become a universal biomarker for cancer. Oncotarget 8 (31), 51946–51962. doi:10.18632/oncotarget.17393
Bartke, T., and Schneider, R. (2020). You are what you eat - how nutrition and metabolism shape the genome through epigenetics. Mol. Metab. 38, 100987. doi:10.1016/j.molmet.2020.100987
Bedossa, P., Poitou, C., Veyrie, N., Bouillot, J. L., Basdevant, A., Paradis, V., et al. (2012). Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology 56 (5), 1751–1759. doi:10.1002/hep.25889
Beier, J. I., and Banales, J. M. (2018). Pyroptosis: an inflammatory link between NAFLD and NASH with potential therapeutic implications. J. Hepatol. 68 (4), 643–645. doi:10.1016/j.jhep.2018.01.017
Berger, S. L., Kouzarides, T., Shiekhattar, R., and Shilatifard, A. (2009). An operational definition of epigenetics: figure 1. Genes Dev. 23 (7), 781–783. doi:10.1101/gad.1787609
Boubia, B., Poupardin, O., Barth, M., Binet, J., Peralba, P., Mounier, L., et al. (2018). Design, synthesis, and evaluation of a novel series of indole sulfonamide peroxisome proliferator activated receptor (PPAR) α/γ/δ triple activators: discovery of lanifibranor, a new antifibrotic clinical candidate. J. Med. Chem. 61 (6), 2246–2265. doi:10.1021/acs.jmedchem.7b01285
Bramhecha, Y. M., Guérard, K. P., Audet-Walsh, É., Rouzbeh, S., Kassem, O., Pernet, E., et al. (2022). Fatty acid oxidation enzyme Δ3, Δ2-enoyl-CoA isomerase 1 (ECI1) drives aggressive tumor phenotype and predicts poor clinical outcome in prostate cancer patients. Oncogene 41 (20), 2798–2810. doi:10.1038/s41388-022-02276-z
Bugge, A., and Mandrup, S. (2010). Molecular mechanisms and genome-wide aspects of PPAR subtype specific transactivation. PPAR Res. 2010, 169506–169512. doi:10.1155/2010/169506
Buzova, D., Braghini, M. R., Bianco, S. D., Lo Re, O., Raffaele, M., Frohlich, J., et al. (2022). Profiling of cell-free DNA methylation and histone signatures in pediatric NAFLD: a pilot study. Hepatol. Commun. 6 (12), 3311–3323. doi:10.1002/hep4.2082
Buzzetti, E., Pinzani, M., and Tsochatzis, E. A. (2016). The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 65 (8), 1038–1048. doi:10.1016/j.metabol.2015.12.012
Byrnes, K., Blessinger, S., Bailey, N. T., Scaife, R., Liu, G., and Khambu, B. (2022). Therapeutic regulation of autophagy in hepatic metabolism. Acta Pharm. Sin. B 12 (1), 33–49. doi:10.1016/j.apsb.2021.07.021
Cariello, M., Piccinin, E., and Moschetta, A. (2021). Transcriptional regulation of metabolic pathways via lipid-sensing nuclear receptors PPARs, FXR, and LXR in NASH. Cell Mol. Gastroenterol. Hepatol. 11 (5), 1519–1539. doi:10.1016/j.jcmgh.2021.01.012
Chalasani, N., Younossi, Z., Lavine, J. E., Charlton, M., Cusi, K., Rinella, M., et al. (2018). The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67 (1), 328–357. doi:10.1002/hep.29367
Chen, J., Li, X., Ge, C., Min, J., and Wang, F. (2022). The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 29 (3), 467–480. doi:10.1038/s41418-022-00941-0
Cheng, R., Wang, L., Le, S., Yang, Y., Zhao, C., Zhang, X., et al. (2022). A randomized controlled trial for response of microbiome network to exercise and diet intervention in patients with nonalcoholic fatty liver disease. Nat. Commun. 13 (1), 2555. doi:10.1038/s41467-022-29968-0
Chiang, J. Y. L. (2017). Targeting bile acids and lipotoxicity for NASH treatment. Hepatol. Commun. 1 (10), 1002–1004. doi:10.1002/hep4.1127
Csak, T., Ganz, M., Pespisa, J., Kodys, K., Dolganiuc, A., and Szabo, G. (2011). Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 54 (1), 133–144. doi:10.1002/hep.24341
Dai, Z., Ramesh, V., and Locasale, J. W. (2020). The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 21 (12), 737–753. doi:10.1038/s41576-020-0270-8
De Backer, J., Maric, D., Zuhra, K., Bogaerts, A., Szabo, C., Vanden Berghe, W., et al. (2022). Cytoglobin silencing promotes melanoma malignancy but sensitizes for ferroptosis and pyroptosis therapy response. Antioxidants (Basel). 11 (8). doi:10.3390/antiox11081548
Delerive, P., De Bosscher, K., Besnard, S., Vanden Berghe, W., Peters, J. M., Gonzalez, F. J., et al. (1999). Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 274 (45), 32048–32054. doi:10.1074/jbc.274.45.32048
Denechaud, P. D., Lopez-Mejia, I. C., Giralt, A., Lai, Q., Blanchet, E., Delacuisine, B., et al. (2015). E2F1 mediates sustained lipogenesis and contributes to hepatic steatosis. J. Clin. Investigation 126 (1), 137–150. doi:10.1172/JCI81542
Deng, M., Sun, S., Zhao, R., Guan, R., Zhang, Z., Li, S., et al. (2022). The pyroptosis-related gene signature predicts prognosis and indicates immune activity in hepatocellular carcinoma. Mol. Med. 28 (1), 16. doi:10.1186/s10020-022-00445-0
Ding, R. B., Bao, J., and Deng, C. X. (2017). Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 13 (7), 852–867. doi:10.7150/ijbs.19370
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29 (1), 15–21. doi:10.1093/bioinformatics/bts635
Dong, X. C. (2023). Sirtuin 6—a key regulator of hepatic lipid metabolism and liver Health. Cells 12 (4), 663. doi:10.3390/cells12040663
Ehara, T., Kamei, Y., Yuan, X., Takahashi, M., Kanai, S., Tamura, E., et al. (2015). Ligand-activated PPARα-dependent DNA demethylation regulates the fatty acid β-oxidation genes in the postnatal liver. Diabetes 64 (3), 775–784. doi:10.2337/db14-0158
Fang, Y. L., Chen, H., Wang, C. L., and Liang, L. (2018). Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: from ‘two hit theory’ to ‘multiple hit model. World J. Gastroenterol. 24 (27), 2974–2983. doi:10.3748/wjg.v24.i27.2974
Francque, S., Szabo, G., Abdelmalek, M. F., Byrne, C. D., Cusi, K., Dufour, J. F., et al. (2021). Nonalcoholic steatohepatitis: the role of peroxisome proliferator-activated receptors. Nat. Rev. Gastroenterol. Hepatol. 18 (1), 24–39. doi:10.1038/s41575-020-00366-5
Francque, S., Verrijken, A., Caron, S., Prawitt, J., Paumelle, R., Derudas, B., et al. (2015). PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 63 (1), 164–173. doi:10.1016/j.jhep.2015.02.019
Gao, Y., Zhang, W., Zeng, L. Q., Bai, H., Li, J., Zhou, J., et al. (2020). Exercise and dietary intervention ameliorate high-fat diet-induced NAFLD and liver aging by inducing lipophagy. Redox Biol. 36, 101635. doi:10.1016/j.redox.2020.101635
Gaul, S., Leszczynska, A., Alegre, F., Kaufmann, B., Johnson, C. D., Adams, L. A., et al. (2021). Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 74 (1), 156–167. doi:10.1016/j.jhep.2020.07.041
Gonzalez-Sanchez, E., Firrincieli, D., Housset, C., and Chignard, N. (2017). Expression patterns of nuclear receptors in parenchymal and non-parenchymal mouse liver cells and their modulation in cholestasis. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1863 (7), 1699–1708. doi:10.1016/j.bbadis.2017.04.004
Grabacka, M., Pierzchalska, M., Płonka, P. M., and Pierzchalski, P. (2021). The role of PPAR alpha in the modulation of innate immunity. Int. J. Mol. Sci. 22 (19).
Guerra, I. M. S., Ferreira, H. B., Melo, T., Rocha, H., Moreira, S., Diogo, L., et al. (2022). Mitochondrial fatty acid β-oxidation disorders: from disease to lipidomic studies-A critical review. Int. J. Mol. Sci. 23 (22).
Hardy, T., Zeybel, M., Day, C. P., Dipper, C., Masson, S., McPherson, S., et al. (2017). Plasma DNA methylation: a potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 66 (7), 1321–1328. doi:10.1136/gutjnl-2016-311526
Hashimoto, K., and Ogawa, Y. (2018). Epigenetic switching and neonatal nutritional environment. Adv. Exp. Med. Biol. 1012, 19–25. doi:10.1007/978-981-10-5526-3_3
Hassannia, B., Wiernicki, B., Ingold, I., Qu, F., Van Herck, S., Tyurina, Y. Y., et al. (2018). Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Invest. 128 (8), 3341–3355. doi:10.1172/JCI99032
Heidenreich, S., Witte, N., Weber, P., Goehring, I., Tolkachov, A., von Loeffelholz, C., et al. (2017). Retinol saturase coordinates liver metabolism by regulating ChREBP activity. Nat. Commun. 8 (1), 384. doi:10.1038/s41467-017-00430-w
Hervouet, E., Vallette, F. M., and Cartron, P. F. (2010). Dnmt1/Transcription factor interactions: an alternative mechanism of DNA methylation inheritance. Genes Cancer 1 (5), 434–443. doi:10.1177/1947601910373794
Hou, T., Tian, Y., Cao, Z., Zhang, J., Feng, T., Tao, W., et al. (2022). Cytoplasmic SIRT6-mediated ACSL5 deacetylation impedes nonalcoholic fatty liver disease by facilitating hepatic fatty acid oxidation. Mol. Cell 82 (21), 4099–4115.e9. doi:10.1016/j.molcel.2022.09.018
Hurtado-Navarro, L., Angosto-Bazarra, D., Pelegrín, P., Baroja-Mazo, A., and Cuevas, S. (2022). NLRP3 inflammasome and pyroptosis in liver pathophysiology: the emerging relevance of Nrf2 inducers. Antioxidants (Basel) 11 (5), 870. doi:10.3390/antiox11050870
Hyun, J., and Jung, Y. (2020). DNA methylation in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 21 (21), 8138. doi:10.3390/ijms21218138
Juanola, O., Martínez-López, S., Francés, R., and Gómez-Hurtado, I. (2021). Non-alcoholic fatty liver disease: metabolic, genetic, epigenetic and environmental risk factors. Int. J. Environ. Res. Public Health 18 (10), 5227. doi:10.3390/ijerph18105227
Kebede, A. F., Nieborak, A., Shahidian, L. Z., Le Gras, S., Richter, F., Gómez, D. A., et al. (2017). Histone propionylation is a mark of active chromatin. Nat. Struct. Mol. Biol. 24 (12), 1048–1056. doi:10.1038/nsmb.3490
Keleher, M. R., Zaidi, R., Hicks, L., Shah, S., Xing, X., Li, D., et al. (2018). A high-fat diet alters genome-wide DNA methylation and gene expression in SM/J mice. BMC Genomics 19 (1), 888. doi:10.1186/s12864-018-5327-0
Kersten, S., and Stienstra, R. (2017). The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 136, 75–84. doi:10.1016/j.biochi.2016.12.019
Kisseleva, T., and Brenner, D. A. (2007). Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J. Gastroenterol. Hepatol. 22 (1), S73–S78. doi:10.1111/j.1440-1746.2006.04658.x
Kitamoto, T., Kitamoto, A., Ogawa, Y., Honda, Y., Imajo, K., Saito, S., et al. (2015). Targeted-bisulfite sequence analysis of the methylation of CpG islands in genes encoding PNPLA3, SAMM50, and PARVB of patients with non-alcoholic fatty liver disease. J. Hepatol. 63 (2), 494–502. doi:10.1016/j.jhep.2015.02.049
Kleiner, D. E., Brunt, E. M., van Natta, M., Behling, C., Contos, M. J., Cummings, O. W., et al. (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41 (6), 1313–1321. doi:10.1002/hep.20701
Knorr, J., Wree, A., and Feldstein, A. E. (2022). Pyroptosis in steatohepatitis and liver diseases. J. Mol. Biol. 434 (4), 167271. doi:10.1016/j.jmb.2021.167271
Kuramoto, J., Arai, E., Fujimoto, M., Tian, Y., Yamada, Y., Yotani, T., et al. (2022). Quantification of DNA methylation for carcinogenic risk estimation in patients with non-alcoholic steatohepatitis. Clin. Epigenetics 14 (1), 168. doi:10.1186/s13148-022-01379-4
Lai, Z., Chen, J., Ding, C., Wong, K., Chen, X., Pu, L., et al. (2020). Association of hepatic global DNA methylation and serum one-carbon metabolites with histological severity in patients with NAFLD. Obes. (Silver Spring. 28 (1), 197–205. doi:10.1002/oby.22667
Lange, N. F., Graf, V., Caussy, C., and Dufour, J. F. (2022). PPAR-targeted therapies in the treatment of non-alcoholic fatty liver disease in diabetic patients. Int. J. Mol. Sci. 23 (8), 4305. doi:10.3390/ijms23084305
Li, L., Fu, J., Liu, D., Sun, J., Hou, Y., Chen, C., et al. (2020). Hepatocyte-specific Nrf2 deficiency mitigates high-fat diet-induced hepatic steatosis: involvement of reduced PPARγ expression. Redox Biol. 30, 101412. doi:10.1016/j.redox.2019.101412
Li, X., Wang, J., Wang, L., Gao, Y., Feng, G., Li, G., et al. (2022). Lipid metabolism dysfunction induced by age-dependent DNA methylation accelerates aging. Signal Transduct. Target Ther. 7 (1), 162. doi:10.1038/s41392-022-00964-6
Li, Y. Y., Tang, D., Du, Y. L., Cao, C. Y., Nie, Y. Q., Cao, J., et al. (2018). Fatty liver mediated by peroxisome proliferator-activated receptor-α DNA methylation can be reversed by a methylation inhibitor and curcumin. J. Dig. Dis. 19 (7), 421–430. doi:10.1111/1751-2980.12610
Loomba, R., Gindin, Y., Jiang, Z., Lawitz, E., Caldwell, S., Djedjos, C. S., et al. (2018). DNA methylation signatures reflect aging in patients with nonalcoholic steatohepatitis. JCI Insight 3 (2), e96685. doi:10.1172/jci.insight.96685
Loomba, R., and Sanyal, A. J. (2013). The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 10 (11), 686–690. doi:10.1038/nrgastro.2013.171
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi:10.1186/s13059-014-0550-8
Luo, Y., Xie, C., Brocker, C. N., Fan, J., Wu, X., Feng, L., et al. (2019). Intestinal PPARα protects against colon carcinogenesis via regulation of methyltransferases DNMT1 and PRMT6. Gastroenterology 157 (3), 744–759.e4. doi:10.1053/j.gastro.2019.05.057
Lyall, M., Thomson, J., Cartier, J., Cameron, K., Hay, D., Meehan, R., et al. (2016). The role of DNA hydroxymethylation in non-alcoholic fatty liver disease. Endocr. Abstr. 2016, 45. doi:10.1530/endoabs.45.oc8.2
Lyall, M. J., Thomson, J. P., Cartier, J., Ottaviano, R., Kendall, T. J., Meehan, R. R., et al. (2020). Non-alcoholic fatty liver disease (NAFLD) is associated with dynamic changes in DNA hydroxymethylation. Epigenetics 15 (1–2), 61–71. doi:10.1080/15592294.2019.1649527
Matsumoto, M., Hada, N., Sakamaki, Y., Uno, A., Shiga, T., Tanaka, C., et al. (2013). An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 94 (2), 93–103. doi:10.1111/iep.12008
Melton, P. E., Burton, M. A., Lillycrop, K. A., Godfrey, K. M., Rauschert, S., Anderson, D., et al. (2023). Differential DNA methylation of steatosis and non-alcoholic fatty liver disease in adolescence. Hepatol. Int. 17 (3), 584–594. doi:10.1007/s12072-022-10469-7
Minami, J. K., Morrow, D., Bayley, N. A., Fernandez, E. G., Salinas, J. J., Tse, C., et al. (2023). CDKN2A deletion remodels lipid metabolism to prime glioblastoma for ferroptosis. Cancer Cell 41, 1048–1060.e9. doi:10.1016/j.ccell.2023.05.001
Mohs, A., Otto, T., Schneider, K. M., Peltzer, M., Boekschoten, M., Holland, C. H., et al. (2021). Hepatocyte-specific NRF2 activation controls fibrogenesis and carcinogenesis in steatohepatitis. J. Hepatol. 74 (3), 638–648. doi:10.1016/j.jhep.2020.09.037
Moody, L., Xu, G. B., Chen, H., and Pan, Y. X. (2019). Epigenetic regulation of carnitine palmitoyltransferase 1 (Cpt1a) by high fat diet. Biochimica Biophysica Acta (BBA) - Gene Regul. Mech. 1862 (2), 141–152. doi:10.1016/j.bbagrm.2018.12.009
Mukai, T., Egawa, M., Takeuchi, T., Yamashita, H., and Kusudo, T. (2017). Silencing of FABP 1 ameliorates hepatic steatosis, inflammation, and oxidative stress in mice with nonalcoholic fatty liver disease. FEBS Open Bio 7 (7), 1009–1016. doi:10.1002/2211-5463.12240
Murphy, S. K., Yang, H., Moylan, C. A., Pang, H., Dellinger, A., Abdelmalek, M. F., et al. (2013). Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 145 (5), 1076–1087. doi:10.1053/j.gastro.2013.07.047
Mwinyi, J., Boström, A. E., Pisanu, C., Murphy, S. K., Erhart, W., Schafmayer, C., et al. (2017). NAFLD is associated with methylation shifts with relevance for the expression of genes involved in lipoprotein particle composition. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862 (3), 314–323. doi:10.1016/j.bbalip.2016.12.005
Ni, Y. H., Wang, R., Wang, W., Li, D. Z., Liu, G., Jiang, C. S., et al. (2023). Tcf21 alleviates pancreatic fibrosis by regulating the epithelial-mesenchymal transformation of pancreatic stellate cells. Dig. Dis. Sci. 68, 3032–3042. doi:10.1007/s10620-023-07849-w
Nieborak, A., and Schneider, R. (2018). Metabolic intermediates - cellular messengers talking to chromatin modifiers. Mol. Metab. 14, 39–52. doi:10.1016/j.molmet.2018.01.007
Niu, L., Xu, Z., and Taylor, J. A. (2016). RCP: a novel probe design bias correction method for Illumina Methylation BeadChip. Bioinformatics 32, 2659–2663. doi:10.1093/bioinformatics/btw285
Ore, A., and Akinloye, O. A. (2021). Phytotherapy as multi-hit therapy to confront the multiple pathophysiology in non-alcoholic fatty liver disease: a systematic review of experimental interventions. Med. Kaunas. 57 (8), 822. doi:10.3390/medicina57080822
Pan, Q., Luo, Y., Xia, Q., and He, K. (2021). Ferroptosis and liver fibrosis. Int. J. Med. Sci. 18 (15), 3361–3366. doi:10.7150/ijms.62903
Pang, X. Y., Wang, S., Jurczak, M. J., Shulman, G. I., and Moise, A. R. (2017). Retinol saturase modulates lipid metabolism and the production of reactive oxygen species. Arch. Biochem. Biophys. 633, 93–102. doi:10.1016/j.abb.2017.09.009
Pougovkina, O., te Brinke, H., Ofman, R., van Cruchten, A. G., Kulik, W., Wanders, R. J. A., et al. (2014). Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum. Mol. Genet. 23 (13), 3513–3522. doi:10.1093/hmg/ddu059
Qiu, Y., Shi, Y. N., Zhu, N., Zhang, S., Zhang, C. J., Gu, J., et al. (2023). A lipid perspective on regulated pyroptosis. Int. J. Biol. Sci. 19 (8), 2333–2348. doi:10.7150/ijbs.81017
Régnier, M., Polizzi, A., Smati, S., Lukowicz, C., Fougerat, A., Lippi, Y., et al. (2020). Hepatocyte-specific deletion of Pparα promotes NAFLD in the context of obesity. Sci. Rep. 10 (1), 6489. doi:10.1038/s41598-020-63579-3
Remely, M., Ferk, F., Sterneder, S., Setayesh, T., Kepcija, T., Roth, S., et al. (2017). Vitamin E modifies high-fat diet-induced increase of DNA strand breaks, and changes in expression and DNA methylation of Dnmt1 and MLH1 in C57bl/6J male mice. Nutrients 9 (6), 607. doi:10.3390/nu9060607
Ren, Y. (2022). Regulatory mechanism and biological function of UHRF1–DNMT1-mediated DNA methylation. Funct. Integr. Genomics 22 (6), 1113–1126. doi:10.1007/s10142-022-00918-9
Rinella, M. E., Lazarus, J. V., Ratziu, V., Francque, S. M., Sanyal, A. J., Kanwal, F., et al. (2023). A multi-society Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 79, 1542–1556. doi:10.1016/j.jhep.2023.06.003
Rom, O., Liu, Y., Liu, Z., Zhao, Y., Wu, J., Ghrayeb, A., et al. (2020). Glycine-based treatment ameliorates NAFLD by modulating fatty acid oxidation, glutathione synthesis, and the gut microbiome. Sci. Transl. Med. 12 (572), eaaz2841. doi:10.1126/scitranslmed.aaz2841
Romero-Gómez, M., Zelber-Sagi, S., and Trenell, M. (2017). Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 67 (4), 829–846. doi:10.1016/j.jhep.2017.05.016
Sabari, B. R., Zhang, D., Allis, C. D., and Zhao, Y. (2017). Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 18 (2), 90–101. doi:10.1038/nrm.2016.140
Sharma, R. S., Harrison, D. J., Kisielewski, D., Cassidy, D. M., McNeilly, A. D., Gallagher, J. R., et al. (2018). Experimental nonalcoholic steatohepatitis and liver fibrosis are ameliorated by pharmacologic activation of Nrf2 (NF-E2 p45-related factor 2). Cell Mol. Gastroenterol. Hepatol. 5 (3), 367–398. doi:10.1016/j.jcmgh.2017.11.016
Sokolowska, K. E., Maciejewska-Markiewicz, D., Bińkowski, J., Palma, J., Taryma-Leśniak, O., Kozlowska-Petriczko, K., et al. (2022). Identified in blood diet-related methylation changes stratify liver biopsies of NAFLD patients according to fibrosis grade. Clin. Epigenetics 14 (1), 157. doi:10.1186/s13148-022-01377-6
Soltis, A. R., Kennedy, N. J., Xin, X., Zhou, F., Ficarro, S. B., Yap, Y. S., et al. (2017). Hepatic dysfunction caused by consumption of a high-fat diet. Cell Rep. 21 (11), 3317–3328. doi:10.1016/j.celrep.2017.11.059
Stepanova, M., and Younossi, Z. M. (2012). Independent association between nonalcoholic fatty liver disease and cardiovascular disease in the US population. Clin. Gastroenterology Hepatology 10 (6), 646–650. doi:10.1016/j.cgh.2011.12.039
Sukur, G., Uysal, F., and Cinar, O. (2023). High-fat diet induced obesity alters Dnmt1 and Dnmt3a levels and global DNA methylation in mouse ovary and testis. Histochem Cell Biol. 159 (4), 339–352. doi:10.1007/s00418-022-02173-2
Sumida, Y., and Yoneda, M. (2018). Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 53 (3), 362–376. doi:10.1007/s00535-017-1415-1
Sun, Q. F., Tang, L. J., Wang, M. J., Zhu, P. W., Li, Y. Y., Ma, H. L., et al. (2022). Potential blood DNA methylation biomarker genes for diagnosis of liver fibrosis in patients with biopsy-proven non-alcoholic fatty liver disease. Front. Med. (Lausanne). 9, 864570. doi:10.3389/fmed.2022.864570
Tayyeb, J. Z., Popeijus, H. E., Mensink, R. P., Konings, MCJM, Mokhtar, F. B. A., and Plat, J. (2020). Short-Chain fatty acids (except hexanoic acid) lower NF-kB transactivation, which rescues inflammation-induced decreased apolipoprotein A-I transcription in HepG2 cells. Int. J. Mol. Sci. 21 (14), 5088. doi:10.3390/ijms21145088
Theys, C., Lauwers, D., Perez-Novo, C., and Vanden Berghe, W. (2022). PPARα in the epigenetic driver seat of NAFLD: new therapeutic opportunities for epigenetic drugs? Biomedicines 10 (12).
Tiffon, C. (2018). The impact of nutrition and environmental epigenetics on human Health and disease. Int. J. Mol. Sci. 19 (11), 3425. doi:10.3390/ijms19113425
Tsurusaki, S., Tsuchiya, Y., Koumura, T., Nakasone, M., Sakamoto, T., Matsuoka, M., et al. (2019). Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 10 (6), 449. doi:10.1038/s41419-019-1678-y
Unoki, M., Nishidate, T., and Nakamura, Y. (2004). ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene 23 (46), 7601–7610. doi:10.1038/sj.onc.1208053
Vilar-Gomez, E., Martinez-Perez, Y., Calzadilla-Bertot, L., Torres-Gonzalez, A., Gra-Oramas, B., Gonzalez-Fabian, L., et al. (2015). Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 149 (2), 367–378. doi:10.1053/j.gastro.2015.04.005
Walczak, R., and Tontonoz, P. (2002). PPARadigms and PPARadoxes: expanding roles for PPARγ in the control of lipid metabolism. J. Lipid Res. 43 (2), 177–186. doi:10.1016/s0022-2275(20)30159-0
Wang, J., Zhang, Y., Zhuo, Q., Tseng, Y., Wang, J., Ma, Y., et al. (2020). TET1 promotes fatty acid oxidation and inhibits NAFLD progression by hydroxymethylation of PPARα promoter. Nutr. Metab. (Lond). 17 (1), 46. doi:10.1186/s12986-020-00466-8
Wang, Z., Bao, A., Liu, S., Dai, F., Gong, Y., and Cheng, Y. (2022). A pyroptosis-related gene signature predicts prognosis and immune microenvironment for breast cancer based on computational biology techniques. Front. Genet. 13, 801056. doi:10.3389/fgene.2022.801056
Watanabe, A., Sohail, M. A., Gomes, D. A., Hashmi, A., Nagata, J., Sutterwala, F. S., et al. (2009). Inflammasome-mediated regulation of hepatic stellate cells. Am. J. Physiology-Gastrointestinal Liver Physiology 296 (6), G1248–G1257. doi:10.1152/ajpgi.90223.2008
Wegermann, K., Henao, R., Diehl, A. M., Murphy, S. K., Abdelmalek, M. F., and Moylan, C. A. (2018). Branched chain amino acid transaminase 1 (BCAT1) is overexpressed and hypomethylated in patients with non-alcoholic fatty liver disease who experience adverse clinical events: a pilot study. PLoS One 13 (9), e0204308. doi:10.1371/journal.pone.0204308
Wree, A., Eguchi, A., McGeough, M. D., Pena, C. A., Johnson, C. D., Canbay, A., et al. (2014a2014a). NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 59 (3), 898–910. doi:10.1002/hep.26592
Wree, A., McGeough, M. D., Peña, C. A., Schlattjan, M., Li, H., Inzaugarat, M. E., et al. (2014b). NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 92 (10), 1069–1082. doi:10.1007/s00109-014-1170-1
Wu, H., Wang, F., Ta, N., Zhang, T., and Gao, W. (2021). The multifaceted regulation of mitochondria in ferroptosis. Life 11 (3), 222. doi:10.3390/life11030222
Wu, J., Lin, S., Wan, B., Velani, B., and Zhu, Y. (2019). Pyroptosis in liver disease: new insights into disease mechanisms. Aging Dis. 10 (5), 1094–1108. doi:10.14336/AD.2019.0116
Xiao, Z., Liu, M., Yang, F., Liu, G., Liu, J., Zhao, W., et al. (2023). Programmed cell death and lipid metabolism of macrophages in NAFLD. Front. Immunol. 14, 1118449. doi:10.3389/fimmu.2023.1118449
Xing, G., Meng, L., Cao, S., Liu, S., Wu, J., Li, Q., et al. (2022). PPARα alleviates iron overload-induced ferroptosis in mouse liver. EMBO Rep. 23 (8), e52280. doi:10.15252/embr.202052280
Xu, Z., Niu, L., Li, L., and Taylor, J. A. (2016). ENmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 44 (3), e20. doi:10.1093/nar/gkv907
Yan, C., Zhang, Y., Zhang, X., Aa, J., Wang, G., and Xie, Y. (2018). Curcumin regulates endogenous and exogenous metabolism via Nrf2-FXR-LXR pathway in NAFLD mice. Biomed. Pharmacother. 105, 274–281. doi:10.1016/j.biopha.2018.05.135
Ye, Y., Dai, Q., and Qi, H. (2021). A novel defined pyroptosis-related gene signature for predicting the prognosis of ovarian cancer. Cell Death Discov. 7 (1), 71. doi:10.1038/s41420-021-00451-x
Younossi, Z. M., Koenig, A. B., Abdelatif, D., Fazel, Y., Henry, L., and Wymer, M. (2016). Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64 (1), 73–84. doi:10.1002/hep.28431
Yuan, X., Tsujimoto, K., Hashimoto, K., Kawahori, K., Hanzawa, N., Hamaguchi, M., et al. (2018). Epigenetic modulation of Fgf21 in the perinatal mouse liver ameliorates diet-induced obesity in adulthood. Nat. Commun. 9 (1), 636. doi:10.1038/s41467-018-03038-w
Zeybel, M., Hardy, T., Robinson, S. M., Fox, C., Anstee, Q. M., Ness, T., et al. (2015). Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin. Epigenetics 7 (1), 25. doi:10.1186/s13148-015-0056-6
Zhang, H., Zhang, E., and Hu, H. (2021). Role of ferroptosis in non-alcoholic fatty liver disease and its implications for therapeutic strategies. Biomedicines 9 (11), 1660. doi:10.3390/biomedicines9111660
Zhou, N., Yuan, X., Du, Q., Zhang, Z., Shi, X., Bao, J., et al. (2023). FerrDb V2: update of the manually curated database of ferroptosis regulators and ferroptosis-disease associations. Nucleic Acids Res. 51 (D1), D571–D582. doi:10.1093/nar/gkac935
Zhou, Y., Zhou, B., Pache, L., Chang, M., Khodabakhshi, A. H., Tanaseichuk, O., et al. (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10 (1), 1523. doi:10.1038/s41467-019-09234-6
Zirath, H., Frenzel, A., Oliynyk, G., Segerström, L., Westermark, U. K., Larsson, K., et al. (2013). MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl. Acad. Sci. 110 (25), 10258–10263. doi:10.1073/pnas.1222404110
Zúñiga, J., Cancino, M., Medina, F., Varela, P., Vargas, R., Tapia, G., et al. (2011). N-3 PUFA supplementation triggers PPAR-α activation and PPAR-α/NF-κB interaction: anti-inflammatory implications in liver ischemia-reperfusion injury. PLoS One 6 (12), e28502. doi:10.1371/journal.pone.0028502
Keywords: PPARα, MASLD, epigenetics, lipid metabolism, bile acid metabolism, NAFLD, ferroptosis, pyroptosis
Citation: Theys C, Vanderhaeghen T, Van Dijck E, Peleman C, Scheepers A, Ibrahim J, Mateiu L, Timmermans S, Vanden Berghe T, Francque SM, Van Hul W, Libert C and Vanden Berghe W (2024) Loss of PPARα function promotes epigenetic dysregulation of lipid homeostasis driving ferroptosis and pyroptosis lipotoxicity in metabolic dysfunction associated Steatotic liver disease (MASLD). Front. Mol. Med. 3:1283170. doi: 10.3389/fmmed.2023.1283170
Received: 25 August 2023; Accepted: 14 December 2023;
Published: 08 January 2024.
Edited by:
Ruben Fernandes, Fernando Pessoa University, PortugalReviewed by:
Kishor Pant, University of Minnesota Twin Cities, United StatesCopyright © 2024 Theys, Vanderhaeghen, Van Dijck, Peleman, Scheepers, Ibrahim, Mateiu, Timmermans, Vanden Berghe, Francque, Van Hul, Libert and Vanden Berghe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Theys, Q2xhdWRpYS50aGV5c0B1YW50d2VycGVuLmJl; Wim Vanden Berghe, d2ltLnZhbmRlbmJlcmdoZUB1YW50d2VycGVuLmJl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.