 Katarina Stoklund Dittlau
Katarina Stoklund Dittlau Ludo Van Den Bosch
Ludo Van Den Bosch
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Med, 23 January 2023
Sec. Molecular Mechanisms of Neurodegeneration
Volume 3 - 2023 | https://doi.org/10.3389/fmmed.2023.1047540
This article is part of the Research TopicThe Role of Glial Cells in NeurodegenerationView all 9 articles
Amyotrophic lateral sclerosis (ALS) is the most common motor neuron disease in adults, causing progressive degeneration of motor neurons, which results in muscle atrophy, respiratory failure and ultimately death of the patients. The pathogenesis of ALS is complex, and extensive efforts have focused on unravelling the underlying molecular mechanisms with a large emphasis on the dying motor neurons. However, a recent shift in focus towards the supporting glial population has revealed a large contribution and influence in ALS, which stresses the need to explore this area in more detail. Especially studies into astrocytes, the residential homeostatic supporter cells of neurons, have revealed a remarkable astrocytic dysfunction in ALS, and therefore could present a target for new and promising therapeutic entry points. In this review, we provide an overview of general astrocyte function and summarize the current literature on the role of astrocytes in ALS by categorizing the potentially underlying molecular mechanisms. We discuss the current efforts in astrocyte-targeted therapy, and highlight the potential and shortcomings of available models.
Amyotrophic lateral sclerosis (ALS) is the most common motor neuron disorder in adults. The annual incidence rate ranges from 0.6–1.8 per 100.000 people, while the annual prevalence rate is around 4-8 per 100.000 persons with a higher occurrence in males compared to females (Adamczyk et al., 2021; Agbas et al., 2021). The number of cases varies demographically and a higher prevalence is predominately found in the Western parts of the world (Adamczyk et al., 2021). ALS causes selective and progressive loss of upper motor neurons present in the motor cortex of the brain and lower motor neurons from the brainstem and the ventral horn of the spinal cord. The loss of the upper motor neurons results in spasticity and hyperreflexia, while degeneration of lower motor neurons leads to spontaneous muscle twitching or fasciculations and ultimately to muscle atrophy and weakness (Brown and Al-Chalabi, 2017). Although the clinical presentation is variable, for the majority of patients symptoms usually start at the periphery affecting distal limbs such as hands and feet, but will soon spread to include larger muscle groups, eventually rendering patients wheelchair bound (Adamczyk et al., 2021). The disorder has a rapid disease progression which limits median survival after symptom onset to 2–5 years mostly due to respiratory failure (Masrori and Van Damme, 2020). ALS is a familial disease in 10% of cases (Figure 1A) and in the majority of familial ALS (fALS) the causative gene is known (Renton et al., 2014). The inheritance pattern is usually autosomal dominant, and mutations in the superoxide dismutase 1 (SOD1) gene, in the TAR DNA binding protein (TARDBP) gene or in the FUS RNA binding protein (FUS) gene are important genetic causes of ALS (Al-Chalabi et al., 2012). The more recently discovered hexanucleotide repeat expansions in the Chromosome 9 open reading frame 72 (C9orf72) gene are by far the most common genetic causes of ALS as these repeat expansions instigate 30%–50% of fALS (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Van Blitterswijk et al., 2012). In 90% of cases, ALS is a sporadic disease clinically indistinguishable from familial forms, but without a familial inheritance pattern. The etiology of sporadic ALS (sALS) remains elusive, but it is thought to arise from a combination of aging, as well as largely undetermined genetic and environmental factors (Al-Chalabi and Hardiman, 2013; Shatunov and Al-Chalabi, 2021). Interestingly, some risk factors such as history of head trauma, electrocution, lead expose and smoking have been associated with a higher chance of developing ALS (Wang et al., 2017; Andrew et al., 2021), but further research is needed to confirm this. Although ALS is considered an adult-onset disease with an average age of onset around middle-to-late 50s, ALS presents itself rather heterogeneous in terms of age of disease onset. As a result, both juvenile and geriatric cases exist (Shang and Huang, 2016). Currently no effective treatment is available for ALS. Despite decades of research into the complex pathophysiology of ALS, dozens of compounds have failed to show any significant impact on disease during clinical trials despite promising preclinical results. Three drugs have been approved by the Food and Drug Administration (FDA); Riluzole, Edaravone and AMX0035 (Petrov et al., 2017; Paganoni et al., 2020; 2021), while only Riluzole is approved by the European Medicines Agency (EMA). Of the three drugs, Riluzole appears to have the strongest albeit limited effect on survival, and most treatment measures therefore rely on symptomatic and palliative care in order to improve the quality of life of patients (Agbas et al., 2021).
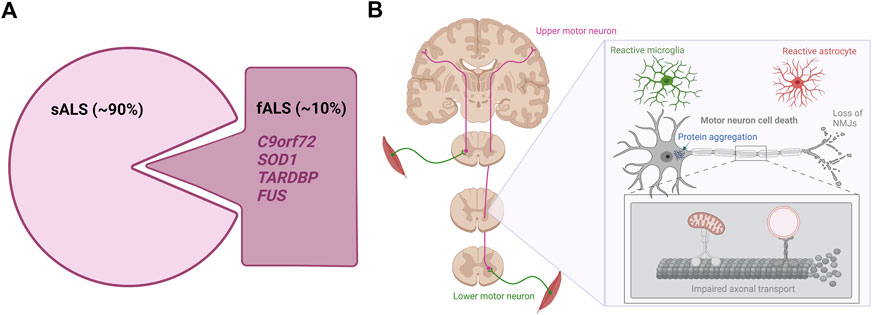
FIGURE 1. The genetics and hallmarks of ALS. (A). In 90% of cases, ALS is a sporadic disease with no family history. The remaining 10% are caused by familial mutations mainly found in the C9orf72, SOD1, TARDBP, and FUS genes (B). ALS affects the upper and lower motor neurons in the brain and spinal cord. Currently hallmarks of ALS disease include motor neuron cell death, protein aggregation, distal motor neuron-muscle disconnection/NMJ loss, axonal transport impairments and reactive gliosis. Figure is created with Biorender.com.
Despite being a complex disorder, certain pathological hallmarks exist (Figure 1B). Apart from the striking motor neuron death evident in all ALS cases, abnormal protein inclusions are found in the cytoplasm of surviving motor neurons in the vast majority of ALS patients. Especially the TDP-43 proteinopathy is one of these hallmarks, as 97% of all ALS cases present histologically with TDP-43-positive inclusions independent of TARDBP mutations (Neumann et al., 2006; Tziortzouda et al., 2021). These inclusions are not limited to the motor neurons, but are likewise found in glial cells throughout the brain and spinal cord emphasizing a broader involvement of different cell types in ALS (Tziortzouda et al., 2021). Additionally, signs of ALS emerge when connections between lower motor neurons and muscle fail. Several studies in animal models and patients show how nerve terminals and neuromuscular junctions (NMJs) are partially degraded in early stages of the disease, while the motor neuron cell bodies in the spinal cord are mostly intact (Fischer et al., 2004; Nair et al., 2010; Walker et al., 2015; Tallon et al., 2016; Martineau et al., 2018; So et al., 2018; Picchiarelli et al., 2019). Upon axonal retraction, the motor neuron initially compensates by sprouting and collateral re-innervation by new axons. However, when the disease progresses, the motor neuron is no longer able to continuously compensate for the retracting axons and it finally dies (Robberecht and Philips, 2013). Symptoms will only occur when large populations of motor neurons are affected, resulting in weakness of muscle groups. Recent findings suggest that axonal transport deficiencies are a common mechanism in ALS as impaired axonal transport of mitochondria, mRNA and endosomes in motor neurons harbouring mutations in FUS, TARDBP and C9orf72 has so far been demonstrated (Alami et al., 2014; Guo, et al., 2017a; Fazal et al., 2021; Fumagalli et al., 2021). Since motor neurons have very long axons, they are more susceptible to axonal retraction and axonal transport dysfunctions than other neurons, further explaining the selective vulnerability in ALS pathogenesis (Robberecht and Philips, 2013). Finally, abnormal glial activation (popularly defined as “gliosis”) is extensively found throughout the brain and spinal cord of both fALS and sALS cases (Agbas et al., 2021; Rossi and Cozzolino, 2021), and especially the involvement of astrocytes will be discussed later in this review.
Despite the fact that ALS is considered a motor neuron disorder, it is increasingly recognized that ALS does not only affect this subpopulation of neurons, but in fact also has a striking influence on other neuronal subtypes as well as on the glial population including astrocytes. Astrocytes are one of the most abundant glial cell types in the central nervous system (CNS) as the glial population is composed of approximately 20%–40% astrocytes (Allen and Lyons, 2018; Verkhratsky and Nedergaard, 2018). This cell type harbours great heterogeneity and is typically organized in highly specialized cellular-level domains, which are focused on the homeostatic needs of its residing region (Verkhratsky and Nedergaard, 2018). The domain diameter varies from <100 to >400 μm (Oberheim et al., 2009), and each astrocytes’ anatomical space is generally non-overlapping, although some interaction through gap junctions between neighbouring domain astrocyte processes is documented at the territorial peripheries (Bushong et al., 2002; 2004). As such, this domain formation could be a reason why atrophy due to neurodegeneration is often seen in specific areas of the brain and it has been hypothesized that neurodegeneration primarily affects astrocytes, which then causes a subsequent spreading to neurons through gap junctions (Sica et al., 2016). Whether this is also the case for ALS is debated.
In order to understand the astrocytic involvement in disease, we first have to understand their role in health. Although astrocytes have been recognized for their important roles in the CNS for decades, research continuously reveals their remarkable functional complexity. Astrocytes are found to be crucial mediators of neurotrophic support, neurotransmitter regulation, synapse plasticity and activity modulation, and for the structural integrity of the brain and spinal cord (Verkhratsky and Nedergaard, 2018). Besides this, astrocytes facilitate the maintenance of an optimal CNS environment through regulation of the blood brain barrier (BBB), water flux, ion and pH homeostasis, and removal of reactive oxygen species (ROS). Astrocytes likewise contribute to the inflammatory and immune response, as they secrete cytokines, phagocytize and facilitate border formation after injury. In the following section, key astrocyte functions will be described in detail.
Astrocytes modulate neuronal synapse plasticity and function through numerous mechanisms. In order to engage with the neuronal network, astrocytes extend their endfeet to form perisynaptic processes (PAPs), which ensheath neuronal synapses and form the so-called “tripartite synapse” (Figure 2). The degree of synapse enwrapping varies between brain regions and depends on synapse morphology. However, it is estimated that at least half of all CNS synapses are covered with astrocytic PAPs (Verkhratsky and Nedergaard, 2018). The astrocytic synapse-enveloping ensures removal of neurotransmitters such as glutamate from the interstitial space through receptor modulation, which counteracts neurotransmitter accumulation and subsequent excitotoxicity (Allen and Eroglu, 2017).
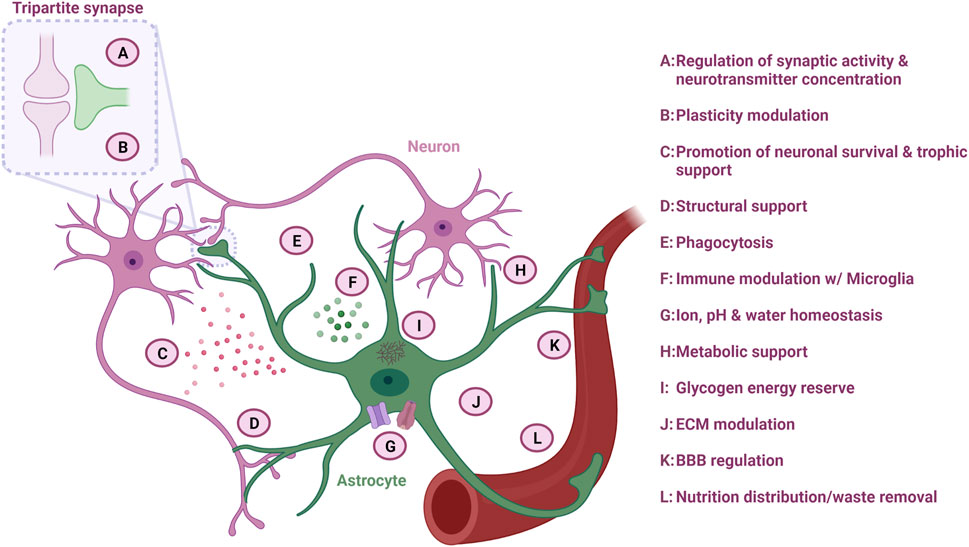
FIGURE 2. Astrocyte function in the CNS. Astrocytes are multifaceted cells with various important functions overall ensuring optimal neuronal homeostasis. Figure created with Biorender.com.
Another mechanism of synapse modulation is through secretion of certain molecules, which is especially important during synaptogenesis. Astrocytes secrete proteins and cytokines such as hevin (SPARCL1), thrombospondins (THBS1/2), glypicans (GPC4/6), transforming growth factor β1 (TGF-β1) and brain-derived neurotrophic factor (BDNF), which through transsynaptic adhesion proteins or receptor modulations promote the formation and maturation of excitatory synapses (Vainchtein and Molofsky, 2020; Liu et al., 2021). TGF-β1 has a dual function as this cytokine likewise promotes the formation of inhibitory synapses in the CNS (Liu et al., 2021). In addition, tumor necrosis factor-α (TNF-α) secreted from astrocytes also plays an important role as it increases the α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors on excitatory synapses, while it downregulates γ-aminobutyric acid (GABA) receptors on inhibitory synapses, overall increasing neuronal activity (Allen and Eroglu, 2017). To negatively regulate synaptogenesis, astrocytes also secrete signals such as SPARC in order to balance the process (Allen and Lyons, 2018).
Synaptic pruning is dependent on neuronal activity (Liu et al., 2021), and astrocytes are also involved in this process (Figure 2). Through direct activation of astrocytic phagocytic receptors Multiple EGF Like domains 10 (MEGF10) and MER proto-oncogene, tyrosine kinase (MERTK) on the astrocyte surface, astrocytes can degrade superfluous synapses in order to improve the precision and efficiency of neuronal networks (Allen and Lyons, 2018). In addition, astrocytes can indirectly modulate synapse pruning by secreting interleukin-33 (IL-33) or by activating the classical complement cascade and C1q expression through TGF-β secretion, which subsequently promotes selective microglia phagocytosis of the synapses destined for degradation (Vainchtein et al., 2018; Liu et al., 2021). As a consequence, astrocytes collaborate with microglia in the dynamic modulation of neuroplasticity, which is especially important for learning and memory.
Astrocytes control the ion homeostasis through multiple ion channels, which is important for maintaining synapse function (Figure 2) (Sibille et al., 2014; Allen and Eroglu, 2017). By regulating the potassium concentration in the extracellular space through clearance via potassium channels such as the main Kir4.1 type, astrocytes control whether neurons become depolarized after an action potential and thereby affects their ability to fire a signal again (Allen and Lyons, 2018). Especially in the spinal cord, astrocytes possess a large amount of Kir4.1 channels, which are important to accommodate the firing rate of large fast motor neurons (Allen and Lyons, 2018; Kelley et al., 2018). Astrocyte can also collaborate with other glial cells such as oligodendrocytes in the formation of a glial syncytium, which controls the potassium concentration and subsequent neuronal activity (Allen and Lyons, 2018).
One of the first recognized functions of astrocytes is their importance in structural support (Figure 2) (Verkhratsky and Nedergaard, 2018). Astrocytes are considered crucial for the integrity of the stroma in the CNS and secrete molecules such as proteoglycans, which are central for the extracellular matrix (ECM) structure within the synaptic cleft and surrounding synapses (Figure 2). This ECM structure ensures the capturing of nutrients and growth factors, as well as buffers the concentration of neurotransmitters to maintain acceptable levels by acting as a diffusion barrier (Liu et al., 2021). Astrocytes also form the glia limitans layer of the pia mater of the CNS through sheets, which contains aquaporin-4 (AQP4) water channels. Through these channels, astrocytes are able to regulate influx and efflux of water within the CNS, and additionally facilitate the glymphatic flow into the perivascular space, which ensures removal of excess fluid and metabolic waste products (Figure 2) (Verkhratsky and Nedergaard, 2018).
Neurons consume a vast amount of energy, and therefore rely on continues supply of energy substrates (Figure 2). In order to accommodate this need, astrocytes provide neurons with energy substrates through one of the key mechanisms: the astrocyte-neuron lactate shuttle. Neurons primarily use oxidative metabolism of glucose to produce large amounts of adenosine triphosphate (ATP), while astrocytes predominantly convert glucose to lactate via aerobic glycolysis resulting in a low ATP production (Verkhratsky and Nedergaard, 2018). The lactate is transferred to adjacent neurons via monocarboxylate transporters (Allen and Lyons, 2018), where the neuronal mitochondria use the lactate in the oxidative cycle for ATP production (Verkhratsky and Nedergaard, 2018). In addition, astrocytes can shuttle lactate from the blood stream directly to neurons (Verkhratsky and Nedergaard, 2018). Astrocytes are also an important player in the glycolytic pathway, as they are the sole processors of brain glycogen (Figure 2) (Verkhratsky and Nedergaard, 2018). During sleep, glycogen is stored, while activity causes glycogen release and metabolism, providing further energy for the cells (Verkhratsky and Nedergaard, 2018). Additionally, astrocytes are the main synthesizers and suppliers of apolipoprotein E (ApoE), and they ensure a sufficient transport of cholesterol to the neurons in the CNS via ApoE receptors (Liu et al., 2021). Cholesterol is an important lipid in membranes and is also essential for the function of glutamatergic neurons as it increases the amount of vesicles and their release probability in the presynapse (Allen and Eroglu, 2017).
Astrocytes are the gatekeepers of the CNS as they maintain the BBB through close interaction with the vasculature (Figure 2). As such, the capillary endothelial cells, which are wrapped by pericytes and the basal lamina, are closely surrounded by astrocytic endfeet processes (Harada et al., 2016). This ability of astrocytes to contact both vessels and neurons facilitates the shuttle of nutrients from the capillaries to the neurons and waste product from the neurons back to the blood stream. Astrocytes are also important in the development of the BBB, as they aid the alignment of endothelial cells and pericytes (Harada et al., 2016). Through the calcium-dependent release of vasodilators or vasoconstrictors such as prostanoids and arachidonic acid, astrocytes can control the blood flow depending on the neuronal energy demand (Liddelow and Barres, 2015).
In contrast to neurons, astrocytes are not able to project excitability through electrical action potentials, but rely on calcium waves to communicate (Liu et al., 2021). Between individual astrocytes and intracellularly, calcium transients move along processes through the Soma via calcium-permeable receptors and ion channels. Although calcium stores within the astrocytes’ endoplasmic reticulum account for a large part of the calcium communication, transport of extracellular calcium into the cells is also mediated by glutamate channels made of AMPA and N-methyl-D-aspartate (NMDA) receptors or voltage-gated calcium channels (Liu et al., 2021). Especially NMDA receptors are important for astrocytic-neuronal communication, as they are required for synapse formation and proper dendritic morphogenesis during development (Harada et al., 2016). In addition, gap junctions composed of connexin-43 (Cx43), which is the predominant connexin protein in astrocytes, allow transport of calcium between adjoining astrocytes (Harada et al., 2016).
Astrocytes express multiple receptors, which are responsive to a large variety of neurotransmitters. Many of these receptors are G protein-coupled, which activates an intracellular signal through calcium (Allen and Lyons, 2018). As such, astrocytes are able to distinguish between glutamatergic and cholinergic synaptic activity through the ability of the receptors to evoke a unique calcium response. Different types of calcium waves exist and the uptake requirements of extracellular calcium is dependent on the subpopulation of astrocytes (Clarke et al., 2021). Most transients are found at the processes, which correlates with the ability of increased calcium concentrations to release astrocytic transmitters such as glutamate, D-serine and ATP, which subsequently can regulate neuronal excitability and synaptic plasticity (Liu et al., 2021). Astrocytes also secrete GABA and glycine, which act as inhibitory transmitters in the brain and spinal cord, respectively (Verkhratsky and Nedergaard, 2018). These “gliotransmitters” are primarily released via vesicular exocytosis or through specific transporters and hemichannels.
Under physiological conditions, non-activated astrocytes are primarily tissue embedded and non-motile, firmly devoted to their catering to neuronal homeostasis within their specific domain (Vainchtein and Molofsky, 2020). Upon injury, disease or infection, some astrocytes become hypertrophic, extend their processes and together with microglia initiate an immune response through cytokine and interleukin secretion prompting a strong collective fight against the harmful agent (Figure 2) (Sica et al., 2016). This inflammatory activation contributes to the remodelling of the BBB, which can allow influx and guidance of peripheral leukocytes further enhancing the immune response (Sica et al., 2016). Activated astrocytes are termed “reactive.” As a consequence, reactive astrocytes might down-prioritize their homeostatic obligations, leaving the neurons under less surveillance (Sica et al., 2016). As such, abnormal and/or chronic astrocyte reactivity observed in neurodegenerative disorders such as ALS likely plays a central role in neuronal distress and cell death.
At least two broad subtypes of reactive astrocytes have been identified, however more are likely present (Sofroniew, 2020). Upon acute focal injuries such as trauma, ischemia, local infection or toxic accumulation, one type of barrier-forming reactive astrocytes will migrate towards the site of injury, proliferate and commence the formation of a physical barrier encircling and containing the damage (Sofroniew, 2020; Vainchtein and Molofsky, 2020). The astrocytic border formation is highly enriched in glial fibrillary acidic protein (GFAP) and also contains large numbers of activated microglia, which remove debris through phagocytosis (Vainchtein and Molofsky, 2020). In the past, this barrier has extensively been referred to as a “glial scar.” However, this nomenclature is now considered somewhat outdated due to the clear difference from scar tissue formed in other tissues (Sofroniew, 2020). The second identified subtype of reactive astrocytes are usually non-proliferative and retain their position and cellular interaction with the CNS, but still respond locally to injury or disease through functional and sometimes morphological changes (Sofroniew, 2020). These cells are shown to undergo hypertrophy within their individual domains, but still maintain their intercellular connections with neurons, glia and the vasculature (Sofroniew, 2020). Emerging studies reveal how this subtype of astrocytes might modulate synapses or alter their homeostatic support. However, little is known about their complex functional alterations in response to injury. Nevertheless, their response is thought to be context-dependent and modulated based on their current state and the incoming reactivity triggers (Sofroniew, 2020). Therefore, it is favourable to discuss both the myriad of potential reactive astrocyte subtypes, as well as the concept of variable reactive states within each subtype. This can be influenced by factors like disease type and its progression over time (Sofroniew, 2020). Importantly, physiologically adaptive reactive astrocytes should not be confused with disease-induced dysfunctional astrocytic reactivity, which contributes to cellular stress and neurodegeneration through cell-autonomous abnormalities. In contrast, the reactive astrocytes constitute a physiological response to injury in order to protect neuronal tissue, re-establish homeostasis and preserve neurological function (Sofroniew, 2020).
In the unravelling of ALS pathogenesis, the primary focus has been mainly on motor neurons. Although, the motor neurons are inarguably the most susceptible or vulnerable cell type in motor neuron disease, hence the name, several studies examining the contributions of astrocytes have been conducted the last couple of decades with remarkable results. And the interest is still growing.
Observations of “bizarre,” “abnormal” or reactive astrocytes as well as “astrocytosis” and “astrogliosis” have been described in several ALS case reports since the 1970’s (Smith et al., 1975; Ghatak and Nochlin, 1982; Kamo et al., 1987; Kushner et al., 1991; Murayama et al., 1991; Nagy et al., 1994; Schiffer et al., 1996). These reports relied primarily on GFAP expression and described increased astrocyte numbers and abnormal morphology throughout both brain and spinal cord tissue. It is now established that astrocyte reactivity is a prominent hallmark in ALS patients (Haidet-Phillips et al., 2011; Qian et al., 2017; Guttenplan et al., 2020), and that the number of reactive astrocytes is shown to correlate with disease progression (Hall et al., 1998).
Most experimental in vivo results have been obtained using mutant SOD1 transgenic mouse models (Gurney et al., 1994; Wong et al., 1995; Bruijn et al., 1997). This model, available since the 1990’s, shows motor dysfunction and motor neuron loss causing a life expectancy varying between 5 and 15 months depending on the copy number and the specific SOD1 mutation. Initially, it was shown that neither (motor) neuron- nor astrocyte specific expression of mutant SOD1 results in motor dysfunction, indicating an interplay between neurons and non-neuronal cells in the disease progression (Gong et al., 2000; Pramatarova et al., 2001; Lino et al., 2002). This interplay was confirmed in chimeric mouse models, where mutant SOD1-containing motor neurons survived longer when surrounded by wild type non-neuronal cells (Clement et al., 2003). A later study reported that neuron-specific expression of human SOD1 did in fact cause degeneration and paralysis in mice, but importantly only in homozygous transgenic mice (Jaarsma et al., 2008). They too did not report any pathology mediated by heterozygous mutant SOD1 expression as in the other studies. Interestingly, transplantation of astrocyte precursor cells harbouring a human SOD1G93A mutation into wild type rodent spinal cords generated mutant reactive astrocytes, which were sufficient to induce motor neuron death, forelimb motor and respiratory dysfunction in addition to reduced glutamate transporter 1 (GLT-1) expression (Papadeas et al., 2011). These findings were in line with previous observations, where SOD1-containing inclusions were extensively found in astrocytes but not in neurons during the initial stages of the disease and that these inclusions became more abundant during disease progression, in addition to increased GFAP expression and loss of GLT-1 (Bruijn et al., 1997). In transgenic SOD1G93A mice, degeneration of astrocytes residing around spinal motor neurons was found to precede the motor symptoms caused by neuronal cell death, while “astrocytosis” continually progressed during the course of the disease (Rossi et al., 2008). Astrocyte involvement is not only evident in SOD1-ALS, as a recent study demonstrated how sALS patient induced pluripotent stem cell (iPSC)-derived astrocytes caused motor neuron degeneration, NMJ denervation and motoric deficits, when transplanted into the spinal cord of immune-deficient mice (Qian et al., 2017). Oppositely, transplantation of healthy astrocyte precursors into spinal cords of human SOD1 transgenic rats resulted in reduced microglial reactivity, extended disease duration and survival, attenuated motor neuron loss and slowed the decline in motor and respiratory functions (Lepore et al., 2008). In addition, astrocyte-specific deletion of mutant SOD1 delayed microglial activation, slowed down the disease progression in mice substantially (Yamanaka et al., 2008; Wang et al., 2011) and restored GLT-1 levels (Wang et al., 2011).
In vitro coculture experiments have likewise confirmed the role of astrocytes in ALS. Cocultures between murine embryonic stem cell (ESC)-derived motor neurons harbouring a human SOD1G93A mutation with primary or ESC-derived SOD1G93A mutant glia, like astrocytes, showed glial toxicity affecting the motor neuron survival (Di Giorgio et al., 2007). These effects were also present in the gene expression changes, supporting the notion that cultured motor neurons and glia affect each other (Phatnani et al., 2013). Importantly, the gene expression profiles showed concordance with in vivo spinal cord tissue, proving the validity of cocultures in recapitulating important aspects of ALS pathogenesis (Phatnani et al., 2013). Similarly, healthy murine (m) or human (h) ESC-derived motor neurons showed survival impairments when directly cocultured with or receiving conditioned media from primary human or rodent SOD1-mutant glia (Nagai et al., 2007; Di Giorgio et al., 2008; Marchetto et al., 2008). These astrocyte-mediated toxic effects were not only apparent in SOD1-ALS, but were also documented in cocultures between astrocytes generated from sALS post-mortem tissue and wild type m- or hESC-derived motor neurons as well as through sALS astrocyte conditioned media (Haidet-Phillips et al., 2011; Re et al., 2014). Relatedly, directly reprogrammed fibroblasts into astrocytes from C9orf72-, SOD1-and sALS patients showed non-cell autonomous toxicity on wild type mESC-derived motor neurons, as motor neuron survival was greatly impaired due to the presence of the ALS astrocytes (Meyer et al., 2014). Despite the multiple overlaps between fALS and sALS presented above, TDP-43-ALS remains controversial. Some studies using astrocytic mutant TDP-43 overexpression or TDP-43-ALS (h)iPSC-derived astrocytes did not report any pathologic effects on motor neurons neither in vitro nor in vivo (Haidet-Phillips et al., 2013; Serio et al., 2013). In contrast, other studies did in fact show astrocyte-specific mutant TDP-43-mediated motor neuron death, muscle denervation, and paralysis in addition to astrocyte glutamate transport GLT-1 and Glutamate/aspartate transporter (GLAST) depletion (Tong et al., 2013; Rojas et al., 2014). These conflicting results suggest that the toxic astrocytic effects might not apply to all forms of ALS, or that caution must be implemented when comparing results obtained from different models. Nevertheless, based on these in vivo and in vitro results, it is difficult to argue against a role of astrocytes in ALS pathology and progression. However, the question is; how much do we know so far about the mechanism(s) involved?
The hypothesis of excitotoxicity being a potential pathologic hallmark of ALS emerged with the report of increased glutamate concentrations in sALS patient cerebrospinal fluids in early disease progression together with the discovery of reduced glutamate transport a few years later (Van Den Bosch et al., 2006). As mentioned above, downregulation or loss of GLAST/GLT-1 transport in astrocytes has been extensively shown in in vivo models, and is likely correlated with abnormal levels of extracellular glutamate, causing excessive neuronal firing, abnormal influx of neuronal calcium and ultimately excitotoxicity (Figure 3) (Fritz et al., 2013). In addition, SOD1-ALS astrocyte-mediated dysregulation of the glutamate receptor 2 (GluR2) subunit of AMPA receptors on motor neurons resulted in increased excitotoxic vulnerability (Van Damme et al., 2007). As a result, efforts went into drug development targeting this mechanism and in 1995 Riluzole was approved for ALS treatment (Bensimon et al., 1994). Riluzole is an anti-glutamatergic agent with anti-excitotoxic properties and inhibits postsynaptic NMDA and AMPA receptors to glutamate as well as its release (Doble, 1996). Life expectancy is prolonged with several months for ALS patients receiving Riluzole treatment, while providing no relieve of symptoms (Jaiswal, 2018). Despite being the only available drug for all ALS patients with a proven effect on survival, its limited therapeutic influence might indicate that ALS pathogenesis is driven by more than one mechanism. Abnormal levels of glutamate clearly have a toxic effect on motor neurons and this could be due to the higher vulnerability of motor neurons to excitotoxicity (Van Den Bosch et al., 2006) in combination with the loss of glutamate transporters and thereby lack of astrocytic support of the homeostatic environment.
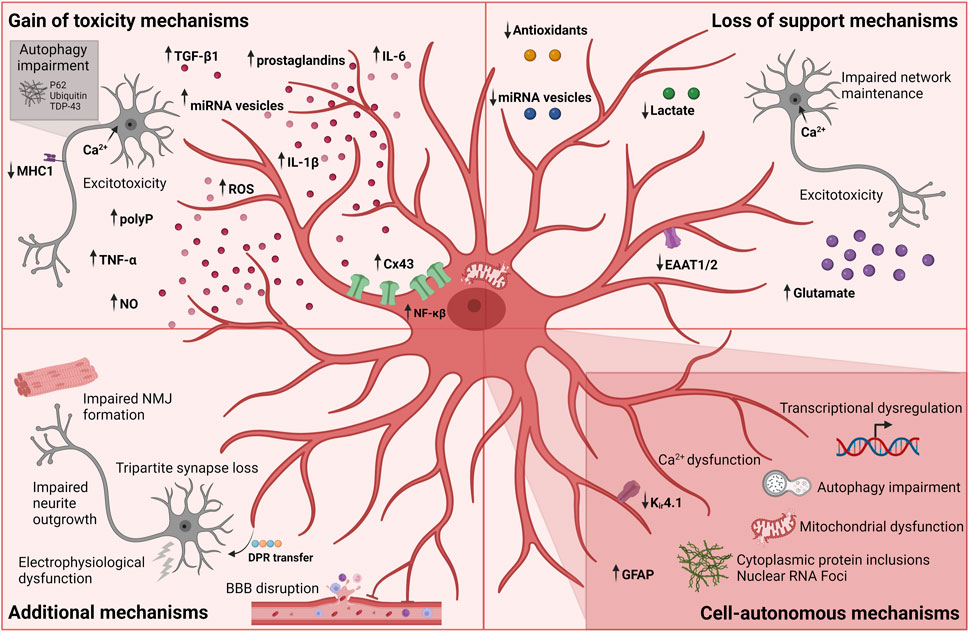
FIGURE 3. Currently known mechanisms in astrocyte-mediated ALS pathology. Each mechanism is divided into broader categories of gain of toxicity, loss of support, additional and cell-autonomous mechanisms. Figure is created with Biorender.com.
Another line of studies explores a similar support mechanism within the astrocyte metabolism: the lactate shuttle. In the context of SOD1-mutations, results from both in vivo, in vitro and ALS patient material showed a decrease in astrocytic intracellular and extracellular lactate levels as well as impairment of the astrocyte lactate efflux transporter (Figure 3) (Ferraiuolo et al., 2011; Madji Hounoum et al., 2017). In addition to the apparent lack of lactate shuttling, low lactate levels could also be caused by a decrease in lactate production due to impaired aerobic glycolysis induced by the downregulation of astrocytic nicotinamide adenine dinucleotide (NAD+) (Madji Hounoum et al., 2017).
Astrocytes have also been shown to harbour mitochondrial dysfunction and oxidative stress resulting in motor neuron degeneration (Cassina et al., 2008; Birger et al., 2019). Mitochondrial dysfunctions including compromised oxygen consumption and decreased membrane potential were observed in rat SOD1G93A astrocytes, which caused astrocyte-induced motor neuron death (Cassina et al., 2008). Interestingly, this toxicity could be rescued by treatment with mitochondria-targeted antioxidants suggesting a loss of astrocytic antioxidant secretion (Figure 3). Similarly, hiPSC-derived astrocytes from C9orf72-ALS patients showed increased oxidative stress as well as a lower secretion of antioxidants (Birger et al., 2019) further confirming this hypothesis. Upon culturing hESC-derived motor neurons in C9orf72-ALS astrocyte conditioned media, a similar increase in ROS levels was found in addition to impaired motor neuron survival. To therapeutically target oxidative stress in ALS, Edaravone was used as a free radical scavenger. However, Edaravone shows a limited effect on disease progression and only in a subset of ALS patients (Cho and Shukla, 2020; Witzel et al., 2022). This potential loss-of-support mechanism mediated through the astrocyte secretome correlates with the lack of microRNA (miRNA)-containing vesicles (Figure 3) (Varcianna et al., 2019; Gomes et al., 2022). C9orf72-ALS astrocytes were shown to have impaired extracellular vesicle formation leading to dysregulated levels of miRNAs (Varcianna et al., 2019). Especially lower levels of secreted miR-494-3p correlated with improper neurite network maintenance, and by restoring miR-494-3p levels, neurite length was restored and motor neuron survival improved (Varcianna et al., 2019). Low levels of mir-494-3p was also found in sALS post-mortem cortico-spinal tracts suggesting a more general mechanism in ALS (Varcianna et al., 2019).
Studies using astrocyte-conditioned medium also tested the hypothesis whether astrocytic release of soluble factors could mediate toxic gain of function reactions (Nagai et al., 2007; Di Giorgio et al., 2008; Rojas et al., 2014). Multiple factors were proposed including the release of cytokines such as TNF-α, interleukin 6 (IL-6) and interleukin 1β (IL-1β) as well as inorganic polyphosphate (polyP), miRNA-containing extracellular vesicles, prostaglandins, nitric oxide (NO), and ROS (Figure 3) (Nagai et al., 2007; Di Giorgio et al., 2008; Marchetto et al., 2008; Mishra et al., 2016; Kia et al., 2018; Chen et al., 2019; Varcianna et al., 2019; Lee et al., 2020; Arredondo et al., 2022; Baofengfeng et al., 2022; Gomes et al., 2022; Jensen et al., 2022; Stoklund Dittlau et al., 2023). Additionally, upregulation of inflammatory genes targeting proinflammatory cytokines, chemokines and components of the complement cascade have been reported in both fALS and sALS (Di Giorgio et al., 2008; Marchetto et al., 2008; Haidet-Phillips et al., 2011; Stoklund Dittlau et al., 2023) in addition to inflammatory NF-κβ pathway upregulation in sALS astrocytes (Haidet-Phillips et al., 2011; Gomes et al., 2019). The involvement of the NF-κβ pathway in astrocyte ALS pathology supports the notion that not only microglia are implicated in the neuroinflammatory response. Interestingly, although NF-κβ upregulation is found in astrocytes, astrocytic inhibition of the pathway did not rescue motor dysfunction or affect disease progression in SOD1 mice (Crosio et al., 2011; Frakes et al., 2014), while having the opposite and beneficial effect on microglia-induced motor neuron toxicity (Frakes et al., 2014). These results argue that microglia are highly reactive and toxic through NF-κβ pathway activation, while astrocytes likely contribute to ALS pathogenesis through a variety of mechanisms. Interestingly, a follow-up study showed how specific NF-κβ suppression in microglia, in combination with SOD1 reduction in motor neurons and astrocytes prolonged the lifespan and motor function in mice (Frakes et al., 2017). This does not only confirm the complex interplay between cell types and their combined influence on ALS pathogenesis, but also the additive effect of targeting multiple cell types in therapy development. In line with these results, overexpression of wild type FUS also provoked an inflammatory response in astrocytes causing motor neuron toxicity and proinflammatory microglial activation (Ajmone-Cat et al., 2019). Similarly, astrocytic NF-κβ activation drove microglial proliferation and leukocyte infiltration in SOD1G93A-mice (Ouali Alami et al., 2018). In contrast, another study reported how microglial activation preceded disease onset in hSOD1G93A mice, while an increase in astrocytic GFAP-expression appeared later at symptom onset (Gerber et al., 2012). Although these results were acquired from overexpression models, they target the ongoing and interesting “chicken and the egg” discussion whether astrocytes activate microglia or vice versa (Liddelow et al., 2017).
Excitotoxicity might also be mediated through a toxic gain of function independent of glutamate levels. Recently, it was shown that astrocyte-secreted polyP mediates motor neuron toxicity in cultures (Arredondo et al., 2022). PolyP is proposed to act as a gliotransmitter by regulating neuronal excitability through ion channel modulation resulting in an increase in action potentials (Stotz et al., 2014). As such, excessive polyP secretion by ALS astrocytes could be linked to excessive influx of calcium causing excitotoxicity in motor neurons (Fritz et al., 2013; Arredondo et al., 2022). Elevated levels of polyP have been found in glial cells from fALS and sALS patient spinal cord sections and in cerebrospinal fluid, further confirming its relevance for ALS (Arredondo et al., 2022).
Astrocyte-induced autophagy impairment has been shown in several studies (Figure 3) (Madill et al., 2017; Tripathi et al., 2017; Allen et al., 2019a). One study showed how ALS-patient iPSC-derived astrocyte conditioned media caused P62 accumulation in HEK293T cells, suggesting an impairment in autophagic flux (Madill et al., 2017). Another study reported how primary reactive mouse astrocytes expressing human SOD1G93A caused the formation of cytoplasmic protein inclusions of P62, ubiquitin and TDP-43, as well as axonal phosphorylated neurofilament heavy chain in hESC-derived motor neurons upon prolonged coculture (Tripathi et al., 2017). Interestingly, this effect was found to be independent of genotype but rather relates to reactivity, as traumatic injury-induced reactivity in wild type astrocytes caused similar motor neuron protein aggregations. The protein aggregations were correlated with reactive astrocytic secretion of TGF-β1, which caused a disruption of motor neuron autophagy through the mTOR-pathway (Figure 3). TGF-β1 has been reported as a neuronal pro-survival factor and, as previously mentioned, it is an important astrocytic regulator of synaptogenesis. However, astrocytic TGF-β1 overexpression or prolonged exposure to motor neurons might have detrimental effects. Indeed another study showed how excessive astrocytic TGF-β1 expression inhibited a neuroprotective microglial inflammatory response and enhanced the disease progression (Endo et al., 2015). Overall, the study by Tripathi and colleagues suggests that the reactive state rather than the SOD1 mutation is driving the pathology in this context.
Abnormal increases in Cx43 expression both at transcription and protein level have been documented in various fALS and sALS in vitro and in vivo models, as well as in sALS patients (Figure 3) (Almad et al., 2016; 2022; Gomes et al., 2022). As previously mentioned, Cx43 gap junctions and hemichannels mitigate calcium waves in addition to facilitating diffusion of ions, metabolites, miRNAs and various second messengers (Giaume et al., 2010). As a consequence of Cx43 upregulation, increases in astrocytic gap junctional coupling and hemichannel activity led to intracellular and possibly intercellular calcium hyperactivity causing motor neuron excitotoxicity and cell death (Almad et al., 2016; 2022). Interestingly, this toxicity could be counteracted by Cx43 gap junction and hemichannel blockers.
Last but not least, ALS astrocyte-mediated downregulation of the expression of major histocompatibility complex 1 (MHC1) on motor neurons was shown to make motor neurons more vulnerable to astrocyte-induced cell death (Figure 3) (Song et al., 2016).
During ALS disease progression, the electrophysiological properties of motor neurons are influenced (Sareen et al., 2013; Wainger et al., 2014; Devlin et al., 2015; Naujock et al., 2016; Guo et al., 2017b), a process also affected by astrocytes (Figure 3). hiPSC-derived C9orf72-astrocytes compromised the electrophysiological output of wild type hiPSC-derived motor neurons in a time-dependent manner, despite having no effect on motor neurons death (Zhao et al., 2020). Another recent study discovered how loss of tripartite synapses in SOD1G93A mice occurred following early onset of motor deficits (Figure 3) (Broadhead et al., 2022). This tripartite loss was confirmed in C9orf72- but not SOD1-patient cervical spinal cord post-mortem tissue, and might be linked to a loss of astrocyte PAPs (Broadhead et al., 2022). Along the same lines, astrocytes were shown to contribute to the disruption of the BBB, which causes a disturbance of the homeostatic environment and an influx of blood leukocytes (Figure 3) (Garbuzova-Davis et al., 2007a; Garbuzova-Davis et al., 2007b). Additionally, astrocytes were shown to take up toxic C9orf72 dipeptide repeats (DPRs) from the medium through endocytosis and redistributing them to motor neurons in vitro, arguing for possible prion-like protein properties as well as an astrocyte-mediated spreading of neurodegeneration (Figure 3) (Marchi et al., 2022). Interestingly, another study showed how TDP-43 aggregates could propagate from motor neurons to astrocytes, suggesting heterogeneity in spreading mechanisms across the ALS mutation spectrum (Smethurst et al., 2020). Further studies are needed to elaborate on the apparent toxic protein propagation between cell types in ALS.
Whether all these effects are due to a loss-of-support or gain-of-toxicity astrocyte mechanism remains unknown. Recently, we found that FUS-ALS hiPSC-derived astrocytes impaired hiPSC-derived motor neuron neurite outgrowth and NMJ formation in a human microfluidic triculture system (Figure 3) (Stoklund Dittlau et al., 2023). These findings correlated with the intriguing observation that mutant astrocytes simultaneously downregulated neuronal support pathways, in addition to upregulating toxic properties. As such, our data argue for a synergistic interplay between both loss-of-support and gain-of-toxicity mechanism in ALS.
Besides the clear influence of ALS astrocytes on motor neuron pathology, many studies confirm perturbed intracellular mechanisms in the astrocytes themselves spanning multiple ALS mutations (Figure 3). In addition to the mechanisms mentioned above, transcriptional dysregulation were reported in several studies (Figure 3) (Baker et al., 2015; Sun et al., 2015; Zhao et al., 2020; Taha et al., 2022; Stoklund Dittlau et al., 2023). These findings include disease stage-dependent upregulation of an immune response, lysosomal and phagocytic pathways and downregulation of ion and cholesterol homeostasis in SOD1G93A mice astrocytes (Baker et al., 2015; Miller et al., 2018). Similarly, fALS-patient iPSC-derived astrocytes showed transcriptional dysregulations causing a cell-autonomous reactive transformation involving activation of inflammatory pathways, as well as a suppression of homeostatic support, albeit in a heterogeneous mutation-dependent manner (Taha et al., 2022; Ziff et al., 2022; Stoklund Dittlau et al., 2023). In addition, cytoplasmic protein inclusions are present in astrocytes spanning various mutations (Bruijn et al., 1997; Serio et al., 2013; Barton et al., 2020) and iPSC-derived C9orf72-ALS astrocytes contain pathogenic nuclear RNA foci (Figure 3) (Zhao et al., 2020). SOD1-ALS patient iPSC-derived astrocytes display downregulation of Kir4.1 expression (Kelley et al., 2018), and murine SOD1-ALS astrocytes were reported to have abnormal intracellular calcium dynamics (Figure 3) (Kawamata et al., 2014). Metabolic screenings on directly from fibroblast-reprogrammed astrocytes of C9orf72 and sALS patients revealed compromised metabolic flexibility under starvation conditions with defects in adenosine, fructose and glycogen metabolism in addition to impaired mitochondrial substrate transport (Figure 3) (Allen et al., 2019a). In addition, low levels of adenosine deaminase hampered astrocyte adenosine metabolism potentially contributing to motor neuron toxicity (Allen et al., 2019b). This likely correlates with the mitochondrial dysfunctions previously mentioned. Finally, cortical organoid-derived astrocytes from C9orf72-ALS patient iPSCs as well as sALS iPSC-derived astrocytes showed perturbed autophagy and P62 accumulations (Figure 3) (Szebényi et al., 2021; Baofengfeng et al., 2022), which argues for a role of autophagy across multiple cell types. Considering the heterogeneous nature of the processes currently known, it is highly likely that many more astrocytic cell-autonomous mechanisms exist in ALS.
Astrocyte-targeted treatment might pose a valid therapeutic strategy. Prostaglandin inhibition reduced the toxic effects of glia on motor neuron survival in vitro (Di Giorgio et al., 2008), while antioxidant treatment decreased the production of ROS from astrocytes and rescued motor neuron death (Marchetto et al., 2008). Neutralizing antibodies targeting TNF-α alleviated FUS-ALS astrocyte-induced motor neuron toxicity (Kia et al., 2018; Jensen et al., 2022), and activation of GLT-1 expression mediated through ß-lactam antibiotic administration in SOD1-mice resulted in delayed disease progression (Rothstein et al., 2005), although this later failed in a clinical trial. Adeno-associated virus (AAV)-based therapy using miRNAs to suppress human SOD1 expression in astrocytes was shown to preserve muscle innervation, prevent muscle atrophy, and sustain motor performance in SOD1G93A mice, although the most prominent effects came from SOD1 silencing in either spinal motor neurons or both cell types with additional rescue of NMJ function, delaying of disease onset and prolonged survival (Dirren et al., 2015; Rochat et al., 2022). Recently, synthetic EphA4 agonists were shown to prevent astrocyte-mediated motor neuron toxicity in astrocyte-motor neuron cocultures in both a SOD1-ALS and sALS context (Dennys et al., 2022), while mesenchymal stem cell-derived miRNA-containing extracellular vesicles as well as transfection with miRNA mimics could ameliorate the neurotoxic profile of ALS-astrocytes (Provenzano et al., 2022). Finally, astrocyte replacement strategies with engraftment of healthy (human) astrocyte progenitors into ALS rodent spinal cords have also proven successful in retaining their gene-expression independent of the environment, albeit with conflicting effects on disease progression (Lepore et al., 2008; 2011; Kondo et al., 2014; Haidet-Phillips et al., 2015; Izrael et al., 2018). Whether astrocyte replacement strategies and/or astrocyte-targeted treatments are viable therapy options for ALS patients is an open question.
The role of astrocytes in ALS pathogenesis is complex. Even with the current knowledge, several aspects related to the pathologic process in ALS are not completely understood. The dynamic interplay between loss-of-support and gain-of-toxicity astrocyte functions complements recent findings in Alzheimer’s disease (Jiwaji et al., 2022), which argue for potential common disease mechanisms across multiple neurodegenerative disorders. However, the intricacy of the current knowledge also poses the question whether we are only just scratching the surface? Astrocytes are impressively multifaceted; in their morphology, their location and their function, which likely transfers into a disease context as well.
To dive further into the exact role of astrocytes in ALS and other neurodegenerative diseases, challenges arise with the available models. No model system is perfect, and much insight has inarguably been gained with past and current ALS models. Traditional mice models have been a corner stone in ALS research for many years and extensively bridged the gap between in vitro and patient material. However, we have to tread lightly when it comes to the impulse of directly transferring rodent results to a human context, as several reports highlight a clear interspecies variability concerning astrocytes' morphology and function. The glial-to-neuron ratio increases as we climb the evolutionary ladder: from ∼0.05–0.1 in invertebrates, 0.3–0.4 in rodents, 0.5–1.0 in rhesus monkeys and 1.5->2 in humans. It is evident that the number of glia grows with the size of the brain and thus with the higher need for neuronal homeostatic support (Verkhratsky and Nedergaard, 2018). Additionally, some subtypes of astrocytes in the human brain are not found in the rodent brain, which complicates the use of these animal models for studies on human physiology and disease (Verkhratsky and Nedergaard, 2018). Astrocytes also differ greatly in size and complexity when comparing human and rodent cells, as human astrocytes are ∼2,6-fold larger in diameter and process length and 10-fold more ramified than their rodent counterparts (Oberheim et al., 2009; Allen and Eroglu, 2017). In addition, human astrocytes have greater overlap of individual astrocyte domains than rodents, communicate through faster and stronger calcium waves and show a different transcriptional output (Oberheim et al., 2009; Zhang et al., 2016; Li et al., 2021). One astrocyte has contact with 20–120.000 synapses in the mouse brain, whereas a human astrocyte interacts with up to 2 million synapses (Bushong et al., 2002; Oberheim et al., 2009). Remarkably, when engrafting human astrocyte progenitors into immune-deficient mice, human astrocytes retained their morphology and calcium communication, while enhancing the synapse plasticity and learning ability of the chimeric mice. This suggests that species-specificities of astrocytes play a central role in the human cognitive superiority (Han et al., 2013). Importantly, many successful results from ALS drug testing in rodents have failed to translate in clinical trials in humans. One reason for this translational gap could be the timing of treatment, since ALS-mutant mice often are treated before the onset of symptoms, while ALS patients usually receive treatment much later into disease progression. However, it could also be caused by the important species differences between mice and man despite an average genomic overlap of 85% of protein-coding regions and 90% of the expressed genes in astrocytes (Chandrasekaran et al., 2016).
One solution to this species-variability problem is the use of patient material. Post-mortem tissue is an obvious and highly valuable resource, but only provide end-stage insights into ALS. On the other hand, newer techniques involving stem cell technology and especially iPSC technology harbour massive potential. From relying on overexpression of human genes in animals, primary (non-)neuronal cultures, and end-stage insight from post-mortem tissue, it became possible to take somatic cell biopsies from patients, reprogram these cells into iPSCs followed by differentiation into any desired neuronal or glial cell type in order to model specific patient pathophysiology. As such, researchers now have a tool to model fALS and maybe even more importantly sALS, which had been impossible before. This technique even bypasses the ethical dilemmas connected to the use of hESCs, which has vastly accelerated the stem cell field over the last decade. With the discovery and application of clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 gene-editing, genetic variability between disease and control lines can be diminished. As described in previous sections, much insight into astrocyte mechanisms in ALS has been gained using stem cell models, and more will likely come. However, despite the clear potential of iPSCs, important limitations remain. These disadvantages include phenotypic variabilities, insufficient maturational state and aging, as well as lack of complexity compared to in vivo models (Agbas et al., 2021). To target these limitations, researchers are exploring the use of standardized protocols, genetic- or chemical-induced aging or direct reprogramming, as well as the development of complex in vitro cocultures through the use of microfluidic devices and organoids (Meyer et al., 2014; Mertens et al., 2015; Guo et al., 2017b; Agbas et al., 2021; Stoklund Dittlau et al., 2021a; 2021b; Gatto et al., 2021; Fathi et al., 2022; Dennys et al., 2023).
Our general understanding of the underlying disease mechanisms of ALS is derived from years of research using both in vivo and in vitro models as well as post-mortem tissue. Important insights have been gained using these approaches, albeit with limited effect on therapeutic drug development. No cure or effective treatment is currently available for ALS, and it is increasingly acknowledged that this is due to large mechanistic complexity, genetic variability and a translational gap between model and patient. Fortunately, the use of in vitro stem cell models has made it easier to obtain mechanistic insight in ALS, and with their growing sophistication, they now propose an important resource for fundamental research as well as for drug discovery. More research is needed to gain the full picture of astrocyte involvement in ALS, but with the increasing interest in the research community and the ongoing improvement of models, there is clear room for optimism. Without a doubt, we should care about astrocytes in motor neuron disease.
KSD planned and wrote the paper. LVDB edited the manuscript. Both authors read and approved the final version of the paper.
Research of the authors was supported by the VIB, KU Leuven (C1 and “Opening the Future” Fund), the Agency for Innovation by Science and Technology (IWT; SBO-iPSCAF), the “Fund for Scientific Research Flanders” (FWO-Vlaanderen), Target ALS, the ALS Liga België (A Cure for ALS), the Belgian Government (Interuniversity Attraction Poles Program P7/16 initiated by the Belgian Federal Science Policy Office), the Thierry Latran Foundation and the “Association Belge contre les Maladies neuro-Musculaires” (ABMM).
The content of this manuscript has previously been published in KSD’s PhD thesis titled “Modelling amyotrophic lateral sclerosis: Human iPSC-derived astrocytes and neuromuscular junctions in microfluidic devices”(Stoklund Dittlau, 2022).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adamczyk, A., Ahmad, A., Amaler, A. S., Aoun, S., Arabambi, B., Babayev, T., et al. (2021). in Public policy in ALS/MND care. Editors R. H. Blank, J. E. Kurent, and D. Oliver (Singapore: Springer Nature Singapore Pte. Ltd). doi:10.1007/978-981-15-5840-5
Agbas, A., Amedei, A., Sebastiao, A. M., Verma, A., Hilaire, C., Raoul, C., et al. (2021). in Amyotrophic lateral sclerosis. Editor Toshiyuki Araki (Brisbane, Australia: Exon publications). doi:10.36255/exonpublications.amyotrophiclateralsclerosis.2021
Ajmone-Cat, M. A., Onori, A., Toselli, C., Stronati, E., Morlando, M., Bozzoni, I., et al. (2019). Increased FUS levels in astrocytes leads to astrocyte and microglia activation and neuronal death. Sci. Rep. 9 (1), 4572. doi:10.1038/s41598-019-41040-4
Al-Chalabi, A., and Hardiman, O. (2013). The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 9 (11), 617–628. doi:10.1038/nrneurol.2013.203
Al-Chalabi, A., Jones, A., Troakes, C., King, A., Al-Sarraj, S., and Van Den Berg, L. H. (2012). The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 124 (3), 339–352. doi:10.1007/s00401-012-1022-4
Alami, N. H., Smith, R. B., Carrasco, M. A., Williams, L. A., Winborn, C. S., Han, S. S. W., et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81 (3), 536–543. doi:10.1016/j.neuron.2013.12.018
Allen, N. J., and Eroglu, C. (2017). Cell biology of astrocyte-synapse interactions. Neuron 96 (3), 697–708. doi:10.1016/j.neuron.2017.09.056
Allen, N. J., and Lyons, D. A. (2018). Glia as architects of central nervous system formation and function. Science 362 (6411), 181–185. doi:10.1126/science.aat0473
Allen, S. P., Hall, B., Castelli, L. M., Francis, L., Woof, R., Siskos, A. P., et al. (2019a). Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 142 (3), 586–605. doi:10.1093/brain/awy353
Allen, S. P., Hall, B., Woof, R., Francis, L., Gatto, N., Shaw, A. C., et al. (2019b). C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 142 (12), 3771–3790. doi:10.1093/brain/awz302
Almad, A. A., Doreswamy, A., Gross, S. K., Richard, J. P., Huo, Y., Haughey, N., et al. (2016). Connexin 43 in astrocytes contributes to motor neuron toxicity in amyotrophic lateral sclerosis. Glia 64 (7), 1154–1169. doi:10.1002/glia.22989
Almad, A. A., Taga, A., Joseph, J., Gross, S. K., Welsh, C., Patankar, A., et al. (2022). Cx43 hemichannels contribute to astrocyte-mediated toxicity in sporadic and familial ALS. PNAS 119 (13), e2107391119. doi:10.1073/pnas.2107391119
Andrew, A. S., Bradley, W. G., Peipert, D., Butt, T., Amoako, K., Pioro, E. P., et al. (2021). Risk factors for amyotrophic lateral sclerosis: A regional United States case-control study. Muscle Nerve 63, 52–59. doi:10.1002/mus.27085
Arredondo, C., Cefaliello, C., Dyrda, A., Jury, N., Martinez, P., Díaz, I., et al. (2022). Excessive release of inorganic polyphosphate by ALS/FTD astrocytes causes non-cell-autonomous toxicity to motoneurons. Neuron 110 (10), 1656–1670.e12. e12. doi:10.1016/j.neuron.2022.02.010
Baker, D. J., Blackburn, D. J., Keatinge, M., Sokhi, D., Viskaitis, P., Heath, P. R., et al. (2015). Lysosomal and phagocytic activity is increased in astrocytes during disease progression in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 9, 410. doi:10.3389/fncel.2015.00410
Baofeng, F., Amponsah, A. E., Guo, R., Liu, X., Zhang, J., Du, X., et al. (2022). Autophagy-mediated inflammatory cytokine secretion in sporadic ALS patient iPSC-derived astrocytes. Oxidative Med. Cell. Longev. 2022, 6483582. doi:10.1155/2022/6483582
Barton, S. K., Lau, C. L., Chiam, M. D. F., Tomas, D., Muyderman, H., Beart, P. M., et al. (2020). Mutant TDP-43 expression triggers TDP-43 pathology and cell autonomous effects on primary astrocytes: Implications for non-cell autonomous pathology in ALS. Neurochem. Res. 45 (6), 1451–1459. doi:10.1007/s11064-020-03048-5
Bensimon, G., Lacomblez, L., and Meininger, V. (1994). A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. New Engl. J. Med. 330 (9), 585–591. doi:10.1056/NEJM199403033300901
Birger, A., Ben-Dor, I., Ottolenghi, M., Turetsky, T., Gil, Y., Sweetat, S., et al. (2019). Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 50, 274–289. doi:10.1016/j.ebiom.2019.11.026
Broadhead, M. J., Bonthron, C., Waddington, J., Smith, W. V., Lopez, M. F., Burley, S., et al. (2022). Selective vulnerability of tripartite synapses in amyotrophic lateral sclerosis. Acta Neuropathol. 143 (4), 471–486. doi:10.1007/s00401-022-02412-9
Brown, R. H., and Al-Chalabi, A. (2017). Amyotrophic lateral sclerosis. N. Engl. J. Med. 377 (2), 162–172. doi:10.1056/NEJMra1603471
Bruijn, L. I., Becher, M. W., Lee, M. K., Anderson, K. L., Jenkins, N. A., Copeland, N. G., et al. (1997). ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18 (2), 327–338. doi:10.1016/S0896-6273(00)80272-X
Bushong, E. A., Martone, M. E., and Ellisman, M. H. (2004). Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int. J. Dev. Neurosci. 22 (2), 73–86. doi:10.1016/j.ijdevneu.2003.12.008
Bushong, E. A., Martone, M. E., Jones, Y. Z., and Ellisman, M. H. (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22 (1), 183–192. doi:10.1523/jneurosci.22-01-00183.2002
Cassina, P., Cassina, A., Pehar, M., Castellanos, R., Gandelman, M., De León, A., et al. (2008). Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: Prevention by mitochondrial-targeted antioxidants. J. Neurosci. 28 (16), 4115–4122. doi:10.1523/JNEUROSCI.5308-07.2008
Chandrasekaran, A., Avci, H. X., Leist, M., Kobolák, J., and Dinnyés, A. (2016). Astrocyte differentiation of human pluripotent stem cells: New tools for neurological disorder research. Front. Cell. Neurosci. 10, 215. doi:10.3389/fncel.2016.00215
Chen, Y., Xia, K., Chen, L., and Fan, D. (2019). Increased interleukin-6 levels in the astrocyte-derived exosomes of sporadic amyotrophic lateral sclerosis patients. Front. Neurosci. 13, 574. doi:10.3389/fnins.2019.00574
Cho, H., and Shukla, S. (2020). Role of edaravone as a treatment option for patients with amyotrophic lateral sclerosis. Pharmaceuticals 14 (1), 29. doi:10.3390/ph14010029
Clarke, B. E., Taha, D. M., Tyzack, G. E., and Patani, R. (2021). Regionally encoded functional heterogeneity of astrocytes in health and disease: A perspective. Glia 69 (1), 20–27. doi:10.1002/glia.23877
Clement, A. M., Nguyen, M. D., Roberts, E. A., Garcia, M. L., Boillee, S., Rule, M., et al. (2003). Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302 (5642), 113–117. doi:10.1126/science.1086071
Crosio, C., Valle, C., Casciati, A., Iaccarino, C., and Carrì, M. T. (2011). Astroglial inhibition of NF-κb does not ameliorate disease onset and progression in a mouse model for amyotrophic lateral sclerosis (ALS). PLoS ONE 6 (3), e17187. doi:10.1371/journal.pone.0017187
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72 (2), 245–256. doi:10.1016/j.neuron.2011.09.011
Dennys, C., Baggio, C., Rodrigo, R., Roussel, F., Kulinich, A., Heintzman, S., et al. (2022). EphA4 targeting agents protect motor neurons from cell death induced by amyotrophic lateral sclerosis -astrocytes. iScience 25, 104877. doi:10.1016/j.isci.2022.104877
Dennys, C. N., Roussel, F., Rodrigo, R., Zhang, X., Sierra Delgado, A., Hartlaub, A., et al. (2023). CuATSM effectively ameliorates ALS patient astrocyte-mediated motor neuron toxicity in human in vitro models of amyotrophic lateral sclerosis. Glia 71 (71), 350–365. doi:10.1002/glia.24278
Devlin, A.-C., Burr, K., Borooah, S., Foster, J. D., Cleary, E. M., Geti, I., et al. (2015). Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 6, 5999. doi:10.1038/ncomms6999
Di Giorgio, F. P., Boulting, G. L., Bobrowicz, S., and Eggan, K. C. (2008). Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 3 (6), 637–648. doi:10.1016/j.stem.2008.09.017
Di Giorgio, F. P., Carrasco, M. A., Siao, M. C., Maniatis, T., and Eggan, K. (2007). Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 10 (5), 608–614. doi:10.1038/nn1885
Dirren, E., Aebischer, J., Rochat, C., Towne, C., Schneider, B. L., and Aebischer, P. (2015). SOD1 silencing in motoneurons or glia rescues neuromuscular function in ALS mice. Ann. Clin. Transl. Neurology 2 (2), 167–184. doi:10.1002/acn3.162
Doble, A. (1996). The pharmacology and mechanism of action of riluzole. Neurology 47, S233–S241. doi:10.1212/WNL.47.6_Suppl_4.233S
Endo, F., Komine, O., Fujimori-Tonou, N., Katsuno, M., Jin, S., Watanabe, S., et al. (2015). Astrocyte-derived TGF-β1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 11 (4), 592–604. doi:10.1016/j.celrep.2015.03.053
Fathi, A., Mathivanan, S., Kong, L., Petersen, A. J., Harder, C. R. K., Block, J., et al. (2022). Chemically induced senescence in human stem cell-derived neurons promotes phenotypic presentation of neurodegeneration. Aging Cell 21, e13541. doi:10.1111/acel.13541
Fazal, R., Boeynaems, S., Swijsen, A., De Decker, M., Fumagalli, L., Moisse, M., et al. (2021). HDAC6 inhibition restores TDP-43 pathology and axonal transport defects in human motor neurons with TARDBP mutations. EMBO J. 40 (7), e106177. doi:10.15252/embj.2020106177
Ferraiuolo, L., Higginbottom, A., Heath, P. R., Barber, S., Greenald, D., Kirby, J., et al. (2011). Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134 (9), 2627–2641. doi:10.1093/brain/awr193
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 185 (2), 232–240. doi:10.1016/j.expneurol.2003.10.004
Frakes, A. E., Braun, L., Ferraiuolo, L., Guttridge, D. C., and Kaspar, B. K. (2017). Additive amelioration of ALS by co-targeting independent pathogenic mechanisms. Ann. Clin. Transl. Neurology 4 (2), 76–86. doi:10.1002/acn3.375
Frakes, A. E., Ferraiuolo, L., Haidet-Phillips, A. M., Schmelzer, L., Braun, L., Miranda, C. J., et al. (2014). Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 81 (5), 1009–1023. doi:10.1016/j.neuron.2014.01.013
Fritz, E., Izaurieta, P., Weiss, A., Mir, F. R., Rojas, P., Gonzalez, D., et al. (2013). Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability. J. Neurophysiology 109 (11), 2803–2814. doi:10.1152/jn.00500.2012
Fumagalli, L., Young, F. L., Boeynaems, S., De Decker, M., Mehta, A. R., Swijsen, A., et al. (2021). C9orf72-derived arginine-containing dipeptide repeats associate with axonal transport machinery and impede microtubule-based motility. Sci. Adv. 7 (15), eabg3013. eabg3013. doi:10.1126/SCIADV.ABG3013
Garbuzova-Davis, S., Haller, E., Saporta, S., Kolomey, I., Nicosia, S. V., and Sanberg, P. R. (2007b). Ultrastructure of blood-brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS. Brain Res. 1157, 126–137. doi:10.1016/j.brainres.2007.04.044
Garbuzova-Davis, S., Saporta, S., Haller, E., Kolomey, I., Bennett, S. P., Potter, H., et al. (2007a). Evidence of compromised blood-spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PLoS ONE 2 (11), e1205. doi:10.1371/journal.pone.0001205
Gatto, N., Dos Santos Souza, C., Shaw, A. C., Bell, S. M., Myszczynska, M. A., Powers, S., et al. (2021). Directly converted astrocytes retain the ageing features of the donor fibroblasts and elucidate the astrocytic contribution to human CNS health and disease. Aging Cell 20, e13281. doi:10.1111/acel.13281
Gerber, Y. N., Sabourin, J. C., Rabano, M., Vivanco, M. d. M., and Perrin, F. E. (2012). Early functional deficit and microglial disturbances in a mouse model of amyotrophic lateral sclerosis. PloS one 7 (4), e36000. doi:10.1371/journal.pone.0036000
Ghatak, N. R., and Nochlin, D. (1982). Glial outgrowth along spinal nerve roots in amyotrophic lateral sclerosis. Ann. neurology 11 (2), 203–206. doi:10.1002/ana.410110219
Giaume, C., Koulakoff, A., Roux, L., Holcman, D., and Rouach, N. (2010). Astroglial networks: A step further in neuroglial and gliovascular interactions. Nat. Rev. Neurosci. 11 (2), 87–99. doi:10.1038/nrn2757
Gomes, C., Cunha, C., Nascimento, F., Ribeiro, J. A., Vaz, A. R., and Brites, D. (2019). Cortical neurotoxic astrocytes with early ALS pathology and miR-146a deficit replicate gliosis markers of symptomatic SOD1G93A mouse model. Mol. Neurobiol. 56 (3), 2137–2158. doi:10.1007/s12035-018-1220-8
Gomes, C., Sequeira, C., Likhite, S., Dennys, C. N., Kolb, S. J., Shaw, P. J., et al. (2022). Neurotoxic astrocytes directly converted from sporadic and familial ALS patient fibroblasts reveal signature diversities and miR-146a theragnostic potential in specific subtypes. Cells 11, 1186. doi:10.3390/cells11071186
Gong, Y. H., Parsadanian, A. S., Andreeva, A., Snider, W. D., and Elliott, J. L. (2000). Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J. Neurosci. 20 (2), 660–665. doi:10.1523/JNEUROSCI.20-02-00660.2000
Guo, W., Fumagalli, L., Prior, R., and Van Den Bosch, L. (2017a). Current advances and limitations in modeling ALS/FTD in a dish using induced pluripotent stem cells. Front. Neurosci. 11, 671. doi:10.3389/fnins.2017.00671
Guo, W., Naujock, M., Fumagalli, L., Vandoorne, T., Baatsen, P., Boon, R., et al. (2017b). HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 8 (1), 861. doi:10.1038/s41467-017-00911-y
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264 (5166), 1772–1775. doi:10.1126/science.8209258
Guttenplan, K. A., Weigel, M. K., Adler, D. I., Couthouis, J., Liddelow, S. A., Gitler, A. D., et al. (2020). Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat. Commun. 11 (1), 3753. doi:10.1038/s41467-020-17514-9
Haidet-Phillips, A. M., Doreswamy, A., Gross, S. K., Tang, X., Campanelli, J. T., and Maragakis, N. J. (2015). Human glial progenitor engraftment and gene expression is independent of the ALS environment. Exp. Neurol. 264, 188–199. doi:10.1016/j.expneurol.2014.12.011
Haidet-Phillips, A. M., Gross, S. K., Williams, T., Tuteja, A., Sherman, A., Ko, M., et al. (2013). Altered astrocytic expression of TDP-43 does not influence motor neuron survival. Exp. Neurol. 250, 250–259. doi:10.1016/j.expneurol.2013.10.004
Haidet-Phillips, A. M., Hester, M. E., Miranda, C. J., Meyer, K., Braun, L., Frakes, A., et al. (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29 (9), 824–828. doi:10.1038/nbt.1957
Hall, E. D., Oostveen, J. A., and Gurney, M. E. (1998). Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. GLIA 23 (3), 249–256. doi:10.1002/(sici)1098-1136(199807)23:3<249:aid-glia7>3.0.co;2-#
Han, X., Chen, M., Wang, F., Windrem, M., Wang, S., Shanz, S., et al. (2013). Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 12 (3), 342–353. doi:10.1016/j.stem.2012.12.015
Harada, K., Kamiya, T., and Tsuboi, T. (2016). Gliotransmitter release from astrocytes: Functional, developmental, and pathological implications in the brain. Front. Neurosci. 9, 499. doi:10.3389/fnins.2015.00499
Izrael, M., Slutsky, S. G., Admoni, T., Cohen, L., Granit, A., Hasson, A., et al. (2018). Safety and efficacy of human embryonic stem cell-derived astrocytes following intrathecal transplantation in SOD1 G93A and NSG animal models. Stem Cell Res. Ther. 9 (1), 152. doi:10.1186/s13287-018-0890-5
Jaarsma, D., Teuling, E., Haasdijk, E. D., Zeeuw, C. I. D., and Hoogenraad, C. C. (2008). Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J. Neurosci. 28 (9), 2075–2088. doi:10.1523/JNEUROSCI.5258-07.2008
Jaiswal, M. K. (2018). Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 39 (2), 733–748. doi:10.1002/med.21528
Jensen, B. K., Mcavoy, K. J., Heinsinger, N. M., Lepore, A. C., Ilieva, H., Haeusler, A. R., et al. (2022). Targeting TNFα produced by astrocytes expressing amyotrophic lateral sclerosis-linked mutant fused in sarcoma prevents neurodegeneration and motor dysfunction in mice. Glia 70 (7), 1426–1449. doi:10.1002/glia.24183
Jiwaji, Z., Tiwari, S. S., Avilés-Reyes, R. X., Hooley, M., Hampton, D., Torvell, M., et al. (2022). Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Aß pathology. Nat. Commun. 13 (1), 135. doi:10.1038/s41467-021-27702-w
Kamo, H., Haebara, H., Akiguchi, I., Kameyama, M., Kimura, H., and McGeer, P. L. (1987). A distinctive distribution of reactive astroglia in the precentral cortex in amyotrophic lateral sclerosis. Acta Neuropathol. 74 (1), 33–38. doi:10.1007/BF00688335
Kawamata, H., Ng, S. K., Diaz, N., Burstein, S., Morel, L., Osgood, A., et al. (2014). Abnormal intracellular calcium signaling and SNARE dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J. Neurosci. 34 (6), 2331–2348. doi:10.1523/JNEUROSCI.2689-13.2014
Kelley, K. W., Ben Haim, L., Schirmer, L., Tyzack, G. E., Tolman, M., Miller, J. G., et al. (2018). Kir4.1-Dependent astrocyte-fast motor neuron interactions are required for peak strength. Neuron 98 (2), 306–319. e7. doi:10.1016/j.neuron.2018.03.010
Kia, A., McAvoy, K., Krishnamurthy, K., Trotti, D., and Pasinelli, P. (2018). Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. GLIA 66 (5), 1016–1033. doi:10.1002/glia.23298
Kondo, T., Funayama, M., Tsukita, K., Hotta, A., Yasuda, A., Nori, S., et al. (2014). Focal transplantation of human iPSC-derived glial-rich neural progenitors improves lifespan of ALS mice. Stem Cell Rep. 3 (2), 242–249. doi:10.1016/j.stemcr.2014.05.017
Kushner, P. D., Stephenson, D. T., and Wright, S. (1991). Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain. J. Neuropathol. Exp. Neurol. 50 (3), 263–277. doi:10.1097/00005072-199105000-00008
Lee, S., Kim, S., Kang, H. Y., Lim, H. R., Kwon, Y., Jo, M., et al. (2020). The overexpression of TDP-43 in astrocytes causes neurodegeneration via a PTP1B-mediated inflammatory response. J. Neuroinflammation 17 (1), 299. doi:10.1186/s12974-020-01963-6
Lepore, A. C., O’Donnell, J., Kim, A. S., Williams, T., Tuteja, A., Rao, M. S., et al. (2011). Human Glial-restricted progenitor transplantation into cervical spinal cord of the SOD1 mouse model of ALS. PLoS ONE 6 (10), e25968. doi:10.1371/journal.pone.0025968
Lepore, A. C., Rauck, B., Dejea, C., Pardo, A. C., Rao, M. S., Rothstein, J. D., et al. (2008). Focal transplantation – based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat. Neurosci. 11 (11), 1294–1301. doi:10.1038/nn.2210
Li, J., Pan, L., Pembroke, W. G., Rexach, J. E., Godoy, M. I., Condro, M. C., et al. (2021). Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nat. Commun. 12 (1), 3958. doi:10.1038/s41467-021-24232-3
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541 (7638), 481–487. doi:10.1038/nature21029
Liddelow, S., and Barres, B. (2015). SnapShot: Astrocytes in health and disease. Cell 162 (5), 1170–1170.e1. doi:10.1016/j.cell.2015.08.029
Lino, M. M., Schneider, C., and Caroni, P. (2002). Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 22 (12), 4825–4832. doi:10.1523/JNEUROSCI.22-12-04825.2002
Liu, X., Ying, J., Wang, X., Zheng, Q., Zhao, T., Yoon, S., et al. (2021). Astrocytes in neural circuits: Key factors in synaptic regulation and potential targets for neurodevelopmental disorders. Front. Mol. Neurosci. 14, 729273. doi:10.3389/fnmol.2021.729273
Madill, M., McDonagh, K., Ma, J., Vajda, A., McLoughlin, P., O’Brien, T., et al. (2017). Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol. Brain 10 (1), 22. doi:10.1186/s13041-017-0300-4
Madji Hounoum, B., Mavel, S., Coque, E., Patin, F., Vourc’h, P., Marouillat, S., et al. (2017). Wildtype motoneurons, ALS-Linked SOD1 mutation and glutamate profoundly modify astrocyte metabolism and lactate shuttling. Glia 65 (4), 592–605. doi:10.1002/glia.23114
Marchetto, M. C. N., Muotri, A. R., Mu, Y., Smith, A. M., Cezar, G. G., and Gage, F. H. (2008). Non-cell-autonomous effect of human SOD1G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 3 (6), 649–657. doi:10.1016/j.stem.2008.10.001
Marchi, P. M., Marrone, L., Brasseur, L., Coens, A., Webster, C. P., Bousset, L., et al. (2022). C9ORF72-derived poly-GA DPRs undergo endocytic uptake in iAstrocytes and spread to motor neurons. Life Sci. alliance 5 (9), e202101276. doi:10.26508/lsa.202101276
Martineau, É., Di Polo, A., Vande Velde, C., and Robitaille, R. (2018). Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. eLife 7, e41973. doi:10.7554/eLife.41973
Masrori, P., and Van Damme, P. (2020). Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurology 27 (10), 1918–1929. doi:10.1111/ene.14393
Mertens, J., Paquola, A. C. M., Ku, M., Hatch, E., Böhnke, L., Ladjevardi, S., et al. (2015). Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17 (6), 705–718. doi:10.1016/j.stem.2015.09.001
Meyer, K., Ferraiuolo, L., Miranda, C. J., Likhite, S., McElroy, S., Renusch, S., et al. (2014). Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. PNAS 111 (2), 829–832. doi:10.1073/pnas.1314085111
Miller, S. J., Glatzer, J. C., Hsieh, Y., and Rothstein, J. D. (2018). Cortical astroglia undergo transcriptomic dysregulation in the G93A SOD1 ALS mouse model. J. Neurogenetics 32 (4), 322–335. doi:10.1080/01677063.2018.1513508
Mishra, P. S., Dhull, D. K., Nalini, A., Vijayalakshmi, K., Sathyaprabha, T. N., Alladi, P. A., et al. (2016). Astroglia acquires a toxic neuroinflammatory role in response to the cerebrospinal fluid from amyotrophic lateral sclerosis patients. J. Neuroinflammation 13 (1), 212. doi:10.1186/s12974-016-0698-0
Murayama, S., Inoue, K., Kawakami, H., Bouldin, T. W., and Suzuki, K. (1991). A unique pattern of astrocytosis in the primary motor area in amyotrophic lateral sclerosis. Acta Neuropathol. 82 (6), 456–461. doi:10.1007/BF00293379
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10 (5), 615–622. doi:10.1038/nn1876
Nagy, D., Kato, T., and Kushner, P. D. (1994). Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J. Neurosci. Res. 38 (3), 336–347. doi:10.1002/jnr.490380312
Nair, G., Carew, J. D., Usher, S., Lu, D., Hu, X. P., and Benatar, M. (2010). Diffusion tensor imaging reveals regional differences in the cervical spinal cord in amyotrophic lateral sclerosis. NeuroImage 53 (2), 576–583. doi:10.1016/j.neuroimage.2010.06.060
Naujock, M., Stanslowsky, N., Bufler, S., Naumann, M., Reinhardt, P., Sterneckert, J., et al. (2016). 4-Aminopyridine induced activity rescues hypoexcitable motor neurons from amyotrophic lateral sclerosis patient-derived induced pluripotent stem cells. Stem Cells 34 (6), 1563–1575. doi:10.1002/stem.2354
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314 (5796), 130–133. doi:10.1126/science.1134108
Oberheim, N. A., Takano, T., Han, X., He, W., Lin, J. H. C., Wang, F., et al. (2009). Uniquely hominid features of adult human astrocytes. J. Neurosci. 29 (10), 3276–3287. doi:10.1523/JNEUROSCI.4707-08.2009
Ouali Alami, N., Schurr, C., Olde Heuvel, F., Tang, L., Li, Q., Tasdogan, A., et al. (2018). NF-κB activation in astrocytes drives a stage-specific beneficial neuroimmunological response in ALS. EMBO J. 37 (16), e98697. doi:10.15252/embj.201798697
Paganoni, S., Hendrix, S., Dickson, S. P., Knowlton, N., Macklin, E. A., Berry, J. D., et al. (2021). Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve 63, 31–39. doi:10.1002/mus.27091
Paganoni, S., Macklin, E. A., Hendrix, S., Berry, J. D., Elliott, M. A., Maiser, S., et al. (2020). Trial of sodium phenylbutyrate–taurursodiol for amyotrophic lateral sclerosis. N. Engl. J. Med. 383, 919–930. doi:10.1056/nejmoa1916945
Papadeas, S. T., Kraig, S. E., Banion, C. O., Lepore, A. C., and Maragakis, N. J. (2011). Astrocytes carrying the superoxide dismutase 1 (SOD1 G93A) mutation induce wild-type motor neuron degeneration in vivo. PNAS 108 (43), 17803–17808. doi:10.1073/pnas.1103141108
Petrov, D., Mansfield, C., Moussy, A., and Hermine, O. (2017). ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front. Aging Neurosci. 9, 68. doi:10.3389/fnagi.2017.00068
Phatnani, H. P., Guarnieri, P., Friedman, B. A., Carrasco, M. A., Muratet, M., O’Keeffe, S., et al. (2013). Intricate interplay between astrocytes and motor neurons in ALS. PNAS 110 (8), E756–E765. doi:10.1073/pnas.1222361110
Picchiarelli, G., Demestre, M., Zuko, A., Been, M., Higelin, J., Dieterlé, S., et al. (2019). FUS-mediated regulation of acetylcholine receptor transcription at neuromuscular junctions is compromised in amyotrophic lateral sclerosis. Nat. Neurosci. 22 (11), 1793–1805. doi:10.1038/s41593-019-0498-9
Pramatarova, A., Laganiere, J., Roussel, J., Brisebois, K., and Rouleau, G. A. (2001). Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. 21 (10), 3369–3374. doi:10.1523/JNEUROSCI.21-10-03369.2001
Provenzano, F., Nyberg, S., Giunti, D., Torazza, C., Parodi, B., Bonifacino, T., et al. (2022). Micro-RNAs shuttled by extracellular vesicles secreted from mesenchymal stem cells dampen astrocyte pathological activation and support neuroprotection in in-vitro models of ALS. Cells 11, 3923. doi:10.3390/cells11233923
Qian, K., Huang, H., Peterson, A., Hu, B., Maragakis, N. J., Ming, G. L., et al. (2017). Sporadic ALS astrocytes induce neuronal degeneration in vivo. Stem Cell Rep. 8 (4), 843–855. doi:10.1016/j.stemcr.2017.03.003
Re, D. B., VercheLeYu, V. C., Amoroso, M. W., Politi, K. A., Phani, S., et al. (2014). Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81 (5), 1001–1008. doi:10.1016/j.neuron.2014.01.011
Renton, A. E., Chio, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17 (1), 17–23. doi:10.1038/nn.3584
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi:10.1016/j.neuron.2011.09.010
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14 (4), 248–264. doi:10.1038/nrn3430
Rochat, C., Bernard-Marissal, N., Källstig, E., Pradervand, S., Perrin, F. E., Aebischer, P., et al. (2022). Astrocyte-targeting RNA interference against mutated superoxide dismutase 1 induces motoneuron plasticity and protects fast-fatigable motor units in a mouse model of amyotrophic lateral sclerosis. Glia 70 (5), 842–857. doi:10.1002/glia.24140
Rojas, F., Cortes, N., Abarzua, S., Dyrda, A., and van Zundert, B. (2014). Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress. Front. Cell. Neurosci. 8, 24. doi:10.3389/fncel.2014.00024
Rossi, D., Brambilla, L., Valori, C. F., Roncoroni, C., Crugnola, A., Yokota, T., et al. (2008). Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 15 (11), 1691–1700. doi:10.1038/cdd.2008.99
Rossi, S., and Cozzolino, M. (2021). Dysfunction of RNA/RNA-binding proteins in ALS astrocytes and microglia. Cells 10 (11), 3005. doi:10.3390/cells10113005
Rothstein, J. D., Patel, S., Regan, M. R., Haenggeli, C., Huang, Y. H., Bergles, D. E., et al. (2005). Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433 (7021), 73–77. doi:10.1038/nature03180
Sareen, D., O’Rourke, J. G., Meera, P., Muhammad, A. K. M. G., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5 (208), 208ra149. 208ra149. doi:10.1126/scitranslmed.3007529
Schiffer, D., Cordera, S., Cavalla, P., and Migheli, A. (1996). Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J. Neurol. Sci. 139, 27–33. doi:10.1016/0022-510x(96)00073-1
Serio, A., Bilican, B., Barmada, S. J., Ando, D. M., Zhao, C., Siller, R., et al. (2013). Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. PNAS 110 (12), 4697–4702. doi:10.1073/pnas.1300398110
Shang, Y., and Huang, E. J. (2016). Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 1647, 65–78. doi:10.1016/j.brainres.2016.03.036
Shatunov, A., and Al-Chalabi, A. (2021). The genetic architecture of ALS. Neurobiol. Dis. 147, 105156. doi:10.1016/J.NBD.2020.105156
Sibille, J., Pannasch, U., and Rouach, N. (2014). Astroglial potassium clearance contributes to short-term plasticity of synaptically evoked currents at the tripartite synapse. J. Physiology 592 (1), 87–102. doi:10.1113/jphysiol.2013.261735
Sica, R. E., Caccuri, R., Quarracino, C., and Capani, F. (2016). Are astrocytes executive cells within the central nervous system? Arq. Neuro-Psiquiatria 74 (8), 671–678. doi:10.1590/0004-282X20160101
Smethurst, P., Risse, E., Tyzack, G. E., Mitchell, J. S., Taha, D. M., Chen, Y. R., et al. (2020). Distinct responses of neurons and astrocytes to TDP-43 proteinopathy in amyotrophic lateral sclerosis. Brain 143 (2), 430–440. doi:10.1093/brain/awz419
Smith, T. W., Tyler, H. R., and Schoene, W. C. (1975). Atypical astrocytes and Rosenthal fibers in a case of amyotrophic lateral sclerosis associated with a cerebral glioblastoma multiforme. Acta Neuropathol. 31 (1), 29–34. doi:10.1007/BF00696884
So, E., Mitchell, J. C., Memmi, C., Chennell, G., Vizcay-Barrena, G., Allison, L., et al. (2018). Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUSWT transgenic mice. Hum. Mol. Genet. 27 (3), 463–474. doi:10.1093/hmg/ddx415
Sofroniew, M. V. (2020). Astrocyte reactivity: Subtypes, states, and functions in CNS innate immunity. Trends Immunol. 41 (9), 758–770. doi:10.1016/j.it.2020.07.004
Song, S., Miranda, C. J., Braun, L., Meyer, K., Frakes, A. E., Ferraiuolo, L., et al. (2016). Major histocompatibility complex class I molecules protect motor neurons from astrocyte-induced toxicity in amyotrophic lateral sclerosis. Nat. Med. 22 (4), 397–403. doi:10.1038/nm.4052
Stoklund Dittlau, K., Krasnow, E. N., Fumagalli, L., Vandoorne, T., Baatsen, P., Kerstens, A., et al. (2021a). Generation of human motor units with functional neuromuscular junctions in microfluidic devices. J. Vis. Exp. 2021 (175), e62959. doi:10.3791/62959
Stoklund Dittlau, K., Krasnow, E. N., Fumagalli, L., Vandoorne, T., Baatsen, P., Kerstens, A., et al. (2021b). Human motor units in microfluidic devices are impaired by FUS mutations and improved by HDAC6 inhibition. Stem Cell Rep. 16 (9), 2213–2227. doi:10.1016/j.stemcr.2021.03.029
Stoklund Dittlau, K. (2022). “Modelling amyotrophic lateral sclerosis: Human iPSC-derived astrocytes and neuromuscular junctions in microfluidic devices,”. [PhD dissertation] (Belgium: KU Leuven).
Stoklund Dittlau, K., Terrie, L., Baatsen, P., Kerstens, A., De Swert, L., Janky, R., et al. (2023). FUS-ALS hiPSC-derived astrocytes impair human motor units through both gain-of-toxicity and loss-of-support mechanisms. Molecular Neurodegeneration. [Accepted]. doi:10.1186/s13024-022-00591-3
Stotz, S. C., Scott, L. O., Drummond-Main, C., Avchalumov, Y., Girotto, F., Davidsen, J., et al. (2014). Inorganic polyphosphate regulates neuronal excitability through modulation of voltage-gated channels. Mol. Brain 7, 42. doi:10.1186/1756-6606-7-42
Sun, S., Sun, Y., Ling, S. C., Ferraiuolo, L., McAlonis-Downes, M., Zou, Y., et al. (2015). Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant sod1-mediated ALS. PNAS 112 (50), E6993–E7002. doi:10.1073/pnas.1520639112
Szebényi, K., Wenger, L. M. D., Sun, Y., Dunn, A. W. E., Limegrover, C. A., Gibbons, G. M., et al. (2021). Human ALS/FTD brain organoid slice cultures display distinct early astrocyte and targetable neuronal pathology. Nat. Neurosci. 24 (11), 1542–1554. doi:10.1038/s41593-021-00923-4
Taha, D. M., Clarke, B. E., Hall, C. E., Tyzack, G. E., Ziff, O. J., Greensmith, L., et al. (2022). Astrocytes display cell autonomous and diverse early reactive states in familial amyotrophic lateral sclerosis. Brain 145 (2), 481–489. doi:10.1093/brain/awab328
Tallon, C., Russell, K. A., Sakhalkar, S., Andrapallayal, N., and Farah, M. H. (2016). Length-dependent axo-terminal degeneration at the neuromuscular synapses of type II muscle in SOD1 mice. Neuroscience 312, 179–189. doi:10.1016/j.neuroscience.2015.11.018
Tong, J., Huang, C., Bi, F., Wu, Q., Huang, B., Liu, X., et al. (2013). Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 32 (13), 1917–1926. doi:10.1038/emboj.2013.122
Tripathi, P., Rodriguez-Muela, N., Klim, J. R., de Boer, A. S., Agrawal, S., Sandoe, J., et al. (2017). Reactive astrocytes promote ALS-like degeneration and intracellular protein aggregation in human motor neurons by disrupting autophagy through TGF-β1. Stem Cell Rep. 9 (2), 667–680. doi:10.1016/j.stemcr.2017.06.008
Tziortzouda, P., Van Den Bosch, L., and Hirth, F. (2021). Triad of TDP43 control in neurodegeneration: Autoregulation, localization and aggregation. Nat. Rev. Neurosci. 22 (4), 197–208. doi:10.1038/s41583-021-00431-1
Vainchtein, I. D., Chin, G., Cho, F. S., Kelley, K. W., Miller, J. G., Chien, E. C., et al. (2018). Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359 (6381), 1269–1273. doi:10.1126/science.aal3589
Vainchtein, I. D., and Molofsky, A. V. (2020). Astrocytes and microglia: In sickness and in health. Trends Neurosci. 43 (3), 144–154. doi:10.1016/j.tins.2020.01.003
Van Blitterswijk, M., Dejesus-Hernandez, M., and Rademakers, R. (2012). How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: Can we learn from other noncoding repeat expansion disorders? Curr. Opin. Neurology 25 (6), 689–700. doi:10.1097/WCO.0b013e32835a3efb
Van Damme, P., Bogaert, E., Dewil, M., Hersmus, N., Kiraly, D., Scheveneels, W., et al. (2007). Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. PNAS 104 (37), 14825–14830. doi:10.1073/pnas.0705046104
Van Den Bosch, L., Van Damme, P., Bogaert, E., and Robberecht, W. (2006). The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochimica Biophysica Acta - Mol. Basis Dis. 1762 (11–12), 1068–1082. doi:10.1016/j.bbadis.2006.05.002
Varcianna, A., Myszczynska, M. A., Castelli, L. M., O’Neill, B., Kim, Y., Talbot, J., et al. (2019). Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. EBioMedicine 40, 626–635. doi:10.1016/j.ebiom.2018.11.067
Verkhratsky, A., and Nedergaard, M. (2018). Physiology of astroglia. Physiol. Rev. 98 (1), 239–389. doi:10.1152/physrev.00042.2016
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S. W., Sandoe, J., et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7 (1), 1–11. doi:10.1016/j.celrep.2014.03.019
Walker, A. K., Spiller, K. J., Ge, G., Zheng, A., Xu, Y., Zhou, M., et al. (2015). Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 130 (5), 643–660. doi:10.1007/s00401-015-1460-x
Wang, L., Gutmann, D. H., and Roos, R. P. (2011). Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum. Mol. Genet. 20 (2), 286–293. doi:10.1093/hmg/ddq463
Wang, M. D., Little, J., Gomes, J., Cashman, N. R., and Krewski, D. (2017). Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. NeuroToxicology 61, 101–130. doi:10.1016/j.neuro.2016.06.015
Witzel, S., Maier, A., Steinbach, R., Grosskreutz, J., Koch, J. C., Sarikidi, A., et al. (2022). Safety and effectiveness of long-term intravenous administration of edaravone for treatment of patients with amyotrophic lateral sclerosis. JAMA Neurol. 79 (2), 121–130. doi:10.1001/jamaneurol.2021.4893
Wong, P. C., Pardo, C. A., Borchelt, D. R., Lee, M. K., Copeland, N. G., Jenkins, N. A., et al. (1995). An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14 (6), 1105–1116. doi:10.1016/0896-6273(95)90259-7
Yamanaka, K., Chun, S. J., Boillee, S., Fujimori-Tonou, N., Yamashita, H., Gutmann, D. H., et al. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11 (3), 251–253. doi:10.1038/nn2047
Zhang, Y., Sloan, S. A., Clarke, L. E., Caneda, C., Plaza, C. A., Blumenthal, P. D., et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89 (1), 37–53. doi:10.1016/j.neuron.2015.11.013
Zhao, C., Devlin, A. C., Chouhan, A. K., Selvaraj, B. T., Stavrou, M., Burr, K., et al. (2020). Mutant C9orf72 human iPSC-derived astrocytes cause non-cell autonomous motor neuron pathophysiology. Glia 68 (5), 1046–1064. doi:10.1002/glia.23761
Keywords: amyotrophic lateral sclerosis, gain-of-toxicity, loss-of-support, disease mechanism, disease model, induced pluripotent stem cell
Citation: Stoklund Dittlau K and Van Den Bosch L (2023) Why should we care about astrocytes in a motor neuron disease?. Front. Mol. Med. 3:1047540. doi: 10.3389/fmmed.2023.1047540
Received: 18 September 2022; Accepted: 13 January 2023;
Published: 23 January 2023.
Edited by:
Kathrin Meyer, Nationwide Children’s Hospital, United StatesReviewed by:
José Ronaldo Dos Santos, Federal University of Sergipe, BrazilCopyright © 2023 Stoklund Dittlau and Van Den Bosch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ludo Van Den Bosch Email, bHVkby52YW5kZW5ib3NjaEBrdWxldXZlbi5iZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.