
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 03 March 2025
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 16 - 2025 | https://doi.org/10.3389/fmicb.2025.1535420
 Myllena Pereira Silverio1,2
Myllena Pereira Silverio1,2 Júnia Schultz3
Júnia Schultz3 Mariana T. D. Parise4
Mariana T. D. Parise4 Doglas Parise4
Doglas Parise4 Marcus Vinicius Canário Viana4
Marcus Vinicius Canário Viana4 Wylerson Nogueira3,4,5
Wylerson Nogueira3,4,5 Rommel Thiago Jucá Ramos5
Rommel Thiago Jucá Ramos5 Aristoteles Góes-Neto4
Aristoteles Góes-Neto4 Vasco Ariston De Carvalho Azevedo4
Vasco Ariston De Carvalho Azevedo4 Bertram Brenig6
Bertram Brenig6 Raquel Regina Bonelli2
Raquel Regina Bonelli2 Alexandre Soares Rosado3,7*
Alexandre Soares Rosado3,7*The genus Pseudomonas includes metabolically versatile microorganisms occupying diverse niches, from environmental habitats to plant pathogens, and has clinically significant strains. For this reason, Pseudomonas spp. might act as a reservoir of antimicrobial resistance genes, which have been detected even in isolated environments. The aim of this study was to report the antimicrobial susceptibility profile of 25 Pseudomonas fluorescens isolates from soil samples collected on King George Island (Antarctic Peninsula), and to select non-clonal isolates with unusual phenotypes for whole genome sequencing (WGS). Six classes of antimicrobials were assessed with disk diffusion and colistin with minimum inhibitory concentration (MIC) by broth microdilution. In order to confirm the discrepant phenotypes, MIC by agar dilution was performed for the beta-lactams aztreonam, ceftazidime, cefepime and the aminoglycoside neomycin. The genus Pseudomonas was confirmed by matrix-assisted laser desorption/ionization – time of flight (MALDI-TOF) and the clonal relationships were examined using repetitive extragenic palindromic polymerase chain reaction (BOX-PCR), from which 14 strains were selected for WGS. Antimicrobial susceptibility testing revealed that all strains were susceptible to neomycin and exhibited varying degrees of intermediate or full resistance to aztreonam and colistin. Additionally, 11 strains demonstrated intermediate resistance to ceftazidime, and six were resistant to cefepime. The genomic analysis identified various efflux pumps, predominantly from the ABC transporter and resistance-nodulation-division families. Resistance genes were detected against eight classes of antimicrobials, listed by prevalence: beta-lactams, tetracyclines, polymyxins, aminoglycosides, fosmidomycin, fosfomycin, quinolones, and chloramphenicol. Genes associated with heavy-metal resistance, prophages, and adaptations to extreme environments were also investigated. One notable isolate exhibited not only the highest number of pathogenicity and resistance islands, but also presented a carbapenemase-encoding gene (blaPFM-2) in its genome. Overall, one plasmid was identified in a distinct isolate, which did not exhibit antimicrobial resistance determinants. The genotypic and phenotypic findings are consistent, suggesting that efflux pumps play a critical role in antimicrobial extrusion. This study offers valuable insight into the evolution of antimicrobial resistance in P. fluorescens, particularly in extreme environments, such as Antarctica. By exploring the antimicrobial resistance mechanisms in P. fluorescens, the study sheds light on how isolated ecosystems drive the natural evolution of resistance genes.
Pseudomonads are ubiquitous and adaptable microorganisms, primarily due to their metabolic versatility and genome plasticity (Craig et al., 2021; Rumbaugh, 2014). The genus is known for its ability to survive cold stress and desiccation (Craig et al., 2021), common environmental conditions in extreme habitats, such as Antarctica. To survive harsh conditions and competition, Pseudomonas has developed an effective response to abiotic stress, including resistance to antimicrobials (Marcoleta et al., 2022; Allen et al., 2009). Therefore, intrinsic resistance in the genus Pseudomonas includes altering membrane permeability and overexpressing efflux pumps or chromosomal resistance genes, such as the beta-lactamase gene blaAmpC (Silverio et al., 2022; Lupo et al., 2018; Chevalier et al., 2017; Olivares Pacheco et al., 2017; Lima et al., 2015).
The aim of this study was to investigate intrinsic resistance mechanisms in isolates belonging to the Pseudomonas fluorescens complex. The isolates were originally from four remote ecosystems in King George Island, Antarctic Peninsula. Antarctica is considered one of the last pristine environments, exhibiting extreme weather conditions, well-preserved ecosystems, and geographical isolation (Cowan et al., 2011). On the other hand, P. fluorescens is an opportunistic pathogen that mainly affects immunocompromised patients. These bacteria behave as reservoirs of antimicrobial resistance genes (ARGs), which makes the treatment challenging (Koh et al., 2004; Rolston et al., 2005; Sader and Jones, 2005; Faccone et al., 2014; Montana et al., 2018). Hitherto, few research papers have focused on the antimicrobial susceptibility profile of Antarctic bacteria using whole genome sequencing (WGS). For example, previous works include the beta-lactam-resistant bacteria Acinetobacter radioresistens A154 from Fildes Peninsula (Opazo-Capurro et al., 2019) and two methicillin-resistant Staphylococci strains from James Ross Island (Pantucek et al., 2018). In this work, 25 P. fluorescens isolates were evaluated with phenotypic antimicrobial susceptibility tests, and 14 were selected for WGS.
Soil samples were collected in four ice-free sites on King George Island (Antarctic Peninsula) during the austral summer of 2007 (Figure 1A). The sampling sites and collected samples include the following:
1. soil under Sanionia uncinata from the North Peak (62°04′849”S, 58°24′024”W; Figure 1B),
2. the rhizosphere of Deschampsia antarctica from Ullmann Point (62°05′015”S, 58°23′987”W; Figure 1C),
3. the rhizosphere of Colobanthus quitensis from Comandante Ferraz Scientific Station (62°05′06”S, 58°24′12”W; Figure 1D), and
4. ornithogenic soil near an Adelie penguin nest from Arctowski Polish Station (62°09′790”S, 58°29′687”W; Figure 1E).

Figure 1. Sample sites at King George Island, Antarctic Peninsula. The island is part of the South Shetland Islands. (A) Map created using ArcGIS Pro v.3.2. (B) Sanionia uncinata. (C) Deschampsia antarctica. (D) Colobanthus quitensis. (E) Ornithogenic soil near an Adelie penguin nest.
The method described by da Silva et al. (2017) was used to isolate culturable bacterial fraction. A preliminary evaluation of the 16S gene rrs indicated that the isolates belong to the genus Pseudomonas. The isolates are part of the Antarctic culture collection at the Microbial Molecular Ecology Laboratory (Federal University of Rio de Janeiro, Brazil).
Twenty-five psychrotolerant isolates affiliated with the genus Pseudomonas were selected for this study. Of these, 13 (52%) were isolated from ornithogenic soil, followed by the rhizosphere of the native plants C. quitensis (n = 6; 24%) and D. antarctica (n = 4; 16%). Two isolates (8%) were isolated from soil covered by the moss S. uncinata. The highest temperature at which we observed growth was 28°C. For this reason, the optimal incubation was 28°C for 24 h (antimicrobial resistance phenotypic screening) to 48 h (DNA extraction).
The genus of each isolate was confirmed using matrix-assisted laser desorption/ionization – time of flight (MALDI-TOF; Microflex LT, Bruker GmbH, Berlin, Germany). This experiment represented the beginning of the trial for Pseudomonas isolates (Figure 2), which was performed using the algorithm provided by the manufacturer. The colonies were transferred in triplicate to the plate “MSP 96 Polished Steel BC,” provided by the manufacturer. The plate was cleaned in accordance with the manufacturer’s instructions, with 70% alcohol followed by 80% trifluoroacetic acid. We added 1 μL of 70% formic acid (Tedia, Fairfield, Ohio, United States) and allowed the plate to fully dry at room temperature. Afterwards, 1 μL of the matrix α-cyano-4-hydroxycinnamic acid (Bruker GmbH, Berlin, Germany) diluted to 10 mg/mL in organic solvent [50% acetonitrile and 2.5% trifluoroacetic acid (Tedia, Fairfield, Ohio, United States)] was applied and dried at room temperature. The calibration strain was Escherichia coli ATCC 25922, which was the reference strain for the peaks in a spectrum of proteins between 2 and 20 kDa, as provided by the manufacturer (software FlexControl v.3.4, Bruker GmbH, Berlin, Germany). The results were compared with the spectra in MALDI Biotyper v 3.1, using the MBT Compass software and the MALDI Biotyper® CA library (Bruker GmbH, Berlin, Germany).
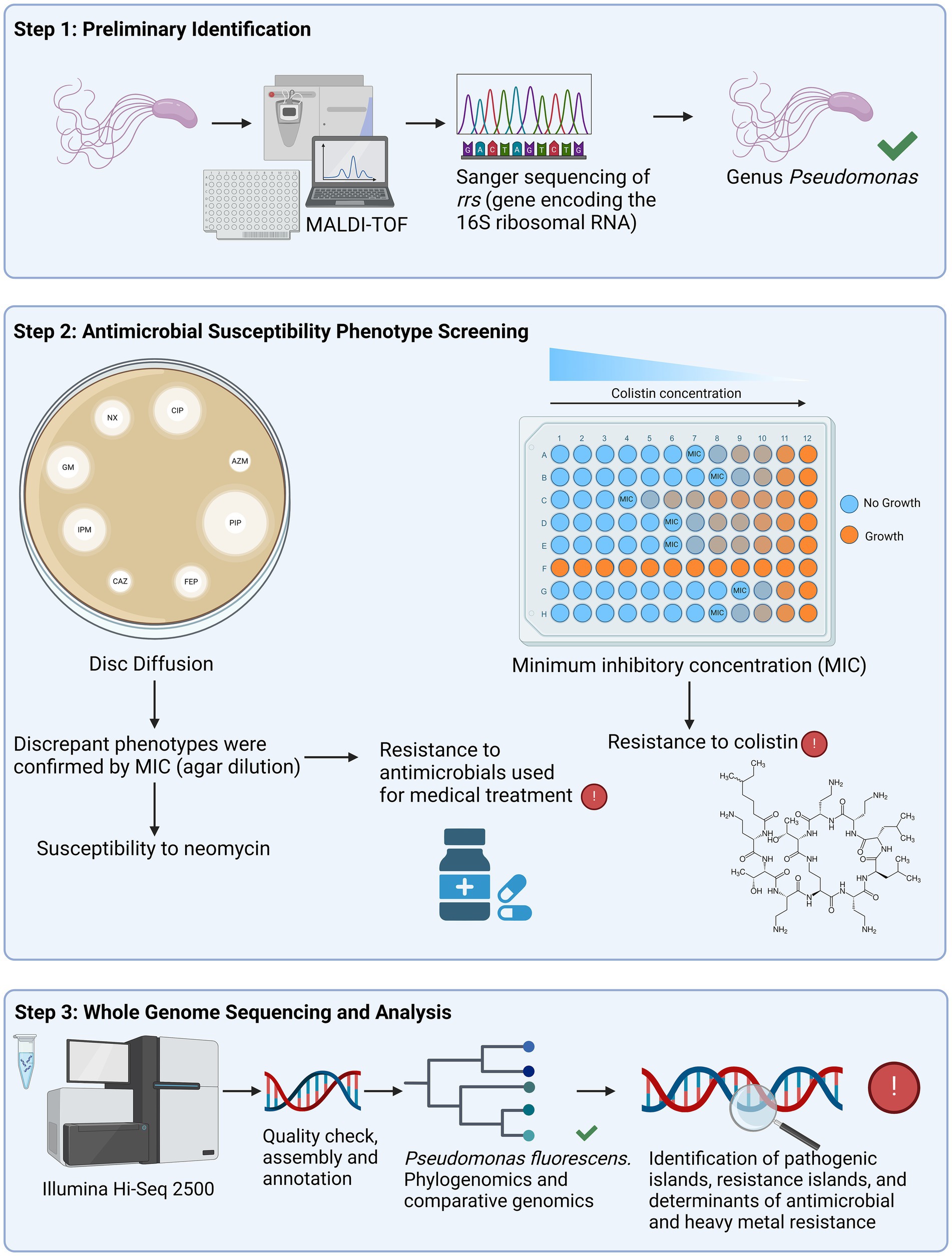
Figure 2. Schematic representation showing the division of the study in three steps. The first step was the preliminary identification using matrix-assisted laser desorption/ionization – time of flight (MALDI-TOF) and Sanger sequencing of the rrs gene, which encodes the 16S rRNA. Twenty-five isolates were confirmed to belong to the genus Pseudomonas and proceeded to the second step, which was the antimicrobial susceptibility screening. Antimicrobial agents were tested using disk diffusion, apart from colistin, which was analyzed with minimum inhibitory concentration (MIC) by broth microdilution [recommended by Clinical Laboratory Standards Institute (CLSI), 2023]. The isolates that displayed susceptibility to neomycin, and resistance to ceftazidime, cefepime and aztreonam were also evaluated with MIC by agar diffusion. The third step consisted of the whole genome sequencing (WGS) of fourteen non-clonal isolates, selected based on their resistance phenotypes. After the quality check and assembly of the raw sequences, we performed the phylogenomics and comparative genomics. Additionally, pathogenic and resistance islands, as well as antimicrobial and heavy metal resistance determinants were annotated. Created in BioRender. Silverio, M.P. https://BioRender.com/p48t351.
The isolates were incubated at 28°C with constant shaking at 150 rpm, until the OD600 reached 1 (approximately 48 h). Bacterial genomic DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega, Madison, Wisconsin, United States), following the manufacturer’s instructions. The DNA was quantified using a Qubit fluorometer (Invitrogen, Waltham, Massachusetts, United States) with the Qubit double-stranded DNA high sensitivity Assay Kit (Life Technologies, Carlsbad, California, United States).
First, the genetic diversity of the isolates was assessed via a repetitive extragenic palindromic polymerase chain reaction (BOX-PCR). The final concentration of each reagent was 1.0 μM of primer BOXA1-R CTACGGCAAGGCGACGCTGACG (Versalovic et al., 1994), 1.25 u of GoTaq® G2 DNA polymerase, 1X Green GoTaq® reaction buffer, 1.5 mM of MgCl2, 0.2 mM of deoxynucleotide triphosphate (Promega, Madison, Wisconsin, United States), and 50 ng/μL of genomic DNA with a final volume of 25 μL. The complete amplification cycle was one cycle of 95°C for 7 min, 30 cycles of 94°C for 1 min, 53°C for 1 min, 65°C for 8 min, and a final extension of one cycle at 65°C for 16 min in a thermocycler (Eppendorf, Hamburg, Germany). The products were analyzed using electrophoresis 1.5% agarose using the 1 kb DNA ladder (Thermo Fisher Scientific, Waltham, Massachusetts, United States). The run took place at 100 V for 30 min. The dendrograms were constructed using the program BioNumerics v.7, with default parameters (Biomérieux, Marcy-l’Étoile, France).
The amplification of the gene rrs was performed with 5 pmol/μL of each primer (27f AGAGTTTGATCATGGCTCAG and 1492r GTTTACCTTGTTACGACT) and a final fragment size of 1,465 base pairs (bp) (Lane, 1991). The reaction had a final volume of 50 μL, using the same concentrations of Taq, reaction buffer, MgCl2, deoxynucleotide triphosphate, and genomic DNA described above. The cycle was performed as follows: one cycle of 94°C for 3 min, 35 cycles of 94°C for 40 s, 55°C for 1 min, 72°C for 2 min, and a final extension of one cycle at 72°C for 10 min (Lane, 1991). The PCR products were analyzed as described above.
Additionally, each amplicon was purified with the enzyme ExoSAP (Exonuclease I, Shrimp Alkaline Phosphatase; Thermo Fisher Scientific, Waltham, Massachusetts, United States). The enzyme was diluted to a ratio of 1:9 in nuclease-free water (Qiagen, Hilden, Germany). Next, the following cycle was performed: one cycle of 37°C for 15 min (enzymatic optimal temperature) and one cycle of 80°C for 15 min (denaturation). In addition, 5 μL of pure amplicon and 5 μL of each primer (at a final concentration of 5 pmol/μL) were inoculated in a microplate and submitted for Sanger sequencing (Macrogen, Seoul, South Korea). The sequences were evaluated, trimmed and aligned using BioEdit v.7.2 (Hall, 1999). The species were defined using the tool Sequence Match, with the nonparametric k-nearest neighbors’ method, available at the Ribosomal Database Project (http://rdp.cme.msu.edu, accessed on March 15, 2019).
Antimicrobial susceptibility was assessed using the disk diffusion method, according to the protocol M02 established by the Clinical Laboratory Standards Institute (CLSI) (2012a). The tests were performed with piperacillin (PIP, 100 μg), aztreonam (AZM, 30 μg), piperacillin-tazobactam (TZP, 110 μg), ceftazidime (CAZ, 30 μg), cefepime (FEP, 30 μg), imipenem (IPM, 10 μg), gentamicin (GM, 10 μg), norfloxacin (10 μg) and ciprofloxacin (5 μg). To check the evolutionary aspects, we also tested antimicrobials known to be ineffective against P. aeruginosa. The list included tetracycline (TE, 30 μg), sulfamethoxazole-trimethoprim (25 μg), NEO (30 μg), chloramphenicol (C, 30 μg), ertapenem (ETP, 10 μg), ampicillin (AM, 10 μg), amoxicillin-clavulanate (AMC, 30 μg), cephalothin (CF, 30 μg), and cefotaxime (CTX, 30 μg). These antimicrobials were selected to evaluate whether the Antarctic P. fluorescens isolates exhibited similar resistance patterns to P. aeruginosa.
To evaluate extended-spectrum beta-lactamase (ESBL) phenotypes, PIP, AZM, CAZ, and FEP disks were positioned 2.5 cm from TZP, while AM, CF, and CTX disks were placed 2.0 cm from AMC. The antimicrobial disks represented the product “sensifar” and were commercially obtained from Cefar (São Paulo, Brazil), except PIP, which was prepared using the lyophilized drug from MilliporeSigma (Burlington, Massachusetts, United States). In addition, P. aeruginosa ATCC 27853 was the positive control strain, and the data were interpreted using CLSI M100 [Clinical Laboratory Standards Institute (CLSI), 2023].
The minimum inhibitory concentration (MIC) of colistin (CL; MilliporeSigma, Burlington, Massachusetts, United States) was accessed using broth microdilution. Each 0.5 McFarland suspension was diluted with a cation-adjusted medium (0.2 mL of Ca2+ and 0.1 mL of Mg2+ to each 100 mL of Mueller Hinton; Difco Laboratories Inc., Detroit, Michigan, United States) [Clinical Laboratory Standards Institute (CLSI), 2012b]. Serial dilutions of CL, with concentrations between 0.032 and 256 μg/mL, were evaluated. The strains P. aeruginosa ATCC 27853 (MIC 0.5–4 μg/mL) and E. coli ATCC 25922 (MIC 0.25–2 μg/mL) were used as susceptible controls, and E. coli C153 (carrying mcr-1, MIC 8 μg/mL) was employed as the CL-resistant control.
The MIC of the beta-lactams CAZ, FEP, AZM, and aminoglycoside NEO (MilliporeSigma, Brulington, Massachusetts, United States) were analyzed using agar dilution [Clinical Laboratory Standards Institute (CLSI), 2012b]. The beta-lactams were dissolved and diluted according to the method in previous work [Clinical Laboratory Standards Institute (CLSI), 2023]. Moreover, NEO was dissolved and diluted with sterile distilled water to a final concentration of 50 mg/mL, following the manufacturer’s instructions. Serial plates with concentrations varying between 1 and 256 μg/mL were analyzed, and P. aeruginosa ATCC 27853 was the control. The results were interpreted using the MIC breakpoints for other non-Enterobacterales [Clinical Laboratory Standards Institute (CLSI), 2023]. When the breakpoints for other non-Enterobacterales were unavailable (as in the case of CL), P. aeruginosa breakpoints were used.
Fourteen non-clonal isolates were selected for WGS. The selection criteria aimed to include isolates with high-level resistance phenotypes toward beta-lactams (specifically CAZ and FEP) and CL, and those with the most susceptible profiles.
Paired-end sequencing libraries (2×150 bp; 450 bp insert size) were constructed using 5 μg/μl of genomic DNA, following the NEBNext Fast DNA Fragmentation and Library Preparation Kit (New England Biolabs Inc., Ipswich, Massachusetts, United States). The quality control analysis of the final libraries was performed using the 2100 bioanalyzer (Agilent Technologies, Santa Clara, California, United States) and was visualized using electrophoresis 1.2% agarose. All samples were sequenced on the Illumina Hi-Seq 2500 platform (Illumina, San Diego, California, United States).
The quality of raw sequences was evaluated with FastQC v.0.11.5 (Andrews, 2010), and the reads and adaptors were trimmed using fastp v.0.23.4 (Chen et al., 2018) with default quality filter of >Q15. The genomes were assembled using Unicycler v.0.5.0 (Wick et al., 2017) with tested k-mer sizes of 27,53,71,87,99,111,119,127. The quality assessment of each assembly was checked with QUAST v.5.2.0 (Gurevich et al., 2013), CheckM2 v.1.0.2 (Chklovski et al., 2024) and GUNC v.1.0.2. As a quality filter, we considered N50 > 70 Kb (QUAST), completeness >90% (CheckM2), contamination <5% (CheckM2) and clade separation score > 0.45 (no chimeric contig) (GUNC). MOB-suite (Robertson and Nash, 2018) was used to identify plasmids in the draft genomes. The plasmid database used is available online at: https://zenodo.org/records/10304948/files/data.tar.gz, accessed on August 25, 2024. Default parameters were used for each software.
To determine the taxonomic classification of each Pseudomonas strain based on their genomes, we performed an analysis using the Genome Taxonomy Database Toolkit (GTDB-Tk) v.2.3.2 (Chaumeil et al., 2022) with the Classify workflow (“classify_wf”) and database r214. In the “ani_screen’ step it uses Mash v.2.3 (Ondov et al., 2016) to find the best hits among the representative genomes in the r214 database, then FastANI v.1.32 (Jain et al., 2018) to identify the species of the query genome using ≥95% as Average Nucleotide Identity (ANI) cutoff (Arahal, 2014). If the ANI analysis does not identify the query genome species the next steps are performed. In the “identify” step it employs Prodigal v.2.6.3 (Hyatt et al., 2010) and HMMER v.3.4 (Finn et al., 2011; Finn et al., 2015) for the identification of 120 bacterial phylogenetic marker genes and performs a multiple sequence alignment. The “align” step concatenates and filters the alignment. Finally, the “classify” step uses pplacer v1.1.alpha19-0-g807f6f3 (Matsen et al., 2010) to determine the place of the genome in the GTDB-Tk reference tree. The genomes were annotated using Prokka v.1.14.6 (Seemann, 2014).
The average nucleotide identity (ANIb) based on Basic Local Alignment Search Tool+ (BLAST+) and the correlation indices of tetra-nucleotide (TETRA) signatures of all analyzed genomes was run using default parameters on Pyani (Pritchard et al., 2016). The results were combined to assess the relationship between the genomes using the R package v.4.0.3, using the Euclidean distance and dist function from the statistics package for distance calculations. The hclust function from the statistics package was applied, using the average method for clustering calculations. Further details are provided in the online manuals (available at: https://www.rdocumentation.org/packages/stats/versions/3.6.2/topics/dist and https://www.rdocumentation.org/packages/stats/versions/3.6.2/topics/hclust, accessed on March 10, 2024).
A scatterplot of the correlation of ANIb and TETRA values was generated using the ggplot2 package (Wickham, 2016), and the correlation was evaluated using Spearman’s correlation and the cor.test function from the statistics package. The cor.test calculates an exact p-value when using “cor.test (clusteredAni$height, clusteredTetra$height, method = “spearman”).” The shapiro.test function from the statistics package was applied to assess the normality of the data, revealing a Gaussian distribution. The shapiro.test calculates an approximate p-value when using “shapiro.test (clusteredAni$height) and shapiro.test (clusteredTetra$height).” Further details are provided in the online manuals (available at: https://www.rdocumentation.org/packages/stats/versions/3.6.2/topics/shapiro.test and https://www.rdocumentation.org/packages/stats/versions/3.6.2/topics/cor.test, accessed on March 10, 2024). Additionally, the confidence of each clade was calculated with a bootstrap of 100 replicates using pvclust 2.2–0 (Suzuki and Shimodaira, 2006), and the factoextra package was employed to generate the dendrogram. For the visualization of the graphs,
We ran Benchmarking Universal Single-Copy Orthologs (BUSCO) (Simao et al., 2015) and BUSCO Phylogenomics (Waterhouse et al., 2018) to create the supermatrix. Besides, MAFFT (Kuraku et al., 2013) was employed to align the sequences of the supermatrix. We used the “--auto” option from MAFFT, which selects the appropriate alignment strategy amongst FFT-NS-2, FFT-NS-i and L-INS-I, according to the size of the input data. Gblocks (Talavera and Castresana, 2007) extracted the best-aligned blocks, using “sequence type equals protein (−t = p)” and “minimum length of a block equals 5 (−b4 = 5)” as the parameters. The output was converted to the Phylogeny Inference Package (PHYLIP) format using ClustalW2 (Larkin et al., 2007). The phylogenomic analysis was performed using RAxML (Stamatakis, 2014), using 100 bootstrap repetitions. The substitution model was defined by “-mPROTGAMMAWAG,” in which the model of heterogeneity is “GAMMA” and the substitution model is “LG.” Further details are provided in the online manual (available at: https://cme.h-its.org/exelixis/resource/download/NewManual.pdf, accessed on March 10, 2024). The tree was visualized and colored using iTOL v.7 (Letunic and Bork, 2024).
Genome Unclutterer (GUNC) v1.0.6 (Orakov et al., 2021) with clade separation score (CSS) > 0.45 was used to detect chimeras, contamination and the annotation of plasmids. For the taxonomy curation, type strain genome server (available at Type Strain Genome Server (dsmz.de); accessed on March 23, 2024) and GTDB-Tk v2 were employed (Chaumeil et al., 2022).
Antimicrobial resistance and further genes of interest were identified using the Genome Annotation tool available at the Pathosystems Resource Integration Center (PATRIC; available at Bacterial and Viral Bioinformatics Resource Center | BV-BRC, accessed on June 24, 2024) (Davis et al., 2020). For the annotation of ARGs, we applied the keywords “resistance,” “beta-lactam,” “penicillin,” “aminoglycoside,” “aph,” “aac,” “chloramphenicol,” “cat,” “polymyxin,” “arn,” “pmrK,” “tetracycline,” “tetR,” “efflux,” “ABC transporter,” and “ABC-type” using default parameters for Bacteria/Archaea. “Isopenicillin N epimerase” was detected for most of the isolates, but it was not included due to its importance on the antimicrobial biosynthesis pathway (not described in this work).
The keywords “heavy metal,” “arsenic” and “prophage” were also investigated. Relevant genes for the adaptation to extreme environments were searched and the function was manually assigned based on the annotation (Wattam et al., 2017).
The following databases were searched for ARGs using the ABRicate v.1.0.1 Pipeline: MEGARes v.3.0 (6635 sequences) (Lakin et al., 2017; Bonin et al., 2023), ResFinder v.4.1 (3077 sequences) (Bortolaia et al., 2020; Florensa et al., 2022), NCBI AMRFinderPlus v3.12.8 (5386 sequences) (Feldgarden et al., 2021; Feldgarden et al., 2022), ARG-ANNOT (2223 sequences) (Gupta et al., 2014), and CARD v.3.2.4 (2631 sequences) (Alcock et al., 2023; McArthur et al., 2013). We used ABRicate v.1.0.1 with a minimum identity threshold of 80% and a minimum coverage threshold of 80%. Databases were queried sequentially and overlapping predictions from multiple databases were considered a validation of results and retained in the final analysis. ABRicate was run on the Galaxy version 24.1.4.dev0 server, where its user-friendly interface allows for parameter configuration and database selection without requiring complex workflows. All databases (DbType nucI) were last updated November 4, 2023.
This study employed the Genomic Island Prediction Software (GIPSy) to check for genomic island availability (Soares et al., 2016). GIPSy uses an eight-step workflow for genomic island prediction, with each step incorporating specific default parameters to identify genomic features: Step 1 processes input files; Step 2 applies a G + C content deviation cutoff of 1.5 standard deviations; Step 3 uses a sensitivity setting of 0.95 for codon usage deviation (Colombo/SigiHMM); Step 4 predicts transposase genes with an HMMer e-value of 0.0001; Step 5 detects virulence or resistance factors using BLASTP with an e-value of 0.000001; Step 6 performs reciprocal BLAST with an e-value of 0.000001; Step 7 identifies tRNA flanking regions with an HMMer e-value of 0.0001; and Step 8 combines results from all previous steps to predict pathogenicity and resistance islands. All steps were executed using default settings provided by the software. In cases where regions were associated with more than one type of genomic island (pathogenic, metabolic, resistance, or symbiotic), PAIs and RIs were retained simultaneously, with overlapping regions plotted together at the same locus on the circular genomic graphs to reflect their dual classification. This research applied BLAST Ring Image Generation (Alikhan et al., 2011) to represent and evaluate the position of genomic islands in various strains of Pseudomonas, and the similarity between strains of the same species. The strain P. antarctica LMG 22709 (NZ_LT629704.1) was selected as the reference genome for GIPSy Island predictions. The strain P. carnis BML-PP010 (BQHE01000001.1) was applied as a reference for pathogenic strain. Genomic ring images were generated, and for groups with more than one sample, the largest genome was selected as the central ring for the circular plots.
The rrs sequences encoding the 16S gene are provided under the nucleotide accession numbers MK681799.1 to MK681824.1. The complete genome sequence data, including raw sequence reads, genome assemblies, and annotations of Pseudomonads applied in this study, were submitted to GenBank under the BioProject accession PRJNA1183857. Supplementary Table S2 displays the nucleotide accession numbers of the isolates evaluated using phylogenomic analysis.
A screening with six distinct classes of antimicrobials was performed using the disk diffusion method in accordance with CLSI standards [Clinical Laboratory Standards Institute (CLSI), 2012a; Clinical Laboratory Standards Institute (CLSI), 2023]. Similar to the control strain P. aeruginosa ATCC 27853, the 25 isolates presented resistance to representatives of four classes of antimicrobials: C, sulfamethoxazole-trimethoprim, TE, and beta-lactams (AM, AMC, CTX, CF, and ETP). The evaluated isolates in this work did not present a halo distortion, suggesting the absence of ESBL.
All isolates presented a putative phenotype susceptible to NEO. The diameter of the inhibition zones varied between 19 and 26 mm, in contrast to the control strain (no observed halo).
Regarding the beta-lactams with breakpoints available for the clinical treatment of other non-Enterobacterales, all isolates presented phenotypes intermediate or resistant to AZM. Moreover, 11 isolates (50%) were intermediate/resistant to CAZ, six (25%) to FEP and one (4%) to IPM (Supplementary Table S1).
The MIC by agar dilution confirmed the low susceptibility to AZM, CAZ, and FEP, with MICs varying from 32 to ≥256 μg/mL, whereas P. aeruginosa ATCC 27853 displayed an MIC of 4 μg/mL for CAZ and AZM and 8 μg/mL for FEP. This method also confirmed the susceptibility of the Antarctic isolates to NEO, with MICs ranging from 2 to 8 μg/mL, whereas the control strain P. aeruginosa ATCC 27853 presented an MIC ≥256 μg/mL.
As stated by Clinical Laboratory Standards Institute (CLSI) (2023), an MIC ≤2 μg/mL is considered intermediate to CL, and a value ≥4 μg/mL is considered resistant. Likewise, all 25 isolates described in this work were intermediate or resistant to this polymyxin. The MIC was 0.5 μg/mL for four isolates, 1 μg/mL for seven, 2 μg/mL for two, and 4 μg/mL for one. Eleven isolates (44%) displayed higher MICs than the positive control E. coli C153 (16 μg/mL): two were 128 μg/mL, and nine were ≥ 256 μg/mL. Table 1 presents the phenotypic data for 14 isolates selected for WGS. Supplementary Table S1 presents the complete data for the 25 phenotypically analyzed isolates.
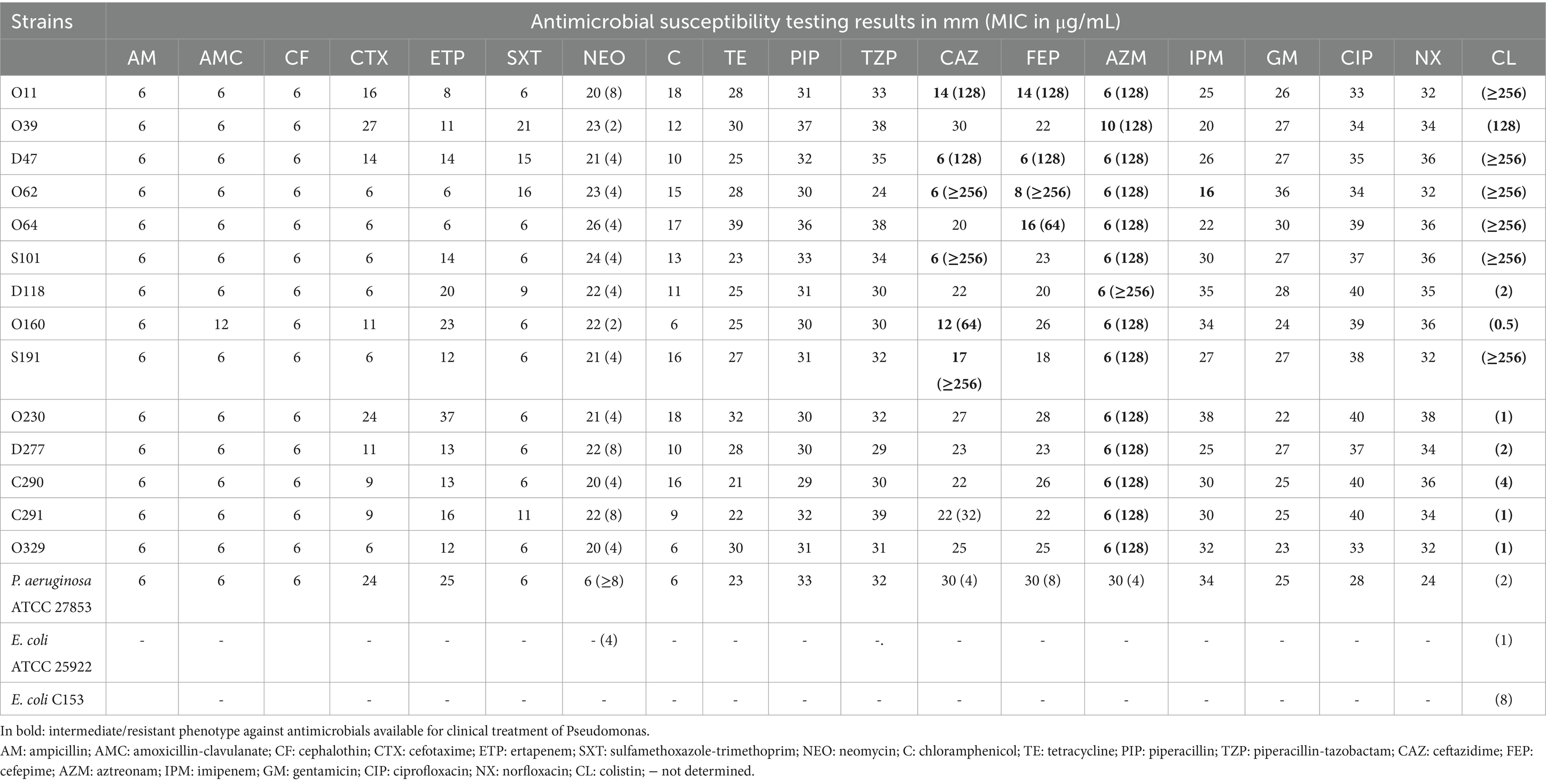
Table 1. Antimicrobial resistance profiles of 14 Pseudomonas sp. strains accessed using disk diffusion, with minimal inhibitory concentrations (MICs) for selected antimicrobial agents.
The MALDI-TOF analysis (scores between 1,703 and 2,259) confirmed that the isolates belong to the genus Pseudomonas. According to BOX-PCR, seven clusters ranged from two to four isolates, represented by clones (Supplementary Figure S1). Isolates representative of such genetic diversity or displaying diverse antimicrobial susceptibility profiles were selected for WGS.
The average genome size of the Pseudomonas was 6.5 Mb (varying between 5.8 and 7.6 Mb) and presented an average GC ratio of 59.7% (between 58 and 60%). In addition, an average of 6,135,071 protein coding sequences were identified. Integrated prophages were found across each genome, in which the most common encoded protein was Gifsy-2, followed by an antirepressor, CP4-57 regulatory, and Lp2 protein 6. Table 2 summarizes the general features of the draft genomes of the Pseudomonas isolates.
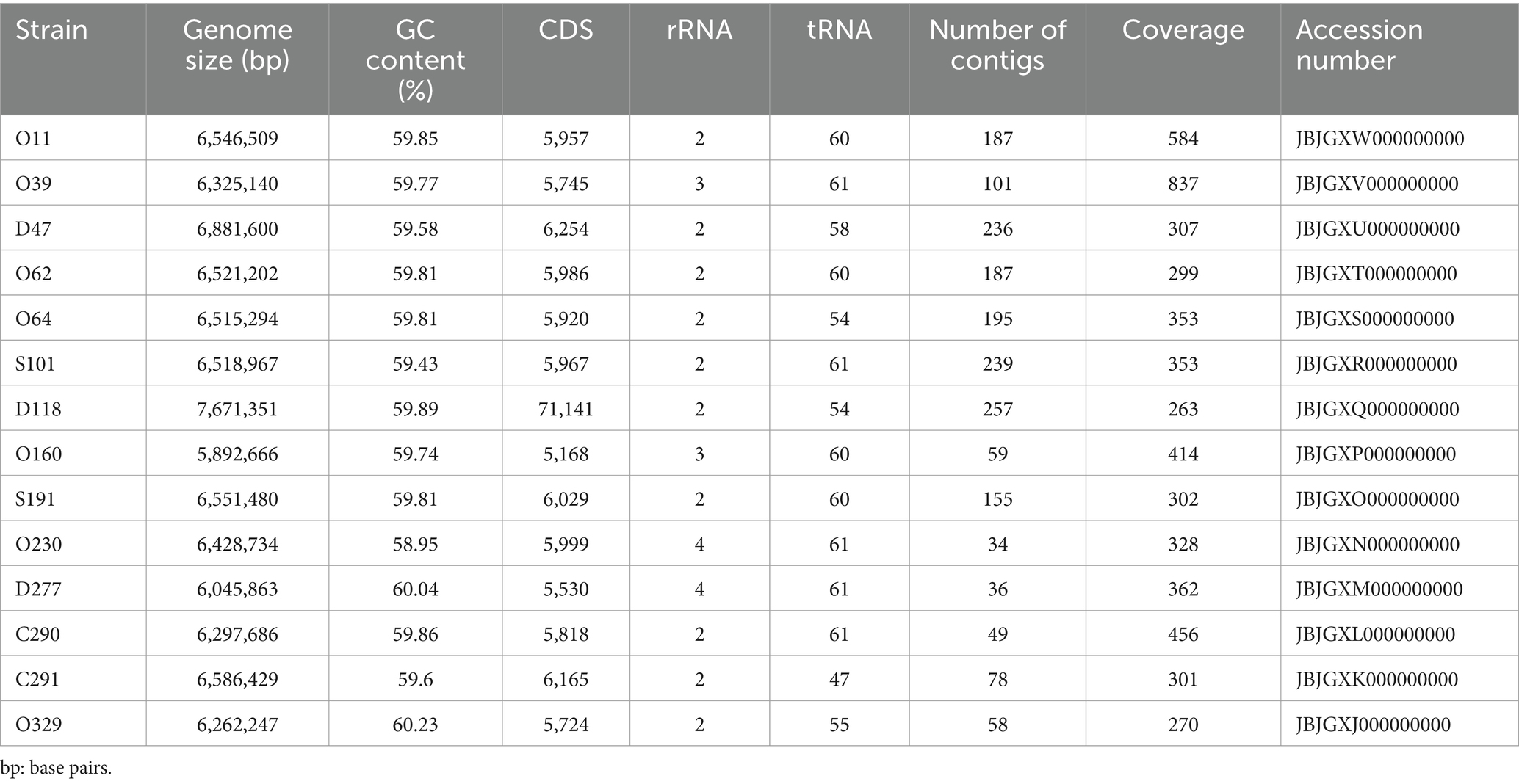
Table 2. Genome features of 14 strains isolated from Antarctic samples submitted to whole gene sequencing.
According to the phylogenomic analyses based on the average nucleotide identity (ANI) derived from the complete genome distance matrix and the correlation of tetra-nucleotide frequency (TETRA), the P. fluorescens isolates from Antarctic samples exhibited high similarity to each other (ANIb >98%). The isolates belong to the P. fluorescens group, forming two distinct clusters (Supplementary Figure S2). The first and largest cluster comprised the isolates C291, O329, and O62 from a recent common ancestor, whereas C290, S191, S101, D118, and O160 were distant from each other. In contrast, the second cluster displayed less divergence between the isolates O230, O39, D277, O11, D47, and O64. Moreover, ANIb and TETRA varied between 0.0 and 0.3, with an outlier after 0.4.
The taxonomy was curated using GTDB-Tk (Chaumeil et al., 2022) and the Type Strain Genome Server, and all isolates belong to the P. fluorescens group. In general, three potential new species were identified (Figure 3). Apart from O62 and O64, presenting a completeness of 99.99%, all genomes presented a completeness of 100%. Furthermore, all presented a very low contamination ratio [between 0 (C290) and 1.49 (D118)], indicating that contaminant contigs from other genomes were not identified.
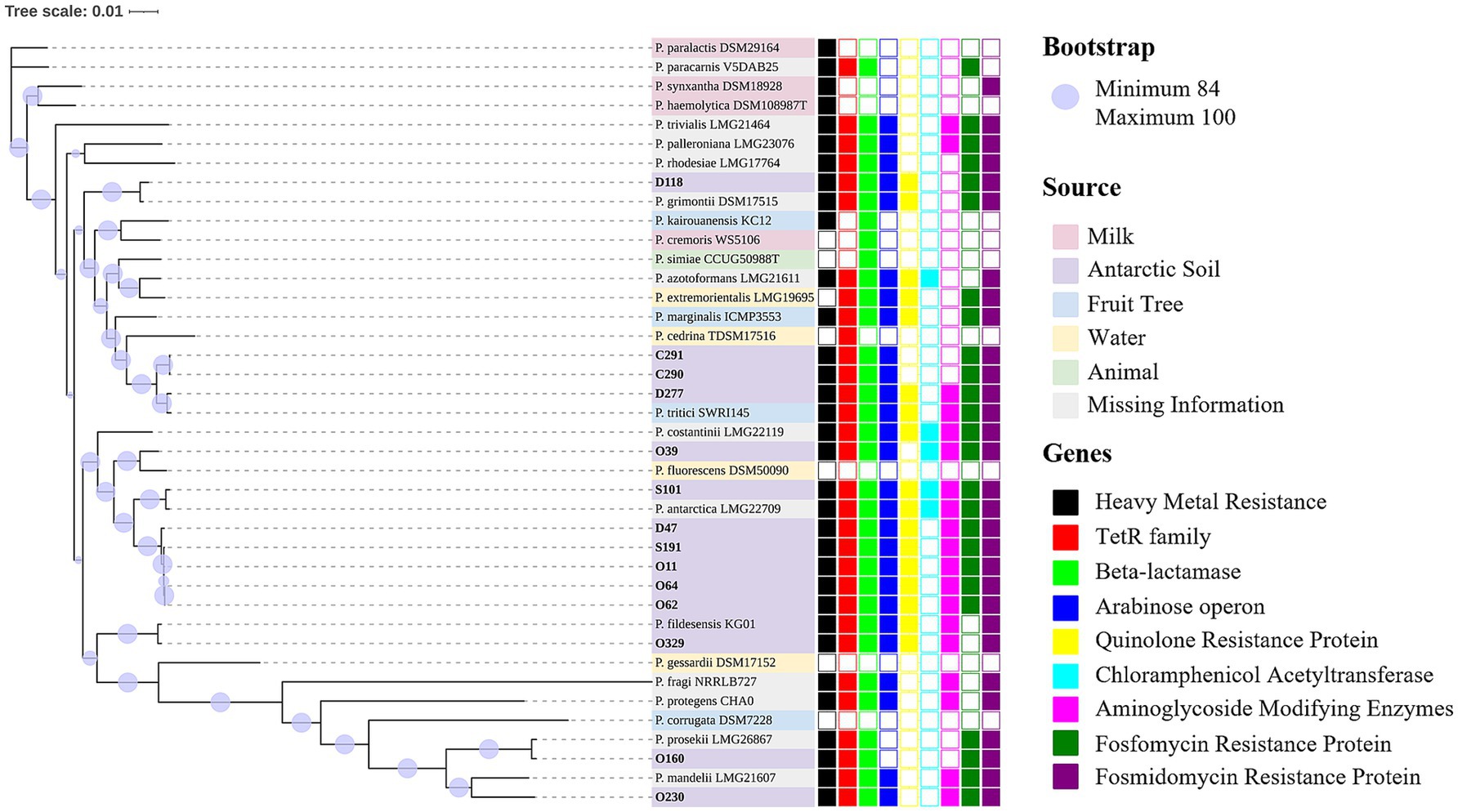
Figure 3. Phylogenomic analysis of Antarctic Pseudomonas fluorescens isolates (in bold). Bootstrap values range from 84 to 100 and are displayed as purple circles. Three potential new species were identified. The first cluster of potential new species isolates was D47, O62, O64, O11, and S191. The second comprises isolate O39, with the closest similarity to P. fluorescens DSM 50090. The third was O230, which is closest to P. mandelii LMG 21607. Regarding previously described species, S101 was identified as P. antarctica and is close to P. antarctica LGM 22709. Strain D118 was identified as P. grimontii, which is close to P. grimontii DSM 17515. Isolate O160 was identified as P. prosekii similar to P. prosekii LMG 26867, whereas O329 (P. fildesensis) is similar to P. fildesensis KG01. The Antarctic isolates D277, C290, and C291 belong to the species P. tritici and formed a cluster with P. tritici SWRI 145. Supplementary Table S2 presents the nucleotide accession numbers of the reference strains. The source is indicated by the label colors: milk (pink), Antarctic soil (purple), fruit tree (blue), water (yellow), animal (green) and missing information (grey). Gene annotation is displayed as squares. The full squares display the presence of the genes, while the empty squares show the absence. Resistance genes were identified for heavy metal (black), tetracycline (red), beta-lactamase (lime), colistin (operon arn; blue), quinolone (yellow), chloramphenicol (cyan), aminoglycoside (magenta), fosfomycin (green) and fosmidomycin (purple). Additionally, genes encoding efflux pumps were frequently detected in large quantities. Genes of adaptation to extreme conditions were detected in each isolate.
This study investigated the genes potentially involved in TE (tetR), aminoglycoside [aph(3′) and aac(6′)], polymyxin (arnC, D, E, F), fosfomycin (fosA) and, to a lesser extent, C (cat). Unknown quinolone and fosmidomycin resistance proteins were also detected. The quinolone resistance protein, which could possibly confer resistance to the first generation (i.e., nalidixic acid; not assayed in this work), was identified in nine isolates and presented 100% coverage and over 99% identity with other P. fluorescens isolates. The penicillinases penicillin-insensitive transglycosylase (EC 2.4.2.-), transpeptidase penicillin-binding protein (PBP-1C), other PBPs, class C beta-lactamases (EC 3.5.2.6) and metallo-beta-lactamases were frequently identified (Figure 3; Table 3).
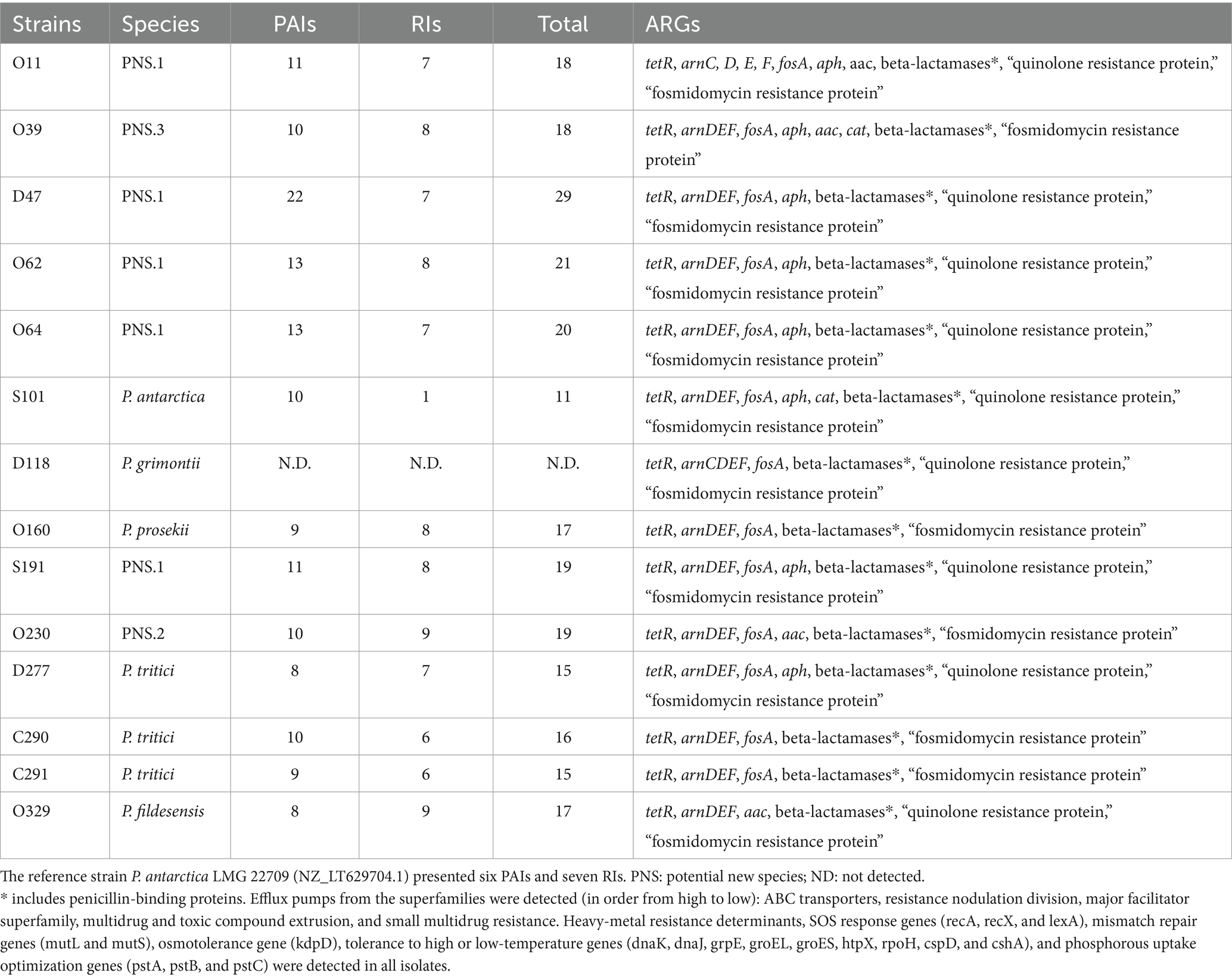
Table 3. Description of Pseudomonas fluorescens spp. and annotation of relevant pathogenicity islands (PAIs), resistance islands (RIs), antimicrobial resistance genes (ARGs), and prophages.
Hundreds of copies of genes encoding efflux pumps were predominantly found in each sample. In order of frequency, all five well-known efflux pump families were detected: ABC transporters (including the gene encoding the macrolide-specific efflux protein MacA), resistance nodulation division, major facilitator superfamily, multidrug and toxic compound extrusion, and small multidrug resistance proteins (Table 3; Supplementary Table S3). The overexpression of efflux pumps, alongside the activity of beta-lactamases, could be responsible for the observed phenotype of resistance towards AZM, CAZ and FEP. Further experiments with efflux pump inhibitors could help in the understanding of the resistance mechanism.
The beta-lactamase blaPFM-2 responsible for carbapenem resistance, which was identified in only one of the tested isolates (strain D47), was identified using NCBI AMRFinderPlus. In the databases CARD and MEGARes, only efflux pump genes from the resistance nodulation division family were detected (Supplementary Table S3). No ARGs were detected using the databases ResFinder and ARG-ANNOT. Moreover, only one plasmid was detected (isolate D118), which did not exhibit antimicrobial resistance determinants (Supplementary Figure S3). Overall, the data suggest that the variety of efflux pumps in the P. fluorescens genomes evaluated in this work plays a significant role in the observed antimicrobial resistance.
Apart from the isolate D118, PAIs and RIs were identified in all characterized genomes. The PAIs were more frequent, varying from six to 22, whereas up to nine RIs were identified. In general, the genomes described in this work presented higher numbers of PAIs and RIs than the pathogenic reference strain (P. carnis BML-PP010; BQHE01000001.1). The isolate D47 presented the highest total PAIs and RIs (n = 29), whereas S101 was even lower than the pathogenic reference strain (11 versus 13; Table 3). Supplementary Figure S4 presents the circular genome comparison plots displaying the islands identified against the genus Pseudomonas, reference strain P. antarctica LMG 22709 (NZ_LT629704.1) and pathogenic reference Pseudomonas carnis BML-PP010 (BQHE01000001.1).
Copies of genes encoding the heavy-metal response regulator, heavy-metal sensor histidine kinase and heavy-metal resistance transcriptional regulator (hmrR) were identified in the genomes. One copy of a membrane-bound cytochrome biogenesis cycz-like domain, annotated as a heavy-metal associated domain, was identified in each genome. In addition, genes encoding arsenic resistance proteins were also found, varying from zero to five copies.
The DNA repair system genes (recA, recX, mutL, and mutS) responsible for the resistance to ionizing radiation were identified in each genome. Likewise, lexA (signaling and regulation) and kdpD (osmotolerance) were also observed. Genes conferring resistance to high pressure or high temperature (dnaK, groEL, dnaJ, grpe, groES, htpX, and rpoH) were also detected, and two genes responsible for the resistance to low temperatures (cspD and cshA) were found. The genes pstA, pstB, and pstC involved in the optimization of phosphorus uptake were detected. The frequency of these bacterial adaptation genes was generally low (from one to three copies), except for dnaJ, where the genomes presented three to five copies each.
Horizontal gene transfer has played a critical role in the appearance of antimicrobial resistance in human-affected sites, which is sometimes also the case for so-called pristine environments. Previous studies have identified members of the P. fluorescens complex harboring ARGs in pristine or human/animal migration-affected areas in Antarctica (Orellana-Saez et al., 2019; Na et al., 2021; Marcoleta et al., 2022). Furthermore, genes conferring resistance to glycopeptides (vanA/vanD and vanB), methilicin (mecA-), and the New Delhi metallo-beta-lactamase (blaNDM) were recently identified in the animal feces of native Antarctic animals (Dimov and Strateva, 2022). Similarly, a study published in 2019 reported the presence of sulfonamide resistance genes (sul1 and sul2) and a quinolone resistance gene (qnrS) in fecal samples collected from animals in the Fildes Peninsula, King George Island, Antarctica (Na et al., 2019). A positive correlation between sul1 and int1 was identified, suggesting that int1 could be involved in spreading ARGs. Sellera and colaborators (Sellera et al., 2017) documented cases of migratory Magellanic penguins (Spheniscus magellanicus) found on the southeast coast of Brazil. These penguins, suffering from pododermatitis, carried E. coli with mcr-1 and blactx-m genes, which confer resistance to colistin and ESBLs, respectively. In the future, research focused on monitoring migratory animals could provide valuable insights into the evolution of antimicrobial resistance and the global dissemination of ARGs.
Antimicrobial resistance is often linked to metal resistance. Metal pollution reaches polar regions through atmospheric and oceanic circulation or through transport by migratory animals (Blais et al., 2005; Mechirackal Balan et al., 2018). Two mechanisms are known to drive the co-selection of metal and antimicrobial resistance. The first, known as “co-resistance,” involves metal- and antimicrobial-resistance determinants being encoded on the same mobile genetic element. The second, referred to as “cross-resistance,” occurs when a single mechanism, such as the overexpression of efflux pumps, confers resistance to both metals and antimicrobial agents (Henriques et al., 2016; Seiler and Berendonk, 2012). Previously, P. frederiksbergensis strain SS18 was highly resistant to mercury and to seven tested antimicrobials (unspecified) (Mechirackal Balan et al., 2018). The strain was isolated from Ny-Ålesund, Svalbard, Arctic, where coal was commercially exploited until the 1960s (Mechirackal Balan et al., 2018). Additionally, mercury and tellurite cross-resistance have previously been identified in three Pseudomonas isolates (Rodriguez-Rojas et al., 2016). The same isolates were resistant to nearly all tested antimicrobials (unspecified, but they were susceptible to amikacin, GM, and ciprofloxacin) (Rodriguez-Rojas et al., 2016).
Efflux pumps play a critical role in the extrusion of toxic compounds (Olivares Pacheco et al., 2017) and have been largely detected in Antarctic microbial isolates. For instance, Pseudomonas sp. strain MPC6, which was isolated from a soil sample on Deception Island (Antarctica), lacks genes encoding aminoglycoside-modifying enzymes, beta-lactamases and chloramphenicol acetyltransferases. Nevertheless, its genome carried genes encoding a variety of efflux pumps (Orellana-Saez et al., 2019). Previously, two P. fluorescens isolates carrying the efflux pump EmhABC presented resistance to C, nalidixic acid, AM, and TE (Hearn et al., 2003; Hearn et al., 2006; Tian et al., 2010). In this work, the observed C-resistant phenotype could be primarily due to the extrusion by efflux pumps, as cat genes were detected in only two of the isolates described here (Figure 3). Additionally, eleven isolates presented a phenotype intermediate/resistant to CAZ. Although this beta-lactam is considered for medical treatment against Pseudomonas infection [Clinical Laboratory Standards Institute (CLSI), 2023], resistance to CAZ in combination with avibactam was previously related to the presence of blaVIM-1 and blaVIM-2 and the overexpression of MexAB-OprM (Castanheira et al., 2019). Recently, Marcoleta et al. (2022) reported that two multidrug-resistant P. fluorescens isolates did not present ARGs in common with the reference strain P. aeruginosa PA7. Instead, these P. fluorescens isolates displayed a higher number of genes associated with ABC transporter and SMR efflux pumps (Marcoleta et al., 2022). Conducting functional assays on the activity of efflux pumps in Antarctic P. fluorescens will offer valuable insights.
In this study, the isolates displayed a multidrug-resistant phenotype, likely due to intrinsic features. The tetR gene, found in all of the isolates, is commonly found in the genus Pseudomonas because of its function of controlling the expression of genes involved in antimicrobial resistance and enzymes from catabolic pathways, the biosynthesis of antimicrobials, osmotic stress, and pathogenicity (Liu et al., 2013; Zhang et al., 2022; Shah and Sorum, 2014). The previous detection of the gene cluster sul2-strA-strB in ice cores from Dome Fuji Station (Eastern Antarctica) highlights the hypothesis that ARGs present distinct functions and may have existed before the preantimicrobial era (Okubo et al., 2019). Additionally, copies of genes encoded by the operon arnBCADTEF (previously known as pmrHFIJKLM), conferring polymyxin resistance in Gram-negative bacteria (Munoz-Escudero et al., 2023), were identified in most of the isolates (except O160, which presented an MIC of 0.5 μg/mL for CL). When the operon arn is induced, a 4-amino-4-deoxy-L-arabinose is added to the lipid A structure (Fernandez et al., 2010; Silverio et al., 2022; Moskowitz and Ernst, 2010; Munoz-Escudero et al., 2023). The isolates that represented a potential new species presented the highest observed MICs for CL (128 and ≥ 256 μg/mL; Table 1).
Pseudomonads are often intrinsically resistant to aminoglycosides due to chromosomal aminoglycoside-modifying enzymes (Zeng and Jin, 2003; Papapetropoulou et al., 1994). Although all isolates were susceptible to NEO, we identified aminoglycoside phosphotransferase [aph(−3′)] and aminoglycoside acetyltransferase [aac-(6′)] genes in most genomes (except for the isolates D118, O160, C290, and C291). The aph(3′) gene encodes phosphotransferases that confer resistance to NEO and kanamycin (Papapetropoulou et al., 1994; Zeng and Jin, 2003), while the aac(6′) gene encodes acetyltransferases that are active against a broad range of aminoglycosides, with the exception of gentamicin (GM) (Kawabe et al., 1975; Kobayashi et al., 2013). Furthermore, the phosphate uptake gene pstB was identified in all isolates, being previously linked to the intrinsic resistance of P. aeruginosa to aminoglycosides (Krahn et al., 2012). Although the isolates were susceptible to NEO, the lack of sequence homology prevented checking for gene mutations. Further transcriptomic analysis is necessary to investigate whether these genes are inactive.
The Antarctic P. fluorescens isolates exhibited resistance to AZM, an antimicrobial agent used in clinical treatment against P. aeruginosa. A previous study also identified AZM and carbapenem resistance in P. fluorescens isolates from chicken meat (Heir et al., 2021). While acquired beta-lactamase genes were absent in these isolates, the authors detected genes encoding efflux pumps, as well as blaAmpC and the PBP-encoding gene mrcA. Additionally, some isolates presented the gene pbpC, encoding a PBP3 homolog that might behave as a target for AZM (Heir et al., 2021; Jorth et al., 2017). In our study, we found that the Antarctic P. fluorescens isolates frequently harbor genes encoding various PBPs. Previous studies have reported that P. fluorescens isolates from pristine environments were resistant to several clinically important antimicrobial agents, including AZM, PIP, CAZ, CL, and various carbapenems (Pavlov et al., 2020; Poblete-Morales et al., 2020; Svec et al., 2020). One isolate, identified as P. fildesensis, was collected from Antarctic soil at the King George Island and exhibited genomic islands and other likely acquired mobile genetic elements (Pavlov et al., 2020). These findings underscore the potential pathogenicity of P. fluorescens isolates from remote environments.
The isolates presented not only a vast amount of efflux pumps, but also antimicrobial- and heavy-metal resistance genes (Figure 3; Table 3; Supplementary Table S3). The determinants hmrR, “DNA binding heavy-metal response regulator,” and “heavy-metal sensor histidine kinase,” alongside genes specifically related to arsenic resistance, were frequently identified. Previously, sodium arsenate and sodium arsenite intrinsic resistance were described in P. fluorescens, encoded by an operon with an arsenite inducible repressor (regulating the expression of arsenate reductase) and an ATP-dependent efflux pump (Prithivirajsingh et al., 2001). However, conducting functional assays to validate resistance to heavy metal is crucial.
When using genomic data for taxonomy assignments, the query genomes were compared to a database of type strains or reference genomes. The cutoff values for considering two genomes from the same species are ANI > 95% (Jain et al., 2018), dDDH >70% and < 1% divergence of G + C content (Meier-Kolthoff and Goker, 2019). Based on our results, all genomes are from the genus Pseudomonas (Supplementary Table S4). Within the genomes, the already described species are P. antarctica (S101), P. fildesensis (O329), P. grimontii (D118), P. prosekii (O160) and P. tritici (D277, C290 and C291). The novel species are PNS 1 (O11, D47, O62, O64 and S191), PNS 2 (O230), and PNS 3 (O39).
In this study, all of the described isolates belonged to the Pseudomonas fluorescens complex. Seven isolates derived from ornithogenic soil, five from the rhizosphere of native Antarctic plants (Deschampsia antarctica and Colobanthus quitensis) and two from soil beneath moss (Sanionia uncinata). One plasmid was detected, but it did not carry ARGs (P. grimontii D118; Supplementary Figure S2). The findings suggest that the observed antimicrobial-resistant phenotypes occurred due to intrinsic features. Nevertheless, we found a beta-lactamase gene encoding a PFM-like carbapenemase in one isolate (D47), which could pose a severe threat to clinical health. This PFM-like metallo-beta-lactamase was previously found in P. synxantha from chicken meat and was described by Poirel et al. (2020). The shared amino acid identity was over 90% for other species belonging to the P. fluorescens complex, indicating that this complex might function similarly to a reservoir (Poirel et al., 2020). Although the isolate did not present a discrepant phenotype when compared to the remaining Antarctic P. fluorescens isolates, further research based on transcriptomic and proteomic approaches need to be conducted, especially because the isolate D47 also presented the highest number of PAIs and RIs.
This study examines the evolutionary characteristics of antimicrobial resistance in P. fluorescens isolates from pristine environments in Antarctica. Resistance was observed to beta-lactams commonly used in clinical treatment, including aztreonam and ceftazidime, while resistance to cefepime and imipenem was detected to a lesser degree. Most of the isolates harbored genes typically considered intrinsic to the Pseudomonas genus, encoding promiscuous enzymes. Interestingly, despite the presence of aminoglycoside-modifying enzymes, the isolates remained susceptible to neomycin, indicating that the corresponding gene was likely inactive. Neomycin, an antimicrobial agent known to be ineffective against P. aeruginosa, was tested as part of an investigation into whether P. fluorescens from Antarctica would exhibit similar resistance patterns. Additionally, several copies of genes related to efflux pumps, heavy metal resistance, prophages, and adaptations to extreme environments were identified. These findings suggest that functional assays, transcriptomics, and proteomics would be crucial for further exploring the roles and functionality of these genes.
The rrs sequences encoding the 16S gene are provided under the nucleotide accession numbers MK681799.1 to MK681824.1. The complete genome sequence data, including raw sequence reads, genome assemblies, and annotations of Pseudomonads applied in this study, were submitted to GenBank under the BioProject accession PRJNA1183857. Supplementary Table S2 displays the nucleotide accession numbers of the isolates evaluated using phylogenomic analysis.
MPS: Writing – original draft, Writing – review & editing, Data curation, Formal analysis, Investigation, Methodology, Validation. JS: Formal analysis, Validation, Writing – original draft, Writing – review & editing. MTDP: Formal analysis, Writing – original draft, Writing – review & editing, Investigation, Methodology. DP: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. MVCV: Formal analysis, Investigation, Writing – original draft, Writing – review & editing. WN: Formal analysis, Investigation, Writing – original draft, Writing – review & editing, Data curation. RTJR: Data curation, Formal analysis, Writing – original draft, Writing – review & editing, Software. AG-N: Data curation, Formal analysis, Software, Writing – original draft, Writing – review & editing. VACA: Software, Writing – original draft, Writing – review & editing, Methodology, Validation, Visualization. BB: Writing – original draft, Writing – review & editing, Resources. RRB: Writing – original draft, Writing – review & editing, Conceptualization, Formal analysis, Investigation, Methodology, Supervision. ASR: Supervision, Writing – original draft, Writing – review & editing, Funding acquisition, Project administration, Resources.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by grants from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Programa Antártico Brasileiro (PROANTAR). This study was also financed in part by INPRA (CNPq 465718/2014-0; FAPERGS17/2551-0000514-7) and the CAPES Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) grant #E-26/211.554/2019. J.S. and A.S.R. were supported by the King Abdullah University of Science and Technology Baseline Grant (BAS/1/1096-01-01).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1535420/full#supplementary-material
Alcock, B. P., Huynh, W., Chalil, R., Smith, K. W., Raphenya, A. R., Wlodarski, M. A., et al. (2023). CARD 2023: expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res. 51, D690–D699. doi: 10.1093/nar/gkac920
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Allen, H. K., Moe, L. A., Rodbumrer, J., Gaarder, A., and Handelsman, J. (2009). Functional metagenomics reveals diverse beta-lactamases in a remote Alaskan soil. ISME J. 3, 243–251. doi: 10.1038/ismej.2008.86
Andrews, S. (2010). 'FastQC: A quality control tool for high throughput sequence Data', Babraham bioinformatics. Cambridge, UK: Babraham Institute.
Arahal, D. R. (2014). “Chapter 6—whole-genome analyses: average nucleotide identity” in Methods in microbiology. eds. M. Goodfellow, I. Sutcliffe, and J. Chun (Academic Press), 41, 103–122.
Blais, J. M., Kimpe, L. E., McMahon, D., Keatley, B. E., Mallory, M. L., Douglas, M. S., et al. (2005). Arctic seabirds transport marine-derived contaminants. Science 309:445. doi: 10.1126/science.1112658
Bonin, N., Doster, E., Worley, H., Pinnell, L. J., Bravo, J. E., Ferm, P., et al. (2023). MEGARes and AMR++, v3.0: an updated comprehensive database of antimicrobial resistance determinants and an improved software pipeline for classification using high-throughput sequencing. Nucleic Acids Res. 51, D744–D752. doi: 10.1093/nar/gkac1047
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. doi: 10.1093/jac/dkaa345
Castanheira, M., Doyle, T. B., Smith, C. J., Mendes, R. E., and Sader, H. S. (2019). Combination of MexAB-OprM overexpression and mutations in efflux regulators, PBPs and chaperone proteins is responsible for ceftazidime/avibactam resistance in Pseudomonas aeruginosa clinical isolates from US hospitals. J. Antimicrob. Chemother. 74, 2588–2595. doi: 10.1093/jac/dkz243
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2022). GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316. doi: 10.1093/bioinformatics/btac672
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chevalier, S., Bouffartigues, E., Bodilis, J., Maillot, O., Lesouhaitier, O., Feuilloley, M. G. J., et al. (2017). Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 41, 698–722. doi: 10.1093/femsre/fux020
Chklovski, A., Parks, D. H., Woodcroft, B. J., and Tyson, G. W. (2024). Author correction: CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 21:735. doi: 10.1038/s41592-024-02248-z
Clinical Laboratory Standards Institute (CLSI). (2012a). Performance standards for antimicrobial disk susceptibility tests; approved standard, M02-A11. Wayne, PA: Clinical and Laboratory Standards Institute.
Clinical Laboratory Standards Institute (CLSI). (2012b). Methods for dilution antimicrobial susceptibility tests for Bacteria that grow aerobically; approved standard, M07-A9. Wayne, PA: Clinical and Laboratory Standards Institute.
Clinical Laboratory Standards Institute (CLSI) (2023). Performance standards for antimicrobial susceptibility testing, M100. Clinical and Laboratory Standards Institute.
Cowan, D. A., Chown, S. L., Convey, P., Tuffin, M., Hughes, K., Pointing, S., et al. (2011). Non-indigenous microorganisms in the Antarctic: assessing the risks. Trends Microbiol. 19, 540–548. doi: 10.1016/j.tim.2011.07.008
Craig, K., Johnson, B. R., and Grunden, A. (2021). Leveraging Pseudomonas stress response mechanisms for industrial applications. Front. Microbiol. 12:660134. doi: 10.3389/fmicb.2021.660134
da Silva, A. C., Rachid, C. T. C. D. C., De Jesus, H. E., Rosado, A. S., and Peixoto, R. S. (2017). Predicting the biotechnological potential of bacteria isolated from Antarctic soils, including the rhizosphere of vascular plants. Polar Biol. 40, 1393–1407. doi: 10.1007/s00300-016-2065-0
Davis, J. J., Wattam, A. R., Aziz, R. K., Brettin, T., Butler, R., Butler, R. M., et al. (2020). The PATRIC bioinformatics resource center: expanding data and analysis capabilities. Nucleic Acids Res. 48, D606–D612. doi: 10.1093/nar/gkz943
Dimov, S. G., and Strateva, T. (2022). Detection of clinically relevant antimicrobial resistance determinants in warm-blooded marine animals in Livingston Island (South Shetland Islands, Antarctica): a field-based molecular genetics study. Mar. Pollut. Bull. 180:113751. doi: 10.1016/j.marpolbul.2022.113751
Faccone, D., Pasteran, F., Albornoz, E., Gonzalez, L., Veliz, O., Prieto, M., et al. (2014). Human infections due to Pseudomonas chlororaphis and Pseudomonas oleovorans harboring new Bla(VIM-2)-borne integrons. Infect. Genet. Evol. 28, 276–277. doi: 10.1016/j.meegid.2014.10.012
Feldgarden, M., Brover, V., Fedorov, B., Haft, D. H., Prasad, A. B., and Klimke, W. (2022). Curation of the AMRFinderPlus databases: applications, functionality and impact. Microb Genom 8:mgen000832. doi: 10.1099/mgen.0.000832
Feldgarden, M., Brover, V., Gonzalez-Escalona, N., Frye, J. G., Haendiges, J., Haft, D. H., et al. (2021). AMRFinderPlus and the reference gene catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 11:12728. doi: 10.1038/s41598-021-91456-0
Fernandez, L., Gooderham, W. J., Bains, M., McPhee, J. B., Wiegand, I., and Hancock, R. E. (2010). Adaptive resistance to the "last hope" antibiotics polymyxin B and colistin in Pseudomonas aeruginosa is mediated by the novel two-component regulatory system ParR-ParS. Antimicrob. Agents Chemother. 54, 3372–3382. doi: 10.1128/AAC.00242-10
Finn, R. D., Clements, J., Arndt, W., Miller, B. L., Wheeler, T. J., Schreiber, F., et al. (2015). HMMER web server: 2015 update. Nucleic Acids Res. 43, W30–W38. doi: 10.1093/nar/gkv397
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Florensa, A. F., Kaas, R. S., Clausen, P. T. L. C., Aytan-Aktug, D., and Aarestrup, F. M. (2022). ResFinder—an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb Genom 8:000748. doi: 10.1099/mgen.0.000748
Gupta, S. K., Padmanabhan, B. R., Diene, S. M., Lopez-Rojas, R., Kempf, M., Landraud, L., et al. (2014). ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58, 212–220. doi: 10.1128/AAC.01310-13
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucl. Acids. Symp. Ser. 41, 95–98.
Hearn, E. M., Dennis, J. J., Gray, M. R., and Foght, J. M. (2003). Identification and characterization of the emhABC efflux system for polycyclic aromatic hydrocarbons in Pseudomonas fluorescens cLP6a. J. Bacteriol. 185, 6233–6240. doi: 10.1128/JB.185.21.6233-6240.2003
Hearn, E. M., Gray, M. R., and Foght, J. M. (2006). Mutations in the central cavity and periplasmic domain affect efflux activity of the resistance-nodulation-division pump EmhB from Pseudomonas fluorescens cLP6a. J. Bacteriol. 188, 115–123. doi: 10.1128/JB.188.1.115-123.2006
Heir, E., Moen, B., Asli, A. W., Sunde, M., and Langsrud, S. (2021). Antibiotic resistance and phylogeny of Pseudomonas spp. isolated over three decades from chicken meat in the Norwegian food chain. Microorganisms 9:207. doi: 10.3390/microorganisms9020207
Henriques, I., Tacao, M., Leite, L., Fidalgo, C., Araujo, S., Oliveira, C., et al. (2016). Co-selection of antibiotic and metal(loid) resistance in gram-negative epiphytic bacteria from contaminated salt marshes. Mar. Pollut. Bull. 109, 427–434. doi: 10.1016/j.marpolbul.2016.05.031
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jain, C., Lm Rodriguez, R., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jorth, P., McLean, K., Ratjen, A., Secor, P. R., Bautista, G. E., Ravishankar, S., et al. (2017). Evolved Aztreonam resistance is multifactorial and can produce Hypervirulence in Pseudomonas aeruginosa. MBio 8:e00517-17. doi: 10.1128/mBio.00517-17
Kawabe, H., Kondo, S., Umezawa, H., and Mitsuhashi, S. (1975). R factor-mediated aminoglycoside antibiotic resistance in Pseudomonas aeruginosa: a new aminoglycoside 6'-N-acetyltransferase. Antimicrob. Agents Chemother. 7, 494–499. doi: 10.1128/AAC.7.5.494
Kobayashi, K., Hayashi, I., Kouda, S., Kato, F., Fujiwara, T., Kayama, S., et al. (2013). Identification and characterization of a novel aac(6′)-Iag associated with the blaIMP-1-integron in a multidrug-resistant Pseudomonas aeruginosa. PLoS One 8:e70557. doi: 10.1371/journal.pone.0070557
Koh, T. H., Wang, G. C., and Sng, L. H. (2004). IMP-1 and a novel metallo-beta-lactamase, VIM-6, in fluorescent pseudomonads isolated in Singapore. Antimicrob. Agents Chemother. 48, 2334–2336. doi: 10.1128/AAC.48.6.2334-2336.2004
Krahn, T., Gilmour, C., Tilak, J., Fraud, S., Kerr, N., Lau, C. H., et al. (2012). Determinants of intrinsic aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 56, 5591–5602. doi: 10.1128/AAC.01446-12
Kuraku, S., Zmasek, C. M., Nishimura, O., and Katoh, K. (2013). aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 41, W22–W28. doi: 10.1093/nar/gkt389
Lakin, S. M., Dean, C., Noyes, N. R., Dettenwanger, A., Ross, A. S., Doster, E., et al. (2017). MEGARes: an antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 45, D574–D580. doi: 10.1093/nar/gkw1009
Lane, D. J. (1991). “Nucleic acid techniques in bacterial systematics” in 16S/23 S rRNA sequencing. eds. E. Stackebrandt and M. Goodfellow (New York: Wiley), 115–175.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Letunic, I., and Bork, P. (2024). Interactive tree of life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82. doi: 10.1093/nar/gkae268
Lima, A. B., Leao-Vasconcelos, L. S., Costa Dde, M., Vilefort, L. O., Andre, M. C., Barbosa, M. A., et al. (2015). Pseudomonas spp. isolated from the oral cavity of healthcare workers from an oncology hospital in midwestern Brazil. Rev Inst Med Trop Sao Paulo 57, 513–514. doi: 10.1590/S0036-46652015000600009
Liu, L., Chen, H., Brecher, M. B., Li, Z., Wei, B., Nandi, B., et al. (2013). Pfit is a structurally novel Crohn's disease-associated superantigen. PLoS Pathog. 9:e1003837. doi: 10.1371/journal.ppat.1003837
Lupo, A., Haenni, M., and Madec, J. Y. (2018). Antimicrobial resistance in Acinetobacter spp. and Pseudomonas spp. Microbiol. Spectr. 6. doi: 10.1128/microbiolspec.ARBA-0007-2017
Marcoleta, A. E., Arros, P., Varas, M. A., Costa, J., Rojas-Salgado, J., Berrios-Pasten, C., et al. (2022). The highly diverse Antarctic peninsula soil microbiota as a source of novel resistance genes. Sci. Total Environ. 810:152003. doi: 10.1016/j.scitotenv.2021.152003
Matsen, F. A., Kodner, R. B., and Armbrust, E. V. (2010). Pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinform. 11:538. doi: 10.1186/1471-2105-11-538
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Mechirackal Balan, B., Shini, S., Krishnan, K. P., and Mohan, M. (2018). Mercury tolerance and biosorption in bacteria isolated from Ny-Alesund, Svalbard, Arctic. J. Basic Microbiol. 58, 286–295. doi: 10.1002/jobm.201700496
Meier-Kolthoff, J. P., and Goker, M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10:2182. doi: 10.1038/s41467-019-10210-3
Montana, S., Lazzaro, T., Uong, S., Place, K., Iriarte, A., Ocampo, C. V., et al. (2018). Genomics helps to decipher the resistance mechanisms present in a Pseudomonas chlororaphis strain recovered in an HIV patient. New Microbes. New Infect. 25, 45–47. doi: 10.1016/j.nmni.2018.07.002
Moskowitz, S. M., and Ernst, R. K. (2010). The role of Pseudomonas lipopolysaccharide in cystic fibrosis airway infection. Subcell. Biochem. 53, 241–253. doi: 10.1007/978-90-481-9078-2_11
Munoz-Escudero, D., Breazeale, S. D., Lee, M., Guan, Z., Raetz, C. R. H., and Sousa, M. C. (2023). Structure and function of ArnD. A Deformylase essential for lipid a modification with 4-Amino-4-deoxy-l-arabinose and Polymyxin resistance. Biochemistry 62, 2970–2981. doi: 10.1021/acs.biochem.3c00293
Na, G., Wang, C., Gao, H., Li, R., Jin, S., Zhang, W., et al. (2019). The occurrence of sulfonamide and quinolone resistance genes at the Fildes peninsula in Antarctica. Mar. Pollut. Bull. 149:110503. doi: 10.1016/j.marpolbul.2019.110503
Na, G., Zhang, W., Gao, H., Wang, C., Li, R., Zhao, F., et al. (2021). Occurrence and antibacterial resistance of culturable antibiotic-resistant bacteria in the Fildes peninsula, Antarctica. Mar. Pollut. Bull. 162:111829. doi: 10.1016/j.marpolbul.2020.111829
Okubo, T., Ae, R., Noda, J., Iizuka, Y., Usui, M., and Tamura, Y. (2019). Detection of the sul2-strA-strB gene cluster in an ice core from dome Fuji Station, East Antarctica. J. Glob. Antimicrob. Resist. 17, 72–78. doi: 10.1016/j.jgar.2018.11.005
Olivares Pacheco, J., Alvarez-Ortega, C., Alcalde Rico, M., and Martinez, J. L. (2017). Metabolic compensation of fitness costs is a general outcome for antibiotic-resistant Pseudomonas aeruginosa mutants overexpressing efflux pumps. MBio 8:e00500-17. doi: 10.1128/mBio.00500-17
Ondov, B. D., Treangen, T. J., Melsted, P., Mallonee, A. B., Bergman, N. H., Koren, S., et al. (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17:132. doi: 10.1186/s13059-016-0997-x
Opazo-Capurro, A., Higgins, P. G., Wille, J., Seifert, H., Cigarroa, C., Gonzalez-Munoz, P., et al. (2019). Genetic features of Antarctic Acinetobacter radioresistens strain A154 harboring multiple antibiotic-resistance genes. Front. Cell. Infect. Microbiol. 9:328. doi: 10.3389/fcimb.2019.00328
Orakov, A., Fullam, A., Coelho, L. P., Khedkar, S., Szklarczyk, D., Mende, D. R., et al. (2021). GUNC: detection of chimerism and contamination in prokaryotic genomes. Genome Biol. 22:178. doi: 10.1186/s13059-021-02393-0
Orellana-Saez, M., Pacheco, N., Costa, J. I., Mendez, K. N., Miossec, M. J., Meneses, C., et al. (2019). In-depth genomic and phenotypic characterization of the Antarctic Psychrotolerant strain Pseudomonas sp. MPC6 Reveals Unique Metabolic Features, Plasticity, and Biotechnological Potential. Front. Microbiol. 10:1154. doi: 10.3389/fmicb.2019.01154
Pantucek, R., Sedlacek, I., Indrakova, A., Vrbovska, V., Maslanova, I., Kovarovic, V., et al. (2018). Staphylococcus edaphicus sp. nov., isolated in Antarctica, harbors the mecC gene and Genomic Islands with a suspected role in adaptation to extreme environments. Appl. Environ. Microbiol. 84:e01746-17. doi: 10.1128/AEM.01746-17
Papapetropoulou, M., Rodopoulou, G., Giannoulaki, E., and Stergiopoulos, P. (1994). Effect of temperature on antimicrobial susceptibilities of Pseudomonas species isolated from drinking water. J. Chemother. 6, 404–407. doi: 10.1080/1120009X.1994.11741174
Pavlov, M. S., Lira, F., Martinez, J. L., Olivares-Pacheco, J., and Marshall, S. H. (2020). Pseudomonas fildesensis sp. nov., a psychrotolerant bacterium isolated from Antarctic soil of King George Island, South Shetland Islands. Int. J. Syst. Evol. Microbiol. 70, 3255–3263. doi: 10.1099/ijsem.0.004165
Poblete-Morales, M., Carvajal, D., Almasia, R., Michea, S., Cantillana, C., Levican, A., et al. (2020). Pseudomonas atacamensis sp. nov., isolated from the rhizosphere of desert bloom plant in the region of Atacama, Chile. Antonie Van Leeuwenhoek 113, 1201–1211. doi: 10.1007/s10482-020-01427-0
Poirel, L., Palmieri, M., Brilhante, M., Masseron, A., Perreten, V., and Nordmann, P. (2020). PFM-like enzymes are a novel family of subclass B2 Metallo-beta-lactamases from Pseudomonas synxantha belonging to the Pseudomonas fluorescens complex. Antimicrob. Agents Chemother. 64:e01700-19. doi: 10.1128/AAC.01700-19
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Prithivirajsingh, S., Mishra, S. K., and Mahadevan, A. (2001). Detection and analysis of chromosomal arsenic resistance in Pseudomonas fluorescens strain MSP3. Biochem. Biophys. Res. Commun. 280, 1393–1401. doi: 10.1006/bbrc.2001.4287
Robertson, J., and Nash, J. H. E. (2018). MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 4:e000206. doi: 10.1099/mgen.0.000206
Rodriguez-Rojas, F., Diaz-Vasquez, W., Undabarrena, A., Munoz-Diaz, P., Arenas, F., and Vasquez, C. (2016). Mercury-mediated cross-resistance to tellurite in Pseudomonas spp. isolated from the Chilean Antarctic territory. Metallomics 8, 108–117. doi: 10.1039/C5MT00256G
Rolston, K. V., Kontoyiannis, D. P., Yadegarynia, D., and Raad, I. I. (2005). Nonfermentative gram-negative bacilli in cancer patients: increasing frequency of infection and antimicrobial susceptibility of clinical isolates to fluoroquinolones. Diagn. Microbiol. Infect. Dis. 51, 215–218. doi: 10.1016/j.diagmicrobio.2004.11.002
Rumbaugh, K. P. (2014). Genomic complexity and plasticity ensure Pseudomonas success. FEMS Microbiol. Lett. 356, 141–143. doi: 10.1111/1574-6968.12517
Sader, H. S., and Jones, R. N. (2005). Antimicrobial susceptibility of uncommonly isolated non-enteric gram-negative bacilli. Int. J. Antimicrob. Agents 25, 95–109. doi: 10.1016/j.ijantimicag.2004.10.002
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seiler, C., and Berendonk, T. U. (2012). Heavy metal driven co-selection of antibiotic resistance in soil and water bodies impacted by agriculture and aquaculture. Front. Microbiol. 3:399. doi: 10.3389/fmicb.2012.00399
Sellera, F. P., Fernandes, M. R., Sartori, L., Carvalho, M. P., Esposito, F., Nascimento, C. L., et al. (2017). Escherichia coli carrying IncX4 plasmid-mediated mcr-1 and blaCTX-M genes in infected migratory Magellanic penguins (Spheniscus magellanicus). J. Antimicrob. Chemother. 72, 1255–1256. doi: 10.1093/jac/dkw543
Shah, S. Q., and Sorum, H. (2014). Genetic localization of a TetR-like transcriptional regulator gene in Pseudomonas fluorescens isolated from farmed fish. J. Appl. Genet. 55, 541–544. doi: 10.1007/s13353-014-0221-1
Silverio, M. P., Kraychete, G. B., Rosado, A. S., and Bonelli, R. R. (2022). Pseudomonas fluorescens complex and its intrinsic, adaptive, and acquired antimicrobial resistance mechanisms in pristine and human-impacted sites. Antibiotics (Basel) 11:985. doi: 10.3390/antibiotics11080985
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Soares, S. C., Geyik, H., Ramos, R. T., de Sa, P. H., Barbosa, E. G., Baumbach, J., et al. (2016). GIPSy: genomic island prediction software. J. Biotechnol. 232, 2–11. doi: 10.1016/j.jbiotec.2015.09.008
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Suzuki, R., and Shimodaira, H. (2006). Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542. doi: 10.1093/bioinformatics/btl117
Svec, P., Kosina, M., Zeman, M., Holochova, P., Kralova, S., Nemcova, E., et al. (2020). Pseudomonas karstica sp. nov. and Pseudomonas spelaei sp. nov., isolated from calcite moonmilk deposits from caves. Int. J. Syst. Evol. Microbiol. 70, 5131–5140. doi: 10.1099/ijsem.0.004393
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tian, T., Wu, X. G., Duan, H. M., and Zhang, L. Q. (2010). The resistance-nodulation-division efflux pump EmhABC influences the production of 2,4-diacetylphloroglucinol in Pseudomonas fluorescens 2P24. Microbiology (Reading) 156, 39–48. doi: 10.1099/mic.0.031161-0
Versalovic, J., Schneider, M., De Bruijn, F. J., and Lupski, J. R. (1994). Genomic fingerprint of Bacteria using repetitive sequence-based polymerase chain reaction. Methods Molec. Cell. Biol. 5, 25–40.
Waterhouse, R. M., Seppey, M., Simao, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2018). BUSCO applications from quality assessments to gene prediction and Phylogenomics. Mol. Biol. Evol. 35, 543–548. doi: 10.1093/molbev/msx319
Wattam, A. R., Davis, J. J., Assaf, R., Boisvert, S., Brettin, T., Bun, C., et al. (2017). Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 45, D535–D542. doi: 10.1093/nar/gkw1017
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Zeng, L., and Jin, S. (2003). Aph(3′)-IIb, a gene encoding an aminoglycoside-modifying enzyme, is under the positive control of surrogate regulator HpaA. Antimicrob. Agents Chemother. 47, 3867–3876. doi: 10.1128/AAC.47.12.3867-3876.2003
Keywords: resistomes, psychrotolerant bacteria, Proteobacteria, Pseudomonadota, antibiotics, acquired resistance, intrinsic resistance, efflux pumps
Citation: Silverio MP, Schultz J, Parise MTD, Parise D, Viana MVC, Nogueira W, Ramos RTJ, Góes-Neto A, Azevedo VADC, Brenig B, Bonelli RR and Rosado AS (2025) Genomic and phenotypic insight into antimicrobial resistance of Pseudomonas fluorescens from King George Island, Antarctica. Front. Microbiol. 16:1535420. doi: 10.3389/fmicb.2025.1535420
Edited by:
Maria Jorge Campos, Polytechnic Institute of Leiria, PortugalReviewed by:
Agustina Natalia Undabarrena, Novo Nordisk Foundation Center for Biosustainability (DTU Biosustain), DenmarkCopyright © 2025 Silverio, Schultz, Parise, Parise, Viana, Nogueira, Ramos, Góes-Neto, Azevedo, Brenig, Bonelli and Rosado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandre Soares Rosado, YWxleGFuZHJlLnJvc2Fkb0BrYXVzdC5lZHUuc2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.