
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 03 February 2025
Sec. Extreme Microbiology
Volume 16 - 2025 | https://doi.org/10.3389/fmicb.2025.1504355
This article is part of the Research TopicDeep Subsurface Microbiology and EnergeticsView all 12 articles
 Paula Rodriguez1*
Paula Rodriguez1* Jasmine S. Berg2
Jasmine S. Berg2 Longhui Deng3,4
Longhui Deng3,4 Hendrik Vogel5Michal Okoniewski6
Hendrik Vogel5Michal Okoniewski6 Mark A. Lever3,7
Mark A. Lever3,7 Cara Magnabosco1
Cara Magnabosco1Most of our knowledge of deep sedimentary life comes from marine environments; however, despite their relatively small volume, lacustrine sediments constitute one of the largest global carbon sinks and their deep sediments are largely unexplored. Here, we reconstruct the microbial functional and taxonomic composition of an 8,000-year Holocene sedimentary succession from meromictic Lake Cadagno (Switzerland) using shotgun metagenomics and 16S rRNA gene amplicon sequencing. While younger sediments (<1,000 years) are dominated by typical anaerobic surface sedimentary bacterial taxa (Deltaproteobacteria, Acidobacteria, and Firmicutes), older layers with lower organic matter concentrations and reduced terminal electron acceptor availability are dominated by taxa previously identified as “persistent populations” within deep anoxic marine sediments (Candidatus Bathyarchaeia, Chloroflexi, and Atribacteria). Despite these dramatic changes in taxonomic community composition and sediment geochemistry throughout the sediment core, higher-order functional categories and metabolic marker gene abundances remain relatively consistent and indicate a microbial community capable of carbon fixation, fermentation, dissimilatory sulfate reduction and dissimilatory nitrate reduction to ammonium. As the conservation of these metabolic pathways through changes in microbial community compositions helps preserve the metabolic pathway connectivity required for nutrient cycling, we hypothesize that the persistence of these functional groups helps enable the Lake Cadagno sedimentary communities persist amidst changing environmental conditions.
Subsurface marine and lacustrine sediments cover approximately 70% of Earth's surface (Oni et al., 2015; Hoshino et al., 2020) and constitute the largest global organic carbon reservoirs (Barber et al., 2017). This substantial habitat hosts a significant fraction of the world's bacterial and archaeal populations (Kallmeyer et al., 2012) and exhibits much slower metabolic rates than their surface sedimentary counterparts (Jorgensen and Marshall, 2016). This is in part due to the low oxygen supply, which often leads to anoxic conditions that promote the preservation of organic matter and environmental signatures in the sediment record (Jessen et al., 2017). Lacustrine sediments are often regarded as time capsules for paleo-reconstructions, as sediment deposition happens continuously and at higher rates, and experiences less reworking, compared to marine sediments (Zolitschka and Enters, 2009). The resulting high-resolution temporal records of environmental change (Zolitschka and Enters, 2009) make lake sediments ideal settings to study the interplay between long-term environmental changes in lake water sheds and biogeochemical processes which slowly alter environmental records after burial (Ariztegui et al., 2015). Despite their unique features, the effects of sediment burial on the community assembly, function, and diversity in deep lacustrine sediments are poorly understood, especially when compared to those of deep marine sediments (Ariztegui et al., 2015). Studies on microbial assemblages in deep lacustrine sediments are thus essential for understanding how the environmental features of lake ecosystems shape sediment microbial communities (Vuillemin et al., 2018).
On a global scale, organic carbon content is one of the main environmental parameters correlated with microbial biogeography patterns in anoxic sediments (Hoshino et al., 2020). In places with high sedimentation rates (e.g., Lake Cadagno, the site of this study), microorganisms living in the water-sediment interface rapidly deplete high-energy terminal electron acceptors (TEAs) such as O2 and nitrate and degrade the chemically most reactive organic matter pools (Orsi, 2018; D'Hondt et al., 2004; Berg et al., 2022). Below this zone, the utilization of terminal electron acceptors in catabolic reactions generates a vertical gradient in dominant respiration reactions (D'Hondt et al., 2004). Following the same trend, organic matter compositions shift toward increasingly degradation-resistant (refractory) species with sediment age and depth, thus limiting the electron donor pool available for microbial oxidation (Gajendra et al., 2023; Deng et al., 2020; Han et al., 2022).
As a general trend, the shift in sediment communities from freshly deposited sediments (Morono et al., 2020; Hoshino et al., 2020) to older “deeper” sediment communities is accompanied by a steep drop in microbial abundance (Chen et al., 2017; Hoshino et al., 2020). It is proposed that despite a reduction in cell numbers with increasing sediment depth, microorganisms continue to drive elemental cycling in deep sediments (Varliero et al., 2019; D'Hondt et al., 2004) and presumably utilize refractory organic matter (Marshall et al., 2019; Hoshino et al., 2020; Hubert et al., 2009). Under these conditions, it is hypothesized that a fraction of the surface microbial populations becomes predominant with sediment depth (Petro et al., 2017), and these are frequently referred to as “persistent sediment populations” (Starnawski et al., 2017). In anoxic marine sediments, these are commonly representatives from the groups Atribacteria, Candidatus Bathyarchaeia (also referred to as Candidatus Bathyarchaeota), and Chloroflexi (Hoshino et al., 2020; Zhou et al., 2018; Lloyd et al., 2013).
Here, we investigated how genome-inferred microbial community composition and metabolic potential relate to the environmental history and organic matter sources of lacustrine sediments from Lake Cadagno, an Alpine meromictic lake in Switzerland with anoxic, sulfidic bottom waters. The high-resolution limnological record of Lake Cadagno reflects these environmental changes, providing an ideal setting to study the co-evolution of anoxic lacustrine sediments and their microbial communities. Our study encompasses deposition throughout the Holocene and after the onset of meromictic conditions in the lake. An initial biological analysis of the sedimentary sequence revealed a weak (R2 = 0.35) correlation between total organic carbon and prokaryotic 16S rRNA gene abundances (Berg et al., 2022); however, the functional and population-level diversity of the microbial communities within these sediments was not investigated. In this study, we combine shotgun metagenomic and 16S rRNA gene sequencing data to investigate the functional and taxonomic microbial community composition across an 8,000-year lacustrine sedimentary covering the period after the establishment of water column stratification and bottom water euxinia in Lake Cadagno.
Lake Cadagno is a meromictic lake located in the Swiss Alps at 1,921 meters above sea level. The lake basin was formed through glacial erosion into bedrock during the Last Glacial Period with lake conditions being established after the retreat of the glacier from the basin ~12,000 years ago (Wirth et al., 2013). Since its formation and throughout its history it has undergone water column redox transitions that are reflected in the sediment record (Wirth et al., 2013). Today, the water column of the lake is stratified into an oxic epilimnion and an anoxic and sulfidic hypolimnion separated by a 0.5–1.0 m thick chemocline populated by purple sulfur bacteria (PSB) and green sulfur bacteria (GSB) (Philippi et al., 2021). The bottom layer of the water column exhibits a high sulfate concentration (~2 mM) relative to other freshwater systems, and surface sediments are similarly enriched in sulfate (up to 1.5 mM) which is rapidly depleted (<0.05 mM) within the first 20 cm below the lake floor (cmblf) (Berg et al., 2022). In the summer of 2019, a 10 m sediment core was retrieved and an initial 16S rRNA gene-based microbial characterization was performed (Berg et al., 2022). In our current study, a new set of sediment samples from this core were independently analyzed for 16S rRNA gene and shotgun metagenomic analyses. A detailed description of the coring, sampling strategy and geochemistry can be found in Berg et al. (2022). In brief, three sediment cores from Lake Cadagno were collected at a water depth of 21 m (46.55060 N and 8.71201 E) using an UWITEC coring platform (Uwitec, TA) and subsampled to biomolecular analytical standards. After retrieval, biomolecular samples were flash-frozen in liquid N2 and stored at −80°C until the DNA extractions described below.
DNA samples from 13 sediment depths between 0 and 738 cmblf were selected for DNA extraction and metagenomic sequencing (Table 1). Sample selection was based on lithostratigraphic and geochemical parameters to make sure the environmental variability of the sedimentary sequence was well represented. The core exhibits layers of organic matter-rich sediments of lacustrine origin and turbidite layers originating from mass-movement events that vary in porewater and solid phase (S0, TOC, C:N ratio, δ13C-TOC) chemistry (Berg et al., 2022) (Table 1). The 13 samples cover the depositional history of the lake in the last 8,000 years after the onset of water column stratification in the lake during the Holocene, as indicated by age-depth modeling based on 14C radiocarbon dating (Berg et al., 2022; Wirth et al., 2013).
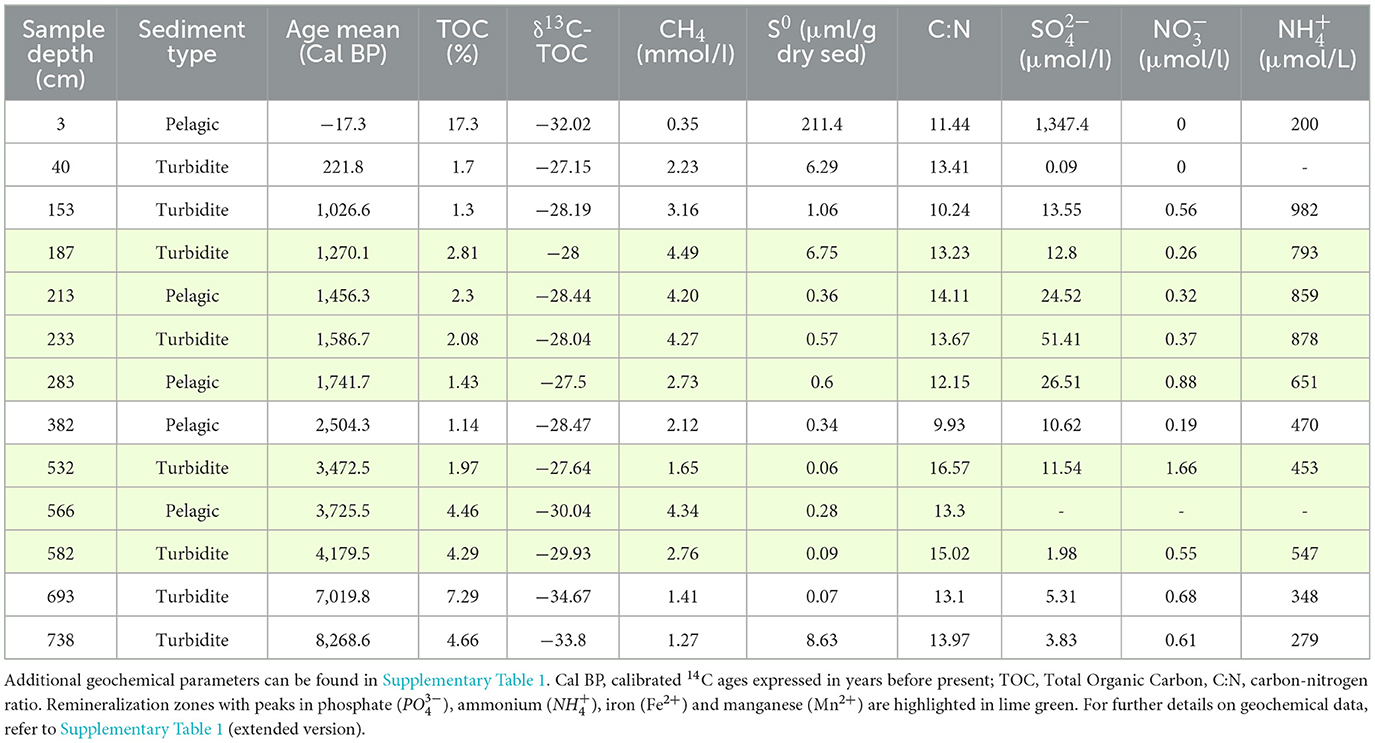
Table 1. General information and geochemical data (Berg et al., 2022) for the 13 sediment samples analyzed this study.
DNA was extracted from three 0.2 g sediment aliquots per sample depth using a modular DNA extraction protocol (Lever et al., 2015) to maximize the DNA yield in deep, old sediments. Briefly, the aliquots were transferred to 2 mL beat-beating tubes and soaked with 10 mM sodium hexametaphosphate solution, and lysis solution I (30 mM Tris-HCl, 30 mM EDTA, 800 mM guanidium hydrochloride, and 0.5% Triton X-100) and then placed in a vortex genie and shaken at maximum speed for 30 s. The samples were then purified using lysis solution II (2% CTAB and 0.1% PVPP) and 24:1 chloroform isoamyl alcohol to remove humic and fluvic acids, residual proteins, and lipids. DNA precipitation was performed using linear polyacrylamide and 70% ethanol. DNA extracts were then purified using magnetic beads (AMPure XP beads 1.0x) followed by a final purification and concentration step using the commercial kit ReliaPrepTM DNA Cleanup and Concentration System (Promega, WI, United States).
After extraction and clean-up, the final DNA concentration of samples ranged between 2.44 ng/μL and 17.15 ng/μL (Supplementary Table 1). Samples were normalized to the minimum concentration of 2 ng/μL, which allowed us to perform metagenomic sequencing using the standard NEBnext Ultra II DNA library preparation kit which uses DNA fragmentation by sonication (New England Biolabs, Massachusetts, United States). The NEBnext Ultra II DNA for Illumina library preparation kit requires an initial DNA input of 50 ng and a three-cycle PCR amplification step. This reduced number of PCR cycles minimizes the impact of PCR on gene duplication rates, unequal amplification of genes, and sequencing artifacts (Rochette et al., 2023). Paired-end high-throughput sequencing was performed on a NovSeq 6000 Sequencing System (Illumina, San Diego, CA, USA) at the Functional Genomics Center Zurich (Zurich, Switzerland).
Sequences were quality-filtered and trimmed using Trimmomatic v.0.35 (Bolger et al., 2014) with the parameters SLIDING WINDOW:4:15 MINLEN:36 and assembled using Spades 3.14.1 (metaSPAdes.py) (Nurk et al., 2017) with k-mer sizes 21, 33,55. Contigs larger than 1,000 bp were retained for further analyses and protein-coding regions were predicted using Prodigal v.2.6.3 (Hyatt et al., 2010). Predicted proteins from all samples were clustered at a 90% amino acid sequence identity threshold using CD-HIT v.4.6.8 (Li and Godzik, 2006). The representative sequence for each predicted protein cluster was functionally and taxonomically annotated as described below. The taxonomic and functional annotation of each predicted protein cluster's representative protein sequence was then assigned to all sequences within the protein cluster.
Functional annotation of protein clusters within the Clusters of Orthologous Groups (COG) broad metabolic categories was made using eggNOG mapper v.2.1.6 with a likelihood threshold of 1e−5. The functional annotation of METABOLIC-derived marker genes (Zhou et al., 2022) for carbon, sulfur, and nitrogen biogeochemical cycling was performed using HMMER v.3.3.2 (Finn et al., 2011, e-value threshold of 1e−15) and the identity of the marker genes was confirmed using BLAST+ v.2.9.0 (e-value threshold of 1e−30, Camacho et al., 2009) against the NCBI-nr database (February 2024). The taxonomic annotation of the RpS3 marker genes was confirmed using BLAST+ v.2.9.0 (e-value threshold of 1e−30; Camacho et al., 2009) against the NCBI-nr database (January and June 2023). The taxonomy was assigned to the last common ancestor using a consensus vote approach with the top 3 hits of the previously described BLASTp search. The functional identities of marker genes involved in nitrogen cycling (hao, narG, nifD, nifH, nifK, norB) and sulfur oxidation (soxB) genes were further confirmed, and the taxonomic identities of the sequences were assigned through phylogenetic placement. Briefly, a “reference” phylogenetic tree for each marker gene was constructed by randomly sampling 100 protein sequences from the original METABOLIC marker gene database using seqtk v.1.3. Protein sequence alignments of METABOLIC marker gene and Lake Cadagno sequences were computed using MUSCLE v.3.8.31 (Edgar, 2004) and de-novo phylogenetic trees were calculated using FastTree v.2.1.11 (Price et al., 2010) using the Le and Gascuel (LG) model (Le et al., 2008). The placement of Lake Cadagno sequences within these marker gene trees was used for marker gene taxonomic identification.
The complete set of protein sequences clustered at 90% amino-acid identity was annotated using the eggNOG mapper v.2.1.6, with a likelihood threshold of 1e−5. Protein clusters within each of the Clusters of Orthologous Groups (COGs) were subsampled for a beta-diversity analysis using complete linkage hierarchical clustering. Based on these results, the category “G” (Carbohydrate transport and metabolism) was selected for an enrichment analysis using the Maaslin2 (Mallick et al., 2021). The enrichment analysis was performed on samples where persistent Amplicon Sequencing Variants (ASVs) from Ca. Bathyarchaeia, Atribacteria, and Chloroflexi constitute more than 10% of the total abundance in the microbial community (samples below 40 cm). Proteins showing significant correlations and anticorrelations to sample depth (q-value <0.05) were further annotated using BLAST (e-value threshold of 1e−30; Camacho et al., 2009) against the NCBI-nr database (April 2024).
Finally, to identify potential cellulases in the complete metagenomic dataset, protein sequences identified as cellulases based on the eggNOG mapper annotation were selected. The taxonomic and functional annotations of these proteins were further confirmed using BLAST (e-value threshold of 1e−30; Camacho et al., 2009) against the NCBI database (April 2024).
Protein sequences clustered at 90% amino-acid identity were annotated using eggNOG mapper v.2.1.6 with a likelihood threshold of 1e−5. Protein clusters annotated within any of the KEGG categories were further annotated using GhostKOALA (Kanehisa et al., 2016). The resulting output file was used as input for KEGGDecoder (Graham et al., 2018) to generate a functional heatmap illustrating the completeness of the metabolic pathways across different sediment depths for the entire metagenomic dataset. The completeness of the metabolic pathways at any given depth is expressed using scores that range from 0.0 (non-present pathway) to 1.0 (indicating a fully present metabolic pathway).
A collection of RpS3 protein sequences was obtained from genomes assigned to Ca. Bathyarchaeia in GenBank (Benson et al., 2017), reported in the Ca. Bathyarchaeia phylogenetic tree of a previous study (Zhou et al., 2018), and identified within the Lake Cadagno metagenomic dataset. A protein sequence alignment was made using MUSCLE (Edgar, 2004). To improve the topological correspondence of our Ca. Bathyarchaeia RpS3 tree and previously reported Ca. Bathyarchaeia phylogenetic trees (Zhou et al., 2018; Hou et al., 2023), a guide tree was calculated using FastTree v.2.1.11 (Price et al., 2010) using the LG substitution model (Le et al., 2008). A maximum likelihood phylogenetic tree was then calculated using the LG protein substitution model (Le et al., 2008), gamma likelihood optimization and guide tree in RAxML v.8.2.12 (Stamatakis, 2014). Bootstrap analysis with 1,000 replicates was performed to assess the robustness of the tree topology.
The V4–V5 hypervariable regions of the 16S rRNA gene were amplified using the universal primer pair 515 F (5′-GTG YCA GCM GCC GCG GTA A-3′) and 926 R (5′-CCG YCA ATT YMT TTR AGT TT-3′) (Quince et al., 2011; Parada et al., 2016). This primer pair covers ~500 bp in the V4–V5 hypervariable regions of the bacterial and archaeal 16S rRNA gene and is different from the universal, bacteria-specific and archaea-specific primer pairs used by Berg et al., 2022 [S-D-Bact-0341-b-S-17/S-D-Bact-0785-a-A-21 (Herlemann et al., 2011) and -D-Arch-0519-a-A-19 (Sørensen and Teske, 2006)/967Rmod (Cadillo-Quiroz et al., 2006)]. Amplicon libraries were prepared using a single-step PCR on the C1000 Touch Thermal Cycler (BioRad, Hercules, California, United States). Samples were run in triplicates, with a reaction volume of 25 μL including 1.0 μL template DNA, 0.5 μL forward primer (10 μM) and reverse primer (10 μM), 10 μL of PlatinumTM Hot Start PCR 2X Master Mix (Thermo Fisher Scientific, Waltham, Massachusetts, United States), and 13 μL of PCR-grade water. The PCR program was run as follows: initial denaturation at 95°C for 3 min, followed by 30 cycles of denaturation at 95°C for 35 s, annealing at 50°C for 45 s, elongation at 68°C for 90 s and a final elongation step at 68°C for 5 min. The DNA amplicons were purified using AMPure XP magnetic beads (Beckman Coulter, Brea, California, United States) at a concentration of 0.8x. After purification, the DNA amplicon concentrations were measured using a Qubit and diluted to create a pool with a final concentration of 4 nM. The quality of the 16S rRNA gene library was determined using the D1000 ScreenTape on the Agilent 4150 TapeStation (Agilent Technologies, Santa Clara, California, United States). Paired-end sequencing was performed on a MiSeq sequencer (Illumina Inc., San Diego, California, United States) using the MiSeq Reagent Kit v2 Nano (500 cycles) (Illumina).
The quality of the 16S rRNA gene sequences was assessed using FastQC (Andrews, 2010). Data analysis was done using the standard the Qiime2 workflow (Bolyen et al., 2019). In brief, the first six nucleotides of the forward reads and 22 of the reverse reads were trimmed using the –p-trim-left parameter in the denoise-paired function of DADA2 (Callahan et al., 2016). Forward and reverse reads were truncated at 200 and 190 bp, respectively, with the –p-trunc-len function. The 16S rRNA gene feature table was generated and summarized using the qiime feature-table summarize function in Qiime2 (Bolyen et al., 2019). Amplicon sequencing variants (ASVs) related identified within the negative control samples were removed from all samples and excluded from all downstream analyses. Remaining ASVs were annotated using the Silva (Quast et al., 2013) nr database version 138.1.
The OTU table was rarefied to the minimum sample read count using the rrarefy function in the Vegan package (Oksanen et al., 2024) in RStudio to normalize sequencing depth across samples. Richness, defined as the number of unique taxa per sample, was calculated by counting non-zero taxa in the rarefied and non-rarefied data. The Shannon diversity index was calculated for each sample using the diversity function from the Vegan R package.
Abundances of prokaryotic 16S rRNA genes were quantified on a LightCycler 480 II (Roche Life Science, Penzberg, Germany) at ETH Zurich's Genetic Diversity Center (Switzerland) using the universal primer pair 515 F (5′- GTG YCA GCM GCC GCG GTA A−3′) and 926 R (5′- CCG YCA ATT YMT TTR AGT TT−3′) (Quince et al., 2011; Parada et al., 2016). Plasmids of 16S rRNA genes from Thermoplasma acidophilum-affiliated archaea and Holophaga foetida were used as standards as described in Han et al. (2020). The samples were run in duplicates with a reaction volume of 10 μL including 2 μL of DNA template, 1 μL of water, 1 μL of BSA, 0.5 μL of forward primer (10 μM) and 0.5 μL of reverse primer (10 μM), and 5 μL SYBR green (SsoFast™ EvaGreen® Supermix with Low ROX 2x) (BioRad, Hercules, California, United States). The quantitative PCR (qPCR) reaction was run as follows: activation of the DNA polymerase at 95°C for 5 min, followed by 35 cycles of initial denaturation at 95°C for 10 s, annealing at 50°C for 30 s, extension at 68°C for 15 s and a melting curve (95°C for 15 s and 55°C for 1 min). The standard curve and melt curve for these reactions are provided in Supplementary Figures 1, 2.
The taxonomic composition of the microbial communities from Lake Cadagno sediments was assessed using the 16S rRNA gene and the ribosomal protein S3 (RpS3) predicted protein clusters at 90% amino acid sequence identity (RpS390%) from targeted and shotgun metagenomic datasets, respectively. At a coarse taxonomic level, the taxonomic profiles reported in this study based on targeted and shotgun metagenomic sequencing data match previously published data, which shows a clear transition from surface sediments dominated by bacteria from groups like Proteobacteria, to deeper sediments dominated by Ca. Bathyarchaeia and other groups such as Planctomycetes, Chloroflexi and Atribacteria (Berg et al., 2022).
The microbial community composition profiles based on 16S rRNA amplicon and RpS390% datasets are generally consistent with the exception of the unique presence of Candidatus Atribacteria (formerly OP9) at relatively high abundances (12–33%) in the 16S rRNA gene dataset from 153 cmblf and 582 cmblf (Figure 1, Supplementary File 2) and the dominance (33%) of Candidatus Acetothermia (Bipolaricaulota) within the youngest (3 cmblf) sediment sample of the RpS390% dataset (Figure 1). Both datasets reveal bacteria-dominated communities composed of Alphaproteobacteria, Gammaproteobacteria, and Deltaproteobacteria with minor populations of Euryarchaeota in the two youngest sediment samples (3 cmblf and 40 cmblf). In deeper and older samples (>225 years, >40 cmblf), the bacteria-dominated communities are replaced by Candidatus Bathyarchaeia-dominated communities (Figure 1). 16S rRNA gene and RpS390% sequences related to Ca. Bathyarchaeia, Ca. Aminicenantes, Deltaproteobacteria, Chloroflexi, Atribacteria, and Planctomycetes are identified in all samples. Finally, higher alpha-diversity metrics [richness (S), Shannon-Wiener Index (H')] are observed within the 16S rRNA gene dataset relative to the RpS390% dataset (Table 2). This discrepancy in dataset-derived S and H' can be explained by the differences in both the sequencing (targeted vs. untargeted) and bioinformatic approach (Amplicon Sequencing Variant vs. RpS390%).
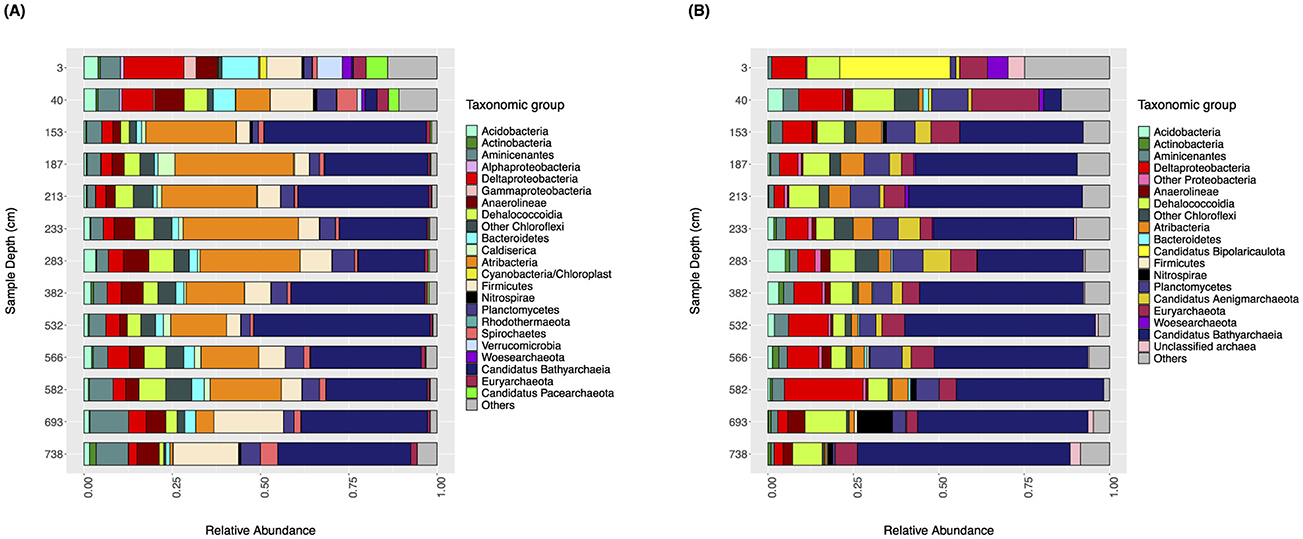
Figure 1. Taxonomic characterization of microbial communities from Lake Cadagno with sediment depth. The relative abundance (x-axis) of bacteria and archaea (bar colors) with respect to sediment depth (y-axis) is shown (A) for 16S rRNA gene sequences and (B) for ribosomal protein S3 (RpS390%) sequence.
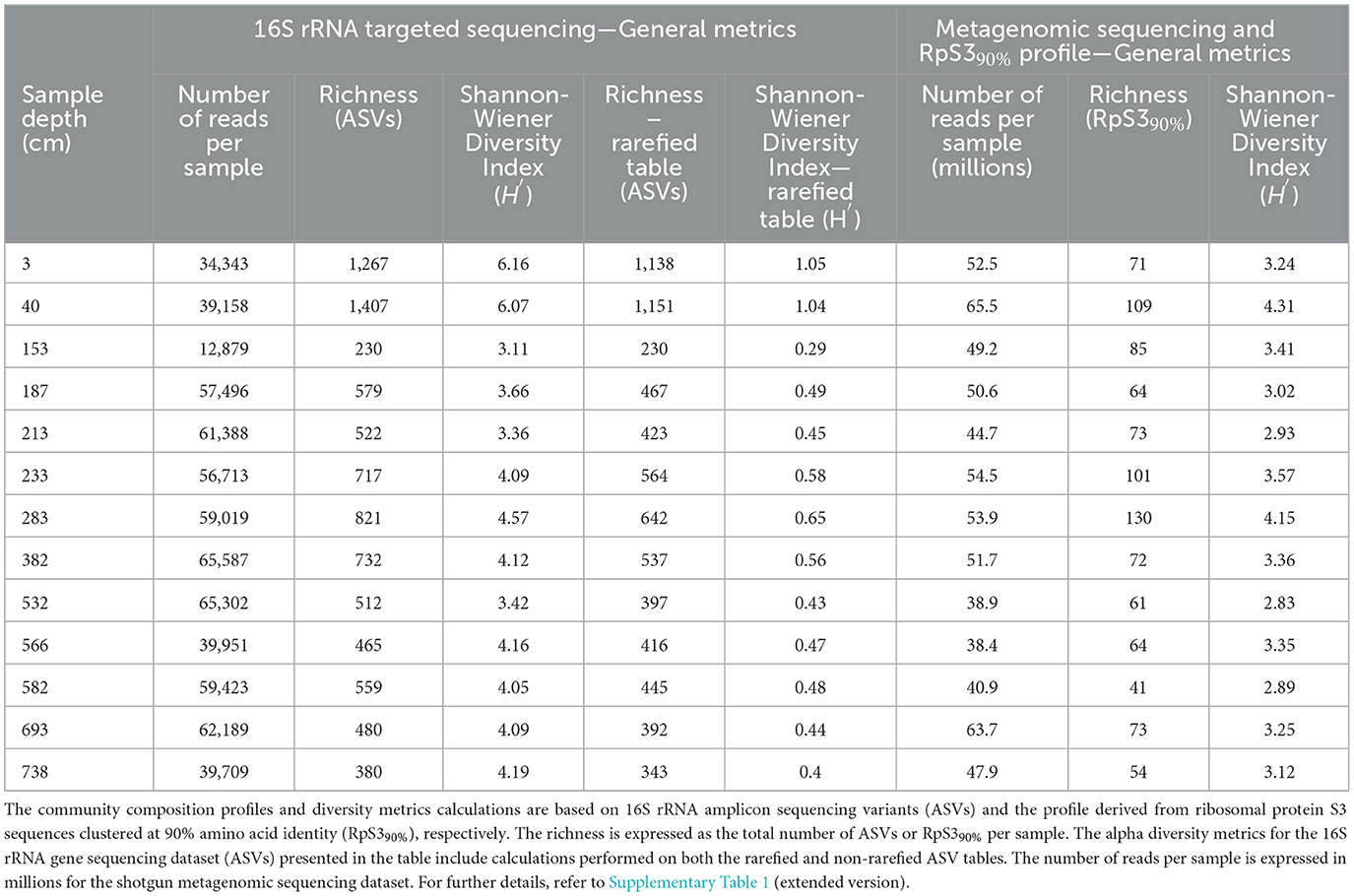
Table 2. General sequencing information and diversity metrics per sample depth for the 16S rRNA gene and shotgun metagenomic sequencing datasets.
In the first 153 cm of sediment, total 16S rRNA gene copy numbers decrease by more than an order of magnitude (2 × 108 to 2 × 107 gene copies per g of wet sediment), and below this depth, community abundances range from 5 × 106 to 8 × 107 16S rRNA gene copies per g of wet sediment (Figure 2B). While almost all samples within the 16S rRNA gene dataset are bacteria-dominated, both bacteria- and archaea-specific abundances vary with depth. Two samples (153 cmblf and 532 cmblf) exhibit nearly equal bacteria-to-archaea ratios as reported by Berg et al. (2022) even though their only shared geochemical traits are similarities in sediment type (turbidite) and phosphate concentrations (30.3 μM) (Table 1).
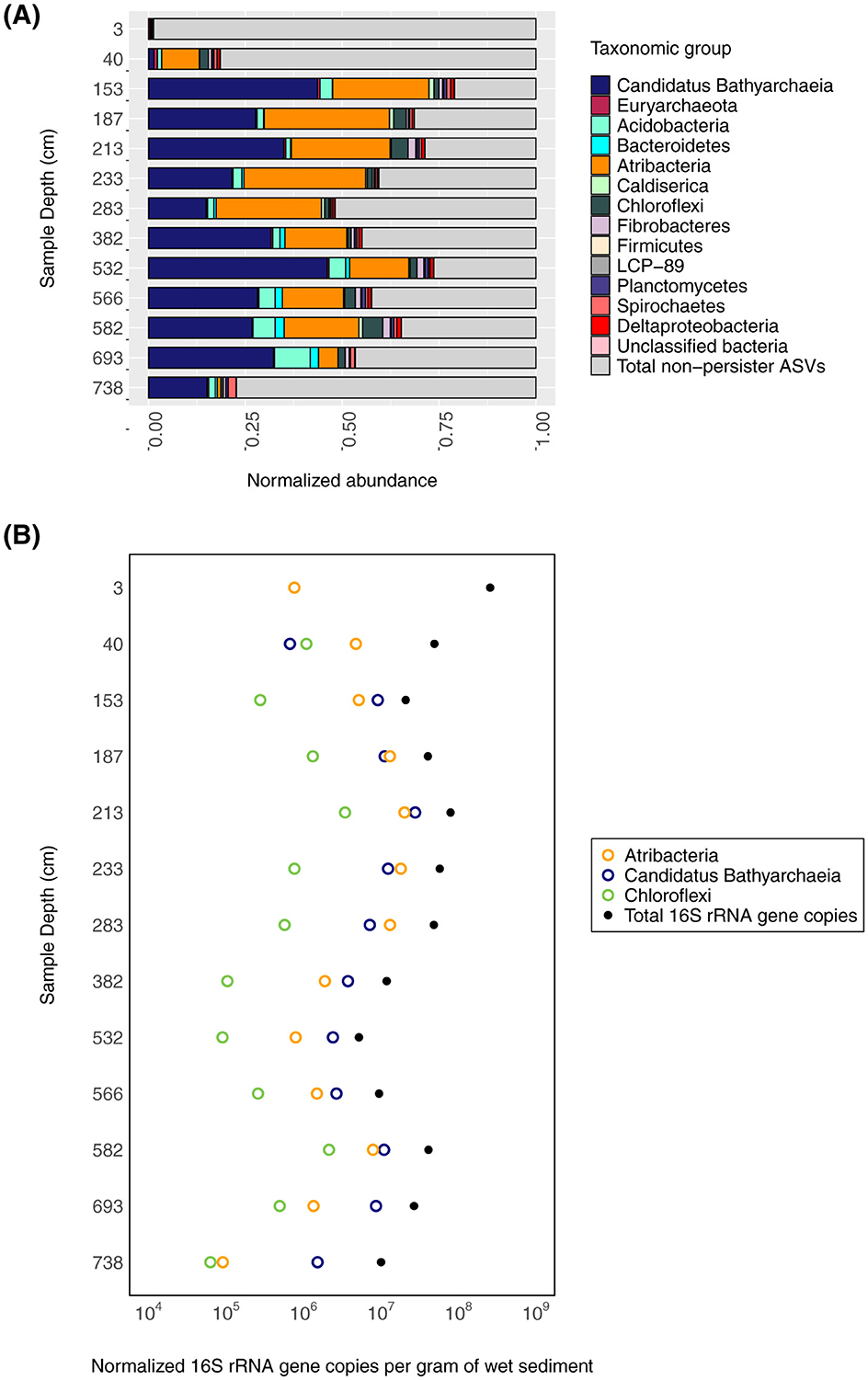
Figure 2. Persistent microbial lineages throughout the sedimentary sequence. (A) The relative abundance of persistent (present in more than 10 sediment samples) 16S rRNA ASVs and non-persistent ASVs (gray bars) are shown with respect to sediment depth. (B) The qPCR-estimated abundance of bacteria and archaea (black dots) relative to estimated persistent Ca. Bathyarchaeia (open blue circles), Atribacteria (open orange circles), and Chloroflexi (open green circles) ASV abundance (x-axis) with depth (y-axis) is shown.
In addition to the changes in Lake Cadagno community composition and abundance, persistent taxonomic groups are also observed throughout the sediment sequence. Persistent groups are generally defined as the members of the community that are present throughout the sediment column (Starnawski et al., 2017). In this study, we define as “persistent groups” the Amplicon Sequencing Variants that are present in more than ten sediment samples in our sedimentary sequence. Out of the 6,148 unique Lake Cadagno Amplicon Sequencing Variants (ASVs), 42 ASVs are observed in at least 11 of the 13 Lake Cadagno sediment sample depths. The persistent ASVs represent a small fraction of the surface sediment community at 3 cmblf (<0.01%) but represent between 33% and 67% of each sample's total 16S rRNA gene-based community below 153 cmblf. The most abundant persistent ASVs are related to Ca. Bathyarchaeia (16/42), Atribacteria (3/42), and Chloroflexi (3/42) (Figure 2B). Additionally, persistent ASVs related to Acidobacteria (5/42), Plantomycetes (2/42), Deltaproteobacteria (2/42), Fibrobacterota (1/42) and Bacteroidota (1/42) were also identified at >2% (SD = 3.51) average abundance below 153 cm. Among these, the most abundant ASV, at an average abundance of 21% of ASVs in sediments below 153 cm, is related to Ca. Bathyarchaeia (ASV_ID= Candidatus Bathyarchaeia ASV 1, Supplementary Figure 3).
Phylogenetic placement of Ca. Bathyarchaeia RpS390% sequences reveals that the Lake Cadagno sediment RpS3 sequence clusters fall within the newly assigned Ca. Bathyarchaeia candidate orders “Baizomonadales” (n = 19; GTDB o__ B26-1; This Study “Cluster 4” and “Cluster 5”), “Houtuarculales” (n = 4; GTDB o__40CM-2-53-6; This Study “Cluster 6”), “Wuzhiqibiales” (n = 4; GTDB o__TCS64; This Study “Cluster 1”), and “Xuanwuarculales” (n = 2; GTDB o__RBG_16_48_13; This Study “Cluster 2”). The majority of these Lake Cadagno-derived Ca. Bathyarchaeia RpS3 sequences are more closely related to other representatives within our sediments, than to other Ca. Bathyarchaeia subgroups reported in previous studies (Zhou et al., 2018; Hou et al., 2023). The most abundant persistent Ca. Bathyarchaeia ASV (ASV_ID= Candidatus Bathyarchaeia ASV 1, Supplementary Figure 3) is significantly correlated to Ca. Bathyarchaeia RpS390% sequence “Cluster 4A” (Figure 3) (r = 0.81, corrected p-value = 6 × 10−4), suggesting that these sequences are derived from the same Ca. Bathyarchaeia genome. A dominant and persistent RpS390% was also identified for each candidate order of Ca. Bathyarchaeia within the Lake Cadagno sedimentary sequence (Figure 3). Noteworthy exceptions appear when two relatively distant RpS390% sequence clusters replace the dominant “Wuzhiqibiales” sequence cluster at 40 cmblf and the dominant “Xuanwuarculales” RpS3 sequence cluster is absent from the 582 cmblf sample.
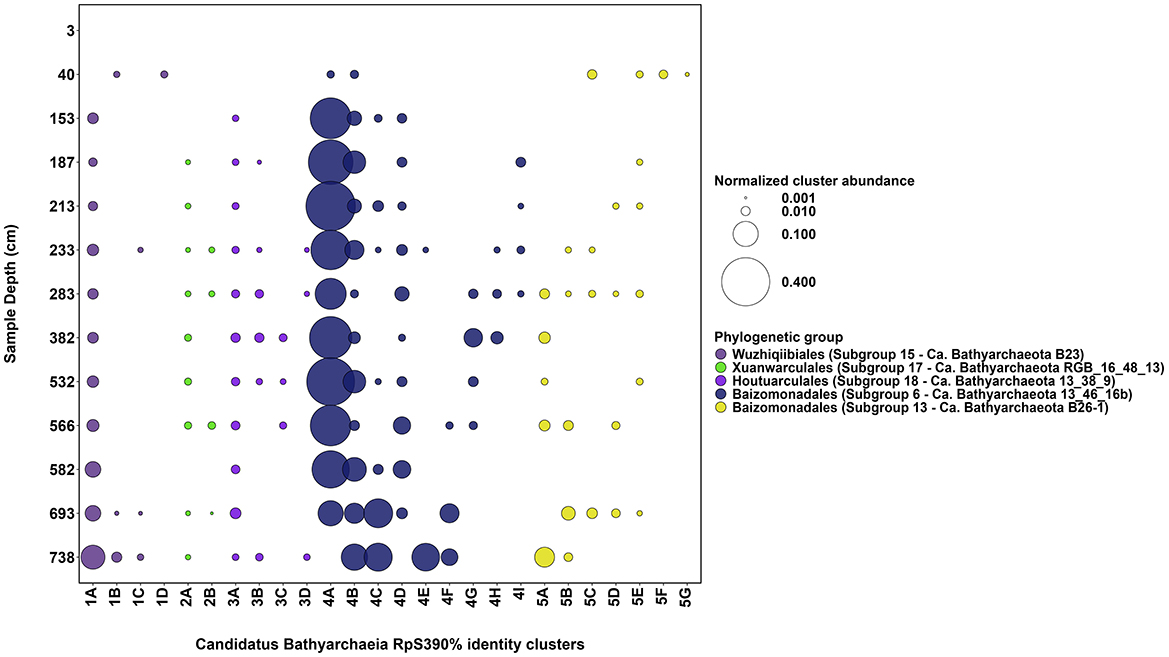
Figure 3. Diversity of Ca. Bathyarchaeia subgroups in Lake Cadagno sediments. The normalized RpS390% cluster abundance (circle size) of the 26 most abundant Ca. Bathyarchaeia RpS390% clusters (x-axis, 1A−5G) per sediment depth (y-axis) is shown. The digits (1–5) associated with each RpS390% cluster ID reflects each cluster's putative phylogenetic clade assignment (circle color).
In our sediments, two Ca. Bathyarchaeia populations (expressed as RpS390% clusters) exhibit significant correlations (corrected p <0.01) with the δ13C-TOC ratios reported in this study (see Table 1) suggesting their potential involvement in carbon cycling in the sediments. These include negative correlations with δ13C-TOC ratios were observed for “Baizomonadaceae” RpS390% sequence cluster 4C (r = −0.84, corrected p = 3.4 × 10−4) and “Wuzhiqibiales” RpS390% sequence cluster 1A (r = −0.71, corrected p = 0.006). The significant negative correlations suggest a link between relatively 13C-depleted organic matter and the presence of these groups.
To evaluate whether specific functional traits are enriched within the Lake Cadagno sediment microbial communities, a characterization of high-order functional categories proposed by the Clusters of Orthologous Genes (COGs) database (Galperin et al., 2021) was performed. Despite the differences in taxonomic composition between samples at 3 cmblf and 40 cmblf compared to sediments below 40 cmblf (Figure 1), the functional profile in terms of COGs remains stable throughout the sediment sequence (Supplementary Figure 4), indicating consistent patterns in the broad functional potential of the microbial community. As biogeochemically relevant gene abundances are more likely to change with changing environmental conditions, a functional profile based on a curated set of biogeochemical marker genes (Zhou et al., 2022) was additionally constructed and analyzed. Within this profile, the per sample carbon-, sulfur-, and nitrogen- cycling marker gene relative abundances are reported as the total coverage of all predicted proteins within a given marker gene protein family (proteintot) divided by the total coverage of all ribosomal protein S3 sequences (RpS3tot) in a sample (Figure 4).

Figure 4. Normalized predicted protein abundance for biological carbon, sulfur and nitrogen cycling marker genes with sediment depth. The circles and their respective sizes indicate the normalized marker gene protein cluster relative abundance to the RpS390% total abundance at a given depth. The circle colors indicate the cycled elements. WLP, Wood Ljungdahl Pathway; CBB cycle, Calvin-Benson-Bassham Cycle; DSR, Canonical dissimilatory sulfate reduction; DNRA, dissimilatory nitrate reduction to ammonia.
The COG category for Carbohydrate Transport and Metabolism (COG category “G”) was the only category related to nutrient acquisition and metabolism with a significant, positive correlation to sample depth (corrected p = 0.039) (Supplementary Table 5). Ten of the “Carbohydrate transport and metabolism” (G COG category) protein clusters were positively enriched with sediment depth (q-value < 0.05, Supplementary File 3). Three of these ten protein clusters are related to Ca. Bathyarchaeia (Supplementary Figure 5) and correspond to a phosphoglucosamine mutase (Normalized enrichment score = 0.81, q-value = 0.04), a 6-phosphofructokinase (Normalized enrichment score = 0.41, q-value = 0.038), and a cofactor-independent phosphoglycerate mutase (Normalized enrichment score = 0.34, q-value = 0.03). The remaining seven protein clusters are related to other bacterial groups including Planctomycetes, Deltaproteobacteria, and Bacteroidetes. On the other hand, 30 protein clusters had negative, significant correlations with sample depth. From these, the 10 proteins with the highest enrichment scores are related to bacterial groups such as Deltaproteobacteria, Planctomycetes, and Chloroflexi, and archaeal groups such as Helarchaeota and Methylomirabilis sp. (Supplementary File 3). Three carbohydrate transport and metabolism predicted proteins derived from Ca. Bathyarchaeia were additionally found to be depleted with sample depth (Supplementary Figure 5). These proteins correspond to Major Facilitator Superfamily (MFS) transporters (Normalized enrichment score = −0.33, q-value = 0.04 and Normalized enrichment score = −0.41, q-value = 0.024) and to a Triose Phosphate Isomerase (TIM) barrel protein (Normalized enrichment score = −0.33, q-value = 0.04).
Predicted proteins of the archaeal carbon monoxide dehydrogenase (CdhC) and bacterial carbon monoxide dehydrogenase (AcsA) were used as markers for microbial carbon fixation via the Wood-Ljundahl pathway (WLP). The associated proteins are part of the five-subunit enzyme complex dehydrogenase/acetyl-CoA synthase (CODH/ACS complex) responsible for the formation of Coenzyme A (CoA) to form acetyl-CoA in the WLP (Adam et al., 2018). These genes have been shown to be also involved in the oxidation of CO to CO2 in methanogenesis and anaerobic oxidation of methane and acetate (Zhou et al., 2022; Bährle et al., 2023) as well as in the reverse direction of the canonical WLP during organo-heterotrophic growth (for a review, see Borrel et al., 2016). A total of 232 unique AcsA and 60 CdhC predicted protein sequence clusters (90% amino acid sequence identity) were observed within the sediments. While a significant anti-correlation between AcsA and CdhC normalized abundances was not observed, the trends indicate that AcsA gene protein cluster abundances decrease below 153 cm and then stabilize, whereas CdhC abundances increase with depth and are linked to a decrease in Methanomicrobiales-related CdhC and an increase in Ca. Bathyarchaeia-related CdhC (Figure 4; Supplementary Figure 6).
A significant correlation (r = 0.80, corrected p = 0.001) between methanogenic CdhC sequence clusters (sequence clusters belonging to Methanomicrobiales) and the methanogenesis marker gene methyl coenzyme M reductase (mcrA) protein cluster abundance was found. In our dataset, McrA protein sequences that are closely related to Methanomicrobiales account for 72% of the total protein sequence abundance in samples between 40 cm and 582 cm and between 693 cm and 738 cm, whereas in the depth interval between 532 cm and 566 cm protein sequences associated with Methanosarcinales are dominant (40%-80% of the total protein sequence abundance) (Supplementary Figure 7). The results from the metabolic pathway completeness analysis suggest that methanogenesis via acetate may be the predominant metabolic pathway in the sediments, as the completeness values are > 0.99 at all depths except at 3 cm and 738 cm (Supplementary File 4). Methanogenesis via trimethylamine is another potentially preferred pathway in sediments between 40 cm and 532 cm (Supplementary File 4).
Archaeal RuBisCO Type III, mainly found in archaea, is the most abundant archaeal C-cycling marker gene observed within the Lake Cadagno sediment sequence. The presence of this protein is frequently associated to the Calvin-Benson-Bassham cycle (CBB cycle); however, it has also been shown to be involved in other processes such as the Reductive Hexulose-Phosphate Pathway (RHP) and the Pentose Phosphate Pathway (Sato et al., 2007; Kono et al., 2017) (Figure 4). Although calculated for the whole-metagenomic dataset rather than an individual genome, the potential prevalence of the CBB cycle in Lake Cadagno sediments is higher (completeness > 0.91) and therefore more likely than the reductive 3-hydroxypropionate (PBP) pathway (completeness <0.50, Supplementary File 4). A significant anticorrelation in the abundance of Type III RuBisCo and AcsA predicted proteins was also observed (r = −0.91, corrected p-value = 1.4 × 10−5) (Supplementary Table 4). In total, 89 Archaeal Type III RuBisCO sequence clusters (90% amino acid sequence identity) were identified with 30 out of 89 of these clusters belonging to Ca. Bathyarchaeia and accounting for 80% of the total Type III RuBisCO abundance. Of these sequences, three sequence clusters related to Ca. Bathyarchaeia are especially abundant within three distinct depth intervals (Supplementary Figure 8); however, a strong correlation between these Type III RuBisCO abundance patterns and a Ca. Bathyarchaeia RpS390% sequence cluster was not identified.
Dissimilatory sulfate reduction (DSR) is an energy-yielding process that reduces sulfate to sulfide (Jorgensen et al., 2019), and frequently used DSR marker genes include the dissimilatory sulfite reductase alpha subunit (dsrA), dissimilatory sulfite reductase beta subunit (dsrB), adenylyl-sulfate reductase alpha subunit (aprA), and adenylyl-sulfate reductase beta subunit (aprB) (Neukirchen et al., 2023). Throughout the sediment sequence, a strong correlation between the abundance of adenylyl sulfate reductase subunits (AprA and AprB, r = 0.92; corrected p = 4.6 x 10−6) and dissimilatory sulfite reduction subunits (DsrA and DsrB, r = 0.97; corrected p = 2.6 x 10−8) provides additional support for continuing DSR at depth (Supplementary Table 4), despite the rapid decrease in sulfate concentrations in the first 20 cm of the sediment core (Table 1, Berg et al., 2022). Dissimilatory sulfate reduction to APS and dissimilatory sulfate reduction to sulfide are the two pathways for sulfate reduction that show highest completeness in the sedimentary sequence (Supplementary File 4), this result is consistent with the presence of marker genes for DSR in the complete set of analyzed samples (Figure 4).
A compositional shift in marker genes for DSR is observed with sediment depth. Forty-seven unique DsrB predicted protein sequence clusters (90% amino acid sequence identity) and 42 unique DsrA protein sequence clusters (90% amino acid sequence identity) were identified, the majority of which are related to Deltaproteobacteria (25/47 and 23/42, respectively). In the youngest samples (3 cmblf and 40 cmblf), DsrB predicted protein sequences related to Chloroflexi, Deltaproteobacteria, and Desulfobacca acetoxidans are abundant but Deltaproteobacteria-related DsrB dominate depths > 40 cmblf and reach a maximal normalized abundance at 582 cmblf (Figure 4, Supplementary Figures 9, 10). Nitrospirae-related DsrB sequences also become prominent in the deepest (693 cm and 738 cm) samples.
Apr is an additional DSR protein that is responsible for catalyzing the oxidative binding of sulfate to AMP to generate APS which is subsequently converted to sulfate by the ATP sulfurylase (Meyer and Kuever, 2007). Although Deltaproteobacteria-related Apr are the dominant Apr subtype from 3 cmblf to 582 cmblf, at depths > 693 cmblf, Nitrospirae-related AprA and AprB protein sequence clusters represent >70% of the per sample sequence abundance for these genes (Supplementary Figures 11, 12).
Protein-encoding genes involved in the re-oxidation of reduced sulfur species were also identified in Lake Cadagno sediments; however, genes (soxA, soxB, and soxC) which are commonly used for sulfur oxidation (although not limited to this metabolic function) were far less prevalent with SoxA and SoxC predicted protein sequences identified in only 3/13 and 2/13 samples respectively, and SoxB predicted protein sequences identified in only the 3 cmblf sample (Supplementary Figures 13–15). Richness of the Sox predicted proteins was also less than the richness of predicted proteins involved in DSR and most of Sox sequences were related to Chlorobi, Betaproteobacteria, and Gemmatimonadetes (Supplementary Figures 13–15).
The complete denitrification pathway involves the reduction of nitrate to nitrogen through a series of N-containing intermediates ( to to NO to N2O to N2). Despite the prevalence of nitric oxide reductase predicted proteins (NorB and NorC) in samples between 40 cm and 693 cm, genomic evidence for the canonical denitrification pathway is sparse (Figure 4). Predicted protein sequences of nitrate reductase subunit alpha (NarG) were identified at relatively low abundances in four of the 13 samples (Figure 4, average NarGtot/RpS3tot = 0.00241 ± 0.00023) and the cytochrome-type dissimilatory nitrite reductase (NirS) was only observed in the 40 cmblf sample (NirStot/RpS3tot= 0.00272). Instead, the enzyme reported to catalyze the reduction of nitrite to ammonia in a process known as denitrification to ammonia (DNRA, Mohan et al., 2004), cytochrome c nitrite reductase (Nrf), was found across all sample depths except 582 cmblf (Figure 4). In total, 50 NrfA predicted protein sequence clusters (90% amino acid sequence identity) and 25 small subunit NrfH predicted protein sequence clusters (90% amino acid sequence identity) were identified at similar abundances and depths (Figure 4). Both predicted proteins show a maximal abundance at 40 cmblf (NrfA/RpS3tot = 0.15 and NrfH/RpS3tot = 0.10) and are mainly related to Planctomycetes, Chloroflexi, Deltaproteobacteria and Bacteroidetes (Supplementary Figures 16, 17).
Microbial cycling of ammonia in the Lake Cadagno sediments may also occur through nitrogen fixation and ammonia oxidation. Although the nitrogenase electron donor subunit (NifH) was identified in the predicted protein pool of all Lake Cadagno sediment samples, the catalytic subunits of nitrogenase, NifD and NifK, were only detected with NifH in four of the thirteen samples (3–158 cmblf and 582 cmblf, Figure 4). The enzyme hydroxylamine oxidoreductase (HAO) which catalyzes the oxidation of NH2OH to and is a marker gene for ammonia oxidation was detected in all samples with an average HAOtot/RpS3tot of 0.063 ± 0.029. HAO is frequently linked to aerobic ammonia oxidation (Junier et al., 2010) but a variant of the enzyme is also involved in anaerobic ammonium oxidation (anammox) (Sui et al., 2020; Kartal and Keltjens, 2016). Taxonomic assignment of Lake Cadagno HAO predicted proteins revealed a close relationship to Planctomycetes and may indicate that anammox occurs within the sediments (Supplementary Figure 18).
Despite the drastic changes in sedimentary sources (lacustrine vs. mass-movement deposits) and a transition from bacteria-dominated communities in the top 40 cm to Ca. Bathyarchaeia-dominated communities in deeper layers of the 8,000-year sediment sequence of Lake Cadagno (Figure 1), dominant organic matter compositions (Berg et al., 2022; Gajendra et al., 2023) and the metabolic potential of the sediment's in situ microbial populations (Figure 4, Supplementary Figure 4) are conserved. Broadly speaking, Lake Cadagno's sedimentary microbial communities have the genomic potential to perform bacterial and archaeal carbon fixation via the Wood-Ljundahl pathway (WLP) and the Calvin-Benson-Bassham cycle (CBB cycle), canonical dissimilatory sulfate reduction (DSR), dissimilatory nitrate reduction to ammonia (DNRA) (Figure 4) and organic matter degradation; however, further studies are needed to confirm the expression and activity of these functions in Lake Cadagno. Interestingly, the taxonomic assignment of persistent marker genes shows strong vertical shifts (Supplementary Figures 6–22) that are not significantly correlated with sediment origin or any of the previously measured environmental parameters for this sedimentary sequence, originally reported in Berg et al. (2022) (Table 1, Supplementary Table 1). Given the absence of significant ecological correlations within this study, we expect that functional taxonomic replacement with depth is most likely driven by neutral processes (Liu et al., 2023), interactions between microbial populations (Martiny et al., 2023), or other non-measured geochemical and physicochemical parameters such as pH, salinity, and porewater content (Hoshino et al., 2020). As previous studies have suggested the preservation of functional traits increases ecosystem stability (Royalty and Steen, 2021), we hypothesize that the stability of functional traits with sediment depth throughout Lake Cadagno likely promotes ecosystem resilience by sustaining the core functions of the community after burial and under energy limitation; however, further activity-based analyses are needed to evaluate this hypothesis.
Although the metagenomic data indicate that a core set of metabolic functions is conserved in the Lake Cadagno sedimentary community, variation within specific metabolic pathways across taxonomic and geochemical gradients is also observed. In other environments, higher amounts of taxonomic variation within the intermediate steps of major metabolic pathways (e.g., hydrogenotrophic vs. acetoclastic methanogenesis) relative to the metabolic endpoint (e.g., methanogenesis) have also been reported (Louca et al., 2018). In Lake Cadagno, the marker gene for methanogenesis (mcrA) is present at relatively similar abundances throughout the sediment core (Figure 4) but a transition between metabolically distinct Methanomicrobiales and Methanosarcinales archaea is also observed (Supplementary Figure 7). Methanomicrobiales persist throughout the sedimentary sequence and typically perform hydrogenotrophic methanogenesis with CO2 (Zhang et al., 2020) while members of Methanosarcinales become the dominant methanogenic group within a local methane peak in Lake Cadagno's sediment (532–566 cmbss, Table 1) and exhibit the potential to perform methylotrophic, hydrogenotrophic and acetoclastic methanogenesis (Evans et al., 2019; Lyautey et al., 2021). Although the complete methanogenesis pathways of Lake Cadagno's methanogens were not recovered in this study, the prevalence of methanogenesis as a core metabolic pathway suggests that it is an essential part of the Lake Cadagno ecological network.
As with the aforementioned methanogens, the vast majority of microbial taxa in our sediments such as Chloroflexi, Proteobacteria, Atribacteria, Aminicenantes, Ca. Bathyarchaeia and Firmicutes are globally widespread in anoxic subsurface sediments of freshwater and marine environments (e.g., D'Hondt et al., 2004; Orsi, 2018; Yu et al., 2018; Orsi et al., 2020; Vuillemin et al., 2018; Suominen et al., 2021; Han et al., 2020). These similarities in microbial community composition can be partially explained by shared sedimentological properties (Hoshino et al., 2020), terminal electron acceptors, substrates, and metabolic products in deep sedimentary environments (D'Hondt et al., 2004; Vuillemin et al., 2018; Zhang et al., 2020; Dong et al., 2023; Jorgensen et al., 2012; He et al., 2022). Interestingly, a relatively small set of these taxa were identified as “persister populations” throughout the Lake Cadagno sedimentary sequence, appearing in ≥80% of the sampled sediment layers. The most abundant Lake Cadagno persister populations are closely related to the groups Ca. Bathyarchaeia, Chloroflexi, and Atribacteria. These groups dominate the taxonomic profiles of carbon fixation marker genes (Supplementary Figures 6, 8, 19–22), contain genes related to carbohydrate metabolism (Supplementary material 1), and have been identified as potentially versatile mixotrophs in sedimentary environments (Mardanov et al., 2020; Hou et al., 2023; Zhou et al., 2018; Nobu et al., 2016; Dodsworth et al., 2013; Fincker et al., 2020; Lee et al., 2018). Based on the prevalence of these groups throughout Lake Cadagno and other sedimentary environments (Orsi, 2018), we hypothesize that the metabolic versatility of these lineages makes them well-adapted for survival in deep marine and lacustrine sedimentary environments.
At the global scale, organic carbon availability is frequently reported as an important control on the diversity and abundance of sedimentary communities (for a review, see Orsi, 2018) and, therefore, it is also worth noting that changes in the relative functional contribution of persister populations to the organic matter pool are observed across the sediment sequence. In Lake Cadagno, the sedimentary carbohydrate content profile shows an increase in the relative contribution of levosugars with sediment depth and it is hypothesized these are potentially cellulose-derived, but their origin may differ depending on sample depth (surface vs. deep sediments) and sedimentary sources (lacustrine vs. mass-movement deposits) (Gajendra et al., 2023). An enrichment analysis of genes involved in carbohydrate cycling, transport, and metabolism shows that Chloroflexi-related gene abundances decrease with sediment depth while deeper sediments are enriched with genes related to Ca. Bathyarchaeia and other bacterial groups (Supplementary Figure 5). These results support previous reports that have identified organic matter as an important driver of sedimentary diversity patterns (e.g., Baker et al., 2015; Orsi, 2018) even though a significant correlation between Lake Cadagno's organic carbon pool and microbial diversity was not observed. Taken together, we interpret the observed persistence of microbial community functionality across the different sediment layers with different organic carbon sources (sediment layers of lacustrine vs. turbidite origin) as a sign of ecosystem resilience (Allison and Martiny, 2008) and the persistence of select taxonomic groups throughout sedimentary environments as an indication that specific niches within the sediment are uniquely filled by well-adapted subsurface lineages. However, it is worth noting that only a subset of the populations may be actively performing these specific functions at the time (Louca et al., 2018) and, therefore, single-cell or community-scale activity levels should be assessed by future studies.
Our findings indicate that microbial diversity patterns within Lake Cadagno sediments follow global sediment biodiversity trends such as a decrease in bacterial diversity and abundance and an increase in the relative abundance of persistent archaeal groups with depth. These changes in microbial community composition align with decreases in total organic carbon availability in the sediment; however, energy-transducing functions related to carbon, sulfur, and nitrogen cycling were preserved independently of the taxonomic composition of the sediment samples.
While many studies have reported evidence for a convergence in metabolic features across larger spatial scales (Chen et al., 2022; Royalty and Steen, 2021; Dopheide et al., 2015; Louca et al., 2016; Nelson et al., 2016), this study indicates that shared functionality is also important at more local scales and across sharp geochemical gradients, irrespective of sediment age and organic matter sources. The absence of evidence supporting deterministic selection based on the geochemical parameters considered in this study suggests the taxonomic diversity of groups encoding the selected marker genes is most likely related to stochastic or ecologically neutral processes (Liu et al., 2023) or a more complex combination of ecological factors such as the interactions and feedbacks between individual microbial populations and/or the physiological acclimation of persisting microorganisms after burial (Martiny et al., 2023). Based on these findings we hypothesize that shared functionality across multiple microbial lineages enables Lake Cadagno sediment-hosted microbial communities to survive in energy limitation and preserve the connectivity of metabolic pathways required for biogeochemical cycling.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
PR: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing – original draft, Writing – review & editing. JB: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – review & editing, Formal analysis. LD: Investigation, Supervision, Validation, Writing – review & editing. HV: Formal analysis, Investigation, Supervision, Writing – review & editing. MO: Data curation, Formal analysis, Writing – review & editing. ML: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review & editing. CM: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by ETH start up-funds to CM, the Swiss National Science Foundation (SNSF) grant no. 182096 to ML which covered the cost of the sampling campaign and of work in the Lever Lab. LD was sponsored by the Shanghai Pujiang Program (22PJ1404800). Open access funding by ETH Zurich.
We acknowledge the Genomics Diversity Center (GDC) Zurich and the Functional Genomics Center Zurich (FGCZ) for use of their research facilities. We thank Lena Bakker for her support with the protein enrichment analyses, Eleanor Georgiadis for her help proofreading the manuscript and Zhe Wang for his assistance with the nitrogen cycling data interpretation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that generative AI was used in the creation of this manuscript. Code was improved in consultation with AI tools. All errors are human.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1504355/full#supplementary-material
The scripts used to create the figures in the manuscript, along with the metagenomic sequencing data table and the 16S rRNA gene targeted sequencing OTU table, and Supplementary Data 1 are stored in the GitHub repository https://github.com/PaulaCat/Lake_Cadagno_PFTGLC.git.
Adam, P. S., Borrel, G., and Gribaldo, S. (2018). Evolutionary history of carbon monoxide dehydrogenase/acetyl-CoA synthase, one of the oldest enzymatic complexes. Proc. Natl. Acad. Sci. USA. 115, E1166–E1173. doi: 10.1073/pnas.1716667115
Allison, S. D., and Martiny, J. B. H. (2008). Resistance, resilience, and redundancy in microbial communities. Proc. Nat. Acad. Sci. 105, 11512–11519. doi: 10.1073/pnas.0801925105
Andrews (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed April 16, 2021).
Ariztegui, D., Thomas, C., and Vuillemin, A. (2015). Present and future of subsurface biosphere studies in lacustrine sediments through scientific drilling. Int. J. Earth Sci. 104, 1655–1665. doi: 10.1007/s00531-015-1148-4
Bährle, R., Böhnke, S., Englhard, J., Bachmann, J., and Perner, M. (2023). Current status of carbon monoxide dehydrogenases (CODH) and their potential for electrochemical applications. Bioresour. Bioproc. 10:84. doi: 10.1186/s40643-023-00705-9
Baker, B. J., Lazar, C. S., Teske, A. P., and Dick, G. J. (2015). Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 3:6. doi: 10.1186/s40168-015-0077-6
Barber, A., Brandes, J., Leri, A., Lalonde, K., Balind, K., Wirick, S., et al. (2017). Preservation of organic matter in marine sediments by inner-sphere interactions with reactive iron. Sci. Rep. 7:366. doi: 10.1038/s41598-017-00494-0
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., et al. (2017). GenBank. Nucleic Acids Res. 45, D37–D42. doi: 10.1093/nar/gkw1070
Berg, J. S., Lepine, M., Laymand, E., Han, X., Vogel, H., Morlock, M. A., et al. (2022). Ancient and modern geochemical signatures in the 13,500-year sedimentary record of Lake Cadagno. Front Earth Sci (Lausanne) 9. doi: 10.3389/feart.2021.754888
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Borrel, G., Adam, P. S., and Gribaldo, S. (2016). Methanogenesis and the wood-ljungdahl pathway: an ancient, versatile, and fragile association. Genome Biol. Evol. 8, 1706–1711. doi: 10.1093/gbe/evw114
Cadillo-Quiroz, H., Bräuer, S., Yashiro, E., Sun, C., Yavitt, J., and Zinder, S. (2006). Vertical profiles of methanogenesis and methanogens in two contrasting acidic peatlands in central New York State, USA. Environ. Microbiol. 8, 1428–1440. doi: 10.1111/j.1462-2920.2006.01036.x
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: Architecture and applications. BMC Bioinform. 10:421. doi: 10.1186/1471-2105-10-421
Chen, H., Ma, K., Lu, C., Fu, Q., Qiu, Y., Zhao, J., et al. (2022). Functional redundancy in soil microbial community based on metagenomics across the globe. Front. Microbiol. 13:878978. doi: 10.3389/fmicb.2022.878978
Chen, X., Andersen, T. J., Morono, Y., Inagaki, F., Jørgensen, B. B., and Lever, M. A. (2017). Bioturbation as a key driver behind the dominance of Bacteria over Archaea in near-surface sediment. Sci. Rep. 7:2400. doi: 10.1038/s41598-017-02295-x
Deng, L., Bölsterli, D., Kristensen, E., Meile, C., Su, C.-C., Bernasconi, S. M., et al. (2020). Macrofaunal control of microbial community structure in continental margin sediments. Proc. Natl. Acad. Sci. U. S. A. 117, 15911–15922. doi: 10.1073/pnas.1917494117
D'Hondt, S., Jorgensen, B. B., Miller, D. J., Batzke, A., Blake, R., Cragg, B. A., et al. (2004). Distributions of microbial activities in deep subseafloor sediments. Science 306, 2216–2221. doi: 10.1126/science.1101155
Dodsworth, J. A., Blainey, P. C., Murugapiran, S. K., Swingley, W. D., Ross, C. A., Tringe, S. G., et al. (2013). Single-cell and metagenomic analyses indicate a fermentative and saccharolytic lifestyle for members of the OP9 lineage. Nat. Commun. 4:2884. doi: 10.1038/ncomms2884
Dong, X., Peng, Y., Wang, M., Woods, L., Wu, W., Wang, Y., et al. (2023). Evolutionary ecology of microbial populations inhabiting deep sea sediments associated with cold seeps. Nat. Commun. 14:1127. doi: 10.1038/s41467-023-36877-3
Dopheide, A., Lear, G., He, Z., Zhou, J., and Lewis, G. D. (2015). Functional gene composition, diversity and redundancy in microbial stream biofilm communities. PLoS ONE 10:e123179. doi: 10.1371/journal.pone.0123179
Edgar, R. C. (2004). MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 5:113. doi: 10.1186/1471-2105-5-113
Evans, P. N., Boyd, J. A., Leu, A. O., Woodcroft, B. J., Parks, D. H., Hugenholtz, P., et al. (2019). An evolving view of methane metabolism in the Archaea. Nat. Rev. Microbiol. 17, 219–232. doi: 10.1038/s41579-018-0136-7
Fincker, M., Huber, J. A., Orphan, V. J., Rapp,é, M. S., Teske, A., and Spormann, A. M. (2020). Metabolic strategies of marine subseafloor Chloroflexi inferred from genome reconstructions. Environ. Microbiol. 22, 3188–3204. doi: 10.1111/1462-2920.15061
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 39:gkr367. doi: 10.1093/nar/gkr367
Gajendra, N., Berg, J. S., Vogel, H., Deng, L., Wolf, S. M., Bernasconi, S. M., et al. (2023). Carbohydrate compositional trends throughout Holocene sediments of an alpine lake (Lake Cadagno). Front. Earth Sci. 11:1047224. doi: 10.3389/feart.2023.1047224
Galperin, M. Y., Wolf, Y. I., Makarova, K. S., Alvarez, R. V., Landsman, D., and Koonin, E. V. (2021). COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 49, D274–D281. doi: 10.1093/nar/gkaa1018
Graham, E. D., Heidelberg, J. F., and Tully, B. J. (2018). Potential for primary productivity in a globally-distributed bacterial phototroph. ISME J. 12, 1861–1866. doi: 10.1038/s41396-018-0091-3
Han, X., Schubert, C. J., Fiskal, A., Dubois, N., and Lever, M. A. (2020). Eutrophication as a driver of microbial community structure in lake sediments. Environ. Microbiol. 22, 3446–3462. doi: 10.1111/1462-2920.15115
Han, X., Tolu, J., Deng, L., Fiskal, A., Schubert, C. J., Winkel, L. H. E., et al. (2022). Long-term preservation of biomolecules in lake sediments: potential importance of physical shielding by recalcitrant cell walls. PNAS Nexus 1:gac076. doi: 10.1093/pnasnexus/pgac076
He, R., Wang, J., Pohlman, J. W., Jia, Z., Chu, Y.-X., Wooller, M. J., et al. (2022). Metabolic flexibility of aerobic methanotrophs under anoxic conditions in Arctic lake sediments. ISME J. 16, 78–90. doi: 10.1038/s41396-021-01049-y
Herlemann, D. P., Labrenz, M., Jürgens, K., Bertilsson, S., Waniek, J. J., and Andersson, A. F. (2011). Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5, 1571–1579. doi: 10.1038/ismej.2011.41
Hoshino, T., Doi, H., Uramoto, G.-I., Wörmer, L., Adhikari, R. R., Xiao, N., et al. (2020). Global diversity of microbial communities in marine sediment. Proc. Natl. Acad. Sci. U. S. A. 117, 27587–27597. doi: 10.1073/pnas.1919139117
Hou, J., Wang, Y., Zhu, P., Yang, N., Liang, L., Yu, T., et al. (2023). Taxonomic and carbon metabolic diversification of Bathyarchaeia during its coevolution history with early Earth surface environment. Sci Adv. 9:eadf5069. doi: 10.1126/sciadv.adf5069
Hubert, C., Loy, A., Nickel, M., Arnosti, C., Baranyi, C., Brüchert, V., et al. (2009). A constant flux of diverse thermophilic bacteria into the cold Arctic seabed. Science 325, 1541–1544. doi: 10.1126/science.1174012
Hyatt, D., Chen, G.-L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119. doi: 10.1186/1471-2105-11-119
Jessen, G. L., Lichtschlag, A., Ramette, A., Pantoja, S., Rossel, P. E., Schubert, C. J., et al. (2017). Hypoxia causes preservation of labile organic matter and changes seafloor microbial community composition (Black Sea). Sci. Adv. 3:e1601897. doi: 10.1126/sciadv.1601897
Jorgensen, B. B., Findlay, A. J., and Pellerin, A. (2019). The biogeochemical sulfur cycle of marine sediments. Front. Microbiol. 10:849. doi: 10.3389/fmicb.2019.00849
Jorgensen, B. B., and Marshall, I. P. G. (2016). Slow microbial life in the seabed. Ann. Rev. Mar. Sci. 8, 311–332. doi: 10.1146/annurev-marine-010814-015535
Jorgensen, S. L., Hannisdal, B., Lanzén, A., Baumberger, T., Flesland, K., Fonseca, R., et al. (2012). Correlating microbial community profiles with geochemical data in highly stratified sediments from the Arctic Mid-Ocean Ridge. Proc. Natl. Acad. Sci. U. S. A. 109, E2846–E2855. doi: 10.1073/pnas.1207574109
Junier, P., Molina, V., Dorador, C., Hadas, O., Kim, O. S., Junier, T., et al. (2010). Phylogenetic and functional marker genes to study ammonia-oxidizing microorganisms (AOM) in the environment. Appl. Microbiol. Biotechnol. 85, 425–440. doi: 10.1007/s00253-009-2228-9
Kallmeyer, J., Pockalny, R., Adhikari, R. R., Smith, D. C., and D'Hondt, S. (2012). Global distribution of microbial abundance and biomass in subseafloor sediment. Proc. Natl. Acad. Sci. U. S. A. 109, 16213–16216. doi: 10.1073/pnas.1203849109
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kartal, B., and Keltjens, J. T. (2016). Anammox biochemistry: a tale of Heme c proteins. Trends Biochem. Sci. 41, 998–1011. doi: 10.1016/j.tibs.2016.08.015
Kono, T., Mehrotra, S., Endo, C., Kizu, N., Matusda, M., Kimura, H., et al. (2017). A RuBisCO-mediated carbon metabolic pathway in methanogenic archaea. Nat. Commun. 8:14007. doi: 10.1038/ncomms14007
Le, S. Q., Gascuel, O., and Quang Le, S. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Lee, Y. M., Hwang, K., Lee, J., Kim, M., Hwang, C. Y., Noh, H. J., et al. (2018). Genomic insight into the predominance of candidate phylum Atribacteria JS1 lineage in marine sediments. Front. Microbiol. 9:02909. doi: 10.3389/fmicb.2018.02909
Lever, M. A., Torti, A., Eickenbusch, P., Michaud, A. B., Šantl-Temkiv, T., and Jorgensen, B. B. (2015). A modular method for the extraction of DNA and RNA, and the separation of DNA pools from diverse environmental sample types. Front. Microbiol. 6:476. doi: 10.3389/fmicb.2015.00476
Li, W., and Godzik, A. (2006). Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Liu, H., Lin, G., Gao, D., Chen, H., He, M., and Lu, J. (2023). Geographic scale influences the interactivities between determinism and stochasticity in the assembly of sedimentary microbial communities on the South China Sea shelf. Microb. Ecol. 85, 121–136. doi: 10.1007/s00248-021-01946-x
Lloyd, K. G., Schreiber, L., Petersen, D. G., Kjeldsen, K. U., Lever, M. A., Steen, A. D., et al. (2013). Predominant archaea in marine sediments degrade detrital proteins. Nature 496, 215–218. doi: 10.1038/nature12033
Louca, S., Jacques, S. M. S., Pires, A. P. F., Leal, J. S., Srivastava, D. S., Parfrey, L. W., et al. (2016). High taxonomic variability despite stable functional structure across microbial communities. Nat. Ecol. Evol. 1:15. doi: 10.1038/s41559-016-0015
Louca, S., Polz, M. F., Mazel, F., Albright, M. B. N., Huber, J. A., O'Connor, M. I., et al. (2018). Function and functional redundancy in microbial systems. Nat. Ecol. Evol. 2, 936–943. doi: 10.1038/s41559-018-0519-1
Lyautey, E., Billard, E., Tissot, N., Jacquet, S., and Domaizon, I. (2021). Seasonal dynamics of abundance, structure, and diversity of methanogens and methanotrophs in lake sediments. Microb. Ecol. 82, 559–571. doi: 10.1007/s00248-021-01689-9
Mallick, H., Rahnavard, A., McIver, L. J., Ma, S., Zhang, Y., Nguyen, L. H., et al. (2021). Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 17:e1009442. doi: 10.1371/journal.pcbi.1009442
Mardanov, A. V., Kadnikov, V. V., Beletsky, A. V., and Ravin, N. V. (2020). Sulfur and methane-oxidizing microbial community in a terrestrial mud volcano revealed by metagenomics. Microorganisms 8, 1–16. doi: 10.3390/microorganisms8091333
Marshall, I. P. G., Ren, G., Jaussi, M., Lomstein, B. A., Jorgensen, B. B., Roy, H., et al. (2019). Environmental filtering determines family-level structure of sulfate-reducing microbial communities in subsurface marine sediments. Isme .J 13, 1920–1932. doi: 10.1038/s41396-019-0387-y
Martiny, J. B. H., Martiny, A. C., Brodie, E., Chase, A. B., Rodríguez-Verdugo, A., Treseder, K. K., et al. (2023). Investigating the eco-evolutionary response of microbiomes to environmental change. Ecol. Lett. 26, S81–S90. doi: 10.1111/ele.14209
Meyer, B., and Kuever, J. (2007). Molecular analysis of the distribution and phylogeny of dissimilatory adenosine-5′-phosphosulfate reductase-encoding genes (aprBA) among sulfur-oxidizing. Microbiology 153, 3478–3498. doi: 10.1099/mic.0.2007/008250-0
Mohan, S. B., Schmid, M., Jetten, M., and Cole, J. (2004). Detection and widespread distribution of the nrfA gene encoding nitrite reduction to ammonia, a short circuit in the biological nitrogen cycle that competes with denitrification. FEMS Microbiol. Ecol. 49, 433–443. doi: 10.1016/j.femsec.2004.04.012
Morono, Y., Ito, M., Hoshino, T., Terada, T., Hori, T., Ikehara, M., et al. (2020). Aerobic microbial life persists in oxic marine sediment as old as 101.5 million years. Nat. Commun. 11:1. doi: 10.1038/s41467-020-17330-1
Nelson, M. B., Martiny, A. C., and Martiny, J. B. H. (2016). Global biogeography of microbial nitrogen-cycling traits in soil. Proc. Natl. Acad. Sci. USA. 113, 8033–8040. doi: 10.1073/pnas.1601070113
Neukirchen, S., Pereira, I. A. C., and Sousa, F. L. (2023). Stepwise pathway for early evolutionary assembly of dissimilatory sulfite and sulfate reduction. ISME J. 17, 1680–1692. doi: 10.1038/s41396-023-01477-y
Nobu, M. K., Dodsworth, J. A., Murugapiran, S. K., Rinke, C., Gies, E. A., Webster, G., et al. (2016). Phylogeny and physiology of candidate phylum “Atribacteria” (OP9/JS1) inferred from cultivation-independent genomics. ISME J. 10, 273–286. doi: 10.1038/ismej.2015.97
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). MetaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Oksanen, J., Simpson, G., Blanchet, F., Kindt, R., Legendre, P., Minchin, P., et al. (2024). vegan: Community Ecology Package. R package version 2.7-0. Available at: https://github.com/vegandevs/vegan
Oni, O. E., Schmidt, F., Miyatake, T., Kasten, S., Witt, M., Hinrichs, K.-U., et al. (2015). Microbial communities and organic matter composition in surface and subsurface sediments of the Helgoland mud area, North Sea. Front. Microbiol. 6:1290. doi: 10.3389/fmicb.2015.01290
Orsi, W. D. (2018). Ecology and evolution of seafloor and subseafloor microbial communities. Nat. Rev. Microbiol. 16, 671–683. doi: 10.1038/s41579-018-0046-8
Orsi, W. D., Schink, B., Buckel, W., and Martin, W. F. (2020). Physiological limits to life in anoxic subseafloor sediment. FEMS Microbiol. Rev. 44, 219–231. doi: 10.1093/femsre/fuaa004
Parada, A. E., Needham, D. M., and Fuhrman, J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414 doi: 10.1111/1462-2920.13023
Petro, C., Starnawski, P., Schramm, A., and Kjeldsen, K. U. (2017). Microbial community assembly in marine sediments. Aquat. Microb. Ecol. 79, 177–195. doi: 10.3354/ame01826
Philippi, M., Kitzinger, K., Berg, J. S., Tschitschko, B., Kidane, A. T., Littmann, S., et al. (2021). Purple sulfur bacteria fix N2 via molybdenum-nitrogenase in a low molybdenum Proterozoic ocean analogue. Nat. Commun. 12:4774. doi: 10.1038/s41467-021-25000-z
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 - Approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e0009490. doi: 10.1371/journal.pone.0009490
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Quince, C., Lanzen, A., Davenport, R. J., and Turnbaugh, P. J. (2011). Removing noise from pyrosequenced amplicons. BMC Bioinformat. 12:38. doi: 10.1186/1471-2105-12-38
Rochette, N. C., Rivera-Colón, A. G., Walsh, J., Sanger, T. J., Campbell-Staton, S. C., and Catchen, J. M. (2023). On the causes, consequences, and avoidance of PCR duplicates: Towards a theory of library complexity. Mol. Ecol. Resour. 23, 1299–1318. doi: 10.1111/1755-0998.13800
Royalty, T. M., and Steen, A. D. (2021). Functional redundancy in ocean microbiomes controls trait stability. bioRxiv. [Preprint]. doi: 10.1101/2021.06.18.448980
Sato, T., Atomi, H., and Imanaka, T. (2007). Archaeal type III RuBisCOs function in a pathway for AMP metabolism. Science 315, 1003–1006. doi: 10.1126/science.1135999
Sørensen, K. B., and Teske, A. (2006). Stratified communities of active archaea in deep marine subsurface sediments. Appl. Environ. Microbiol. 72, 4596–4603. doi: 10.1128/AEM.00562-06
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Starnawski, P., Bataillon, T., Ettema, T. J. G., Jochum, L. M., Schreiber, L., Chen, X., et al. (2017). Microbial community assembly and evolution in subseafloor sediment. Proc. Natl. Acad. Sci. USA. 114, 2940–2945. doi: 10.1073/pnas.1614190114
Sui, Q., Wang, Y., Wang, H., Yue, W., Chen, Y., Yu, D., et al. (2020). Roles of hydroxylamine and hydrazine in the in-situ recovery of one-stage partial nitritation-anammox process: Characteristics and mechanisms. Sci. Total Environm. 707:135648. doi: 10.1016/j.scitotenv.2019.135648
Suominen, S., van Vliet, D. M., Sánchez-Andrea, I., van der Meer, M. T. J., Sinninghe Damsté, J. S., and Villanueva, L. (2021). Organic matter type defines the composition of active microbial communities originating from anoxic baltic sea sediments. Front. Microbiol. 12:628301. doi: 10.3389/fmicb.2021.628301
Varliero, G., Bienhold, C., Schmid, F., Boetius, A., and Molari, M. (2019). Microbial diversity and connectivity in deep-sea sediments of the South Atlantic Polar Front. Front. Microbiol. 10:665. doi: 10.3389/fmicb.2019.00665
Vuillemin, A., Ariztegui, D., Horn, F., Kallmeyer, J., Orsi, W. D., Anselmetti, F., et al. (2018). Microbial community composition along a 50 000-year lacustrine sediment sequence. FEMS Microbiol. Ecol. 94:fiy029. doi: 10.1093/femsec/fiy029
Wirth, S. B., Gilli, A., Niemann, H., Dahl, T. W., Ravasi, D., Sax, N., et al. (2013). Combining sedimentological, trace metal (Mn, Mo) and molecular evidence for reconstructing past water-column redox conditions: The example of meromictic Lake Cadagno (Swiss Alps). Geochim. Cosmochim. Acta 120, 220–238. doi: 10.1016/j.gca.2013.06.017
Yu, T., Wu, W., Liang, W., Lever, M. A., Hinrichs, K. U., and Wang, F. (2018). Growth of sedimentary Bathyarchaeota on lignin as an energy source. Proc. Natl. Acad. Sci. USA. 115, 6022–6027. doi: 10.1073/pnas.1718854115
Zhang, C.-J., Pan, J., Liu, Y., Duan, C.-H., and Li, M. (2020). Genomic and transcriptomic insights into methanogenesis potential of novel methanogens from mangrove sediments. Microbiome 8:94. doi: 10.1186/s40168-020-00876-z
Zhou, Z., Pan, J., Wang, F., Gu, J. D., and Li, M. (2018). Bathyarchaeota: globally distributed metabolic generalists in anoxic environments. FEMS Microbiol. Rev. 42, 639–655. doi: 10.1093/femsre/fuy023
Zhou, Z., Tran, P. Q., Breister, A. M., Liu, Y., Kieft, K., Cowley, E. S., et al. (2022). METABOLIC: high-throughput profiling of microbial genomes for functional traits, metabolism, biogeochemistry, and community-scale functional networks. Microbiome 10:8. doi: 10.1186/s40168-021-01213-8
Keywords: deep lacustrine sediments, functional potential, microbial communities, biogeochemical cycling, metagenomics
Citation: Rodriguez P, Berg JS, Deng L, Vogel H, Okoniewski M, Lever MA and Magnabosco C (2025) Persistent functional and taxonomic groups dominate an 8,000-year sedimentary sequence from Lake Cadagno, Switzerland. Front. Microbiol. 16:1504355. doi: 10.3389/fmicb.2025.1504355
Received: 30 September 2024; Accepted: 06 January 2025;
Published: 03 February 2025.
Edited by:
Alberto Robador, University of Southern California, United StatesReviewed by:
Carl-Eric Wegner, Heinrich Heine University of Düsseldorf, GermanyCopyright © 2025 Rodriguez, Berg, Deng, Vogel, Okoniewski, Lever and Magnabosco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paula Rodriguez, cHJhbWlyZXpAZXRoei5jaA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.