
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 21 February 2025
Sec. Microbe and Virus Interactions with Plants
Volume 16 - 2025 | https://doi.org/10.3389/fmicb.2025.1501741
This article is part of the Research Topic Metagenomic Insights into Microbial Communities in Fruits and Vegetable Plants View all articles
 Serafina Serena Amoia1,2
Serafina Serena Amoia1,2 Maria Saponari1*
Maria Saponari1* Pasquale Saldarelli1
Pasquale Saldarelli1 Angela Maria Ligorio1
Angela Maria Ligorio1 Carmine Del Grosso1
Carmine Del Grosso1 Giuliana Loconsole1
Giuliana Loconsole1 Giusy D’Attoma1
Giusy D’Attoma1 Donato Boscia1
Donato Boscia1 Annalisa Giampetruzzi1*
Annalisa Giampetruzzi1*Introduction: Xylella fastidiosa is a phytopathogenic bacterium of worldwide importance causing detrimental diseases in several crops. Recent reports from European and Mediterranean countries raised great concerns and have given impetus to new studies investigating both the pathogenicity of the newly emerged strains and the susceptibility and vulnerability of Mediterranean agroecosystems, with the outbreak in olive trees in southern Italy being the most investigated new pathosystem. The complexity of this pathogen makes difficult to understand its interaction mechanisms with host plants and plant microbial communities.
Materials and methods: In this study, we performed a pilot dual RNA-seq analysis on a diseased olive tree infected by Xylella fastidiosa subspecies pauca, to gather information about bacterial infection dynamics and reciprocal interactions between plant host and the bacterium. Adopting a mRNA enrichment protocol allowed to better probe bacterial sequences by increasing the resolution of differential gene expressions.
Results: The overexpression of a bacteriocin (cvaC-1), as the major result gained by the transcriptomic analysis, led us to validate its potential application as a marker of Xylella fastidiosa multiplication in olive, citrus and periwinkle artificially inoculated plants. Transcriptomic analysis of in vitro cultured strains of Xylella fastidiosa subspecies pauca, while confirming that bacteriocin-related genes are the most abundant transcripts, unraveled strain differences in the cvaC-1 and cvaC-2 ratio.
Discussion: Our findings suggest that the cvaC-1-related transcript can be employed in RT-qPCR/RT-PCR to improve the detectability of actively growing Xylella fastidiosa cells in vitro and in host’s xylem vessels. Indeed, being the most expressed component of bacterial weapons, novel studies focusing on its functions and role in the bacterial pathogenic life cycle should be envisioned.
Xylella fastidiosa (Xf), Lysobacteraceae family, Gammaproteobacteria class (Wells et al., 1987) is a fastidious, non-flagellate, Gram-negative, xylem-limited bacterium, known to cause destructive diseases worldwide in a broad range of crops (grapevine, citrus, stone fruit, olive, pecan, blueberry, alfa alfa, and coffee) but also ornamental and forestry plant species (elm, oak, sycamore, and oleander) (Henneberger et al., 2004; Rapicavoli et al., 2018). The updated Xf host plant database lists over 712 species across 312 genera and 89 families in which the bacterium causes symptomatic or latent infections (Cavalieri et al., 2024). The Xf long-distance spread and its associated diseases are primarily attributed to human activities related to the movement and exchange of infected plant material while locally, the bacterium is dispersed through xylem-sap feeding insects (Almeida and Nunney, 2015). Xf was originally discovered in the late 1800’s in the Americas, where is now endemically distributed and more recently, in Asia and Europe, being reported, up to now, in more than 20 countries across America, Asia, and Europe [PM 7/24 (5) Xylella fastidiosa, 2023]. The first Xf finding in Europe occurred in 2013, associated with the Olive Quick Decline Syndrome (OQDS) (Saponari et al., 2013, 2014; Cariddi et al., 2014), a severe epidemic decimating olive trees in the Salento peninsula (Apulia, Italy), although phylogenetic and genomic investigations dated the first introduction of the pathogen in the European continent back to the 80’s of the last century (Soubeyrand et al., 2018; Landa et al., 2020).
The plant-bacterial interactions enhancing systemic host colonization and modulating plant defense response are still largely unknown. It is well-known that Xf has a dual lifestyle encompassing a planktonic motile form to systemically colonize the plant vessels, and a steady condition, where cells are sticky and organized in aggregates embedded in an exopolysaccharide (EPS) matrix, which is required by the insect vector for the acquisition and transmission (Chatterjee et al., 2008), passing through a phase transition that is finely regulated by a mechanism called “quorum sensing.”
A further process regulating the cell growth dynamics, the bacterium survival to various stresses, and the virulence is based on bacterial toxin-antitoxin (TA) systems (Merfa et al., 2016; de Souza-Neto et al., 2022). TA loci are organized in operons and encode for two neighboring genes, a stable protein-based “toxin,” that could exert harmful effects on the cells inhibiting their growth, and a specific cognate labile “antitoxin” (either RNA or protein), that acts as a transcriptional repressor, preventing self-toxicity (Maisonneuve and Gerdes, 2014). This auto-inhibition, called “conditional cooperativity,” relies on the proper toxin: antitoxin stoichiometric ratio in the cells under normal conditions (Cataudella et al., 2012), while the uneven co-expression of TA genes have been associated with cell growth dynamics under stress conditions (Burbank and Stenger, 2017; Gupta et al., 2017). Xf has multiple TA modules on its chromosome involved both in virulence and stress survival (Van Sluys et al., 2003; Lee et al., 2014). One such TA system, named cvaC/cvi, represents a putative component of the virulence process (Zaini et al., 2008). The cvaC toxin belongs to the bacteriocin class, sharing homologies with the RTX protein recovered in Rizobium leguminosarum, a hemolysin and leukotoxin (Oresnik et al., 1999; Holtsmark et al., 2008) that possibly plays a structural role in biofilm maturation (de Souza et al., 2020). Xf bacteriocin has a comparable structure with colicin V (Col-V) secreted by E. coli and other Enterobacteriaceae members (Waters and Crosa, 1991). The peptide, synthesized as a 103 amino acids precursor molecule, endowed with a leader 15-amino acid motif, is successively post-translationally modified during its secretion by the proteolytic cleavage of the leader to give rise to the 88 amino acid long mature protein. This “core peptide” represents the bioactive molecule. Indeed, the abundant levels of transcript of this gene, under standard growth conditions (De Souza et al., 2003) and in microbiological media supplemented with glucose (Pashalidis et al., 2005), iron (Zaini et al., 2008), calcium (Chen and De La Fuente, 2020) and grape sap (Nunes et al., 2010; Surano et al., 2023) confirms that the toxin encoded by cvaC gene is likely a prevalent weapon employed by Xf to efficiently compete with other closely-related sensitive endophytes in the xylem and insect foregut (Zaini et al., 2008). In most of Xf genomes publicly available, cvaC gene has a tripled-tandem organization, with the first copy (cvaC-1) in antisense and the other two (cvaC-2 and cvaC-3) in sense direction. Located upstream of this module is the immunity protein cvi, which self-protects the producing strain from the toxic effects of its own antibacterial product (Pashalidis et al., 2005; Baquero et al., 2019; Figure 1). Because of the molecular mass of cvaC below 10 kDa, and the existence of a dedicated export system for the translocation, it should fall in the microcin group (MccV-like) (Duquesne et al., 2007) likely acting as a pore-forming toxin (PFT), causing membrane permeabilization and destabilization, i.e., the formation of pores and channels in the lipid bilayers, through the binding of specific receptors, and therefore, the dissipation of the protonmotive force of sensitive bacteria (Chatterjee et al., 2005; Hasper et al., 2006).
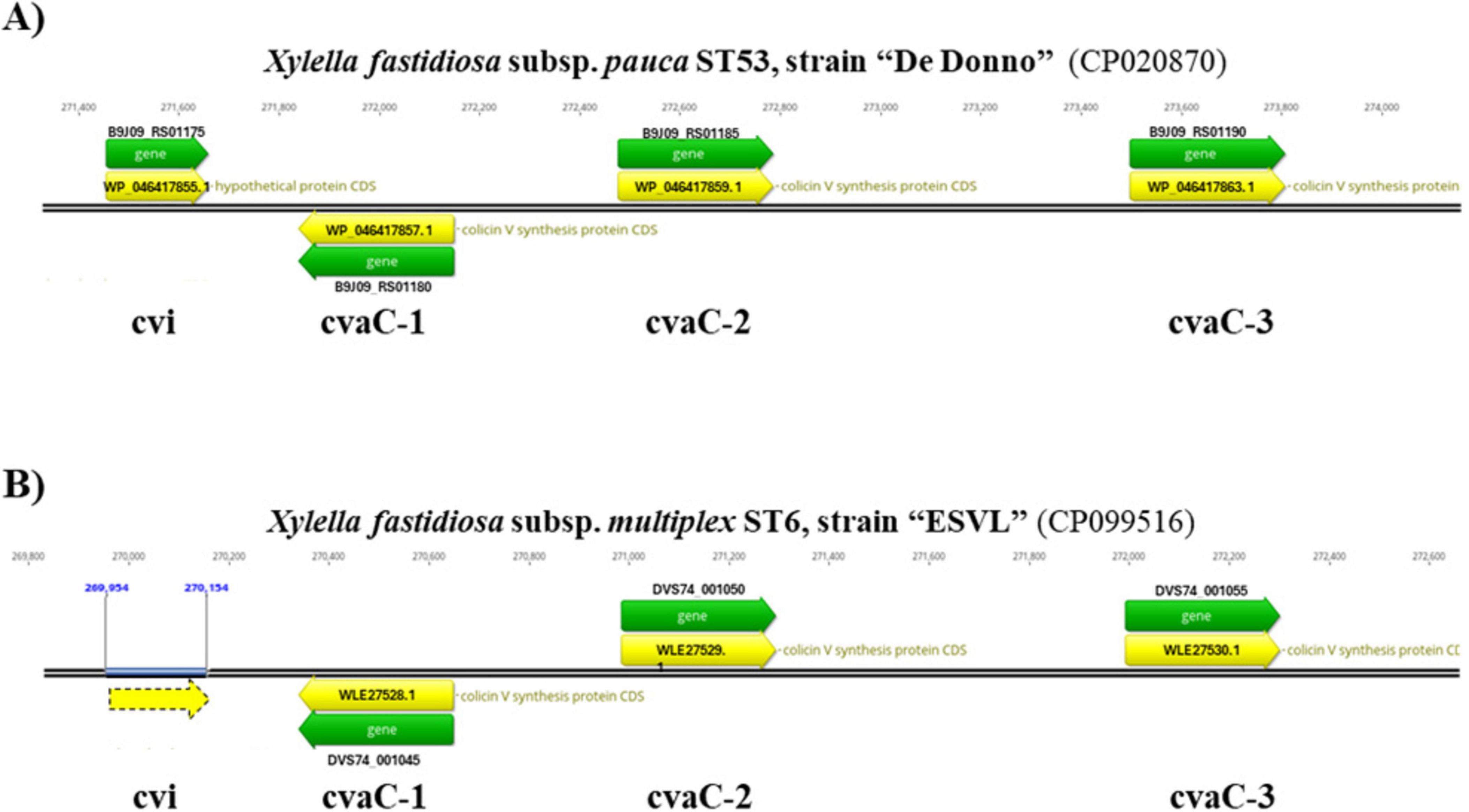
Figure 1. Tripled-tandem organization of the cvaC operon. The correspondent genome regions in the Xfp De Donno (Acc. Nr. CP020870.1) (A) and Xfm ESVL (Acc. Nr. CP099516.1) genome (B) are displayed. In both genomes, cvaC-1 gene is in antisense direction and the self-immunity protein, encoded by cvi gene, is placed upstream of the cvaC operon. In the ESVL genome, the gene coding for the cvi protein is not annotated, even if an ORF (dashed yellow arrow) is present in the sequence. Gene ID (Locus tag) and Protein IDs are also reported.
While several transcriptome studies provided insights into the plant response to Xf infection (Rodrigues et al., 2013; Giampetruzzi et al., 2016; Zaini et al., 2018), the bacterial gene expression in infected plants has been poorly investigated (Surano et al., 2023) or limited to the condition mimicking the xylem environment (De Souza et al., 2003; Parker et al., 2016; Chen and De La Fuente, 2020; Wei et al., 2021). The application of dual RNA sequencing (dRNA-seq) allows for the simultaneous study of the host and the pathogen transcriptomes, detecting pathogen-specific transcripts and host responses in the same tissues, thus providing a comprehensive understanding of their interaction (Westermann et al., 2012) and revealing novel aspects of the infective process. Besides shedding light on the Xf/host interaction, such studies could identify genes of the pathogen to exploit as biomarkers activated at the early stage of the infection, allowing to develop new molecular approaches based on the replication activity and physiological/viability state of the microorganism in a sample (Kovalchuk et al., 2019; Kunjeti et al., 2012; Meyer et al., 2016). Indeed, the identification of biomarkers linked to the primary phase of the infection, prior the symptoms onset and before the pathogen population reaches detectable level, is crucial for successful early detection.
In the present work, a pilot dRNA-seq experiment was carried out to profile the Xf subsp. pauca (Xfp) gene expression in a naturally infected olive tree and the data, compared to the transcriptome profile of in vitro grown Xf, allowed us to identify a bacterial transcript highly expressed in planta during the infection process and conserved across strains belonging to different subspecies. The transcript, annotated as a “Colicin V synthesis protein” (cvaC-1) coding for a virulence-related microcin, resulted in an effective molecular target for a timely diagnosis of Xf active growing cells in tissues of different hosts, by quantitative reverse transcription PCR (RT-qPCR) or conventional two-step RT-PCR.
The developed assay proved to be more sensitive (with higher analytical sensitivity) than the official real-time quantitative PCR (qPCR, Harper et al., 2010) test widely used for bacterial detection. Indeed, when the results of the RT-qPCR assay were compared with those of the standard qPCR targeting the bacterial DNA, it was possible to retrieve additional useful indications about the status of the infection in the host plants, i.e., active replicating and colonizing the plant vs non-growing bacterial cells.
Xf strains used in our experiments belonged to subspecies pauca and multiplex (Xfm). More specifically, Xfp strains “De Donno” (later on DD) (CFBP 8402), APL-64 and APL-69, all belonging to sequence type (ST) 53, were isolated between 2013 and 2017 from OQDS affected olive trees in the Salento area (Apulia, Italy) (Giampetruzzi et al., 2017a; Saponari et al., 2017; Sicard et al., 2021). Xfm strain ESVL (CFBP 8684) ST6 was recovered from an almond tree in Guadalest Valley (Alicante, Spain) with notable almond leaf scorch disease signs (ALSD) (Giampetruzzi et al., 2019b). Moreover, Xfm strain “TOS4” having ST87 genotype, was recovered from an infected almond in the Italian region of Tuscany (northern Italy) (Giampetruzzi et al., 2019a) All strains were cultured for 10 days on PD3 agar plates (Davis, 1992) at 28°C from −80°C glycerol stock (50%), re-streaked onto new PD3 plates and cultured for another 7 days before use.
Tissues for the dual RNA-seq were sampled from a Xfp-infected olive of the susceptible cultivar Ogliarola salentina grown in the Salento epidemic area. Wooden shoots were collected from the sections of the canopy close to branches with severe symptoms of desiccation of 1 or 2 years-old twigs (ca. 0.5 cm in diameter and 1–1.5 cm long) were debarked with a scalpel to obtain ca. 1 g of xylem-enriched tissues that was used to extract total DNAs and RNAs fractions. Total DNAs were extracted using the PureFood GMO and Authentication Kit on the dedicated automatic platform Maxwell® RSC (Promega Corporation, United States) (Loconsole et al., 2021) and processed to assess the Xf presence using a qPCR assay following the protocol of (Harper et al., 2010), with some minor modifications (Amoia et al., 2023).
Total RNAs extraction was carried out on xylem-enriched tissues of olive obtained as described above. One g of twigs was grinded in a mortar using 10 mL of cold McKenzie grinding buffer (Foissac et al., 2001) plus 2% sodium metabisulfite. The homogenate was added with 20% N-Lauroylsarcosine Sodium salt and centrifuged (13.000 × g) to throw out the plant debris. The clear slurry was extracted twice with TRIzol™ (Invitrogen™) and chloroform in a proportion of 1:1 (v:v) by 5.000 × g centrifugation, and the final supernatant was added with 2.5 volumes of 70% ethanol and stored overnight at −20°C. Total RNAs were recovered by centrifugation at 20.000 × g and resuspended in 450 μL RTL buffer (RNeasy Plant Mini Kit, Qiagen) to be further purified according to the manufacturer’s instructions. RNA concentration and quality were evaluated by measuring the absorbance ratio 260/280 nm with the spectrophotometer NanoPhotometer™ N60 UV/Vis (Implen, München Germany). RNA quality was assessed by evaluating the integrity of 28S and 18S rRNA bands on 1.2% TBE (Tris boric EDTA) agarose gel and by capillary electrophoresis (Supplementary Figure 1).
Total RNAs were used for the dRNA-seq library construction which was outsourced to Vertis Biotechnology AG, Germany1, according to a custom protocol based on depletion of ribosomal RNA (rRNA) molecules (plant plus bacteria). The ribodepleted RNAs were first fragmented by ultrasounds (2 pulses of 30 s each at 4°C). Then, an oligonucleotide adapter was ligated to the 3′ end of the fragmented RNA molecules. First-strand cDNA synthesis was primed by an oligonucleotide complementary to the 3′ adapter using M-MLV reverse transcriptase. The first strand cDNA was purified, and the 5’ Illumina TruSeq sequencing adapter was ligated to the 3′ end of the antisense cDNA. The resulting cDNA was PCR-amplified (16 cycles) to achieve about 10–20 ng/μL of DNA using a high-fidelity DNA polymerase and the TruSeq barcode sequences. The cDNA was purified using the Agencourt AMPure XP kit (Beckman Coulter Genomics) and analyzed by capillary electrophoresis (Supplementary Figure 3). For the Illumina NextSeq sequencing, the DNA sample was size-fractionated in the range of 200–550 bp using a preparative agarose gel. An aliquot was also analyzed by capillary electrophoresis (Supplementary Figure 3). The DNA pool was sequenced on an Illumina NextSeq 500 system using 1 × 75 bp read length.
Three different Xfp ST53 strains (Xfp DD, APL-64, APL-69) grown in vitro were used for RNASeq analysis. Upon scraping bacterial colonies and dissolving them in sterile water to obtain a turbid suspension, cells were centrifugated at 10.000 × g for 10 min at 4°C. Total RNAs were extracted from the three bacterial pellets according to the TRIzol™ Reagent protocol (Invitrogen™) and successively DNAse digested with RNase-free DNase I following the manufacturer’s instruction (Thermo Scientific™), to avoid contamination by genomic DNA. Purity, concentration and integrity of RNA samples were evaluated by agarose gel electrophoresis as described above. RNA samples with a RNA:DNA ratio > 15 and RNA integrity number (RIN) > 7 were employed for libraries construction. Libraries were prepared with the TruSeq stranded total RNA kit construction for microbe with NEBNext® rRNA Depletion Kit (Bacteria). Paired-end sequencing (100 × 2 bp) was performed on an Illumina Novaseq 6000 platform at the Macrogen Europe B.V. facilities (Amsterdam, NE).
The FastQC tool software2 was used to assess the quality of the raw reads obtained by the two different RNASeq sequencing approaches. The reference genome data of Olea europaea L. subsp. europaea var. europaea cv Farga v9 (Julca et al., 2020) and Xfp DD (Giampetruzzi et al., 2017b) were downloaded from NCBI database3. Raw reads obtained from the dRNA-seq, preliminarily checked for quality and trimmed of the adapters, were first mapped to the cv. Farga genome v9 using TopHat2 (Kim et al., 2013). The remaining unmapped reads were then mapped to the Xfp DD genome. In parallel, raw reads obtained from the RNASeq of the in vitro cultured bacteria were directly mapped to the Xfp DD bacterial genome. Reads count data after mapping on the reference genome (Xfp DD bacterial genome) were determined using SeqMonk software version 1.48.14. Gene expression levels of both RNASeq and dRNASeq were measured as raw count reads per annotated transcript by SeqMonk. Therefore, two abundance matrices of reads to the annotated bacterial genome were obtained and reported in the Excel file for further comparison between the two. The number of counts per annotated transcript was also normalized to 20,000, which was the lowest amount of total mapped reads obtained from the dRNA-seq library.
A Sybr-green-based two-step RT-qPCR assay was developed to target three out of four genes forming the cvaC operon in Xf genome. Primers for cvaC-1, cvaC-2 and cvi genes were designed using Primer-Blast tool version 4.1.0, available at NCBI website, and checked in silico for their specificity to target transcripts. The tandem arrangement of the three cvaC copies and the putative self-immunity protein cvi were found to be similarly, organized and annotated in two reference strains Xfp DD and Xfm ESVL (Figure 1).
The three cvaC genes are annotated as “colicin V synthesis protein” in both genomes (Xfm ESVL: Acc. Nr. CP099516.1; protein_ID: WLE27528.1, WLE27529.1, WLE27530.1; Xfp DD: Acc. Nr. CP020870.1; protein_ID: WP_046417857.1, WP_046417859.1, WP_046417863.1) (Giampetruzzi et al., 2017a,2019b; Román-Écija et al., 2023). The cvi gene is functionally annotated as a “hypothetical protein” in Xfp DD (WP_046417855.1) and it is not annotated in Xfm ESVL, although an open reading frame (ORF), underlined with a dashed yellow arrow, is present (Figure 1).
RNA extraction and DNase treatment were performed following the TRIzol™ Reagent protocol (Invitrogen™) and DNase I treatment (Thermo Scientific), respectively; cDNA synthesis was carried out using the M-MLV Reverse Transcriptase protocol (Invitrogen™). RNA fractions were recovered from 7 days old bacterial colonies grown on PD3 medium. The reaction was set up using a 2x Fast SYBR™ Green Master Mix (Applied Biosystems™) according to manufacturer’s recommendations. The following thermal cycling profile was used: 50°C for 2 min, 95°C for 2 min, followed by 40 cycles of 95°C for 15 s, 58°C for 30 s and 72°C for 30 s. Primer sets specificities were confirmed by checking the absence of non-specific amplifications in melting curve analysis, consisting of a continuous 0.5°C increase in temperature from 65 to 95°C. Experiments were repeated in two independent times and each sample was amplified in three technical replicates. The efficiencies of the newly designed primers and the one of housekeeping gene were preliminarily assessed using 10-fold serial dilution of purified bacterial DNA with a known concentration. The stable transcript of the dnaQ gene, encoding a DNA polymerase III subunit epsilon, was used as an internal control to normalize the expression of the other genes (Zaini et al., 2008; Wei et al., 2021). The change of the expression levels of the three target genes (cvaC-1, cvaC-2 and cvi), compared to the dnaQ gene control, was calculated using the 2–ΔCt method (Livak and Schmittgen, 2001). To verify the potential presence of residual DNA after digestion, qPCR reactions without reverse transcriptase enzyme were also set up using the dnaQ specific primers.
Based on multiple alignments of representative Xfp and Xfm genomes deposited in GenBank (Supplementary Figure 2), performed with Geneious version 2023.2.1, of the first copy (cvaC-1) of cvaC operon, a primer pair (col_F/R) and a 6′-carboxyfluorescein/Black Hole Quencher-1-labeled probe (6′FAM/BHQ) TaqMan (col_Prb) were specifically designed using NCBI Primer-Blast tool v. 4.1.0 and PrimerQuest™ Tool (Integrate DNA Technology) for a TaqMan based RT-qPCR. Protocols used for plant RNA extraction and RT-qPCR conditions are described in the paragraph below.
To assess the diagnostic sensitivity of the new cvaC-1 RT-qPCR assay and establish its limit of detection (LoD), total RNA recovered from xylem tissues of a Xfp infected olive, either with the traditional CTAB method (Loconsole et al., 2014) followed by DNase I-treatment or by using the Maxwell® RSC Plant RNA Kit (as described in paragraph 2.7.1) was used. Primer efficiency and analytical sensitivity were determined by calculating the slope value of the standard curves generated with five ten-fold (from 1 to 10–5) serial dilutions of the two digested RNA samples. Each dilution was analyzed in duplicate and the average Cq was calculated. The experiment was repeated in two independent runs. The primer efficiency was computed using the following formula: E = 100 × (10–1/slope – 1). The associated Pearson’s product-moment correlation coefficients (PMCC) were also reported. To further support RT-qPCR results, a primer pair for end-point RT-PCR, covering the entire length of the gene, was manually designed for the amplification of the whole cvaC-1 gene. The diagnostic sensitivity of this assay was assessed by using the same serial dilutions used for RT-qPCR. All primer and probe sequences used in this study are listed in Table 1.
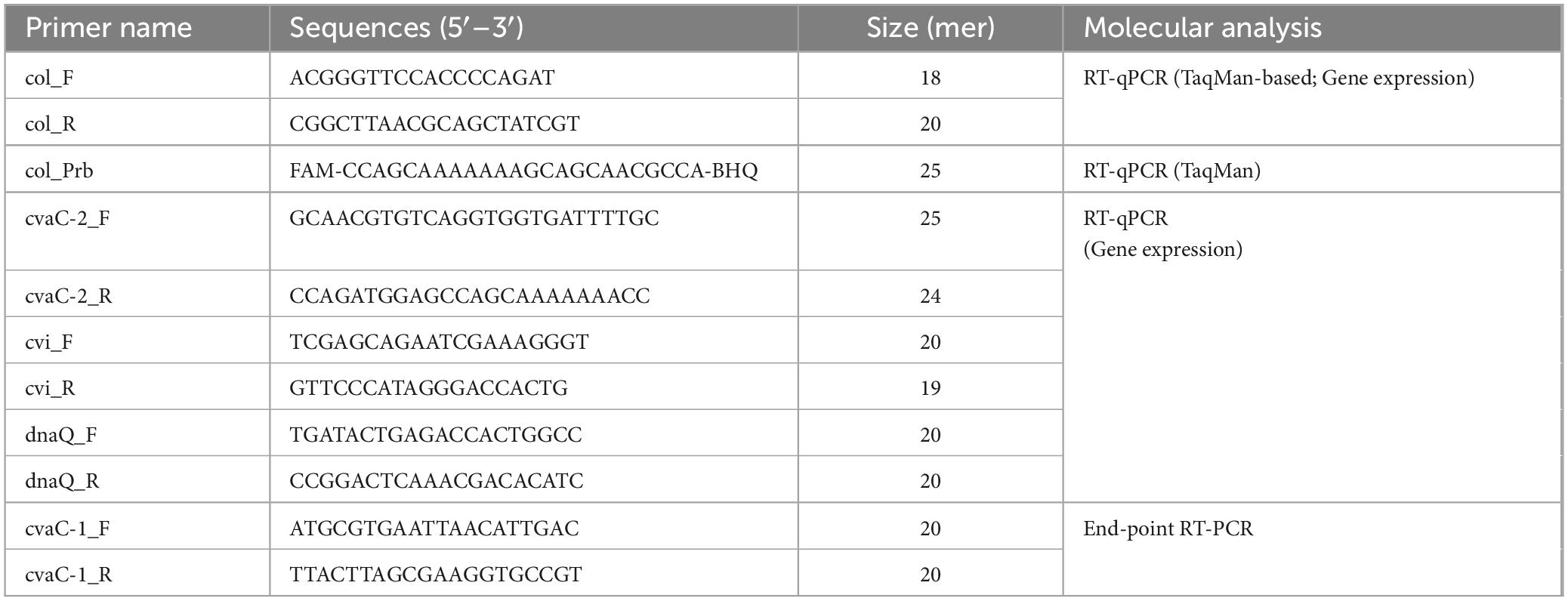
Table 1. RT-qPCR and RT-PCR primer/probe sequences used in this study.
Two bacterial strains, Xfp DD and Xfm ESLV were used to inoculate three different plant species and subsequently monitor the bacterial host colonization by standard qPCR assay (Harper et al., 2010) targeting bacterial DNA (Amoia et al., 2023) in comparison with the newly developed TaqMan-based RT-qPCR assay targeting the cvaC-1 gene. More specifically, the test included 24 olive trees cultivar Ogliarola salentina, 72 periwinkle plants (Catharanthus roseus (L.) G.Don, 1837) cultivar Madagascar and 72 citrus plants (Citrus × sinensis (L.) Osbeck, 1,765) cultivar Madam Vinous. Olive is known to be highly susceptible to Xfp DD infections while, in previous greenhouse tests, the strain ESLV needle-inoculated in olives proved to be able to colonize the plants to a lower extent than Xfp DD and without inducing typical shoot dieback and desiccations (Saponari, personal communication). Two-years-old olive plants with several ramifications were used, which allowed for the inoculation of multiple twigs, thus requiring a lower number of plants compared to the other species. These latter consisted of seedlings grown as single stems. Inoculated plants were grown in a quarantine greenhouse at 25°C ± 2 and relative humidity (RH) of 65% ± 10. For needle inoculation, bacterial suspensions were prepared by dispersing scraped colonies in sterile distilled water and their titers were adjusted to an optical density (OD600) of 1.0. Two small drops (8–10 μL each) of the suspension were placed at the level of two consecutive leaf nodes in the basal part of the main stem for citrus and periwinkle and multiple twigs for olive. Drops were pricked 4–5 times with a sterile entomological needle. Overall, experiments on olive and citrus lasted 12 months, while on periwinkle 6 months. Sampling and testing were performed on a monthly basis, starting 1 month after the inoculation, as follows: for the first three (in periwinkle) and 6 months (in olive and citrus) a total of six stem portions, each comprising an inoculation point (samples identified with the suffix PI), were collected and tested. For the remaining experimental period, in addition to the six inoculation points, the portions of the stem located 10 cm above the last inoculation point (samples identified with the suffix UP) were also harvested and tested (Figures 2A, B, C). Conclusively, 54 samples for each strain/species combination were tested (Figure 2D).
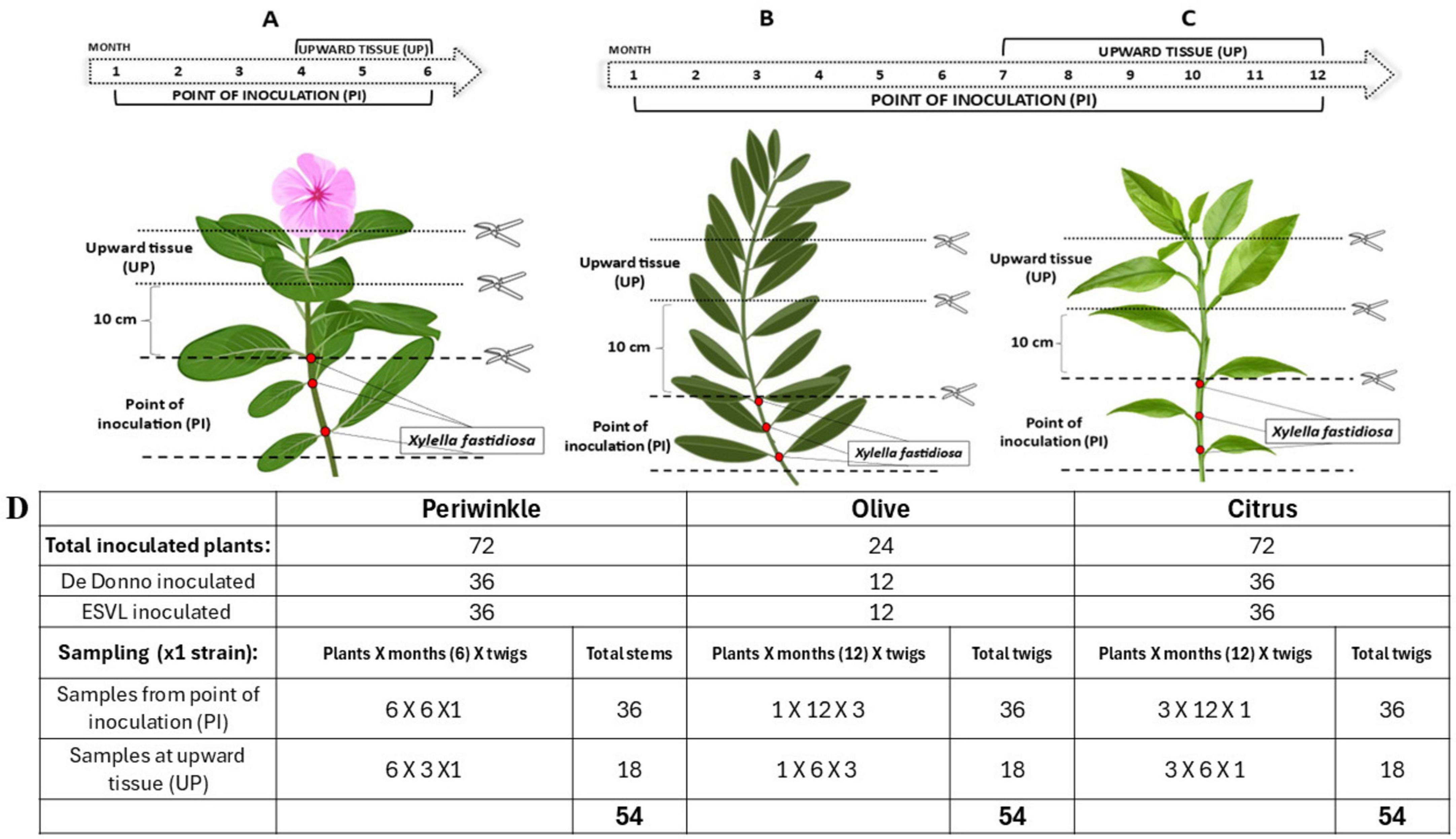
Figure 2. Schematic cartoon representing the time course of Xf inoculation experiment. For all the three species (periwinkle, olive and citrus), two consecutive needle-inoculation points (red dots) were made at the basal part of the stem. Six and three stem/twigs, harboring one or two inoculation points (PIs) were collected once per month for the first three and 6 months, respectively from the periwinkle (A) and from the olive (B) and citrus (C) plants. The remaining inoculated stems/twigs were monthly collected, by sampling the PIs and the 10 cm portions above the last inoculation point (UP) during the last 3 and 6 months of the trial for periwinkle and olive and citrus plants, respectively. Overall, 54 tissue samples per species per Xf-strain were analyzed. (D) Summary table of sampled tissue for each species throughout the whole trial.
Small pieces of stems/twigs were used to extract total DNAs with the commercial PureFood GMO Authentication Kit and total RNAs with the Maxwell® RSC Plant RNA Kit on an automated extraction platform. Total DNAs were employed to set up qPCR reactions for the amplification of the bacterial rimM gene (Harper et al., 2010), using the conditions described by (Amoia et al., 2023). Whereas total RNAs, previously digested with DNAse I/RNAse free, were tested by RT-qPCR and RT-PCR using the primers designed in this work (Table 1).
Briefly, 500 mg of tissue was homogenized in the presence of commercial CTAB (Cetyltrimethylammonium bromide) buffer added with chilled 2% 1-thioglycerol and heated at 65°C for 15 min; the clear homogenate was loaded in the cartridge according to manufacturer’s instructions, including the step of the DNase I treatment. The eluted RNAs (50 μL) were stored at −80°C until processing. For cvaC-1 quantitative amplification, the optimal primer/probe concentrations in the reaction mix were determined as those yielding the highest reporter fluorescence (RFUs) and the lowest threshold cycle (Ct). To establish the best annealing temperature of the new primer/probe set, a temperature gradient was carried out on a range spanning from 58 to 63°C. One-step RT-qPCR reactions were performed in a final volume of 11 μL containing 5.5 μL 2x iTaq Universal Probes Reaction Mix (Bio-Rad, Hercules, CA, USA), 0.3 μL each forward and reverse primer (10 μM; col_F/R), 0.2 μL FAM-labeled probe (col_Prb), 0.2 μL iScript Reverse Transcriptase (RNAse H ++ MMLV enzyme), 1 μL (up to circa 100 ng μL–1) of digested RNA sample and nuclease-free water to the final volume. Each sample was amplified in duplicate and the average Cq was considered. All qPCR and RT-qPCR analyses were run in a Bio-Rad CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Data acquisition and analysis were performed using the CFX Maestro 1.1 version N. 4.1 software (Bio-Rad, Hercules). The threshold line was automatically set by the software or manually adjusted when necessary. To carry out RT-PCR, cDNA was first synthesized using the above-mentioned standard reverse transcription protocol. Then, PCR reaction mix was set up as follows: 2x GoTaq® Green Master Mix, 0.5 μL of each primer (cvaC-1_F/R), 1 μL of cDNA and nuclease-free H2O to 20 μL final volume. The PCR program included: one cycle at 95°C for 3 min, 35 cycles at 94°C for 30 s, 58°C for 30 s, and 72°C for 30 s, followed by a final extension at 72°C for 5 min.
A few of the periwinkle plants displaying systemic infections, upon the inoculation of both strains (Xfp DD and Xfm ESLV), were used for re-isolation of the inoculated bacterial strains 6 months after the inoculations, before ending the experiment. Systemic infection was assessed by testing tissues distant from the inoculation points that gave positive results in qPCR for the detection of Xf (Montilon et al., 2022). Stem pieces were washed under tap water, surface-sterilized in 2% sodium hypochlorite for 3 min, soaked in 70% ethanol for 3 min and rinsed three times in sterile water. Cutted smaller pieces were automatically macerated in an extraction bag (Bioreba) in presence of sterile 1x Phosphate Buffered Saline (PBS) buffer (0.05 M NaCl, pH 7.2) and three consecutive ten-fold dilutions of the recovered sap were plated on periwinkle wilt gelrite (PWG) agar plates (Hill and Purcell, 1995). Plates were incubated for 3–4 weeks at 28°C and periodically inspected for the growth of Xf colonies.
Systemically infected olives (i.e., olives where Xf was detected by qPCR also in tissues far from the points of inoculations) were used to test the reliability of the RT-PCR and RT-qPCR detection assays for the identification of bacterial RNA and discriminate between tissues supporting active bacterial multiplication (i.e., infected vegetating shoots) and those harboring non-growing cells, likewise infected desiccated plant tissues in which the bacterial cells are not sustained any multiplication process, as Xf is not a saprophytic bacterium and requires living plants, specifically the xylematic system, to survive and cause disease. Infected plants were obtained by grafting onto healthy 2 years old olive plants (cv Ogliarola salentina), cuttings collected from field naturally infected trees of the same cultivar. Upon graft taking and sprouting, the occurrence of the bacterium in the new shoots was assessed, and 48 plants yielding qPCR-positive reactions were retained for the experiment, along with 6 Xf-free plants of the same cultivar. Six of the 48 plants were treated with tetracycline by trunk injection, delivering the antibiotic directly to the xylem where Xf multiplies, to assess the impact of the antibiotic on the bacterial multiplication rate as well as the efficiency of the newly developed cvaC-1-based assays to monitor the effect of such applications. Previous in vitro and in planta studies have shown that tetracycline could inhibit Xf growth leading to a reduction or temporal suppression of symptoms in treated plants, but none of the treatments eliminated the bacterium permanently (EFSA Panel on Plant Health (PLH), 2016). Briefly, at 12 months after grafting, corresponding to 6 months after the application of tetracycline for the treated plants, when approximately half of the plants started to show mild to severe shoot dieback and desiccation (Supplementary Figure 3), samples consisting of small pieces of twigs were collected. In the case of symptomatic plants, these consisted of desiccated, withered, and dried tissues. The DNA and RNA fractions were recovered as previously described. An aliquot of the DNA was used to set up the qPCR assay for the conventional detection of the bacterium, whereas an aliquot of the purified and DNA-digested RNA was used to arrange the two-step RT-PCR and the one-step RT-qPCR reactions, both targeting the cvaC-1 transcript.
Regression analysis for analytical sensitivity was performed in R environment, using Rstudio version 4.3.1. According to the normal distribution of results, parametric (Paired t-test) or non-parametric (Wilcoxon Signed Rank Test) analysis was performed between qPCR and RT-qPCR Cq data in SigmaPlot v.15.0. P-values connected to the significance of both tests are also reported. GraphPAD Prism 9.3.0 software was also employed to perform the correlation analysis and the Principal Component Analysis (PCA) of qPCR/RT-qPCR results for tetracycline-treated/non-treated samples, and generate whisker plots with Cq data obtained from qPCR/RT-qPCR on artificially inoculated plants. The correlation between RT-qPCR cvaC-1 and qPCR by Harper et al., 2010 was determined using Spearman’s correlation coefficient.
The olive tree S2, displaying the lowest quantification cycle (Cq) value (< 20, corresponding to a bacterial population in the range of 107–106 CFU/mL) among the 12 screened by qPCR, was selected for the total RNAs extraction and the construction of the dRNA-seq library. The sequenced library generated 40,261,113 raw reads. After the quality check, 31,407,417 (78%) reads were aligned to the olive genome (cv. Farga v9) and the resulted unmapped reads (8,853,696) were aligned to the Xfp De Donno (Xfp DD) reference genome (CP020870), resulting in 21,411 aligned reads to the bacterial genome (sequencing reads available from NCBI BioProject ID PRJNA1141272) (Table 2). Reads mapped to the Xfp DD genome were used to determine the abundance of annotated bacterial transcripts which consisted of 2,319 genes (GCF_002117875.1_ASM211787v1_genomic.gff.gz) including 6 ribosomal RNAs (rRNA), 49 transfer RNAs (tRNAs), 3 non-coding RNAs (ncRNAs), 1 transfer-messenger RNA (tmRNA) and 2,260 coding protein sequence (CDS). The raw counts were normalized to 20,000 mapped reads to obtain expression values comparable to the bacterial RNASeq data set obtained from the in vitro grown Xf cultures. Only 125 out of the 2,319 annotated genes were covered by a number of mapped reads greater than 10, and only 10 genes were covered by over 100 reads (Supplementary Table 1, mapped reads in dual RNA-seq; S2 column). Unexpectedly, the gene with the highest number of mapped reads (6,770 normalized counts) codes for a “colicin V precursor” (B9J09_RS01180, i.e., the first gene in the cvaC operon of Xf, indicated in the text as cvaC-1), and was much more expressed than the rnpB gene (B9J09_RS09380), an essential bacterial catalytic RNA (RNase P) known to be highly transcribed (Parker et al., 2016; Ellis, 2020) (Figure 3 and Supplementary Table 1).

Table 2. Statistics on RNASeq sequencing libraries (DualRNA-seq and bacterial RNASeq).
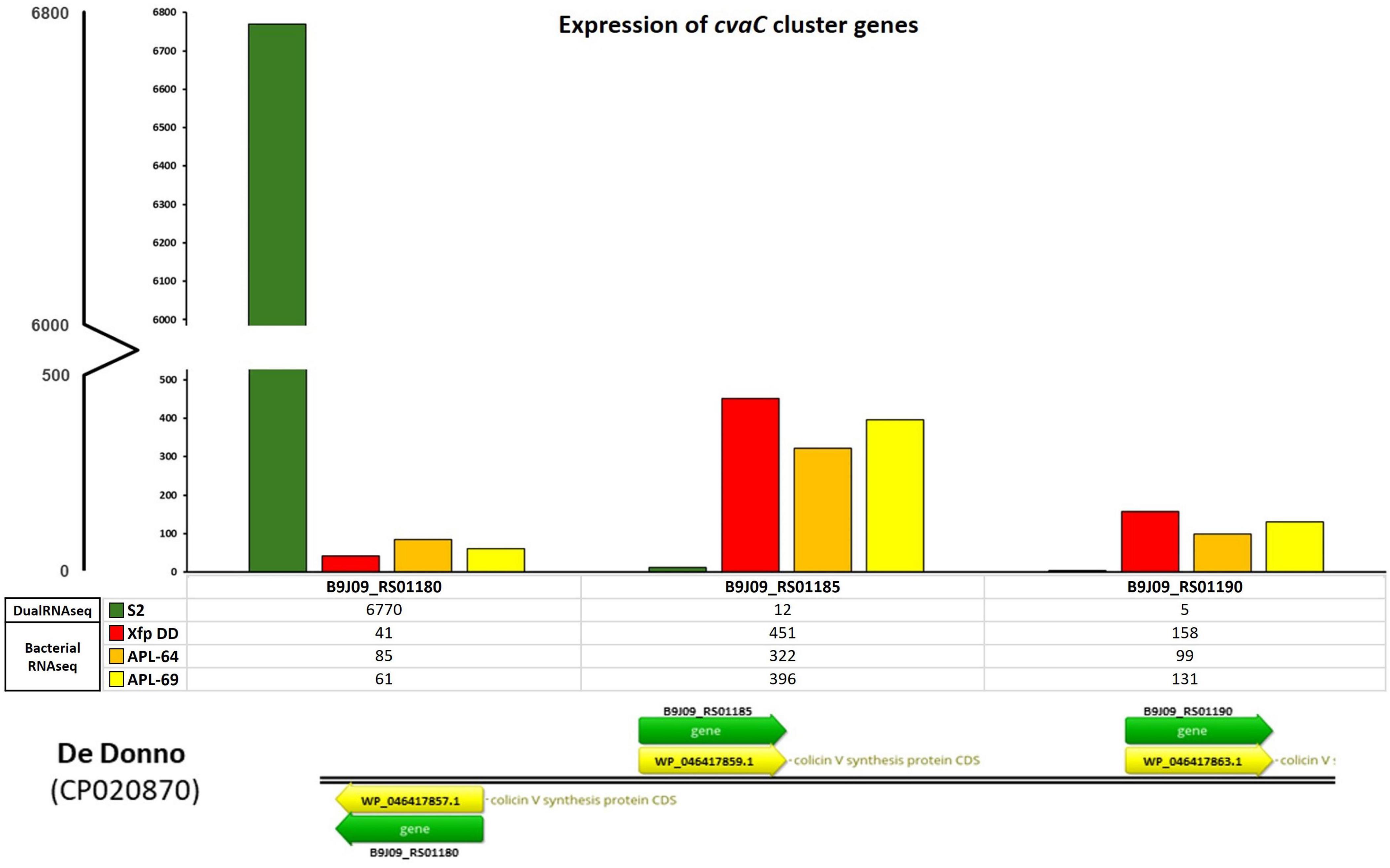
Figure 3. cvaC-operon transcripts from bacterial RNASeq and dual RNA-seq. Expression of cvaC genes (cvaC-1, cvaC-2, cvaC-3) in planta from dual RNA-seq (S2) and in vitro from the RNASeq of Xf cultured strains (Xfp DD, APL-64 and APL-69). The numbers of normalized reads of the three genes in the operon and the legenda are reported on the y-axis
To extend the dataset of bacterial gene expression profiles, three RNASeq libraries were constructed and sequenced starting from total RNAs extracted from three cultured strains of Xfp ST53 (Xfp DD, APL-64, APL-69; NCBI BioProject ID PRJNA505446) grown in vitro on PD3 agar plates. High-throughput sequencing of these three libraries generated 44,798,814, 40,463,402 and 46,980,188 raw reads, respectively for Xfp DD, APL-64, APL-69. After the quality check, 31,721,799 (Xfp DD), 28,505,044 (APL-64) and 33,586,808 (APL-69) reads were mapped to the Xfp DD reference genome (CP020870) with an average coverage of 1000X (sequencing mapped reads available from NCBI BioProject ID PRJNA1141272) (Supplementary Table 1, mapped reads in Bacterial RNASeq columns). Beside the rnpB (B9J09_RS09380) and ssrA (small stable RNA A; B9J09_RS11595) genes, the gene B9J09_RS06930 annotated as Blp family class II bacteriocin, was the highest expressed transcript with 505, 476 and 455 normalized reads in the three RNASeq libraries (Supplementary Table 1, mapped reads in Bacterial RNASeq columns). The number of reads mapping cvaC-1 “colicin V precursor gene” (B9J09_RS01180), found to be overexpressed in planta, was approximately ten times lower, ranging from 41 to 85. By contrast, the number of reads mapping the cvaC-2 gene, the second gene of the cvaC operon, was much higher and similar to that of the most expressed in vitro genes (between 322 and 451 reads) (Supplementary Table 1, mapped reads in Bacterial RNASeq columns, Figure 3).
The RT-qPCR results showed that cvaC-1 transcript is upregulated under the in vitro culturing conditions for the three strains, although its expression level (fold changes) was low in Xfp DD and Xfm strain TOS4 and higher in Xfm strain ESVL (Figure 4). Conversely, cvaC-2 transcript was over-expressed in Xfp DD compared to both Xfm cultured strains. The RT-qPCR results for these target genes confirmed the RNASeq data on the Xfp strains, with the expression (2–ΔCt) ratio between cvaC-2 and cvaC-1 (7,6653) almost overlapping the average ratio of the reads cvaC-2: cvaC-1 recovered from RNASeq (7,0722) for the three Xfp-cultured strains Xfp DD, APL-64 and APL-69. For the strains tested, a low expression level was consistently detected by RT-qPCR for cvi gene (Figure 4). Direct qPCR amplification of the RNA extracts produced high quantification cycle (Cq) values (> 35), indicating that the RNA templates did not contain any residual DNA traces.
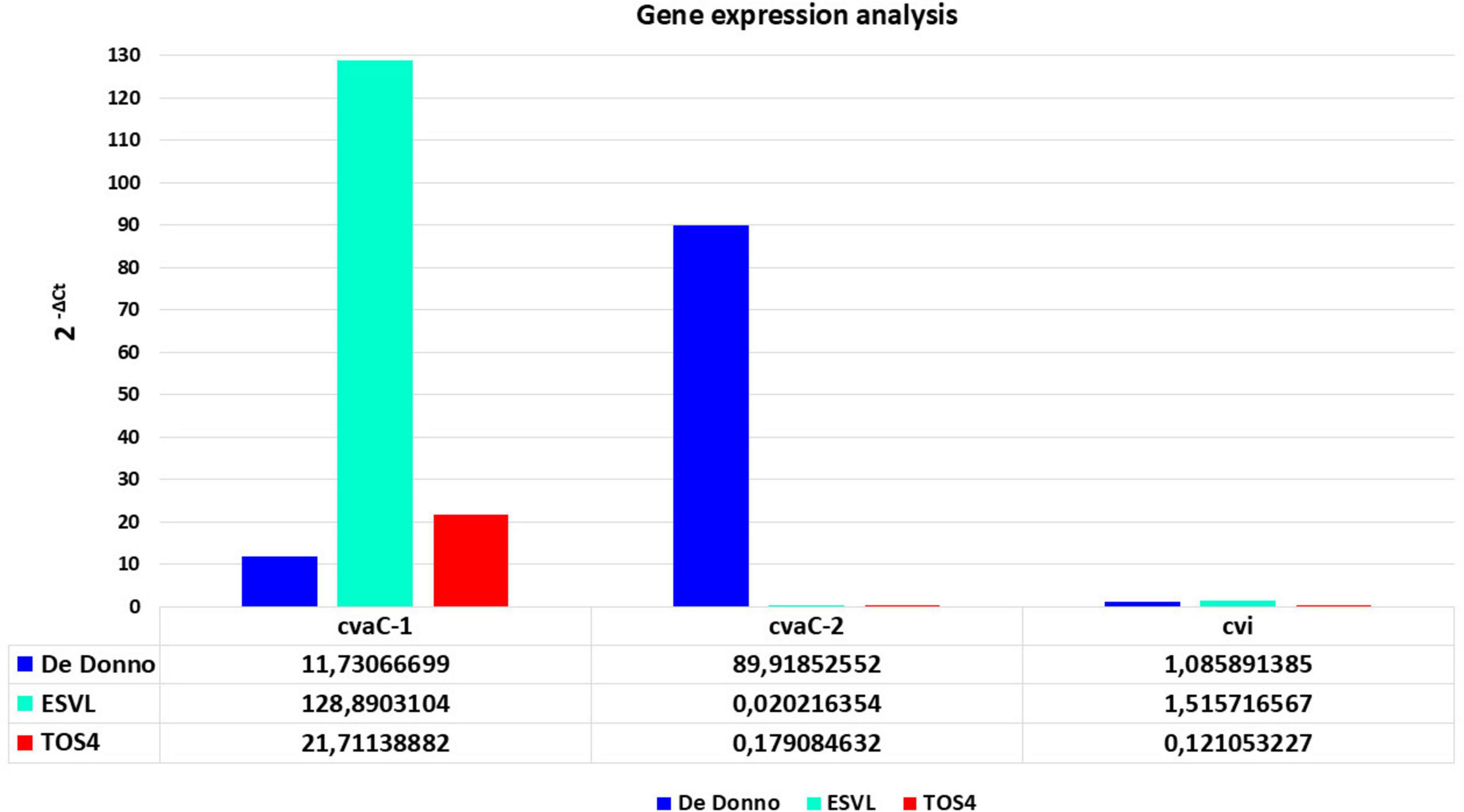
Figure 4. RT-qPCR gene expression analysisof cvaC-1, cvaC-2 and cvi,of in vitro grown bacterial strains of Xf (Xfp DD, Xfm ESVL and TOS4). The 2– ΔCt expression values of the three selected genes, compared to the stable one of housekeeping gene dnaQ, are shown on y-axis. RT-qPCR reactions were done in triplicate.
The primer pair col_F/R, amplifying a 69 bp fragment, coupled with a TaqMan probe (col_Prb) successfully detected the cvaC-1 transcript by RT-qPCR assays in artificially inoculated plants at different time points post-inoculation. The optimal annealing temperature was defined at 59°C. Thus, the selected RT-qPCR parameters were as follows: an initial reverse transcription step at 50°C for 10 min; polymerase activation and DNA denaturation at 95°C for 3 min followed by 40 cycles of denaturation at 94°C for 15 s and annealing at 59°C for 30 s. No reaction amplifications were observed in negative internal control (NIC) and no template control (NTC). When Cq values were plotted against the logarithm of the dilutions, standard curves with very good linearity and high confidence level, having R2 values of 0.9845 and 0.9972, respectively using the RNA recovered by the CTAB-based method and with the Maxwell® kit, were generated (Figures 5A, B). In both cases, the efficiencies recorded (103.91 and 107.62%) fit in the optimal range of 90–110% (Bustin et al., 2009). These values are similar to those previously reported for the qPCR assay (Amoia et al., 2023). High Pearson’s correlation values (0.9922 and 0.9986, for RNA CTAB-based and Maxwell® RSC Plant RNA kit extracted, respectively) were achieved. The linear regression equations associated with calibration curves are displayed in Figures 5A, B. In addition, primers cvaC-1_F/R designed to amplify the whole gene in RT-PCR, yielding a 309-bp amplicon, successfully identified the entire transcript in systemically infected plant tissues, although as expected, the diagnostic sensitivity of the assay was lower than the RT-qPCR test (Supplementary Figure 4). Even so, this assay proved to be highly specific in identifying cvaC-1 intact and complete transcripts, such as those associated with bacterial multiplication under optimal conditions. Indeed, negative results (no amplifications) were consistently obtained with RT-PCR (i.e., amplifying the whole gene) even if the same total RNAs purified from desiccated tissues, where the bacterial growth and multiplication could be suffering, generated very high Cq values (> 30) in RT-qPCR.
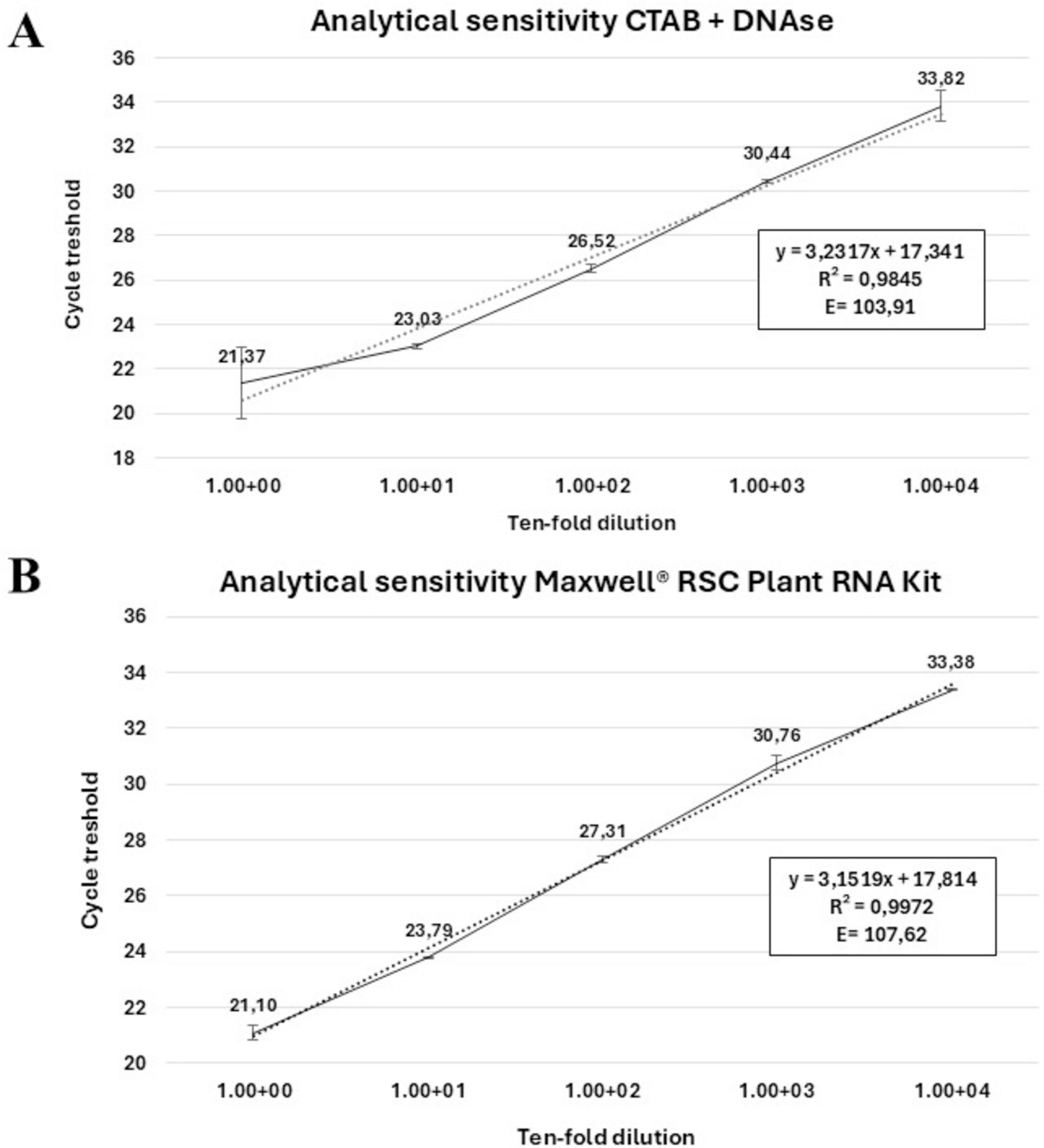
Figure 5. Analytical sensitivity of TaqMan-based RT-qPCR. Linear regression curves of the RT-qPCR assay targeting the cvaC-1 transcript. Standard curves were generated using 10-fold dilution series of the total RNA extracted from a Xfp infected olive twig purified with (A) a CTAB-based protocol with DNA digestion and (B) the RSC Plant RNA kit. Quantification cycles on the y-axis are correlated to the log of dilutions on the x-axis. In both figures, the equation regression, Pearson’s correlation values (R2) and % primer efficiency are also reported.
Altogether, 54 samples/strain (in total 108 for each species) were analyzed during the 6 and 12 months of the trials, respectively for periwinkle, and olive and citrus. Results of the qPCR and RT-qPCR assays for each plant species (olive, citrus and periwinkle) and samples (PI and UP) are reported in detail in Supplementary Figure 5, while Cq values, cumulative from all sampling times, are graphically reported in Figure 6. In Xfp DD-infected olives (Figure 6A), the average Cq values obtained by RT-qPCR were always lower than those recovered by qPCR assay, both for the samples collected at the point of inoculation (samples identified with the suffix PI) (25.69 ± 3.54 vs 28.01 ± 1.88; P-value < 0.001) and 10 cm above the last inoculation point (samples identified with the suffix UP) (27.74 ± 4.64 vs 31.36 ± 3.92; P-value = 0.00161). The opposite was observed in Xfm ESVL-infected olives where the average Cq values of infected sample “PI” were 31.38 ± 2.98 vs 30.38 ± 1.72, with no significant difference between them, and those from “UP” samples corresponding to 32.27 ± 1.74 vs 34.30 ± 2.88 (P-value = 0.008), respectively in the RT-qPCR cvaC-1 amplification vs qPCR (Figure 6). These findings denote that Xfp DD replicates at higher rate than Xfm ESVL in olive trees and corroborate the results of previous works (Nunney et al., 2019); Saponari, personal communication) indicating a reduced rate of olive colonization and pathogenicity of Xfm ESVL in olives. Indeed, Nunney et al. (2019) reported that enzyme-linked immunosorbent assay (ELISA) does not detect any positive olive inoculated with Xfm strain “Dixon,” which belongs to the same ST (ST6) of ESVL used in this study. Citrus species have been proved to be immune to Xfp ST53-isolate and no report of infections caused by strains of the subspecies multiplex have been so far recorded. The results of our experiments are consistent with the predicted immunity to both bacterial strains. More specifically, samples collected at the PI yielded low Cq values in conventional qPCR, i.e., 25.76 ± 2.10 and 27.51 ± 2.00 for Xfp DD and Xfm ESVL inoculated plants, respectively, while generated high Cq values by RT-qPCR targeting cvaC-1 (Xfp DD 30.89 ± 2.60 vs Xfm ESVL 30.20 ± 2.21), indicating that no active bacterial multiplication occurred at the inoculation points following the needle inoculations and that the low Cq values generated in qPCR corresponded to the detection of the cells of the inoculated bacterial suspensions. When the portions collected 10 cm above the point of inoculations were tested, Cq values close to the detection limit were obtained both by qPCR (31.95 ± 2.91 for Xfp DD and 32.86 ± 2.21 for Xfm ESVL) and RT-qPCR (32.87 ± 2.07 for Xfp DD and 33.60 ± 1.12 for Xfm ESVL). Again, the Cq values recorded in RT-qPCR for cvaC-1 were higher than those obtained for the conventional qPCR assay, suggesting the lack of an active multiplication of the bacteria. Moreover, the observed high Cq could be ascribed to the passive translocation of the inoculated bacterium through the xylem flux. Periwinkle confirmed its high susceptibility to the bacterium. For both strains, Cq values obtained at the inoculation points in qPCR were close or lower than 26; while the Cq values generated in RT-qPCR were close or lower than 28, with a P-value = 0.029 vs < 0.001. Tests on the UP portions of the plants showed different results for the two bacterial strains. Plants inoculated with Xfm ESVL strain generated low and equivalent Cq values in qPCR (24.36 ± 2.73) and RT-qPCR (24.94 ± 1.91). Conversely, the comparison of qPCR and RT-qPCR results (32.03 ± 5.01 vs 29.89 ± 3.84; P-value = 0.010) showed that periwinkle is a less conducive host for Xfp DD (Figure 5). Indeed, re-isolation of both bacterial strains was successfully achieved from the distal part of the inoculated plants (UP), obtaining 1.05 × 105 and 5.9 × 103 CFU/mL, respectively from Xfm ESVL and Xfp DD infected periwinkles.
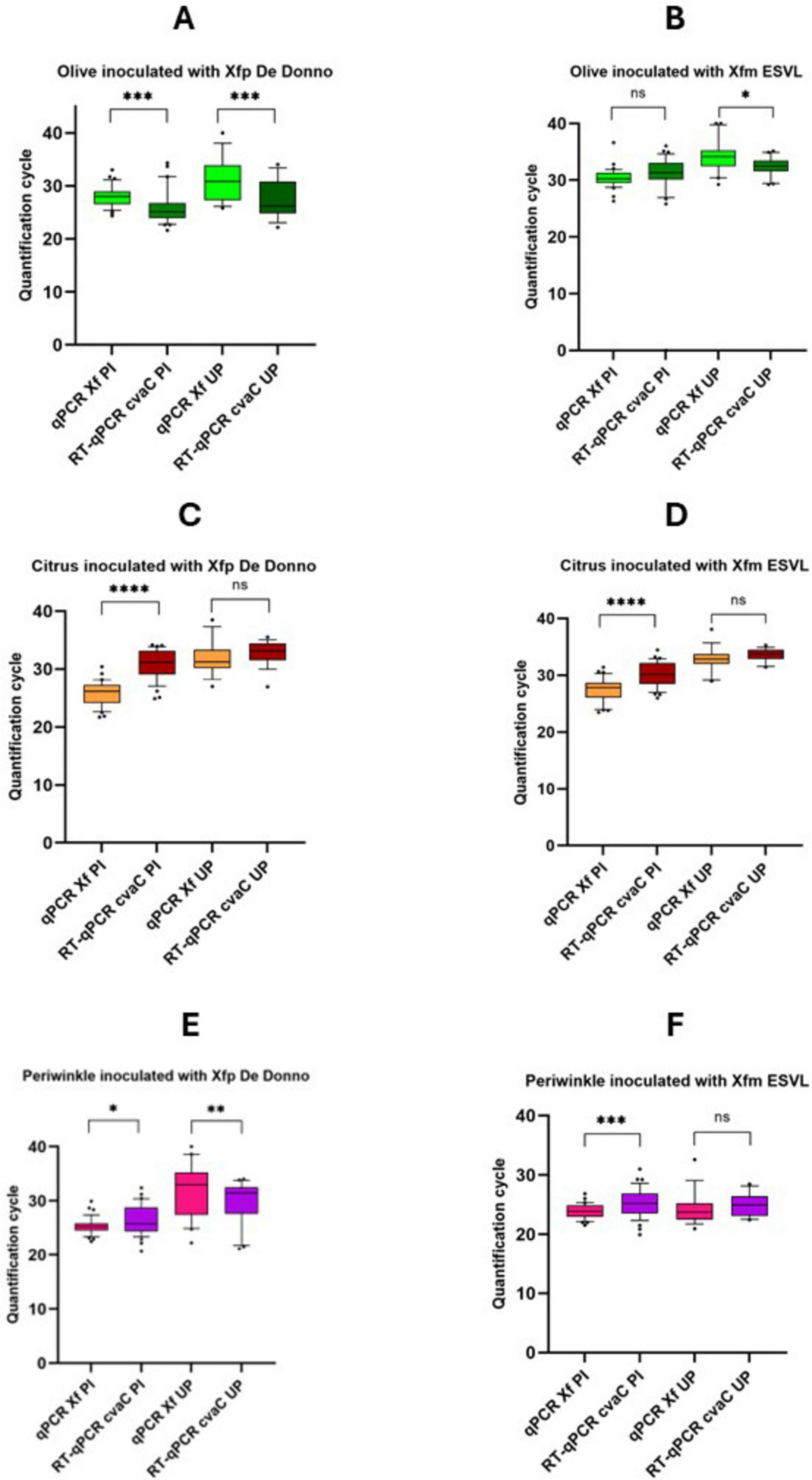
Figure 6. Whisker plots of qPCR and RT-qPCR results. Data are recovered from olive (A,B), citrus (C,D), and periwinkle (E,F) plants artificially inoculated with Xfp DD and Xfm ESVL bacterial suspension (OD600 = 1). Molecular analyses were performed using purified nucleic acid extracts recovered both from stem tissue comprised by two consecutive inoculation points (PI) and twig/stem portions 10 cm far from the last inoculation point (UP). Quantification cycles are indicated on the y-axis. The statistical significance level is also reported above each thesis (****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant).
Regardless of the vitality of the plant tissues (desiccated or vegetating twigs), positive qPCR reactions, targeting the bacterial DNA, were generated for all 42 infected olive plants. More than 50% of the non-tetracycline-treated plants (19 out of 36) had developed shoot dieback and desiccation (Supplementary Figure 3, Supplementary Table 2). As expected, in the samples collected from these plants, Cq values were higher, but still indicating a relevant bacterial DNA accumulation for the withered/desiccated tissues than for the green asymptomatic cuttings (25.18 ± 1.38 vs 20.70 ± 0.91). Values above the Cq negativity threshold were observed in the six Xf-free control plants.
In contrast, the Cq values generated by the cvaC-1 RT-qPCR were significantly different, with an average of 16.65 ± 1.14 for the symptomless plants (i.e., viable and green twigs) and an average Cq of 29.80 ± 1.43 for the desiccated twigs (Supplementary Tables 2, 3). Remarkable results were recorded on the six tetracycline-treated plants which showed broader variation compared to the untreated ones for conventional qPCR and RT-qPCR, respectively (Supplementary Table 3), suggesting that the treatment impacted cell vitality in some plants. Indeed, this issue is also supported by the associated coefficients of variation, which were 13.04% for standard qPCR and 25.35% for RT-qPCR. Only one of these plants showed shoot dieback, and consistently with the results on the non-treated plants, high Cq values (qPCR: 30.56; RT-qPCR: 31.87) were obtained in both assays on the sampled tissues. The remaining five treated and asymptomatic plants could be differentiated into two groups. The first included three plants in which the results were in line with those recorded in vegetating non-treated plants (qPCR: 22.52 ± 0.95; RT-qPCR: 19.05 ± 0.72), thus supporting high bacterial populations and intact cvaC-1 transcript, as demonstrated by the low Cq values in both assays and the positive RT-PCR results with a clear band in agarose electrophoresis gel. The second group encompassed the remaining two asymptomatic antibiotic-treated plants, whose diagnostic results were more similar to those recorded for the desiccated tissues harboring non-growing bacterial cells (qPCR: 25.94 ± 0.31; RT-qPCR: 29.05 ± 2.73), including the lack of amplification in RT-PCR, indicating that the bacterial growth was affected by the antibiotic treatment and its effect could be measured with the tests herein developed.
These results confirm that cvaC-1 transcript accumulates in host tissues with active bacterial multiplication, while desiccated tissues, in which the bacterial multiplication is compromised, generate high Cq values, most likely related to low levels of residual and degraded RNAs. By comparing the qPCR and RT-qPCR results, it appears that when the bacterium is actively colonizing the plant tissues, the Cq values of the RT-qPCR are consistently lower than those obtained using the conventional qPCR targeting the bacterial DNA (rimM gene). Whereas under conditions not favorable for bacterial multiplication and survival, such as in withered and desiccated tissues, RT-qPCR generated Cq values higher than those recorded in qPCR.
Clear-cut results were obtained through RT-PCR. A single DNA band of the expected size was observed in all vegetating shoots tested (Supplementary Table 2), while samples consisting of desiccated and dead tissues did not show any band in the electrophoresed agarose gel. More precisely, no DNA bands were detected in samples that generated Cq values higher than 25 in RT-qPCR assays. Such discrepancy can be attributed most likely to the occurrence of degraded and fragmentary RNA in these tissues, whose small fragments were still detected by RT-qPCR, while the amplification of the whole transcript was impaired, rather than to the lower analytical sensitivity of the RT-PCR compared to the RT-qPCR (Supplementary Figure 4). Further statistical evaluation of the relationships between conventional qPCR and cvaC-1 RT-qPCR results occurring among the analyzed olives are shown by Spearman’s correlation and PCA (Figures 7A, B). The correlation graph (Figure 7A) shows a strong relationship (r = 0.90; P < 0.001) between the Cq values of RT-qPCR for the cvaC-1 gene and those of the conventional qPCR. The PCA clearly clusters Cq data from green-twigs (vegetating infected twigs), withered-twigs (withered infected twigs), dead-twigs (dead infected twigs), treated (twigs from infected plants treated with tetracycline HCl), and healthy control (twigs from non-infected plants). The results show that the first principal component (PC1) has an eigenvalue of 62.49, explaining about 91.75% of the total dataset variance. The second principal component (PC2) has an eigenvalue of 5.62, explaining 8.25% of the variance, meaning that a single direction (PC1) captures almost all the information present in the data, indicating an excellent simplification of the variables present in the data set (Figure 7B).
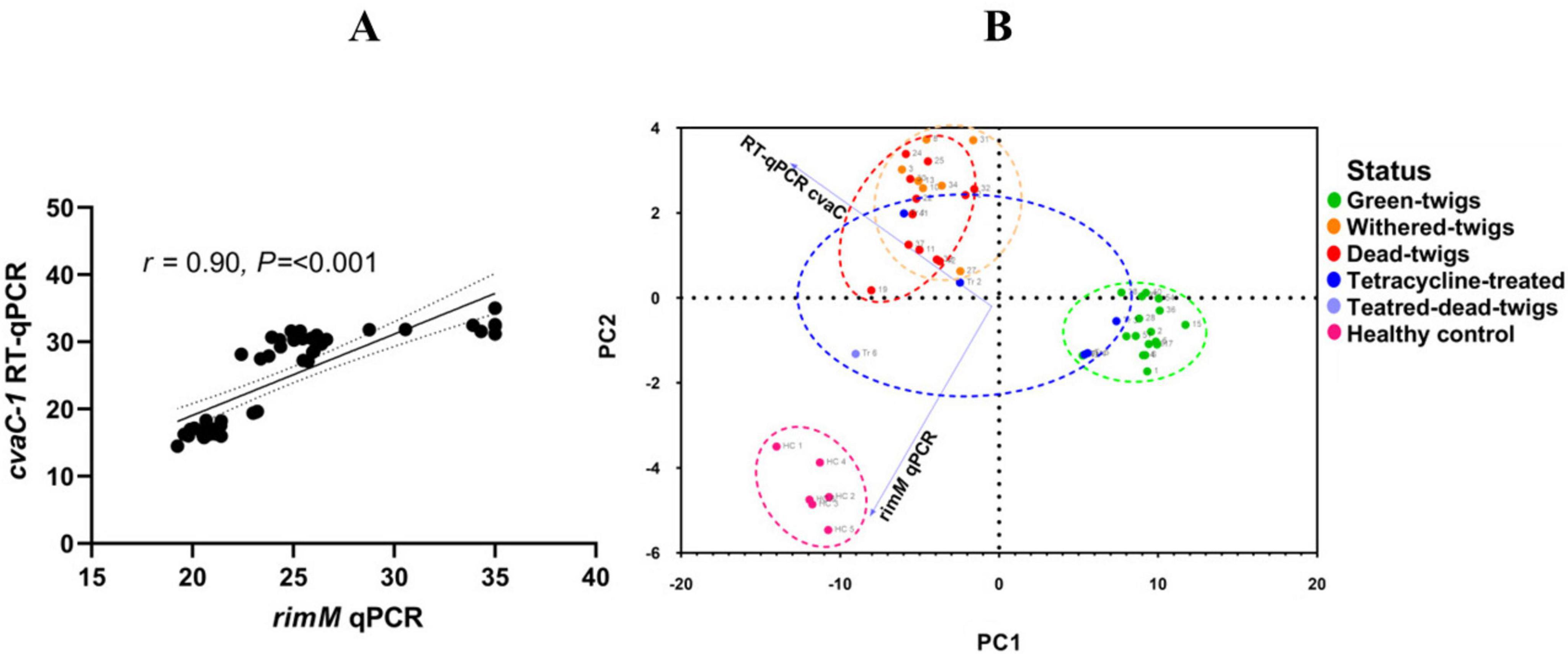
Figure 7. Principal component analyses (PCA) of Xfp infected olives. (A) Scatter plot depicting the Spearman correlation between the RT-qPCR cvaC-1 and conventional qPCR assays. The strong positive correlation indicates that both methods consistently detect and quantify bacterial genetic material across different sample types. (B) PCA biplot representing the clustering of olive samples based on Cq values from RT-qPCR cvaC-1 and qPCR. Samples are color-coded according to their condition: green-twigs (green), withered-twigs (orange), dead-twigs (red), treated (blue), treated-dead-twigs (light blue), and healthy control (pink). The first principal component (PCA1) accounts for 91.75% of the total variance, highlighting distinct group separation based on bacterial viability and treatment effects.
Overall, the estimation of the accumulation of the cvaC-1 transcript in host plants throughout the infection process (Supplementary Figure 5) and in chronically and symptomatic tissues, consistently showed that: (i) with highly susceptible host-bacterial strain combinations (i.e., olive - Xfp DD; periwinkle - Xfm ESLV) or moderately susceptible combinations (i.e., olive - Xfm ESLV; periwinkle - Xfp DD) detection of cvaC-1 transcripts consistently displayed equal or, more frequently, lower Cq values than those generated by qPCR targeting the bacterial DNA; (ii) when incompatible host-bacterial strain combinations (i.e., citrus - Xfp DD; citrus - Xfm ESLV) were examined, the average Cq values recorded for the cvaC-1 transcript where > 30, differently from those observed in the aforementioned combinations, and consistently higher than those generated in the qPCR for the DNA amplification of the rimM gene. These results emphasize that this host species was not conducive to the bacterial multiplication, with amplifications being correlated to the initial inoculum source or to the initial colonization of a few primary xylem vessels in which Xf was entrapped after inoculation as reported in Niza et al. (2015).
Transcriptomic approaches are widely exploited to investigate either the host or the pathogen gene expression profiles to gather insights into the infection process and the host defense mechanisms. Preliminary RNASeq studies, using total RNAs from Xfp-infected olives (Giampetruzzi et al., 2016), did not allow to obtain data from the bacterial gene expression in planta, since bacterial mRNAs have short polyA tails which represent a small fraction of the plant total RNAs (Sarkar, 1997), thus not detectable using the conventional RNASeq approach. The dual RNASeq technology, by depleting the large amount of plant and prokaryotic ribosomal RNAs, allows the enrichment of the remaining total RNAs, including the bacterial mRNAs. In the present work, we took advantage of this technology to study the gene expression profile of Xfp ST53 colonizing olive trees. In addition, gene expression profiles were also produced for three in vitro cultured strains previously recovered from OQDS-affected olives (Sicard et al., 2021). The analysis of the combined transcriptome datasets unraveled novel features of the bacterial lifestyle in real and artificial environments. As expected, the coverage of the bacterial genome was much higher for the transcriptomes from pure bacterial cultures than that obtained by dRNA-seq from total RNAs performed on a selected highly infected olive tree.
The gene expression profile of the bacteria grown in vitro showed an elevated expression level of gene B9J09_RS06930, annotated as Blp family class II bacteriocin (GO:0042742 - defense response to bacterium). Class II bacteriocins, most likely the best characterized and the most distributed group in food-associated lactic acid bacteria (Zhang et al., 2022), are synthesized as pre-bacteriocins containing an N-terminal leader sequence that is cleaved during secretion resulting in a small protein (< 60 amino acids) which is heat-stable. A typical gene cluster involved in the biosynthesis of class II bacteriocins consists of genes that encode a bacteriocin precursor peptide, an immunity protein, and an ATP-binding cassette (ABC) transporter. We did not find the associated immunity and ABC genes in the Xfp DD genome, where this bacteriocin gene is embedded in a genomic region surrounded by prophage-related genes (data not shown), likely originating from an event of phage-mediated horizontal gene transfer (Touchon et al., 2017).
In planta, the dual transcriptome profile unraveled abundant levels of transcript of a colicin-like precursor protein (colicin V), encoded by the cvaC-1 gene. This protein belongs to the large family of bacteriocins, the most abundant antimicrobial molecules largely conserved across the main bacterial lineages (Riley and Wertz, 2002). Unlike other antimicrobial molecules and antibiotics, they exhibit a narrow spectrum of activity, often limited to members of the same species as the producer, with which it competes in the ecological niche (Riley, 2011). Because of their limited range of activity and moderately few side effects, bacteriocins could represent an attractive option for plant disease control and inspire the design of antimicrobial molecules for medical and agricultural use. Notably, those found in Xf genomes are mainly PFT and constitute a putative virulence factor of the bacterium, thus contributing to pathogen invasion, systemic spread and biofilm formation (Pashalidis et al., 2005; Zaini et al., 2008) in xylem vessels. From the structural point of view, the C-terminal domain harbors both the cytotoxic activity and the specific site for the immunity protein cvi, whereas the presence of a double-glycine-type leader sequence, often associated with some bacterial peptide antibiotics, most likely could be engaged in the quorum sensing mechanism. Our work unveiled that cvaC-1 and cvaC-2 microcins have a major role in the Xf life cycle, being among the most expressed genes either in planta or in vitro. Indeed, the differential transcription levels detected between the two investigated conditions indicate that the expression of the two genes is modulated by different environmental conditions like pH and temperature, and factors e.g., nutritional stimuli or the competition with other sensitive bacteria as a strategy to eliminate competitors and improve survival. Moreover, their high expression and toxin production rate in vitro may explain the low number of contaminants (i.e., endophytic bacteria) growing on the artificial media when isolating Xf in axenic conditions from plant matrices, with Xf having a competitive advantage over other bacterial species thriving in the xylem environment. Interestingly, the two Xfm strains (ESVL and TOS4) showed a different level of expression of the two cvaC genes when grown in vitro, underlining different biological features for strains belonging to the same subspecies but harboring different sequence types. A differential transcription level has been also reported in the three in vitro grown Xfp cultures (Xfp DD, APL-64, APL-69) for the Blp bacteriocin class (Dawid et al., 2007; Fontaine et al., 2007).
The high cvaC-1 transcription level detected in Xfp-infected olives prompted to explore its use as a replication biomarker, i.e., an indicator of the presence of actively growing cells and Xf multiplication in plants. To test this hypothesis, we monitored the level of cvaC-1 transcript in time course artificial infection experiments using strains of two Xf subspecies (i.e., Xfp DD and Xfm ESVL) in three different plant species (olive, citrus and periwinkle). According to previous studies, citrus is renowned for being immune to both strains; on the contrary, periwinkle is susceptible to both, with the Spanish isolate being more aggressive on this host (Delbianco et al., 2021). The olive cultivar Ogliarola salentina is highly susceptible to Xfp DD, developing severe canopy desiccations, often leading to the death of the infected trees; while needle-inoculation of the Xfm ESVL strain, even if resulted in systemic infections, did not cause shoot dieback and desiccation phenomena, whose transcriptomes profiles did not display overexpression of defense related-genes (Giampetruzzi personal communication). As reported by Nunney et al. (2019), also the Xfm strain “Dixon,” which shares the same ST as ESVL used in this study (i.e., ST6), does not induce leaf symptoms and is not able to colonize olive stems, producing negative results in ELISA (Nunney et al., 2019).
Experiments aimed to compare the performances of qPCR targeting the rimM region (Harper et al., 2010) and RT-qPCR assay to detect the cvaC-1 bacterial transcript in the same Xf-infected plants, Currently, qPCR represents the golden standard approach for bacterial detection, being the official regulated method in the EU. However, this assay while detecting with high diagnostic sensitivity the bacterial DNA, fails to discriminate between living and dead bacterial cells. Therefore, the developed RT-qPCR assay, targeting the cvaC-1 transcript, was optimized to overcome this significant limitation.
In host plants where the inoculated strains did not develop systemic infections, low levels of cvaC-1 transcript were detected, with high Cq consistently above the values recorded in qPCR targeting the rimM gene, suggesting that these positive qPCR reactions most likely were related to residual non-growing bacteria cells, i.e., those inoculated but not able to initiate the infection process due to the incompatibility with the host species or the activation of strong defense responses, or unfavorable environmental conditions occurred upon the inoculation events.
Conversely, when host plants harbor active growing colonies, the estimated level of expression of the cvaC-1 transcript either overlapped the bacterial DNA content or was consistently higher, confirming the overexpression of the target gene during the bacterial host colonization regardless of the strain and the plant species. However, a further feature characterizing this susceptible condition, is the range of Cq values consistently below 30 for both assays, indicating an increased bacterial titer compared to the initial inoculum used to infect the plants.
As expected, in Xfp DD-infected olives, both qPCR and RT-qPCR detected a more copious presence of Xfp active cells at PI and UP portions compared to the plants infected by Xfm ESVL. Conversely, for periwinkle a higher prevalence of growing Xfm ESVL cells was detected in all sampled tissues compared to those of Xfp DD inoculated plants.
The lack of primary infections in the PI sites of the inoculated citrus plants was evident from the comparison of the Cq values generated by qPCR which were almost ten times (10x) lower than those generated by RT-qPCR, for Xfp DD and Xfm ESVL strains. As reported in other studies, the lignification process of primary xylem cells is a phenomenon characterizing the resistant genotypes (Coletta-Filho et al., 2007; Niza et al., 2015) with the bacterium remaining confined in the inoculation sites and triggering the secretion of defense compounds that lead to bacterial death (Rodrigues et al., 2013).
Tests performed on the chronically infected plants, showing desiccated olive shoots highlighted two important aspects: (i) conventional qPCR produced strong positive reactions (i.e., Cq < 25) even with dead bacterial cells and processing non-optimal plant tissues, indicating that bacterial DNA persists for prolonged periods, also in dead plant tissues remaining detectable most likely as small fragments of degraded DNA sequences; (ii) although bacterial mRNAs have a short half-life spanning from seconds to hours (Sheridan et al., 1998; Rauhut and Klug, 1999; Laalami et al., 2014), because the RT-qPCR targeted a very short sequence of the cvaC-1 transcript (69bp), positive reactions occurred even with degraded RNA fractions, as showed by the Cq values recorded for the desiccated shoots tested. Conversely, when these samples were tested by RT-PCR amplifying the whole cvaC-1 transcript, none of the RNA fractions recovered from dead twigs produced detectable amplification bands. This discrepancy further confirms that RNA is still detectable by RT-qPCR as small fragments even in non-growing cells. However, this was not the case for the whole target transcript, whose integrity was compromised in the desiccated tissues, and as expected, the lack of bacterial multiplication resulted in negative amplification reactions. Although very preliminary and based on a limited number of plants, the efficacy of the application of an antibiotic product on the bacterial multiplication in the plants was clearly detected with our tests, being able to differentiate plants in which a positive effect could be recorded from those in which most likely, the application did not have any impact on the targeted bacterium.
Even if RT-qPCR detected cvaC-1 traces in desiccated tissues, the PCA analysis distinctly separated the samples into different clusters, based on their infection status and treatment, providing different significant insights on the efficacy of the assays herein developed.
The Spearman correlation coefficient (r = 0.90) indicates a strong positive correlation between the Cq values obtained with RT-qPCR for the cvaC-1 transcript and those from the qPCR targeting the rimM gene (Amoia et al., 2023). This further supports that RT-qPCR assay for cvaC-1 transcript is reliable in detecting and quantifying the presence of bacterial genetic material. The lower Cq values observed in “green-twigs” samples confirm as expected, a more elevated bacterial viability in this tissue, while higher values, such as those recorded in the “withered-twigs” and “dead-twigs” samples, indicate a reduced presence of bacterial genetic material, suggesting lower bacterial activity or the presence of non-growing bacterial cells.
A critical aspect emerging from the results concerns the impact of tetracycline HCl treatment. Treated samples show more considerable variability of Cq values compared to untreated ones, suggesting that the antibiotic treatment significantly affected bacterial cell viability. Specifically, in treated samples, the increased Cq values of RT-qPCR compared to those of conventional qPCR suggest that the antibiotic negatively impacted bacterial multiplication. Quantification cycles in “withered-twigs,” “dead-twigs” and those treated with tetracycline HCl were consistently higher in RT-qPCR than the qPCR, remarking that conventional detection approaches, targeting bacterial DNA, are not suitable to retrieve information about the viability and growing stage of the bacterium in the infected tissues, thus not suitable for example, to estimate the effect and the efficacy of anti-bacterial treatments in planta.
In conclusion, our work sheds light on molecular pathways in the Xf life cycle, both in vitro and in planta, disclosing that bacteriocins may play a critical role in the successful bacterial host colonization and to conquer the xylem niche by successfully competing with endophytes. However, it still remains unclear how these interactions may result in severe diseases or latent infections in other host-strain combinations.
On the other hand, the high level of cvaC-1 expression in planta can be exploited as a diagnostic marker, either to improve the sensitivity of the current and classical diagnostic tests or to assess if the detectable bacterial DNA is linked to an active Xf multiplication stage of the infection or to non-growing bacterial cells unable to further support the infection process in the host plant. This represents a crucial aspect for estimating the phytosanitary risks linked to the detection of the bacterium, especially for those host plants from which it is challenging to recover active growing colonies on artificial media and make an assessment of the effect of control means on the viability of the bacterium upon the treatments. Nonetheless, the cvaC-1-based protocols proved to be a promising tool for monitoring the effect of the applications of antimicrobial compounds to control the bacterium in infected plants.
Previously, attempts have been made to develop molecular assays able to quantify viable cells (v-qPCR) i.e., with intact membranes, from dead ones displaying membrane damages (Sicard et al., 2019; Baró et al., 2020). However, these approaches require pretreatment with DNA-binding dyes and complex bacterial isolation protocols from the host plants, which ultimately are not suitable for routine detection. Indeed, the main v-qPCR disadvantage is the possible overestimation of cell mortality, as the exposure and photoactivation conditions, required to bind the DNA from dead cells to the dyes, could also cause damage to viable cells. The high throughput RT-qPCR assay herein developed allows for adapting the standard total nucleic extraction protocols to rapidly assess, based on the Cq values and their ratio (Cq recovered for DNA vs Cq yielded for the RNA transcript), if an active bacterial multiplication occurs in the infected plants or this is impaired and only non-growing bacterial cells are detected. Indeed, the two-step RT-PCR protocol herein developed, even if slightly less sensitive than the RT-qPCR test and more time-consuming, proved to selectively produce amplicon only in host tissues supporting active bacterial multiplication. RNA-based PCR assays for specific detection and quantification of viable bacterial cells have been previously developed for culturable and unculturable bacteria (Golmohammadi et al., 2012; Fujikawa et al., 2020; Wang et al., 2023), providing an efficient alternative diagnostic tool to indirectly estimate viable bacterial cells. The work herein described, while contributing to discover the highly transcribed genes of the target pathogen in the infected plants, represents the first attempt to use a RNA-based approach to improve the detection of this emergent plant pathogen upon performing several in planta assessments. This newly developed assay could be useful not only to evaluate the effects of the application of antimicrobial compounds to control Xf but also in research context, for monitoring the progression of Xf infection in relation to the host plant response, and for predicting the pathogen-host compatibility.
The raw data supporting the conclusions of this article are included in this article/Supplementary material. Further inquiries can be directed to the corresponding authors. The sequencing dataset presented in this study was deposited in the National Center for Biotechnology Information (NCBI) BioProject repository https://www.ncbi.nlm.nih.gov under accession number PRJNA1141272.
SSA: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review and editing. MS: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review and editing. PS: Conceptualization, Investigation, Methodology, Writing – original draft, Writing – review and editing. AML: Formal analysis, Resources, Writing – review and editing. CDG: Data curation, Resources, Writing – original draft, Writing – review and editing. GL: Funding acquisition, Methodology, Project administration, Supervision, Writing – review and editing. GD: Formal analysis, Investigation, Writing – review and editing. DB: Funding acquisition, Project administration, Supervision, Writing – review and editing. AG: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Writing – original draft, Writing – review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was partially supported by the Regione Puglia (DD no. 495 del 14/10/2015 and no. 279 del 9/8/2016, Progetto di ricerca Linea B-STIPXYT and by Ministero dell’agricoltura, della sovranità alimentare e delle foreste in the framework of the Research Projects 1LIVEXYLELLA - ID01- Trasmissione D.M. n. 664519 del 28/12/2022 (Tecnologie portatili e protocolli innovativi per la diagnosi ultrasensibile di Xylella fastidiosa direttamente in piante e vettori). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We wish to thank Prof. Leonardo de La Fuente (University of Auburn, United States) for the critical discussion on the research plan.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1501741/full#supplementary-material
Almeida, R. P. P., and Nunney, L. (2015). How do plant diseases caused by Xylella fastidiosa emerge? Plant Dis. 99, 1457–1467. doi: 10.1094/PDIS-02-15-0159-FE
Amoia, S. S., Minafra, A., Ligorio, A., Cavalieri, V., Boscia, D., Saponari, M., et al. (2023). Detection of Xylella fastidiosa in host plants and insect vectors by droplet digital PCR. Agriculture 13:716. doi: 10.3390/agriculture13030716
Baquero, F., Lanza, V. F., Baquero, M. R., del Campo, R., and Bravo-Vázquez, D. A. (2019). Microcins in Enterobacteriaceae: Peptide antimicrobials in the eco-active intestinal chemosphere. Front. Microbiol. 10:2261. doi: 10.3389/fmicb.2019.02261
Baró, A., Badosa, E., Montesinos, L., Feliu, L., Planas, M., Montesinos, E., et al. (2020). Screening and identification of BP100 peptide conjugates active against Xylella fastidiosa using a viability-qPCR method. BMC Microbiol. 20:229. doi: 10.1186/s12866-020-01915-3
Burbank, L. P., and Stenger, D. C. (2017). The DinJ/RelE toxin-antitoxin system suppresses bacterial proliferation and virulence of xylella fastidiosa in grapevine. Phytopathology 107, 388–394. doi: 10.1094/PHYTO-10-16-0374-R
Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., et al. (2009). The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. doi: 10.1373/clinchem.2008.112797
Cariddi, C., Saponari, M., Boscia, D., De Stradis, A., Loconsole, G., Nigro, F., et al. (2014). Isolation of a Xylella fastidiosa strain infecting olive and oleander in Apulia. Italy. J. Plant Pathol. 96, 425–429. doi: 10.4454/JPP.V96I2.024
Cataudella, I., Trusina, A., Sneppen, K., Gerdes, K., and Mitarai, N. (2012). Conditional cooperativity in toxin-antitoxin regulation prevents random toxin activation and promotes fast translational recovery. Nucleic Acids Res. 40, 6424–6434. doi: 10.1093/nar/gks297
Cavalieri, V., Fasanelli, E., Gibin, D., Gutierrez Linares, A., La Notte, P., Pasinato, L., et al. (2024). Update of the Xylella spp. host plant database - Systematic literature search up to 31 December 2023. EFSA J. Eur. Food Saf. Auth. 22:e8898. doi: 10.2903/j.efsa.2024.8898
Chatterjee, C., Paul, M., Xie, L., and van der Donk, W. A. (2005). Biosynthesis and mode of action of lantibiotics. Chem. Rev. 105, 633–684. doi: 10.1021/cr030105v
Chatterjee, S., Wistrom, C., and Lindow, S. E. (2008). A cell-cell signaling sensor is required for virulence and insect transmission of Xylella fastidiosa. Proc. Natl. Acad. Sci. U. S. A. 105, 2670–2675. doi: 10.1073/pnas.0712236105
Chen, H., and De La Fuente, L. (2020). Calcium transcriptionally regulates movement, recombination and other functions of Xylella fastidiosa under constant flow inside microfluidic chambers. Microb. Biotechnol. 13, 548–561. doi: 10.1111/1751-7915.13512
Coletta-Filho, H. D., Pereira, E. O., Souza, A. A., Takita, M. A., Cristofani-Yale, M., and Machado, M. A. (2007). Analysis of resistance to Xylella fastidiosa within a hybrid population of Pera sweet orange × Murcott tangor. Plant Pathol. 56, 661–668. doi: 10.1111/j.1365-3059.2007.01605.x
Davis, M. J. (1992). “Fastidious bacteria of plant vascular tissues and their invertebrate vectors,” in The Prokaryotes: A Handbook on the Biology of Bacteria: Ecophysiology, Isolation, Identification, Applications, eds A. Balows, H. G. Trüper, M. Dworkin, W. Harder, and K.-H. Schleifer (New York, NY: Springer New York), 4030–4049. doi: 10.1007/978-1-4757-2191-1_66
Dawid, S., Roche, A. M., and Weiser, J. N. (2007). The blp bacteriocins of Streptococcus pneumoniae mediate intraspecies competition both in vitro and in vivo. Infect. Immun. 75, 443–451. doi: 10.1128/IAI.01775-05
De Souza, A. A., Takita, M. A., Coletta-Filho, H. D., Caldana, C., Goldman, G. H., Yanai, G. M., et al. (2003). Analysis of gene expression in two growth states of Xylella fastidiosa and its relationship with pathogenicity. Mol. Plant Microbe Interact. 16, 867–875. doi: 10.1094/MPMI.2003.16.10.867
de Souza, J. B., Almeida-Souza, H. O., Zaini, P. A., Alves, M. N., de Souza, A. G., Pierry, P. M., et al. (2020). Xylella fastidiosa subsp. Pauca strains fb7 and 9a5c from citrus display differential behavior, secretome, and plant virulence. Int. J. Mol. Sci. 21:6769. doi: 10.3390/ijms21186769
de Souza-Neto, R. R., Carvalho, I. G. B., Martins, P. M. M., Picchi, S. C., Tomaz, J. P., Caserta, R., et al. (2022). MqsR toxin as a biotechnological tool for plant pathogen bacterial control. Sci. Rep. 12:2794. doi: 10.1038/s41598-022-06690-x
Delbianco, A., Gibin, D., Pasinato, L., and Morelli, M. (2021). Update of the Xylella spp. host plant database – systematic literature search up to 31 December 2020. EFSA J. 19:e06674. doi: 10.2903/j.efsa.2021.6674
Duquesne, S., Petit, V., Peduzzi, J., and Rebuffat, S. (2007). Structural and functional diversity of microcins, gene-encoded antibacterial peptides from enterobacteria. J. Mol. Microbiol. Biotechnol. 13, 200–209. doi: 10.1159/000104748
EFSA Panel on Plant Health (PLH) (2016). Treatment solutions to cure Xylella fastidiosa diseased plants. EFSA J. 14:e04456. doi: 10.2903/j.efsa.2016.4456
Ellis, J. C. (2020). P finder: Genomic and metagenomic annotation of RNase P RNA gene (rnpB). BMC Genomics 21:334. doi: 10.1186/s12864-020-6615-z
European and Mediterranean Plant Protection Organization (EPPO) (2023). PM 7/24 (5) Xylella fastidiosa. EPPO Bull. 53, 205–276.
Foissac, X., Svanella-Dumas, L., Dulucq, M. J., Candresse, T., and Gentit, P. (2001). Polyvalent detection of fruit tree tricho, capillo and foveaviruses by nested rt-pcr using degenerated and inosine containing primers (pdo rt-pcr). Acta Hortic. 550, 37–44. doi: 10.17660/actahortic.2001.550.2
Fontaine, L., Boutry, C., Guédon, E., Guillot, A., Ibrahim, M., Grossiord, B., et al. (2007). Quorum-sensing regulation of the production of Blp bacteriocins in Streptococcus thermophilus. J. Bacteriol. 189, 7195–7205. doi: 10.1128/JB.00966-07
Fujikawa, T., Taguchi, M., Kido, K., Kusano, S., Enya, J., Mihara, T., et al. (2020). Evaluation of an RNA-based PCR assay to detect viable Candidatus Liberibacter solanacearum (Lso) in Lso-contaminated carrot seeds using different disinfection methods. J. Plant Pathol. 102, 205–211. doi: 10.1007/s42161-019-00405-4
Giampetruzzi, A., D’Attoma, G., Zicca, S., Kubaa, R. A., Rizzo, D., Boscia, D., et al. (2019a). Draft genome sequence resources of three strains (tos4, tos5, and tos14) of xylella fastidiosa infecting different host plants in the newly discovered outbreak in tuscany, Italy. Phytopathology 109, 1516–1518. doi: 10.1094/PHYTO-04-19-0108-A
Giampetruzzi, A., Morelli, M., Saponari, M., Loconsole, G., Chiumenti, M., Boscia, D., et al. (2016). Transcriptome profiling of two olive cultivars in response to infection by the CoDiRO strain of Xylella fastidiosa subsp. pauca. BMC Genomics 17:475. doi: 10.1186/s12864-016-2833-9
Giampetruzzi, A., Saponari, M., Almeida, R. P. P., Essakhi, S., Boscia, D., Loconsole, G., et al. (2017a). Complete genome sequence of the olive-infecting strain Xylella fastidiosa subsp. pauca De Donno. Genome Announc. 5:e00569-17. doi: 10.1128/genomeA.00569-17
Giampetruzzi, A., Saponari, M., Loconsole, G., Boscia, D., Savino, V. N., Almeida, R. P. P., et al. (2017b). Genome-wide analysis provides evidence on the genetic relatedness of the emergent Xylella fastidiosa genotype in Italy to isolates from Central America. Phytopathology 107, 816–827. doi: 10.1094/PHYTO-12-16-0420-R
Giampetruzzi, A., Velasco-Amo, M. P., Marco-Noales, E., Montes-Borrego, M., Román-Écija, M., Navarro, I., et al. (2019b). Draft genome resources of two strains (“ESVL” and “IVIA5901”) of Xylella fastidiosa associated with almond leaf scorch disease in alicante, Spain. Phytopathology 109, 219–221. doi: 10.1094/PHYTO-09-18-0328-A
Golmohammadi, M., Llop, P., Scuderi, G., Gell, I., Graham, J. H., and Cubero, J. (2012). mRNA from selected genes is useful for specific detection and quantification of viable Xanthomonas citri subsp. citri. Plant Pathol. 61, 479–488. doi: 10.1111/j.1365-3059.2011.02526.x
Gupta, A., Venkataraman, B., Vasudevan, M., and Gopinath Bankar, K. (2017). Co-expression network analysis of toxin-antitoxin loci in Mycobacterium tuberculosis reveals key modulators of cellular stress. Sci. Rep. 7:5868. doi: 10.1038/s41598-017-06003-7
Harper, S. J., Ward, L. I., and Clover, G. R. G. (2010). Development of LAMP and real-time PCR methods for the rapid detection of Xylella fastidiosa for quarantine and field applications. Phytopathology 100, 1282–1288. doi: 10.1094/PHYTO-06-10-0168
Hasper, H. E., Kramer, N. E., Smith, J. L., Hillman, J. D., Zachariah, C., Kuipers, O. P., et al. (2006). An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science 313, 1636–1637. doi: 10.1126/science.1129818
Henneberger, T. S. M., Stevenson, K. L., Britton, K. O., and Chang, C. J. (2004). Distribution of Xylella fastidiosa in sycamore associated with low temperature and host resistance. Plant Dis. 88, 951–958. doi: 10.1094/PDIS.2004.88.9.951
Hill, B. L., and Purcell, A. H. (1995). Acquisition and retention of Xylella fastidiosa by an efficient vector, Graphocephala atropunctata. Phytopathology 85, 209–212.
Holtsmark, I., Eijsink, V. G. H., and Brurberg, M. B. (2008). Bacteriocins from plant pathogenic bacteria. FEMS Microbiol. Lett. 280, 1–17. doi: 10.1111/j.1574-6968.2007.01010.x
Julca, I., Marcet-Houben, M., Cruz, F., Gómez-Garrido, J., Gaut, B. S., Díez, C. M., et al. (2020). Genomic evidence for recurrent genetic admixture during the domestication of Mediterranean olive trees (Olea europaea L.). BMC Biol. 18:148. doi: 10.1186/s12915-020-00881-6
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Kovalchuk, A., Zeng, Z., Ghimire, R. P., Kivimäenpää, M., Raffaello, T., Liu, M., et al. (2019). Dual RNA-seq analysis provides new insights into interactions between Norway spruce and necrotrophic pathogen Heterobasidion annosum s.l. BMC Plant Biol. 19:2. doi: 10.1186/s12870-018-1602-0
Kunjeti, S. G., Evans, T. A., Marsh, A. G., Gregory, N. F., Kunjeti, S., Meyers, B. C., et al. (2012). RNA-Seq reveals infection-related global gene changes in Phytophthora phaseoli, the causal agent of lima bean downy mildew. Mol. Plant Pathol. 13, 454–466. doi: 10.1111/j.1364-3703.2011.00761.x
Laalami, S., Zig, L., and Putzer, H. (2014). Initiation of mRNA decay in bacteria. Cell. Mol. Life Sci. 71, 1799–1828. doi: 10.1007/s00018-013-1472-4
Landa, B. B., Castillo, A. I., Giampetruzzi, A., Kahn, A., Román-Écija, M., Velasco-Amo, M. P., et al. (2020). Emergence of a plant pathogen in europe associated with multiple intercontinental introductions. Appl. Environ. Microbiol. 86:e01521–19. doi: 10.1128/AEM.01521-19
Lee, M. W., Tan, C. C., Rogers, E. E., and Stenger, D. C. (2014). Toxin-antitoxin systems mqsR/ygiT and dinJ/relE of Xylella fastidiosa. Physiol. Mol. Plant Pathol. 87, 59–68. doi: 10.1016/j.pmpp.2014.07.001
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Loconsole, G., Potere, O., Boscia, D., Altamura, G., Djelouah, K., Elbeaino, T., et al. (2014). Detection of Xylella fastidiosa in olive trees by molecular and serological methods. J. Plant Pathol. 96, 1–8. doi: 10.4454/JPP.V96I1.041
Loconsole, G., Zicca, S., Manco, L., El Hatib, O., Altamura, G., Potere, O., et al. (2021). Diagnostic procedures to detect xylella fastidiosa in nursery stocks and consignments of plants for planting. Agriculture 11:922. doi: 10.3390/agriculture11100922
Maisonneuve, E., and Gerdes, K. (2014). Molecular mechanisms underlying bacterial persisters. Cell 157, 539–548. doi: 10.1016/j.cell.2014.02.050
Merfa, M. V., Niza, B., Takita, M. A., and De Souza, A. A. (2016). The MqsRA toxin-antitoxin system from Xylella fastidiosa plays a key role in bacterial fitness, pathogenicity, and persister cell formation. Front. Microbiol. 7:904. doi: 10.3389/fmicb.2016.00904
Meyer, F. E., Shuey, L. S., Naidoo, S., Mamni, T., Berger, D. K., Myburg, A. A., et al. (2016). Dual RNA-sequencing of Eucalyptus nitens during Phytophthora cinnamomi challenge reveals pathogen and host factors influencing compatibility. Front. Plant Sci. 7:191. doi: 10.3389/fpls.2016.00191
Montilon, V., De Stradis, A., Saponari, M., Abou Kubaa, R., Giampetruzzi, A., D’Attoma, G., et al. (2022). Xylella fastidiosa subsp. pauca ST53 exploits pit membranes of susceptible olive cultivars to spread systemically in the xylem. Plant Pathol. 72, 144–153. doi: 10.1111/ppa.13646
Niza, B., Coletta-Filho, H. D., Merfa, M. V., Takita, M. A., and de Souza, A. A. (2015). Differential colonization patterns of Xylella fastidiosa infecting citrus genotypes. Plant Pathol. 64, 1259–1269. doi: 10.1111/ppa.12381
Nunes, L. R., Ciraulo, M. B., Santos, D. S., Rodrigues, A. C. D. F. O., Oliveira, M. V., and De, et al. (2010). Transcriptome analysis of the phytobacterium xylella fastidiosa growing under xylem-based chemical conditions. J. Biomed. Biotechnol. 2010:781365. doi: 10.1155/2010/781365
Nunney, L., Azad, H., and Stouthamer, R. (2019). An experimental test of the host-plant range of nonrecombinant strains of north American Xylella fastidiosa subsp. multiplex. Phytopathology 109, 294–300. doi: 10.1094/PHYTO-07-18-0252-FI
Oresnik, I. J., Twelker, S., and Hynes, M. F. (1999). Cloning and characterization of a Rhizobium leguminosarum gene encoding a bacteriocin with similarities to RTX toxins. Appl. Environ. Microbiol. 65, 2833–2840. doi: 10.1128/aem.65.7.2833-2840.1999
Parker, J. K., Chen, H., Mccarty, S. E., Liu, L. Y., and De La Fuente, L. (2016). Calcium transcriptionally regulates the biofilm machinery of Xylella fastidiosa to promote continued biofilm development in batch cultures. Environ. Microbiol. 18:13242. doi: 10.1111/1462-2920.13242
Pashalidis, S., Moreira, L. M., Zaini, P. A., Campanharo, J. C., Alves, L. M. C., Ciapina, L. P., et al. (2005). Whole-genome expression profiling of Xylella fastidiosa in response to growth on glucose. Omi. A J. Integr. Biol. 9, 77–90. doi: 10.1089/omi.2005.9.77
Rapicavoli, J., Ingel, B., Blanco-Ulate, B., Cantu, D., and Roper, C. (2018). Xylella fastidiosa: An examination of a re-emerging plant pathogen. Mol. Plant Pathol. 19, 786–800. doi: 10.1111/mpp.12585
Rauhut, R., and Klug, G. (1999). mRNA degradation in bacteria. FEMS Microbiol. Rev. 23, 353–370. doi: 10.1111/j.1574-6976.1999.tb00404.x
Riley, M. A., and Wertz, J. E. (2002). Bacteriocins: Evolution, ecology, and application. Annu. Rev. Microbiol. 56, 117–137. doi: 10.1146/annurev.micro.56.012302.161024
Rodrigues, C. M., de Souza, A. A., Takita, M. A., Kishi, L. T., and Machado, M. A. (2013). RNA-Seq analysis of Citrus reticulata in the early stages of Xylella fastidiosa infection reveals auxin-related genes as a defense response. BMC Genomics 14:676. doi: 10.1186/1471-2164-14-676
Román-Écija, M., Navas-Cortés, J. A., Velasco-Amo, M. P., Arias-Giraldo, L. F., Gómez, L. M., De La Fuente, L., et al. (2023). Two Xylella fastidiosa subsp. multiplex strains isolated from almond in spain differ in plasmid content and virulence traits. Phytopathology 113, 960–974. doi: 10.1094/PHYTO-06-22-0234-R
Saponari, M., Boscia, D., Altamura, G., Loconsole, G., Zicca, S., D’Attoma, G., et al. (2017). Isolation and pathogenicity of Xylella fastidiosa associated to the olive quick decline syndrome in southern Italy. Sci. Rep. 7:17723. doi: 10.1038/s41598-017-17957-z
Saponari, M., Boscia, D., Nigro, F., and Martelli, G. P. (2013). Identification of dna sequences related to Xylella fastidiosa in oleander, almond and olive trees exhibiting leaf scorch symptoms in Apulia (Southern Italy). J. Plant Pathol. 95:668. doi: 10.4454/JPP.V95I3.035
Saponari, M., Loconsole, G., Cornara, D., Yokomi, R. K., Stradis, A., and De, et al. (2014). Infectivity and transmission of Xylella fastidiosa by Philaenus spumarius (Hemiptera: Aphrophoridae) in Apulia. Italy. J. Econ. Entomol. 107, 1316–1319. doi: 10.1603/EC14142
Sarkar, N. (1997). Polyadenylation of mRNA in prokaryotes. Annu. Rev. Biochem. 66, 173–197. doi: 10.1146/annurev.biochem.66.1.173
Sheridan, G. E. C., Masters, C. I., Shallcross, J. A., and Mackey, B. M. (1998). Detection of mRNA by reverse transcription-PCR as indicator of viability in Escherichia coli cells. Appl. Environ. Microbiol. 64, 1313–1318. doi: 10.1128/aem.64.4.1313-1318.1998
Sicard, A., Merfa, M. V., Voeltz, M., Zeilinger, A. R., De La Fuente, L., and Almeida, R. P. P. (2019). Discriminating between viable and membrane-damaged cells of the plant pathogen Xylella fastidiosa. PLoS One 14:e0221119. doi: 10.1371/journal.pone.0221119
Sicard, A., Saponari, M., Vanhove, M., Castillo, A. I., Giampetruzzi, A., Loconsole, G., et al. (2021). Introduction and adaptation of an emerging pathogen to olive trees in Italy. Microb. Genomics 7:000735. doi: 10.1099/mgen.0.000735
Soubeyrand, S., de Jerphanion, P., Martin, O., Saussac, M., Manceau, C., Hendrikx, P., et al. (2018). Inferring pathogen dynamics from temporal count data: The emergence of Xylella fastidiosa in France is probably not recent. New Phytol. 219, 824–836. doi: 10.1111/nph.15177
Surano, A., del Grosso, C., Musio, B., Todisco, S., Giampetruzzi, A., Altamura, G., et al. (2023). Exploring the xylem-sap to unravel biological features of Xylella fastidiosa subspecies pauca ST53 in immune, resistant and susceptible crop species through metabolomics and in vitro studies. Front. Plant Sci. 14:1343876. doi: 10.3389/fpls.2023.1343876
Touchon, M., Moura, de Sousa, J. A., and Rocha, E. P. (2017). Embracing the enemy: The diversification of microbial gene repertoires by phage-mediated horizontal gene transfer. Curr. Opin. Microbiol. 38, 66–73. doi: 10.1016/j.mib.2017.04.010
Van Sluys, M. A., De Oliveira, M. C., Monteiro-Vitorello, C. B., Miyaki, C. Y., Furlan, L. R., Camargo, L. E. A., et al. (2003). Comparative analyses of the complete genome sequences of Pierce’s disease and citrus variegated chlorosis strains of Xylella fastidiosa. J. Bacteriol. 185, 1018–1026. doi: 10.1128/JB.185.3.1018-1026.2003
Wang, G., Nie, X., Yang, L., and Liao, H. (2023). A comparative analysis of quantitative detection methods for viable food-borne pathogens using RT-qPCR and PMA-qPCR. Lett. Appl. Microbiol. 76:ovad120. doi: 10.1093/lambio/ovad120
Waters, V. L., and Crosa, J. H. (1991). Colicin V virulence plasmids. Microbiol. Rev. 55, 437–450. doi: 10.1128/mmbr.55.3.437-450.1991
Wei, W., Sawyer, T., and Burbank, L. (2021). Csp1, a cold shock protein homolog in Xylella fastidiosa influences cell attachment, pili formation, and gene expression. Microbiol. Spectr. 9:e0159121. doi: 10.1128/spectrum.01591-21
Wells, J. M., Raju, B. C., Hung, H.-Y., Weisburg, W. G., Mandelco-Paul, L., and Brenner, D. J. (1987). Xylella fastidiosa gen. nov., sp. nov: Gram-negative, xylem-limited, fastidious plant bacteria related to Xanthomonas spp. Int. J. Syst. Bacteriol. 37, 136–143. doi: 10.1099/00207713-37-2-136
Westermann, A. J., Gorski, S. A., and Vogel, J. (2012). Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 10, 618–630. doi: 10.1038/nrmicro2852
Zaini, P. A., Fogaça, A. C., Lupo, F. G. N., Nakaya, H. I., Vêncio, R. Z. N., and Da Silva, A. M. (2008). The iron stimulon of Xylella fastidiosa includes genes for type IV pilus and colicin V-Like bacteriocins. J. Bacteriol. 190, 2368–2378. doi: 10.1128/JB.01495-07
Zaini, P. A., Nascimento, R., Gouran, H., Cantu, D., Chakraborty, S., Phu, M., et al. (2018). Molecular profiling of Pierce’s disease outlines the response circuitry of vitis vinifera to xylella fastidiosa infection. Front. Plant Sci. 9:771. doi: 10.3389/fpls.2018.00771
Keywords: transcriptomics, dual RNA-seq, host-pathogen interaction, bacteriocin, replication biomarker, early detection, Xylella fastidiosa
Citation: Amoia SS, Saponari M, Saldarelli P, Ligorio AM, Grosso CD, Loconsole G, D’Attoma G, Boscia D and Giampetruzzi A (2025) Preliminary transcriptomic analyses reveal in vitro and in planta overexpression of various bacteriocins in Xylella fastidiosa. Front. Microbiol. 16:1501741. doi: 10.3389/fmicb.2025.1501741
Received: 25 September 2024; Accepted: 31 January 2025;
Published: 21 February 2025.
Edited by:
Alina S. Puig, United States Department of Agriculture, United StatesReviewed by:
Blanca B. Landa, Spanish National Research Council (CSIC), SpainCopyright © 2025 Amoia, Saponari, Saldarelli, Ligorio, Grosso, Loconsole, D’Attoma, Boscia and Giampetruzzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annalisa Giampetruzzi, YW5uYWxpc2EuZ2lhbXBldHJ1enppQGlwc3AuY25yLml0; Maria Saponari, bWFyaWEuc2Fwb25hcmlAY25yLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.