 Lisa Zierke
Lisa Zierke Rodi Mourad
Rodi Mourad Thomas P. Kohler
Thomas P. Kohler Mathias Müsken
Mathias Müsken Sven Hammerschmidt
Sven Hammerschmidt- 1Department of Molecular Genetics and Infection Biology, Interfaculty Institute for Genetics and Functional Genomics, Center for Functional Genomics of Microbes, University of Greifswald, Greifswald, Germany
- 2Central Facility for Microscopy, Helmholtz Center for Infection Research (HZI), Braunschweig, Germany
Introduction: The capsular polysaccharide (CPS) of pathogenic bacteria is a critical virulence factor, often evading phagocytosis by host immune cells, while also interfering with the contact of the pathogen with host cells and contributing to biofilm formation. Klebsiella pneumoniae, a Gram-negative human pathogen associated with high antimicrobial resistances, produces 77 CPS serotypes. The CPS masks proteinaceous factors but also protects K. pneumoniae from uptake by host phagocytic cells and activation of the complement system. In addition to nosocomial, urinary tract and bloodstream infections or pneumonia hypervirulent strains have a highly mucoid phenotype and can cause soft tissue infections, liver abscesses, and meningitis as well. The CPS is therefore crucial for both escaping detection by the immune system and enhancing the virulence potential.
Methods: In this study, we generated a non-encapsulated mutant (Kpn2146∆wza) to observe how the CPS interferes with K. pneumoniae adhesion, survival in blood, and invasiveness in an experimental infection model.
Results: Infection of A549 lung epithelial cells showed similar adherence levels for the wild-type and non-capsulated strain, while our data showed a moderately higher internalization of Kpn2146Δwza when compared to the wild-type. In whole blood killing assays, we demonstrate that the K. pneumoniae capsule is essential for survival in human blood, protecting K. pneumoniae against recognition and clearance by the human immune system, as well as complement-mediated opsonization and killing. The non-encapsulated mutant, in contrast, was unable to survive in either whole blood or human plasma. Infections of Galleria mellonella larvae showed a significantly decreased virulence potential of the CPS-deficient mutant.
Discussion: In conclusion, our data indicate a crucial role of CPS in vivo.
1 Introduction
Klebsiella pneumoniae is a major opportunistic human pathogen responsible for a wide range of nosocomial infections and an important causative agent of healthcare- and community-acquired infections. Typical clinical manifestations are urinary tract infections, liver abscesses, bloodstream infections, and respiratory tract infections resulting in severe pneumonia (Montgomerie, 1979; Ullmann, 1998; Pastagia and Arumugam, 2008; Richelsen et al., 2020; Wang et al., 1998). The rapid spread of multidrug-resistant K. pneumoniae (Kpn) strains in recent years is a major threat to human health (Paczosa and Mecsas, 2016). Due to global spread and high morbidity and mortality rates (40–50%), carbapenemase-producing Kpn strains (KPC) are one of the most clinically relevant pathogens among Enterobacterales (Patel et al., 2008; Bowers et al., 2015; Adeolu et al., 2016; McAdam, 2020). The dominant multi-locus sequence types (ST) of KPC belong to ST258, which has emerged worldwide since the early 2000s, along with its variant ST11, the dominant strain in Asia and South America (Munoz-Price et al., 2013; Chen et al., 2014). The bacterial polysaccharide capsule (CPS) and lipopolysaccharides (LPS) are important virulence factors of Gram-negative bacterial pathogens (Holmes et al., 2021). According to their strain-specific CPS (K-antigen), Kpn can be differentiated into 77 capsule serotypes. To differentiate between hypervirulent and classical Klebsiella pneumoniae strains, various biomarkers are used. Examples include the genes peg-344, iroB, iucA, and rmpA and rmpA2 as plasmid-based genes, which serve as biomarkers for identifying hypervirulent strains. Another biomarker of hypervirulence is the increased production of siderophores (30 μg/mL) (Harada and Doi, 2018). The CPS confers resistance to antimicrobial peptides and provides protection against recognition and uptake by professional phagocytes (Patro and Rathinavelan, 2019). Furthermore, the CPS protects against complement-mediated opsonization and killing and has been shown to modulate the host’s innate immune defense mechanisms (Simoons-Smit et al., 1986; Merino et al., 1992; Yoshida et al., 2000; March et al., 2013; Lee et al., 2014). In experimental mouse pneumonia and septicemia infection models, non-encapsulated Kpn strains are dramatically attenuated compared to encapsulated strains (Cortes et al., 2002; Lawlor et al., 2005; Paczosa and Mecsas, 2016). Remarkably, phagocytosed hypervirulent Kpn strains and other clinical strains have the capability to survive within host macrophages and neutrophils (Li et al., 2014; Cano et al., 2015). Recently, it was shown that the production of a thick capsule provides resistance against phagocytosis and provides increased virulence in vivo (Ernst et al., 2020).
The synthesis of CPS is a complex process involving various enzymes. Genes encoding the required enzymes are clustered in the so-called cps locus. The genes galF, wzi, wza, wzc, gnd, and ugd encoding the capsule biosynthesis enzymes of the cps locus are highly conserved. In contrast, genes encoding specific sugar synthesis proteins are highly variable (Pan et al., 2015). In general, the cps locus and the lipopolysaccharide locus (lps locus) are transcriptionally separated; however, strain-dependent fusion of both loci can occur. It was previously shown that in Kpn ATCC BAA2146 (Kpn2146), the fusion of the cps- with the lps locus is mediated through the deletion of the terminal cps P3 unit. The lps locus was inserted into a nearby area containing a high number of insertion sequences (Figure 1) (Shu et al., 2009; Hudson et al., 2014; Wyres et al., 2016).
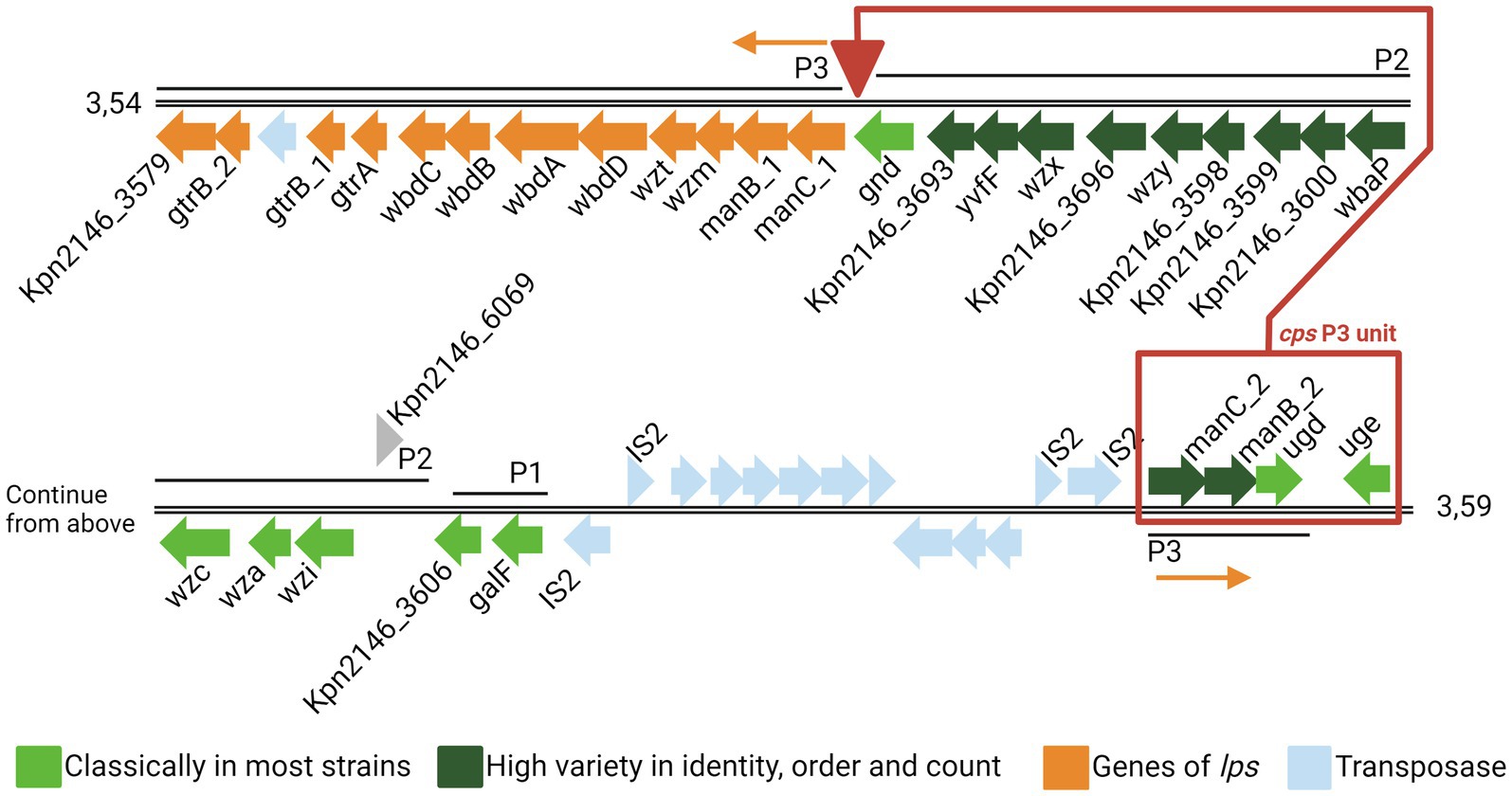
Figure 1. Genomic organization of the capsule locus of Klebsiella pneumoniae ATCC BAA2146. Coding sequences (CDSs) are represented as arrows. The color is based on the predicted functions of the resulting proteins. The three cps promotors P1, P2, and P3 are taken from Arakawa et al. (1995). Genes of the cps are in light (highly conserved) or dark green (highly variable), and genes of lps are in orange. Exceptionally, the cps P3 unit (red box) is deleted from its original position (downstream gnd) and inserted into the transposase region upstream of the cps genes cluster. The figure was adapted from Hudson et al. (2014) and NCBI GenBank CP006659.2 (Hudson et al., 2014).
The capsule production of Kpn2146 is, as shown in Figure 2, initiated by WbaP, a undecaprenyl-phosphate galactose phosphotransferase that links galactose to an undecaprenyl phosphate (Und-P). Other glycosyltransferases (GTs) add other oligosaccharides, which are at the non-reducing end of the glycosyl-Und-P. The flippase Wzx flips the oligosaccharide across the inner membrane into the periplasmic space where the polymerase Wzy polymerizes other trisaccharide units to the residues. The tyrosine kinase Wzc regulates the length of capsule polysaccharide chains, which are transferred to the outer membrane by the exporter Wza. In the final step, Wzi anchors the capsule to the cell surface (Whitfield, 2006; Rendueles, 2020). It has been demonstrated in Escherichia coli that the absence of Wza does not lead to the accumulation of capsule polymers in the periplasm, suggesting that the export of capsule polysaccharides is coupled to polymerization (Drummelsmith and Whitfield, 2000; Nesper et al., 2003).
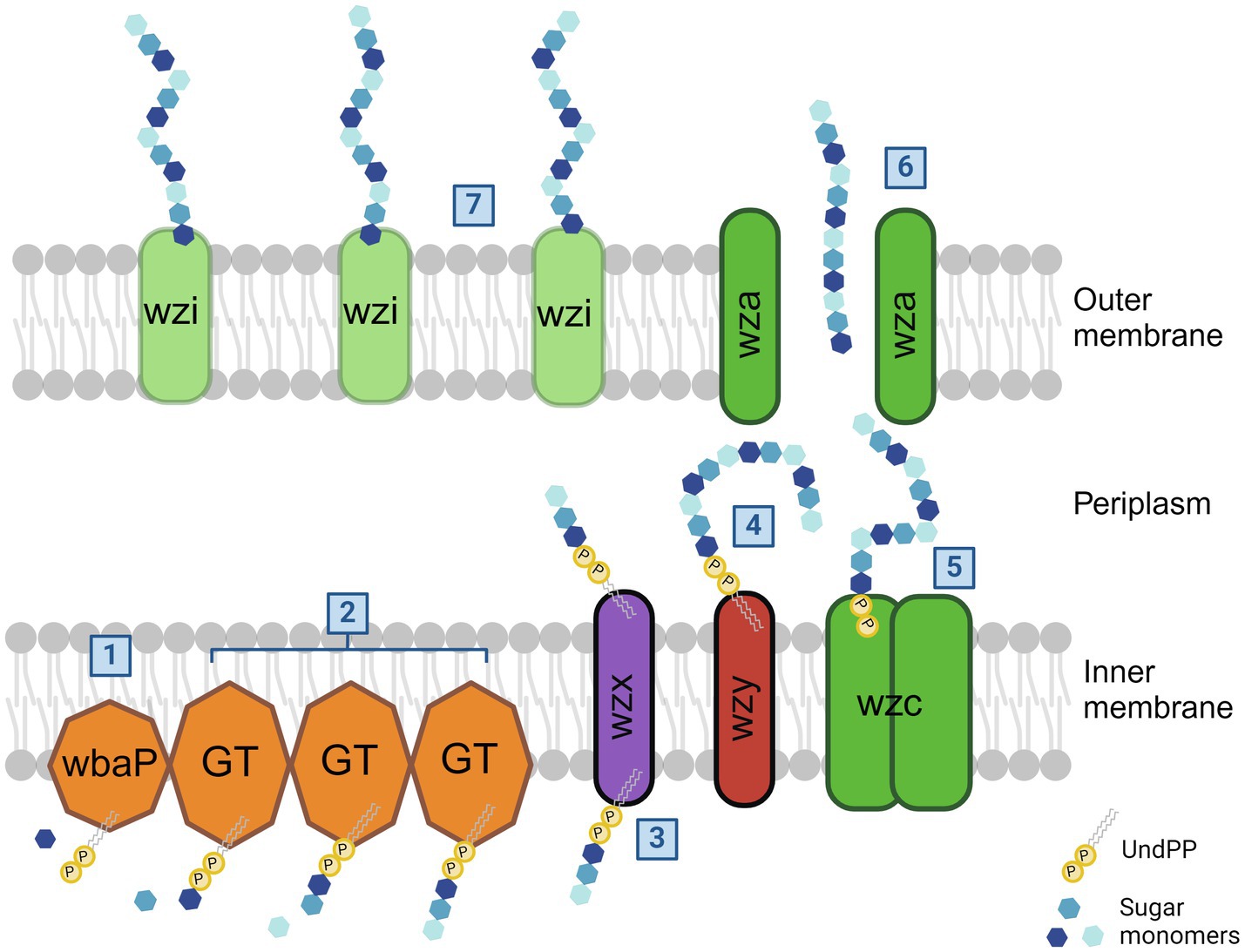
Figure 2. Klebsiella pneumoniae capsule biosynthesis pathway. In the case of Kpn2146, the capsule synthesis is initiated by undecaprenyl-phosphate galactose phosphotransferase WbaP by linking the first galactose to an undecaprenyl phosphate (1). Additional glycosytransferases bind additional oligosaccharides to the initial galactose (2). After flipping across the inner membrane by flippase Wzx (3), the polymerase Wzy then adds other trisaccharide units to the sugar residues (4). The length of capsule polysaccharides is then regulated by tyrosine-protein kinase Wzc (5). The outer membrane exporter Wza transfers the capsule to the cell surface (6). Wzi is then responsible for the linkage of CPS to the outer membrane (7). The figure was adapted from Olaya Rendueles, 2020 and created with BioRender (Rendueles, 2020).
In 2010, Kpn2146 was the first clinical isolate found to encode for the so-called New Delhi metallo-β-lactamase NDM-1, which presents a high resistance to β-lactam antibiotics in the United States and belongs to ST11 (CDC, 2013; Hudson et al., 2014). The non-profit organization American Type Culture Collection (ATCC) has conducted an extensive investigation regarding antimicrobial resistance, in which Kpn2146 showed resistance to all tested antimicrobial and antimicrobial inhibitor combinations including antibiotics and antimicrobial peptides (ATCC, 2024).
In this study, the highly conserved wza gene encoding the polysaccharide export protein was deleted in Kpn2146 by insertion–deletion mutagenesis. The pathophenotype of the generated non-encapsulated mutant was compared to the wild-type. We therefore assessed the phenotype and the capacity to adhere and invade in non-professional and professional phagocytes, and, importantly, we investigated the role of the CPS for survival under in vitro and in vivo conditions.
2 Methods
2.1 Ethics statement
The use of whole blood from healthy adult individuals was approved by the Ethics Committee of the University Medicine Greifswald (BB 117/23). All volunteers gave written informed consent in accordance with the Declaration of Helsinki. All experiments were carried out in accordance with the approved guidelines.
2.2 Bacterial strains
Bacterial strains used in this study are listed in Table 1. The cultivation of Kpn and E. coli strains was performed in lysogeny broth (LB) medium at 37°C. If necessary, LB was supplemented with 200 μg/mL chloramphenicol or 200 μg/mL spectinomycin.
Table 1. Bacterial strains.
2.3 Construction of a K. pneumoniae capsule-deficient mutant
To construct an isogenic capsule-deficient mutant of Kpn, the capsule polysaccharide exporter gene wza and 500 bp up- and downstream regions were amplified using primers 2187 and 2188 (see all primers in Table 2). The purified PCR product was ligated with pMiniT (all plasmids are listed in Table 3), and E. coli 10β chemically competent cells were transformed with the resulting plasmid pMiniT::wza_up_down. The purified plasmid was digested with NotI and BamHI. Afterward, the purified DNA insert containing the wza gene region was ligated with similarly digested pKOV. E. coli DH5α chemically competent cells were transformed with the resulting plasmid pKOV::wza_up_down. After plasmid purification, pKOV::wza_up_down was used as a template for an inverse PCR reaction using primers 2189 and 2190. To replace the wza gene with an antibiotic resistance marker, the spectinomycin resistance gene (aad9) was amplified by PCR by using pGEM_specr (Table 3) and primers 2185 and 2186. The final plasmid construct for mutagenesis pKOV::Δwza was used to transform competent Kpn2146 via electroporation. Therefore, Kpn2146 was incubated by shaking at 37°C until an OD600 of 0.5–0.7 was reached. After cell harvesting, the bacteria were washed twice with 15 mL of ice-cold 10% glycerol and finally resuspended in 1 mL of 10% ice-cold glycerol. Then, 50 μL of these electro-competent bacteria were transferred to a pre-cooled electroporation cuvette and mixed with 500 ng plasmid DNA. An electric pulse was applied with 25 μF, 200 ohms, and 1,800 V. Transformants were cultured on LB agar plates containing 200 μg/mL chloramphenicol at 37°C and 5% CO2, and positive clones were selected after overnight culture. For plasmid curing, positive clones were cultivated in LB medium containing 200 μg/mL spectinomycin and 5% sucrose at 40°C for 4 h. Cured clones were selected on LB agar plates containing 200 μg/mL spectinomycin at 37°C and 5% CO2 overnight. The successful chromosomal deletion of wza was verified using PCR with primers specforNheI (2185) and cpsKO_Reverse_BamHI (2188) and sequencing.
Table 2. Primers.

Table 3. Plasmids.
2.4 Growth analysis
Growth of Kpn2146 and its isogenic capsule mutant Kpn2146Δwza was analyzed in complex tryptic soy broth (TSB) and chemically defined medium (CDM), which is the non-supplemented cell culture medium RPMI-1640 (Capricorn™). Overnight cultures of Kpn grown on blood agar plates were used to inoculate TSB or CDM with a starting OD600 of 0.010 to 0.020 followed by an incubation at 37°C and 5% CO2. The optical density was measured hourly for 12 h, and one sample was measured after 24 h. Four biological replicates were performed.
2.5 Electron microscopy
To preserve the capsular polysaccharide during the EM sample preparation, bacteria were fixed with fixation solution 1 (0.15% ruthenium red dissolved in 0.2 M cacodylate buffer, 2% paraformaldehyde, 2.5% glutaraldehyde, 75 mM L-lysine) for 20 min on ice followed by two washing steps (0.15% ruthenium red dissolved in 0.2 M cacodylate buffer). The bacteria were further fixed using fixation solution 2 (0.15% ruthenium red dissolved in 0.2 M cacodylate buffer, 2% paraformaldehyde, 2.5% glutaraldehyde) for 2 h on ice and washed three times (0.15% ruthenium red dissolved in 0.2 M cacodylate buffer). The final fixation step with 1% osmium was performed at room temperature for 1 h followed by washing steps with 0.1 M EM-HEPES buffer. For SEM, samples were pipetted onto a 12 mm poly-l-lysine coated glass coverslip and fixed with glutaraldehyde. After washing with HEPES buffer, dehydration took place in a graded series of acetone (10, 30, 50, 70, 90%, and 2× 100%) for 15 min at each step. Afterward, the samples were subjected to critical point drying with liquid CO2 (CPD 300, Leica Microsystems, Wetzlar) and sputter-coated with a gold–palladium film (SCD 500, Bal-Tec, Lichtenstein). Examination was performed with a field emission scanning electron microscope FESEM Merlin (Zeiss, Oberkochen, Germany) using the Everhart-Thornley SE detector and the SE InLens detector at a 75:25 ratio, with an acceleration voltage of 5 kV. For TEM, bacterial samples were immobilized in agarose, dehydrated in a series of ethanol solutions, followed by infiltration with the resin LR White (1:1, 2:1, 100% LR White/EtOH) and polymerization was carried out at 55°C for 2 days. Ultrathin sections were generated using an Ultramicrotome Ultracut (Reichelt/Leica) and further counterstained with 4% aqueous uranyl acetate. Images were acquired using a transmission electron microscope Libra 120 (Zeiss) at an acceleration voltage of 120 kV and at calibrated magnifications.
2.6 Epithelial adhesion and invasion assay
Adherence to and invasion of K. pneumoniae into epithelial cells was analyzed using human A549 lung epithelial cells (ATCC® CCl-185™). A549 cells were cultured in DMEM (HyClone) supplemented with 10% heat-inactivated fetal calf serum (FCS, Sigma-Aldrich) at 37°C and 5% CO2. One day prior to the infection with Kpn, the A549 cells were seeded in wells of a 24-cell culture plate (Greiner Bio-One). A total of 80 to 90% confluent A549 cell layers (~1 × 105 cells per well) were infected with an MOI 10 or MOI 50 of early exponentially grown wild-type and capsule-deficient bacteria in infection medium (DMEM, HyClone) in the presence of 1% heat-inactivated fetal calf serum. The infection was conducted at 37°C and 5% CO2 for 1 h, 2 h, or 3 h. For adhesion, infected A549 cells were washed three times with phosphate-buffered saline (PBS) to remove extracellular unbound bacteria. Afterward, the cells were lysed using DMEM containing 1% saponin. To determine the number of adherent and intracellular bacteria, the suspension was plated on Columbia blood agar. To calculate the number of recovered intracellular Kpn, the A549 cells were washed three times with PBS and then incubated for 1 h with an infection medium supplemented with 200 μg/mL apramycin (Biozol) to kill extracellular bacteria. Afterward, the infected A549 cells were washed again three times with PBS to remove antibiotics and lysed using DMEM containing 1% saponin (Sigma-Aldrich) to release intracellular bacteria. Colony-forming units (CFUs) of intracellular bacteria were enumerated by plating the lysed host cells containing the bacteria on Columbia blood agar. All experiments were repeated three times as duplicates. All assays were analyzed using an unpaired t-test (GraphPad Prism version 8).
2.7 Phagocytosis assay
THP-1 monocytes (ATCC® TIB-202™) were cultivated in 24-well plates (2 × 105 per well) using RPMI-1640 medium supplemented with 10% heat-inactivated FCS (Sigma-Aldrich) and differentiated into macrophages using 100 ng/mL phorbol 12-myristate 13-acetate (PMA Roth) for 72 h at 37°C and 5% CO2. Afterward, the cells were washed with RPMI-1640 medium supplemented with 10% heat-inactivated FCS and incubated for another 24 h at 37°C and 5% CO2. For the phagocytosis assay, Kpn2146 and Kpn2146Δwza were cultured in TSB until the early exponential phase (A600 = 0.7–1), then centrifuged and resuspended in the infection medium (RPMI-1640 + 1% FCS). THP-1 cells, washed with the infection medium, were infected with Kpn2146 or Kpn2146Δwza (MOI 10) in 1 mL infection medium. Infections were carried out for 30 min, 60 min, and 120 min at 37°C and 5% CO2. After infection, THP-1 cells were washed three times with phosphate-buffered saline (PBS) and incubated for another hour in the infection medium supplemented with 200 μg/mL apramycin (Biozol) at 37°C and 5% CO2 to remove and kill extracellular bacteria. Finally, phagocytes were washed three times with PBS and lysed with 1% saponin (Sigma-Aldrich) to permeabilize the THP-1 cells. Recovered intracellular bacteria were plated on Columbia blood agar plates. In extended infection assays, the intracellular survival of Kpn2146 and Kpn2146Δwza was analyzed. Here, the THP-1 cells were washed three times with PBS 1 h post-infection and continued to be incubated in the infection medium supplemented with 200 μg/mL apramycin for 6 h and 24 h. After 6 h or 24 h, phagocytes were washed three times with PBS, lysed with 1% saponin (Sigma-Aldrich), and suspensions plated on Columbia blood agar to determine the survival rate or number of recovered intracellular bacteria. All experiments were repeated six times as biological replicates. All assays were analyzed using an unpaired t-test (GraphPad Prism version 8).
2.8 Whole blood killing assay
Blood was collected from healthy male and female human donors from the median cubital vein using BD Vacutainer® sodium citrate tubes (BD). Until usage, the blood was stored at 37°C.
(i) Kpn2146 WT and Kpn2146∆wza were grown at 37°C on an orbital shaker until reaching an A600 of 0.7–1, harvested and resuspended in 0.9% NaCl. For the whole blood killing assay, 200 μL of citrate-anticoagulated blood was added per well in a 96-well plate. Bacteria were added to the blood with 1×106 colony-forming units (cfu) in 10 μL and incubated for 10, 30, or 60 min at 37°C. To calculate the number of survived bacteria, the cfu was determined by plating a serial 10-fold dilution on blood agar plates.
(ii) For plasma replacement, 200 μL of citrate-anticoagulated blood was added per well in a 96-well plate and centrifuged for 5 min at 700×g. The plasma was removed, and the sedimented blood cells were resuspended with 5% or 10% pooled plasma of 21 donors (blood type AB) in 0.9% NaCl to a final volume of 200 μL. The wild-type Kpn2146 WT and the capsule-deficient mutant Kpn2146∆wza were added to the blood and incubated for 10, 30, or 60 min at 37°C. Bacterial survival was calculated as described above.
(iii) For plasma inactivation, pooled plasma of 21 donors (blood type AB) was heat-inactivated at 56°C for 20 min, leading to an inactivation of the complement binding capacity. Again, 200 μL of citrate-anticoagulated blood was added per well in a 96-well plate and centrifuged for 5 min at 700×g. The plasma was removed, and sedimented blood cells were mixed with 10% or 5% inactivated pooled plasma of 21 donors in 0.9% NaCl. Wild-type Kpn2146 WT and the capsule-deficient mutant Kpn2146∆wza were added to the blood containing inactivated pooled plasma and incubated for 10, 30, or 60 min at 37°C. Bacterial survival was calculated as described above.
(iv) To examine the effect of the complement in the absence of blood cells on bacterial survival, 200 μL pooled plasma and heat-inactivated plasma was added per well in a 96-well plate. For 50, 30, 10, and 5% active plasma, active plasma was mixed with heat-inactivated plasma accordingly. The wild-type Kpn2146 and the capsule-deficient mutant Kpn2146∆wza were added to the plasma and incubated for 10, 30, or 60 min at 37°C. Bacterial survival was calculated as described above.
2.9 Galleria mellonella infection model
Galleria mellonella larvae between 0.3 to 0.4 g (PROINSECTS, Minden, Germany) were kept in groups of 10 larvae at room temperature (21°C) in the dark for 1–2 days before infection. For survival analysis, Kpn2146 wild-type and non-encapsulated Kpn2146Δwza were cultured in TSB medium until the early exponential phase (A600 = 0.7–1), centrifuged and resuspended in 0.9% sodium chloride. The larvae were infected via the intrahemocelic route by injecting 10 μL (2 × 106 bacteria) into the right hind proleg of the larvae using a gastight microliter syringe (Hamilton) coupled with a repeating dispenser (Hamilton). After infection, the larvae were incubated in the dark with food at 37°C for up to 7 days. The larvae were monitored daily, and their survival was documented. Statistics were performed using the Gehan–Breslow–Wilcoxon test.
2.10 Statistics
Statistical analysis was performed using GraphPad Prism 8 software (La Jolla, CA, United States), with significant differences determined using the (unpaired) t-test in combination with the Mann–Whitney test. A p-value of less than 0.05 was considered statistically significant.
3 Results
3.1 Impact of the CPS on the physiology and morphology of K. pneumoniae
To investigate the impact of the CPS on the physiology and morphology of Kpn strain ATCC BAA2146 (Kpn2146), we have constructed a capsule-deficient strain by allelic replacement of the capsule polysaccharide exporter gene wza. The loss of CPS was phenotypically confirmed using electron microscopy. For both transmission electron microscopy (TEM) and field emission scanning electron microscopy (FESEM), the lysine ruthenium red fixation (LRR) method was applied to preserve the capsule of the wild-type (Fassel and Edmiston, 1999; Hammerschmidt et al., 2005). The capsule-deficient mutant Kpn2146∆wza showed a complete loss of the capsular polysaccharide material in comparison with the wild-type as indicated by TEM and FESEM (Figure 3A). The illustrations by scanning electron microscopy show the CPS in wild-type bacteria, whose colonies appear smooth on blood agar. In contrast, the capsule-deficient mutant with rough colonies on blood agar completely lacks CPS material surrounding the rod-shaped bacteria, thereby allowing the septum to be seen (Figures 3A,B; Supplementary Figure S1).
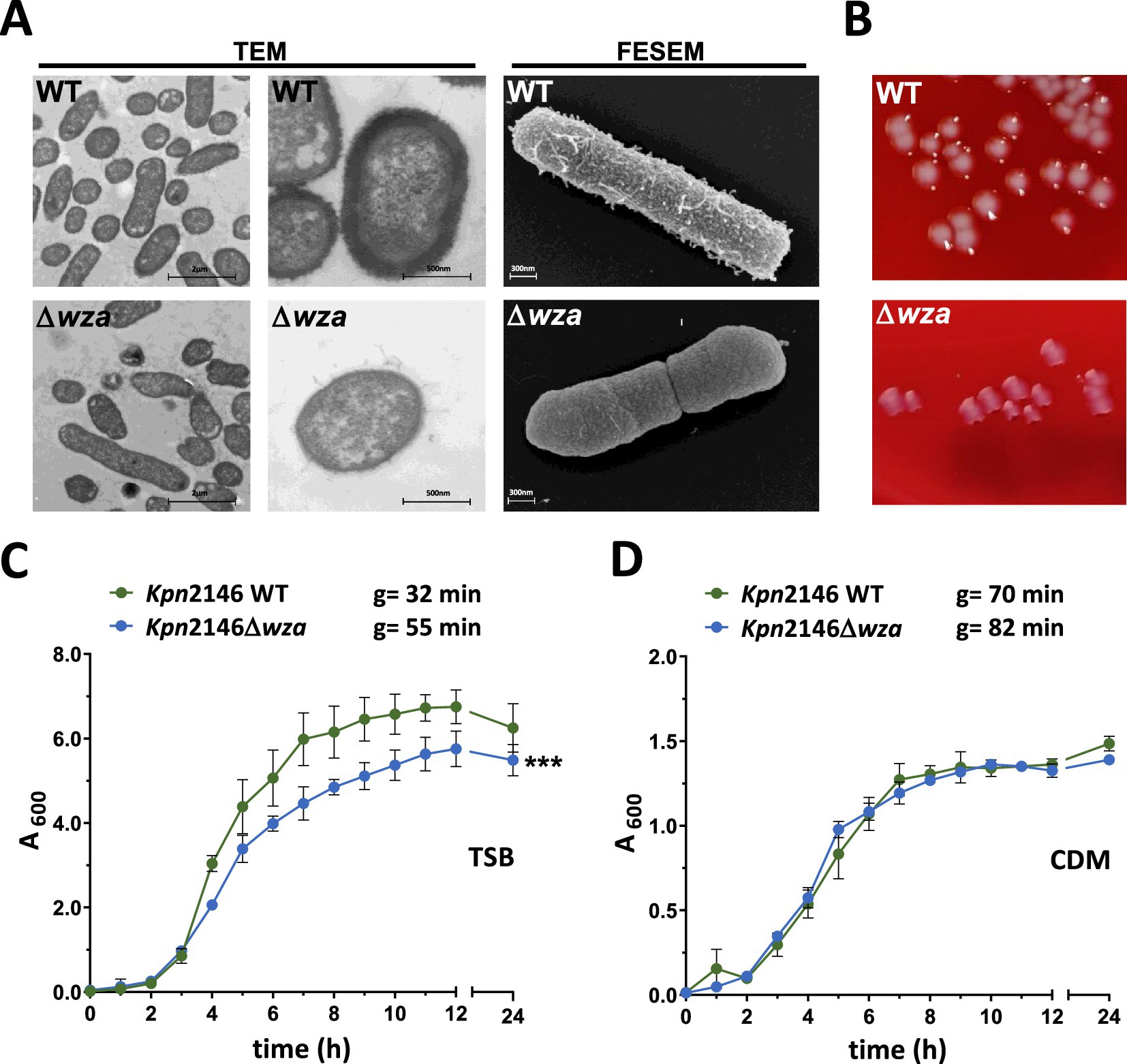
Figure 3. Analysis of K. pneumoniae cell morphology and growth behavior. (A) TEM and FESEM of LRR-fixed bacteria revealed a complete loss of the polysaccharide capsule. Bacteria were cultured in TSB. No difference in cell shape or cell division could be observed. (B) Representative colony phenotypes of Kpn2146 und Kpn2146Δwza on blood agar plates (see also Supplementary Figure S1). (C,D) Growth of Kpn2146 wild-type (green) and Kpn2146Δwza (blue) in complex TSB medium and chemically defined medium (CDM). Bacterial growth was monitored hourly for 12 h and after 24 h. g = generation time. The results are expressed as mean ± SD. Statistics: unpaired t-test, n = 4, ***p < 0.0005.
CPS production is an energy-intensive process. To evaluate the impact of the impaired CPS production on Kpn physiology, we analyzed the growth of Kpn2146 wild-type and its isogenic mutant Kpn2146Δwza under two different physiological culture conditions. Bacterial growth was monitored in a complex medium (TSB) and a chemically defined medium (CDM) (Figures 3C,D). The generation times of the wild-type and its isogenic mutant were calculated and compared. In TSB, the capsule-negative mutant Kpn2146Δwza showed a significantly reduced growth compared to the isogenic wild-type. The mutant reached the stationary phase at a lower optical density and showed an enhanced generation time of 52.2 min compared to 32.8 min for the wild-type. In general, Kpn growth in CDM is slower and shows generation times of 70.4 min for the wild-type and 82.2 min for Kpn2146Δwza. However, the wild-type and isogenic wza-mutant exhibited similar growth behavior without statistically significant differences when cultured in CDM.
3.2 Capsule deficiency enhances internalization by human epithelial cells but not K. pneumoniae adhesion
The initial step of invasive bacterial host infections is often associated with the ability of pathogens to adhere to host epithelial or endothelial cells (Pizarro-Cerda and Cossart, 2006). We hypothesized that the CPS of Kpn, similar to other Gram-negative pathogens such as Neisseria meningitidis or Acinetobacter baumannii, masks adhesins or factors facilitating bacterial adherence to host cells (Hammerschmidt et al., 1996a; Craig et al., 2004; Pizarro-Cerda and Cossart, 2006; Holmes et al., 2021). We therefore investigated whether and how the CPS influences Kpn adherence to or invasion into human epithelial cells using the lung epithelial cell line A549. The host cells were infected with Kpn2146 WT or its capsule-deficient mutant Kpn2146Δwza with an MOI 10 (multiplicity of infection) or MOI 50 for 1 h, 2 h, and 3 h, respectively. Our results showed a time-dependent increase of adherent Kpn irrespective of the phenotype at a low infection dose, but not when a high infection dose was used. However, at identical time points post-infection, similar numbers of host cell adherent Kpn were determined for wild-type and wza-mutant bacteria, when comparing identical MOIs (Figure 4A). Regarding uptake and internalization of Kpn, our data demonstrate a time-dependent increase in intracellular bacteria during infection. After 2 h and 3 h incubation of A549 cells with a low infection dose (MOI 10), the number of recovered intracellular bacteria was significantly higher for the capsule-deficient mutant in comparison with the wild-type. When using a high infection dose (MOI 50), no significant differences in recovered intracellular bacteria were observed at time points 1 h or 3 h post-infection, whereas there was a significant difference 2 h post-infection of A549 cells (Figure 4B). These results suggest that the CPS of Kpn plays only a minor role in adherence to lung epithelial cells but is probably important to prevent internalization by non-professional host cells.
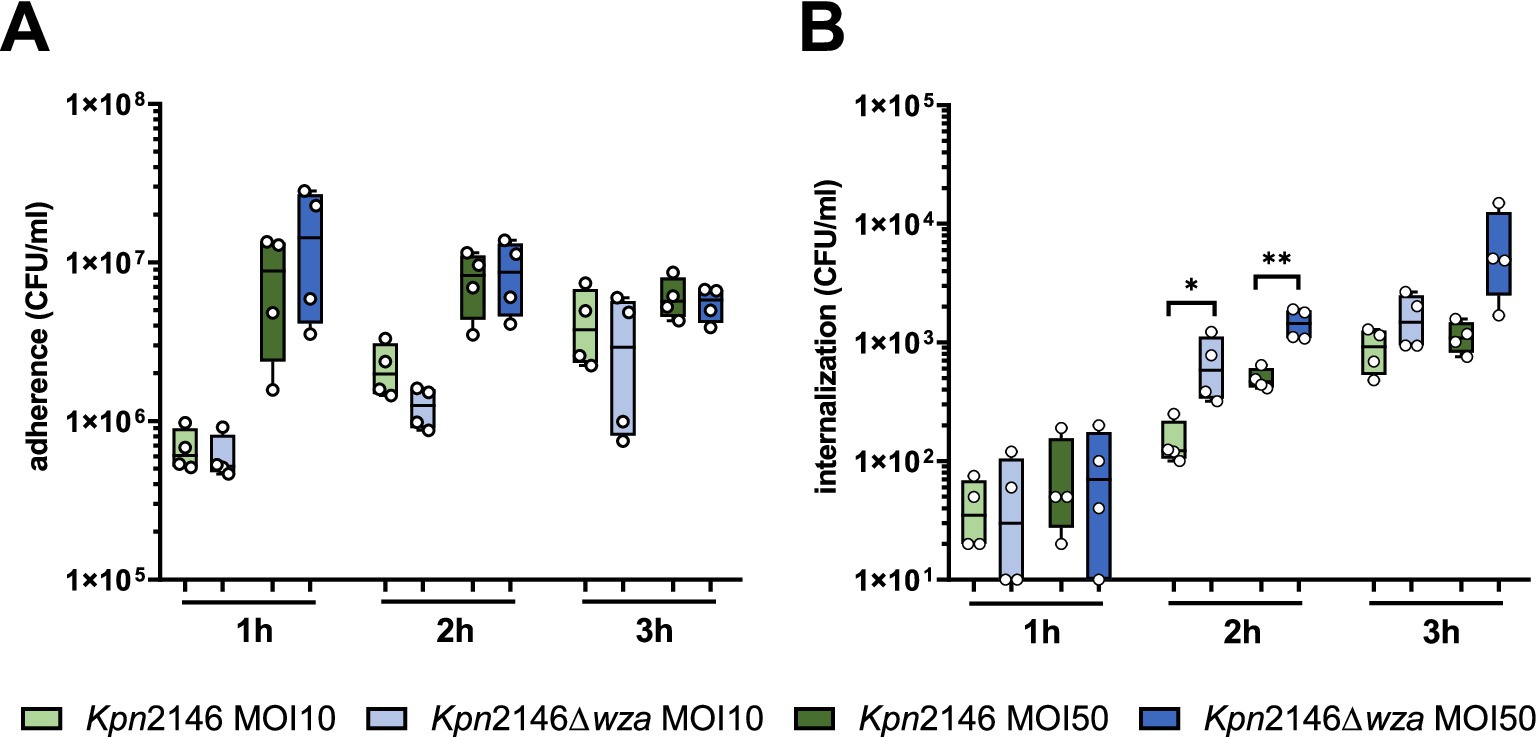
Figure 4. Influence of K. pneumoniae CPS on adherence to and internalization by human lung epithelial cells (A549). Lung cells were infected with the encapsulated Kpn2146 wild-type and isogenic non-encapsulated isogenic mutant Kpn2146∆wza for 1 h, 2 h, or 3 h with an MOI of 10 or 50 bacteria per lung cell. (A) Adherence of K. pneumoniae to A549 lung cells. Post-infection, colony-forming units (CFU) were enumerated by plating host cell adherent bacteria on blood agar plates after removing non-adherent bacteria. (B) Number of recovered intracellular K. pneumoniae. After infecting A549 cells, extracellular bacteria were killed at indicated time points by antibiotic treatment, host cells permeabilized, and surviving intracellular bacteria plated on blood agar plates to determine the recovered CFU of intracellular K. pneumoniae wild-type or CPS-deficient bacteria. Statistical analysis: unpaired t-test, n = 4, *p < 0.05, **p < 0.005.
3.3 The capsular polysaccharide protects against phagocytosis by macrophages
Next, we investigated the role of the Kpn CPS on phagocytosis by human THP-1 macrophages. Prior to infection with Kpn, THP-1 monocytes were treated with PMA (phorbol 12-myristate 13-acetate) to differentiate into mature macrophages. After infecting differentiated THP-1 cells with an MOI 10 of Kpn wild-type or wza-mutant bacteria, we determined the number of intracellular surviving bacteria. We killed extracellular bacteria with antibiotic treatment and permeabilized THP-1 macrophages containing intracellular bacteria, which were then plated on blood agar plates. Our results indicated a time-dependent uptake of both the Kpn2146 wild-type and its capsule-deficient mutant Kpn2146Δwza. After 30 min of incubation, we measured a significant increase in the number of phagocytosed capsule-deficient Kpn compared to the wild-type (Figure 5A). When THP-1 cells were incubated for 1 h or 2 h with Kpn, our results did not show differences between the wild-type and the non-encapsulated wza-mutant (Figure 5A). We further assessed the capacity of Kpn to survive intracellularly. Extracellular bacteria were removed after killing with antibiotics 1 h after infection, and intracellular survivors were determined immediately, or after 6 h and 24 h, respectively. At time points 1 h and 6 h post-infection, we determined no differences between the wild-type and CPS-negative mutant (Figure 5B). Strikingly, the intracellular survival of the mutant lacking the CPS was significantly increased after 24 h in comparison with the wild-type (Figure 5B). Taken together, our data indicate a crucial role of the CPS in protecting Kpn against phagocytosis by macrophages at the early stage of infection. Remarkably, our data suggest that the loss of the CPS has a positive effect on the intracellular survival of Kpn.
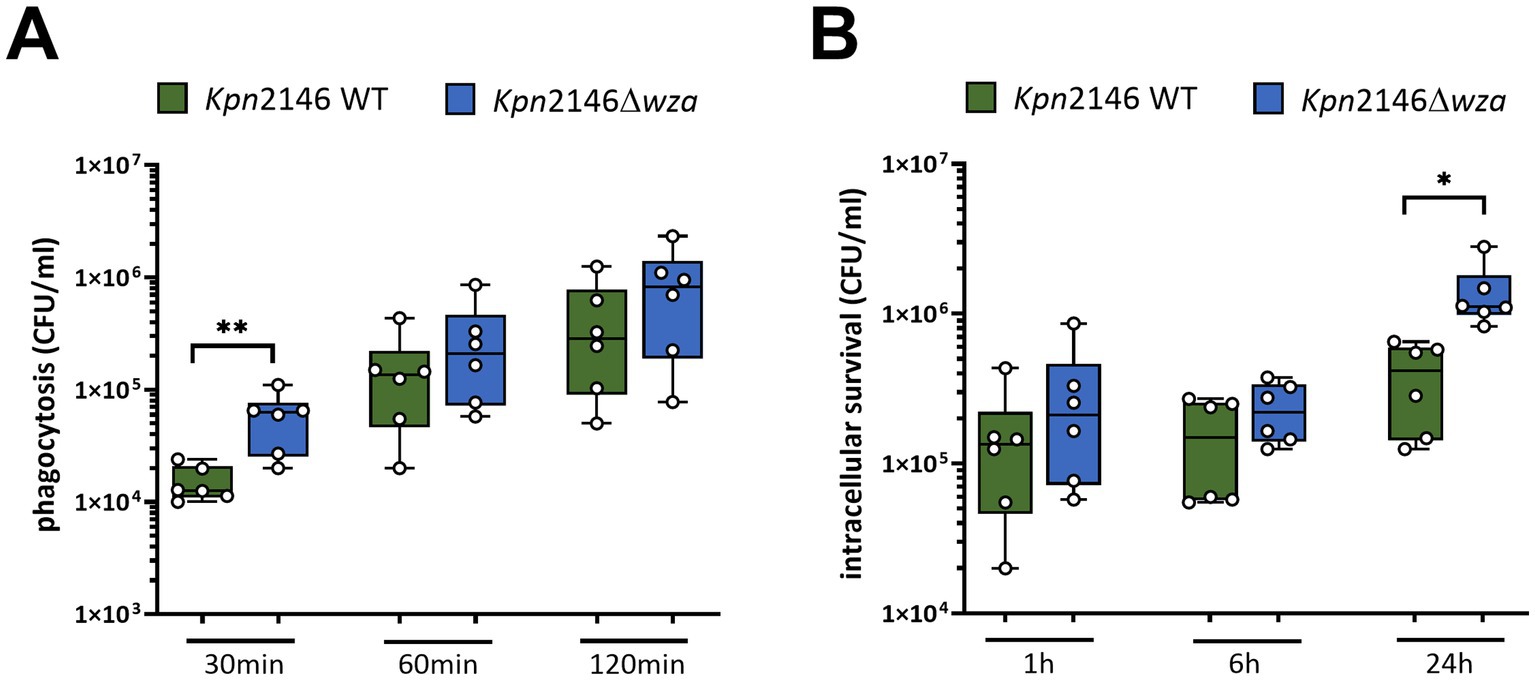
Figure 5. Impact of the polysaccharide capsule of K. pneumoniae on phagocytosis by macrophages. (A) PMA-differentiated THP-1 macrophages were infected with an MOI of 10 with wild-type or non-encapsulated K. pneumoniae for 30 min, 60 min, or 120 min. Post-infection, CFUs were determined by plating intracellular bacteria on blood agar plates. (B) THP-1 macrophages were infected with wild-type or non-encapsulated K. pneumoniae for 1 h. After 1 h, extracellular bacteria were removed by washing and further incubated in the presence of 200 μg/mL apramycin to kill remaining extracellular bacteria. After 6 h and 24 h, intracellular K. pneumoniae were recovered and enumerated by plating on blood agar plates. The results were presented as mean ± SD of four independent experiments. Statistical analysis: unpaired t-test, n = 6, *p < 0.05, **p < 0.005.
3.4 The polysaccharide capsule protects against killing in whole blood
The production of a bacterial CPS is one of the most important protection strategies against recognition by the host immune system. To determine the effect of the CPS on bacterial survival in human blood, a whole blood killing assay was performed. Because of the inter-individual variability of human blood plasma, we replaced the plasma of individual donors with pooled plasma, which is the mixture of plasma sampled from several (n = 21) healthy donors. In a further experiment, we assessed Kpn killing using our pooled plasma as well as heat-inactivated pooled plasma in the absence of blood cells. We incubated Kpn2146 wild-type or its isogenic mutant Kpn2146Δwza in 200 μL of (i) whole human blood (without plasma replacement) or in 200 μL (ii) human blood cells with replaced pooled active and (iii) inactivated plasma for 10 or 30 min. After incubation, bacterial survival was quantified by plating wild-type and wza-mutant Kpn on blood agar plates. The results indicated a rapid clearing of the non-encapsulated wza-mutant in whole human blood. Similarly, we measured a rapid killing of our non-encapsulated mutant when incubating the bacteria in human blood cells with replaced 5% (in 0.9% NaCl) active and heat-inactivated plasma. Already after 10 min of incubation, no living mutant Kpn2146Δwza could be recovered. For Kpn wild-type bacteria, we measured a slight drop in the number of bacteria when using whole blood, which was more pronounced after 30 min of incubation (Figures 6A,B). The effects were even stronger when using 10% human plasma as a replacement as shown in Supplementary Figure S2.
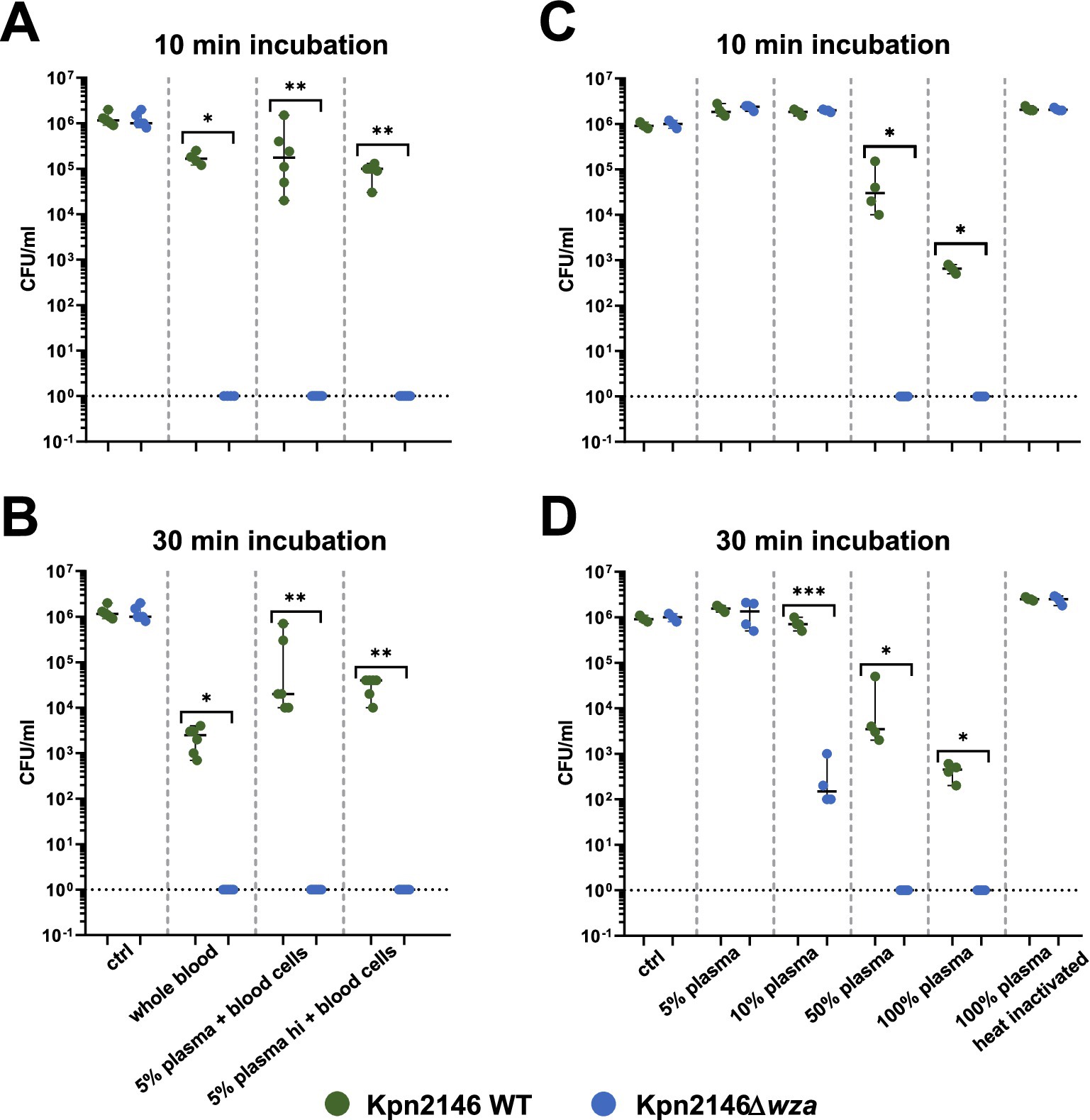
Figure 6. Impact of capsule deficiency on killing of K. pneumoniae in human (whole) blood. (A) Bacterial survival was determined in (i) citrate-anticoagulated whole blood (n = 4), (ii) blood cells with pooled 5% plasma (n = 6), and (iii) blood cells with pooled heat-inactivated (hi) 5% plasma (n = 6) in 0.9% NaCl after 10 min of incubation. (B) Bacterial survival was determined in (i) citrate-anticoagulated whole blood (n = 4), (ii) blood cells with pooled 5% plasma (n = 6), and (iii) blood cells with pooled heat-inactivated (hi) 5% plasma (n = 6) in 0.9% NaCl after 30 min. (C) Bacterial survival was determined with (iv) 5, 10, 50, and 100% active pooled human plasma filled with the appropriate amount of heat-inactivated plasma and 100% heat-inactivated pooled human plasma after 10 min of incubation (n = 4). (D) Bacterial survival was determined with (iv) 5, 10, 50, and 100% active pooled human plasma filled with the appropriate amount of heat-inactivated plasma and 100% heat-inactivated pooled human plasma after 30 min of incubation (n = 4); Mann–Whitney test and an unpaired t-test were used for statistics. *p < 0.05; **p < 0.005; ***p < 0.0005.
We further tested bacterial survival in plasma and in the absence of human blood cells. In pooled plasma, we observed a concentration-dependent decrease in survival for both the wild-type and the capsule-deficient mutant. After 10 min of incubation in 50% or 100% active plasma, we did not find a single wza-mutant grown on blood agar, whereas the growth of the wild-type bacteria was only moderately impaired. The plasma effects were even more dramatic after 30 min of incubation. We measured a reduction in survival of the capsule-deficient Kpn strain when using an active plasma concentration of 10%, which was not visible after 10 min incubation. For 100% heat-inactivated plasma, no decrease in survival could be observed for the wild-type as well as the non-encapsulated mutant Kpn2146Δwza (Figures 6B,C). In conclusion, these data demonstrate that the CPS protects Kpn against the recognition and clearing by the human immune system and against complement-mediated opsonization and killing.
3.5 Capsule deficiency attenuates K. pneumoniae virulence in the Galleria mellonella infection model
The G. mellonella larvae model was applied to analyze the impact of the CPS on Kpn virulence in vivo. G. mellonella larvae were infected via the intrahemocelic route with Kpn2146 wild-type or the isogenic capsule-deficient mutant Kpn2146Δwza. First, the infectious dose was determined (Supplementary Figure S3). Using an infection dose of 2 × 106 bacteria, eight out of ten infected larvae died 24 h post-infection when infected with the wild-type (Figure 7A). In contrast, only two of the larvae infected with the capsule-deficient mutant Kpn2146Δwza died 24 h post-infection, although the infection was clearly visible due to the darker color of the larvae (Figure 7B). Even 7 days post-infection, 30% of the larvae infected with Kpn2146Δwza were still alive, while only 10% of wild-type-infected larvae survived. The control group showed a time-dependent death of larvae starting at day 4 post-injection of sodium chloride. This time course is comparable to that of larvae infected with Kpn2146Δwza. Taken together, our data suggest a significant attenuation of capsule-deficient Kpn indicated by the decreased capability to kill G. mellonella larvae.
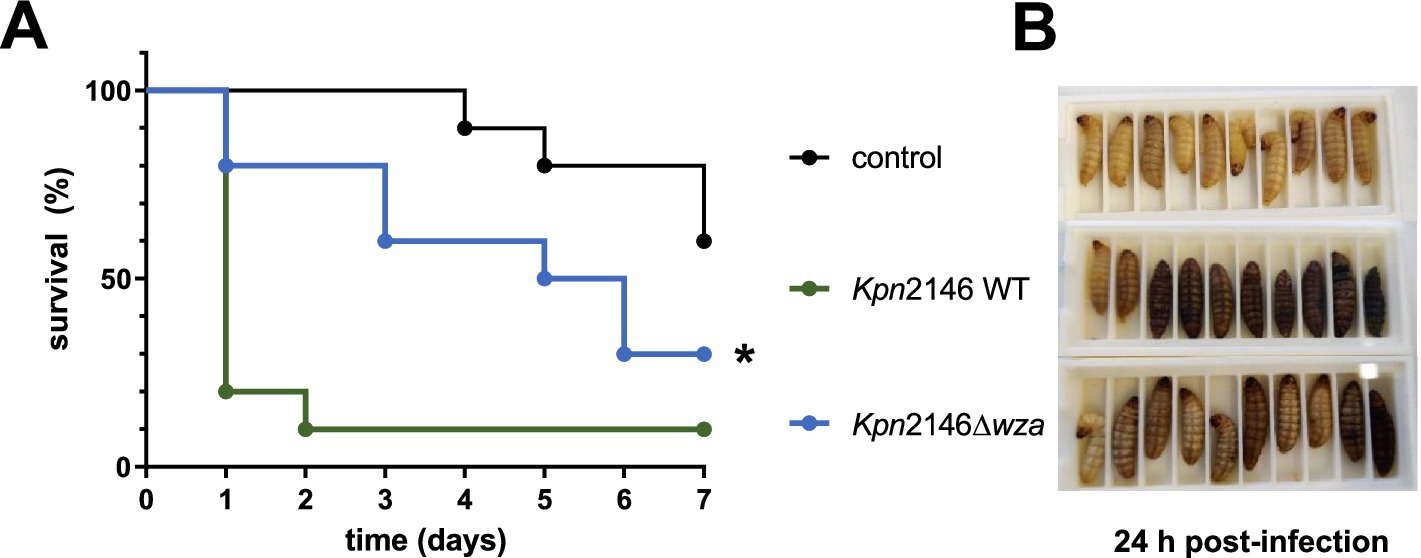
Figure 7. Impact of capsule deficiency on K. pneumoniae virulence. (A) Kaplan–Meier survival curve of G. mellonella after infection with Kpn2146 wild-type (green) and the capsule-deficient Kpn2146Δwza (blue). Bacteria were grown in TSB medium to A600 = 0.7–1, washed with 0.9% sodium chloride, and diluted to 2 × 106 bacteria per infection dose. Groups of 10 larvae (0.3–0.4 g) were infected via the intrahemocelic route and incubated for up to 7 days at 37°C with sufficient food. The survival rate was monitored daily. (B) Groups of larvae (Control, Kpn2146 WT infected and Kpn2146Δwza infected) 24 h post-infection. All larvae of the control group were alive, and eight out of ten larvae were infected with the wild-type strain died during the first 24 h. For the capsule-deficient mutant, only two out of ten died in the first 24 h, but the infection is clearly visible. *p < 0.05.
4 Discussion
Klebsiella pneumoniae is a major threat to human health due to the high number of infections and the limited therapy options (WHO, 2024). In addition to the type 1 and 3 fimbriae, siderophores, and LPS, the capsule is one of the most important virulence factors of Kpn (Paczosa and Mecsas, 2016). The CPS is known to protect the bacterium against killing by antibiotics and antimicrobial peptides and recognition by the host immune system (Domenico et al., 1999; March et al., 2013; Lee et al., 2014). To systematically analyze the impact of a capsule loss on bacterial fitness, pathogenicity, and survival in the host, we generated an isogenic capsule deletion mutant of Kpn ATCC BAA2146. The wza-mutant was also tested for its sensitivity against polymyxin and colistin. The results showed killing of both the wild-type and wza-mutant, suggesting that our mutant Kpn2146Δwza has not undergone changes such as LPS modification or alterations in the phospholipid membrane leading to higher resistance (Supplementary Figure S4).
A prerequisite for successful bacterial colonization, which is often followed by an infection, is the ability of the pathogen to attach to non-professional host cells (Pizarro-Cerda and Cossart, 2006). It was previously shown that the bacterial CPS masks surface-associated bacterial adhesins like the self-recognizing protein antigen 43 and the type I fimbriae resulting in a reduced adhesion of wild-type bacteria to host cells in comparison with non-encapsulated mutants (Schembri et al., 2004; Schembri et al., 2005; Clements et al., 2008; Tan et al., 2020). In contrast to these studies, we found that the CPS loss does not affect the bacterial adhesion to human lung epithelial cells (A549 cells). We speculate that among others, the tip protein of the type 1 fimbriae FimH probably extending out of the CPS could be mainly responsible for the adhesion capability of Kpn2146 wild-type bacteria. In 2008, Stahlhut et al. performed an allelic diversity analysis of fimH in Kpn strains. It was assumed that a mutation in the signal peptide of Kpn FimH could result in the synthesis of prolonged fimbriae, which could maybe protrude beyond the CPS (Stahlhut et al., 2009). The FimH adhesion is mainly mediated via trimannosyl residues. For E. coli, it was found that FimH variants are also capable of mediating an additional adhesion via monomannosyl residues (Sokurenko et al., 1997). These variants are closely related to E. coli strains causing urinary tract infections (Sokurenko et al., 1997; Rosen et al., 2008). Because of its origin as a urinary tract isolate, Kpn2146 could also possess an FimH variant related to urinary E. coli strains. However, this is only a speculation because we have not investigated this in detail. In addition to the type 1 fimbriae, Kpn possesses type 3 fimbriae, which were shown to mediate adhesion to human respiratory tissues (Hornick et al., 1992; Alcantar-Curiel et al., 2013).
In some cases, the invasion of host cells is an advantage for a successful infection (Pizarro-Cerda and Cossart, 2006). The capability of Kpn to invade host epithelial cells was previously shown (Oelschlaeger and Tall, 1997; Sahly et al., 2000). We measured a significantly increased uptake of the capsule-deficient mutant into lung epithelial cells after 2 h of infection, indicating that the CPS could prevent the intimate contact of the pathogen with the host cells and thereby reducing the probability of Kpn uptake into these non-professional host cells. Ernst et al. (2020) and Kaszowska et al. (2021) described a similar effect when incubating host epithelial cells with a wild-type Kpn strain and an non-encapsulated deficient mutant. In contrast to our study, Ernst et al. used capsule-deficient mutants of clinical isolates (UCI_38, BWH_36 and BWH_45) based on the deletion of wbaP, the initial glycosyltransferase of capsule synthesis. For invasion assays, bladder epithelia cells were infected for 4 h, 24 h, and 48 h, and an increase in intracellular mutant bacteria was observed. In addition, Ernst et al. (2020) showed an increased persistence of capsule-deficient mutants, which resided in LAMP1-positive vacuoles. Kaszowska et al. also used a capsule-deficient mutant based on a knockout wbaP. Here, an infection of lung epithelial cells (A549) for 2 h showed a significantly higher level of internalized mutant bacteria than wild-type bacteria, which confirms our data (Kaszowska et al., 2021). Our study would have benefited from a complemented mutant expressing Wza in trans. However, when using vector systems such as pBAD33 for complementation, we were able to clone the wza gene into the vector systems, but we did not obtain any in trans-complementation. Wza is an exporter and is highly hydrophobic; therefore, we hypothesize that in trans-complementation and its expression from a plasmid might have toxic effects on the cell. After successfully gaining access to the blood, e.g., by transcytosis of host cell barriers, bacterial survival in the bloodstream is essential for further spreading within the host during infection. This requires the evasion from host phagocytes such as macrophages and complement, which both play an important role in the clearance of Kpn (Broug-Holub et al., 1997). Our results indicate that the bacterial Kpn CPS plays an important role during the early stage of phagocytosis. Both encapsulated wild-type bacteria and capsule-deficient Kpn were able to persist within the macrophages, but strikingly we found that the loss of the capsule increased the viable amount of bacteria over time. The persistence of Kpn within host cells was also confirmed and verified by others (Maisonneuve et al., 2017; Grubwieser et al., 2023). Kpn interferes with the iron metabolism of the host cells and thus survives intracellularly (Grubwieser et al., 2023). In 2015, Cano et al. found an accumulation of Kpn within a vacuolar compartment associated with the endocytic pathway in which the downregulation of the cps was triggered. This study suggested that downregulation is beneficial for better survival in a nutrient-poor environment (Cano et al., 2015). Taken together, our findings suggest that the loss of the capsule is advantageous for Kpn survival in host macrophages.
Already in the 1980s, it was found that LPS is the main factor for protecting Kpn against complement-mediated killing (Tomás et al., 1986). In addition, Merino et al. found that the exposed LPS, e.g., through the loss of the CPS can activate the complement system. However, at the same time, resistance against complement-mediated killing is provided (Merino et al., 1992). In this study, we suggest that the CPS but not LPS is a main factor for resistance against complement-mediated killing and our data are in accordance with another study (Clements et al., 2008). Clements et al. showed that both a non-encapsulated Kpn mutant and a double knockout, deficient for CPS and LPS, had a reduced viability in human serum after 30 min of incubation, suggesting that the CPS is necessary to prevent complement-mediated killing, while LPS is not sufficient to protect the bacteria (Clements et al., 2008).
Galleria mellonella had become a popular model for in vivo bacterial infections in the last couple of years. Here, we demonstrated that the loss of the CPS impairs the virulence of Kpn during host infection, further indicating the importance of the CPS under in vivo infections as a sine qua non-virulence factor. The wild-type was able to kill 90% of the larvae within the first 24 h post-infection, whereas only 20% of larvae died after 24 h after infection with the capsule-deficient Kpn. Our data are in agreement with an earlier study, showing that capsule-deficient manC mutants of Kpn exhibit reduced virulence than the encapsulated wild-type (Insua et al., 2013).
5 Conclusion
We demonstrated the importance of the CPS for the bacterial fitness and pathogenicity of Kpn. Interestingly, and in contrast to other pathogenic bacteria such as Streptococcus pneumoniae or N. meningitidis (Hammerschmidt et al., 1996b, 2005), the lack of the CPS enhances the uptake of Kpn into host cells. Strikingly, intracellular survival appears to be facilitated in the absence of CPS, which may link Kpn pathophysiology with intracellular fitness. Finally, whole blood killing assays indicated the importance of the CPS for a lower recognition of Kpn by blood cells and a higher complement resistance.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethics Committee of the University Medicine Greifswald, Greifswald, Germany (BB 117/23). The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from primarily isolated as part of your previous study for which ethical approval was obtained. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
LZ: Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft. RM: Formal analysis, Investigation, Methodology, Writing – review & editing. TK: Investigation, Methodology, Supervision, Writing – review & editing. MM: Investigation, Methodology, Resources, Visualization, Writing – review & editing. SH: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The project was funded by the intermural funding of the University of Greifswald and the Landesgraduiertenförderung Mecklenburg-Pomerania, Germany.
Acknowledgments
The authors thank Ina Brentrop (HZI) and Kristine Sievert-Giermann (University of Greifswald) for their technical support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the preparation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1450984/full#supplementary-material
References
Adeolu, M., Alnajar, S., Naushad, S., and Gupta, R. S. (2016). Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: proposal for Enterobacterales Ord. Nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. Nov., Pectobacteriaceae fam. Nov., Yersiniaceae fam. Nov., Hafniaceae fam. Nov., Morganellaceae fam. Nov., and Budviciaceae fam. Nov. Int. J. Syst. Evol. Microbiol. 66, 5575–5599. doi: 10.1099/ijsem.0.001485
Alcantar-Curiel, M. D., Blackburn, D., Saldana, Z., Gayosso-Vazquez, C., Iovine, N. M., De la Cruz, M. A., et al. (2013). Multi-functional analysis of Klebsiella pneumoniae fimbrial types in adherence and biofilm formation. Virulence 4, 129–138. doi: 10.4161/viru.22974
Arakawa, Y., Wacharotayankun, R., Nagatsuka, T., Ito, H., Kato, N., and Ohta, M. (1995). Genomic organization of the Klebsiella pneumoniae cps region responsible for serotype K2 capsular polysaccharide synthesis in the virulent strain Chedid. J. Bacteriol. 177, 1788–1796. doi: 10.1128/jb.177.7.1788-1796.1995
Bowers, J. R., Kitchel, B., Driebe, E. M., MacCannell, D. R., Roe, C., Lemmer, D., et al. (2015). Genomic analysis of the emergence and rapid global dissemination of the clonal group 258 Klebsiella pneumoniae pandemic. PLoS One 10:e0133727. doi: 10.1371/journal.pone.0133727
Broug-Holub, E., Toews, G. B., van Iwaarden, J. F., Strieter, R. M., Kunkel, S. L., Paine, R. 3rd, et al. (1997). Alveolar macrophages are required for protective pulmonary defenses in murine Klebsiella pneumonia-elimination of alveolar macrophages increases neutrophil recruitment but decreases bacterial clearance and survival. Infect. Immun. doi: 10.1128/iai.65.4.1139-1146.1997, 65, 1139–1146
Cano, V., March, C., Insua, J. L., Aguilo, N., Llobet, E., Moranta, D., et al. (2015). Klebsiella pneumoniae survives within macrophages by avoiding delivery to lysosomes. Cell. Microbiol. 17, 1537–1560. doi: 10.1111/cmi.12466
CDC (2013). Vital signs: carbapenem-resistant Enterobacteriaceae. MMWR Morb. Mortal Wkly. Rep. 62, 165–170.
Chen, L., Mathema, B., Chavda, K. D., DeLeo, F. R., Bonomo, R. A., and Kreiswirth, B. N. (2014). Carbapenemase-producing Klebsiella pneumoniae: molecular and genetic decoding. Trends Microbiol. 22, 686–696. doi: 10.1016/j.tim.2014.09.003
Clements, A., Gaboriaud, F., Duval, J. F., Farn, J. L., Jenney, A. W., Lithgow, T., et al. (2008). The major surface-associated saccharides of Klebsiella pneumoniae contribute to host cell association. PLoS One 3:e3817. doi: 10.1371/journal.pone.0003817
Cortes, G., Borrell, N., de Astorza, B., Gomez, C., Sauleda, J., and Alberti, S. (2002). Molecular analysis of the contribution of the capsular polysaccharide and the lipopolysaccharide O side chain to the virulence of Klebsiella pneumoniae in a murine model of pneumonia. Infect. Immun. 70, 2583–2590. doi: 10.1128/IAI.70.5.2583-2590.2002
Craig, L., Pique, M. E., and Tainer, J. A. (2004). Type IV pilus structure and bacterial pathogenicity. Nat. Rev. Microbiol. 2, 363–378. doi: 10.1038/nrmicro885
Domenico, P., Tomas, J. M., Merino, S., Rubires, X., and Cunha, B. A. (1999). Surface antigen exposure by bismuth dimercaprol suppression of Klebsiella pneumoniae capsular polysaccharide. Infect. Immun. 67, 664–669. doi: 10.1128/IAI.67.2.664-669.1999
Drummelsmith, J., and Whitfield, C. (2000). Translocation of group 1 capsular polysaccharide to the surface of Escherichia coli requires a multimeric complex in the outer membrane. EMBO J. 19, 57–66. doi: 10.1093/emboj/19.1.57
Ernst, C. M., Braxton, J. R., Rodriguez-Osorio, C. A., Zagieboylo, A. P., Li, L., Pironti, A., et al. (2020). Adaptive evolution of virulence and persistence in carbapenem-resistant Klebsiella pneumoniae. Nat. Med. 26, 705–711. doi: 10.1038/s41591-020-0825-4
Fassel, T. A., and Edmiston, C. E. Jr. (1999). Ruthenium red and the bacterial glycocalyx. Biotech. Histochem. 74, 194–212. doi: 10.3109/10520299909047974
Grubwieser, P., Hilbe, R., Gehrer, C. M., Grander, M., Brigo, N., Hoffmann, A., et al. (2023). Klebsiella pneumoniae manipulates human macrophages to acquire iron. Front. Microbiol. 14:1223113. doi: 10.3389/fmicb.2023.1223113
Hammerschmidt, S., Hilse, R., Putten, J. P. M. V., Gerardy-Schahn, R., Unkmeir, A., and And Frosch, M. (1996a). Modulation of cell surface sialic acid expression in Neisseria meningitidis via a transposable genetic element. EMBO J. 15, 192–198. doi: 10.1002/j.1460-2075.1996.tb00347.x
Hammerschmidt, S., Muller, A., Sillmann, H., Muhlenhoff, M., Borrow, R., Fox, A., et al. (1996b). Capsule phase variation in Neisseria meningitidis serogroup B by slipped-strand mispairing in the polysialyltransferase gene (siaD): correlation with bacterial invasion and the outbreak of meningococcal disease. Mol. Microbiol. 20, 1211–1220. doi: 10.1111/j.1365-2958.1996.tb02641.x
Hammerschmidt, S., Wolff, S., Hocke, A., Rosseau, S., Muller, E., and Rohde, M. (2005). Illustration of pneumococcal polysaccharide capsule during adherence and invasion of epithelial cells. Infect. Immun. 73, 4653–4667. doi: 10.1128/IAI.73.8.4653-4667.2005
Harada, S., and Doi, Y. (2018). Hypervirulent Klebsiella pneumoniae: a call for consensus definition and international collaboration. J. Clin. Microbiol. 56:e00959-18. doi: 10.1128/JCM.00959-18
Holmes, C. L., Anderson, M. T., Mobley, H. L. T., and Bachman, M. A. (2021). Pathogenesis of gram-negative bacteremia. Clin. Microbiol. Rev. 34:e00234-20. doi: 10.1128/CMR.00234-20
Hornick, D. B., Allen, B. L., Horn, M. A., and Clegg, S. (1992). Adherence to respiratory epithelia by recombinant Escherichia coli expressing kp type 3 fimbriae gene products. Infect. Immun. 60, 1577–1588. doi: 10.1128/iai.60.4.1577-1588.1992
Hudson, C. M., Bent, Z. W., Meagher, R. J., and Williams, K. P. (2014). Resistance determinants and mobile genetic elements of an NDM-1-encoding Klebsiella pneumoniae strain. PLoS One 9:e99209. doi: 10.1371/journal.pone.0099209
Insua, J. L., Llobet, E., Moranta, D., Pérez-Gutiérrez, C., Tomás, A., Garmendia, J., et al. (2013). Modeling Klebsiella pneumoniae pathogenesis by infection of the wax moth galleria mellonella. Infect. Immun. 81, 3552–3565. doi: 10.1128/IAI.00391-13
Kaszowska, M., Majkowska-Skrobek, G., Markwitz, P., Lood, C., Jachymek, W., Maciejewska, A., et al. (2021). The mutation in wbaP cps gene cluster selected by phage-borne Depolymerase abolishes capsule production and diminishes the virulence of Klebsiella pneumoniae. Int. J. Mol. Sci. 22. doi: 10.3390/ijms222111562
Lawlor, M. S., Hsu, J., Rick, P. D., and Miller, V. L. (2005). Identification of Klebsiella pneumoniae virulence determinants using an intranasal infection model. Mol. Microbiol. 58, 1054–1073. doi: 10.1111/j.1365-2958.2005.04918.x
Lee, C. H., Chang, C. C., Liu, J. W., Chen, R. F., and Yang, K. D. (2014). Sialic acid involved in hypermucoviscosity phenotype of Klebsiella pneumoniae and associated with resistance to neutrophil phagocytosis. Virulence 5, 673–679. doi: 10.4161/viru.32076
Li, J., Liu, F., Wang, Q., Ge, P., Woo, P. C., Yan, J., et al. (2014). Genomic and transcriptomic analysis of NDM-1 Klebsiella pneumoniae in spaceflight reveal mechanisms underlying environmental adaptability. Sci. Rep. 4:6216. doi: 10.1038/srep06216
Maisonneuve, E., Cateau, E., Delouche, M., Quellard, N., and Rodier, M. H. (2017). An observational study of phagocytes and Klebsiella pneumoniae relationships: different behaviors. Microbes Infect. 19, 259–266. doi: 10.1016/j.micinf.2016.12.005
March, C., Cano, V., Moranta, D., Llobet, E., Perez-Gutierrez, C., Tomas, J. M., et al. (2013). Role of bacterial surface structures on the interaction of Klebsiella pneumoniae with phagocytes. PLoS One 8:e56847. doi: 10.1371/journal.pone.0056847
McAdam, A. J. (2020). Enterobacteriaceae? Enterobacterales? What should we call enteric gram-negative Bacilli? A Micro-comic strip. J. Clin. Microbiol. 58. doi: 10.1128/JCM.01888-19
Merino, S., Camprubí, S., Albertí, S., Benedí, V. J., and Tomás, J. M. (1992). Mechanisms of Klebsiella pneumoniae resistance to complement-mediated killing. Infect. Immun. 60, 2529–2535. doi: 10.1128/iai.60.6.2529-2535.1992
Montgomerie, J. Z. (1979). Epidemiology of Klebsiella and hospital-associated infections. Rev. Infect. Dis. 1, 736–753. doi: 10.1093/clinids/1.5.736
Munoz-Price, L. S., Poirel, L., Bonomo, R. A., Schwaber, M. J., Daikos, G. L., Cormican, M., et al. (2013). Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect. Dis. 13, 785–796. doi: 10.1016/S1473-3099(13)70190-7
Nesper, J., Hill, C. M., Paiment, A., Harauz, G., Beis, K., Naismith, J. H., et al. (2003). Translocation of group 1 capsular polysaccharide in Escherichia coli serotype K30. Structural and functional analysis of the outer membrane lipoprotein Wza. J. Biol. Chem. 278, 49763–49772. doi: 10.1074/jbc.M308775200
Oelschlaeger, T., and Tall, B. D. (1997). Invasion of cultured human epithelial cells by Klebsiella pneumoniae isolated from the urinary tract. Infect. Immun. 65, 2950–2958. doi: 10.1128/iai.65.7.2950-2958.1997
Paczosa, M. K., and Mecsas, J. (2016). Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 80, 629–661. doi: 10.1128/MMBR.00078-15
Pan, Y. J., Lin, T. L., Chen, C. T., Chen, Y. Y., Hsieh, P. F., Hsu, C. R., et al. (2015). Genetic analysis of capsular polysaccharide synthesis gene clusters in 79 capsular types of Klebsiella spp. Sci. Rep. 5:15573. doi: 10.1038/srep15573
Pastagia, M., and Arumugam, V. (2008). Klebsiella pneumoniae liver abscesses in a public hospital in Queens, New York. Travel Med. Infect. Dis. 6, 228–233. doi: 10.1016/j.tmaid.2008.02.005
Patel, G., Huprikar, S., Factor, S. H., Jenkins, S. G., and Calfee, D. P. (2008). Outcomes of carbapenem-resistant Klebsiella pneumoniae infection and the impact of antimicrobial and adjunctive therapies. Infect. Control Hosp. Epidemiol. 29, 1099–1106. doi: 10.1086/592412
Patro, L. P. P., and Rathinavelan, T. (2019). Targeting the sugary armor of Klebsiella species. Front. Cell. Infect. Microbiol. 9:367. doi: 10.3389/fcimb.2019.00367
Pizarro-Cerda, J., and Cossart, P. (2006). Bacterial adhesion and entry into host cells. Cell 124, 715–727. doi: 10.1016/j.cell.2006.02.012
Rendueles, O. (2020). Deciphering the role of the capsule of Klebsiella pneumoniae during pathogenesis: a cautionary tale. Mol. Microbiol. 113, 883–888. doi: 10.1111/mmi.14474
Richelsen, R., Smit, J., Anru, P. L., Schonheyder, H. C., and Nielsen, H. (2020). Incidence of community-onset extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae infections: an 11-year population-based study in Denmark. Infect Dis (Lond) 52, 547–556. doi: 10.1080/23744235.2020.1763452
Rosen, D. A., Pinkner, J. S., Walker, J. N., Elam, J. S., Jones, J. M., and Hultgren, S. J. (2008). Molecular variations in Klebsiella pneumoniae and Escherichia coli FimH affect function and pathogenesis in the urinary tract. Infect. Immun. 76, 3346–3356. doi: 10.1128/IAI.00340-08
Sahly, H., Podschun, R., Oelschlaeger, T. A., Greiwe, M., Parolis, H., Hasty, D., et al. (2000). Capsule impedes adhesion to and invasion of epithelial cells by Klebsiella pneumoniae. Infect. Immun. 68, 6744–6749. doi: 10.1128/IAI.68.12.6744-6749.2000
Schembri, M. A., Blom, J., Krogfelt, K. A., and Klemm, P. (2005). Capsule and fimbria interaction in Klebsiella pneumoniae. Infect. Immun. 73, 4626–4633. doi: 10.1128/IAI.73.8.4626-4633.2005
Schembri, M. A., Dalsgaard, D., and Klemm, P. (2004). Capsule shields the function of short bacterial adhesins. J. Bacteriol. 186, 1249–1257. doi: 10.1128/JB.186.5.1249-1257.2004
Shu, H. Y., Fung, C. P., Liu, Y. M., Wu, K. M., Chen, Y. T., Li, L. H., et al. (2009). Genetic diversity of capsular polysaccharide biosynthesis in Klebsiella pneumoniae clinical isolates. Microbiology (Reading) 155, 4170–4183. doi: 10.1099/mic.0.029017-0
Simoons-Smit, A. M., Vught, A. M. J. J. V.-V., and Maclaren, D. M. (1986). The role of K antigens as virulence factors in Klebsiella. J. Med. Microbiol. 21, 133–137. doi: 10.1099/00222615-21-2-133
Sokurenko, E. V., Chesnokova, V., Doyle, R. J., and Hasty, D. L. (1997). Diversity of the Escherichia coli type 1 fimbrial lectin. Differential binding to mannosides and uroepithelial cells. J. Biol. Chem. 272, 17880–17886. doi: 10.1074/jbc.272.28.17880
Stahlhut, S. G., Chattopadhyay, S., Struve, C., Weissman, S. J., Aprikian, P., Libby, S. J., et al. (2009). Population variability of the FimH type 1 fimbrial adhesin in Klebsiella pneumoniae. J. Bacteriol. 191, 1941–1950. doi: 10.1128/JB.00601-08
Tan, Y. H., Chen, Y., Chu, W. H. W., Sham, L. T., and Gan, Y. H. (2020). Cell envelope defects of different capsule-null mutants in K1 hypervirulent Klebsiella pneumoniae can affect bacterial pathogenesis. Mol. Microbiol. 113, 889–905. doi: 10.1111/mmi.14447
Tomás, J. M., Benedí, V. J., Ciurana, B., and Jofre, J. (1986). Role of capsule and O antigen in resistance of Klebsiella pneumoniae to serum bactericidal activity. Infect. Immun. 54, 85–89. doi: 10.1128/iai.54.1.85-89.1986
Ullmann, P. A. (1998). Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 11, 589–603. doi: 10.1128/CMR.11.4.589
Wang, J.-H., Liu, Y.-C., Lee, S. S.-J., Yen, M.-Y., Chen, Y.-S., Wang, J.-H., et al. (1998). Primary liver abscess due to Klebsiella pneumoniae in Taiwan. Clin. Infect. Dis. 26, 1434–1438. doi: 10.1086/516369
Whitfield, C. (2006). Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu. Rev. Biochem. 75, 39–68. doi: 10.1146/annurev.biochem.75.103004.142545
WHO (2024). WHO bacterial priority pathogens list, 2024: bacterial pathogens of public health importance to guide research, development and strategies to prevent and control antimicrobial resistance. Geneva: Word Health Organization, 1–56.
Wyres, K. L., Wick, R. R., Gorrie, C., Jenney, A., Follador, R., Thomson, N. R., et al. (2016). Identification of Klebsiella capsule synthesis loci from whole genome data. Microb. Genom. 2:e000102. doi: 10.5281/zenodo.55773
Keywords: Klebsiella pneumoniae, capsule, adherence, phagocytosis, infection, Galleria mellonella
Citation: Zierke L, Mourad R, Kohler TP, Müsken M and Hammerschmidt S (2025) Influence of the polysaccharide capsule on virulence and fitness of Klebsiella pneumoniae. Front. Microbiol. 16:1450984. doi: 10.3389/fmicb.2025.1450984
Edited by:
Axel Cloeckaert, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceReviewed by:
William T. Doerrler, Louisiana State University, United StatesCostas C. Papagiannitsis, University of Thessaly, Greece
Adriano de Souza Santos Monteiro, Federal University of Bahia (UFBA), Brazil
Copyright © 2025 Zierke, Mourad, Kohler, Müsken and Hammerschmidt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sven Hammerschmidt, c3Zlbi5oYW1tZXJzY2htaWR0QHVuaS1ncmVpZnN3YWxkLmRl