 Jia Yee Ho1,2†
Jia Yee Ho1,2† Xiu Qi Koh3†
Xiu Qi Koh3† Deborah Yebon Kang2
Deborah Yebon Kang2 Adrian Low3
Adrian Low3 Dalong Hu1,2
Dalong Hu1,2 Mindia A. S. Haryono2
Mindia A. S. Haryono2 Rohan B. H. Williams2
Rohan B. H. Williams2 Rebecca J. Case4,5
Rebecca J. Case4,5 Yann Felix Boucher1,2,6*
Yann Felix Boucher1,2,6*- 1Saw Swee Hock School of Public Health, National University of Singapore and National University Health System and National University Hospital System, Singapore, Singapore
- 2Singapore Centre for Environmental Life Sciences Engineering (SCELSE), National University of Singapore, Singapore, Singapore
- 3Centre for Translational Medicine, Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
- 4Singapore Centre for Environmental Life Science Engineering, Nanyang Technological University, Singapore, Singapore
- 5School of Biological Sciences, Nanyang Technological University, Singapore, Singapore
- 6Infectious Diseases Translational Research Program, Department of Microbiology and Immunology, Yong Loo Lin School of Medicine, National University of Singapore and National University Hospital System, Singapore, Singapore
Marine heterotrophic bacteria in coastal waters respond to the influx of carbon from natural and anthropogenic sources. We identified two nearly identical, (99.9% average nucleotide identity; 100% amino acid identity; same DNA G + C content of 52.3 mol%) high-quality (≥99% CheckM completeness and ≤ 1.3% contamination) draft metagenome-assembled genomes (MAGs; SJ0813 and SJ0972) from seawater microbiomes of a southern island of Singapore that is in a protected marine park. The MAGs were only assigned to the Cellvibrionaceae family according to Genome Taxonomy Database. Overall genome related indices to Pseudomaricurvus alkylphenolicus KU41GT as the closest phylogenetic relative revealed no more than 70.45% average nucleotide identity (ANIcutoff < 95%), below the 50% percentage of conserved proteins (POCPcutoff = 43.54%) for genera cutoff and low digital DNA–DNA hybridization values (DDH = 20.6 and 20.8%). The major respiratory quinone is predicted to be ubiquinone-9 from the annotation of 3-demethylubiquinone-9 3-methyltransferase (ubiG, K00568) involved in the last step of the ubiquinone biosynthesis pathway (M00117), which differed from the ubiquinone-8 utilized by known members of Cellvibrionaceae. Both MAGs contained a complete pathway for dissimilatory nitrate reduction to ammonia, which increases bioavailability of nitrogen in seawater. An identical choline dehydrogenase found in both MAGs have a low amino-acid identity (≤64.47%) compared to existing GMC family oxidoreductases, expanding on the diversity of this family of enzymes. The MAGs meet nearly all the minimum requirements but lack a 16S rRNA gene of sufficient length required for the proposed novel genus and species under SeqCode. Nevertheless, phylogenetic trees based on core-genome and RpoB as an alternative phylogenetic marker are congruent with the taxon standing as a monophyletic clade to other taxa of the order Cellvibrionales. Taken together, the MAGs (SJ0813 and SJ0972) represent an uncultured, undescribed genus and species in which we tentatively propose the name Candidatus Pelagadaptatus aseana gen. nov., sp. nov. and strain SJ0813TS (=BAABNI000000000.1TS) as type sequence. Phylogenetic inference from core-genome and RpoB phylogenetic trees placed Umboniibacter marinipuniceus KMM 3891T outside Cellvibrionaceae. We, therefore, propose the transfer of the genus Umboniibacter from the family Cellvibrionaceae to a new family Umboniibacteraceae according to the International Code of Nomenclature of Prokaryotes.
1 Introduction
Coastal waters are dynamic environments subjected to nutrient influx from terrestrial and mixing from the open sea, and marine bacteria are proposed as indicators of environmental change (Pinhassi et al., 2022; Lønborg et al., 2021). The marine Gammaproteobacteria is a highly diverse class of bacteria consisting of distinct metabolic traits and growth strategies (Liu and Liu, 2020; Spring et al., 2015; Cho and Giovannoni, 2004). Over the years, the phylogeny of the Gammaproteobacteria class and particularly the Orders Alteromonadales, Cellvibrionales, Oceanospirillales, and Pseudomonadales have undergone several shifts and reclassifications based on core-proteins and-genes phylogenomic analyses (Liao et al., 2020; Williams et al., 2010). Within the Cellvibrionales order, are the families Cellvibrionaceae, Halieaceae, Microbulbiferaceae, Porticoccaceae, and Spongiibacteraceae. Most bacterial species of the Cellvibrionaceae characterized to date are found in the marine environment and are crucial players in the global carbon cycle as heterotrophic bacteria since they break down complex polysaccharides such as agarose, chitin, and cellulose, which constitute macroalgae and detritus fallen from coastal plants (Spring et al., 2015). The current Cellvibrionaceae family comprises 17 validly published genera of Agaribacterium, Agarilytica, Cellvibrio, Eionea, Exilibacterium, Gilvimarinus, Halioxenophilus, Maricurvus, Marinimicrobium, Pseudomaricurvus, Pseudoteredinibacter, Saccharophagus, Sessilibacter, Simiduia, Teredinibacter, Thalassocella and Umboniibacter with many representative species isolated from seawater or host-associated marine environments (Reimer et al., 2022; Spring et al., 2015; Parte et al., 2020). Within these, Cellvibrionaceae species Cellvibrio japonicus, Gilvimarinus chinensis, and Marinimicrobium agarilyticum have been shown to exhibit r-strategist traits that respond quicker to nutrient influx compared to k-strategists bacterial species in the Halieaceae and Spongiibacteraceae families, which form an oligotrophic marine gammaproteobacterial clade (Spring et al., 2015). The marine microbiota in Singapore is largely uncultured and surveys have mostly been conducted through 16S rRNA gene amplicon sequencing or microarray-based compositional analyses, albeit with limited phylogenetic resolution (Teramoto et al., 2013; Wijaya et al., 2023; Lau et al., 2013; Chenard et al., 2019; Deignan et al., 2023; Soh et al., 2024). It has been shown that the distribution of Gammaproteobacteria and Cellvibrionales differ around the coastal waters of Singapore and may be influenced by anthropogenic effects such as eutrophication and petroleum contamination (Teramoto et al., 2013; Chenard et al., 2019). Here, we describe two highly similar metagenome-assembled genomes (MAGs) belonging to a novel genus in the marine Gammaproteobacteria. The MAGs were from a spatial microbiome study of coastal seawater surrounding an island that is part of a marine conservation zone at South of Singapore. Furthermore, phylogenetic analysis of the order Cellvibronales suggests the creation of a novel family for the transfer of the validly recognized genus Umboniibacter, which is currently classified under the family Cellvibronaceae.
2 Materials and methods
2.1 Sample collection and processing
Surface water samples were collected from two different locations through grab sampling (<1 m) along the coastal areas of St. John’s Island, Singapore (map grid reference: 1.216595 N, 103.850151 E) in August 2022, namely “Lagoon” (SJ0813; sample ID WLagoon_S196) and “Pier” (SJ0972; sample ID WPier_S191).” “Lagoon” serves as a frequented location where numerous visitors engage in swimming activities, and “Pier” serves as the designated area for the berthing of incoming and outgoing ferries. In-situ parameters of the water samples were measured using YSI ProQuatro Multiparameter Meter. Two liters of the collected water samples were filtered onto 0.22 μm sterile Cellulose Nitrate membrane filters (Sartorius, Göttingen, Germany) and extracted using the DNeasy® PowerWater® Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions.
2.2 Metagenomic sequencing, assembly, binning, and quality checks
Metagenomics sequencing of the extracted DNA samples was performed at the Singapore Centre for Environmental Life Sciences Engineering (SCELSE). The DNA libraries were constructed using TruSeq Nano DNA Library Kit and dual barcoded TruSeq DNA UD Indexes (Illumina, San Diego, USA) before 150 bp paired-end sequencing reads were generated using the Illumina HiSeqX platform. An average of 18.3 Mbp raw sequencing reads per sample were obtained (350 Mbp for 19 samples). Raw reads were filtered using the Kneaddata pipeline v0.7.71 where human contamination and low-quality reads were filtered before constructing the initial assembly via metaSPAdes v3.13.0 with default parameters (Nurk et al., 2017). Additionally, after initial raw reads quality step, MetaPhlAn 4 (database: vJan21_CHOCOPhlAnSGB_202103) was used for profiling the composition of microbial communities to draft taxonomic assignments and estimation of relative abundance (Blanco-Míguez et al., 2023). Subsequently, the binning, bin refinement, bin reassembly, and initial bin taxonomy classification were performed using the metaWRAP pipeline v1.3 with the quality threshold for the bins set at >50% completeness and < 10% contamination rate (Uritskiy et al., 2018). The taxonomy of the final bins was further assessed using Genome Taxonomy Database toolkit (GTDB-Tk) v2.1.1 using the Genome Taxonomy Database release 214 (Chaumeil et al., 2022). MAGs were accessed for completeness and contamination using CheckM v1.2.2 (Parks et al., 2015). To estimate the abundance of our novel Cellvibrionaceae MAGs in the metagenomic reads, trimmed raw reads of each sample were mapped back to the Candidatus Pelagadaptatus aseana MAGs using Bowtie2 v2.5.3 with default settings (Langmead and Salzberg, 2012). The relative abundance of the MAGs was calculated by dividing the number of mapped reads by the total number of reads for the sample.
2.3 Genome annotation and phylogenetic analysis
For all software, default parameters were selected unless otherwise specified. Prokka v1.14.5 was used to predict protein-coding sequences (CDS) and RNA, and the resulting protein and nucleotide sequences were applied to downstream applications (Seemann, 2014). The predicted protein sequences were annotated using Kyoto Encyclopedia of Genes and Genomes (KEGG) database release 108.1 and BlastKOALA (accessed 24 Jan 2024) (Kanehisa et al., 2016; Kanehisa et al., 2023). Panaroo v1.3.2 was used to find and align (MAFFT) the core genes among the RefSeq genomes with changes to the following parameters (−-clean-mode strict, −f 0.5, −c 0.85, −-len_dif_percent 0.8, and --core_threshold 1.0, −-aligner mafft) (Tonkin-Hill et al., 2020; Katoh and Standley, 2013; Katoh et al., 2002). Genomes selected for comparison are those of the earliest published species for each genus. The aligned concatenated core genes was used to build a maximum-likelihood tree with RAxML-NG using the GTR + G model, 12,345 as seed with bootstrapping performed using Transfer Bootstrap Expectation (TBE) method (Kozlov et al., 2019). RpoB protein sequences were aligned using MUSCLE in MEGA-X followed by RAxML-NG using the LG + G model, 12,345 as seed with bootstrapping performed using TBE method (Kozlov et al., 2019; Edgar, 2004; Kumar et al., 2018). Average nucleotide identity (ANI) using BLAST algorithm was performed using JSpeciesWS (Richter et al., 2016). Average amino acid identity (AAI) was performed using CompareM v0.1.22. The Percentage of Conserved Proteins (POCP) was analysed using POCP calculator3 (Qin et al., 2014). DNA–DNA hybridization values were calculated using formula 2 (identities/high scoring segment pair length) in the Genome-to-Genome Distance Calculator v3.04 (Meier-Kolthoff et al., 2021). Taxonomic nomenclature and etymology were based on suggestions using Protologger v1.0 on Galaxy (accessed 24 Jan 2024) and published guidelines (Hitch et al., 2021; Trüper, 1999; Galaxy, 2022). Protein BLAST (blastp) of the RpoB sequence and choline dehydrogenase was performed against NCBI metagenomic proteins (10,979,702 sequences) and non-redundant protein databases (717,423,220 sequences) (both accessed 25 March 2024) (Altschul et al., 1997; Altschul et al., 2005). The accession numbers for all the genomes and RpoB sequences are provided in Supplementary Table S1. Proksee was used to generate the plot for genome comparison (Grant et al., 2023).
2.4 SSU rRNA and rpoB identification from binned and unbinned reads
Infernal v1.1.5 was used (cmsearch --tblout) to identify SSU rRNA genes from assembled contigs (binned or unbinned) (Nawrocki and Eddy, 2013). Sequences with length > = 1,125 bp (SeqCode 16S rRNA gene requirement >75% complete) were shortlisted. The start (seq from) and end (seq to) position of 16S rRNA sequence detected by Infernal was used to extract the 16S rRNA sequence from the MAGs with samtools faidx (samtools v1.6) (Danecek et al., 2021). Extracted sequences were then aligned to 16S_ribosomal_RNA database downloaded from NCBI (modified 27-08-2024, downloaded 06-11-2024) using BlastN v2.15.0+ (−outfmt 7), and taxonomic information (Organism and Family) were manually added according to the NCBI accession ID (Sayers et al., 2021). To find the rpoB sequence of SJ0972 (WPier_S191), we used the rpoB gene sequence of SJ0813 as a template to search the unbinned reads using BlastN v2.16.0 (−outfmt 6) (Sayers et al., 2021).
3 Results
3.1 Phylogenomic analysis of phylogenetically novel MAGs
Two high-quality (≥99% completeness and ≤ 1.3% contamination) draft MAGs of 3.46 Mbp (SJ0813) and 3.45 Mbp (SJ0972) assembled with read coverage (≥61×) were sequenced from seawater collected from two locations around St. John’s Island, Singapore. The MAGs are highly similar to each other with 99.99% ANI and AAI (Table 1 and Supplementary Figure S1). Phylogenetic analysis using the GTDB classifier only assigned the MAGs to the family Cellvibrionaceae with multiple sequence alignments showing ≤83.22% protein identity to the closest genome and relative evolutionary divergence (≤0.883) (Table 1). A maximum-likelihood tree was constructed using 20 single copy core genes (100%) of type strains and/or representative sequence genomes (RefSeq) (n = 16) in Cellvibrionaceae as well as one type species from the remaining four families within the order Cellvibronales: Halieaceae, Microbulbiferaceae, Porticoccaceae, and Spongiibacteraceae (n = 4). The phylogeny supports that the MAGs belong to the Cellvibrionaceae lineage but are yet distinct from their closest relative, Pseudomaricurvus alkylphenolicus KU41GT (Figure 1A). Moreover, each MAG (SJ0813 and SJ0972) contained an identical 143 bp segment belonging to the start of the 16S rRNA gene (V1 regions) with closest BLAST hit (95.1% identity) to Cellvibrio japonicus Ueda107 of the family Cellvibrionaceae (Supplementary Table S2). Comparative alignment of the short SSU rRNA genes to C. japonicus Ueda107, Cellvibrio mixtus ACM 2601T, and P. alkylphenolicus KU41G revealed identities of 93.9, 90.5 and 90.5%, which are less than the threshold (94.5%) for genus cutoff for full-length SSU rRNA gene, as previously proposed (Yarza et al., 2014). Further attempts to find a SSU rRNA gene to fulfill the requirement of SeqCode (~1,250 bp or > 75% of complete SSU rRNA gene) in the binned and unbinned contigs using the Infernal pipeline, yielded 21 sequences that do not belong to any bacterial species of Cellvibrionaceae (Supplementary Table S3). As an alternative marker, we utilized RpoB to build a maximum-likelihood tree to show that the MAGs are indeed their own monophyletic clade (Figure 1B). This relationship is congruent to the RpoB maximum-likelihood tree as shown in Figure 1A. It should be noted that the SJ0972 rpoB gene was only partial (3,318 bp) albeit with 100% similarity to the rpoB of SJ0813 (4,085 bp), and both (1,361 aa for SJ0813TS; 1,106 aa for SJ0972) were included in the RpoB phylogenetic tree. The POCP comparison to P. alkykphenolicus is no more than 43.54%, ANI comparison to P. alkylphenolicus KU41GT shows no more than 70.45% identity and 39.80% coverage and low digital DDH of 20.6 and 20.8% (Table 1).
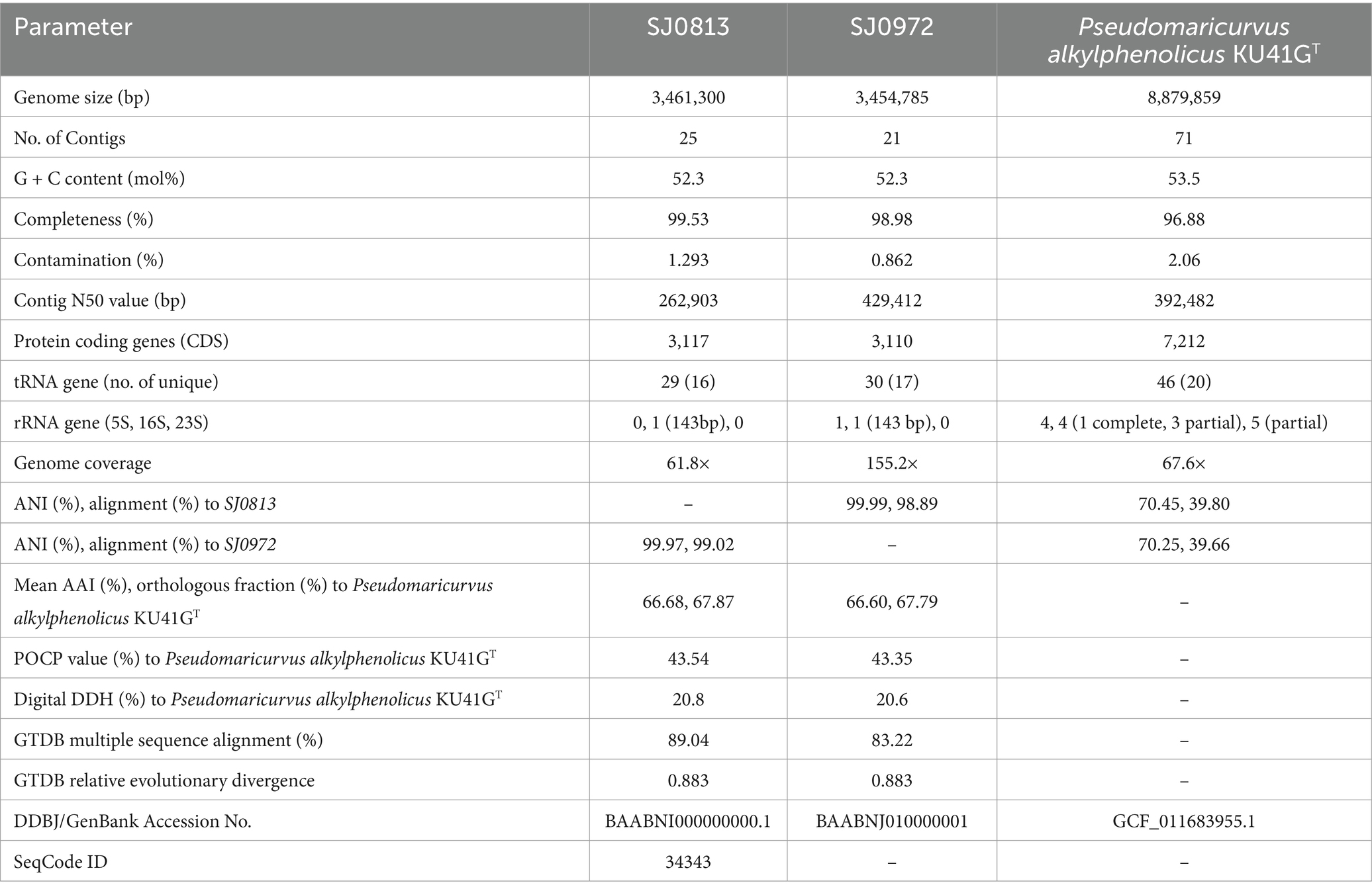
Table 1. Statistics and quality of metagenome-assembled genomes compared to the closest known bacterial species.
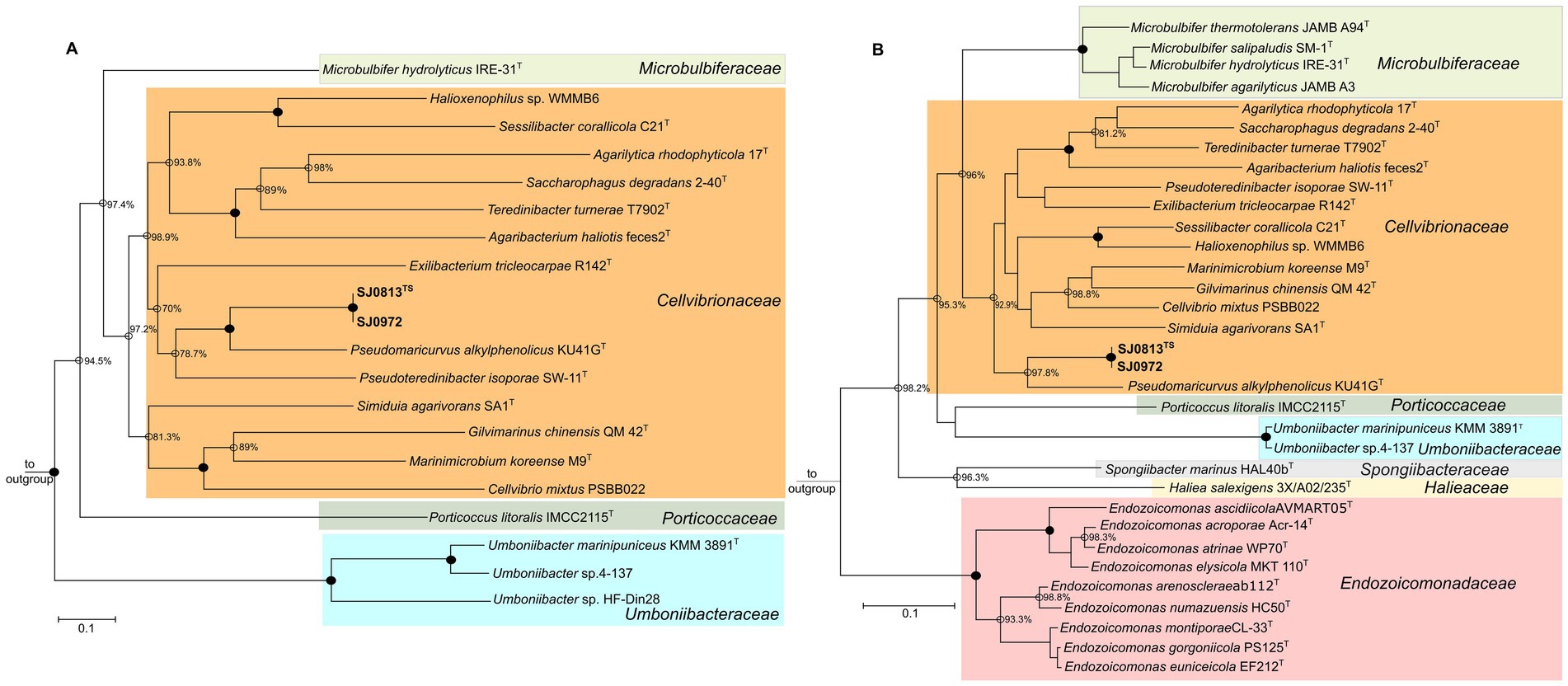
Figure 1. Maximum-likelihood trees of selected Gammaproteobacteria. (A) Core-genome tree of on 20 core genes belonging to 22 genomes of the type species for each genus including two addition reference genomes of Umboniibacteraceae of the Cellvibrionales order. The phylogenome tree is rooted to Haliea salexigens and Microbulbifer hydrolyticus as outgroup. (B) A phylogenetic tree of RpoB sequences to resolve Umboniibacter phylogeny. RpoB protein sequence of SJ0972 is partial (1,106 aa), while Umboniibacter sp.4–137 lacked the RpoB protein sequence. The RpoB tree is rooted to Escherichia coli ATCC 11775T (RefSeq WP_000263098.1) as outgroup and is not shown. For both trees, the scale bar indicates 0.1 nucleotide or amino acid substitution per site. Family lineages are color coded. Filled nodes denote 100% bootstrap (transfer bootstrap expectation) values and open nodes are bootstrap values of at least 70%. NCBI/DDBJ RefSeq or GenBank accession numbers are provided in Supplementary Table S1.
3.2 Description of characteristic respiratory, flagella and metabolic proteins
We annotated the CDS of the novel MAGs against the KEGG orthology (KO). Similar proportion of CDS of both MAGs matched a KO protein (Figure 2A) and the major subsystems are proteins related to genetic information processing and signaling and cellular processes (Figure 2B). The complete KEGG modules and KOs of both MAGs are listed in Supplementary Table S4. The presence of the citrate cycle pathway and the dissimilatory nitrate reduction to ammonia may be indicative of both aerobic and anerobic respiratory mechanisms. Other features include ubiG encoding for a 3-demethylubiquinone-9 3-methyltransferase in the ubiquinone biosynthesis pathway (M00117) may express ubiquinone-9 (Q9) as the predominant lipoquinone. Proteins that encode for chemotaxis (KEGG pathway map02030, 15 proteins) and flagella assembly (KEGG proteins map02040; 38 proteins) suggest motility of planktonic cells. Additionally, both our MAGs had unique protein sequence of choline dehydrogenase, which is identical to each other but with only 64.47% amino acid identity to a Glucose-Methanol-Choline (GMC) family oxidoreductase belonging to an uncultured Colwelliaceae sp. BS250 (GenBank MEW6996986.1). When matched against choline dehydrogenase sequence of P. alkylphenolicus KU41GT (GenBank WP_166985401.1), the identity was only 43.28%. Moreover, both genomes have complete carbohydrate metabolic pathways, glycolysis (M0002), pyruvate oxidation (M00307), pentose phosphate pathway (M00007), and PRPP biosynthesis (M00004).
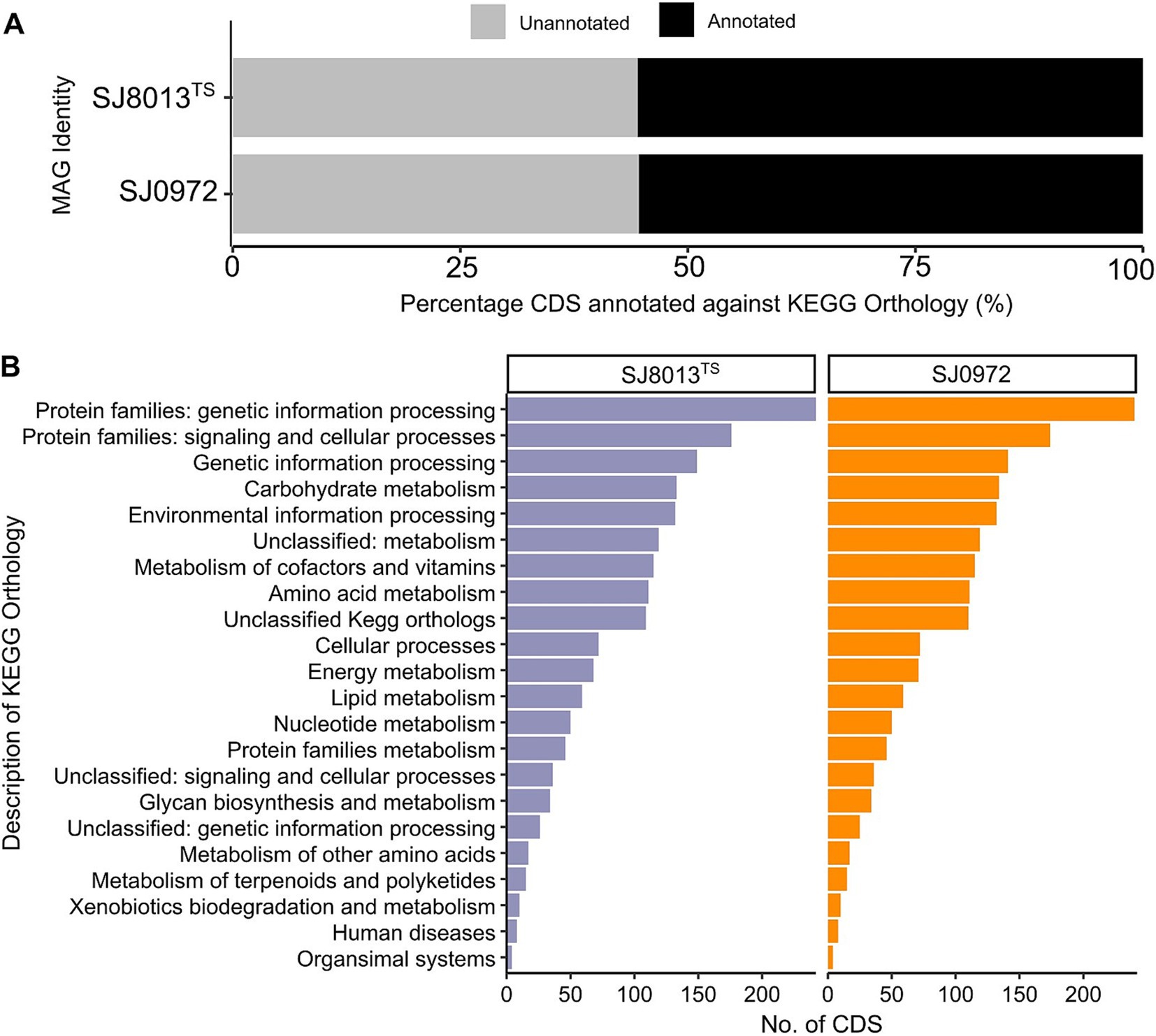
Figure 2. Annotation of coding sequence (CDS) using KEGG Orthology (KO) for both the MAGs (SJ0813 and SJ0972). (A) Percentage of CDS annotated and unannotated by KO. (B) Number of CDS assigned to major subsystems of KEGG modules, including proteins related to genetic information processing and signaling and cellular processes, genetic information processing, carbohydrate metabolism, environmental information processing, metabolism of cofactors and vitamins, amino acid metabolism, cellular processes, energy metabolism, lipid metabolism, nucleotide metabolism, protein families metabolism, glycan biosynthesis and metabolism, metabolism of other amino acids, metabolism of terpenoids and polyketides, xenobiotics biodegradation and metabolism, human diseases and organismal systems as well as some unclassified KO.
3.3 Local and global distribution of the novel MAGs
To ascertain the global distribution of the novel MAGs, we ran protein BLAST of the RpoB sequence (SJ0813TS) against the NCBI metagenomic proteins (env_nr) and non-redundant protein sequences (nr) to leverage global sequence archive of the International Nucleotide Sequence Database Collaboration. The RpoB sequence matched 80.66% identity (99% query coverage) to a RpoB of a deep marine sediments metagenome (GenBank accession no. KKN45857.1) and 90.15% identity (99% query coverage) to a RpoB (GenBank accession no. MAZ90271.1) of a MAG (TOBG_SAT-84) sequenced from marine surface water, respectively (Spang et al., 2015; Tully et al., 2018). However, the MAG (TOBG_SAT-84) was too low in quality (draft genome of 1.17 Mbp, 64.32% completeness, 1.15% contamination) to make a further comparisons (Tully et al., 2018). From the BLAST Atlas comparison, the rpoB gene of SJ0813 showed no similarity to that of P. alkylphenolicus (Supplementary Figure S1).
3.4 Diversity of seawater microbiome
The in-situ parameters of seawater reflect in the lagoon and pier are similar at the point of sampling for Temperature (°C), dissolved oxygen (mg/L), conductivity (mS/cm), total dissolved solid (TDS) (mg/L), salinity (ppt) and pH (Table 2). The water temperature is similar to those recorded for St. John’s Island in the Marine Environment Sensing Network’s Ombak database, ranging from 24.2°C to 32.0°C (December 2022 to September 2024 except August 2022 where data was not available). We characterized the microbiomes of seawater of a lagoon (SJ0813) and a pier (SJ0972) and obtained a taxonomic profile using MetaPhlAn 4. A large proportion of the sequencing reads were not assigned to either bacteria or archaea for both the samples, lagoon (88.11%, relative abundance of MetaPhlAn cluster to total MetaPhlAn cluster) and pier (83.58%) sites. At the phylum level, Pseudomonadota (previously known as Proteobacteria) have the highest relative abundance (5.68% at the lagoon and 9.69% at the pier), followed by Firmicutes at the lagoon (2.34%) and Cyanobacteria at the pier (1.94%) (Figure 3A). Within Proteobacteria, Alphaproteobacteria (lagoon = 2.36% and pier = 4.72%) and Gammaproteobacteria (lagoon = 1.34% and pier = 3.36%) are the predominant class in both microbiomes (Figure 3B). Within Gammaproteobacteria, the most abundant Order is Oceanospirillales (lagoon = 0.87% and pier = 1.55%) followed by Cellvibrionales (0.21%) at the lagoon or Alteromonadales (1.11%) at the pier (Figure 3C). Within Cellvibrionales, the family Halieaceae was the most abundant with Cellvibrionaceae ≤ 0.001% as a minor population (Figure 3D). The relative abundance of our novel Cellvibrionaceae MAGs was estimated by mapping the raw reads onto Candidatus Pelagadaptatus aseana. In the lagoon and pier, the relative abundance of Candidatus Pelagadaptus aseana MAGs to total metagenomic reads was 2.72 and 6.25%, respectively.
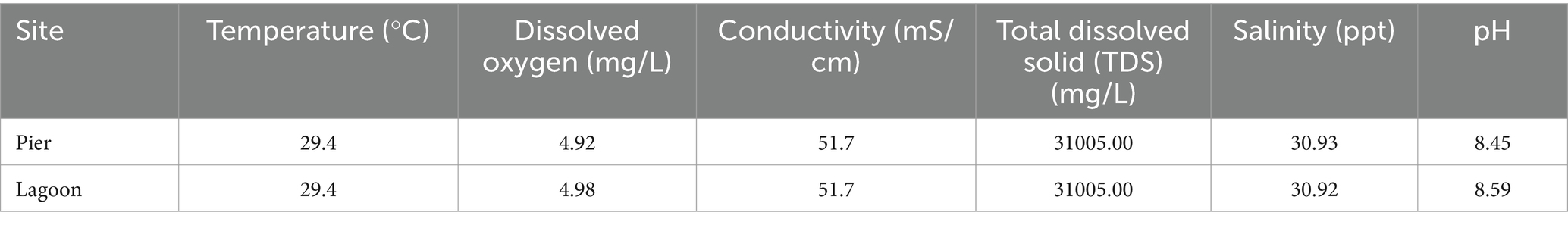
Table 2. In situ physiochemical parameter measurements at the sampling locations.
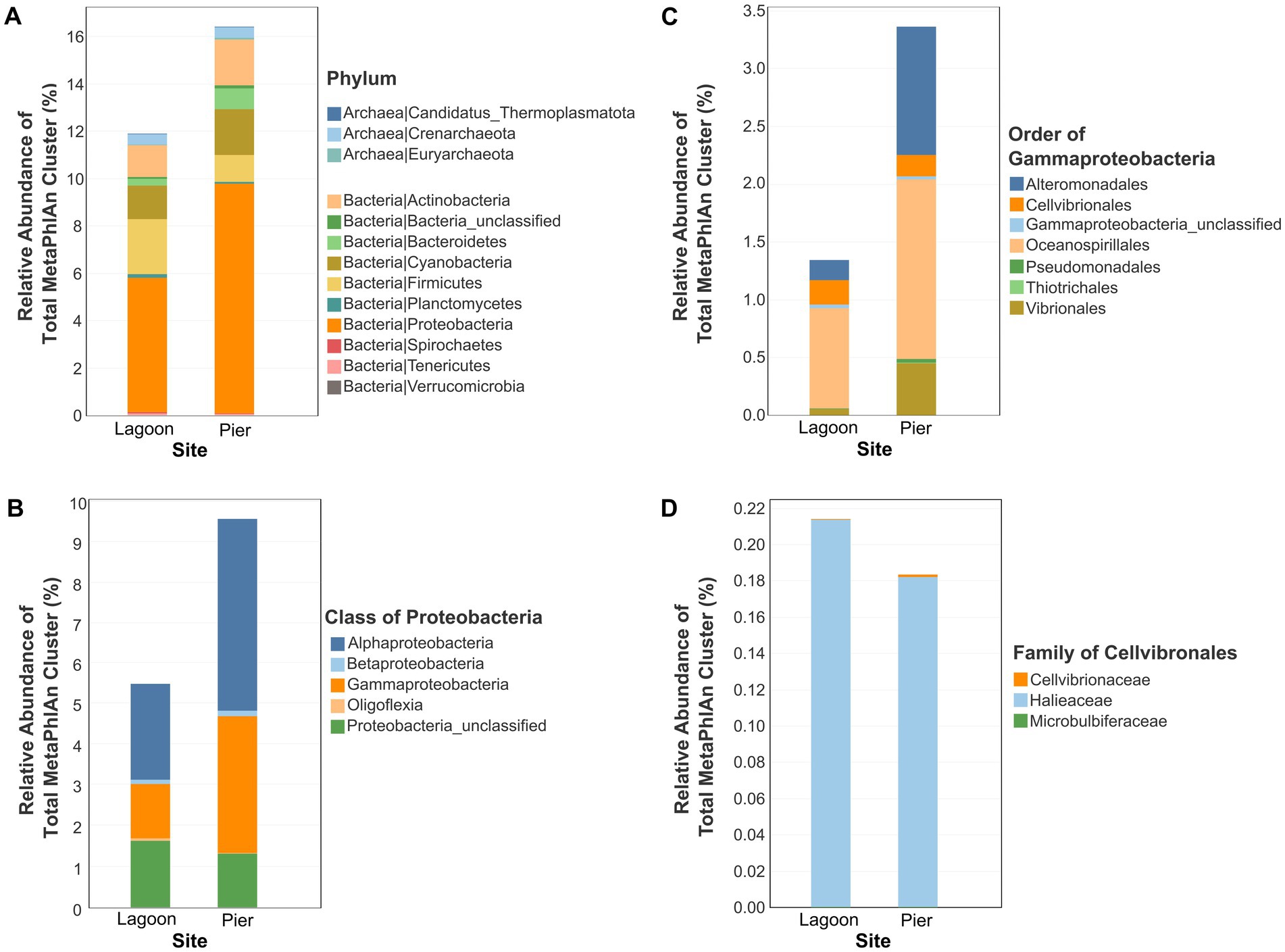
Figure 3. The relative abundance of a MetaPhlAn cluster to total MetaPhlAn cluster for lagoon and pier samples at different taxonomic levels analyzed using MetaPhlAn 4. (A) An overall microbial community at the phylum level for archaea and bacteria. (B) Diversity within the Proteobacteria at the class level. (C) Diversity within Gammaproteobacteria at the order level. (D) Diversity within Cellvibronales at the family level. Barplots depicting taxa with low relative abundance is diminutive to discern.
3.5 The proposed transfer of Umboniibacter from Cellvibrionaceae to a new family Umboniibacteraceae
Interestingly, the maximum-likelihood phylogenetic tree based on core-genome showed that the formerly described Umboniibacter marinipuniceus KMM 3891T and undescribed Umboniibacter spp. strains 4–137 and HF-Din28 fell outside of Cellvibrionaceae clade and are more closely related and yet distinct from Porticoccus litoralis IMCC 2115T (Porticoccaceae), Haliea salexigens 3X/A02/235T (Halieaceae), and Spongiibacter marinus HAL40bT (Spongiibacteraceae) (Figure 1A). We constructed the maximum-likelihood tree using RpoB and included nine Endozoicomonas spp. and four Microbulbifer species, alongside the taxa from the core-genome tree. Like the core-genome phylogenetic tree, U. marinipuniceus KMM 3891T and Umboniibacter sp. strain 4–137 formed a distinct clade from outside of the families Cellvibrionaceae and Porticoccaceae, Halieaceae, Spongiibacteraceae and Endozoicomonadaceae (Figure 1B). The AAI (57.2, 46.31% orthologous fraction) and ANI 65.46, 20.24% coverage of P. litoralis IMCC 2115T and U. marinipuniceus KMM 3891T support their delineation belonging to different genera and species, respectively. Based on the phylogeny of U. marinipuniceus KMM 3891T and its close relatives, Umboniibacter spp. strains 4–137 and HF-Din28, which form a monophyletic clade and are distinct from other members in the Order Cellvibrionales. We proposed transfer of Umboniibacter from the family Cellvibrionaceae to a newly proposed family, Umboniibacteraceae based on the International Code of Nomenclature of Prokaryotes (ICNP).
4 Discussion
The tropical marine microbiota diversity of South-East Asia is still largely uncharacterized and previous studies of Singapore waters using the 16S rRNA gene lacked phylogenetic resolution (Teramoto et al., 2013; Wijaya et al., 2023; Lau et al., 2013; Chenard et al., 2019; Deignan et al., 2023; Soh et al., 2024). In this study, we used metagenomics to describe the genome and phylogeny of an uncultured marine bacteria, which was found in two separate locations surrounding an island at South of Singapore, the St. John’s Island. This island is part of a marine protection zone that has a rich diversity of corals, which serve as spawning sites and nursery grounds for marine life and thus are economically important for the fishery industry (NG and CHOU, 2017).
The MAGs are phylogenetically novel based on the different phylogenomic measurements, genome assembly quality criteria, and predicted biochemical pathways. POCP and ANI comparison of our MAGs to P. alkylphenolicus was well below the 50% genus boundary (Qin et al., 2014) and 95% bacterial species ANI threshold (Goris et al., 2007; Parks et al., 2020). A majority of species within Cellvibrionales order have ubiquinone-8 (Q8) as the major respiratory lipoquinone (Spring et al., 2015) in contrast with our novel MAGs, which may express ubiquinone-9 (Q9) as the predominant lipoquinone. This lipoquinone is synthesized by 3-demethylubiquinone-9 3-methyltransferase (ubiG, K00568) in the ubiquinone biosynthesis pathway (M00117; 9 proteins), which includes the three ubiquinone flavin-dependent monooxygenases, UbiH, UbiI, and UbiF (Kazemzadeh et al., 2023; Kanehisa et al., 2023). Notably, Pseudoteredinibacter isoporae, which is basal to the clade grouping P. alkylphenolicus and our novel MAG (Figure 1) also predominantly express Q9 lipoquinone (Spring et al., 2015; Chen et al., 2011). It should be noted that the respiratory quinones are inferred from in silico annotations and would still require biochemical analysis to validate the phenotype (Vieira et al., 2021). The carbohydrate and nitrogen metabolism based on complete KEGG modules suggest a chemoheterotrophic lifestyle capable of utilizing glucose through the citrate cycle but may be capable of anaerobic respiration through dissimilatory nitrate reduction (Einsle et al., 1999). The gene, nitrate reductase (NapAB) and nitrite reductase (NirBD) which is essential for the reduction are present in both our MAGs and P. alkylphenolicus. Incidentally, while P. alkylphenolicus KU41GT also possess the genes for NapAB and NirBD, it did not exhibit nitrate reduction to nitrite when biochemically tested (Seo et al., 2015). Hence, the biochemical pathways described here remain as predictions until there is empirical evidence. Dissimilatory nitrate reduction is a major pathway in coastal zones for the preservation of nitrogen in the form of ammonium that is source of nitrogen for sea plants and is an advantageous trait during low oxygen levels as a result of eutrophication, which are common around coastal waters and off-shore fisheries (Gillis et al., 2017). The discovery of a choline dehydrogenase with low sequence identity to existing GMC oxidoreductase of an uncultured Colwelliaceae marine bacterium expands the sequence diversity of the GMC superfamily, which is found in all kingdoms of life (Ivanova et al., 2004; Aleksenko et al., 2020). Choline dehydrogenase catalyzes the oxidation of choline to glycine-betaine, which has several biochemical functions, including being an osmolyte for marine bacterial adaptation to salinity in seawater and a precursor in the methionine cycle (Mausz et al., 2022; Gadda and McAllister-Wilkins, 2003; Yancey, 2005). Taken together, we tentatively proposed a novel bacterial genus and species called Candidatus Pelagadaptatus aseana gen. nov., sp. nov. (Pe.la.ga.dap.ta’tus Gr. neut. n. pelagos, the sea; adaptatus L. adjectival noun masc., adapted to; Pelagadaptatus: a microbe of the sea; a.se.a’na N. L. n. asean, derived from the acronym for Association of Southeast Asian Nations) and SJ0813TS as the sequence type for future SeqCode consideration.
While the ecological importance of Candidatus Pelagadaptatus aseana is unclear until an isolate is obtained, several bacterial species of Cellvibrionaceae such as Sessilibacter corallicola and Pseudoteredinibacter isoporae are known for their close association with corals (Chen et al., 2011; Liu et al., 2022) and other species are known for their metabolic traits of nonylphenol-degrading and xylene-utilizing (Iwaki et al., 2012; Iwaki et al., 2018). From the BLAST (blastp) searches of the RpoB, Candidatus Pelagadaptatus aseana is currently only detected in marine waters of the Southern Island, Singapore. Further sampling and metagenomic sequencing of coastal waters of Southeast Asia could verify its distribution in the region.
Cellvibrionaceae detected in MetaPhlAn was ≤0.001% (relative abundance of MetaPhlAn cluster to total MetaPhlAn cluster); while the relative abundance of our Candidatus Pelagadaptatus aseana MAGs to total metagenomic reads was 6.25 and 2.72%. Our finding suggests that MetaPhlAn 4, a reference-based method for taxonomic profiling grossly underestimates the abundance of Cellvibrionaceae taxa in the marine environment, despite incorporating MAGs of marine communities from best available sources (e.g., CAMI 2 Taxonomic profiling challenge) (Blanco-Míguez et al., 2023; Nickols et al., 2024). This highlights the need to expand the metagenome database for more environments including the marine metagenome of Southeast Asia. Hence, the approach used to estimate relative abundance of our novel MAG is to map metagenomic reads onto the MAG genome (Duru et al., 2024). The short 16S rRNA gene limited the formal proposal of our novel MAG, Candidatus Pelagadaptatus aseana SJ0813TS under SeqCode (Hedlund et al., 2022). The 16S rRNA gene is often underrepresented in MAGs, being present in only 7 to 17.3% of them (Hiseni et al., 2022; Parks et al., 2017). Short-read assemblers do not reliably reconstruct the 16S rRNA gene owing to multiple 16S rRNA gene copies and sequence heterogeneity that hinders its correct binning with the rest of the genome (Yuan et al., 2015; Song et al., 2022). Previous studies have shown the RpoB protein as a better marker at resolving the relationships in the Gammaproteobacteria taxa than the 16S rRNA gene, hence we constructed a maximum-likelihood tree using this protein (Spring et al., 2015; Case et al., 2007).
The initial phylogeny of U. marinipuniceus KMM 3891T, an isolate from a marine mollusc, was performed using the 16S rRNA gene and subsequent proposed inclusion into Cellvibrionaceae was not re-evaluated using core-genome phylogenetic analysis (Spring et al., 2015; Romanenko et al., 2010). Based on the 16S rRNA gene phylogenetic tree by Romanenko et al. (2010), U. marinipuniceus phylogeny was unresolved with less than 90% boot-strap confidence to Endozoicomonas elysicola MKT 110T (family Endozoicomonadaceae, order Oceanospirillales), Microbulbifer spp. (family Microbulbiferaceae, order Cellvibrionales) and Simiduia agarivorans (family Cellvibrionaceae, order Cellvibrionales) (Romanenko et al., 2010). This is congruent in subsequent 16S rRNA gene phylogenetic trees of the two other Umboniibacter species, Umboniibacter caenipelagi and Umboniibacter roseus (Sung et al., 2015; Park et al., 2017). As there is no RpoB or genome assemblies available for U. caenipelagi and U. roseus, we included the sequences of two currently undescribed Umboniibacter spp. strains 4–137 and HF-Din28 represented in GTDB release 220, to show that the Umboniibacter spp. are indeed phylogenetically distinct from members of the Cellvibrionaceae and Porticoccaceae (Sung et al., 2015, Park et al., 2017). Physiologically, U. marinipuniceus is the only species known within Cellvibrionales to have Q7 as the major ubiquinone, which suggests physiological difference to the other species of Cellvibrionales (Spring et al., 2015). Unfortunately, the major respiratory lipoquinone of P. litoralis IMCC 2115T has not been characterized (Oh et al., 2010). The congruence between the core-genome, RpoB phylogenetic trees and the monophyletic relationship of the three Umboniibacter species with the 16S rRNA gene supports the proposed transfer of Umboniibacter from the family Cellvibrionaceae to a newly proposed family, Umboniibacteraceae based on the International Code of Nomenclature of Prokaryotes (ICNP). Currently, Umboniibacter spp. have been isolated from benthic environments off Korea and Japan (Sung et al., 2015; Park et al., 2017; Romanenko et al., 2010). Its distribution may span to Mexico as Umboniibacter spp. 16S amplicons have been sequenced from oysters there (Trabal Fernández et al., 2014). The clarification of its phylogenetic placement would help future culture-dependent and -independent research in delineating the phylogeny of Umboniibacter species.
5 Description of Umboniibacteraceae fam. nov.
Umboniibacteraceae (Um.bo.ni.i.bac’ter.a.ce’ae N.L. n. Umbonium scientific name of a genus of marine mollusc; N.L. masc. Referring to the isolation of the first strains from the sand snail U. costatum; −aceae ending to denote a family; N.L. fem. pl. n. Umboniibacteraceae the Umboniibacter family).
This family is circumscribed based on core-genome phylogeny, the family belongs to the order Cellvibrionales, and the class Gammaproteobacteria. At present, the family only contains three species, Umboniibacter marinipuniceus, Umboniibacter roseus and Umboniibacter caenipelagi, all of which were isolated from marine environments such as benthic mollusks and sediments. A distinctive feature of this family is the utilization of Q7 as the major respiratory quinone. Phosphatidylethanolamine, and phosphatidylglycerol are dominant polar lipids within these species. DNA G + C content ranges from 48.9 mol% to 51.7 mol%. Major cellular fatty acid patterns are C13:0, C16:0 or C17:1ω8c. The type genus of the family is Umboniibacter.
Data availability statement
Sequencing raw reads and the two assembled MAGs were submitted to DNA Data Bank of Japan (DDBJ) using the DDBJ Fast Annotation and Submission Tool v. 1.6.0 (2022.3.24) under the BioProject number PRJDB17097 (Tanizawa et al., 2022). The raw reads for which the MAGs were assembled have the BioSample numbers SAMD00732050 (SJ0813; sample ID: WLagoon_S196) and SAMD00732052 (SJ0972; sample ID: WPier_S191). MAGs have the following DDBJ/GenBank accession numbers =BAABNI000000000.1 (BAABNI010000001-BAABNI010000025) for SJ0813 contigs (Biosample SAMD00738017) and =BAABNJ010000001 (BAABNJ010000001 to BAABNJ010000021) for SJ0972 contigs (Biosample SAMD00738018). The proposed nomenclature has been registered onto the SeqCode registry https://disc-genomics.uibk.ac.at/seqcode/registers/r:7vdf8rw3.
Author contributions
JH: Writing – review & editing, Data curation, Investigation, Methodology, Project administration, Software, Visualization. XK: Formal analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. DK: Data curation, Formal analysis, Writing – review & editing. AL: Formal analysis, Methodology, Software, Writing – original draft, Writing – review & editing. DH: Data curation, Software, Writing – review & editing, Formal analysis. MH: Data curation, Software, Writing – review & editing. RW: Data curation, Software, Writing – review & editing, Resources. RC: Funding acquisition, Writing – review & editing, Resources. YB: Conceptualization, Funding acquisition, Supervision, Writing – review & editing, Resources.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research is supported by the partners under Singapore’s One Health National Strategic Action Plan: Ministry of Health, National Environment Agency, National Parks Board, PUB, Singapore’s National Water Agency, and Singapore Food Agency, and administered by the National Centre for Infectious Diseases (OHARP Grant No: OHARP-002) awarded to YF Boucher. This work was also supported by A*STAR SFS IAF-PP grant (A20H7a0152) awarded to Rebecca J. Case.
Acknowledgments
The Singapore Centre for Environmental Life Sciences Engineering (SCELSE) is funded by the Ministry of Education, Singapore, the National Research Foundation of Singapore, Nanyang Technological University Singapore (NTU) and National University of Singapore (NUS). We would like to thank the National University of Singapore Information Technology for their support in management of the high-performance computing, the St. John’s Island National Marine Laboratory for providing the facility necessary for conducting the research; the Laboratory is a National Research Infrastructure under the National Research Foundation Singapore. We would also like to acknowledge the Marine Environmental Sensing Network (MESN) for providing the St. John’s Island water temperature datasets. MESN is funded by the National Research Foundation of Singapore. We would like to extend our gratitude to Mr. Sebastian Jie Hong Yeo for his immense help in the fieldwork.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1437936/full#supplementary-material
Footnotes
1. ^https://github.com/biobakery/kneaddata
2. ^https://github.com/donovan-h-parks/CompareM
3. ^https://github.com/SilentGene/Bio-py/tree/master/POCP-calculator
References
Aleksenko, V. A., Anand, D., Remeeva, A., Nazarenko, V. V., Gordeliy, V., Jaeger, K.-E., et al. (2020). Phylogeny and structure of fatty acid Photodecarboxylases and glucose-methanol-choline oxidoreductases. Catalysts 10:1072. doi: 10.3390/catal10091072
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Altschul, S. F., Wootton, J. C., Gertz, E. M., Agarwala, R., Morgulis, A., Schaffer, A. A., et al. (2005). Protein database searches using compositionally adjusted substitution matrices. FEBS J. 272, 5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x
Blanco-Míguez, A., Beghini, F., Cumbo, F., Mciver, L. J., Thompson, K. N., Zolfo, M., et al. (2023). Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 41, 1633–1644. doi: 10.1038/s41587-023-01688-w
Case, R. J., Boucher, Y., Dahllöf, I., Holmström, C., Doolittle, W. F., and Kjelleberg, S. (2007). Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Appl. Environ. Microbiol. 73, 278–288. doi: 10.1128/AEM.01177-06
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2022). Gtdb-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316. doi: 10.1093/bioinformatics/btac672
Chen, M. H., Sheu, S. Y., Arun, A. B., Young, C. C., Chen, C. A., Wang, J. T., et al. (2011). Pseudoteredinibacter isoporae gen. Nov., sp. nov., a marine bacterium isolated from the reef-building coral Isopora palifera. Int. J. Syst. Evol. Microbiol. 61, 1887–1893. doi: 10.1099/ijs.0.024190-0
Chenard, C., Wijaya, W., Vaulot, D., Lopes Dos Santos, A., Martin, P., Kaur, A., et al. (2019). Temporal and spatial dynamics of Bacteria, Archaea and protists in equatorial coastal waters. Sci. Rep. 9:16390. doi: 10.1038/s41598-019-52648-x
Cho, J. C., and Giovannoni, S. J. (2004). Cultivation and growth characteristics of a diverse group of oligotrophic marine Gammaproteobacteria. Appl. Environ. Microbiol. 70, 432–440. doi: 10.1128/AEM.70.1.432-440.2004
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of Samtools and Bcftools. GigaScience 10, giab008. doi: 10.1093/gigascience/giab008
Deignan, L. K., Pwa, K. H., Loh, A. A. R., Rice, S. A., and Mcdougald, D. (2023). The microbiomes of two Singaporean corals show site-specific differentiation and variability that correlates with the seasonal monsoons. Coral Reefs. 42, 1–15. doi: 10.1007/s00338-023-02376-6
Duru, I. C., Lecomte, A., Shishido, T. K., Laine, P., Suppula, J., Paulin, L., et al. (2024). Metagenome-assembled microbial genomes from Parkinson’s disease fecal samples. Sci. Rep. 14:18906. doi: 10.1038/s41598-024-69742-4
Edgar, R. C. (2004). Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Einsle, O., Messerschmidt, A., Stach, P., Bourenkov, G. P., Bartunik, H. D., Huber, R., et al. (1999). Structure of cytochrome c nitrite reductase. Nature 400, 476–480. doi: 10.1038/22802
Gadda, G., and Mcallister-Wilkins, E. E. (2003). Cloning, expression, and purification of choline dehydrogenase from the moderate halophile Halomonas elongata. Appl. Environ. Microbiol. 69, 2126–2132. doi: 10.1128/AEM.69.4.2126-2132.2003
Galaxy, C. (2022). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 50, W345–W351. doi: 10.1093/nar/gkac247
Gillis, L. G., Paul, M., and Bouma, T. (2017). Ammonium uptake rates in a seagrass bed under combined waves and currents. Front. Mar. Sci. 4, 1–7. doi: 10.3389/fmars.2017.00207
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Grant, J. R., Enns, E., Marinier, E., Mandal, A., Herman, E. K., Chen, C.-Y., et al. (2023). Proksee: in-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 51, W484–W492. doi: 10.1093/nar/gkad326
Hedlund, B. P., Chuvochina, M., Hugenholtz, P., Konstantinidis, K. T., Murray, A. E., Palmer, M., et al. (2022). SeqCode: a nomenclatural code for prokaryotes described from sequence data. Nat. Microbiol. 7, 1702–1708. doi: 10.1038/s41564-022-01214-9
Hiseni, P., Snipen, L., Wilson, R. C., Furu, K., and Rudi, K. (2022). Questioning the quality of 16S rRNA gene sequences derived from human gut metagenome-assembled genomes. Front. Microbiol. 12:822301. doi: 10.3389/fmicb.2021.822301
Hitch, T. C. A., Riedel, T., Oren, A., Overmann, J., Lawley, T. D., and Clavel, T. (2021). Automated analysis of genomic sequences facilitates high-throughput and comprehensive description of bacteria. ISME Commun. 1:16. doi: 10.1038/s43705-021-00017-z
Ivanova, E. P., Flavier, S., and Christen, R. (2004). Phylogenetic relationships among marine Alteromonas-like proteobacteria: emended description of the family Alteromonadaceae and proposal of Pseudoalteromonadaceae fam. Nov., Colwelliaceae fam. Nov., Shewanellaceae fam. Nov., Moritellaceae fam. Nov., Ferrimonadaceae fam. Nov., Idiomarinaceae fam. Nov. and Psychromonadaceae fam. Nov. Int. J. Syst. Evol. Microbiol. 54, 1773–1788. doi: 10.1099/ijs.0.02997-0
Iwaki, H., Takada, K., and Hasegawa, Y. (2012). Maricurvus nonylphenolicus gen. Nov., sp. nov., a nonylphenol-degrading bacterium isolated from seawater. FEMS Microbiol. Lett. 327, 142–147. doi: 10.1111/j.1574-6968.2011.02471.x
Iwaki, H., Yamamoto, T., and Hasegawa, Y. (2018). Isolation of marine xylene-utilizing bacteria and characterization of Halioxenophilus aromaticivorans gen. Nov., sp. nov. and its xylene degradation gene cluster. FEMS Microbiol. Lett. 365, fny042. doi: 10.1093/femsle/fny042
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M., and Ishiguro-Watanabe, M. (2023). Kegg for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. doi: 10.1093/nar/gkac963
Kanehisa, M., Sato, Y., and Morishima, K. (2016). Blastkoala and Ghostkoala: Kegg tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). Mafft: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Katoh, K., and Standley, D. M. (2013). Mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kazemzadeh, K., Pelosi, L., Chenal, C., Chobert, S. C., Hajj Chehade, M., Jullien, M., et al. (2023). Diversification of ubiquinone biosynthesis via gene duplications, transfers, losses, and parallel evolution, 40:msad219. doi: 10.1093/molbev/msad219
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). Raxml-Ng: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). Mega X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lau, S. C., Zhang, R., Brodie, E. L., Piceno, Y. M., Andersen, G., and Liu, W. T. (2013). Biogeography of bacterioplankton in the tropical seawaters of Singapore. FEMS Microbiol. Ecol. 84, 259–269. doi: 10.1111/1574-6941.12057
Liao, H., Lin, X., Li, Y., Qu, M., and Tian, Y. (2020). Reclassification of the taxonomic framework of orders Cellvibrionales, Oceanospirillales, Pseudomonadales, and Alteromonadales in Class Gammaproteobacteria through Phylogenomic Tree Analysis, mSystems. 5:10.1128/msystems.00543-20. doi: 10.1128/msystems.00543-20
Liu, J., Li, J., Luo, J., Li, Y., Yang, Z., Huang, C., et al. (2022). Sessilibacter corallicola gen. Nov., sp. nov., a sessile bacterium isolated from coral Porites lutea. Int. J. Syst. Evol. Microbiol. 72, 005401. doi: 10.1099/ijsem.0.005401
Liu, S., and Liu, Z. (2020). Distinct capabilities of different Gammaproteobacterial strains on utilizing small peptides in seawater. Sci. Rep. 10:464. doi: 10.1038/s41598-019-57189-x
Lønborg, C., Müller, M., Butler, E. C., Jiang, S., Ooi, S. K., Trinh, D. H., et al. (2021). Nutrient cycling in tropical and temperate coastal waters: is latitude making a difference? Estuarine Coastal Shelf Sci. 262:107571. doi: 10.1016/j.ecss.2021.107571
Mausz, M. A., Airs, R. L., Dixon, J. L., Widdicombe, C. E., Tarran, G. A., Polimene, L., et al. (2022). Microbial uptake dynamics of choline and glycine betaine in coastal seawater. Limnol. Oceanogr. 67, 1052–1064. doi: 10.1002/lno.12056
Meier-Kolthoff, J. P., Carbasse, J. S., Peinado-Olarte, R. L., and Göker, M. (2021). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50, D801–D807. doi: 10.1093/nar/gkab902
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster Rna homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Ng, C.-S. L., and Chou, L.-M. (2017). Coral reef restoration in Singapore — Past, Present and Future. Sustain. Matters. 3–23. doi: 10.1142/9789813230620_0001
Nickols, W. A., Mciver, L. J., Walsh, A., Zhang, Y., Nearing, J. T., Asnicar, F., et al. (2024). Evaluating metagenomic analyses for undercharacterized environments: what's needed to light up the microbial dark matter?. bioRxiv [Preprint]. 9:2024.11.08.622677 doi: 10.1101/2024.11.08.622677
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). Metaspades: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Oh, H. M., Kim, H., Kim, K. M., Min, G. S., and Cho, J. C. (2010). Porticoccus litoralis gen. Nov., sp. nov., a Gammaproteobacterium isolated from the Yellow Sea. Int. J. Syst. Evol. Microbiol. 60, 727–732. doi: 10.1099/ijs.0.013938-0
Park, S., Choi, S. J., Park, J. M., and Yoon, J. H. (2017). Umboniibacter caenipelagi sp. nov., isolated from a tidal flat. Int. J. Syst. Evol. Microbiol. 67, 2959–2963. doi: 10.1099/ijsem.0.002056
Parks, D. H., Chuvochina, M., Chaumeil, P.-A., Rinke, C., Mussig, A. J., and Hugenholtz, P. (2020). A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086. doi: 10.1038/s41587-020-0501-8
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parks, D. H., Rinke, C., Chuvochina, M., Chaumeil, P.-A., Woodcroft, B. J., Evans, P. N., et al. (2017). Recovery of nearly 8, 000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542. doi: 10.1038/s41564-017-0012-7
Parte, A. C., Sarda Carbasse, J., Meier-Kolthoff, J. P., Reimer, L. C., and Goker, M. (2020). List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70, 5607–5612. doi: 10.1099/ijsem.0.004332
Pinhassi, J., Farnelid, H., Garcia, S. M., Teira, E., Galand, P. E., Obernosterer, I., et al. (2022). Functional responses of key marine bacteria to environmental change - toward genetic counselling for coastal waters. Front. Microbiol. 13:869093. doi: 10.3389/fmicb.2022.869093
Qin, Q. L., Xie, B. B., Zhang, X. Y., Chen, X. L., Zhou, B. C., Zhou, J., et al. (2014). A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 196, 2210–2215. doi: 10.1128/JB.01688-14
Reimer, L. C., Sarda Carbasse, J., Koblitz, J., Ebeling, C., Podstawka, A., and Overmann, J. (2022). Bac dive in 2022: the knowledge base for standardized bacterial and archaeal data. Nucleic Acids Res. 50, D741–D746. doi: 10.1093/nar/gkab961
Richter, M., Rossello-Mora, R., Oliver Glockner, F., and Peplies, J. (2016). Jspeciesws: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Romanenko, L. A., Tanaka, N., and Frolova, G. M. (2010). Umboniibacter marinipuniceus gen. Nov., sp. nov., a marine Gammaproteobacterium isolated from the mollusc Umbonium costatum from the sea of Japan. Int. J. Syst. Evol. Microbiol. 60, 603–609. doi: 10.1099/ijs.0.010728-0
Sayers, E. W., Bolton, E. E., Brister, J. R., Canese, K., Chan, J., Comeau, D. C., et al. (2021). Database resources of the national center for biotechnology information. Nucleic Acids Res. 50, D20–D26. doi: 10.1093/nar/gkab1112
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seo, H.-S., Yang, S.-H., Oh, J. H., Lee, J.-H., and Kwon, K. K. (2015). Pseudomaricurvus alcaniphilus sp. nov., a marine bacterium isolated from tidal flat sediment and emended descriptions of the genus Pseudomaricurvus, Pseudomaricurvus alkylphenolicusIwaki et al. 2014 and Maricurvus nonylphenolicus Iwaki et al. 2012. Int. J. Syst. Evol. Microbiol. 65, 3591–3596. doi: 10.1099/ijsem.0.000463
Soh, M., Tay, Y. C., Lee, C. S., Low, A., Orban, L., Jafaar, Z., et al. (2024). The gut microbiota of tropical marine fish is largely uncultured and distinct from surrounding water microbiota. NPJ Biofilms Microbiomes 10:11. doi: 10.1038/s41522-024-00484-x
Song, W., Zhang, S., and Thomas, T. (2022). Markermag: linking metagenome-assembled genomes (Mags) with 16S rRNA marker genes using paired-end short reads. Bioinformatics 38, 3684–3688. doi: 10.1093/bioinformatics/btac398
Spang, A., Saw, J. H., Jørgensen, S. L., Zaremba-Niedzwiedzka, K., Martijn, J., Lind, A. E., et al. (2015). Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521, 173–179. doi: 10.1038/nature14447
Spring, S., Scheuner, C., Göker, M., and Klenk, H.-P. (2015). A taxonomic framework for emerging groups of ecologically important marine gammaproteobacteria based on the reconstruction of evolutionary relationships using genome-scale data. Front. Microbiol. 6:281. doi: 10.3389/fmicb.2015.00281
Sung, H. R., Kim, M., and Shin, K. S. (2015). Umboniibacter roseus sp. nov., isolated from coastal seawater. Int. J. Syst. Evol. Microbiol. 65, 4221–4226. doi: 10.1099/ijsem.0.000564
Tanizawa, Y., Fujisawa, T., Kodama, Y., Kosuge, T., Mashima, J., Tanjo, T., et al. (2022). DNA data bank of Japan (DDBJ) update report 2022. Nucleic Acids Res. 51, D101– D105. doi: 10.1093/nar/gkac1083
Teramoto, M., Queck, S. Y., and Ohnishi, K. (2013). Specialized Hydrocarbonoclastic Bacteria prevailing in seawater around a port in the strait of Malacca. PLoS One 8:e66594. doi: 10.1371/journal.pone.0066594
Tonkin-Hill, G., Macalasdair, N., Ruis, C., Weimann, A., Horesh, G., Lees, J. A., et al. (2020). Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 21:180. doi: 10.1186/s13059-020-02090-4
Trabal Fernández, N., Mazón-Suástegui, J. M., Vázquez-Juárez, R., Ascencio-Valle, F., and Romero, J. (2014). Changes in the composition and diversity of the bacterial microbiota associated with oysters (Crassostrea corteziensis, Crassostrea gigas and Crassostrea sikamea) during commercial production. FEMS Microbiol. Ecol. 88, 69–83. doi: 10.1111/1574-6941.12270
Trüper, H. G. (1999). How to name a prokaryote?: etymological considerations, proposals and practical advice in prokaryote nomenclature. FEMS Microbiol. Rev. 23, 231–249. doi: 10.1111/j.1574-6976.1999.tb00397.x
Tully, B. J., Graham, E. D., and Heidelberg, J. F. (2018). The reconstruction of 2, 631 draft metagenome-assembled genomes from the global oceans. Sci Data 5:170203. doi: 10.1038/sdata.2017.203
Uritskiy, G. V., Diruggiero, J., and Taylor, J. (2018). Metawrap-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6:158. doi: 10.1186/s40168-018-0541-1
Vieira, S., Huber, K. J., Neumann-Schaal, M., Geppert, A., Luckner, M., Wanner, G., et al. (2021). Usitatibacter rugosus gen. Nov., sp. nov. and Usitatibacter palustris sp. nov., novel members of Usitatibacteraceae fam. Nov. within the order Nitrosomonadales isolated from soil. Int. J. Syst. Evol. Microbiol. 71, 004631. doi: 10.1099/ijsem.0.004631
Wijaya, W., Suhaimi, Z., Chua, C. X., Sunil, R. S., Kolundzija, S., Rohaizat, A. M. B., et al. (2023). Frequent pulse disturbances shape resistance and resilience in tropical marine microbial communities. ISME Commun 3:55. doi: 10.1038/s43705-023-00260-6
Williams, K. P., Gillespie, J. J., Sobral, B. W., Nordberg, E. K., Snyder, E. E., Shallom, J. M., et al. (2010). Phylogeny of gammaproteobacteria. J. Bacteriol. 192, 2305–2314. doi: 10.1128/JB.01480-09
Yancey, P. H. (2005). Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 208, 2819–2830. doi: 10.1242/jeb.01730
Yarza, P., Yilmaz, P., Pruesse, E., Glöckner, F. O., Ludwig, W., Schleifer, K.-H., et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12, 635–645. doi: 10.1038/nrmicro3330
Keywords: marine Gammaproteobacteria, a novel bacterial species, dissimilatory nitrate reduction, choline dehydrogenase, Singapore coast
Citation: Ho JY, Koh XQ, Kang DY, Low A, Hu D, Haryono MAS, Williams RBH, Case RJ and Boucher YF (2025) Discovery of a phylogenetically novel tropical marine Gammaproteobacteria elucidated from assembled genomes and the proposed transfer of the genus Umboniibacter from the family Cellvibrionaceae to Umboniibacteraceae fam. nov. Front. Microbiol. 16:1437936. doi: 10.3389/fmicb.2025.1437936
Edited by:
Maher Gtari, Carthage University, TunisiaReviewed by:
Judith Maria Braganca, Birla Institute of Technology and Science, IndiaXingkun Jin, Hohai University, China
Copyright © 2025 Ho, Koh, Kang, Low, Hu, Haryono, Williams, Case and Boucher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yann Felix Boucher, ZXBoeWJAbnVzLmVkdS5zZw==
†These authors have contributed equally to this work and share first authorship