 Maria Isabel Nogueira Di Azevedo1*
Maria Isabel Nogueira Di Azevedo1* Frederico Kremer2
Frederico Kremer2 Camila Ezepha1
Camila Ezepha1 João Pedro Gomes Greco2
João Pedro Gomes Greco2 Isadora Cosenza Vieira da Silva2
Isadora Cosenza Vieira da Silva2 Pascale Bourhy3
Pascale Bourhy3 Walter Lilenbaum1
Walter Lilenbaum1- 1Laboratory of Veterinary Bacteriology, Biomedical Institute, Fluminense Federal University, Niterói, Rio de Janeiro, Brazil
- 2Laboratory of Bioinformatics - Omixlab, Technological Development Center, Federal University of Pelotas, Capão do Leão, Rio Grande do Sul, Brazil
- 3Institut Pasteur, Biology of Spirochetes Unit, National Reference Center for Leptospirosis, Paris, France
Bovine genital leptospirosis (BGL) is a silent and chronic reproductive syndrome associated with reproductive failures that result in animal suffering and substantial financial losses for farmers. Important aspects of the interactions between the host and the pathogen during chronic leptospirosis have been well described in the kidney, but little is known about the genital infection mechanisms. The present study sheds light on the pathophysiology of BGL based on comparative genomic analysis of renal versus genital isolates of Leptospira santarosai genomes, an endemic species on Latin America. A significant number of genes were exclusive of the genital strains, with emphasis on genes associated with cell wall/membrane/envelope biogenesis, mobilome: prophages and transposons, and signal transduction mechanisms. Overall, these gene clusters play crucial roles in bacterial colonization and evasion of the immune response, which can reflect leptospiral tissue tropism to the genital niche. We provide new insights into the pathophysiology of an important and neglected syndrome in bovine, helping to elucidate the evolution of adaptation of leptospires in the genital tract of cows.
1 Introduction
Leptospira sp. is a highly diverse spirochete bacteria that comprises several taxa based on a genetic classification subdivided into 64 genomic species, including saprophytic, intermediate and pathogenic species. Only eight pathogenic species are responsible for human and animal infections: L. interrogans, L. kirschneri, L. noguchi, L. santarosai, L. mayottensis, L. borgpetersenii, L. alexanderi, and L. weilli (Vincent et al., 2019). This group includes the etiologic agents of the globally neglected zoonotic disease leptospirosis (Picardeau, 2020). Serological classification is important for epidemiological purposes. It defines serovars based on the lipopolysaccharides of the outer membrane, and, according to its homogeneity, groups the serovars into serogroups. Using this approach, approximately 24 serogroups containing over 250 serovars have thus far been recognized (Hagedoorn et al., 2024). Notably, host-serogroup adaptability has been demonstrated and associated with asymptomatic and or chronic/silent disease (Xu et al., 2016). The two classifications are independent and their integrative use is recommended.
In bovine, leptospirosis is one of the major causes of reproductive failures and is associated with abortions, estrus repetition, stillbirths, the birth of weak calves, a decrease in growth parameters, and a drop in milk production (Fávero et al., 2017), all factors that result in substantial financial losses for farmers. This silent and chronic reproductive syndrome is formally called bovine genital leptospirosis (BGL). Currently, strains from the Sejroe serogroup are recognized as the main agents of BGL due to the syndrome’s adaptability to the bovine host (Loureiro and Lilenbaum, 2020).
Within the Sejroe serogroup, the main serovars are Hardjoprajitno (L. interrogans) and Hardjobovis (L. borgpetersenii) yet the serovar Guaricura (L. santarosai) has also been frequently identified in cattle, especially in Latin America (Kremer et al., 2015; Loureiro et al., 2016; Aymée et al., 2022b; Chinchilla et al., 2023) and its significance as a BGL agent has grown.
Important aspects of the interactions between the host and the pathogen during chronic leptospirosis have been well described in the kidney (Monahan et al., 2009). For years, studies have focused on urine/renal tissues, with kidneys being considered an immune-privileged and primary site of infection that facilitates persistent colonization by leptospires. Leptospiral infection in the reproductive tract has traditionally been considered secondary to renal pathogenesis. However, recent studies involving reproductive tract samples have demonstrated the genital tract as an important extra-renal site of infection by Leptospira spp. The spirochaete has been identified on the molecular level in the uterus (Cabral Pires et al., 2018; Di Azevedo et al., 2020; Aymée et al., 2021, 2022a, 2023; Borges et al., 2024), vaginal fluid (Loureiro et al., 2016; Aymée et al., 2022b), follicular fluid (Di Azevedo et al., 2021; Dos Santos Pereira et al., 2022), and cervicovaginal mucus (Aymée et al., 2021, 2022a, 2023; Borges et al., 2024). Most of the cows studied had a history of reproductive problems, including abortion, chronic subfertility, and embryonic mortality.
Although Leptospira spp. infections in the urinary and genital tract have a distinct pathophysiology with different clinical manifestations, the dynamics of genital infection, adaptability to the uterine environment, and mechanisms of pathogenesis are not fully understood. The use of Whole Genome Sequencing (WGS) has helped to further elucidate the evolution of the virulence of the pathogenic species of Leptospira associated with the urinary tract in several hosts (Xu et al., 2016; Santos et al., 2018; Koizumi et al., 2020; Ramli et al., 2021; de Oliveira et al., 2022). However, studies applying WGS to genital Leptospira strains have never been conducted. Recently, our group provided the first draft genomes of three L. santarosai strains isolated from the vaginal fluid of Brazilian cows with reproductive disorders (Di Azevedo et al., 2023). As urinary L. santarosai genomes are also available (Kremer et al., 2015; Di Azevedo et al., 2023), the present study aims to shed light on the pathophysiology of BGL using comparative genomic analyses of renal versus genital isolates.
2 Materials and methods
2.1 Samples, culturing, and initial genetic characterization of strains
The Leptospira santarosai strains used in the present study included urinary (2014_U76, 2013_U160, 2013_U164, and 2013_U233) and genital isolates (2013_VF52, 2014_VF66, and VF237), all associated with bovine genital leptospirosis. They were characterized using the serology and secY gene sequencing conducted by our group in previous studies on cows from the State of Rio de Janeiro, Brazil (Hamond et al., 2015; Loureiro et al., 2016; Aymée et al., 2022b). Information regarding the strains included in the present study is summarized in Table 1.
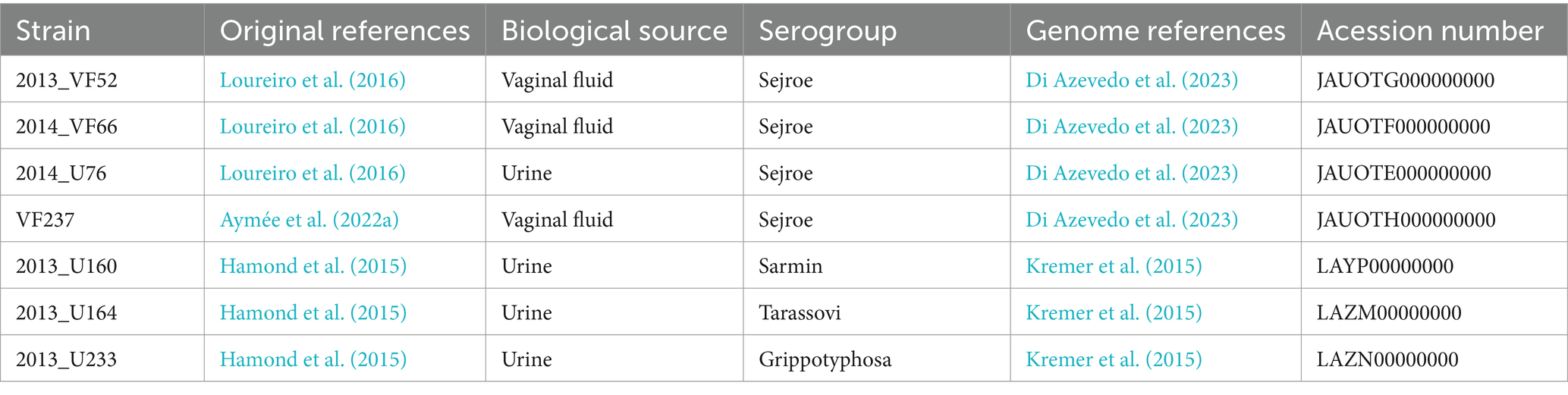
Table 1. Summarized information regarding Leptospira santarosai strains isolated from bovine included in the present study.
Briefly, after collection, the samples were immediately seeded into tubes with EMJH medium supplemented with the antimicrobial cocktail STAFF (Loureiro et al., 2015). The tubes were kept at room temperature and transported to the laboratory. All cultures were maintained at 29°C and examined weekly for 16 weeks using dark field microscopy. As soon as pure cultures were obtained, with few passages (average 4), they were frozen at liquid nitrogen (−196°C) in the Collection of Bacterial Cultures of Veterinary Interest (Federal Fluminense University, Brazil). Cultures were thawed exclusively further for the analyses of the present study.
The isolates were tested by Microscopic Agglutination Test (MAT) against a panel of rabbit antisera of 32 reference serovars representing 24 serogroups [provided by KIT (formerly the Royal Tropical Institute), Amsterdam], as recommended (Haake and Levett, 2015). DNA was extracted using the Wizard SV Genomic DNA Purification System® (Promega, Madison, USA) according to the manufacturer’s instructions. Genotyping was performed using the secY locus as described by Ahmed et al. (2006). PCR products were directly sequenced in both directions using the Big Dye Terminator 3.1 Cycle Sequencing Kit (Life Technologies, Foster City, USA) on an ABI 3730XL Genetic Analyzer (Life Technologies, Carlsbad, CA, USA) on the PDTIS/Fiocruz Bioinformatics Platform RPT01A. After Pairwise/Blast/NCBI comparisons were performed, the sequences were taxonomically identified as L. santarosai (>98%).
2.2 Genome sequencing, assembly, and annotation
Whole genome sequences from the strains 2013_U160, 2013_U164, and 2013_U233 were previously sequenced by Kremer et al. (2015), as were those from the strains 2014_U76, 2013_VF52, 2014_VF66, and VF237, which were sequenced by Di Azevedo et al. (2023) (Table 1). Briefly, based on the high-quality genomic DNA obtained from 5 × 1010 to 2 × 1011 pelleted L. santarosai cultures, WGS was performed on Illumina’s (San Diego, California, USA) MiSeq Sequencing System applying a 150 × 2-bp run and starting with a genomic library prepared using Illumina DNA Prep (previously known as Nextera DNA Flex). The quality of the reads was evaluated using fastQC, and the genomes were assembled de novo using the Unicycler 0.4.8 (Wick et al., 2017) workflow under default parameters. The annotation was added using the NCBI Prokaryotic Genome Annotation Pipeline. Additionally, for comparative analyses involving the pangenome of L. santarosai, a reference sequence of human origin, the L. santarosai serovar Shermani strain LT 821, was also included, as it is a pioneer strain already well characterized, being useful for visualizing the distribution and location of genes and for comparative analyses purposes (Chou et al., 2014).
As L. santarosai str. 2013_FV52 is a pioneer strain in several other studies (Loureiro et al., 2016; Martins et al., 2022; Barbosa et al., 2023), it was chosen for the circular representation of the genome, with predicted Coding DNA Sequences (CDSs). For this purpose, its draft genome (GenBank: JAUOTE010000000) was aligned and ordered based on the genome of the L. santarosai serovar Shermani str. LT 821 (GenBank: NZ_CP006694.1, NZ_CP006695.1) using CONTIGuator (Galardini et al., 2011), and the circular genome plot was produced using the program DNAPlotter (Carver et al., 2009) from the Artemis package (Berriman and Rutherford, 2003).
2.3 Comparative genomic analysis
Genomes were obtained from NCBI using the NCBI Datasets service.1 They were retrieved in GenBank File Format (GBFF) and converted to GFF3 files using BioPerl.2 The GFF3 files for all genomes were then analyzed using Roary (Page et al., 2015) under default settings to ascertain the pangenome and core-genome estimations. Roary operates by first clustering genes from all input genomes using a sequence identity threshold, typically set at 95%, to group similar genes together. This clustering is crucial for distinguishing between core and accessory genes. The software then generates a matrix of gene presence and absence, which is used to define the core genome as the set of genes present in all strains (Page et al., 2015).
A maximum-likelihood (ML) tree based on L. santarosai genomes was constructed using the Hasegawa-Kishino-Yano 85 model (HKY85) on PhyML 3.0 software (Guindon et al., 2010). Based on the Roary outputs, the core genomes of the urinary and genital strains were defined as the genes present in all members of each group. Functional annotations of the pangenome and each Cluster of Orthologous Groups (COG) category (Galperin et al., 2021) were performed using COGClassifier.3 A Fisher exact test followed by a Bonferroni correction of the p-values were performed to identify the differences in the distribution of COG categories in the genes conserved in each group of strains. All data analysis was performed using Python and the libraries BioPython,4 Pandas,5 SciPy,6 and Statsmodels.7 An additional functional annotation was performed using the InterProScan webserver.8
Additionally, to provide more informative results, we compared the identities of the genomes and genes identified in the strains of the present study with other L. santarosai genomes from other hosts. For this purpose, a BLAST search was performed using the NCBI-BLAST+ package to identify genes homologous to the urinary and genital strain core gene sets in the dataset of L. santarosai genomes obtained from the NCBI RefSeq. Homologous genes were identified based on the following criteria: an e-value threshold of 1e-10 (Cai et al., 2009), a minimum identity of 70%, and a minimum query coverage of 70%, cutoff points already widely used in the search for homology (Janaki et al., 2021; Martins et al., 2023). The percentages of genes in the genital and urinary gene sets were computed for each genome.
3 Results
3.1 Descriptive genomic analysis
Based on the characteristics of the reference genome L. santarosai serovar Shermani strain LT 821, it was possible to infer that the genome of L. santarosai serovar Sejroe strain 2013_VF52 consists of two chromosomes, namely, the larger chromosome I (3.48 Mb, GC% 42.05) and the smaller chromosome II (286.99Kb, GC% 42.30) (Figure 1). It is possible to observe that chromosome 1 presents a higher number of genes conserved in the genital strains, with emphasis on a region between 17.000 and 34.000 bp, containing genes related to cell wall/membrane/envelope biogenesis. This structure was very similar to the L. santarosai strains obtained from vaginal fluid (2014_VF66 and VF237). Further details of the Leptospira santarosai genomes from bovine released to NCBI can be found in Kremer et al. (2015) and Di Azevedo et al. (2023).
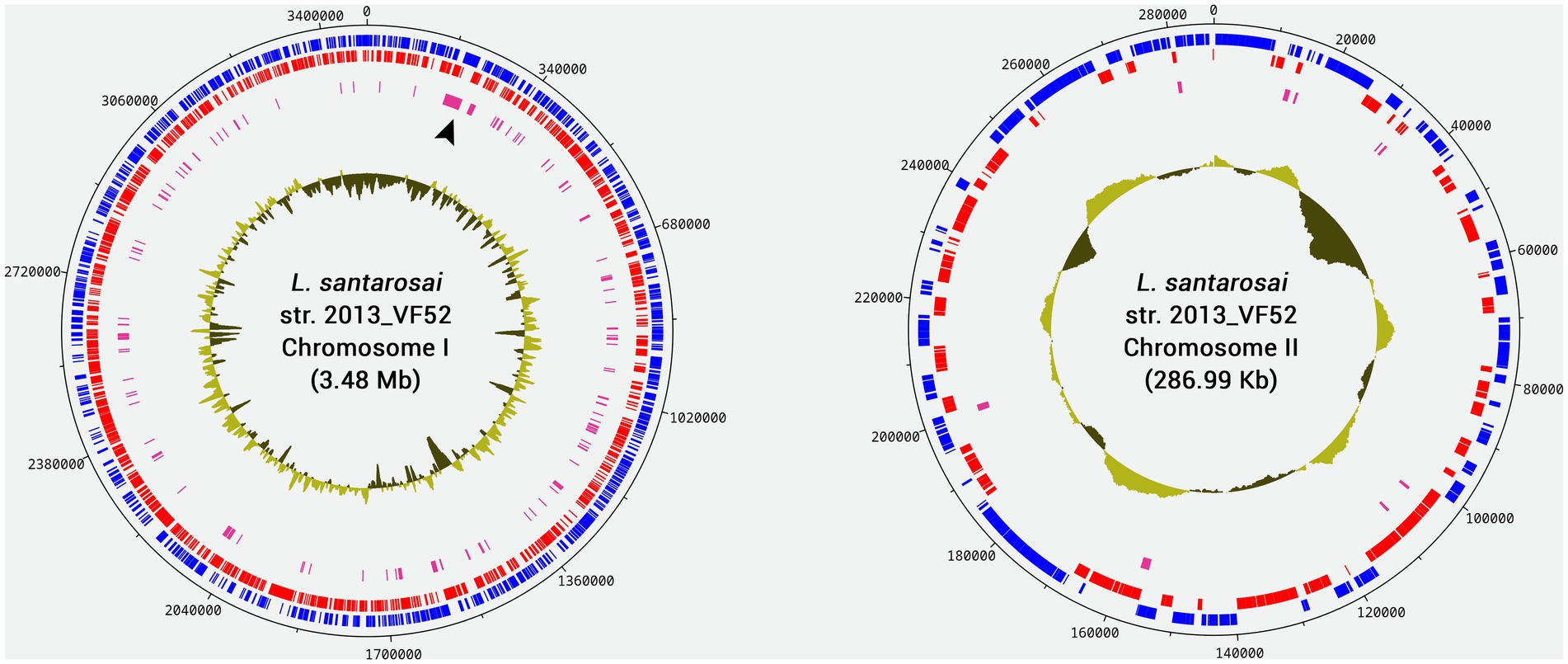
Figure 1. Circular genome diagram of the Leptospira santarosai strain 2013_VF52 genome generated based on chromosomes I and II of L. santarosai serovar Shermani str. LT 821 using CONTIGuator and DNAPlotter. The blue marks indicate the Coding DNA Sequences (CDSs) present in the forward strand, while the red marks indicate CDSs in the reverse strand. Pink marks indicate genes that are conserved exclusively in genital strains. The location indicated with the black arrowhead in chromosome I represents an extensive region highly conserved only in the genital strains. Deviations in the CG content are plotted inside each chromosome.
3.2 Comparative genomic analysis
An ML-phylogenetic tree created using the WGS of the L. santarosai strains revealed the polyphyletic origin of the urinary strains, while genital strains formed a monophyletic group separate from urinary strains (Figure 2). Importantly, strains VF237 and 2013_VF52 are identical.
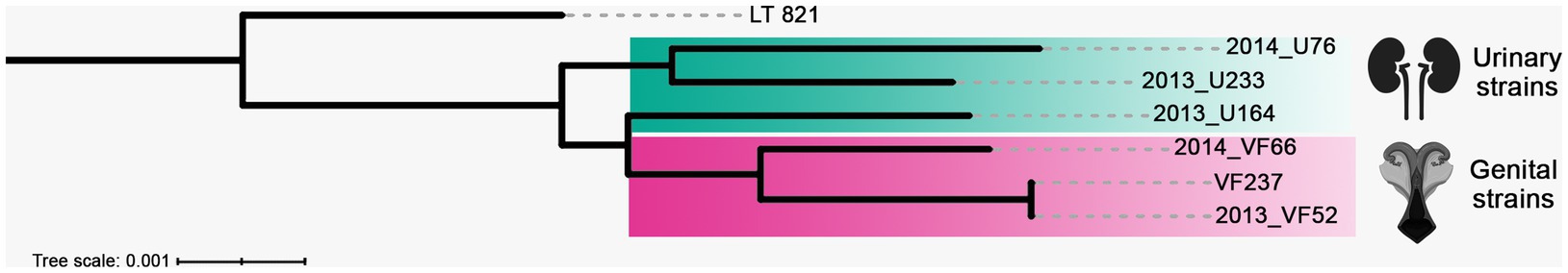
Figure 2. A maximum-likelihood (ML)-phylogenetic tree inferred from whole genome sequences of L. santarosai Sejroe serogroup strains of urinary (yellow rectangle) and genital (pink rectangle) origin. The Leptospira santarosai serovar Shermani strain LT 821 is the outgroup taxa.
Based on the Roary analysis, the pangenome of the selected strains was estimated in 4,969 genes, while the core genomes of the urinary and genital strains were estimated, respectively, in 2,912 and 3,206 genes. The differences between the core genomes are displayed in Figure 3, indicating that the 377 genes conserved in the genital strains were not conserved in the urinary strains, while 83 genes were conserved in the urinary strains but not in the genital strains. These subsets of genes were named the “urinary strain-specific” (USS) core genome and the “genital strain-specific” (GSS) core genome.
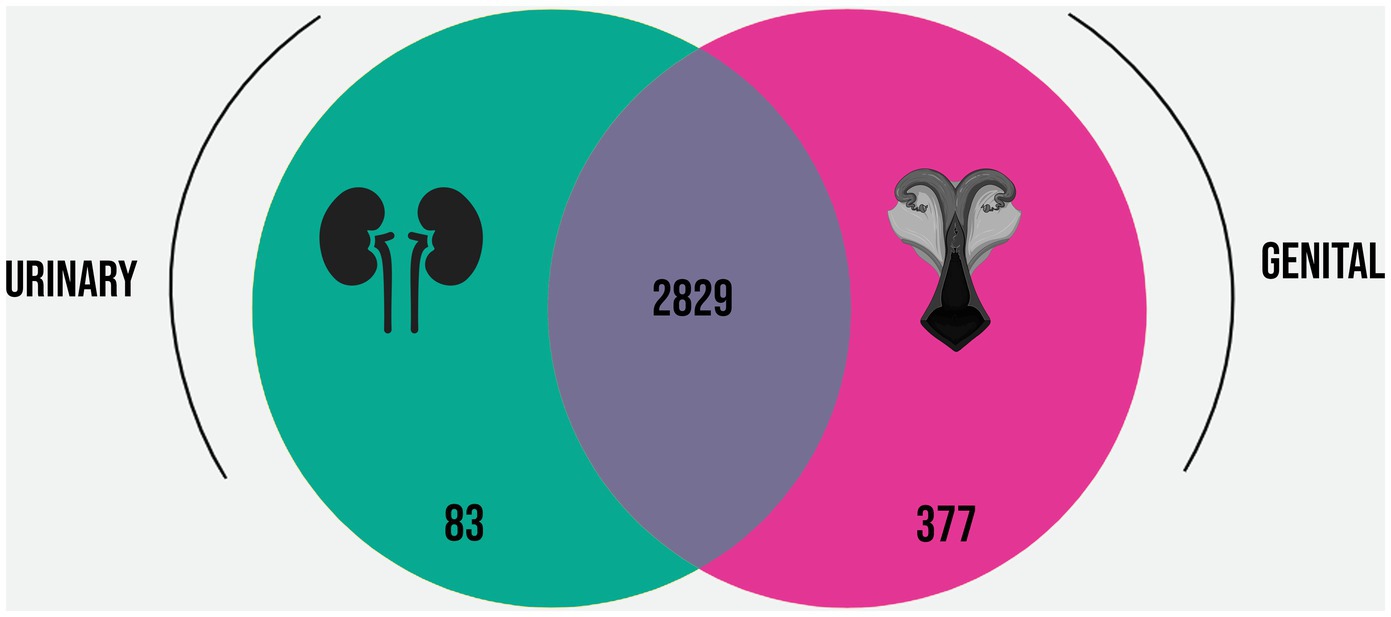
Figure 3. Venn diagram representing the overlap and differences in the core genomes of the L. santarosai strains derived from urinary (yellow) and genital (pink) samples.
Based on the functional annotation of the pangenome, the encoded proteins were mapped to COGs categories. The distribution of these categories in the pangenome of L. santarosai and the USS and GSS core genomes are illustrated in Figure 4. The functional enrichment analysis of the USS core genome as compared with the GSS core genome is shown in Figure 5. These results identify three COG categories as enriched in the GSS core genome (cell wall/membrane/envelope biogenesis, mobilome: prophages and transposons, and signal transduction mechanisms), with significant differences observed as regards the USS core genome (Supplementary material 1), indicating that proteins related to these biological processes are more present and/or conserved on genital strains. No enriched COG category was observed in the distribution of COG categories in the USS core genome analysis. The genes conserved in the GSS core genome and absent in the USS core genome relating to the aforementioned biological processes are presented in Supplementary material 2. Moreover, 24, 18, and 13 COGs were found to be related to cell wall/membrane/envelope biogenesis, mobilome, and signal transduction mechanisms, respectively, totaling 55 exclusive genital strain protein domains (Figure 5 and Supplementary material 2).
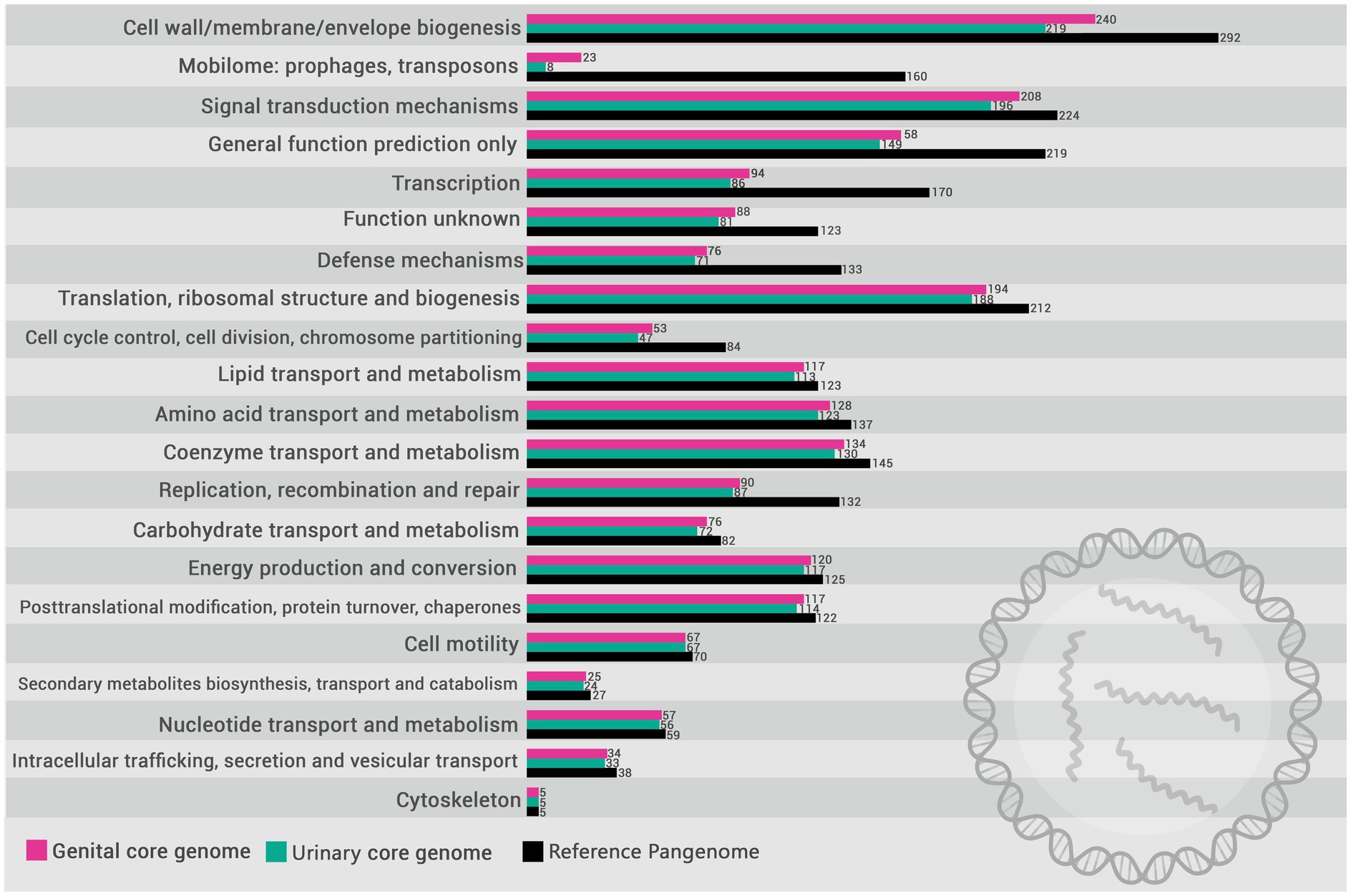
Figure 4. Distribution of COG (Cluster of Orthologous Groups) categories in the pangenome of L. santarosai (gray) and the core genomes of urinary (yellow) and genital strains (pink).
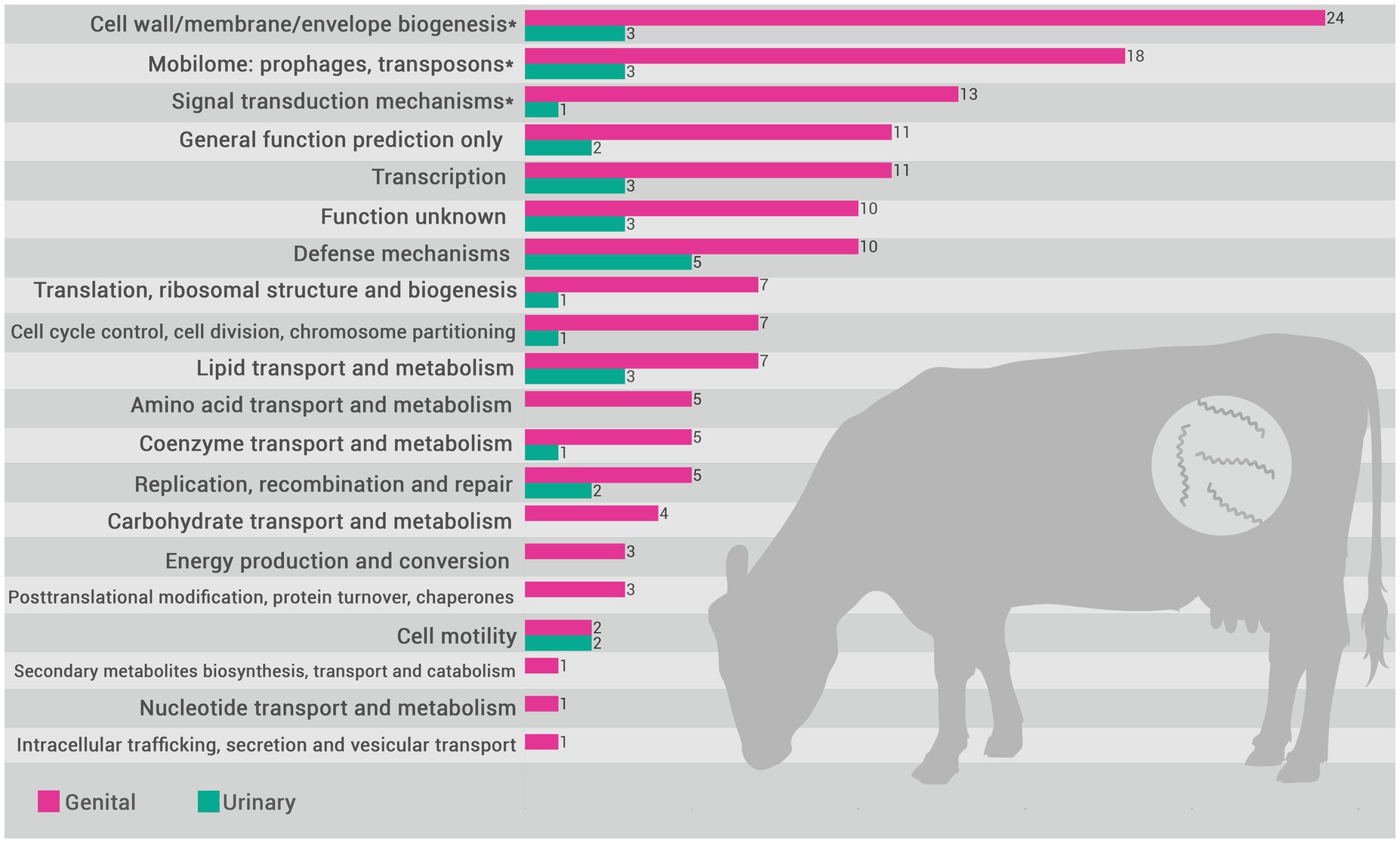
Figure 5. Distribution of COG (Cluster of Orthologous Groups) categories in the L. santarosai urinary strain specific (USS) (yellow) and genital strain specific (GSS) (pink) core genomes. Categories indicated with an asterisk (*) are those where a significant difference between categories (USS vs. GSS) was observed (p < 0.05).
Regarding the percentages of genes in the USS and GSS gene sets as compared with the L. santarosai genomes obtained from the NCBI RefSeq, a maximum identity of 95% was found for urinary gene orthologues (L. santarosai strain M72/6–6 and serovar Grippotyphosa) and 71.9% for genital gene orthologues (L. santarosai strain M4/98 from serovar Guaricura) (Supplementary material 3).
4 Discussion
Although BGL is a well-recognized and characterized syndrome in many respects, the mechanisms involved in its pathophysiology in the genital tract and the adaptability profile of Sejroe strains to this site remain enigmatic. In this context, the results of the present study deliver new and important insights based on comparative genomics analyses of genital versus urinary strains. Notwithstanding that the strains are from the same host species (bovine) and geographic location (southeastern Brazil), phylogenetic analysis based on L. santarosai WGS revealed a monophyletic group including only genital strains (Figure 2) separate from urinary strains, suggesting important differences in the genome based on the biological origin of the leptospires. These evident genomic differences between genital and urinary strains may have important practical applications, such as optimizing diagnosis and developing an effective vaccine for BGL, based on the exploration of exclusive and conserved regions of genital strains. Importantly, it has been demonstrated that vaccination with bacterins cannot prevent colonization in the genital tract, and infected animals keep shedding leptospires (Martins et al., 2022), which can be explained by this high difference between urinary and genital strains, added to the peculiarities of the humoral response triggered in these two distinct sites.
Of note, a specific cluster including the strains 2013_VF52, 2013_VF66, and VF237 was observed in secY gene analysis (Aymée et al., 2022b; Aymée et al., 2022a), confirming the good taxonomic resolution of this genetic marker and its proper application for molecular epidemiological purposes, which is particularly useful for uncultured clinical samples when WGS cannot be performed. The strains 2013_VF52 and 2013_VF66 were first identified and characterized in asymptomatic cattle (Loureiro et al., 2016), suggesting the traditional “silent disease” associated with these strains. Importantly, it was demonstrated in vivo that the strains VF237 and 2013_VF52, which were demonstrated to be identical in the present study, determine chronic genital leptospirosis in the hamster model (Barbosa et al., 2023). Moreover, based on secY gene analysis, all strains were closely genetically associated with another strain from the Sejroe serogroup serovar Guaricura (UT27), which was isolated from a cow presenting embryonic loss, estrus repetition, and subfertility (Aymée et al., 2022b). This high genetic identity between strains is relevant in several scientific and practical perspectives, including: (i) the existence of a potential common source of infection; (ii) clonal expansion within bovine population; (iii) presence of exclusive characteristics that enhance its ability to cause chronic disease; (iv) supports the development of targeted strategies, such as vaccines; and (v) provides insights into the genetic stability of the strains and helps identify genetic variants linked to virulence or antimicrobial resistance.
Analysis of the urinary and genital L. santarosai core genomes revealed the presence of 377 genes exclusive of genital strains, a greater number than found exclusively in urinary strains (Figure 3). This may indicate the complexity of the genetic mechanisms involved in the colonization, maintenance, and survival of pathogenic leptospires from the Sejroe serogroup over long periods in the reproductive tract of cows. Additionally, when compared with another strain of bovine L. santarosai also from the Guaricura serovar but isolated from urine, the percentage of orthologous genital genes was only 71% (Supplementary material 3), indicating differences in gene constitution even within the same serovar.
Investigating the COGs (Figure 5 and Supplementary material 2), we identified that 55 GSS genes were significantly enriched compared with the USS genes, including three metabolic categories: cell wall/membrane/envelope biogenesis (n = 24), mobilome (n = 18), and signal transduction mechanisms (n = 13). Overall, these gene clusters play crucial roles in bacterial colonization and evasion of the immune response, and may contribute significantly to leptospiral tissue tropism and adaptation to the genital niche. It is unsurprising that genes related to cell wall/membrane/envelope biogenesis are significantly more prevalent in genital strains (n = 24 in GSS vs. n = 3 in USS). Cell wall biogenesis is essential for maintaining the structural and functional integrity of bacteria, allowing them to resist adverse environmental conditions, in addition to facilitating the colonization of specific niches in the host (Mayer and Dörr, 2019). Both kidney and genital infections caused by Sejroe serogroup strains are characterized by a host-pathogen biological equilibrium, with leptospires successfully evading the immune response, causing a silent and chronic disease (Loureiro and Lilenbaum, 2020). However, it is recognized that the kidney is an immune-privileged site that facilitates Leptospira colonization, with the absence of “complement one” a main hypothesis for how the leptospires evade the renal immune response (Monahan et al., 2009). Conversely, although the immunopathogenesis of the leptospiral infection in the genital tract remains unclear, it has been demonstrated that bovine cervicovaginal mucus contains high levels of immune-active proteins such as immunoglobulin A (IgA), lactoferrin, and lysozyme, which protect against infection by blocking adhesion and mediating microbial killing (Adnane et al., 2018). Therefore, the mechanisms of adhesion and maintenance of cell wall integrity for long periods in a hostile environment are essential for leptospiral survival. Similarly, COG analysis revealed that cell wall/membrane/envelope biogenesis genes were also differentially expressed in Brucella abortus, a bacterium also associated with reproductive failure in cattle (Sun et al., 2023).
Biofilm supports cell growth and protects cells from a variety of environmental stresses (Flemming et al., 2016), playing a crucial role in the maintenance of chronic bacterial infections (Dsouza et al., 2024). While its formation has been documented in pathogenic Leptospira in various biotic environments (Ristow et al., 2008; Ackermann et al., 2021) and in natural reservoirs (Santos et al., 2021), it is not clear whether it is formed in the bovine genital environment. Nonetheless, the presence of the wca family genes strongly suggests this as it is associated with biofilm formation in other Gram-negative bacteria (Yin et al., 2018; Zheng, 2018; Pal et al., 2019). It was demonstrated in Klebsiella pneumoniae that wcaJ gene encodes a glycosyltransferase that is crucial for the initiation of colanic acid synthesis, a polysaccharide component of the bacterial extracellular matrix. This enzyme facilitates the loading of the first sugar, glucose-1-phosphate, onto the lipid carrier undecaprenyl phosphate, which is a critical step in the biosynthesis of colanic acid (Pal et al., 2019). Although there is no direct evidence of wca genes in Leptospira, other genomic analyses reveal the presence of analogous genes potentially related to biofilm formation, such as capsular polysaccharide biosynthesis genes and extracellular matrix assembly and transport genes (Davignon et al., 2023). Thibeaux and colleagues (Thibeaux et al., 2020) demonstrated that biofilm production in L. interrogans is regulated by intracellular levels of bis-(3′-5′)-cyclic dimeric guanosine monophosphate (c-di-GMP), which underpins the bacterium’s ability to withstand a wide variety of simulated environmental stresses. It is important to note that biofilm formation is associated with a significant increase in antibiotic resistance, which can complicate the treatment of leptospiral infections (Kumar et al., 2016).
Signal transduction in Leptospira also plays an important role in adaptation and immune evasion. It has been showed that genes involved in signal transduction mechanisms are abundant in the accessory parts of the Leptospira pangenome, suggesting their importance in adaptation and survival in different environments (Lata et al., 2022). These mechanisms may help the bacterium respond rapidly to changes in the host environment, facilitating colonization and evasion of the immune system. In this context, chemotaxis is crucial for the survival of bacteria (Yang and Briegel, 2020). As genital strains of Leptospira move toward locations that are considerably more hostile than renal sites, the bacteria must constantly sense its surroundings, respond to different nutrient gradients, and coexist with a greater number of competitors. Consequently, it can be argued that the group of genes associated with signal transduction mechanisms plays a major role in the adaptability of genital strains in the reproductive tract.
The mobilome, defined as all the mobile genetic elements (MGEs) of the microbiome, seems to be another important factor for survival and adaptation of leptospires in the reproductive tract. It has been previously demonstrated that LPS-associated genes is frequent, which can promote the survival of recombinant strains and contribute to antigenic diversity, hindering the host immune response and the development of effective vaccines (Mejía et al., 2022). Moreover, the mobilome influences the composition of microbial communities and the spread of antimicrobial resistance genes and virulence factors via horizontal gene transfer (Carr et al., 2021). Triple the number of genes related to mobilomes, including phages, plasmids, and transposons, was found in genital strains compared with urinary strains (Figure 5). This is unsurprising since the vaginal environment presents greater microbial diversity that facilitates bacterial communication and the exchange of genetic elements. This inter-microbial exchange of genetic elements, including resistance genes, may provide a plausible explanation for the inefficiency of the traditional therapeutic scheme for bovine leptospirosis in eliminating infection in the genital tract. On the other hand, we need to consider that the function of these MGEs in the pathogen has not yet been fully demonstrated, but insights into mobilome-associated genes may inform strategies for limiting genetic exchange that contributes to virulence and resistance.
The present study provides important and impactful initial results based on in silico genomic analyses of L. santarosai. It is important to note that in vivo and in vitro studies are also needed to deeply understand how the host-pathogen interaction may influence gene expression by the strains. CRISPR interference (CRISPRi) technology can be used to silence specific genes in Leptospira, as demonstrated in the investigation of the role of proteins such as LipL32, LipL41, LipL21 and OmpL1 (Fernandes et al., 2023). The creation of knockdown mutants allows the evaluation of the impact of these genes on virulence and host interaction, using animal models such as the hamster to observe changes in pathogenicity and colonization (Fernandes et al., 2022). Characterization of membrane proteins can be performed to understand their role in adhesion to host tissues and complement evasion, as well demonstrated for TolC Efflux Protein (Hota and Kumar, 2024). Binding and inhibition experiments with specific antibodies can help elucidate functions. Experiments that assess the interaction of Leptospira mutants with host molecules, such as extracellular matrix components and component factors, may help identify mechanisms of adhesion and immune evasion, as demonstrate by Zhu et al. (2023).
Future analyses, including that of further genomes from other pathogenic Leptospira species from the Sejroe serogroup, such as L. interrogans and L. borgpetersenii, are necessary to determine whether these results can be replicated in other species with adaptability to the genital site. Transcriptomics studies are also essential to obtain a more comprehensive view of the genes being expressed in the reproductive tract to better elucidate pathogen-host interactions and the mechanisms of pathogenicity by genital strains. Furthermore, targeted and random mutagenesis techniques achieved through the insertion of transposons and genetic complementation of bacteria transformed by synthetic plasmids will enable the exploration of the individual roles of genes. Finally, in vitro tests, such as those exploring interaction with defense cells, behavior when exposed to the complement system, and biofilm formation evaluation, and in vivo studies, such as those analyzing the effects of mutant strains in the animal model, will be of great importance to this area of study and will be pursued in the future by our scientific team.
Based on comparative genomics analyses, we provide new insights into the pathophysiology of bovine genital leptospirosis, elucidating the evolution of virulence in Leptospira santarosai in the genital tract of cows, which, until now, was unknown. Importantly, we find significant differences both at the phylogenetic level and in the functional analysis of the genome when comparing urinary versus genital strains. These latter strains present a greater diversity of genes, with a higher number of genes observed in relation to adhesion, mobile genetic elements, and signal transduction, showing that these processes and factors are key parts of the pathogenesis of BGL. Our results inform the development of a more precise diagnosis of the syndrome, novel therapy proposals, and, importantly, targeted prevention and control strategies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repositories and accession number(s) can be found in the article and in Supplementary material.
Author contributions
MA: Conceptualization, Formal analysis, Investigation, Validation, Visualization, Writing – original draft, Writing – review & editing. FK: Data curation, Formal analysis, Methodology, Software, Visualization, Writing – review & editing. CE: Data curation, Investigation, Methodology, Visualization, Writing – original draft. JG: Data curation, Formal analysis, Methodology, Software, Writing – review & editing. IS: Data curation, Formal analysis, Investigation, Methodology, Software, Writing – review & editing. PB: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – review & editing. WL: Conceptualization, Funding acquisition, Investigation, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1517151/full#supplementary-material
Footnotes
1. ^https://www.ncbi.nlm.nih.gov/datasets/
3. ^https://github.com/moshi4/COGclassifier
5. ^https://pandas.pydata.org/
References
Ackermann, K., Kenngott, R., Settles, M., Gerhards, H., Maierl, J., and Wollanke, B. (2021). In vivo biofilm formation of pathogenic Leptospira spp. in the vitreous humor of horses with recurrent uveitis. Microorganisms 9:1915. doi: 10.3390/microorganisms9091915
Adnane, M., Meade, K. G., and O’Farrelly, C. (2018). Cervico-vaginal mucus (CVM) - an accessible source of immunologically informative biomolecules. Vet. Res. Commun. 42, 255–263. doi: 10.1007/s11259-018-9734-0
Ahmed, N., Devi, S. M., Valverde, M. D. L. A., Vijayachari, P., Machang’u, R. S., Ellis, W. A., et al. (2006). Multilocus sequence typing method for identification and genotypic classification of pathogenic Leptospira species. Ann. Clin. Microbiol. Antimicrob. 5:28. doi: 10.1186/1476-0711-5-28
Aymée, L., Di Azevedo, M. I. N., Borges, A. L. D. S. B., Carvalho-Costa, F. A., and Lilenbaum, W. (2022a). Leptospira spp. strains associated with bovine genital leptospirosis (BGL). Microb. Pathog. 173:105841. doi: 10.1016/j.micpath.2022.105841
Aymée, L., Gregg, W. R. R., Loureiro, A. P., Di Azevedo, M. I. N., Pedrosa, J. S., Melo, J. D. S. L., et al. (2021). Bovine genital leptospirosis and reproductive disorders of live subfertile cows under field conditions. Vet. Microbiol. 261:109213. doi: 10.1016/j.vetmic.2021.109213
Aymée, L., Di Azevedo, M. I. N., Reis, L., Mendes, J., FFA, C., Carvalho-Costa, F. A., et al. (2023). Unconventional sites for diagnosis ofl in bovine anicteric fetuses. Animals 13:2832. doi: 10.3390/ani13182832
Aymée, L., Di Azevedo, M. I. N., de Souza Pedrosa, J., Loria de Melo, J. D. S., Carvalho-Costa, F. A., and Lilenbaum, W. (2022b). The role of Leptospira santarosai serovar Guaricura as agent of bovine genital leptospirosis. Vet. Microbiol. 268:109413. doi: 10.1016/j.vetmic.2022.109413
Barbosa, C., Oliveira, A. D. S., Mendes, J. S., Ristow, P., and Lilenbaum, W. (2023). Standardization of chronic uterine infection of hamsters by Leptospira santarosai serovar Guaricura, from serogroup Sejroe. Vet. Res. Commun. 47, 2215–2219. doi: 10.1007/s11259-023-10108-y
Berriman, M., and Rutherford, K. (2003). Viewing and annotating sequence data with Artemis. Brief. Bioinform. 4, 124–132. doi: 10.1093/bib/4.2.124
Borges, A. L. D. S. B., Aymée, L., Carvalho-Costa, F. A., Lilenbaum, W., and Di Azevedo, M. I. N. (2024). Molecular epidemiology of Leptospira spp. serogroup Sejroe associated with chronic bovine leptospirosis. Vet. Microbiol. 298:110238. doi: 10.1016/j.vetmic.2024.110238
Cabral Pires, B., Berzin Grapiglia, J., Moreira, L., Jaeger, L. H., Carvalho-Costa, F. A., and Lilenbaum, W. (2018). Occurrence of uterine carriers for Leptospira interrogans on slaughtered cows. Microb. Pathog. 114, 163–165. doi: 10.1016/j.micpath.2017.11.056
Cai, X., Hu, H., and Li, X. (2009). A new measurement of sequence conservation. BMC Genomics 10:623. doi: 10.1186/1471-2164-10-623
Carr, V. R., Shkoporov, A., Hill, C., Mullany, P., and Moyes, D. L. (2021). Probing the mobilome: discoveries in the dynamic microbiome. Trends Microbiol. 29, 158–170. doi: 10.1016/j.tim.2020.05.003
Carver, T., Thomson, N., Bleasby, A., Berriman, M., and Parkhill, J. (2009). DNAPlotter: circular and linear interactive genome visualization. Bioinformatics 25, 119–120. doi: 10.1093/bioinformatics/btn578
Chinchilla, D., Nieves, C., Gutiérrez, R., Sordoillet, V., Veyrier, F. J., and Picardeau, M. (2023). Phylogenomics of Leptospira santarosai, a prevalent pathogenic species in the Americas. PLoS Negl. Trop. Dis. 17:e0011733. doi: 10.1371/journal.pntd.0011733
Chou, L.-F., Chen, T.-W., Ko, Y.-C., Pan, M.-J., Tian, Y.-C., Chiu, C.-H., et al. (2014). Potential impact on kidney infection: a whole-genome analysis of Leptospira santarosai serovar Shermani. Emerg. Microbes Infect. 3:e82. doi: 10.1038/emi.2014.78
Davignon, G., Cagliero, J., Guentas, L., Bierque, E., Genthon, P., Gunkel-Grillon, P., et al. (2023). Leptospirosis: toward a better understanding of the environmental lifestyle of Leptospira. Front. Water 5:1195094. doi: 10.3389/frwa.2023.1195094
de Oliveira, N. R., Kremer, F. S., de Brito, R. S. A., Zamboni, R., Dellagostin, O. A., and Jorge, S. (2022). Pathogenesis and genomic analysis of a virulent Leptospira interrogans rerovar Copenhageni isolated from a dog with lethal infection. Trop. Med. Infect. Dis. 7:333. doi: 10.3390/tropicalmed7110333
Di Azevedo, M. I. N., Kremer, F., Ezepha, C., Kraychete, G. B., Bourhy, P., and Lilenbaum, W. (2023). Draft-genome sequences of Leptospira santarosai strains isolated from urogenital tract of cows. Microbiol. Resour. Announc. 12:e0068623. doi: 10.1128/MRA.00686-23
Di Azevedo, M. I. N., Pires, B. C., Barbosa, L. F. C., Carvalho-Costa, F. A., and Lilenbaum, W. (2021). Characterization of leptospiral DNA in the follicular fluid of non-pregnant cows. Vet. Rec. 188:e143. doi: 10.1002/vetr.143
Di Azevedo, M. I. N., Pires, B. C., Libonati, H., Pinto, P. S., Cardoso Barbosa, L. F., Carvalho-Costa, F. A., et al. (2020). Extra-renal bovine leptospirosis: molecular characterization of the Leptospira interrogans Sejroe serogroup on the uterus of non-pregnant cows. Vet. Microbiol. 250:108869. doi: 10.1016/j.vetmic.2020.108869
Dos Santos Pereira, P. V., Di Azevedo, M. I. N., Dos Santos Baptista Borges, A. L., Loureiro, A. P., Martins, G., Carvalho-Costa, F. A., et al. (2022). Bovine genital leptospirosis: evidence of ovarian infection by Leptospira interrogans. Vet. Microbiol. 271:109489. doi: 10.1016/j.vetmic.2022.109489
Dsouza, F. P., Dinesh, S., and Sharma, S. (2024). Understanding the intricacies of microbial biofilm formation and its endurance in chronic infections: a key to advancing biofilm-targeted therapeutic strategies. Arch. Microbiol. 206:85. doi: 10.1007/s00203-023-03802-7
Fávero, J. F., de Araújo, H. L., Lilenbaum, W., Machado, G., Tonin, A. A., Baldissera, M. D., et al. (2017). Bovine leptospirosis: prevalence, associated risk factors for infection and their cause-effect relation. Microb. Pathog. 107, 149–154. doi: 10.1016/j.micpath.2017.03.032
Fernandes, L. G. V., Putz, E. J., Stasko, J., Lippolis, J. D., Nascimento, A. L. T. O., and Nally, J. E. (2022). Evaluation of LipL32 and LigA/LigB knockdown mutants in Leptospira interrogans serovar Copenhageni: impacts to proteome and virulence. Front. Microbiol. 12:799012. doi: 10.3389/fmicb.2021.799012
Fernandes, L. G. V., Teixeira, A. F., and Nascimento, A. L. T. O. (2023). Evaluation of Leptospira interrogans knockdown mutants for LipL32, LipL41, LipL21, and OmpL1 proteins. Front. Microbiol. 14:1199660. doi: 10.3389/fmicb.2023.1199660
Flemming, H. C., Wingender, J., Szewzyk, U., Steinberg, P., Rice, S. A., and Kjelleberg, S. (2016). Biofilms: an emergent form of bacterial life. Nat. Rev. Microbiol. 14, 563–575. doi: 10.1038/nrmicro.2016.94
Galardini, M., Biondi, E. G., Bazzicalupo, M., and Mengoni, A. (2011). CONTIGuator: a bacterial genomes finishing tool for structural insights on draft genomes. Source Code Biol. Med. 6:11. doi: 10.1186/1751-0473-6-11
Galperin, M. Y., Wolf, Y. I., Makarova, K. S., Vera Alvarez, R., Landsman, D., and Koonin, E. V. (2021). COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 49, D274–D281. doi: 10.1093/nar/gkaa1018
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Haake, D. A., and Levett, P. N. (2015, 2015). Leptospirosis in humans. Curr. Top. Microbiol. Immunol. 387, 65–97. doi: 10.1007/978-3-662-45059-8_5
Hagedoorn, N. N., Maze, M. J., Carugati, M., Cash-Goldwasser, S., Allan, K. J., Chen, K., et al. (2024). Global distribution of Leptospira serovar isolations and detections from animal host species: a systematic review and online database. Trop. Med. Int. Health 29, 161–172. doi: 10.1111/tmi.13965
Hamond, C., Pinna, M., Medeiros, M. A., Bourhy, P., Lilenbaum, W., and Picardeau, M. (2015). A multilocus variable number tandem repeat analysis assay provides high discrimination for genotyping Leptospira santarosai strains. J. Med. Microbiol. 64, 507–512. doi: 10.1099/jmm.0.000045
Hota, S., and Kumar, M. (2024). Unveiling the impact of Leptospira TolC efflux protein on host tissue adherence, complement evasion, and diagnostic potential. Infect. Immun. 92:e0041924. doi: 10.1128/iai.00419-24
Janaki, C., Gowri, V. S., and Srinivasan, N. (2021). Master blaster: an approach to sensitive identification of remotely related proteins. Sci. Rep. 11:8746. doi: 10.1038/s41598-021-87833-4
Koizumi, N., Wada, T., Morita, M., Mu, J.-J., and Ohnishi, M. (2020). Comparative genomic analysis of Leptospira borgpetersenii serogroup Javanica isolated from Rattus species in southern Japan, Philippines, and Taiwan. Infect. Genet. Evol. 85:104447. doi: 10.1016/j.meegid.2020.104447
Kremer, F. S., Eslabão, M. R., Provisor, M., Woloski, R. D., Ramires, O. V., Moreno, L. Z., et al. (2015). Draft genome sequences of Leptospira santarosai strains U160, U164, and U233, isolated from asymptomatic cattle. Genome Announc. 3, e00910–e00915. doi: 10.1128/genomeA.00910-15
Kumar, K. V., Lall, C., Raj, R. V., Vedhagiri, K., Sunish, I. P., and Vijayachari, P. (2016). In vitro antimicrobial susceptibility of pathogenic Leptospira biofilm. Microb. Drug Resist. 22, 511–514. doi: 10.1089/mdr.2015.0284
Lata, K. S., Kumar, S., Vindal, V., Patel, S., and Das, J. (2022). A core and pan gene map of Leptospira genus and its interactions with human host. Microb. Pathog. 162:105347. doi: 10.1016/j.micpath.2021.105347
Loureiro, A. P., Hamond, C., Pinto, P., Bremont, S., Bourhy, P., and Lilenbaum, W. (2016). Molecular analysis of leptospires from serogroup Sejroe obtained from asymptomatic cattle in Rio de Janeiro - Brazil reveals genetic proximity to serovar Guaricura. Res. Vet. Sci. 105, 249–253. doi: 10.1016/j.rvsc.2016.02.012
Loureiro, A. P., and Lilenbaum, W. (2020). Genital bovine leptospirosis: a new look for an old disease. Theriogenology 141, 41–47. doi: 10.1016/j.theriogenology.2019.09.011
Loureiro, A. P., Martins, G., Pinto, P., Narduche, L., Teixeira, R. C., and Lilenbaum, W. (2015). Usage of a selective media (EMJH-STAFF) in primary culturing of pathogenic leptospires from bovine clinical samples. Lett. Appl. Microbiol. 61, 603–606. doi: 10.1111/lam.12501
Martins, G., Guadelupe, B., Aymée, L., Balaro, M. F. A., Pinto, P. H., Di Azevedo, M. I. N., et al. (2022). The efficacy of vaccination in the prevention of renal and genital leptospirosis in experimentally infected sheep. Trop. Med. Infect. Dis. 7:321. doi: 10.3390/tropicalmed7100321
Martins, Y. C., Jurberg, A. D., and Daniel-Ribeiro, C. T. (2023). Visiting molecular mimicry once more: pathogenicity, virulence, and autoimmunity. Microorganisms 11:1472. doi: 10.3390/microorganisms11061472
Mayer, C., and Dörr, T. (2019). Editorial: bacterial cell wall structure and dynamics. Front. Microbiol. 10:2051. doi: 10.3389/fmicb.2019.02051
Mejía, L., Prado, B., Cárdenas, P., Trueba, G., and González-Candelas, F. (2022). The impact of genetic recombination on pathogenic Leptospira. Infect. Genet. Evol. 102:105313. doi: 10.1016/j.meegid.2022.105313
Monahan, A. M., Callanan, J. J., and Nally, J. E. (2009). Review paper: host-pathogen interactions in the kidney during chronic leptospirosis. Vet. Pathol. 46, 792–799. doi: 10.1354/vp.08-VP-0265-N-REV
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pal, S., Verma, J., Mallick, S., Rastogi, S. K., Kumar, A., and Ghosh, A. S. (2019). Absence of the glycosyltransferase WcaJ in Klebsiella pneumoniae ATCC13883 affects biofilm formation, increases polymyxin resistance and reduces murine macrophage activation. Microbiology 165, 891–904. doi: 10.1099/mic.0.000827
Picardeau, M. (2020). Leptospira and Leptospirosis. Methods Mol. Biol. 2134, 271–275. doi: 10.1007/978-1-0716-0459-5_24
Ramli, S. R., Bunk, B., Spröer, C., Geffers, R., Jarek, M., Bhuju, S., et al. (2021). Complete genome sequencing of Leptospira interrogans isolates from Malaysia reveals massive genome rearrangement but high conservation of virulence-associated genes. Pathogens 10:1198. doi: 10.3390/pathogens10091198
Ristow, P., Bourhy, P., Kerneis, S., Schmitt, C., Prevost, M.-C., Lilenbaum, W., et al. (2008). Biofilm formation by saprophytic and pathogenic leptospires. Microbiology 154, 1309–1317. doi: 10.1099/mic.0.2007/014746-0
Santos, L. A., Adhikarla, H., Yan, X., Wang, Z., Fouts, D. E., Vinetz, J. M., et al. (2018). Genomic comparison among global isolates of L. interrogans Serovars Copenhageni and Icterohaemorrhagiae identified natural genetic variation caused by an Indel. Front. Cell. Infect. Microbiol. 8:193. doi: 10.3389/fcimb.2018.00193
Santos, A. A. N., Ribeiro, P. D. S., da França, G. V., Souza, F. N., Ramos, E. A. G., Figueira, C. P., et al. (2021). Leptospira interrogans biofilm formation in Rattus norvegicus (Norway rats) natural reservoirs. PLoS Negl. Trop. Dis. 15:e0009736. doi: 10.1371/journal.pntd.0009736
Sun, D., Liu, Y., Peng, X., Dong, H., Jiang, H., Fan, X., et al. (2023). ClpP protease modulates bacterial growth, stress response, and bacterial virulence in Brucella abortus. Vet. Res. 54:68. doi: 10.1186/s13567-023-01200-x
Thibeaux, R., Soupé-Gilbert, M.-E., Kainiu, M., Girault, D., Bierque, E., Fernandes, J., et al. (2020). The zoonotic pathogen Leptospira interrogans mitigates environmental stress through cyclic-di-GMP-controlled biofilm production. NPJ Biofilms Microbiomes 6:24. doi: 10.1038/s41522-020-0134-1
Vincent, A. T., Schiettekatte, O., Goarant, C., Neela, V. K., Bernet, E., Thibeaux, R., et al. (2019). Revisiting the taxonomy and evolution of pathogenicity of the genus Leptospira through the prism of genomics. PLoS Negl. Trop. Dis. 13:e0007270. doi: 10.1371/journal.pntd.0007270
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Xu, Y., Zhu, Y., Wang, Y., Chang, Y.-F., Zhang, Y., Jiang, X., et al. (2016). Whole genome sequencing revealed host adaptation-focused genomic plasticity of pathogenic Leptospira. Sci. Rep. 6:20020. doi: 10.1038/srep20020
Yang, W., and Briegel, A. (2020). Diversity of bacterial chemosensory arrays. Trends Microbiol. 28, 68–80. doi: 10.1016/j.tim.2019.08.002
Yin, B., Zhu, L., Zhang, Y., Dong, P., Mao, Y., Liang, R., et al. (2018). The characterization of biofilm formation and detection of biofilm-related genes in Salmonella isolated from beef processing plants. Foodborne Pathog. Dis. 15, 660–667. doi: 10.1089/fpd.2018.2466
Zheng, J. X. (2018). Biofilm formation in Klebsiella pneumoniae bacteremia strains was found to be associated with CC23 and the presence of wcaG. Front. Cell. Infect. Microbiol. 8:21. doi: 10.3389/fcimb.2018.00021
Keywords: leptospirosis, BGL, veterinary microbiology, WGS, infectious disease
Citation: Di Azevedo MIN, Kremer F, Ezepha C, Greco JPG, da Silva ICV, Bourhy P and Lilenbaum W (2025) Comparative genomics of Leptospira santarosai reveals genomic adaptations in bovine genital strains. Front. Microbiol. 15:1517151. doi: 10.3389/fmicb.2024.1517151
Edited by:
Jens Andre Hammerl, Bundesinstitut für Risikobewertung, GermanyReviewed by:
Mônica Larucci Vieira, Federal University of Minas Gerais, BrazilShuichi Nakamura, Tohoku University, Japan
Sridhar Kavela, Chaitanya (Deemed to be University), India
Luan Gavião Prado, Cleveland Clinic, United States
Copyright © 2025 Di Azevedo, Kremer, Ezepha, Greco, da Silva, Bourhy and Lilenbaum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Isabel Nogueira Di Azevedo, aXNhYmVsZGlhemV2ZWRvQGdtYWlsLmNvbQ==