 Adriana Freitas Moraes Monteiro1,2
Adriana Freitas Moraes Monteiro1,2 Fábio Silva da Silva2
Fábio Silva da Silva2 Ana Cecília Ribeiro Cruz1,2Sandro Patroca da Silva2Alice Louize Nunes Queiroz2
Ana Cecília Ribeiro Cruz1,2Sandro Patroca da Silva2Alice Louize Nunes Queiroz2 Livia Medeiros Neves Casseb1,2
Livia Medeiros Neves Casseb1,2 Livia Carício Martins1,2Daniele Barbosa de Almeida Medeiros1,2*
Livia Carício Martins1,2Daniele Barbosa de Almeida Medeiros1,2*- 1Graduate Program in Virology, Evandro Chagas Institute — IEC/MS/SVSA, Ananindeua, Brazil
- 2Evandro Chagas Institute — IEC/MS/SVSA, Department of Arbovirology and Hemorragic Fevers, Ananindeua, Brazil
Wild rodents serve as crucial reservoirs for zoonotic viruses. Anthropogenic and environmental disruptions, particularly those induced by mining activities, can destabilize rodent populations and facilitate the emergence of viral agents. In the Canaã dos Carajás and Curionópolis regions of Brazil, significant environmental changes have occurred due to mining expansion, potentially creating conditions conducive to the emergence of rodent-associated viral diseases. This study aimed to investigate the viral diversity in wild rodents captured in Canaã dos Carajás and Curionópolis, Pará, between 2017 and 2019. A total of 102 rodent samples were taxonomically identified through karyotyping and screened for anti-Orthohantavirus antibodies using the ELISA method. Subsequently, nucleotide sequencing and bioinformatics analyses were conducted on 14 selected samples to characterize the virome. This selection was based on the most commonly associated rodent genera as reservoirs of Orthohantavirus and Mammarenavirus. Of the 102 samples tested via ELISA, 100 were negative, and two showed optical density at the cutoff point. Sequencing of the 14 samples generated approximately 520 million reads, with 409 million retained after quality control. These reads were categorized into 53 viral families, including both DNA and RNA viruses, with Retroviridae, Baculoviridae, and Microviridae being the most abundant. Viral contigs were identified, including one fragment related to Arenaviridae and three to Filoviridae. Metagenomic analysis revealed high viral diversity in the sampled rodents, with the presence of viral families of public health concern, such as Arenaviridae and Filoviridae. The findings suggest that increased human activities associated with mining may contribute to the emergence of these viruses, underscoring the need for ongoing surveillance to prevent potential outbreaks.
1 Introduction
Emerging infectious diseases, particularly those of zoonotic origin, have become a significant public health challenge in recent decades (Rahman et al., 2020). Among animals, rodents play a crucial role as reservoirs for many viruses due to their species diversity, varied habitats, and tendency to live close to human-inhabited areas (Luis et al., 2013; Han et al., 2015; Carlson et al., 2019). Viral diversity in rodents can be influenced by anthropogenic factors such as deforestation, changes in land use, and climate change (Aguirre, 2017; Plowright et al., 2021; Rupasinghe et al., 2022), which can disrupt ecological relationships, alter host dynamics, and promote the increase and/or emergence of viral agents (Thompson, 2013; Horefti, 2023).
Advances in next-generation sequencing (NGS) technologies combined with bioinformatics have revealed a vast viral diversity among rodent populations, whether or not they are associated with human diseases (Yin et al., 2022; Zhao et al., 2022). Among the most commonly described viruses in these reservoirs are hantaviruses and arenaviruses (Families Hantaviridae and Arenaviridae), known to cause serious diseases in humans, such as Hantavirus pulmonary syndrome (HPS) and hemorrhagic fevers. However, other viral families have also been identified, including Astroviridae (Phan et al., 2011; Yin et al., 2022), Togaviridae, Reoviridae (He et al., 2022), Coronaviridae (Tsoleridis and Ball, 2020; Wang et al., 2020), Flaviviridae (Meerburg et al., 2009; Han et al., 2015; Tirera et al., 2021), Arteriviridae, Anelloviridae, and Circoviridae, among others (Wu et al., 2021; He et al., 2022). These discoveries are crucial for the identification and surveillance of viral pathogens as well as for the prevention of potential outbreaks (Wu et al., 2016).
In Brazil, regions such as Canaã dos Carajás and Curionópolis, in the southwest of the State of Pará, are notable for their rich biodiversity and significant mineral reserves. However, these areas have faced intense environmental impacts due to deforestation, largely driven by the expansion of mining projects, road construction, agricultural growth, and the emergence of small villages. These environmental changes create favorable conditions for the emergence and spread of viral diseases associated with rodents (Ferreira et al., 2005).
In this study, we used a metagenomic approach to characterize viral diversity in wild rodents from the regions of Canaã dos Carajás and Curionópolis. Our objective is to investigate the viral composition in these animals and contribute to a better understanding of the viral dynamics in these reservoirs and their potential risks to public health.
2 Materials and methods
2.1 Rodent samples
To conduct this study, 102 biological samples of wild rodents were used, captured during eco-epidemiological expeditions in the municipalities of Canaã dos Carajás and Curionópolis (Figure 1), located in the State of Pará, between 2017 and 2019. All samples are stored in the Arbovirology Section of the Evandro Chagas Institute. The rodent samples were identified using the code RO, followed by a specific number, designated as registered identification. This code served as a unique reference for each sample throughout the analysis process, ensuring the traceability and organization of the data obtained during the study. The taxonomic identification of the captured rodents was performed by means of karyotyping (Bovincino et al., 2008).
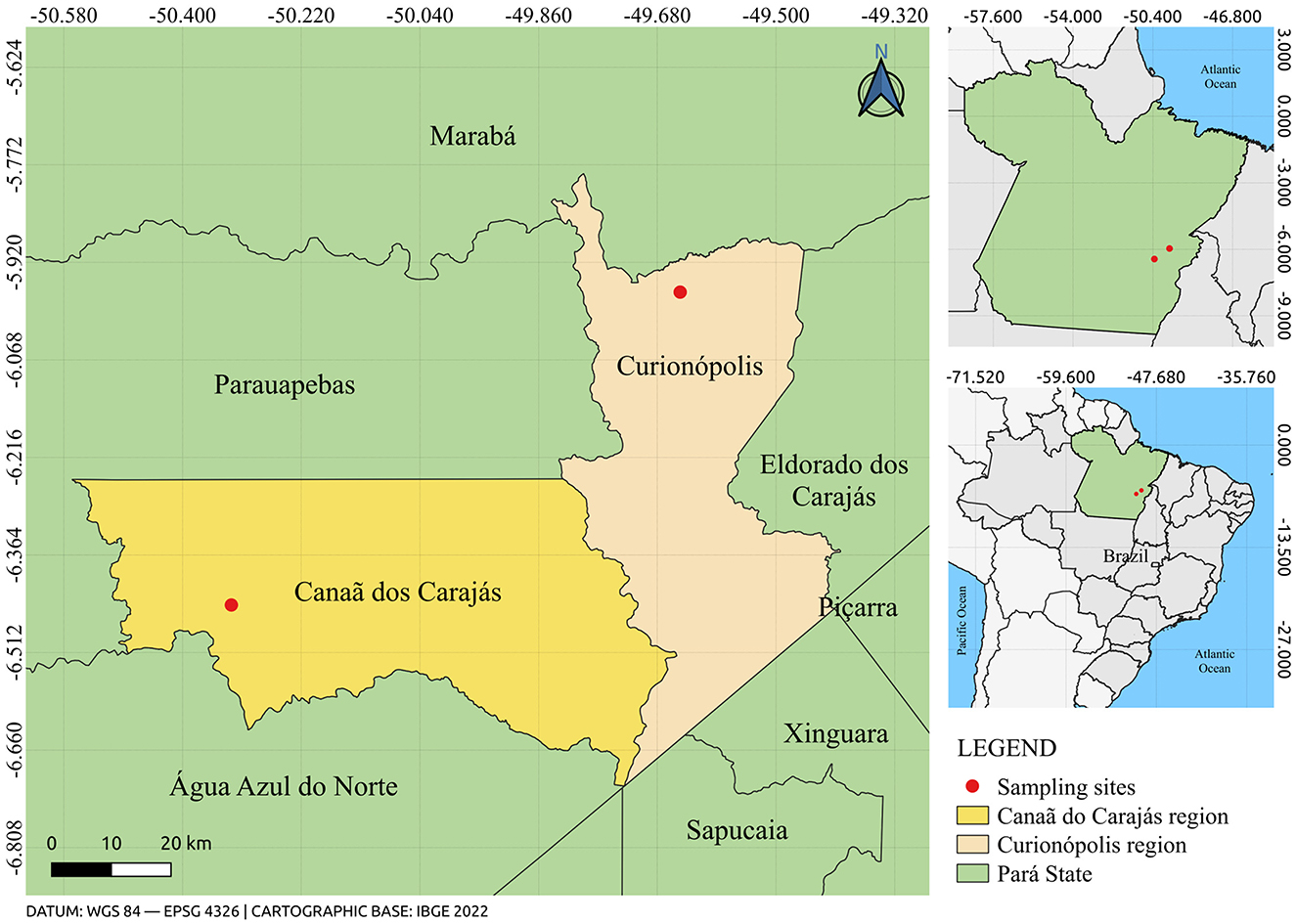
Figure 1. Map showing the location of rodent capture sites in the municipalities of Canaã dos Carajás and Curionópolis, State of Pará, Brazil. The figure was created using QGIS v.3.10.4 software (available at https://qgis.org/pt_BR/site), in conjunction with the IBGE 2022 database (available at: https://www.ibge.gov.br/).
2.2 Enzyme-linked immunosorbent assay for detection of anti-orthohantavirus antibodies
The ELISA was conducted as a screening method using 102 selected rodent samples. For this purpose, a truncated recombinant protein, HTN-120, based on the N gene of orthohantaviruses circulating in the Amazon and developed by the Evandro Chagas Institute, was used. ELISA microplates were coated with 4 μg of the recombinant HTN-120 antigen and a negative antigen control, then incubated overnight at 4°C. The plates were washed four times with PBS-Tween buffer, blocked with PBS-Tween and lectin solution, and incubated for 1 h at 37°C. After blocking, rodent sera/blood samples were added at 1:100 and 1:400 dilutions and incubated for an additional hour at 37°C. The plates were washed again, and an anti-Peromyscus IgG conjugate, diluted 1:500, was added and incubated for 1 h at 37°C. The plates were then washed and developed with TMB One Solution for 15 min at 37°C. The reaction was stopped with 1M HCl, and the absorbance was read at 450 nm. Samples with an optical density (OD) below 0.2 were considered negative.
2.3 RNA extraction
For RNA extraction, fragments of viscera (brain, lung, kidney, spleen, and heart) were used, with pools formed for each species of captured rodent. After preparing the pools, 1 mL of Trizol RNA isolation reagent was added for every 10 mg of tissue per microtube, along with a 5 mm diameter stainless steel sphere. The samples were macerated in the Tyssueliser II (Qiagen) equipment for 2 min at a frequency of 25 Hz in the level 3 biosafety laboratory (NB3) of the Arbovirology section of the Evandro Chagas Institute. Immediately afterward, the lysed content was transferred to a tube containing a phase separation polymer (phasemaker) and incubated for 5 min at room temperature (RT). Next, 200 μL of chloroform was added, and the microtubes were vortexed for 15 s and incubated for 15 min at RT. Subsequently, the samples were centrifuged in a refrigerated centrifuge (4°C) for 5 min at 12,000 xg. The aqueous phase was removed and transferred to a new tube containing an equal volume of 70% ethanol and homogenized by vortexing for 15 s.
For purification, the PureLink RNA Mini kit (Invitrogen) was used. The aqueous solution was transferred to the column and centrifuged at 12,000 × g for 15 s. Washes were performed with wash buffers I and II, followed by another centrifugation at 12,000 × g for 15 s. After this process, the samples were eluted with 60 μL of ultrapure water (Invitrogen), incubated for 1 min at RT, and centrifuged again at 12,000 × g for 2 min. Finally, the extracted RNA was quantified using the Qubit 4.0 equipment (Invitrogen) with the Qubit RNA HS assay kit (Invitrogen), following the manufacturer's recommendations, and stored in a freezer at −70°C.
2.4 Double-stranded cDNA synthesis, genomic library construction and sequencing
The preparation of cDNA from RNA began with the synthesis of the first and second cDNA strands, using the SuperScript™ IV VILO™ Master Mix (Invitrogen) and NEBNext mRNA Second Strand Synthesis Module (New England BioLabs), respectively. The cDNA purification was performed using the PureLink® PCR Purification Kit (Invitrogen). The synthesized cDNA was then quantified using the Assay DNA HS Kit (Invitrogen) on the Qubit 4.0 equipment (Invitrogen). The genomic library was constructed according to the Nextera XT DNA kit (Illumina) protocol guidelines and sequenced using the NextSeq 500 platform (Illumina, Inc) with the NextSeq 500/550 High Output Kit v2.5 (Illumina) in a 300-cycle reaction (paired-end, 2 × 150), following the manufacturer's recommendations.
2.5 Computational analysis
The files obtained after sequencing were initially subjected to quality assessment using Fastp v.0.23.4 (Chen et al., 2018). This tool was configured to remove adapter sequences and reads with a Phred quality score < 20 and a length < 50 nt. Additional filtering of ribosomal reads was performed using SortMeRNA v.2.1 (Kopylova et al., 2012) with the default sequence database provided by the tool.
The assessment of potential viral read diversity, targeting the Family taxonomic level, was performed using DIAMOND tool v.2.1.9.163 (Buchfink et al., 2021) (Blastx, e-value of 10−5, taxonlist 10239) with a non-redundant protein database (NR—NCBI, available at https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/). The results were compiled into table files to calculate general metrics of Alpha diversity (abundance heatmaps and diversity indices) and Beta diversity (Bray-Curtis dissimilarity matrix and PCoA), using the R language along with the pheatmap, vegan, and ggplot2 libraries.
After quality control, the data were subjected to genomic assembly using the de novo method with MEGAHIT software v.1.2.9 (Li et al., 2015) in its default configuration (k-mers ranging from 21 to 141 nt). The assembled contigs were then compared with a non-redundant protein database (NR—NCBI), targeting viral sequences using DIAMOND tool v.2.1.9.163 (Blastx, e-value of 10−5, taxonlist 10239), followed by manual inspection with Geneious software v.11.1.5 (Kearse et al., 2012). Viral contigs were identified and annotated based on homology searches in databases such as NCBI (using the BlastX tool) and EMBL (using the InterProScan tool to verify functional domains). Additionally, confirmation of host species, initially identified based on morphological aspects, was verified through the analysis of mitochondrial contigs, with a particular focus on the Cytochrome B (CytB) subunit, considering a species confirmation threshold of 98%. Functional annotation of the obtained mtDNAs was performed using the Mitochondrial Genome Annotation Server (MITOS) (Bernt et al., 2013) (available at https://usegalaxy.eu/).
The reconstruction of the phylogenies of the identified viruses was performed by considering their respective contigs along with other sequences from their respective groups available in the NCBI database. The sequences were initially aligned using the MAFFT v.7.520 algorithm (Katoh et al., 2002). The best amino acid and nucleotide substitution models were determined, followed by phylogeny reconstruction using the Maximum Likelihood method with bootstrapping values (BPP) set to 1,000 replicates, performed using IQtree v.1.6.12 (Nguyen et al., 2015). Finally, the obtained topologies were visualized using Figtree v.1.4.496 (available at http://tree.bio.ed.ac.uk/software/figtree/) and edited using Inkscape v.0.92 (available at https://inkscape.org/pt-br/).
3 Results
3.1 Enzymatic immunoassay for detection of anti-Orthohantavirus antibodies
The 102 rodents collected and considered for this study were taxonomically identified in nine genera, as shown in Supplementary Table 1. Due to the observation of very similar morphological and chromosomal characteristics, some species of rodents collected were identified only to the taxonomic level of genus. Samples from all 102 collected specimens were tested by ELISA for detection of IgG antibodies to Orthohantavirus. Of these, 100 presented OD lower than 0.2, and therefore were considered negative, while two samples presented OD at the cut-off: 0.239 for a rodent captured in Canaã dos Carajás (Oligoryzomys microtis, sample RO23088) and 0.261 for another captured in Curionópolis (Necromys lasiurus, sample RO23100).
3.2 Samples submitted for metagenomic analysis
The selection of samples for metagenomic study was carried out based on the rodent genera already described as potential reservoirs of Orthohantavirus and Mammarenavirus: Proechimys (2), Oligoryzomys (3), Oryzomys (1), Necromys (2), Calomys (1), Oxymycterus (2) and Oecomys (1) (Levis et al., 1998; Johnson et al., 1999; Moncayo et al., 2001; Travassos da Rosa et al., 2005; Milazzo et al., 2010; Oliveira et al., 2011; Travassos da Rosa et al., 2011; Firth et al., 2012; Travassos da Rosa et al., 2012; Oliveira et al., 2014; Nunes et al., 2015; Tapia-Ramírez et al., 2022). Rodents from different genera were selected to expand the knowledge about their viral diversities. Furthermore, among the rodents selected for metagenomic study, some were identified, taxonomically, only up to the genus level, with their identification at the species level being confirmed through the characterization of mtDNA (CytB subunit). Thus, a total of ~520 million reads were generated from genomic sequencing. Of these, 409 million reads (78.6%) remained after quality control, as detailed in Supplementary Table 2, together with the identification and confirmation of rodent species based on the CytB subunit.
3.3 Diversity analysis
Based on the analysis of the diversity of viral readings obtained, from the comparison with the reference database considered, it was possible to identify a wide variety of viruses, both DNA and RNA, potentially present in the evaluated samples, which were distributed in 53 families, with the most abundant being Retroviridae, Baculoviridae and Microviridae (Figure 2; Supplementary Table 3), corresponding to approximately 96.7% of the classified readings.
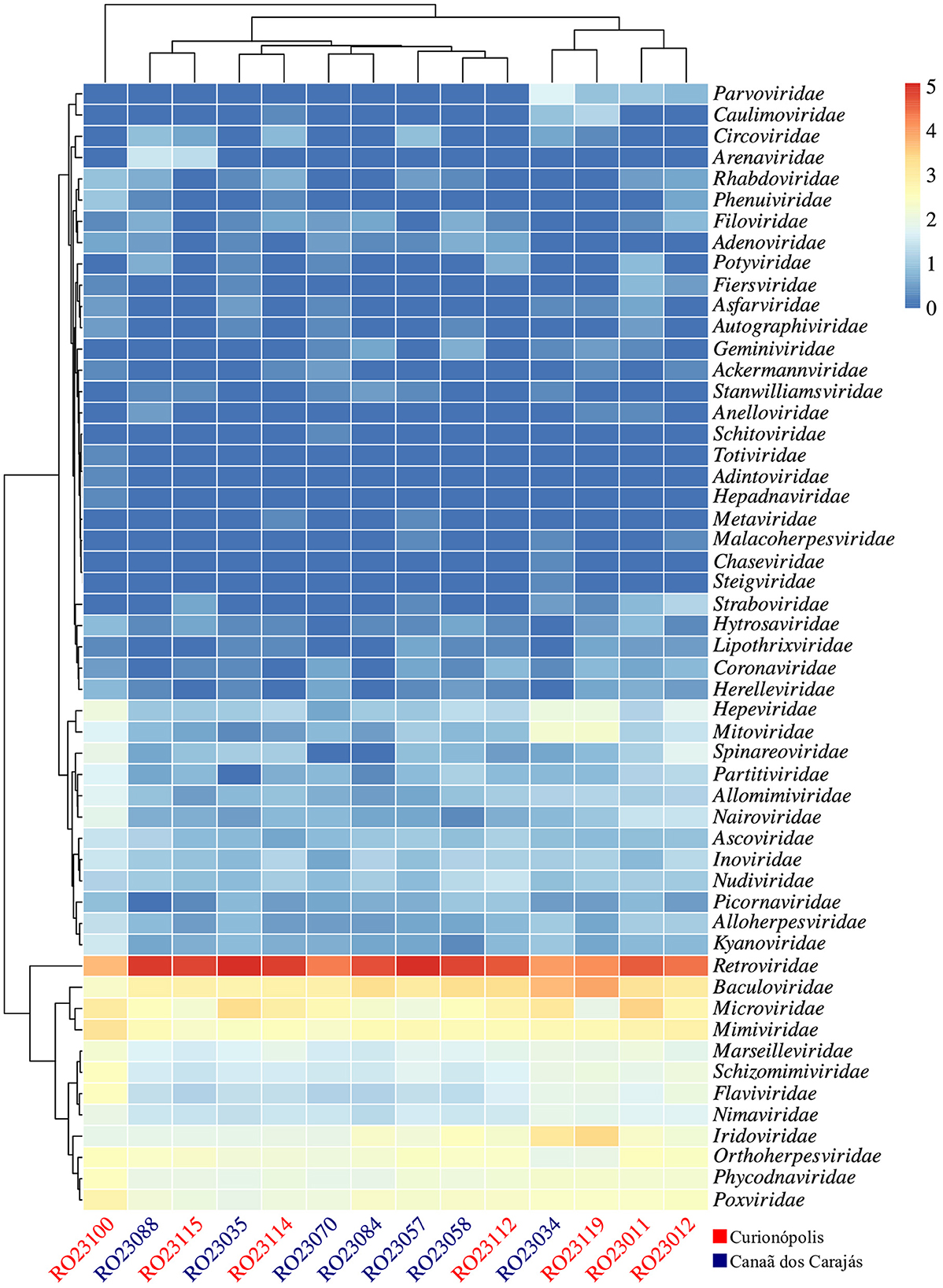
Figure 2. Heatmap of viral reads, normalized on a log10 scale, showing 53 viral families detected across the evaluated samples. The clusters indicate groupings based on abundance levels. The samples are displayed on the x-axis, and the potentially detected viral families are displayed on the y-axis. Samples from the regions of Canaã dos Carajás and Curionópolis are highlighted in blue and red, respectively.
Samples RO23035 (Oecomys bicolor) and RO23057 (Oryzomys megacephalus) exhibited the highest abundance of classified viral reads, particularly related to the Retroviridae family, while sample RO23100 (N. lasiurus) had the lowest abundance index (Supplementary Figure 1A). However, when evaluating the diversity indices, particularly the richness of viral families, sample RO23100 showed the highest records, as indicated by the Shannon and Simpson indices. This result reflects a greater equitability (distribution of reads among represented viral families) in this sample. Despite being collected from different locations, the metagenomic profiles of the evaluated samples showed relative similarity, grouping them according to the diversity indices and as demonstrated by the principal component plot (PCoA) based on the Bray-Curtis dissimilarity matrix (Supplementary Figures 1B, 2).
3.4 Potential viral contigs identified
3.4.1 Arenaviridae
A viral fragment related to the Arenaviridae family was obtained from a rodent sample of the species O. microtis (sample RO23115), captured during ecoepidemiological expeditions in the municipality of Curionópolis. The identified contig was 441 nt long and included functional domains associated with the glycoprotein region of Arenaviridae (Figure 3A), sharing an amino acid identity of 64% with Mammarenavirus piritalense.
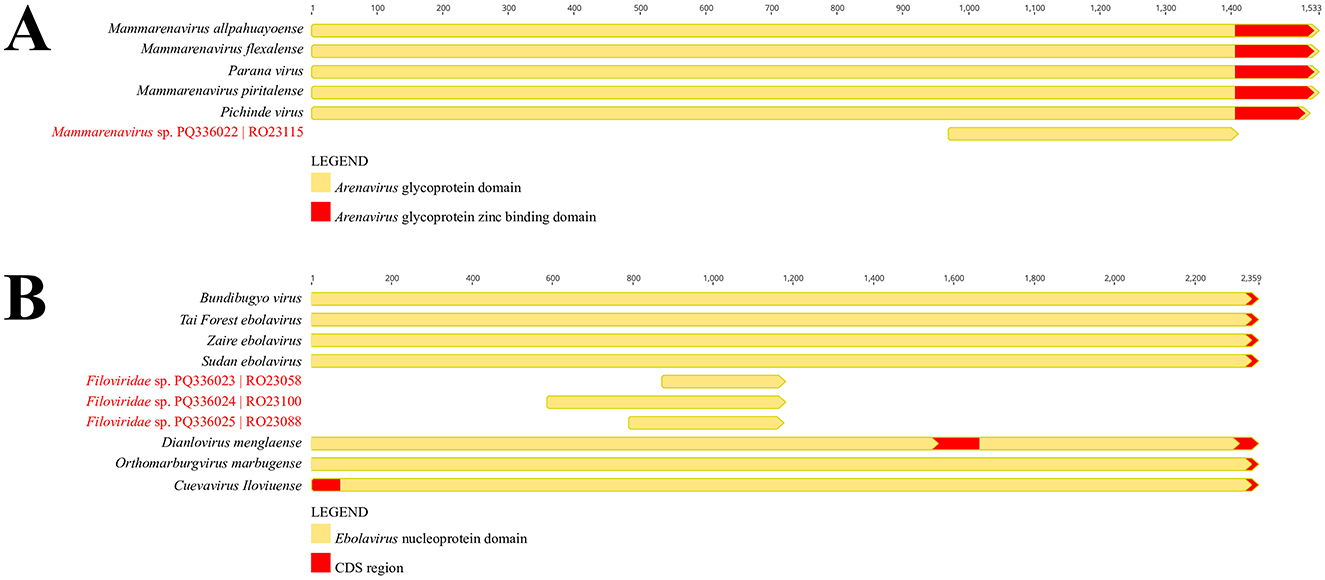
Figure 3. (A) Representation of the functional domains of the Arenaviridae glycoprotein region, comparing the identified contig with other members of the viral family. (B) Representation of the functional domains of the Filoviridae nucleoprotein region, comparing the identified contigs with other members of the viral family (BioProject PRJNA1157879).
Based on the phylogenetic reconstruction using the Maximum Likelihood method, incorporating amino acid and nucleotide information from a set of 45 Arenaviridae sequences representing New and Old World species, it was observed that the identified contig clustered with taxa belonging to clade A of New World Arenaviruses. This clustering was supported by 90.2% (LG+I+G4 substitution model) and 81.6% (GTR+F++R6 substitution model) of quartets resolved during the reconstruction of the amino acid and nucleotide phylogenies, respectively (Figures 4A, B; Supplementary Figure 3). The nucleotide and amino acid identity percentages, particularly between representatives of clades A and C of Arenaviridae in the New World, are shown in Figure 4C.
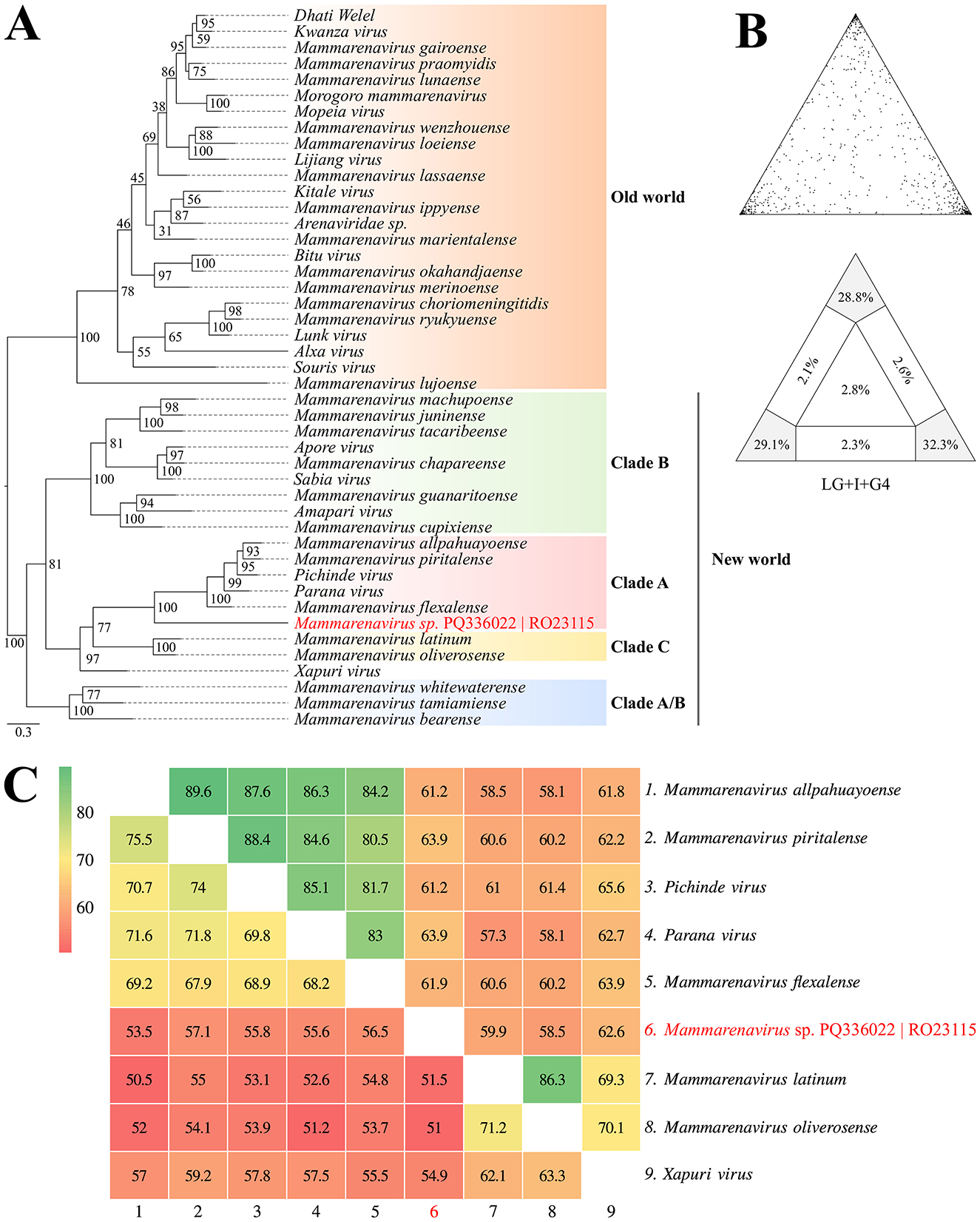
Figure 4. (A) Phylogenetic reconstruction using the Maximum Likelihood method based on amino acid sequences from the glycoprotein region of New and Old World Arenaviruses, including the identified contig highlighted in red. (B) Maximum likelihood mapping diagram showing the quality of the phylogenetic signal from the quartet analysis, with 90.2% of quartets resolved. (C) Heatmap of nucleotide (lower triangle) and amino acid (upper triangle) identities between representatives of clades (A, C) of New World Arenaviridae, including the identified contig (BioProject PRJNA1157879).
3.4.2. Filoviridae
Three viral contigs potentially associated with the Filoviridae family were identified from rodent samples of the species N. lasiurus (samples RO23058 and RO23100) and O. microtis (sample RO23088), identified based on the CytB gene. Of these samples, two were captured in the Canaã dos Carajás region (RO23058 and RO23088), and one was from the Curionópolis region (RO23100). The contigs recovered from rodents captured in Canaã dos Carajás were 303 nt (RO23058) and 393 nt (RO23088) long, featuring functional domains associated with the Filoviridae nucleoprotein region, and shared amino acid identities of 61.8 and 60.7% with Bundibugyo virus and Zaire Ebolavirus, respectively (Figure 3B). Similarly, the contig from sample RO23100, collected in Curionópolis, was 588 nt long, also associated with the Filoviridae nucleoprotein region, and shared an amino acid identity of 53.8% with Bundibugyo virus (Figure 3B).
Based on the phylogenetic reconstruction using the Maximum Likelihood method, incorporating amino acid and nucleotide information from a set of 16 Filoviridae family representatives, it was observed that while the identified contigs were positioned in the same general region of the phylogenetic tree, their exact placement varied depending on the sequence model analyzed. When using the amino acid alignment, the contigs clustered closely with the clade comprising taxa from the genera Orthomarbugvirus and Dianlovirus (Figures 5A, B). Conversely, when evaluated using nucleotide alignment, the contigs were associated as a sister group to the clade containing Orthoebolavirus and Cuevavirus (Supplementary Figure 4). Despite the variable positioning, the contigs, along with the cited genera, formed a monophyletic and well-supported group (BSamino = 99% and BSnucleo = 93%). Although the topologies align with current molecular taxonomic classifications for this viral group, the number of unresolved quartets in each phylogenetic analysis approached or exceeded the 30% threshold, underscoring the need for a larger sample set to accurately define the observed groupings. Additionally, the percentages of nucleotide and amino acid identity between the potential Filoviridae viral fragments and representatives of the genera Orthoebolavirus, Cuevavirus, Orthomarbugvirus, and Dianlovirus are shown in Figure 5C.

Figure 5. (A) Phylogenetic reconstruction using the Maximum Likelihood method based on amino acid sequences of Filoviridae. (B) Maximum Likelihood mapping diagram showing the quality of the phylogenetic signal from the quartet analysis, with 64.8% of quartets resolved. (C) Heatmap of nucleotide (lower triangle) and amino acid (upper triangle) identities between the identified contigs and representatives of the genera Orthoebolavirus, Cuevavirus, Orthomarbugvirus, and Dianlovirus (BioProject PRJNA1157879).
4 Discussion
The municipalities of Canaã dos Carajás and Curionópolis, located in the southwest of the state of Pará, Brazil, are known for their rich biodiversity of Amazonian flora and fauna (Palheta et al., 2015). These areas are also recognized for their mineral reserves, including copper, iron, and nickel (Dos Santos et al., 2018). However, intense mining activities in these regions can lead to habitat destruction for various animals, including rodents (Furtado et al., 2023). This study utilized metagenomic analysis to explore the rodent virome and enhance our understanding of the viral diversity present in these regions.
Initially, 102 rodent samples were used to develop the study. These samples were screened by ELISA as a strategic way to verify whether any of these rodents had anti-Orthohantavirus antibodies and to direct the selection of samples for metagenomic study. Thus, of the 102 rodent samples, 100 presented OD values below 0.2, being considered negative, while two samples were defined as indeterminate, presenting OD at the cutoff point: one from O. microtis (sample RO23088), captured in Canaã dos Carajás, and another from N. lasiurus (sample RO23100), captured in Curionópolis.
In Brazil, the rodent O. microtis has been described as a reservoir of the Rio Mamoré orthohantavirus (RIOMV) in the states of Amazonas (Santos et al., 2006) and Acre (Nunes et al., 2015), while the rodent N. lasiurus was associated as the main reservoir of the Araraquara virus (ARAV) in the states of São Paulo, Minas Gerais, Mato Grosso do Sul, and Paraná (Suzuki et al., 2004; Santos et al., 2006; Raboni et al., 2009; Oliveira et al., 2011; dos Santos et al., 2011; Travassos da Rosa et al., 2012). In the state of Pará, in the municipality of Altamira, human cases of Hantavirus Cardiopulmonary Syndrome (HCPS) were associated with the Castelo dos Sonhos virus (CASV) (Johnson et al., 1999). Subsequently, studies carried out in Campo Novo do Parecis, in the state of Mato Grosso, identified the rodent Oligorizomys utiaritensis as the reservoir of CASV (Travassos da Rosa et al., 2011).
Later, ~520 million reads were identified in the metagenomic profile, with 409 million remaining after quality control. These reads were assigned to 53 distinct viral families. Notably, around 43.75% of these reads were related to 14 families that infect vertebrates, such as Arenaviridae, Coronaviridae, Filoviridae, Flaviviridae, Picornaviridae, and Retroviridae. These families are particularly relevant to public health, as many of their members can cause diseases in humans and other vertebrate animals. Furthermore, 46.88% of the reads were associated with 15 viral families infecting invertebrates (e.g., Ascoviridae, Baculoviridae, Iridoviridae), plants (e.g., Caulimoviridae, Potyviridae), and bacteria (e.g., Myoviridae, Siphoviridae). The remaining 9.38% of the reads were linked to other hosts, including fungi (e.g., Partitiviridae) and amoebas (e.g., Marseilleviridae). These results underscore the capacity of these hosts to harbor a wide variety of viruses.
Analysis of the alpha and beta diversity indices revealed that samples RO23035 (O. microtis) and RO23057 (Oryzomys sp.), collected from rodents in Canaã dos Carajás, had the highest abundance of viral reads, predominantly associated with the Retroviridae family. Conversely, sample RO23100 (N. lasiurus), from a rodent captured in Curionópolis, showed the lowest abundance of viral reads but exhibited the highest diversity index, indicating a greater richness of viral families. This finding was supported by the Shannon and Simpson indices, which demonstrated a better distribution of viral families in sample RO23100. Despite this high diversity index, the metagenomic profiles of samples from the two regions showed similarities, with abundant reads for the families Baculoviridae, Iridoviridae, Marseilleviridae, Microviridae, Mimiviridae, Phycodnaviridae, Poxviridae, and Retroviridae. This distribution may relate to the feeding habits of some rodent species (e.g., consumption of plants and insects), environmental contamination of habitats, or changes in the ecological dynamics due to human activities in these areas (Zhao et al., 2022).
Kane et al. (2024) observed that viral diversity in rodents is positively correlated with habitat disturbance. Their studies revealed that rodents such as Rattus tanezumi, captured in homes and agricultural fields, exhibited high viral diversity in their metagenomic profiles. Conversely, Rattus andamanensis found in rubber tree plantations had higher viral diversity compared to individuals of the same species from less disturbed tropical forests. These findings suggest that increased habitat disturbance due to human activities may lead to greater virome diversity in rodents, highlighting the impact of environmental changes on viral composition.
In this study, metagenomic analysis identified four virus-like fragments: one associated with the Arenaviridae family and three with the Filoviridae family. The Arenaviridae family consists of ambisense RNA viruses with genomes approximately 10.5 kb in size. These viruses infect mammals (mammarenaviruses), snakes (hartmaniviruses and reptarenaviruses), and fish (antennaviruses) (Radoshitzky et al., 2019).
The genome of Arenaviridae is divided into two main segments: the L (large) segment, encoding the RNA-dependent RNA polymerase (RdRP) and the matrix protein (Z), and the S (small) segment, encoding the nucleoprotein (NP) and the glycoprotein (GP) (Radoshitzky et al., 2023). Among the recovered fragments, one associated with the Arenaviridae family was identified from a rodent of the genus Oligoryzomys (sample RO23115), captured in the Curionópolis region. This fragment contained functional domains within the glycoprotein region and shared 64% amino acid identity with Mammarenavirus piritalense, a virus discovered in Venezuela associated with the rodent Sigmodon alstoni (Fulhorst et al., 1997). Additionally, the rodent O. microtis has been described as a reservoir for Chapare virus (CHPV), which causes Bolivian hemorrhagic fever (Loayza et al., 2022). These findings suggest possible circulation of arenaviruses in rodents in the Curionópolis region, similar to viruses known to infect humans in other parts of South America. However, further sampling is needed to enhance the robustness of these results (Fulhorst et al., 1997; Loayza et al., 2022).
In Brazil, the natural reservoir for the Brazilian Mammarenavirus (Sabiá virus), which causes Brazilian hemorrhagic fever, remains unidentified (Nastri et al., 2022). It is suspected that a rodent species may be the host, underscoring the importance of identifying potential natural hosts for epidemiological surveillance and reinforcing the significance of metagenomic studies (Mello et al., 2020; Lendino et al., 2024).
The Filoviridae family comprises negative-sense, linear, non-segmented RNA viruses, ~13.1 to 20.9 kb in size, that infect fish, mammals, and reptiles. This family contains five open reading frames (ORFs) encoding a nucleoprotein (NP), a polymerase cofactor (VP35), a glycoprotein (GP), a transcriptional activator (VP30), and a large protein (L) with an RNA-directed RNA polymerase (RdRP) domain (Biedenkopf et al., 2023). It is divided into six genera: Cuevavirus, Dianlovirus, Orthoebolavirus, Orthomarburgvirus, Striavirus, and Thamnovirus (Kuhn et al., 2019). The genera Orthoebolavirus and Orthomarburgvirus include viruses known to cause human diseases, such as Ebola virus (EBOV), Sudan virus (SUDV), Bundibugyo virus (BDBV), Tai Forest virus (TAFV), Reston virus (RESTV), Marburg virus (MARV), and Ravn virus (RAVV). Other viruses like Bombali virus (BOMV) and Lloviu virus (LLOV) (genus Cuevavirus) and Mêngla virus (MLAV) (genus Dianlovirus) are known members of this family but have not been associated with human diseases.
Peterson et al. (2007) propose that some rodent groups might act as potential reservoirs for filoviruses due to their wide geographic distribution, which overlaps with known areas of Ebola and Marburg outbreaks in Africa. However, they stress the necessity for further research, particularly in filovirus-endemic regions, to confirm this hypothesis.
Taylor et al. (2010, 2011) have highlighted a significant evolutionary association between rodents and filoviruses. Their studies revealed the presence of integrated non-retroviral viral elements (NIRVs) in rodent genomes, which are similar to the nucleoprotein (NP) and polymerase cofactor (VP35) proteins of filoviruses. In 2010, they identified these NIRVs in mice and rats, suggesting an ancient coevolution that may affect the biological functions of these elements, potentially related to immune responses. In 2011, they confirmed the conservation of these genes in rodents over millions of years, indicating that these elements might have been co-opted for essential biological functions rather than being mere viral remnants. Although the absence of live viruses or antibodies in rodents raises questions about their role as current reservoirs, the persistence of these NIRVs points to a long-standing evolutionary interaction between filoviruses and rodents, with possible implications for the ecology and evolution of filoviruses.
Recent research by Taylor and Barnhart (2023) identified an orthologous Filovirus-like nucleoprotein in murid and spalacid rodents. Structural and phylogenetic analyses suggest that these integrated elements may serve important biological functions, preserved for over 20 million years, possibly through purifying selection. This evidence supports a long history of interaction between rodents and filoviruses, indicating that these animals may have been ancient reservoirs. Additionally, the relationship between NIRVs in Australian marsupials and African filoviruses suggests that regions of the New World may harbor previously undetected filoviruses or could be sources for existing filoviruses.
Dupuy et al. (2023) emphasize the need to investigate potential natural reservoirs for filoviruses, including rodents, despite most current evidence focusing on bats. Their study underscores the importance of further research to clarify the role of rodents in Filovirus ecology, particularly in regions where the coevolution of these viruses with their hosts remains poorly understood. The presence of Filovirus-like fragments in N. lasiurus and O. microtis suggests that these rodents might play a role in maintaining these viruses in nature. However, these findings, along with existing literature, highlight the need for additional studies to better understand the role of these rodents in Filovirus ecology and to assess potential transmission risks to other species, including humans. Further collections in the studied areas are essential to obtain more sequences and elucidate the relationship between rodents and these viruses.
Data availability statement
The sequences obtained here was deposited in the GenBank database under accessions PQ336022 (Mammarenavirus sp.), PQ336023 - PQ336025 (Filoviridae sp.), and the raw sequence reads generated are available in the NCBI Sequence Read Archive (SRA) database under BioProject PRJNA1157879. The deposit number of the sequences can be seen in the images.
Ethics statement
The animal study was approved by Evandro Chagas Institute CEUA/IEC (protocol no. 06/2014, certificate no. 43/2015). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
AM: Conceptualization, Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. FS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. AC: Resources, Writing – review & editing. SS: Data curation, Methodology, Writing – review & editing. AQ: Methodology, Writing – review & editing. LC: Resources, Writing – review & editing. LM: Funding acquisition, Project administration, Resources, Writing – review & editing. DM: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Coordination for the Improvement of Higher Education Personnel (CAPES) (process No. 88887.799456/2022-00) and by the Evandro Chagas Institute (IEC/SVSA/MS).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1502462/full#supplementary-material
References
Aguirre, A. A. (2017). Changing patterns of emerging zoonotic diseases in wildlife, domestic animals, and humans linked to biodiversity loss and globalization. ILAR J. 58, 315–318. doi: 10.1093/ilar/ilx035
Bernt, M., Donath, A., Jühling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Biedenkopf, N., Bukreyev, A., Chandran, K., Di Paola, N., Formenty, P. B. H., Griffiths, A., et al. (2023). Renaming of genera Ebolavirus and Marburgvirus to Orthoebolavirus and Orthomarburgvirus, respectively, and introduction of binomial species names within family Filoviridae. Arch. Virol. 168:220. doi: 10.1007/s00705-023-05834-2
Bovincino, C. R., Oliveira, J. A., and Andrea, P. S. (2008). Guia dos roedores do Brasil, com chave para gêneros baseadas em caracteres externos. Rio de Janeiro: Centro Pan–Americano de Febre Aftosa, OPAS/OMS, 122.
Buchfink, B., Reuter, K., and Drost, H.-G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368. doi: 10.1038/s41592-021-01101-x
Carlson, C. J., Zipfel, C. M., Garnier, R., and Bansal, S. (2019). Global estimates of mammalian viral diversity accounting for host sharing. Nat. Ecol. Evol. 3, 1070–1075. doi: 10.1038/s41559-019-0910-6
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
dos Santos, J. P., Steinke, E. T., and García-Zapata, M. T. A. (2011). Land use and occupation and hantavirosis dissemination in the São Sebastião region, Federal District: 2004 - 2008. Rev. Soc. Bras. Med. Trop. 44, 53–57. doi: 10.1590/S0037-86822011000100013
Dos Santos, M. M., Rodrigues, R. L., and da Cruz, T. M. (2018). Mineração e conflitos pela posse da terra em Canaã dos Carajás: O caso do acampamento Planalto Serra Dourada. Revista De Extensão E Estudos Rurais 7, 117–140. doi: 10.36363/rever722018117-140
Dupuy, L. C., Spiropoulou, C. F., Towner, J. S., Spengler, J. R., Sullivan, N. J., and Montgomery, J. M. (2023). Filoviruses: scientific gaps and prototype pathogen recommendation. J. Infect. Dis. 228, S446–S459. doi: 10.1093/infdis/jiad362
Ferreira, L. V., Venticinque, E., and Almeida, S. (2005). O desmatamento na Amazônia e a importância das áreas protegidas. Estud. Av. 19, 157–166. doi: 10.1590/S0103-40142005000100010
Firth, C., Tokarz, R., Simith, D. B., Nunes, M. R. T., Bhat, M., Rosa, E. S. T., et al. (2012). Diversity and distribution of hantaviruses in South America. J. Virol. 86, 13756–13766. doi: 10.1128/JVI.02341-12
Fulhorst, C. E., Bowen, M. D., Salas, R. A., de Manzione, N. M., Duno, G., Utrera, A., et al. (1997). Isolation and characterization of pirital virus, a newly discovered South American arenavirus. Am. J. Trop. Med. Hyg. 56, 548–553. doi: 10.4269/ajtmh.1997.56.548
Furtado, L. G., Braga Pereira, C., Silva, D. F., Belato, L. S., and Pereira, B. W. (2023). Detecção de Mudanças do Uso e Cobertura do Solo no Município de Canaã dos Carajás. Pará Revista Verde Grande. 5, 116–131. doi: 10.46551/rvg2675239520232116131
Han, B. A., Schmidt, J. P., Bowden, S. E., and Drake, J. M. (2015). Rodent reservoirs of future zoonotic diseases. Proc. Natl. Acad. Sci. U. S. A. 112, 7039–7044. doi: 10.1073/pnas.1501598112
He, X., Wang, X., Fan, G., Li, F., Wu, W., Wang, Z., et al. (2022). Metagenomic analysis of viromes in tissues of wild Qinghai vole from the eastern Tibetan Plateau. Sci. Rep. 12:17239. doi: 10.1038/s41598-022-22134-y
Horefti, E. (2023). The importance of the One Health concept in combating zoonoses. Pathogens 12:977. doi: 10.3390/pathogens12080977
Johnson, A. M., de Souza, L. T. M., Ferreira, I. B., Pereira, L. E., Ksiazek, T. G., Rollin, P. E., et al. (1999). Genetic investigation of novel hantaviruses causing fatal HPS in Brazil. J. Med. Virol. 59, 527–535. doi: 10.1002/(SICI)1096-9071(199912)59:4<527::AID-JMV17>3.0.CO;2-Y
Kane, Y., Tendu, A., Li, R., Chen, Y., Mastriani, E., Lan, J., et al. (2024). Viral diversity in wild and urban rodents of Yunnan Province, China. Emerg. Microbes Infect. 13:2290842. doi: 10.1080/22221751.2023.2290842
Katoh, K., Misawa, K., Kuma, K.-I., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucl. Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kopylova, E., Noé, L., and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. doi: 10.1093/bioinformatics/bts611
Kuhn, J. H., Amarasinghe, G. K., Basler, C. F., Bavari, S., Bukreyev, A., Chandran, K., et al. (2019). ICTV virus taxonomy profile: Filoviridae. J. Gen. Virol. 100, 911–912. doi: 10.1099/jgv.0.001252
Lendino, A., Castellanos, A. A., Pigott, D. M., and Han, B. A. (2024). A review of emerging health threats from zoonotic New World mammarenaviruses. BMC Microbiol. 24:115. doi: 10.1186/s12866-024-03257-w
Levis, S., Morzunov, S. P., Rowe, J. E., Enria, D., Pini, N., Calderon, G., et al. (1998). Genetic diversity and epidemiology of hantaviruses in Argentina. J. Infect. Dis. 177, 529–538. doi: 10.1086/514221
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Loayza, M. R., Morales-Betoulle, M. E., Romero, C., Cossaboom, C. M., Whitmer, S., Alvarez, A. C. E., et al. (2022). Chapare hemorrhagic fever and virus detection in rodents in Bolivia in 2019. N. Engl. J. Med. 386, 2283–2294. doi: 10.1056/NEJMoa2110339
Luis, A. D., Hayman, D. T. S., O'Shea, T. J., Cryan, P. M., Gilbert, A. T., Pulliam, J. R. C., et al. (2013). A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc. Biol. Sci. 280:20122753. doi: 10.1098/rspb.2012.2753
Meerburg, B. G., Singleton, G. R., and Kijlstra, A. (2009). Rodent-borne diseases and their risks for public health. Crit. Rev. Microbiol. 35, 221–270. doi: 10.1080/10408410902989837
Mello, M. F., Amgarten, D., Nastri, A. C., Ho, S. S., Boas, C. Y. L., and Basqueira, L. V. (2020). Sabiá virus-like mammarenavirus in patient with fatal hemorrhagic fever, Brazil. Brazil. Emerg. Infect. Dis. 26, 1332–1334 doi: 10.3201/eid2606.200099
Milazzo, M. L., Barragán-Gomez, A., Hanson, J. D., Estrada-Franco, J. G., Arellano, E., González-Cózatl, F. X., et al. (2010). Antibodies to Tacaribe serocomplex viruses (familyarenaviridae, genusArenavirus) in cricetid rodents from New Mexico, Texas, and Mexico. Vect. Borne Zoon. Dis. 10, 629–637. doi: 10.1089/vbz.2009.0206
Moncayo, A. C., Hice, C. L., Watts, D. M., Travassos de Rosa, A. P. A., Guzman, H., Russell, K. L., et al. (2001). Allpahuayo virus: a newly recognized Arenavirus (arenaviridae) from arboreal rice rats (Oecomys bicolor and Oecomys paricola) in northeastern Peru. Virology 284, 277–286. doi: 10.1006/viro.2000.0803
Nastri, A. C., Duarte-Neto, A. N., Casadio, L. V. B., Souza, W. M., de Claro, I. M., Manuli, E. R., et al. (2022). Understanding Sabiá virus infections (Brazilian mammarenavirus). Travel Med. Infect. Dis. 48:102351. doi: 10.1016/j.tmaid.2022.102351
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nunes, M. L., de Oliveira, S. V., da Elkhoury, M. R., Fonseca, L. X., Pereira, S. V. C., et al. (2015). Evidência de circulação de hantavirus em área silenciosa da Região Amazônica. Rev. Panamazonica Saude 6, 63–67. doi: 10.5123/S2176-62232015000400009
Oliveira, R. C., Guterres, A., Fernandes, J., D'Andrea, P. S., Bonvicino, C. R., and de Lemos, E. R. S. (2014). Hantavirus reservoirs: current status with an emphasis on data from Brazil. Viruses 6, 1929–1973. doi: 10.3390/v6051929
Oliveira, R. C., Padula, P. J., Gomes, R., Martinez, V. P., Bellomo, C., Bonvicino, C. R., et al. (2011). Genetic characterization of hantaviruses associated with sigmodontine rodents in an endemic area for hantavirus pulmonary syndrome in southern Brazil. Vect. Borne Zoon. Dis. 11, 301–314. doi: 10.1089/vbz.2010.0001
Palheta, J. M., Silva, C. N., and Medeiros, G. N. (2015). Territórios com mineração na região sudeste do Pará-Norte do Brasil. Revista Anpege 11, 281–308. doi: 10.5418/RA2015.1115.0011
Peterson, A. T., Papeş, M., Carroll, D. S., Leirs, H., and Johnson, K. M. (2007). Mammal taxa constituting potential coevolved reservoirs of filoviruses. J. Mammal. 88, 1544–1554. doi: 10.1644/06-MAMM-A-280R1.1
Phan, T. G., Kapusinszky, B., Wang, C., Rose, R. K., Lipton, H. L., and Delwart, E. L. (2011). The fecal viral flora of wild rodents. PLoS Pathog. 7:e1002218. doi: 10.1371/journal.ppat.1002218
Plowright, R. K., Reaser, J. K., Locke, H., Woodley, S. J., Patz, J. A., Becker, D. J., et al. (2021). Land use-induced spillover: a call to action to safeguard environmental, animal, and human health. Lancet Planet. Health 5, e237–e245. doi: 10.1016/S2542-5196(21)00031-0
Raboni, S. M., de Borba, L., Hoffmann, F. G., de Noronha, L., Azevedo, M. L. V., Carstensen, S., et al. (2009). Evidence of circulation of Laguna Negra-like hantavirus in the Central West of Brazil: case report. J. Clin. Virol. 45, 153–156. doi: 10.1016/j.jcv.2009.03.015
Radoshitzky, S. R., Buchmeier, M. J., Charrel, R. N., Clegg, J. C. S., Gonzalez, J.-P. J., Günther, S., et al. (2019). ICTV virus Taxonomy profile: Arenaviridae. J. Gen. Virol. 100, 1200–1201. doi: 10.1099/jgv.0.001280
Radoshitzky, S. R., Buchmeier, M. J., Charrel, R. N., Gonzalez, J.-P. J., Günther, S., Hepojoki, J., et al. (2023). ICTV virus Taxonomy profile: Arenaviridae 2023. J. Gen. Virol. 104:1891. doi: 10.1099/jgv.0.001891
Rahman, M. T., Sobur, M. A., Islam, M. S., Ievy, S., Hossain, M. J., El Zowalaty, M. E., et al. (2020). Zoonotic diseases: etiology, impact, and control. Microorganisms 8:1405. doi: 10.3390/microorganisms8091405
Rupasinghe, R., Chomel, B. B., and Martínez-López, B. (2022). Climate change and zoonoses: a review of the current status, knowledge gaps, and future trends. Acta Trop. 226:106225. doi: 10.1016/j.actatropica.2021.106225
Santos, M. C., de Lacerda, M. V. G., Benedetti, S. M., Albuquerque, B. C., de Aguiar Filho, A. A. B. V., da Rosa Elkhoury, M., et al. (2006). Human hantavirus infection, Brazilian Amazon. Emerg. Infect. Dis. 12, 1165–1167. doi: 10.3201/eid1207.060074
Suzuki, A., Bisordi, I., Levis, S., Garcia, J., Pereira, L. E., Souza, R. P., et al. (2004). Identifying rodent hantavirus reservoirs, Brazil. Emerg. Infect. Dis. 10, 2127–2134. doi: 10.3201/eid1012.040295
Tapia-Ramírez, G., Lorenzo, C., Navarrete, D., Carrillo-Reyes, A., Retana, Ó., and Carrasco-Hernández, R. (2022). A review of mammarenaviruses and rodent reservoirs in the Americas. Ecohealth 19, 22–39. doi: 10.1007/s10393-022-01580-0
Taylor, D. J., and Barnhart, M. H. (2023). Genomic transfers help to decipher the ancient evolution of filoviruses and interactions with vertebrate hosts. bioRxiv [preprint]. doi: 10.1101/2023.11.29.569234
Taylor, D. J., Dittmar, K., Ballinger, M. J., and Bruenn, J. A. (2011). Evolutionary maintenance of filovirus-like genes in bat genomes. BMC Evol. Biol. 11:336. doi: 10.1186/1471-2148-11-336
Taylor, D. J., Leach, R. W., and Bruenn, J. (2010). Filoviruses are ancient and integrated into mammalian genomes. BMC Evol. Biol. 10:193. doi: 10.1186/1471-2148-10-193
Thompson, R. C. A. (2013). Parasite zoonoses and wildlife: One health, spillover and human activity. Int. J. Parasitol. 43, 1079–1088. doi: 10.1016/j.ijpara.2013.06.007
Tirera, S., de Thoisy, B., Donato, D., Bouchier, C., Lacoste, V., Franc, A., et al. (2021). The influence of habitat on viral diversity in neotropical rodent hosts. Viruses 13:1690. doi: 10.3390/v13091690
Travassos da Rosa, E. S., Medeiros, D. B. A., Nunes, M. R. T., Simith, D. B., de Souza Pereira, A., Elkhoury, M. R., et al. (2011). Pygmy rice rat as potential host of Castelo dos Sonhos Hantavirus. Emerg. Infect. Dis. 17, 1527–1530. doi: 10.3201/eid1708.101547
Travassos da Rosa, E. S., Medeiros, D. B. A., Nunes, M. R. T., Simith, D. B., Pereira, A., de, S., et al. (2012). Molecular epidemiology of Laguna Negra virus, Mato Grosso State, Brazil. Emerg. Infect. Dis. 18, 982–985. doi: 10.3201/eid1806.110948
Travassos da Rosa, E. S., Mills, J. N., Padula, P. J., Elkhoury, M. R., Ksiazek, T. G., Mendes, W. S., et al. (2005). Newly recognized hantaviruses associated with hantavirus pulmonary syndrome in northern Brazil: partial genetic characterization of viruses and serologic implication of likely reservoirs. Vect. Borne Zoon. Dis. 5, 11–19. doi: 10.1089/vbz.2005.5.11
Tsoleridis T Ball J. K. (2020). Discovery of novel coronaviruses in rodents. Methods. Mol. Biol. 2203, 33–40. doi: 10.1007/978-1-0716-0900-2_2
Wang, W., Lin, X.-D., Zhang, H.-L., Wang, M.-R., Guan, X.-Q., Holmes, E. C., et al. (2020). Extensive genetic diversity and host range of rodent-borne coronaviruses. Virus Evol. 6:veaa078. doi: 10.1093/ve/veaa078
Wu, Z., Han, Y., Liu, B., Li, H., Zhu, G., Latinne, A., et al. (2021). Decoding the RNA viromes in rodent lungs provides new insight into the origin and evolutionary patterns of rodent-borne pathogens in Mainland Southeast Asia. Microbiome 9:18. doi: 10.1186/s40168-020-00965-z
Wu, Z., Yang, L., Ren, X., He, G., Zhang, J., Yang, J., et al. (2016). Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 10, 609–620. doi: 10.1038/ismej.2015.138
Yin, H.-C., Wan, D.-C., and Chen, H.-Y. (2022). Metagenomic analysis of viral diversity and a novel astroviruse of forest rodent. Virol. J. 19:138. doi: 10.1186/s12985-022-01847-6
Keywords: Arenaviridae, epidemiologic vigilance, Filoviridae, metagenomics, rodent
Citation: Monteiro AFM, Silva FSd, Cruz ACR, Silva SPd, Queiroz ALN, Casseb LMN, Martins LC and Medeiros DBdA (2025) Viral diversity in wild rodents in the regions of Canaã de Carajás and Curionopólis, State of Pará, Brazil. Front. Microbiol. 15:1502462. doi: 10.3389/fmicb.2024.1502462
Received: 26 September 2024; Accepted: 10 December 2024;
Published: 07 January 2025.
Edited by:
Tiong Gim Aw, Tulane University, United StatesReviewed by:
Jing Yang, National Institute for Communicable Disease Control and Prevention (China CDC), ChinaBeatriz Cancino-Faure, Universidad Católica del Maule, Chile
Copyright © 2025 Monteiro, Silva, Cruz, Silva, Queiroz, Casseb, Martins and Medeiros. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniele Barbosa de Almeida Medeiros, ZGFuaWVsZW1lZGVpcm9zQGllYy5nb3YuYnI=