 Jing An1
Jing An1 Jizhong Zhou
Jizhong Zhou Dashuai Mu
Dashuai Mu- 1Marine College, Shandong University, Weihai, China
- 2Institute of Ecology, Key Laboratory for Earth Surface Processes of the Ministry of Education, College of Urban and Environmental Sciences, Peking University, Beijing, China
- 3Department of Microbiology and Plant Biology, University of Oklahoma, Norman, OK, United States
- 4Institute for Environmental Genomics, University of Oklahoma, Norman, OK, United States
- 5State Key Joint Laboratory of Environment Simulation and Pollution Control, School of Environment, Tsinghua University, Beijing, China
- 6School of Civil Engineering and Environmental Sciences, University of Oklahoma, Norman, OK, United States
- 7School Key Laboratory of Microbial Technology, Shandong University, Qingdao, China
- 8Weihai Research Institute of Industrial Technology of Shandong University, Weihai, China
The strain designated as Y139T is a novel Gram-stain-negative, aerobic, and non-motile bacterium, was isolated from a soil sample in McClain County, Oklahoma, United States. The cells of strain Y139T were a rod-shaped, with the width of 0.4–0.7
1 Introduction
The genus Luteolibacter is classified within the phylum Verrucomicrobiota. Luteolibacter pohnpeiensis is the type species of the genus Luteolibacter which was first described in 2008 by Yoon et al. (2008). This genus includes 11 validly named species listed in LPSN.1 The genus Luteolibacter has been found in various environments, including marine environments (Zhang et al., 2017; Xie et al., 2022; Zhou et al., 2023), tundra soil (Jiang et al., 2012; Kim et al., 2015), activated sludge (Park et al., 2013), soil (Pascual et al., 2017; Dahal et al., 2021), skin of Anderson’s salamander (Busse et al., 2021) and Hirudo medicinalis (Glaeser et al., 2012). The major respiratory quinone present in Luteolibacter species is Menaquinone-9 (MK-9). The genomic DNA G + C content of the DNA in these species varies from 53.5 to 65.0% (Yoon et al., 2008; Xie et al., 2022).
The significance of global climate change is undeniable, as it has widespread impacts on ecosystems. Microorganisms are crucial in climate change due to their wide distribution, short generation times and larger populations (Zhang and Xu, 2008; Collins, 2011; Thomas et al., 2012). In terrestrial ecosystems, soil serves as the largest carbon sink, small changes in soil microorganisms can cause significant changes in climate feedback. The phylum Verrucomicrobiota is commonly found in soil environments, especially in grasslands and soil horizons. This suggests that members of this phylum may play significant roles in the soil environment (Kielak et al., 2009; Bergmann et al., 2011). The phylum Verrucomicrobiota has a significant impact on the global biogeochemical cycling process, particularly in terms of their contribution to nutrient cycling (Banerjee et al., 2016). Despite the acknowledged significance of Verrucomicrobiota in the environment, research on their responses to climate change remains limited.
The composition and functionality of microbial communities may shift due to climate change (Zhou et al., 2012; Xu et al., 2013; Guo et al., 2018; Wu et al., 2022). The phylum Verrucomicrobiota has been revealed to be sensitive to climate warming and precipitation. This sensitivity could have several implications. (a) Distribution: changes in temperature and precipitation patterns could affect the geographic distribution (Oliverio et al., 2017); (b) Abundance: population sizes might fluctuate in response to climatic variations, a rise in temperature triggered a significant drop in both species diversity and relative abundance. Increased precipitation can enhance the relative abundance, indicating their favorable survival in environments with higher moisture content (Zhang et al., 2018; Zhao et al., 2024); (c) Activity: metabolic rates and ecological functions could be impacted by changing environmental conditions. Our research focused on an aerobic soil isolate, recognized as a novel species of genus Luteolibacter from the phylum Verrucomicrobiota. Enhancing the genus Luteolibacter will support future research on microbial responses to climate variability.
2 Materials and methods
2.1 Study site and sampling
The Kessler Atmospheric and Ecological Field Station (KAEFS) is located in McClain County, Oklahoma, in the US Great Plains (34° 59′ N, 97° 31′ W) (Guo et al., 2018). The site is mainly composed of C3 and C4 herbaceous plants (Xu et al., 2013). From 1948 to 1999, the average temperature in January was 3.3°C and that in July was 28.1°C, with an annual average of 16.3°C. Precipitation ranged from 82 to 240 mm, with an average annual total of 914 mm. The soils are classified as a Pulaski-Port Keokuk complex, which is characterized by a loamy texture (Li et al., 2013). In July 2009, a field experiment was built. A blocked split-plot experiment was built to analyze the impact of three climate factors on ecosystems: warming, precipitation, and clipping. The warming experimental plots were heated continuously to maintain a temperature of 3°C above ambient. Precipitation adjustment consisted of setting the target level to +100% of ambient precipitation to simulate increased rainfall. Also, aboveground biomass was clipped annually during the peak growing season (Guo et al., 2018; Wu et al., 2022).
2.2 Amplicon sequencing and data preprocessing
The construction of the 16S rRNA gene (V4 region) library involved a two-step PCR amplification method, followed by Wu et al. (2022). The paired-end sequences obtained were subjected to primer sequence trimming. After trimming, the sequences were merged using FLASH. Merged sequences with ambiguous bases or a length of less than 245 bp for the 16S rRNA gene were not included in the subsequent analysis. High-quality sequences of the 16S rRNA gene that remained were then processed to create amplicon sequence variants (ASVs). This processing step was performed using UNOISE3 (Edgar, 2018).
2.3 Bacterial isolation and cultivation
A soil sample was obtained from a grassland site at KAEFS in August 2009. The sampling depth was established at approximately 0–15 cm below the surface, then soil samples were stored at −80°C (Guo et al., 2018). In 2020, 1 g of soil sample was serially diluted to 10−2–10−4 in sterilized water and 0.1 mL aliquots of each dilution were spread onto the surface of 1/3R2A agar medium. The R2A agar medium contained (g/L): tryptone 0.5, yeast 0.5, casein 0.5, starch 0.5, glucose 0.5, K2HPO4 0.3, sodium pyruvate 0.3, and MgSO4∙7H2O 0.05, agar 15 g, all w/v, pH 7.2. The incubation of the agar plates was carried out aerobically at 28°C for around 10 days. A yellow colony was isolated from the incubated plates and subsequently purified through successive subcultures on R2A agar identified as strain Y139T. Pure cultures of the strains were preserved at −80°C supplemented with 15% (v/v) glycerol.
2.4 16S rRNA gene sequence analysis
PCR amplification was performed using the universal primers 27F and 1492R (Liu et al., 2014). The ContEst16S algorithm was used to extract the complete sequence from the genome and submit it to the GenBank database (Lee et al., 2017). The 16S rRNA sequences were compared using BLAST algorithms on GenBank2 and the EzTaxon-e server3 to identify the phylogenetic position of strain Y139T (Kim et al., 2012). The phylogenetic trees were reconstructed in MEGA 11 using neighbor-joining (NJ), maximum-parsimony (MP), and maximum-likelihood (ML) methods (Felsenstein, 1981; Saitou and Nei, 1987; Kannan and Wheeler, 2012; Kumar et al., 2018). The ML tree reconstruction employed the model GTR + G + I. To ascertain the confidence level of the branch nodes, a bootstrap test with 1,000 replicates was performed.
2.5 Genomic analyses
Using a bacterial genomic DNA kit (TaKaRa Biotechnology, Japan), the genomic DNA of strain Y139T was extracted and purified, and subsequently sequenced by Beijing Novogene Bioinformatics Technology (Beijing, China) on the NovaSeq 6,000 sequencing platform. Genome sequences of Luteolibacter flavescens MCCC 1K03193T (GCA_025950085.1), Luteolibacter. pohnpeiensis CCTCC AB 2011006T (GCA_016595435.1) and Luteolibacter arcticus CCTCC AB 2014275T (GCA_025950235.1) were downloaded from the NCBI database. Completeness and contamination levels of genomes were evaluated with CheckM (Parks et al., 2015). To annotate the genome information, the NCBI prokaryotic genome annotation pipeline (PGAP) was used. The average amino acid identity (AAI) value was computed with CompareM. The average nucleotide identity (ANI) value was calculated using an online ANI tool4 (Figueras et al., 2014). The digital DNA–DNA hybridization (dDDH) values were calculated using the Genome-to- Genome Distance Calculator.5 The JTT + CAT parameters were used in FastTree and the LG + F + I + G4 model was used in IQ-Tree to reconstruct phylogenetic trees (Price et al., 2010; Trifinopoulos et al., 2016). For tree support, 1,000 bootstrap replicates were used in both approaches.
2.6 Comparative genomics of the genus Luteolibacter
Genomes of type species within the genus Luteolibacter were obtained from NCBI. The genomes were annotated, and comparative genomics analysis was performed of the genus Luteolibacter through three databases: KEGG database6 (Kanehisa et al., 2023), RAST database7 (Aziz et al., 2008) and Anti-SMASH database8 (Blin et al., 2019). To assess genomic diversity and identify orthologous groups among the members of the genus Luteolibacter, pan-genome analysis using the bacterial pan-genome analysis (BPGA) tool was performed with default parameters (50% amino acid sequence identity) (Chaudhari et al., 2016).
2.7 Physiological and biochemical characteristics
Cells were grown on R2A agar at 30°C for 3 days to investigate the morphological features of strain Y139T. A Gram stain kit (Hopebio, Qingdao, China) was used to perform Gram staining. The motility characteristics were assessed by examining gliding motility (Bernardet et al., 2002). Light microscopy (Nikon E600) and scanning electron microscopy (Nova NanoSEM 450; FEI) were used to determine the morphology and size of the cells. The growth conditions for strain Y139T were evaluated across a range of temperatures (0, 4, 10, 15, 20, 25, 28, 30, 33, 37, 40, 42, and 45°C) on R2A agar. To investigate the salt tolerance of cells, they were cultured in R2A medium with NaCl added at concentrations of 0, 0.5, 1, 1.5, 2, 3, 4 and 5% (w/v). The range of pH values (5.5–9.5) was assessed by adding specific buffers into the R2A medium: MES (pH 5.5–6.0), PIPES (pH 6.5–7.0), HEPES (pH 7.5–8.0), Tricine (pH 8.5), and CAPSO (pH 9.0–9.5) at a concentration of 20 mM. Using a reagent kit from bioMérieux, oxidase activity was assessed. Catalase activity was determined by adding 3% (v/v) hydrogen peroxide solution and observing the bubbles formed. Anaerobic growth was evaluated on R2A plates with or without 0.1% (w/v) KNO3, incubated in an anaerobic jar (containing 10% H2, 10% CO2 and 80% N2) at 30°C for 14 days. Enzymatic activity toward substrates including alginate, CM-cellulose, starch and lipids (Tweens 20, 40, 60, and 80) were investigated. After incubating for 3 days at 30°C, the sizes of the inhibition zones surrounding the antibiotic-impregnated disks were tested to determine antibiotic susceptibility (Jorgensen and Turnidge, 2015). Subsequently, they were cultured under optimal conditions, using API ZYM, API 20E, API 50CHB (bioMérieux) strips, and Biolog GEN III systems. The physiological and biochemical traits of strain Y139T and experimental strains were analyzed, followed by the manufacturer’s directions. Experimental strains of L. pohnpeiensis CCTCC AB 2011006T and L. arcticus CCTCC AB 2014275T were acquired from the China Center for Type Culture Collection (CCTCC) (Yoon et al., 2008; Kim et al., 2015), L. flavescens MCCC 1K03193T was obtained from Marine Culture Collection of China (MCCC) (Zhang et al., 2017).
2.8 Chemotaxonomy
Strain Y139T was cultured for 2 days on R2A at 30°C at a pH of 7.0, then harvested during the exponential growth phase and freeze-dried. Fatty acids, polar lipids, and respiratory quinones were extracted using these preparations. Menaquinone was extracted following the methods described by Minnikin et al. (1984), and then analyzed by high-performance liquid chromatography (HPLC) (Kroppenstedt, 1982). The Microbial Identification System (Sherlock version 4.5; database: TSBA40; MIDI) was used to identify cellular fatty acid methyl esters (FAMEs) (Chou et al., 1990). Additionally, polar lipids were extracted with a chloroform/methanol system and analyzed by two-dimensional thin-layer chromatography (Du et al., 2014).
2.9 Biogeographic distribution of genus Luteolibacter
The distribution and habitat preferences of Luteolibacter globally were assessed using the analytical tools from the Microbe Atlas Project (MAP).9 The study was conducted with a rigorous 96% sequence similarity threshold. Microbial community abundance was assessed through MAPseq, a closed reference method for analyzing ribosomal RNA sequences (Matias Rodrigues et al., 2017).
2.10 Data analysis
The trends of relative abundances of the phylum Verrucomicrobiota and the genus Luteolibacter were statistically analyzed using different plots as replicates. The t-test was conducted with the R package stats (v4.4.1, 2024).
3 Results and discussion
3.1 Phenotypic properties
Yellow colonies formed by strain Y139T were observed during cultivation on R2A agar plates. The width and length of the cells were found to be 0.4–0.7 μm and 1.5–2.0 μm, respectively (Supplementary Figure S1). Strain Y139T exhibited growth at temperatures ranging from 20 to 37°C (optimum 30°C); tolerated NaCl concentrations from 0 to 1.0% (w/v) (optimum 0%); and pH conditions from 5.5 to 9.5 (optimum 7.0). It exhibited the capacity to hydrolyze cellulose and starch, but could not hydrolyze Tweens 20, 40, 60, 80, alginate, or casein. All strains were positive for tryptophan deaminase, arabinose, oxidase and catalase; negative for casein, nitrite reduction and Tween 20. Same as experimental strains, Y139T was positive for alkaline phosphatase, leucine arylamidase, acid phosphatase, naphthol-AS-BI-phosphohydrolase, β-galactosidase, esterase lipase (C8) and esterase (C4). Negative for lipase (C14), cystine arylamidase, α-chymotrypsin, β-glucuronidase, α-glucosidase and N-acetyl-β-glucosaminidase. Moreover, negative activities were observed for α-galactosidase, β-glucosidase and α-mannosidase of strain Y139T. In API 50CHB strips, acids were produced from D-mannose, glycerol, D-glucose, D-fructose, D-sorbitol, amygdalin, esculin ferric citrate, salicin, D-saccharose, D-melibiose, D-lactose, D-maltose, D-trehalose, inulin, D-raffinose, glycogen, gentiobiose, methyl-α-D-mannopyranoside, methyl-α-D-glucopyranoside, arbutin, D-melezitose and potassium 5-ketogluconate of all strains. Strain Y139T displayed positive results for D-xylose, D-galactose, D-cellobiose, D-turanose and potassium gluconate, distinguishing it from the experimental strains. In the Biolog GEN III systems, all strains were negative for propionic acid, β-hydroxy-D, α-keto-butyric acid, α-hydroxy-butyric acid, D-malic acid, D-aspartic acid, L-butyric acid and γ-amino-butyric acid. Unlike the experimental strains, strain Y139T was negative for fusidic acid, ρ-hydroxy-phenylacetic acid and L-malic acid. Table 1 outlines the main features of strain Y139T and experimental strains. Strain Y139T displayed resistance to streptomycin (10 μg per disk), chloramphenicol (30 μg per disk), neomycin (30 μg per disk), carbenicillin (100 μg per disk), vancomycin (30 μg per disk), norfloxacin (30 μg per disk), penicillin (10 μg per disk), ampicillin (10 μg per disk), erythromycin (15 μg per disk), tobramycin (10 μg per disk), and exhibited intermediate resistance to kanamycin (30 μg per disk) and gentamycin (10 μg per disk). Conversely, it was susceptible to ofloxacin (5 μg per disk), ceftriaxone (30 μg per disk), cefotaxime sodium (30 μg per disk), polymyxin B (300 μg per disk), tetracycline (30 μg per disk) and rifampin (5 μg per disk).
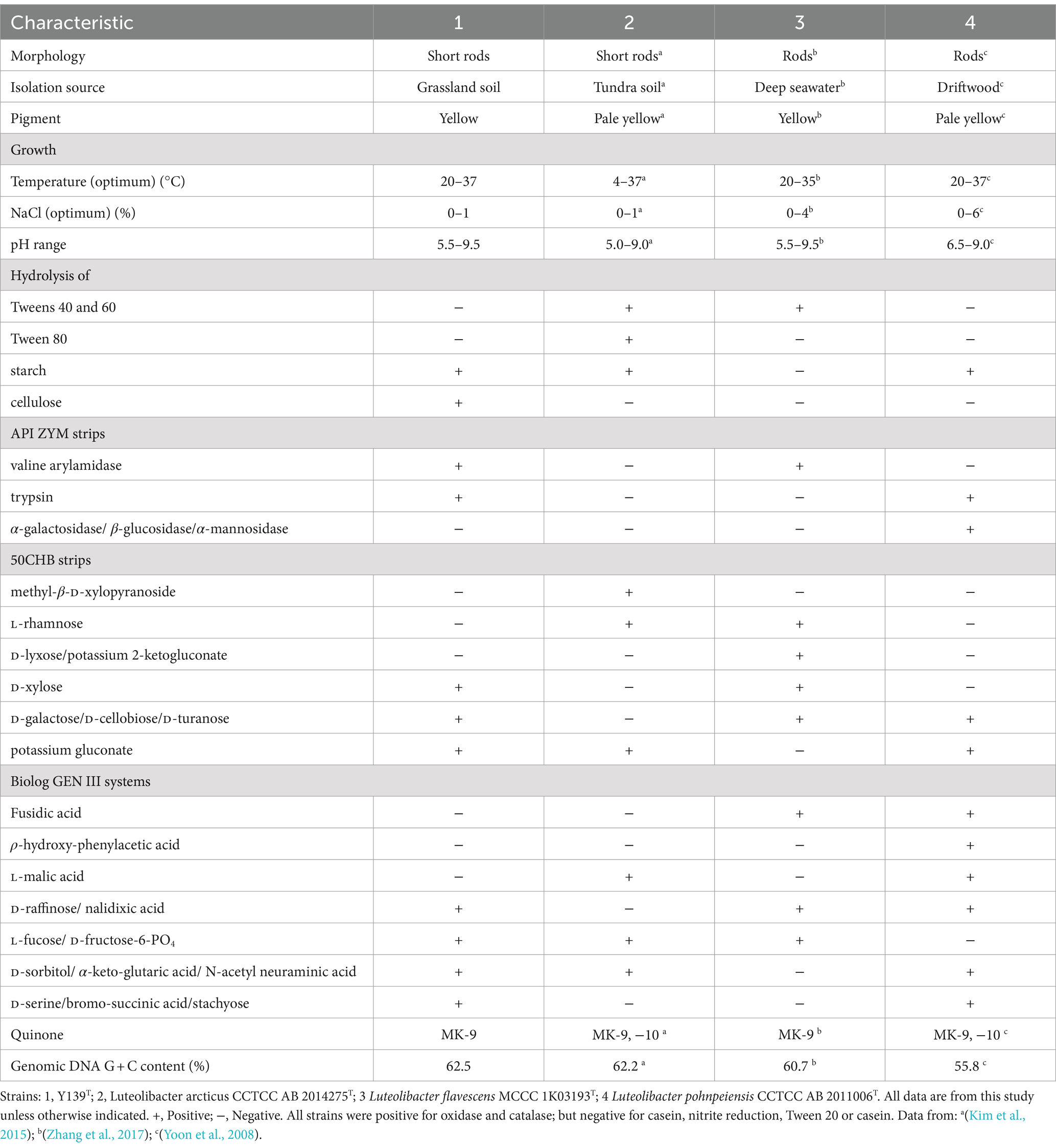
Table 1. Differential characteristics of strain Y139T and experimental strains.
3.2 Chemotaxonomic features
Consistent with strains of the genus Luteolibacter, MK-9 was the only respiratory quinone of strain Y139T (Yoon et al., 2008). C16:0, iso-C14:0, and C16:1 ω9c are the major cellular fatty acids that were similar to the profiles of the experimental strains. Unlike strain Y139T, C16:0 is the main fatty acid of L. flavescens MCCC 1K03193T. C17:0 3-OH was identified in strain Y139T, but it was not found in other strains. The differences in the proportions of some fatty acids are shown in Table 2. Five polar lipids were identified of strain Y139T, including phosphatidylglycerol (PG), phosphatidylethanolamine (PE), phosphatidyldimethylethanolamine (PME), diphosphatidylglycerol (DPG), and an unidentified lipid (L). The presence of diphosphatidylglycerol (DPG), phosphatidylglycerol (PG) and phosphatidylethanolamine (PE) were conserved in all strains. Phosphatidyldimethylethanolamine (PME) was found in L. pohnpeiensis CCTCC AB 2011006T, but not in other experimental strains. Further detailed polar lipids with different specific strains are given in Supplementary Figure S2.
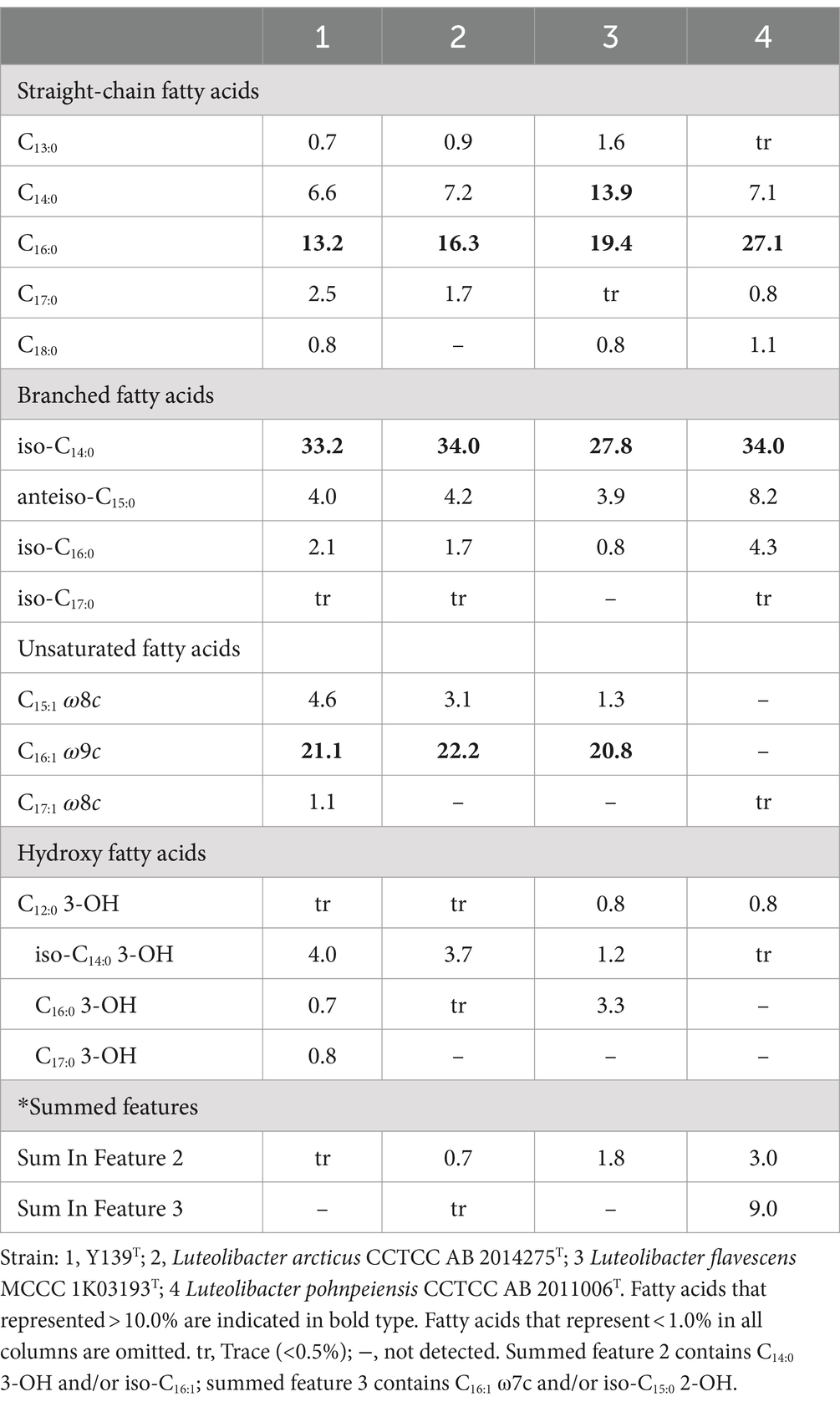
Table 2. Major cellular fatty acid comparison of strain Y139T and experimental strains of the genus Luteolibacter.
3.3 16S rRNA gene sequence and phylogenetic analysis
To align the 16S rRNA gene sequence of strain Y139T (1,518 bp), the EzBioCloud database was utilized. The sequence similarity to the genus Luteolibacter ranged from 92.1 to 98.3%. The results showed that strain Y139T was affiliated with the genus Luteolibacter and closely related to L. flavescens MCCC 1K03193T and L. arcticus CCTCC AB 2014275T with 98.3 and 98.2% 16S rRNA gene similarities, respectively. This is below the threshold for distinguishing between two species (98.6%) (Kim et al., 2014). To explore its evolutionary position, phylogenetic tree based on 16S rRNA gene sequences was constructed. The result shows that strain Y139T was most closely related to L. arcticus CCTCC AB 2014275T, and should be classified within the genus Luteolibacter (Figure 1A). Similar topologies were also obtained with three algorithms (NJ, ML and MP). In addition, the phylogenomic tree reconstructed using protein-coding genes can also lead to the same conclusions. (Figure 1B).
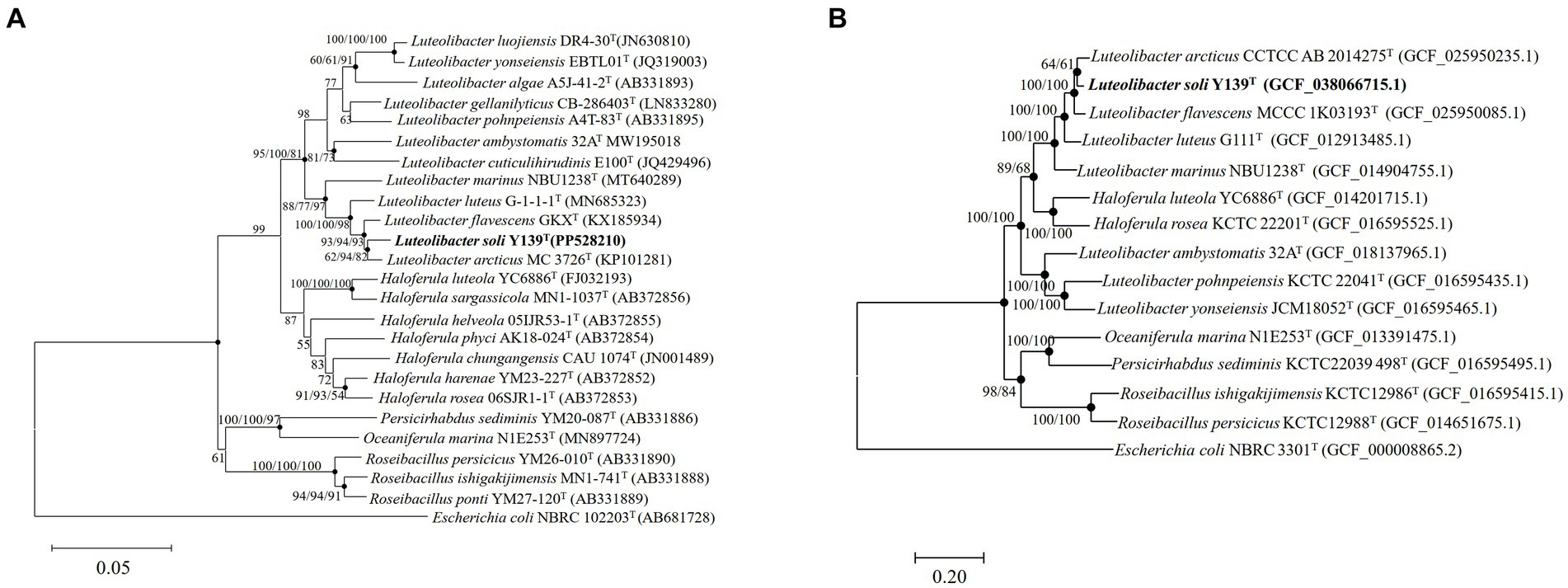
Figure 1. The phylogenetic tree of strain Y139T and other related species, Escherichia coli NBRC 102203T was used as the outgroup. (A) Neighbor-joining phylogenetic tree based on 16S rRNA gene sequences of strain Y139T and other closely related species. Filled circles indicate branches that were recovered with neighbor-joining, maximum-likelihood, and minimum-evolution methods. Bootstrap values above 50% (1,000 replicates) are shown at branch nodes (NJ/ML/MP). Bar: 0.05 substitutions per nucleotide position; (B) The FastTree is based on 120 ubiquitous single-copy proteins. Bootstrap values above 50% (1,000 replicates) are shown at branch nodes. Filled circles indicate that the same topology is also obtained using the IQ-Tree algorithm. Escherichia coli NBRC 102203T was used as the outgroup. Bar: 0.20 substitutions per nucleotide position.
3.4 Genomic characteristics
For strain Y139T, the draft genome measured 7,106,054 bp in length and consisted of 31 scaffolds, with a genomic DNA G + C content of 62.5%, consistent with the range of other species in the genus Luteolibacter (Yoon et al., 2008; Xie et al., 2022). The results of the genome analysis showed a completeness of 98.8%. The draft genome of strain Y139T contains 5,715 genes, which consist of 5,651 protein-coding genes, 5 pseudogenes, and 59 RNA genes (3 rRNA genes, 53 tRNA genes, and 3 non-coding RNA genes). The antiSMASH analysis revealed biosynthetic gene clusters responsible for secondary metabolites such as betalactone, terpenes, type I polyketide synthase (PKS), type III polyketide synthase (PKS), and non-ribosomal peptide synthetase (NRPS) (Supplementary Figure S3). The AAI values were 82.2 and 82.4% between strain Y139T and L. flavescens MCCC 1K03193T and L. arcticus CCTCC AB 2014275T, respectively, exceeding the 62–72% threshold for genus delimitation (Nicholson et al., 2020). The ANI values were 80.6 and 82.1% for strain Y139T compared to L. flavescens MCCC 1K03193T and L. arcticus CCTCC AB 2014275T, respectively, which are below the 95% threshold for species delimitation (Jain et al., 2018). The dDDH values were 23.7 and 25.5% between strain Y139T and L. flavescens MCCC 1K03193T and L. arcticus CCTCC AB 2014275T, respectively, these values are all below the 70% threshold, which is commonly used to delineate a novel species (Goris et al., 2007; Figure 2). These findings indicate that strain Y139T should be a novel species within the genus Luteolibacter.
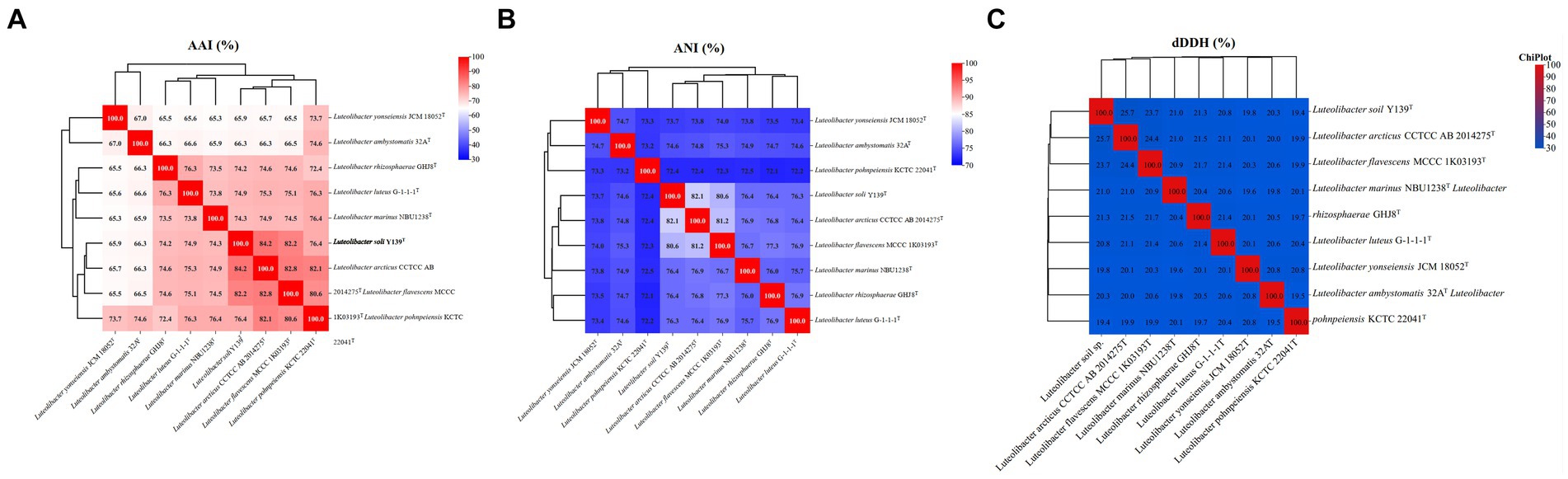
Figure 2. Genomic similarities of strain Y139T to some members of the genus Luteolibacter. (A) The AAI values between isolates of the genus; (B) The ANI values between isolates of the genus; (C) The dDDH values between isolates of the genus.
3.5 Comparative genomic analysis
In the genus Luteolibacter, including strain Y139T, genomes ranged from 4,667,805 to 7,486,770 bp, with the genomic DNA G + C content varying from 53.5 to 65.0% (Supplementary Table S1). Analysis of the pan-genome was conducted using orthologous protein groups, showing that the average number was 4,882, with a total of 1,177 core genes accounting for 24% of the genomes in the genus Luteolibacter (Supplementary Figure S4A). Each genome of Luteolibacter has 23.3–55.2% accessory genes and 21.9–46.7% unique genes (Supplementary Figure S4B). Unique genes exhibited a broader distribution across various metabolic pathways including carbohydrate metabolism, energy metabolism, amino acid metabolism, signal transduction and cellular community (Supplementary Figure S5).
The KEGG distribution analysis of the genus Luteolibacter species revealed the significant role of core genes in fundamental metabolic pathways crucial for sustaining life, such as amino acid metabolism, carbohydrate metabolism, and translation. The completion of key pathways like the TCA cycle pathway (M00009), glycolysis pathway (M00001), and pentose phosphate pathway (M00004) was observed, while cytochrome c oxidase (M00156) was not complete (Figure 3). The genus Luteolibacter species demonstrated conservatism in carbohydrate metabolism, amino acid metabolism and nucleotide metabolism, with notable variations observed primarily in energy metabolism. All strains within the Luteolibacter genus exhibited completeness in assimilatory sulfate reduction (M00176). Most amino acids biosynthesis pathways were complete, including proline biosynthesis (M00015), methionine biosynthesis (M00017), threonine biosynthesis (M00018), valine/isoleucine biosynthesis (M00019), cysteine biosynthesis (M00021), tryptophan biosynthesis (M00023), ornithine biosynthesis (M00028), leucine biosynthesis (M00432), lysine biosynthesis (M00527), isoleucine biosynthesis (M00570) and arginine biosynthesis (M00844) in nine members of the genus Luteolibacter. Notably, proline has been identified to enhance bacterial growth under stress conditions, particularly aiding in osmotic stress tolerance through cellular accumulation of amino acids. The survival and growth of bacteria are influenced by their capacity to regulate osmotic pressure by intracellular amino acid accumulation. Based on the RAST genome annotation, gene distribution associated with subsystems falls into 24 different categories, as shown in Supplementary Figure S6.
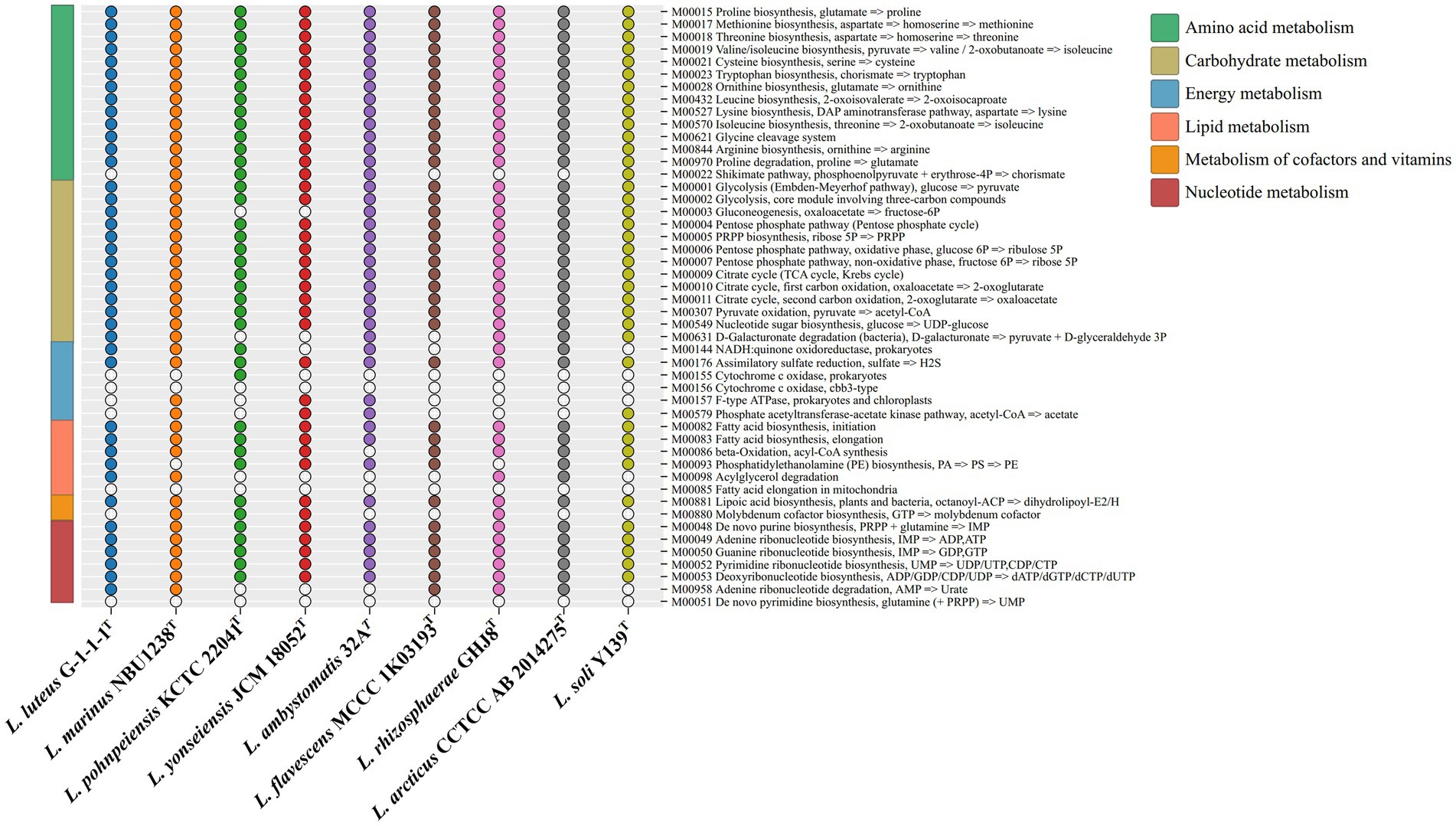
Figure 3. The metabolic module integrity of the genus Luteolibacter. The solid circles and hollow circles indicate that the metabolic pathways were complete and incomplete, respectively.
3.6 Biogeographic distribution
The MAP database10 was used to determine the worldwide distribution of the genus Luteolibacter. A total of 79,176 samples from 8,000 projects were analyzed to identify the representative sequence. The results of the biogeographic distribution analysis indicated that Luteolibacter is widespread across different habitats, including aquatic, soil, animal, and plant environments. Specifically, Luteolibacter bacteria were found in 22,332 soil samples (28.2%), 10,034 aquatic samples (12.7%), 5,673 plant samples (7.2%), and 4,239 animal samples (5.4%). Among the known environments, the primary habitats for Luteolibacter were identified as the rhizosphere (3.2%), field (2.9%), agricultural areas (2.1%), river (2.7%) and lake ecosystems (2.1%) (Figure 4A). The mapping of database sequencing reads to the standard OTU sequence showed that the lake environment had a predominant proportion (22.3%) of reads from the genus Luteolibacter (Figure 4B).

Figure 4. Biogeographic distribution analysis of genus Luteolibacte. (A) Frequency of samples with representative OTU sequence, by habitat and sub-habitat; (B) Abundance of sequencing reads mapping to the representative OTU sequence, by habitat and sub-habitat.
3.7 Impact of climate change on genus Luteolibacter
The genus Luteolibacter, belonging to the phylum Verrucomicrobiota, is widely distributed in soil environments. Relative abundances were calculated at the amplicon sequence variant (ASV) level, revealing that the relative abundances of the phylum Verrucomicrobiota and the genus Luteolibacter in soil samples ranged from 0.3 to 3.3% and from 0 to 0.1%, respectively, from 2009 and 2020. The relative abundances of the genus Luteolibacter and the phylum Verrucomicrobiota were tracked over several years under three climate change factors: warming, precipitation, and clipping. The genus Luteolibacter exhibited higher relative abundances under conditions of increased temperature and double precipitation over time, suggesting that these climate change factors have a substantial impact on its population. Clipping also influenced the relative abundance of the genus Luteolibacter, but the effect was irregular and fluctuating (Figures 5A–C). This consistent increase in relative abundance under specific conditions highlights the potential resilience and adaptability of Luteolibacter to changing environmental parameters. In contrast, we did not observe similar changes in the phylum Verrucomicrobiota. Precipitation had a greater effect on the phylum Verrucomicrobiota than warming and clipping, but this effect was variable and fluctuated over time (Figures 5D–F).
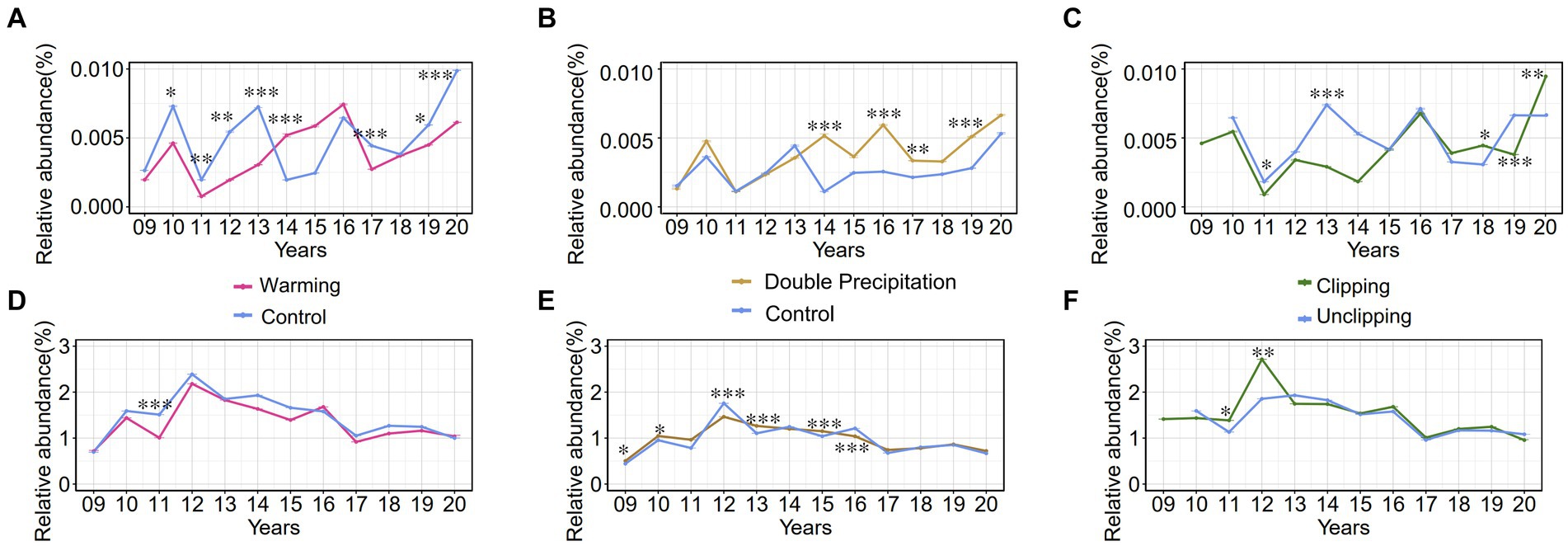
Figure 5. The relative abundance (%) trends of phylum Verrucomicrobia and genus Luteolibacter from 2009 to 2020 under various conditions. 09 to 20 represent the years 2009–2020, respectively. (A–C) Effect of warming/precipitation/clipping on the relative abundance of genus Luteolibacter; (D-F) Effect of warming/precipitation/clipping on the relative abundance of phylum Verrucomicrobia. Data are presented as mean ± SD of the estimated effect sizes. Statistical significance is based on t test, asterisks indicate statistical significance, with the number of asterisks representing the level of significance (* for p < 0.05; ** for p < 0.01; *** for p < 0.001).
4 Description of Luteolibacter soli sp. nov
4.1 Luteolibacter soli (so′li. L. gen. Neut. n. soli of soil)
Cells of strain Y139T are Gram-stain-negative, non-motile, aerobic, rod-shaped, with the width of 0.4–0.7 and the length of 1.5–2.0 . They exhibit growth between 20 and 37°C (optimum, 30°C), with pH tolerance from 5.5 to 9.5 (optimum, pH 7.0), and 0–1.0% NaCl concentration (optimum, 0%). The cells display oxidase and catalase activity and can hydrolyze cellulose and starch, but not alginate, casein, or Tweens 20, 40, 60, and 80. The only respiratory quinone is menaquinone-9 (MK-9). The dominant fatty acids are C16:0, iso-C14:0, and C16:1 ω9c. The major polar lipids include phosphatidylethanolamine (PE), phosphatidylglycerol (PG), phosphatidyldimethylethanolamine (PME), diphosphatidylglycerol (DPG), and an unidentified lipid (L). Cells test positive for β-galactosidase, esterase lipase (C8), esterase (C4), acid phosphatase, naphthol-AS-BI-phosphohydrolase, leucine arylamidase and alkaline phosphatase. In carbon source oxidation tests, negative results are obtained for fusidic acid, ρ-hydroxy-phenylacetic acid, D-malic acid, L-malic acid, γ-amino-butyric acid, α-hydroxy-butyric acid, α-keto-butyric acid, β-hydroxy-D, L-butyric acid and propionic acid. Cells are also positive for the utilization of tryptophan, sucrose, maltose and L-arabinose. Acid is produced from methyl-α-D-mannopyranoside, methyl-α-D-glucopyranoside, D-melezitose, glycerol, D-xylose, D-galactose, D-glucose, D-fructose, D-mannose, D-sorbitol, amygdalin, salicin, arbutin, D-cellobiose, D-maltose, D-lactose, D-melibiose, D-saccharose, D-trehalose, inulin, D-raffinose, amidon, esculin ferric citrate, glycogen, gentiobiose, D-turanose, potassium 5-ketogluconate, and potassium gluconate.
Type strain Y139T (= KCTC 92029T = MCCC 1H00491T) was isolated from a soil sample in McClain County, Oklahoma, United States. GenBank numbers for the 16S rRNA gene sequence and draft genome of Luteolibacter Y139T are PP528210 and JBBUKT000000000.1, respectively.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
JA: Formal analysis, Writing – original draft, Writing – review & editing, Data curation, Methodology, Resources, Software, Visualization. XX: Writing – review & editing, Supervision, Validation. YW: Supervision, Validation, Writing – review & editing. LW: Data curation, Methodology, Writing – review & editing. JZ: Project administration, Supervision, Writing – review & editing. DM: Writing – review & editing, Funding acquisition, Project administration, Visualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (32470010); Science and Technology Fundamental Resources Investigation Program (Grant nos. 2019FY100700 and 2022FY101100) and the National Natural Science Foundation of China (41876166).
Acknowledgments
This work of scanning electron microscope (Nova NanoSEM 450, FEI) was supported by the Physical–Chemical Materials Analytical and Testing Center of Shandong University at Weihai. Luteolibacter arcticus CCTCC AB 2014275T was provided by China Center for Type Culture Collection (CCTCC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1483195/full#supplementary-material
Footnotes
1. ^https://www.bacterio.net/, 27 March 2024.
2. ^https://blast.ncbi.nlm.nih.gov/Blast.cgi
3. ^https://www.ezbiocloud.net/
4. ^https://www.ezbiocloud.net/tools/ani
5. ^https://ggdc.dsmz.de/distcalc2.php
6. ^https://www.genome.jp/kegg/
References
Aziz, R. K., Bartels, D., Best, A. A., Dejongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9, 1–15. doi: 10.1186/1471-2164-9-75
Banerjee, S., Kirkby, C. A., Schmutter, D., Bissett, A., Kirkegaard, J. A., and Richardson, A. E. (2016). Network analysis reveals functional redundancy and keystone taxa amongst bacterial and fungal communities during organic matter decomposition in an arable soil. Soil Biol. Biochem. 97, 188–198. doi: 10.1016/j.soilbio.2016.03.017
Bergmann, G. T., Bates, S. T., Eilers, K. G., Lauber, C. L., Caporaso, J. G., Walters, W. A., et al. (2011). The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol. Biochem. 43, 1450–1455. doi: 10.1016/j.soilbio.2011.03.012
Bernardet, J.-F., Nakagawa, Y., Holmes, B., Flavobacterium, S. O. T. T. O., and Prokaryotes, C. L. B. (2002). Proposed minimal standards for describing new taxa of the family Flavobacteriaceae and emended description of the family. Int. J. Syst. Evol. Microbiol. 52, 1049–1070. doi: 10.1099/00207713-52-3-1049
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Busse, H.-J., Kämpfer, P., Szostak, M. P., and Spergser, J. (2021). Luteolibacter ambystomatis sp. nov., isolated from the skin of an Anderson’s salamander (Ambystoma andersoni). Int. J. Syst. Evol. Microbiol. 71:005043. doi: 10.1099/ijsem.0.005043
Chaudhari, N., Gupta, V., and Dutta, C. (2016). BPGA-an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Chou, S., Kasatiya, S., and Irvine, N. (1990). Cellular fatty acid composition of Oerskovia species, CDC Coryneform groups A-3, A-4, A-5, Corynebacterium aquaticum, listeria denitrificans and Brevibacterium acetylicum. Antonie Van Leeuwenhoek 58, 115–119. doi: 10.1007/bf00422727
Collins, S. (2011). Many possible worlds: expanding the ecological scenarios in experimental evolution. Evol. Biol. 38, 3–14. doi: 10.1007/s11692-010-9106-3
Dahal, R. H., Chaudhary, D. K., Kim, D.-U., and Kim, J. (2021). Luteolibacter luteus sp. nov., isolated from stream bank soil. Arch. Microbiol. 203, 377–382. doi: 10.1007/s00203-020-02048-x
Du, Z.-J., Wang, Y., Dunlap, C., Rooney, A. P., and Chen, G.-J. (2014). Draconibacterium Orientale gen. Nov., sp. nov., isolated from two distinct marine environments, and proposal of Draconibacteriaceae fam. Nov. Int. J. Syst. Evol. Microbiol. 64, 1690–1696. doi: 10.1099/ijs.0.056812-0
Edgar, R. C. (2018). Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics 34, 2371–2375. doi: 10.1093/bioinformatics/bty113
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Figueras, M. J., Beaz-Hidalgo, R., Hossain, M. J., and Liles, M. R. (2014). Taxonomic affiliation of new genomes should be verified using average nucleotide identity and multilocus phylogenetic analysis. Genome Announc. 2, e00927–e00914. doi: 10.1128/genomeA.00927-14
Glaeser, S. P., Galatis, H., Martin, K., and Kämpfer, P. (2012). Luteolibacter cuticulihirudinis sp. nov., isolated from Hirudo medicinalis. Antonie Van Leeuwenhoek 102, 319–324. doi: 10.1007/s10482-012-9741-z
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Guo, X., Feng, J., Shi, Z., Zhou, X., Yuan, M., Tao, X., et al. (2018). Climate warming leads to divergent succession of grassland microbial communities. Nat. Clim. Chang. 8, 813–818. doi: 10.1038/s41558-018-0254-2
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jiang, F., Li, W., Xiao, M., Dai, J., Kan, W., Chen, L., et al. (2012). Luteolibacter luojiensis sp. nov., isolated from Arctic tundra soil, and emended description of the genus Luteolibacter. Int. J. Syst. Evol. Microbiol. 62, 2259–2263. doi: 10.1099/ijs.0.037309-0
Jorgensen, J. H., and Turnidge, J. D. (2015). “Susceptibility test methods: dilution and disk diffusion methods” in Manual clinical microbiology. eds. J. H. Jorgensen, K. C. Carroll, G. Funke, M. A. Pfaller, M. L. Landry, and S. S. Richter (United States: American Society of Microbiology), 1253–1273.
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M., and Ishiguro-Watanabe, M. (2023). KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. doi: 10.1093/nar/gkac963
Kannan, L., and Wheeler, W. C. (2012). Maximum parsimony on phylogenetic networks. Algorithm Mol. Biol. 7, 1–10. doi: 10.1186/1748-7188-7-9
Kielak, A., Rodrigues, J. L. M., Kuramae, E. E., Chain, P. S. G., Van Veen, J. A., and Kowalchuk, G. A. (2009). Phylogenetic and metagenomic analysis of Verrucomicrobia in former agricultural grassland soil. FEMS Microbiol. Ecol. 71, 23–33. doi: 10.1111/j.1574-6941.2009.00785.x
Kim, O.-S., Cho, Y.-J., Lee, K., Yoon, S.-H., Kim, M., Na, H., et al. (2012). Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int. J. Syst. Evol. Microbiol. 62, 716–721. doi: 10.1099/ijs.0.038075-0
Kim, M., Oh, H.-S., Park, S.-C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Kim, M., Pak, S., Rim, S., Ren, L., Jiang, F., Chang, X., et al. (2015). Luteolibacter arcticus sp. nov., isolated from high Arctic tundra soil, and emended description of the genus Luteolibacter. Int. J. Syst. Evol. Microbiol. 65, 1922–1928. doi: 10.1099/ijs.0.000202
Kroppenstedt, R. M. (1982). Separation of bacterial menaquinones by HPLC using reverse phase (RP18) and a silver loaded ion exchanger as stationary phases. J. Liq. Chromatogr. R. T. 5, 2359–2367. doi: 10.1080/01483918208067640
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lee, I., Chalita, M., Ha, S.-M., Na, S.-I., Yoon, S.-H., and Chun, J. (2017). ContEst16S: an algorithm that identifies contaminated prokaryotic genomes using 16S RNA gene sequences. Int. J. Syst. Evol. Microbiol. 67, 2053–2057. doi: 10.1099/ijsem.0.001872
Li, D., Zhou, X., Wu, L., Zhou, J., and Luo, Y. (2013). Contrasting responses of heterotrophic and autotrophic respiration to experimental warming in a winter annual-dominated prairie. Glob. Chang. Biol. 19, 3553–3564. doi: 10.1111/gcb.12273
Liu, Q.-Q., Wang, Y., Li, J., Du, Z.-J., and Chen, G.-J. (2014). Saccharicrinis carchari sp. nov., isolated from a shark, and emended descriptions of the genus Saccharicrinis and Saccharicrinis fermentans. Int. J. Syst. Evol. Microbiol. 64, 2204–2209. doi: 10.1099/ijs.0.061986-0
Matias Rodrigues, J. F., Schmidt, T. S., Tackmann, J., and Von Mering, C. (2017). MAPseq: highly efficient k-mer search with confidence estimates, for rRNA sequence analysis. Bioinformatics 33, 3808–3810. doi: 10.1093/bioinformatics/btx517
Minnikin, D., O’Donnell, A., Goodfellow, M., Alderson, G., Athalye, M., Schaal, A., et al. (1984). An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiol. Methods 2, 233–241. doi: 10.1016/0167-7012(84)90018-6
Nicholson, A. C., Gulvik, C. A., Whitney, A. M., Humrighouse, B. W., Bell, M. E., Holmes, B., et al. (2020). Division of the genus chryseobacterium: observation of discontinuities in amino acid identity values, a possible consequence of major extinction events, guides transfer of nine species to the genus epilithonimonas, eleven species to the genus kaistella, and three species to the genus halpernia gen. Nov., with description of kaistella daneshvariae sp. nov. and epilithonimonas vandammei sp. nov. derived from clinical specimens. Int. J. Syst. Evol. Microbiol. 70, 4432–4450. doi: 10.1099/ijsem.0.003935
Oliverio, A. M., Bradford, M. A., and Fierer, N. (2017). Identifying the microbial taxa that consistently respond to soil warming across time and space. Glob. Chang. Biol. 23, 2117–2129. doi: 10.1111/gcb.13557
Park, J., Baek, G. S., Woo, S.-G., Lee, J., Yang, J., and Lee, J. (2013). Luteolibacter yonseiensis sp. nov., isolated from activated sludge using algal metabolites. Int. J. Syst. Evol. Microbiol. 63, 1891–1895. doi: 10.1099/ijs.0.046664-0
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pascual, J., García-López, M., González, I., and Genilloud, O. (2017). Luteolibacter gellanilyticus sp. nov., a gellan-gum-degrading bacterium of the phylum Verrucomicrobia isolated from miniaturized diffusion chambers. Int. J. Syst. Evol. Microbiol. 67, 3951–3959. doi: 10.1099/ijsem.0.002227
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Thomas, M. K., Kremer, C. T., Klausmeier, C. A., and Litchman, E. (2012). A global pattern of thermal adaptation in marine phytoplankton. Science 338, 1085–1088. doi: 10.1126/science.1224836
Trifinopoulos, J., Nguyen, L.-T., Von Haeseler, A., and Minh, B. Q. (2016). W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235. doi: 10.1093/nar/gkw256
Wu, L., Zhang, Y., Guo, X., Ning, D., Zhou, X., Feng, J., et al. (2022). Reduction of microbial diversity in grassland soil is driven by long-term climate warming. Nat. Microbiol. 7, 1054–1062. doi: 10.1038/s41564-022-01147-3
Xie, F., Zhu, S., Guo, C., Liu, X., He, S., and Zhang, W. (2022). Luteolibacter marinus sp. nov., a novel bacterium in the family Verrucomicrobiaceae, isolated from marine sediment. Int. J. Syst. Evol. Microbiol. 72:005544. doi: 10.1099/ijsem.0.005544
Xu, X., Sherry, R. A., Niu, S., Li, D., and Luo, Y. (2013). Net primary productivity and rain-use efficiency as affected by warming, altered precipitation, and clipping in a mixed-grass prairie. Glob. Chang. Biol. 19, 2753–2764. doi: 10.1111/gcb.12248
Yoon, J., Matsuo, Y., Adachi, K., Nozawa, M., Matsuda, S., Kasai, H., et al. (2008). Description of Persicirhabdus sediminis gen. Nov., sp. nov., Roseibacillus ishigakijimensis gen. Nov., sp. nov., Roseibacillus ponti sp. nov., Roseibacillus persicicus sp. nov., Luteolibacter pohnpeiensis gen. Nov., sp. nov. and Luteolibacter algae sp. nov., six marine members of the phylum ‘Verrucomicrobia’, and emended descriptions of the class Verrucomicrobiae, the order Verrucomicrobiales and the family Verrucomicrobiaceae. Int. J. Syst. Evol. Microbiol. 58, 998–1007. doi: 10.1099/ijs.0.65520-0
Zhang, C., Dong, B., Wang, R., Su, Y., Han, S., Yu, X., et al. (2017). Luteolibacter flavescens sp. nov., isolated from deep seawater. Int. J. Syst. Evol. Microbiol. 67, 729–735. doi: 10.1099/ijsem.0.001713
Zhang, H., Liu, H., Zhao, J., Li, G., Lai, X., and Li, J. (2018). Effects of simulated nitrogen deposition and precipitation change on soil bacterial community structure in a Stipa baicalensis steppe. Acta Ecol. Sin. 15, 244–253. doi: 10.3389/fmicb.2024.1414724
Zhang, L., and Xu, Z. (2008). Assessing bacterial diversity in soil: a brief review. J. Soils Sediments 8, 379–388. doi: 10.1007/s11368-008-0043-z
Zhao, J., Xie, X., Jiang, Y., Li, J., Fu, Q., Qiu, Y., et al. (2024). Effects of simulated warming on soil microbial community diversity and composition across diverse ecosystems. Sci. Total Environ. 911:168793. doi: 10.1016/j.scitotenv.2023.168793
Zhou, S. Y. D., Lie, Z., Liu, X., Zhu, Y. G., Penuelas, J., Neilson, R., et al. (2023). Distinct patterns of soil bacterial and fungal community assemblages in subtropical forest ecosystems under warming. Glob. Chang. Biol. 29, 1501–1513. doi: 10.1111/gcb.16541
Keywords: Luteolibacter, 16S rRNA gene, genome, phylogenetic, climate change
Citation: An J, Xuan X, Wang Y, Wu L, Zhou J and Mu D (2024) Analysis of genomic and characterization features of Luteolibacter soli sp. nov., isolated from soil. Front. Microbiol. 15:1483195. doi: 10.3389/fmicb.2024.1483195
Edited by:
Jinbo Xiong, Ningbo University, ChinaReviewed by:
Keshao Liu, Institute of Tibetan Plateau Research (CAS), ChinaYang Liu, Guangdong Academy of Science, China
Copyright © 2024 An, Xuan, Wang, Wu, Zhou and Mu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dashuai Mu, ZGFzaHVhaS5tdUBzZHUuZWR1LmNu; Jizhong Zhou, anpob3VAb3UuZWR1