 Zhi-Xin Xu1
Zhi-Xin Xu1 Le-Xing You
Le-Xing You- 1High Voltage Branch of State Grid Fujian Electric Power Co., Ltd., Fuzhou, China
- 2College of Geography and Environmental Sciences, Zhejiang Normal University, Jinhua, China
The surge in global energy demand mandates a significant expansion of electric power substations. Nevertheless, the ecological consequences of electric power substation operation, particularly concerning the electromagnetic field, on soil microbial communities and nitrogen enrichment remain unexplored. In this study, we collected soil samples from six distinct sites at varying distances from an electric power substation in Xintang village, southeastern China, and investigated the impacts of electromagnetic field on the microbial diversity and community structures employing metagenomic sequencing technique. Our results showed discernible dissimilarities in the fungal community across the six distinct sites, each characterized by unique magnetic and electric intensities, whereas comparable variations were not evident within bacterial communities. Correlation analysis revealed a diminished nitrogen fixation capacity at the site nearest to the substation, characterized by low moisture content, elevated pH, and robust magnetic induction intensity and electric field intensity. Conversely, heightened nitrification processes were observed at this location compared to others. These findings were substantiated by the relative abundance of key genes associated with ammonium nitrogen and nitrate nitrogen production. This study provides insights into the relationships between soil microbial communities and the enduring operation of electric power substations, thereby contributing fundamental information essential for the rigorous environmental impact assessments of these facilities.
1 Introduction
The relentless advance of modernization has brought about an increasing demand for electric energy. This demand necessitates the efficient transportation of electricity from power generation facilities. This intricate process relies on a vast network comprising power substations, transmission lines, and distribution lines. Among them, power substations serve as crucial installations of equipment designed for voltage control and adjustment (Biasotto and Kindel, 2018). The Chinese landscape reflects a proliferation of power substations and the extensive deployment of electrical power lines, traversing diverse terrains such as farmland and highways. This remarkable infrastructural landscape is, however, compelled to evolve in response to the escalating need for electricity, necessitating the establishment of additional electric power substations. Nevertheless, it is essential to recognize that electric power substations, throughout their construction and operational phases, pose potential environmental considerations (Bagli et al., 2011).
In many countries, rigorous vegetation management is required in the vicinity beneath power cables to avert interference with line structural integrity and energy transmission. Furthermore, the operation of power lines and electrical equipment engenders the generation of electromagnetic fields (Biasotto and Kindel, 2018). As crucial components of terrestrial ecosystems, soil inevitably bear the imprint of electric power substations. Soil microorganisms, as guardians of soil health, regulate soil nutrient cycles, including the vital nitrogen cycle, and exert a profound influence on plant productivity (Gadd, 2010; Faust and Raes, 2012; Whalen et al., 2022). In response to environmental stressors, soil microorganisms exhibit distinctive adaptive responses, such as the production of diverse enzymes like superoxide dismutase (SOD), which provide effective resistance against the harmful effects of reactive oxygen species (Roubalová et al., 2018) and safeguards plant well-being (Huseynova et al., 2014). Despite substantial progress in environmental impact assessment pertaining to electric power substations, notable gaps persist in representing distinct biological strata, such as genes, and integrating a wide range of biodiversity metrics encompassing composition, structure, and function into impact prediction (Biasotto and Kindel, 2018). Of particular note is the dearth of scholarly exploration into the effects of electromagnetic fields generated by electric power substations on soil microorganisms, despite the uncertain biological implications at times (Beretta et al., 2019).
Aligned with the aforementioned context, the principal objective of this study was to delineate the effects of magnetic induction intensity (MII) and electric field intensity (EFI) emanating from the operation of electric power substations on soil physicochemical properties, alongside the structural and functional aspects of microbial communities across and shifts in functional genes related to nitrogen cycling. A specific focus was placed on elucidating correlations between soil physicochemical properties and microbial communities, as well as genes associated with the nitrogen cycle in the proximity of the substation.
2 Materials and methods
2.1 Site description and soil sample collection
Soil specimens were systematically obtained from the environs of an electric power substation (1,000 kV, 16.3 hm2) located at coordinates 119°08′N latitude and 26°34′E longitude, situated within Xintang village, Minhou county, Fujian province, southeastern China (as delineated in Supplementary Table S1). Xintang village occupies a locale characterized by a subtropical oceanic monsoon climate, typified by annual mean temperatures ranging between 17 and 21°C, and annual average precipitation levels spanning 1,400 to 2000 mm. The topography of the study site comprises a modest, low-lying basin with predominant rainfall occurring between May and September. Soil samples were diligently collected from distinct designated regions within the 0–20 cm stratum of the topsoil (Figure 1). It is noteworthy that the selected areas exhibited minimal vegetation cover. For each of the sampling locales (denoted as S1 through S6), an array of triplicate sampling plots, each measuring 2 meters by 2 meters, was judiciously established in a randomized fashion. Within the confines of each sampling plot, three discrete soil samples were haphazardly extracted (Guo et al., 2020; Chen et al., 2023), sealed within aseptic ziplock bags and expeditiously transported, under cryogenic conditions facilitated by dry ice, to the laboratory facility. Each soil sample underwent a meticulous sieving process, utilizing a 2 mm mesh screen, with the dual objectives of homogenizing the soil matrix and eliminating larger particles, soil-dwelling organisms, and vegetative debris.
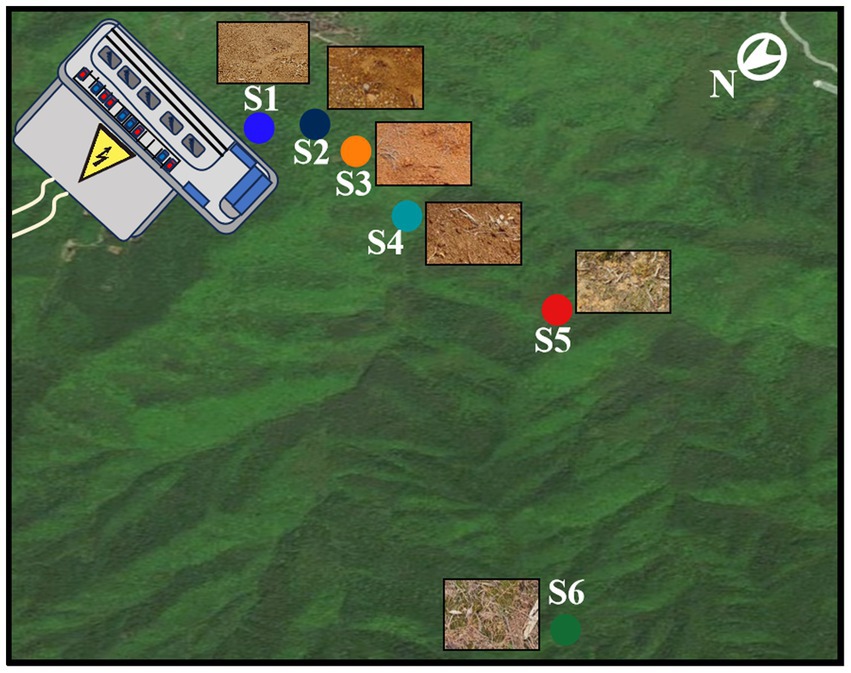
Figure 1. Schematic of soil collection sites distance from an electric power substation.
2.2 Soil physicochemical properties measurement
Fresh soil (100 g) was collected in each designated sampling plot using ring knives to determine soil bulk density, a parameter assessed through weight method (Zhang et al., 2023). The determination of soil dry matter and moisture content was undertaken employing the classical gravimetric method, while soil particle density was ascertained through the volume replacement technique. The quantification of soil organic matter (SOM) content was executed following the classical potassium dichromate method. The MII and EFI of sampling sites were measured using a Low-frequency electromagnetic field analyzer (Narda EFA-300, Germany). Moreover, the pH value of each soil sample, prepared at a soil-water ratio: 1:2.5, was diligently measured utilizing a pH meter (Sartorius PB-10, Germany). Specifically, for the determination of nitrite nitrogen (NO2−-N) and ammonium nitrogen (NH4+-N) concentrations, the soil samples (soil-water ratio: 1:5) were intimately mixed with KCl solution at a final concentration of 1 M. Subsequently, this mixture underwent agitation at 200 rpm for a duration of 1 h and was then subjected to centrifugation (3,000 rpm, 10 min) to facilitate the retrieval of the supernatant. The contents of NO2−-N and NH4+-N within the supernatant were subsequently quantified utilizing a UV–vis spectrophotometer (TU-1810, Bejing Purkinje General Instrument Co. Ltd., China) (see details in Supplementary Section S1), in strict accordance with their respective standard curves. The content of nitrate nitrogen (NO3−-N) within the filtered supernatant which was determined using a flow injection auto-analyzer (Skalar Analytical, AACE, Germany) (Chen et al., 2023). Furthermore, the overall content of total nitrogen (TN), soil organic nitrogen (SON), total carton (TC) and total organic carbon (TOC) was comprehensively determined (see Supplementary Sections S1, S2).
2.3 Determination of enzyme content in soil
The levels of SOD, malondialdehyde (MDA), glutathione (GSH), lactate dehydrogenase (LDH), acid protease, acid phosphatase and soil sucrase were quantified utilizing an enzyme assay kit (Gene Hunter, HongKong, China) (see the details in Supplementary Section S3) to evaluate the response of soil microorganisms to environmental stressors, including electromagnetic fields.
2.4 Soil DNA extraction and sequencing
Microbial genomic DNA were extracted from individual soil samples (0.5 g) using the Omega E.Z.N.A Stool DNA Kit for Soil (Omega Bio-tek, Inc., United States), following the manufacturer’s instructions. The purity and quality of the genomic DNA were assessed via 1% agarose gels electrophoresis, and their concentration were accurately quantified using the Qubit 4.0 Fluorometer (Thermo Fisher Scientific Inc., United States). Subsequently, the DNA was fragmented to 300 bp employing the Covaris ultrasonic crusher, and the resulting fragments underwent further processing, including end repair, A tailing, and ligation of Illumina compatible adapters. Finally, sequencing were conducted on an Illumina NovaSeq PE150 platform at Allwegene Company (Beijing, China) (Supplementary Table S2; Supplementary Section S4). The Illumina NovaSeq sequencing data were deposited in the Sequence Read Archive (SRA) database of the National Center for Biotechnology Information (NCBI), under accession number PRJNA1037611 (SUB13936859). The abundance of functional genes relevant to nitrogen metabolism were examined based on sequencing data combined with Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
2.5 Statistical analyses
Significant differences were assessed through one-way analysis of variance (SPSS 19) with ρ < 0.05 considered as statistically significant. Non-metric multidimensional scaling (NMDS) and principal coordinate analysis (PCoA) based on the Bray–Curtis dissimilarity were conducted using the R (Version 4.0.2) package ggplot2. Vegan ggplot2 package was employed for redundancy analyses (RDA) to analyze the correlation between environmental factors and the abundance of microbial community phyla. A Pearson correlation test was executed to examine the relationships between soil physicochemical properties and the relative abundance of microbial community, utilizing the R (Version 3.6) packages “psych” and “pheatmap.”
3 Results
3.1 Soil physicochemical properties of six different collection sites
The physicochemical characteristics of soil samples from each collection site were comprehensively detailed in Table 1. Variations in soil pH were observed across locations, ranging from 4.9 to 7.1, while SOM and bulk density spanned from 8.84 to 27.25 g·Kg−1 and 1.01 to 1.51 g·cm−3, respectively. Both MII and EFI exhibited the highest values at the S1 site, diminishing with increasing distance from the electric power substation The TC and TOC content in the soil samples from S1 were lower than those at other collection sites, with the TN content (5.50 g·Kg−1) at S1 surpassing that observed at other sites. The NO3−-N content at the S1 and S2 sites exceeded that at the remaining sampling sites. Conversely, for NH4+-N, the lowest content was observed at the S1 site compared to other collection points, with its content being lower than that of NO3−-N. Furthermore, we assessed several types of soil enzyme content as key indicators for microbial functioning controlling the decomposition rate of SOM and nutrient cycling processes, evaluating the influence of the operation of the electric power substation on the soil. The levels of MDA, SOD, GSH, LDH, acid protease, acid phosphatase and soil sucrase in the soil samples from S1 were marginally higher than those at other collection sites (Table 1).
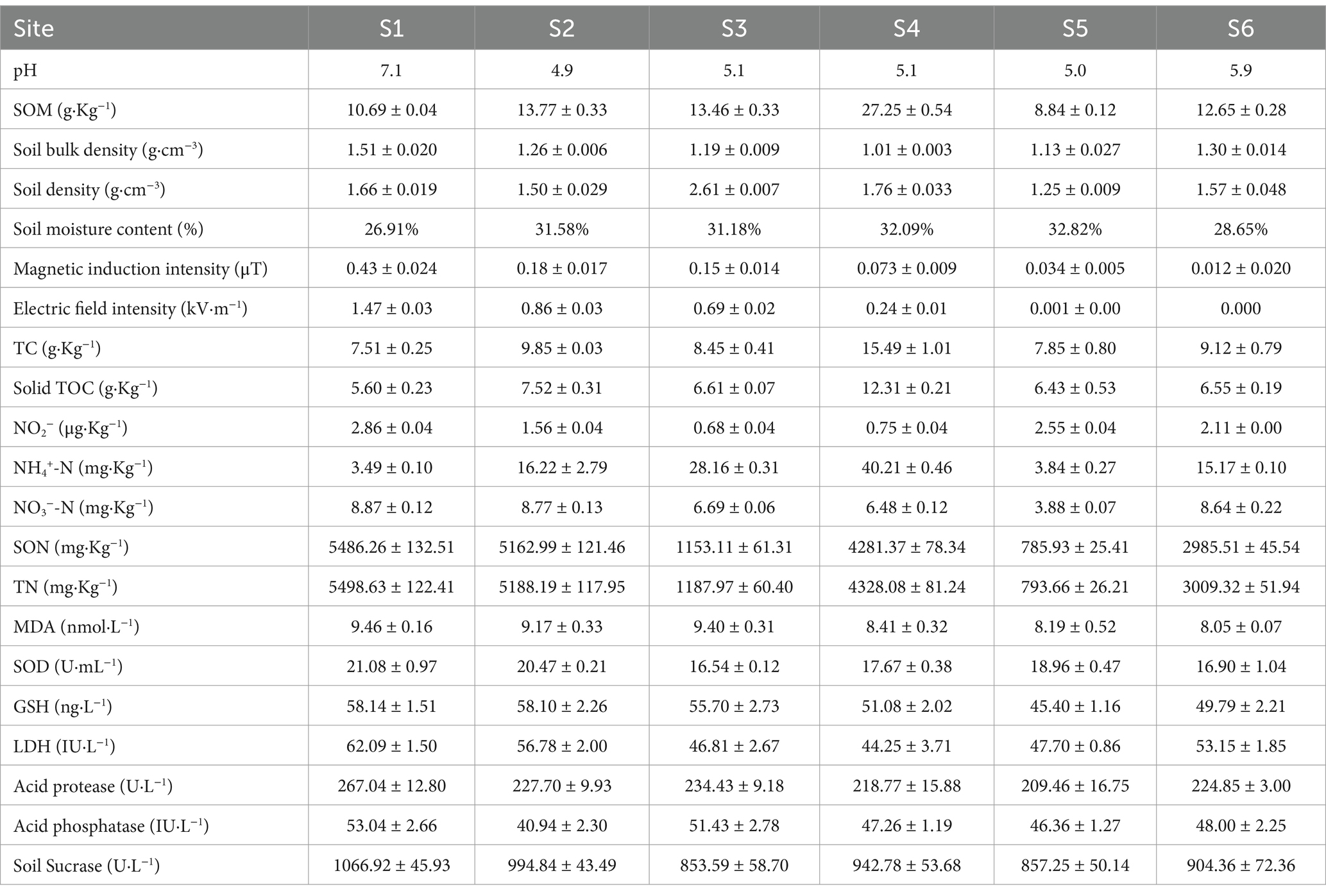
Table 1. Soil physicochemical properties and soil enzymes content from each collection site.
3.2 Soil microbial characteristics of six different collection sites
Metagenomic sequencing was conducted on these six sampling sites, yielding an average of 7.21 million reads per sample (Supplementary Table S2). From these metagenomes, 5,694,546 non-redundant catalog genes were identified, characterized by an average length of 637.2 bp. Among them, there were 1,648,487 genes annotated by KEEG with an annotation rate of 0.289, suggesting that the existence of numerous genes with functions yet to be elucidated. Representative sequences of the non-redundant gene catalog were annotated using the NCBI NR database (Version: 2021.11) through BLASTP implemented in Diamond (Buchfink et al., 2014) (http://www.diamondsearch.org/index.php, version 0.8.35), with an e-value cutoff of 1e−5 for taxonomic annotations. In the Initial phase of analysis, an alpha diversity assessment was undertaken to evaluate species richness within the soil samples. As illustrated in Figures 2A,B, a substantial difference in the richness and alpha diversity of soil microbes was evident among the 6 sites, as indicated by the Shannon index (ρ < 0.05) and Simpson index (ρ < 0.05). Notably, soil samples from S1 and S6, representing the sites nearest and furthest from the power substation, respectively, exhibited the highest values of Simpson index.
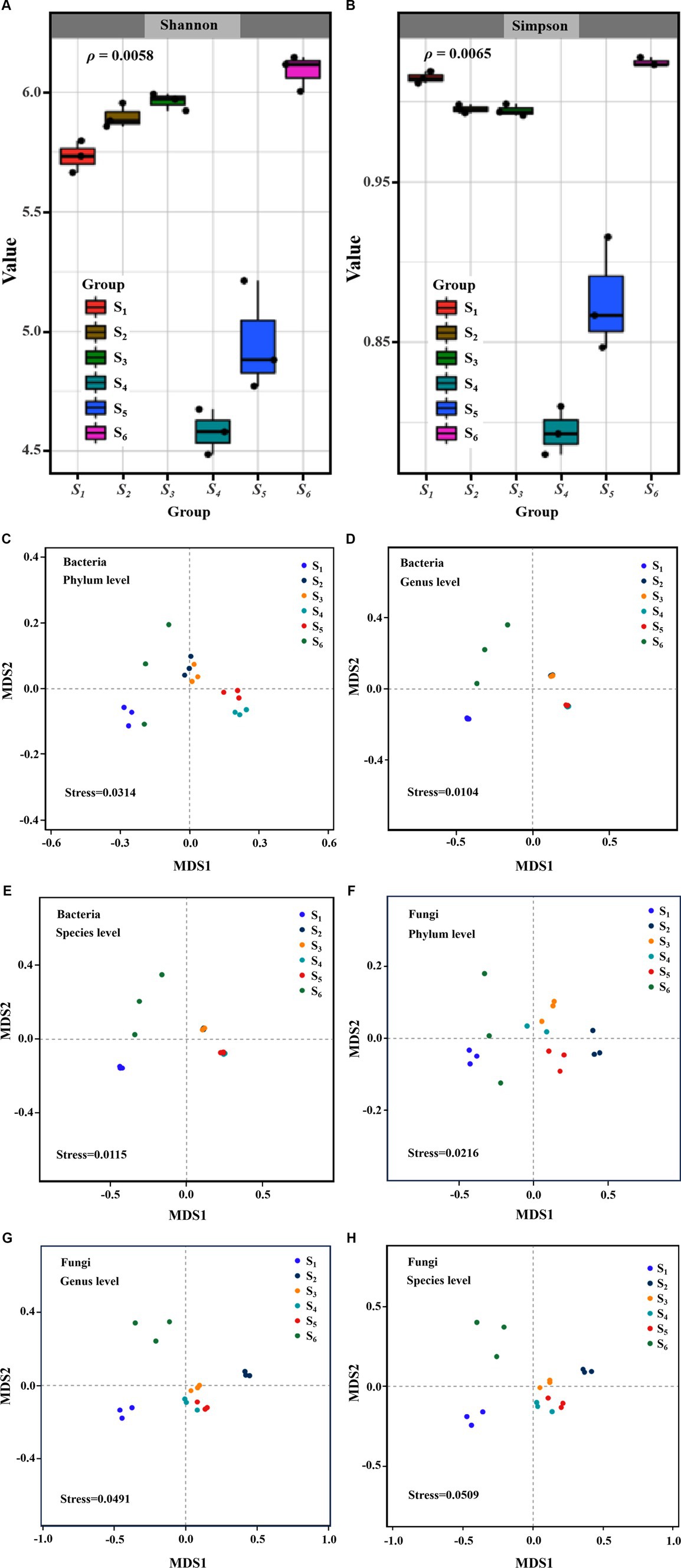
Figure 2. Alpha diversity Shannon (A) and (B) Simpson index of soil microbial communities collected from 6 points distance from an electric power substation; NMDS analysis of the differences in bacterial and fungus communities structures at phylum (C,F), genus (D,G) and species (E,H) based on Bray-Curtis distance.
Furthermore, the analysis of beta diversity in soil microbial communities, employing NMDS and PCoA to elucidate differences in species composition, unveiled significant variation among the collection sites. Utilizing Bray–Curtis distance measurements, both NMDS (Figures 2C–E) and PCoA (Supplementary Figures S1A–C) highlighted noteworthy dissimilarities in the community composition of bacteria (stress<0.05; PERMANOVA, ρ = 0.001) across the 6 sites. The microbial communities of sites (S2–S3) and (S4–S5) exhibited tight clustering, indicating a relatively consistent microbial community composition at the S2 and S3 sites, as well as the S4 and S5 sites. However, sites S1 and S6 were distinctly separated, signifying inconsistency in the microbial community composition at these locations. In contrast, the fungal community structure did not demonstrate representativeness (stress>0.05) at the species level across the 6 sites (Figures 2F–H), but exhibited significant differences (PERMANOVA, ρ = 0.001) (Supplementary Figures S1D–F).
3.3 Soil microbial communities of six different collection sites
3.3.1 Community composition of bacteria
Bacterial phyla within soil samples from each collection site were analyzed at the phylum level (Figure 3A). Legends presenting relative abundance below the top 30 were excluded and categorized into other groups. The predominant bacterial populations, encompassing Proteobacteria, Acidobacteria, Actinobacteria, and Chloroflexi, collectively constituted over 65% of the total microbial population across all soil samples. The average relative abundance of Proteobacteria at the S1 site (42.64%) surpassed that of other sites. Acidobacteria, the most abundant bacterial phylum, exhibited reduced abundance at the S1 (13.03%) and S7 (11.31%) sites relative to other collection points. The phylum Actinobacteria attained its highest relative abundance at the S2 site (29.26%) and its lowest at the S5 site (13.48%). Chloroflexi exhibited notably lower relative abundance at the S1 site (1.55%) compared to the S5 (11.66%), S4 (5.89%), S3 (10.94%), S2 (8.77%) and S6 (9.86%) sites (Figure 3A). The relative notable enrichment of Gemmatimonadetes was observed at the S1 (10.35%) sites, exceeding that at the S5 (1.34%), S4 (1.15%), S3 (1.14%), S2 (1.60%) and S6 (6.34%).
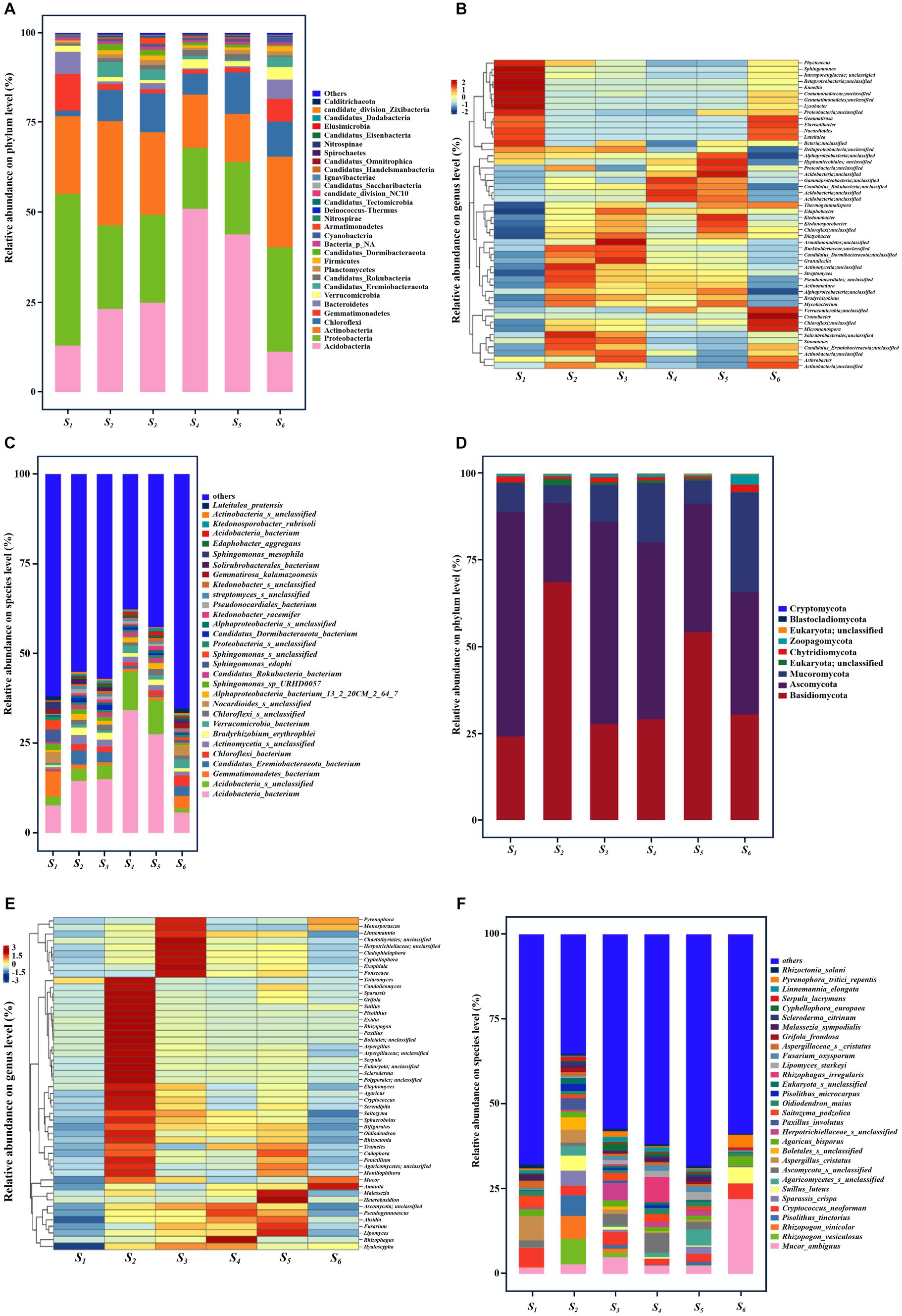
Figure 3. Composition of soil microbial communities in six collection sites. Composition of (A–C) bacteria and (D–F) fungal at the (A,D) phylum, (B,E) genus and (C,F) species level.
The dissimilarities in soil microbial community composition across different sites were further elucidated through a detailed analysis employing a heatmap of bacteria genera, showcasing the top 50 species in overall abundances. As illustrated in Figure 3B, the relative abundances of these bacteria, including Phycicoccus (Actinobacteria), Sphingomonas (Proteobacteria), Unclassified_Comamonadaceae (Proteobacteria), Knoellia (Actinobacteria), Unclassified_Gemmatimonadetes, Lysobacter (Proteobacteria), Flavisolibacter (Bacteroidetes), Nocardioides (Actinobacteria), Luteitalea (Acidobacteria), exhibited significantly higher abundances at the S1 site compared to other sites. At the species level (Figure 3C), Sphingomonas mesophila and Sphingomonas edaphi, individually constituting 2.72 and 4.85% of the total bacterial community, manifested a manifold increase in abundance at the S1 site relative to other sites. Notably, there was a heightened abundance of Luteitalea_pratensis at the S1 site.
3.3.2 Community composition of fungi
Metagenomic sequence taxonomic analysis revealed nine phyla, with Basidiomycota, Ascomycota and Mucoromycota constituting the most abundant, collectively averaging over 95% of all sequences in the fungi community (Figure 3D). Ascomycota emerged as the most abundant phylum, with an average relative abundance exceeding 60% at the S1 sites and surpassing 50% at the (S3, S4) sites, while its relative abundance at the S2 site was comparatively lower. The relative abundance of Basidiomycota at the (S2 and S5) exceeded that at the other points, whereas Mucoromycota exhibited the highest relative abundance at the (S4 and S6) sites. The Heatmap of fungal genera with the top 50 species in overall abundances were presented in Figure 3E. The genera of detected fungal species, showing greater richness, displayed distinct distribution characteristics at the 6 sites. It was observed that a relative lower abundant was noted for most all species at the S1 site compared to other points. The most representative fungal genus was Talaromyces (Ascomycota) and Aspergillus (Ascomycota), individually constituting 4.63 and 12.75% of the total fungal community. Furthermore, Figure 3F delineated the distinct community composition at the species level for different collection sites. Notably, for the S6 site, the most representative species with greater abundance was Aspergillus_cristatus, which accounted for 8.16% of the total fungal community.
3.4 Nitrogen processing of soil microbial communities at different collection site
The functions annotated under KEGG level 1 for the 6 types of soil surrounding the electric power substation encompassed diverse categories: metabolism (ave. 51.73%), genetic information processing (ave. 14.59%), environmental information processing (ave. 13.08%), cellular processes (ave. 10.91%), human diseases (ave. 5.30%), and organic systems (ave. 4.39%) (Supplementary Figure S2A). Significantly enriched functional pathways at level 2 (>4%) included carbohydrate metabolism, amino acid metabolism, energy metabolism, metabolism of cofactors and vitamins, cellular community-prokaryotes, signal transduction, and membrane transport (Supplementary Figure S2B). Among these, the S1 site exhibited a higher proportion of amino acid metabolism, and a lower proportion of carbohydrate metabolism and cellular community-prokaryotes compared to all other sites. Relatively more abundant functional pathways at level 3 (>5%), such as two-component system, quorum sensing, ABC transporters, Oxidative phosphorylation, pyruvate metabolism, and ribosome, were observed across the 6 points (Supplementary Figure S2C), with no significant difference.
Noteworthy were the nitrogen metabolism pathways, including nitrogen fixation, nitrification and denitrification. The relative abundance of the genes, such as nifA, nifD, nifH, nifK, and nifV (Kuypers et al., 2018; Bellés-Sancho et al., 2021), was lower at the S1 site than at other sites (Figure 4), suggesting a lower capacity for soil microbial nitrogen fixation (N2 → NH3). In contrast, the relative abundance of two genes, amoA and amoB (Wang et al., 2022), was higher at the S1 site than at other sites, indicating increased nitrification (NH4+ → NO3−). This observation aligns with the finding of low NH4+-N and high NO3−-N at the S1 site (Table 1). The total abundance of nitrogen fixation genes exhibited and increase with distance from the electric power substation (from S1 to S4), followed by a decline at S5 and S6 sites primarily attributed to the lower content of SOM, findings that corresponded with NH4+-N levels. Furthermore, the relative abundance of genes associated with NO2−, nitric oxide and nitrous oxide, as reported by Li et al. (2023), generally mirrored the levels of NO2− in each soil sample, with the relative abundances of nirK, nirS, norB, norC and nosZ also depicted in Figure 4.
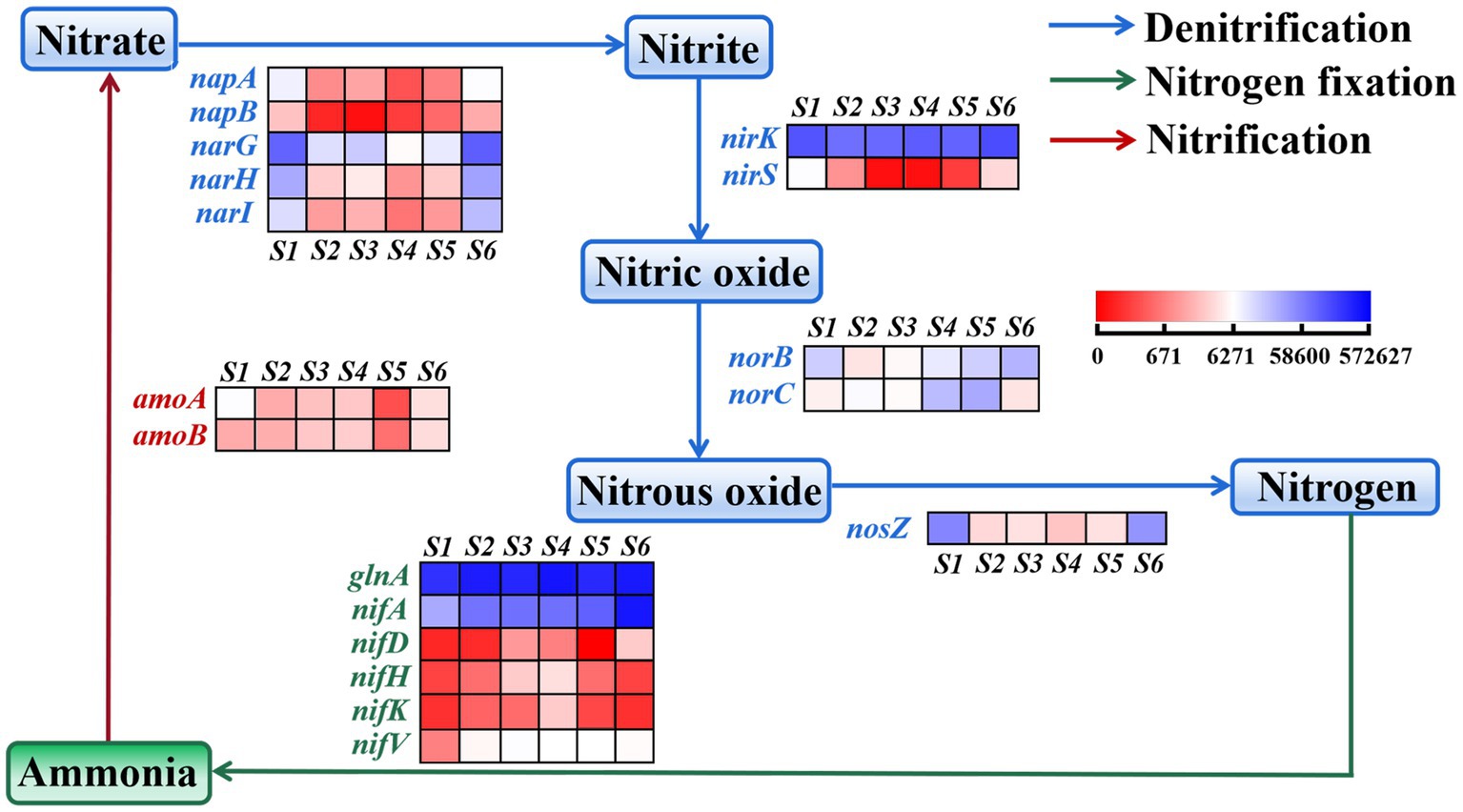
Figure 4. Heatmap representing the relative abundance of nitrogen functional genes in soil community in the S1–S6 samples.
3.5 Correlation between soil microbial communities and environmental factors
Environmental factors exerted a substantial influence on microbial communities and their functions. RDA served as a valuable tool for expressing the correlation between environmental factors and microbial communities. As illustrated in Figure 5A, the RDA1 and RDA2 axis accounted for 63.61 and 21.76%, respectively, of the total variation in microbial community composition and soil properties at the phylum level. The RDA model, based on soil microbial phylum-level data, effectively differentiated soils from various collection sites, corroborating our NMDS results. Specifically, the S1 site, situated nearest to the electric power substation, exhibited discernible associations with NO3−-N, SOM, EFI, MII, pH and soil enzymes, including SOD, GSH, soil sucrase, LDH, acid phosphatase, acid protease, MDA, but was negatively correlated with distance, moisture content and NH4+-N. However, the S3 and S4 sites were opposite. The S4 and S5 sites exhibited negatively correlated with EFI, NO3−-N, MDA and GSH. Cluster analysis revealed notable correlations between (Acidobacteria, Cyanobacteria) and TOC, SOM, moisture content, distance, MII, pH and SOD. Actinobacteria was positively correlations with NH4+-N, NO3−-N, EFI, GSH, MDA, LDH and distance. Proteobacteria and Bacteroidetes demonstrated positive correlations with pH, SOD, NO3−-N, MII, EFI and the aforementioned soil enzymes. Additionally, RDA of the correlation between environmental factors and microbial communities at the genus was provided in Supplementary Figure S3.
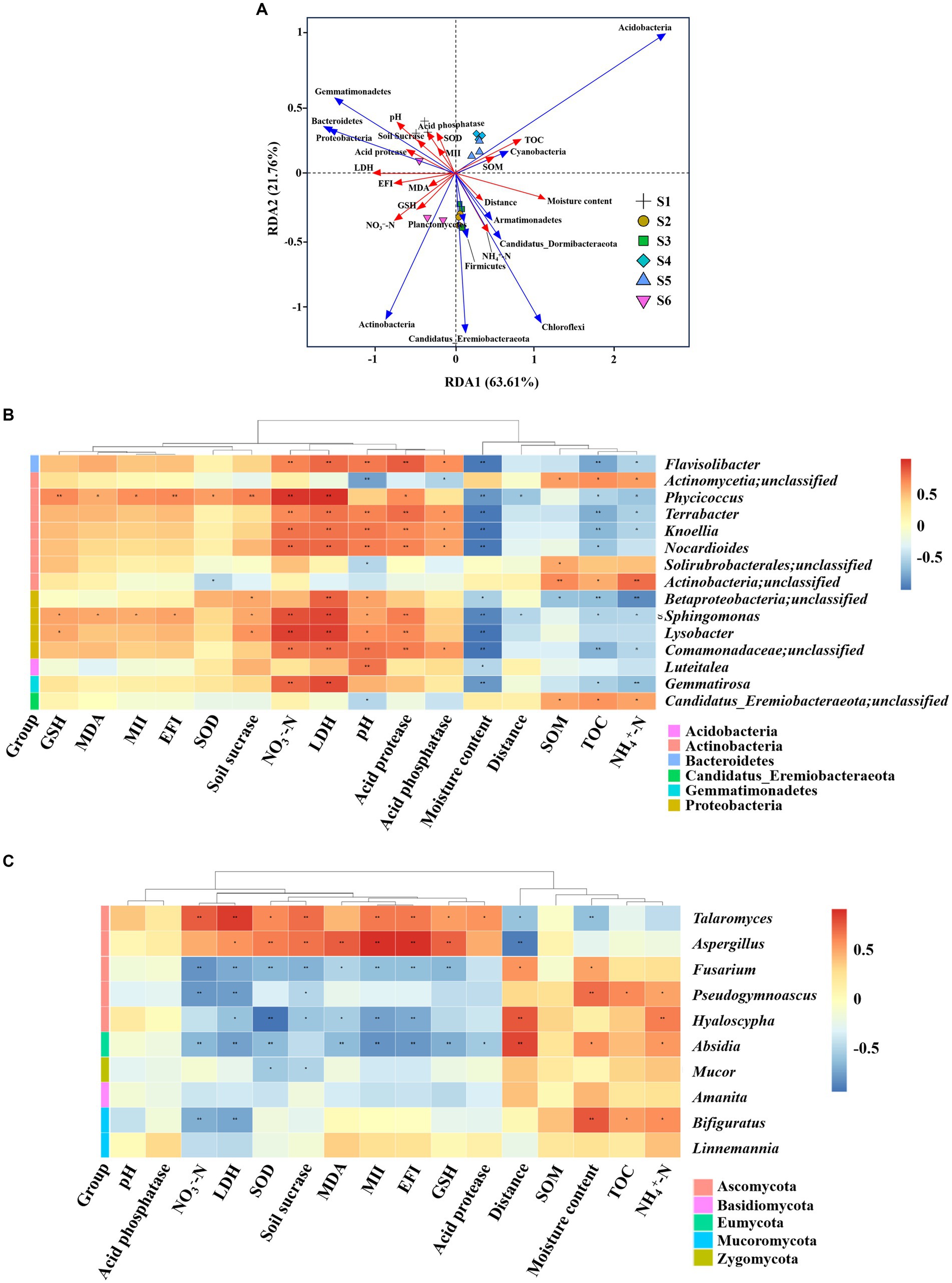
Figure 5. Correlation analysis between microorganisms and soil physicochemical properties across sites S1–S6. (A) Redundancy analysis of microbial communities with environmental factors. (B,C) Pearson correlation analyses between microbial communities and environmental factors. The distance between soil sample points signifies the similarities and differences in functional composition among samples. The projected distance from the sample point to the environmental factor indicated the extent to which the sample was influenced by the environmental factor. The closer the projection line, the more similar the impact of the environmental factor on the two samples. The angle between environmental factors/species denotes the positive and negative correlations between environmental factors/species. NH4+-N, ammonium nitrogen; MII, magnetic induction intensity; SOM, soil organic matter; TOC, total organic carbon; NO3−-N, nitrate nitrogen; EFI, electric field intensity; MDA, malondialdehyde; GSH, glutathione; SOD, superoxide dismutase; LDH, lactate dehydrogenase. Significance levels: *ρ < 0.05; **ρ < 0.01.
Pearson’s correlation analyses were further conducted to elucidate the effects of the 16 environmental factors on the microbial communities at the genus level (see the phylum level in Supplementary Figure S4), showcasing distinct abundances at the S1 site compared to other sites. It was observed that Cyanobacteria had a higher abundance at the S1 site, showing significant negative correlations with MII and EFI (ρ < 0.05) and significant positive correlations with distance (ρ < 0.05). Among the 15 bacteria genera (Figure 5B), four Actinobacteria genera, three Proteobacteria genera, and one Gemmatimonadetes genus demonstrated significant positive correlations with NO3−-N (ρ < 0.05), whereas one unidentified Actinobacteria genus, one unidentified Candidaus_Eremiobacteraeota genus, and one unidentified Actinobacteria genus exhibited significant positive correlations with NH4+-N (ρ < 0.05). Notably, bacteria that were significantly negatively correlated with moisture content included four Actinobacteria genera, four Proteobacteria genera, one unidentified Gemmatimonadetes genera, one Acidobacteria genus and one Bacteroidetes genus. Bacteria showing significant positive associations with pH included four Proteobacteria genera, one Actinobacteria genera, one unidentified Gemmatimonadetes genus, one Bacteroidetes genus. Additionally, Four Actinobacteria genera, one Bacteroidetes genus, three Proteobacteria genera, and one Gemmatimonadetes genus (Gemmatirosa) displayed a negative correlation with TOC (ρ < 0.05). Most importantly, Actinobacteria genus (phycicoccus) and Proteobacteria genera (Sphingomonas) exhibited a significant positive correlation (ρ < 0.05) with MII, EFI and distance. Regarding soil fungi (Figure 5C), those displaying a positive correlation with NH4+-N and moisture content included one Ascomycota genus, one Mucoromycota (Bifiguratus) and one Eumycota genus (Absidia), while those with a negative correlation with NO3−-N included two Ascomycota genus, one Mucoromycota (Bifiguratus) and one Eumycota genus (Absidia). Interestingly, Absidia, Hyaloscypha, and Fusarium demonstrated a significantly negative correlation with MII and EFI, while Aspergillus exhibited the opposite trend.
4 Discussion
4.1 Influence of soil collection site on soil properties
Soil, a vital component of an ecosystems, serves as the material foundation for plant and microbial survival. Its physicochemical attributes dictate the structural compositions of plant and microbial communities (Yang and Hu, 2021). As indicated by Wu et al. (2019), sites with lower moisture content and higher pH could experience upward movement of soluble salts from the deeper soil layers. Higher soil pH through reduced H+ release by roots and organic matter lead to lower soil TOC level (Hong et al., 2018), which was further accelerated by soil moisture content, which was known to enhance soil microbial activities influencing SOM mineralization and decomposition (Huang et al., 2013; Zhang et al., 2019; Cao et al., 2021). This was all exemplified by the conditions at the S1 site and other sites (Table 1). Notably, TN levels at the S1 site exceeded those at other sites (Table 1), possible attributed to the presence of strong MII and EFI, significant factors influencing microbial community assembly (Aksoy et al., 2010). Despite the absence of waste discharge typical of coal-fired power plants (Sun et al., 2024), the operation of electric power substation impacted soil physicochemical properties.
4.2 Influence of collection site on the composition of soil microbial communities
Across all soil samples, Acidobacteria, Proteobacteria, Actinobacteria and Chloroflexi emerged as dominant phyla (Figure 3A), aligning with findings from soils neighboring coal-fired power plants (Sun et al., 2024) as well as mining and smelting areas (Liu B. et al., 2021). However, NMDS and PCoA analysis indicated of distinct distribution patterns for soil bacterial community (Figure 2; Supplementary Figure S1). Particularly, the S1 site exhibited significantly higher abundances of Proteobacteria and Acidobacteria and lower abundances of Chloroflexi compared to other sites (Supplementary Table S1), which contradicted observations for coal-fired power plants (Sun et al., 2024). These characteristics indicated a higher capacity of reducing nitrogen loss and a diminished capability for autotrophic denitrification (Zhou et al., 2019; Ge et al., 2020; Tao et al., 2022; Wang et al., 2023; Wei et al., 2023). At the bacterial genus level, soil properties explained 85.5% of the variation in soil microorganism composition (Supplementary Figure S3), and discernible differences were evident among the 6 sites (Figure 3B). Bacterial taxa that were more enriched at the S1 site relative to other sites (Figure 3C), contained: (1) the genus Unclassified_Comamonadaceae (Sotres et al., 2016), Lysobacter (Iwata et al., 2010), and Sphingomonas (Yang et al., 2014), known for their proficiency in facilitating superior nitrogen conversion. (2) Other microbial entities such as Flavisolibacter (Hernández-Guzmán et al., 2022), Gemmatirosa (Liu B. et al., 2021; Liu Z. et al., 2021; Sun et al., 2023), Terrabacter (Kruglova et al., 2017), and Phycicoccus (Yang et al., 2023), associated with nitrogen metabolism, along with the genera like Nocardioides, the functions of which were yet to be elucidated in the biogeochemical cycle. At the species level, despite an enrichment of Sphingomonas mesophila (Li et al., 2019) and Sphingomonas edaphi (Kim et al., 2020) was observed at the S1 site, their precise roles remained ambiguous. Nevertheless, most bacteria did not show statistically significant correlations with distance, as well as MII and EFI.
Reports regarding the composition and structure of soil fungal communities in the vicinity of electric power substation are notably limited. In this investigation, the fungal communities at the phylum level were predominantly constituted by Basidiomycota and Ascomycota in all soil samples (Figure 3D), a pattern consistent with their ubiquity in most soils (Shary et al., 2007). The prevalence of Ascomycota might be associated with its capacity to degrade cellulose and hemicellulose (Shary et al., 2007; Yang et al., 2023), while the phyla Basidiomycota and Mucoromycota had been reported to be linked to the degradation of complex lignocelluloses (Lundell et al., 2010; Huhe et al., 2017). Interestingly, in contrast to bacteria, several fungal genera such as Talaromyces, Aspergillus, Fusarium, Hyaloscypha and Absidia exhibited significant associations with MII, EFI and distance, suggesting that the influence of electric power substation operations on soil fungal communities surpasses that on bacterial populations.
4.3 Influence of soil collection site on the nitrogen processing
Nitrogen, a crucial limiting nutrient in ecosystems, plays a fundamental role in ecosystem productivity, where nitrogen fixation serves as a key mechanism for microorganisms to acquire nitrogen resources. The phyla Proteobacteria (42.64%) and Gemmatimonadetes (10.35%) from Supplementary Figure S4A demonstrated significant negative correlations with NH4+-N, displaying higher abundance at the S1 site characterized by lower soil moisture content (26.91%) and higher levels of MII and EFI. As revealed by RDA analysis, NH4+-N levels were negatively correlated with MII and EFI but positively correlated with distance. Given that nitrogen fixation predominantly occurs in anaerobic environments, the relative lower moisture content at the S1 site may impede soil nitrogen fixation (Zhang et al., 2020), which could explain the observed lower relative abundance of nitrogen fixation genes shown in Figure 4 and the reduction in NH4+-N content at this site. While nitrification, a key microbial nitrogen-loss pathway, displayed a positive correlation with MII and EFI, with genes associated with nitrification function showing increased abundance at the S1 site, likely contributing to the observed rise in NO3−-N levels post-operation of the electric power substation, although the mechanisms governing bacterial responses to electromagnetic fields of varying intensities remain unclear, impeding predictions of microbial behavior (Beretta et al., 2019).
5 Conclusion
This study employed metagenomic sequencing to investigate the relationships between microbial communities and environmental variables in soils located at varying distances from an electric power substation, which generated MII and EFI. Noteworthy discrepancies in soil physicochemical properties, as well as in the diversity, composition and structure of soil fungal communities, were evident under the influence of the electric power substation, while such distinctions were not observed in soil bacterial communities. Moisture content and pH influenced the soil bacterial communities, whereas the impact of anthropogenic disturbance linked to MII and EFI was pronounced in the immediate vicinity of the electric power substation, yet minimal as the sampling sites were distanced from the substation. Moreover, the genes associated with nitrogen fixation and nitrification functions were found to correlate with the observed levels of NH4+-N and NO3−-N. The soil fungal communities displayed greater sensitivity to the electric power substation operation compared to bacteria, and their dynamics had a direct and significant impact on microbial community diversity within the substation ecosystem.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
Z-XX: Data curation, Investigation, Methodology, Writing – original draft. BZ: Investigation, Visualization, Writing – original draft. SC: Data curation, Methodology, Writing – original draft. SX: Data curation, Formal analysis, Validation, Writing – original draft. L-GJ: Formal analysis, Validation, Writing – original draft. XL: Data curation, Validation, Visualization, Writing – review & editing. Y-FW: Validation, Visualization, Writing – original draft. L-XY: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (grant number 22072017).
Conflict of interest
Z-XX, BZ, SC, SX, L-GJ, XL, and Y-FW were employed by High Voltage Branch of State Grid Fujian Electric Power Co., Ltd.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1453162/full#supplementary-material
References
Aksoy, H., Unal, F., and Ozcan, S. (2010). Genotoxic effects of electromagnetic fields from high voltage power lines on some plants. Int. J. Environ. Res. 4, 595–606. Available at: https://sid.ir/paper/300825/en
Bagli, S., Geneletti, D., and Orsi, F. (2011). Routeing of power lines through least-cost path analysis and multicriteria evaluation to minimise environmental impacts. Environ. Impact Assess. Rev. 31, 234–239. doi: 10.1016/j.eiar.2010.10.003
Bellés-Sancho, P., Lardi, M., Liu, Y., Hug, S., Pinto-Carbó, M. A., Zamboni, N., et al. (2021). Paraburkholderia phymatum homocitrate synthase NifV plays a key role for nitrogenase activity during symbiosis with papilionoids and in free-living growth conditions. Cells 10:952. doi: 10.3390/cells10040952
Beretta, G., Mastorgio, A. F., Pedrali, L., Saponaro, S., and Sezenna, E. (2019). The effects of electric, magnetic and electromagnetic fields on microorganisms in the perspective of bioremediation. Rev. Environ. Sci. Biotechnol. 18, 29–75. doi: 10.1007/s11157-018-09491-9
Biasotto, L. D., and Kindel, A. (2018). Power lines and impacts on biodiversity: a systematic review. Environ. Impact Assess. Rev. 71, 110–119. doi: 10.1016/j.eiar.2018.04.010
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Cao, J. J., Wang, H. R., Holden, N. M., Adamowski, J. F., Biswas, A., Zhang, X. F., et al. (2021). Soil properties and microbiome of annual and perennial cultivated grasslands on the Qinghai–Tibetan plateau. Land Degrad. Dev. 32, 5306–5321. doi: 10.1002/ldr.4110
Chen, J., Xiao, Q. C., Xu, D. L., Li, Z. S., Chao, L. M., Li, X. Y., et al. (2023). Soil microbial community composition and co-occurrence network responses to mild and severe disturbances in volcanic areas. Sci. Total Environ. 901:165889. doi: 10.1016/j.scitotenv.2023.165889
Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10, 538–550. doi: 10.1038/nrmicro2832
Gadd, G. M. (2010). Metals, minerals and microbes: geomicrobiology and bioremediation. Microbiology-(UK) 156, 609–643. doi: 10.1099/mic.0.037143-0
Ge, X. Y., Cao, X., Song, X. S., Wang, Y. H., Si, Z. H., Zhao, Y. F., et al. (2020). Bioenergy generation and simultaneous nitrate and phosphorus removal in a pyrite-based constructed wetland-microbial fuel cell. Bioresour. Technol. 296:122350. doi: 10.1016/j.biortech.2019.122350
Guo, Y. N., Liu, X. H., Tsolmon, B., Chen, J., Wei, W., Lei, S. G., et al. (2020). The influence of transplanted trees on soil microbial diversity in coal mine subsidence areas in the loess plateau of China. Glob. Ecol. Conserv. 21:e00877. doi: 10.1016/j.gecco.2019.e00877
Hernández-Guzmán, M., Pérez-Hernández, V., Navarro-Noya, Y. E., Luna-Guido, M. L., Verhulst, N., Govaerts, B., et al. (2022). Application of ammonium to a N limited arable soil enriches a succession of bacteria typically found in the rhizosphere. Sci. Rep. 12:4110. doi: 10.1038/s41598-022-07623-4
Hong, S. B., Piao, S. L., Chen, A. P., Liu, Y. W., Liu, L. L., Peng, S. S., et al. (2018). Afforestation neutralizes soil pH. Nat. Commun. 9:520. doi: 10.1038/s41467-018-02970-1
Huang, L. B., Bai, J. H., Gao, H. F., Xiao, R., Liu, P. P., and Chen, B. (2013). Soil organic carbon content and storage of raised field wetlands in different functional zones of a typical shallow freshwater lake, China. Soil Res. 50, 664–671. doi: 10.1071/SR12236
Huhe, C., Chen, X., Hou, F., Wu, Y., and Cheng, Y. (2017). Bacterial and fungal community structures in loess plateau grasslands with different grazing intensities. Front. Microbiol. 8, 1–10. doi: 10.3389/fmicb.2017.00606
Huseynova, I. M., Aliyeva, D. R., and Aliyev, J. A. (2014). Subcellular localization and responses of superoxide dismutase isoforms in local wheat varieties subjected to continuous soil drought. Plant Physiol. Biochem. 81, 54–60. doi: 10.1016/j.plaphy.2014.01.018
Iwata, K., Azlan, A., Yamakawa, H., and Omori, T. (2010). Ammonia accumulation in culture broth by the novel nitrogen-fixing bacterium, Lysobacter sp. E4. J. Biosci. Bioeng. 110, 415–418. doi: 10.1016/j.jbiosc.2010.05.006
Kim, H., Chhetri, G., and Seo, T. (2020). Sphingomonas deaphi sp. nov., a novel species isolated from beach soil in the Republic of Korea. Int. J. Syst. Evol. Microbiol. 70, 522–529. doi: 10.1099/ijsem.0.003780
Kruglova, A., Gonzalez-Martinez, A., Kråkström, M., Mikola, A., and Vahala, A. (2017). Bacterial diversity and population shifts driven by spotlight wastewater micropollutants in low-temperature highly nitrifying activated sludge. Sci. Total Environ. 605, 291–299. doi: 10.1016/j.scitotenv.2017.06.191
Kuypers, M. M. M., Marchant, H. K., and Kartal, B. (2018). The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 16, 263–276. doi: 10.1038/nrmicro.2018.9
Li, Z., Qiu, Y., Yu, Y. L., Ji, Y. L., Li, H. N., Liao, M. L., et al. (2023). Long-term operation of cathode-enhanced ecological floating bed coupled with microbial electrochemical system for urban surface water remediation: from lab-scale research to engineering application. Water Res. 237:119967. doi: 10.1016/j.watres.2023.119967
Li, Y. Q., Rao, M. P. N., Zhang, H., Guo, Y. M., Dong, Z. Y., Alkhalifah, D. H. M., et al. (2019). Description of Sphingomonas mesophila sp. nov., isolated from Gastrodia elata Blume. Int. J. Syst. Evol. Microbiol. 69, 1030–1034. doi: 10.1099/ijsem.0.003263
Liu, Z., Tang, J., Ren, X., and Schaeffer, S. M. (2021). Effects of phosphorus modified nZVI-biochar composite on emission of greenhouse gases and changes of microbial community in soil. Environ. Pollut. 274:116483. doi: 10.1016/j.envpol.2021.116483
Liu, B., Yao, J., Ma, B., Chen, Z. H., Zhao, C. C., Zhu, X. Z., et al. (2021). Microbial community profiles in soils adjacent to mining and smelting areas: contrasting potentially toxic metals and co-occurrence patterns. Chemosphere 282:130992. doi: 10.1016/j.chemosphere.2021.130992
Lundell, T. K., Mäkelä, M. R., and Hildén, K. (2010). Lignin-modifying enzymes in filamentous Basidiomycetes–ecological, functional and phylogenetic review. J. Basic Microbiol. 50, 5–20. doi: 10.1002/jobm.200900338
Roubalová, R., Dvořák, J., Procházková, P., Škanta, F., Pacheco, N. I. N., Semerád, J., et al. (2018). The role of CuZn-and Mn-superoxide dismutases in earthworm Eisenia andrei kept in two distinct field-contaminated soils. Ecotox. Environ. Safe. 159, 363–371. doi: 10.1016/j.ecoenv.2018.04.056
Shary, S., Ralph, S. A., and Hammel, K. E. (2007). New insights into the ligninolytic capability of a wood decay ascomycete. Appl. Environ. Microbiol. 73, 6691–6694. doi: 10.1128/AEM.01361-07
Sotres, A., Cerrillo, M., Vinas, M., and Bonmatí, A. (2016). Nitrogen removal in a two-chambered microbial fuel cell: establishment of a nitrifying–denitrifying microbial community on an intermittent aerated cathode. Chem. Eng. J. 284, 905–916. doi: 10.1016/j.cej.2015.08.100
Sun, X., Tao, R. D., Xu, D. Q., Qu, M. J., Zheng, M. M., Zhang, M., et al. (2023). Role of polyamide microplastic in altering microbial consortium and carbon and nitrogen cycles in a simulated agricultural soil microcosm. Chemosphere 312:137155. doi: 10.1016/j.chemosphere.2022.137155
Sun, B. W., Zhu, R. B., Shi, Y., Zhang, W. Y., Zhou, Z. M., Ma, D. W., et al. (2024). Effects of coal-fired power plants on soil microbial diversity and community structures. J. Environ. Sci. 137, 206–223. doi: 10.1016/j.jes.2023.02.014
Tao, Z. K., Jing, Z. Q., Tao, M. N., and Chen, R. J. (2022). Recycled utilization of ryegrass litter in constructed wetland coupled microbial fuel cell for carbon-limited wastewater treatment. Chemosphere 302:134882. doi: 10.1016/j.chemosphere.2022.134882
Wang, N. Y., Awasthi, M. K., Pan, J. T., Jiang, S. L., Wan, F. C., Lin, X., et al. (2022). Effects of biochar and biogas residue amendments on N2O emission, enzyme activities and functional genes related with nitrification and denitrification during rice straw composting. Bioresour. Technol. 357:127359. doi: 10.1016/j.biortech.2022.127359
Wang, Z., Xu, Y. L., Yang, T., Liu, Y. Q., Zheng, T. T., and Zheng, C. L. (2023). Effects of biochar carried microbial agent on compost quality, greenhouse gas emission and bacterial community during sheep manure composting. Biochar 5:3. doi: 10.1007/s42773-022-00202-w
Wei, C. F., Liu, S. T., Li, Q., He, J., Sun, Z. J., and Pan, X. Y. (2023). Diversity analysis of vineyards soil bacterial community in different planting years at eastern foot of Helan Mountain, Ningxia. Rhizosphere 25:100650. doi: 10.1016/j.rhisph.2022.100650
Whalen, E. D., Stuart Grandy, A., Sokol, N. W., Keiluweit, M., Ernakovich, J., Smith, R. G., et al. (2022). Clarifying the evidence for microbial-and plant-derived soil organic matter, and the path toward a more quantitative understanding. Glob. Chang. Biol. 28, 7167–7185. doi: 10.1111/gcb.16413
Wu, Y., Wang, Q., Wang, H. M., Wang, W. J., and Han, S. J. (2019). Shelter-belt poplar forests induced soil changes in deep soil profiles and climates contributed their inter-site variations in dry land regions, northeastern China. Front. Plant Sci. 10:220. doi: 10.3389/fpls.2019.00220
Yang, H., and Hu, C. (2021). Soil chemistry and nutrients influence the distribution of aerobic anoxygenic phototrophic bacteria and eukaryotic phototrophic microorganisms of physical soil crusts at different elevations on the Tibetan plateau. Microb. Ecol. 83, 100–113. doi: 10.1007/s00248-021-01734-7
Yang, S. J., Zhang, X. H., Cao, Z. Y., Zhao, K. P., Wang, S., Chen, M. X., et al. (2014). Growth-promoting Sphingomonas paucimobilis ZJSH1 associated with Dendrobium officinale through phytohormone production and nitrogen fixation. Microb. Biotechnol. 7, 611–620. doi: 10.1111/1751-7915.12148
Yang, C., Zhang, H., Zhao, X. Q., Liu, P., Wang, L. S., and Wang, W. Y. (2023). A functional metagenomics study of soil carbon and nitrogen degradation networks and limiting factors on the Tibetan plateau. Front. Microbiol. 14:1170806. doi: 10.3389/fmicb.2023.1170806
Zhang, X. D., Jia, X., Wu, H. D., Li, J., Yan, L., Wang, J. Z., et al. (2020). Depression of soil nitrogen fixation by drying soil in a degraded alpine peatland. Sci. Total Environ. 747:141084. doi: 10.1016/j.scitotenv.2020.141084
Zhang, K., Shi, Y., Cui, X., Yue, P., Li, K., Liu, X., et al. (2019). Salinity is a key determinant for soil microbial communities in a desert ecosystem. mSystems 4, e00225–e00218. doi: 10.1128/mSystems.00225-18
Zhang, Z., You, L. X., Lin, J. S., Wu, Y. B., Zhong, H. L., Chen, J., et al. (2023). Impact of soil surface properties on soil swelling of different soil layers in collapsing wall of Benggang. PLoS One 18:e0280729. doi: 10.1371/journal.pone.0280729
Keywords: soil microbial community, electric power substation, nitrogen processing, distance, metagenomic sequencing technique
Citation: Xu Z-X, Zeng B, Chen S, Xiao S, Jiang L-G, Li X, Wu Y-F and You L-X (2024) Soil microbial community composition and nitrogen enrichment responses to the operation of electric power substation. Front. Microbiol. 15:1453162. doi: 10.3389/fmicb.2024.1453162
Edited by:
Yong Yuan, Guangdong University of Technology, ChinaReviewed by:
Zhongbing Chen, Czech University of Life Sciences Prague, CzechiaXixi Cai, Fujian Agriculture and Forestry University, China
Copyright © 2024 Xu, Zeng, Chen, Xiao, Jiang, Li, Wu and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Le-Xing You, bGVveHlvdUB6am51LmVkdS5jbg==